
Abstract
EGFR inhibitors have revolutionized the treatment of advanced non-small-cell lung cancer (NSCLC). Mefatinib is a novel, bioavailable, second-generation, irreversible pan-EGFR inhibitor. This phase Ib/II open-label, single-arm, multi-center study investigated the efficacy, safety, biomarker, and resistance mechanisms of mefatinib in the first-line treatment of patients with advanced EGFR-mutant NSCLC. This study included 106 patients with EGFR-mutant stage IIIB-IV NSCLC who received first-line mefatinib at a daily dose of either 60 mg (n = 51) or 80 mg (n = 55). The primary endpoint was progression-free survival (PFS). Secondary endpoints were overall response rate (ORR), disease control rate (DCR), overall survival (OS), and safety. The cohort achieved an ORR of 84.9% and DCR of 97.2%. The median PFS was 15.4 months and the median OS was 31.6 months. Brain metastasis was detected in 29% of patients (n = 31) at diagnosis and demonstrated an ORR of 87.1%, PFS of 12.8 months, and OS of 25.2 months. Adverse events primarily involved skin and gastrointestinal toxicities, which were well-tolerated and manageable. Analyses of mutation profiles were performed using targeted sequencing of plasma samples at baseline, first follow-up 6 weeks from starting mefatinib therapy (F1), and at progression. Patients with concurrent TP53 mutations had comparable PFS as wild-type TP53 (14.0 vs 15.4 months; p = 0.315). Furthermore, circulating tumor DNA clearance was associated with longer PFS (p = 0.040) and OS (p = 0.002). EGFR T790M was the predominant molecular mechanism of mefatinib resistance (42.1%, 16/38). First-line mefatinib provides durable PFS and an acceptable toxicity profile in patients with advanced EGFR-mutant NSCLC.
Similar content being viewed by others
Introduction
Mutations in epidermal growth factor receptor (EGFR) has transformed the standard of care and improved the prognosis of a subset of patients whose lung cancers are driven primarily by EGFR.1,2,3,4,5,6,7,8 Numerous clinical trials have established the superiority of first-, second-, and third-generation EGFR-TKI in managing advanced EGFR-mutant non-small-cell lung cancer (NSCLC) compared with conventional chemotherapy regimens.9,10,11,12,13 In 2016, gefitinib was the first EGFR-TKI approved as first-line therapy of metastatic NSCLC harboring EGFR exon 19 deletion (19del) and exon 21 L858R.9,13 Secondary EGFR mutations, particularly T790M, are the most common mechanism of resistance acquired during EGFR-TKI treatment;6,14,15 however, some EGFR-TKI-resistant tumors that do not develop EGFR T790M mutations still retain their dependence on the EGFR pathway.16,17 Irreversible second-generation EGFR-TKIs, including afatinib and dacomitinib, were developed to overcome this resistance from reversible first-generation EGFR-TKIs gefitinib and erlotinib.16,17,18 Afatinib potently inhibits various ErbB receptor tyrosine kinase family members, including HER2 (ErbB2), ErbB3, and ErbB4, and was approved in the first-line treatment of patients with EGFR-mutant NSCLCs since 2013.11,12,17,19,20 As compared to gefitinib, afatinib as front-line therapy demonstrated a higher objective response rate (ORR, 72.5% vs 56%) and longer median time to treatment failure (13.7 vs 11.5 months) but no difference in overall survival (OS).21,22 On the other hand, dacomitinib as front-line therapy demonstrated a significantly improved progression-free survival (PFS, 14.7 vs. 9.2 months) and OS (OS, 34.1 vs. 27.0 months) compared with gefitinib.18,23 However, the presence of concurrent mutations, including TP53 mutations and EGFR amplification, could severely limit the efficacy of currently available EGFR-TKI therapy, including afatinib, and lead to poorer clinical outcomes of patients harboring these concurrent mutations treated with EGFR-TKI.24,25,26,27,28
Mefatinib (MET306) is a novel, second-generation EGFR-TKI, which was designed to irreversibly bind to mutated tyrosine kinase domain of EGFR and HER2. Unpublished preclinical studies using xenograft mouse models have demonstrated the strong inhibitory activity of single-agent mefatinib on lung cancer harboring EGFR- or HER2-overexpression, or EGFR mutations.
In this article, we report the combined results of the phases Ib and II open-label, single-arm, multi-center studies on the clinical efficacy and safety of mefatinib in the first-line treatment of patients with advanced EGFR-mutant NSCLC. We also explored genetic biomarkers associated with the efficacy of mefatinib therapy and the molecular mechanisms mediating acquired resistance to mefatinib therapy.
Results
Patient characteristics
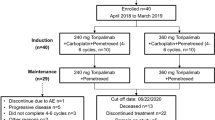
A total of 106 patients with advanced EGFR-mutant NSCLC were enrolled. The cohort comprised 43% males and 57% females with a median age of 64 years (range: 27–78 years). A majority of the cohort was never smokers (63.2%, 67/106) and 34.0% (36/106) were current and former smokers. All the patients were diagnosed with stage IIIB-IV lung adenocarcinoma and were EGFR-TKI-naive before receiving mefatinib therapy. The cohort also included 29.2% (31/106) patients who had brain metastasis at presentation. Before study inclusion, the EGFR mutation status of the patients was confirmed by ARMS-PCR of tissue biopsy samples, with 56.6% (60/106) of the patients harboring EGFR 19del, 40.6% (43/106) of the patients having EGFR L858R, and 3 (2.8%) patients having both mutations. Table 1 summarizes the baseline clinical characteristics of the patients.
Efficacy
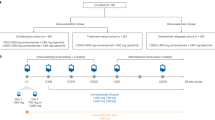
Supplementary Figure S1 summarizes the study design. The phase Ib study enrolled 34 patients who were non-randomized/assigned to receive 60 mg mefatinib (n = 15) or 80 mg mefatinib (n = 19). The phase II study enrolled a total of 72 patients who were randomized to receive either 60 mg mefatinib (n = 36) or 80 mg mefatinib (n = 36). In total, 51 patients (48.1%, 51/106) received a lower dose (60 mg) and 55 patients (51.9%, 55/106) received a higher dose (80 mg) of mefatinib. The patients who received 60 mg had an ORR of 80.4%, disease control rate (DCR) of 96.1%, PFS of 15.1 months, and OS of 30.6 months. As compared to 60 mg, the patients who received 80 mg had a numerically higher ORR (89.1%) and DCR (98.2%) but a close PFS (15.4 months) and OS (32.1 months) (Fig. 1a, b). Collectively, the cohort achieved an ORR of 84.9%, DCR of 97.2%, median PFS of 15.4 months, and median OS of 31.6 months. Table 2 summarizes the clinical outcomes of mefatinib.

Survival outcomes with mefatinib were similar for patients who received mefatinib at 60 and 80 mg but were significantly different based on brain metastatic status at presentation. Kaplan–Meier curves comparing the progression-free survival (a, c) and overall survival (b, d) of patients with advanced EGFR–mutant non-small-cell lung cancer who received mefatinib as first-line therapy at a daily dose of either 60 mg or 80 mg (a, b) or based on brain metastatic status at presentation (c, d)
We also analyzed the clinical outcomes of the patients according to their brain metastasis status at presentation. With first-line mefatinib, patients with brain metastasis achieved an ORR of 87.1% and DCR of 96.8%, while patients with no brain metastasis at baseline achieved an ORR of 82.4% and DCR of 97.3% (Supplementary Table S1). Although treatment outcomes were similar, patients with brain metastasis (n = 31) had significantly shorter PFS (12.8 vs. 18.5 months; HR: 0.442; p = 0.002; Fig. 1c) and OS (25.2 vs. 32.2 months; HR: 0.552; p = 0.03; Fig. 1d), suggesting that brain metastasis at baseline is a negative prognostic factor.
Safety profile
All the 106 patients were evaluable for safety profile. Mefatinib at both doses tested was generally well-tolerated at the first-line setting. In patients treated with 60 mg of mefatinib, common grade ≥ 3 treatment-related adverse events observed were rash (13.8%; n = 7), diarrhea (11.8%; n = 6), decreased appetite (5.9%; n = 3), ureterolithiasis (3.9%; n = 2), and anemia (3.9%; n = 2). Among the patients treated with 80 mg of mefatinib, diarrhea (27.3%; n = 15), rash (20.0%; n = 11), mouth ulceration (7.3%; n = 4), and stomatitis (7.3%; n = 4) were the commonly observed grade ≥ 3 treatment-related adverse events. Mefatinib dose reduction was necessary on 31 events for 16 patients (15.1%; 16/106) due to rash (5.7%; n = 6; 7 events), diarrhea (3.8%; n = 4; 8 events); paronychia (2.8%; n = 3; 5 events), and stomatitis (2.8%; n = 3; 3 events). Only two patients who received the initial mefatinib dose of 60 mg required dose reduction (3.9%; 2/51; 5 events), while dose reductions were necessary for 14 patients (25.5%; 14/55; 26 events) who received 80 mg mefatinib. Two patients (1.9%; 2/106) who each received 60 mg and 80 mg had grade IV rash that resulted in treatment termination. No fatal adverse events and unexpected drug-related toxicity were observed in both cohorts. Table 3 summarizes the adverse events reported by >10% of the study cohort.
Potential predictive molecular biomarkers
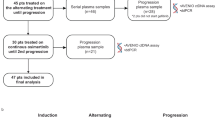
We first investigated the survival outcomes based on specific baseline EGFR sensitizing mutations. Supplementary Figure S2 summarizes the mutation profile of the 69 patients who had baseline plasma samples submitted for NGS-based mutation profiling. Of them, 59.4% (41/69) had EGFR 19del and 40.6% (28/69) had L858R. Patients harboring EGFR 19del had similar PFS (p = 0.899, HR: 0.961, 95% CI: [0.518–1.783]) and OS (p = 0.083, HR: 0.555, 95% CI: [0.285–1.08]) as compared to patients harboring EGFR L858R (Supplementary Fig. S3a). Among the patients harboring EGFR 19del, PFS (p = 0.72) and OS (p = 0.37) were comparable for those with common E746_A750del (n = 30) and uncommon non-E746_A750del (n = 11) (Supplementary Fig. S3b).
We then investigated the impact of certain concurrent mutations at baseline on survival outcomes. The most common concurrent mutations in the cohort were TP53 and EGFR amplifications, identified from 47.8% (33/69) and 20.3% (14/69) of the patients, respectively (Supplementary Fig. S2). In our cohort, patients harboring any concurrent TP53 mutations (n = 33) (14.0 vs 15.4 months; p = 0.315, HR: 0.736, 95% CI: [0.405–1.337]; Fig. 2a) or those harboring TP53 mutations in exons 5–8 (n = 27) (p = 0.49, HR: 0.802, 95% CI: [0.428–1.502]; Supplementary Fig. S4a) had a comparable PFS as patients having wild-type TP53. The OS for both groups have not been reached (p = 0.034, HR: 0.483, 95% CI: [0.246–0.946]; Fig. 2b; p = 0.067, HR: 0.514, 95% CI: [0.252–1.048]; Supplementary Fig. S4a). Patients with concurrent mutations in genes involved in DNA damage repair (DDR) at baseline had similar PFS for first-line mefatinib therapy (p = 0.18, HR: 0.515, 95% CI: [0.197–1.347]) but significantly shorter OS (p = 0.004, HR: 0.265, 95% CI: [0.108–0.649]) as compared to those who were wild-type for these genes (Supplementary Fig. S4b). Supplementary Table S2 summarizes the list of genes related to the DDR pathway included in the analysis. Moreover, patients with or without concurrent copy number variations (CNV), including EGFR amplifications, had statistically similar PFS (CNV, p = 0.092, HR: 0.571, 95% CI: [0.297–1.096]; Supplementary Fig. S4c; EGFRamp, p = 0.14, HR: 0.595, 95% CI: [0.298–1.187]; Supplementary Fig. S4d) but significantly shorter OS (CNV, p = 0.008, HR: 0.389, 95% CI: [0.193–0.786]; Supplementary Fig. S4c; EGFR amplifications, p = 0.018, HR: 0.412, 95% CI: [0.197–0.856]; Supplementary Fig. S4d). These data suggest that despite harboring concurrent mutations that are correlated with shorter EGFR-TKI response and/or poor overall prognosis, the patients who harbor these concurrent mutations derive similar survival benefit from first-line mefatinib therapy as their wild-type counterpart.

Kaplan–Meier curves comparing the PFS (a) and OS (b) of patients with advanced EGFR–mutant non-small-cell lung cancer who had concomitant TP53 mutations as compared to patients who had wild-type TP53. Patients who experienced ctDNA clearance at first follow-up had significantly longer survival outcomes. Kaplan–Meier curves comparing the PFS (c) and OS (d) of patients with EGFR–mutant non-small-cell lung cancer who experienced ctDNA clearance at first follow-up compared with patients who had detectable mutations
In addition to baseline molecular factors, we further investigated the dynamic changes in mutations from baseline until the first follow-up 6 weeks after initiating mefatinib therapy (F1) to understand whether their detection or the lack thereof could predict efficacy and survival outcomes. The mutation detection rates and the maximum allelic fraction (MaxAF) at F1 were dramatically reduced compared with baseline levels (Supplementary Fig. S5). MaxAF is defined as the highest allelic fraction among all the mutations detected from the patient sample. At F1, patients with ctDNA clearance, defined as having no mutation detected from the panel used, were associated with significantly longer PFS (p = 0.04, HR: 0.486, 95% CI: [0.245–0.968]; Fig. 2c) and OS (p = 0.0024, HR: 0.318, 95% CI: [0.152–0.667]; Fig. 2d) as compared to patients who had mutations detected. Consistently, patients who experienced clearance of EGFR sensitizing mutations at F1 had a trend of longer PFS (p = 0.094, HR: 0.465, 95% CI: [0.189–1.14]) and significantly longer OS (p < 0.001, HR: 0.219, 95% CI: [0.092–0.522] than patients who retained their EGFR sensitizing mutations (Supplementary Fig. S6a). Notably, 50 (75.8%) and 55 (87.3%) patients had ctDNA clearance and clearance of EGFR mutation at F1, respectively. Among the patients who achieved PR, the number of patients who had ctDNA clearance was numerically higher than those who remained ctDNA positive; however, it did not reach statistical significance (72.7% (40/55) vs 27.3% (15/55); p = 0.27). The patients who achieved PR as best response and experienced ctDNA clearance at first follow-up had statistically similar PFS (p = 0.064, HR: 0.513, 95% CI: [0.253–1.039]) but had significantly longer OS (p = 0.007, HR: 0.334, 95% CI: [0.151–0.739]) than patients who were ctDNA positive (Supplementary Fig. S6b).
Molecular mechanisms of acquired resistance to mefatinib
Finally, the mutation profiles at baseline and disease progression were compared to elucidate the molecular mechanisms of acquired mefatinib resistance. EGFR-TKI progression was associated with the emergence of new mutations or the increase in the allelic fraction of EGFR sensitizing mutation or MaxAF.29 Consistently, in our cohort, the mutation detection rate and MaxAF had an increasing pattern when PD was confirmed (Supplementary Fig. S5). A total of 38 patients experienced PD, with most of them detected with EGFR T790M (42.1%, 16/38). Three patients who acquired EGFR T790M also had concurrent bypass resistance mechanisms such as BRAF V600E (n = 2) and MET amplification (n = 1). Other acquired resistance mechanisms detected from the cohort included ERBB2 amplification (n = 1), MET amplification (n = 1), and TP53 mutations (n = 2). The remaining 18 patients had no known resistance mechanisms detected. Figure 3 illustrates the distribution of molecular mechanisms of acquired resistance for mefatinib detected from our cohort.

Distribution of molecular mechanisms of acquired resistance to mefatinib. Pie chart illustrating the distribution of acquired mutations identified in the cohort at the progression from mefatinib therapy
Discussion
Mefatinib, with chemical formula: C29H27ClF3N5O10 and molecular weight of 698 g/mol, is a second-generation, irreversible, highly effective dual inhibitor of EGFR/HER2 (Supplementary Fig. S7). The half-maximal inhibitory concentration (IC50) of mefatinib for EGFR kinase is 0.4 nanomolar (nM) and for HER2 kinase is 11.7 nM. Using nude mouse xenograft models of erlotinib-resistant human NSCLC NCI-H1975 harboring EGFR L858R/T790M double mutation, mefatinib demonstrated similar or even better anti-tumor activity as compared with afatinib at the same dose. In the nude mouse xenograft model of human lung adenocarcinoma HCC827, the lowest effective dose of 0.2 mg/kg for mefatinib achieved 99.6% suppression of tumor growth, while the comparator group treated with erlotinib at a dose of 12.5 mg/kg only achieved 73.1% tumor growth inhibition. Mefatinib has good drug metabolism, pharmacokinetics (PK), and oral bioavailability. After oral administration, mefatinib is quickly distributed to various tissues of the body, and highly distributed in the lung, small intestine, and stomach. The PK/pharmacodynamic test in rats showed that mefatinib is quickly distributed into tumor tissues after oral administration. Mefatinib metabolism undergoes N-demethylation, sulfation, and glutathione conjugation, with the metabolites concentrated in feces and undetected in plasma, bile, and urine. Mefatinib has no obvious inducing effect on rat liver microsomal cytochrome P450 enzymes. Safety pharmacology shows that mefatinib has no significant effect on the central nervous system function of mice, the cardiovascular, respiratory system, and body temperature of awake cynomolgus monkeys. At the same time, the IC50 value of hERG potassium channel inhibition is 27.86 mM, which is ~100 times higher than the expected maximum serum concentration (Cmax) (daily dose of 100 mg). Toxicity tests of repeated administration in rats, dogs, and cynomolgus monkeys showed that the gastrointestinal tract and kidney are the main target organs for toxicity, consistent with other approved EGFR inhibitors. In addition, in vivo and in vitro studies with mefatinib have shown no teratogenicity and reproductive toxicity.
Our present study reports the results of the phase Ib/II clinical trial of mefatinib as first-line treatment of patients with EGFR-mutant locally advanced/advanced NSCLC. Our study achieved its primary and secondary objectives. Mefatinib at either dose of 60 or 80 mg once daily achieved an ORR of 84.9%, DCR of 97.2%, median PFS of 15.4 months, and median OS of 31.6 months in the first-line treatment of patients with EGFR-mutant stage IIIB-IV NSCLC. Our study has demonstrated a better efficacy for mefatinib than the reported clinical outcomes of afatinib in two phase III studies, LUX-Lung 3 (n = 345; ORR 56%, median PFS 11.1 months, median OS 28.2 months) and LUX-Lung 6 (n = 364; ORR 67%, median PFS 11.0 months, median OS 23.1 months).11,12 Moreover, mefatinib also showed better ORR and PFS than the reported outcomes for first-line dacomitinib from the ARCHER 1050 study (n = 227; ORR 75%, median PFS 14.7 months, median OS 34.1 months).18,23 Similar to the drug-related toxicity profile of afatinib,11,12 the adverse events observed for mefatinib even at a higher dose (80 mg) primarily involved skin (i.e., rash and stomatitis) and gastrointestinal (i.e., diarrhea) toxicities, which were well-tolerated and manageable. As expected, the higher dose of mefatinib had more events of drug-related toxicity than the lower dose. Hence, based on its better safety profile, 60 mg is the recommended daily dose for mefatinib.
Brain metastasis was frequently observed among patients with EGFR-mutant than EGFR-wild-type NSCLC (29.4% vs 19.7%).30 Subgroup analyses of patients with brain metastasis from the LUX-Lung 3 and LUX-Lung 6 have demonstrated a PFS of 11.1 months and 8.2 months, respectively, for first-line afatinib therapy.31 As compared to afatinib, our cohort demonstrated better clinical outcomes with mefatinib therapy with a PFS of 12.8 months, suggesting that first-line mefatinib is a good treatment option for patients who presented with brain metastasis.
Anti-tumor activity of first- and second-generation EGFR-TKI could vary depending on the specific EGFR sensitizing mutation, wherein NSCLCs harboring EGFR L858R demonstrate poorer clinical outcomes to gefitinib, erlotinib, and afatinib than those harboring 19del.4,6,22,32 Meanwhile, patients with common EGFR 19del E746_A750del and uncommon non-E746_A750del also demonstrate distinct EGFR-TKI sensitivity and survival outcomes.33,34 Interestingly, with mefatinib therapy, common EGFR sensitizing mutations did not affect the efficacy as shown by the statistically similar survival outcomes of patients with EGFR 19del and L858R and patients with different EGFR 19del subtypes (Supplementary Fig. S3). These findings suggest that mefatinib effectively and non-selectively inhibits various subtypes of EGFR-mutant lung cancer resulting in similar clinical benefits for patients regardless of EGFR sensitizing mutation types. The main advantage of using NGS in mutational profiling at baseline is its ability to reflect intratumor genetic heterogeneity by simultaneously detecting concurrent somatic mutations associated with treatment response prediction. TP53 mutations are the most common concurrent mutations in NSCLC, accounting for 30–72%, and are associated with poor clinical outcomes with EGFR-TKI therapy.24,25,26,27,28 In our cohort, we found statistically similar PFS outcomes between patients with wild-type TP53 and those who harbor TP53 mutations, even those located in exons 5–8 (Supplementary Fig. S5a), indicating that mefatinib might be a more suitable therapeutic option for patients with concurrent TP53 mutation. In addition to TP53 mutations, other concurrent mutations associated with poor prognosis, including mutations in the DDR pathway, copy number amplifications in any genes, and EGFR amplification, also did not affect mefatinib efficacy. This finding suggests that patients with concurrent gene mutations benefit from mefatinib therapy and therapeutic decisions on their subsequent-line therapy after mefatinib progression are crucial in improving their prognosis. In addition to baseline mutation profiling, serial analysis of plasma samples during treatment could also provide information on the molecular changes associated with therapeutic response or resistance. Numerous studies have demonstrated the association between better clinical response and survival outcomes of patients who experienced ctDNA clearance and either decrease or clearance of EGFR sensitizing mutations during EGFR-TKI therapy.29,35,36,37,38,39 Consistently, the patients in our cohort who had ctDNA and EGFR sensitizing mutation clearance within 6 weeks after initiating mefatinib therapy had a better overall prognosis than patients who remained ctDNA mutation-positive.
At progression, the emergence of additional mutations is implicated in resistance to therapy and tumor progression.29,35,40 The molecular mechanisms of acquired resistance with mefatinib were predominantly EGFR T790M, consistent with other commercially available first- and second-generation EGFR-TKI.6,14,15,40,41 Other common bypass mechanisms, including MET and ERBB2 amplifications, were also identified in our cohort. In general, our analysis demonstrated that mefatinib had a similar profile of acquired resistance as compared to first- and second-generation EGFR-TKIs, particularly the acquisition rate of EGFR T790M. This finding indicates that after developing resistance from first-line mefatinib, osimertinib, and other therapeutic strategies or guidelines currently in use for managing NSCLC after progression from prior generations of EGFR-TKI remains applicable after mefatinib progression. However, this investigation on resistance mechanisms was only limited to the molecular mechanisms of acquired resistance detected from plasma samples and did not include the analysis of tissue rebiopsy samples. An analysis of the rate of histological transformations after mefatinib progression, particularly among the subset of patients who had unknown molecular resistance mechanisms, could be clinically relevant and should be explored in future studies.
Recent reports have demonstrated the superior clinical activity of afatinib in targeting uncommon EGFR mutations, including single G719X, L861Q, and S7681, and compound mutations.42,43 A phase II trial is currently ongoing to explore the activity of mefatinib in targeting uncommon EGFR mutations (ChiCTR2000029058). Our present study is limited to investigating the clinical outcomes of patients with common EGFR sensitizing mutations such as 19del and L858R. A prospective, double-blind, randomized, controlled phase III study with a larger cohort that compares the efficacy of mefatinib with gefitinib is currently ongoing (ChiCTR2000028763). We anticipate the results from these two clinical studies to further establish the efficacy of mefatinib as first-line therapy of patients with advanced EGFR-mutant NSCLC.
In conclusion, our study provides clinical evidence that mefatinib is an effective and well-tolerable therapeutic option for the first-line treatment of patients with advanced EGFR-mutant NSCLC. Mefatinib might benefit patients with concurrent TP53 mutations as well as those with brain metastasis at presentation. CtDNA clearance within six weeks of mefatinib treatment was associated with better survival outcomes. Furthermore, mefatinib had similar molecular mechanisms of acquired resistance than other EGFR-TKI, indicating the applicability of currently available therapeutic strategies and guidelines for managing advanced NSCLCs after EGFR-TKI progression.
Patients and methods
Clinical trial registration
The Phase Ib (ChiCTR2000029062) and phase II (ChiCTR2000029059) studies on mefatinib were registered on the Chinese Clinical Trial Registry.
Patient inclusion and exclusion criteria
Patients newly diagnosed with EGFR-mutant stage IIIB-IV NSCLC from five participating centers from May 2017 to June 2018 for the phase Ib study and July 2018 to December 2018 for the phase II study were enrolled in this study. The phases Ib and II aimed to investigate and compare two mefatinib doses (60 and 80 mg), which were selected based on the results of the phase Ia dose-escalation study. The main objective of the phase Ib study was to investigate and compare the ORR and safety between the two mefatinib doses and included 34 patients who were non-randomly assigned to each dose group, with 15 patients assigned to receive 60 mg and 19 patients assigned to receive 80 mg. Meanwhile, the main objective of the phase II part of this study was to investigate the efficacy and safety of long-term administration of mefatinib and compare the PFS between the two mefatinib dose groups. A total of 72 patients were enrolled and randomized to receive either of the two mefatinib doses. EGFR status was assessed using amplification refractory mutation system polymerase chain reaction (ARMS-PCR). Inclusion criteria included: (1) Having cytologically or histologically confirmed NSCLC; (2) Unresectable, locally advanced (stage IIIB/IIIC) or metastatic disease (stage IV); (3) Having confirmed EGFR mutation status (i.e., 19del or L858R) of tissue biopsy samples using ARMS-PCR; (4) Having at least 1 unidimensional measurable lesion as per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1; (5) Having an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1; (6) Have not received previous systemic therapy; (7) Signed informed consent. Exclusion criteria included: (1) Concurrent mutation in oncogenic drivers including ALK, BRAF, ERBB2, KRAS, MET, RET, and ROS1; (2) Prior local and systemic anti-tumor therapy including chemotherapy, EGFR-TKIs, angiogenesis inhibitors, and immunotherapy; (3) Other clinical factors that were deemed unsuitable for this study. All patients were required to provide written informed consent before enrollment. The study was performed in accordance with the Declaration of Helsinki and its amendments. The study protocol was approved by the Ethics committee of all the participating hospitals.
Treatment schedule and evaluation of treatment response and adverse events
Patients were instructed to take mefatinib at either 60 mg or 80 mg orally once daily. Comprehensive examination, including history taking, physical evaluation, blood and urine laboratory testing, and radiologic imaging (computed tomography or magnetic resonance imaging), was performed at screening, at follow-up visits every six weeks, and at treatment termination. Treatment response was assessed by the investigators according to the RECIST version 1.1 criteria.44 Treatment-related adverse events were evaluated according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.3. Reported adverse events were graded based on severity, with grade 1 as the mildest and grade 5 as death. Dose reductions were permitted for intolerable drug-related toxicities.
Study endpoints
For the phase Ib part of the study, ORR was the primary efficacy endpoint and treatment-related adverse events were the secondary endpoint. Based on the promising efficacy and manageable toxicity of both mefatinib doses in the phase Ib study, we pursued to investigate the efficacy and safety of long-term administration of both doses of mefatinib in a multi-center, randomized, open-label phase II study. The primary objective of the phase II study was to assess PFS. The secondary endpoints were ORR, DCR, OS, and adverse events. ORR was defined as the proportion of patients who achieved complete response (CR) and partial response (PR). DCR was defined as the proportion of the patients who achieved CR, PR, and stable disease (SD). PFS was defined as the time from the start of mefatinib treatment until the treatment termination or the day of the last follow-up. OS was defined as the time from the start of mefatinib treatment until the date of death or the last day of follow-up. Surviving patients at the time of data cutoff were censored for OS as of the last follow-up date. The data cutoff date was 24 March 2021.
Next-generation sequencing (NGS)-based mutation profiling
For the biomarker analyses, longitudinal blood samples were obtained from the patients to elucidate the somatic mutation profile at each time point using NGS. Approximately 10 ml of blood samples were obtained from the patients at each of the three-time points: at baseline before receiving mefatinib therapy, at first follow-up 6 weeks after starting mefatinib therapy (cycle 1), and at the time when disease progression is radiologically confirmed (PD). Plasma samples were submitted for capture-based targeted genomic sequencing to Burning Rock Biotech, a commercial clinical laboratory accredited by the College of American Pathologists and certified by the Clinical Laboratory Improvement Amendments. Targeted genomic sequencing was performed using a panel consisting of 168 lung cancer-related genes (Lung Plasma, Burning Rock Biotech, Guangzhou, China) following optimized protocols as described previously.45,46 The data for the exploratory biomarker study are included as Supplementary data S1.
Statistical methods
Statistical analyses were performed using R software (R version 3.5.3; Vienna, Austria; RRID:SCR_001905). Descriptive statistical analysis was performed to evaluate the safety and efficacy without statistical testing. The quantitative parameters were described as the number and percentage of cases with the categorical variables described as median with range. All analyses were summarized by dose. The DCR and ORR were presented as means and 95% confidence intervals (CI) for each dose group. The Kaplan–Meier method was used to estimate the median PFS and OS and their corresponding 95% CI. Cox proportional hazards regression analyses were applied to the survival analyses to calculate the corresponding hazard ratios (HR) and 95% CI and compare the survival outcomes between groups using baseline brain metastatic status as the adjustment covariate. P values <0.05 were defined as statistically significant.
Data availability
All authors confirm adherence to the policy. The data that support the findings of the exploratory biomarker study are included as Supplementary data S1. Supplementary data S1 includes individual participant data that underlie the results of the biomarker study reported in the article after de-identification. The data will be available upon publication with no end date to anyone who wishes to access the data. Correspondence and requests for additional data should be addressed to KW.
References
Lynch, T. J. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 (2004).
Paez, J. G. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004).
Pao, W. et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl Acad. Sci. USA 101, 13306–13311 (2004).
Mitsudomi, T. et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J. Clin. Oncol. 23, 2513–2520 (2005).
Ji, H. et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 9, 485–495 (2006).
Sharma, S. V., Bell, D. W., Settleman, J. & Haber, D. A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 7, 169–181 (2007).
Rosell, R. et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 361, 958–967 (2009).
Karachaliou, N., Fernandez-Bruno, M., Paulina Bracht, J. W. & Rosell, R. EGFR first-and second-generation TKIs—there is still place for them in EGFR-mutant NSCLC patients. Transl. Cancer Res. 8(Suppl 1), S23–S47 (2019).
Mitsudomi, T. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 11, 121–128 (2010).
Zhou, C. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 12, 735–742 (2011).
Wu, Y. L. et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 15, 213–222 (2014).
Yang, J. C.-H. et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 16, 141–151 (2015).
Kazandjian, D. et al. FDA approval of gefitinib for the treatment of patients with metastatic EGFR mutation-positive non-small cell lung cancer. Clin. Cancer Res. 22, 1307–1312 (2016).
Arcila, M. E. et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin. Cancer Res. 17, 1169–1180 (2011).
Kobayashi, S. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 (2005).
Kwak, E. L. et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl Acad. Sci. USA 102, 7665–7670 (2005).
Li, D. et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27, 4702–4711 (2008).
Wu, Y. L. et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 18, 1454–1466 (2017).
Dungo, R. T. & Keating, G. M. Afatinib: first global approval. Drugs 73, 1503–1515 (2013).
Ricciuti, B., Baglivo, S., De Giglio, A. & Chiari, R. Afatinib in the first-line treatment of patients with non-small cell lung cancer: clinical evidence and experience. Ther. Adv. Respir. Dis. 12, 1753466618808659 (2018).
Park, K. et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 17, 577–589 (2016).
Paz-Ares, L. et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-Lung 7 trial. Ann. Oncol. 28, 270–277 (2017).
Mok, T. S. K. et al. Updated overall survival (OS) from extended follow up in ARCHER 1050: a randomized phase III study comparing dacomitinib with gefitinib as first-line therapy for patients (pts) with EGFR mutations. Ann. Oncol. 30, ix200–ix201 (2019).
Scoccianti, C. et al. Prognostic value of TP53, KRAS and EGFR mutations in nonsmall cell lung cancer: the EUELC cohort. Eur. Respir. J. 40, 177–184 (2012).
Canale, M. et al. Impact of TP53 mutations on outcome in EGFR-mutated patients treated with first-line tyrosine kinase inhibitors. Clin. Cancer Res. 23, 2195–2202 (2017).
Hong, S. et al. Concomitant genetic alterations with response to treatment and epidermal growth factor receptor tyrosine kinase inhibitors in patients with EGFR-mutant advanced non-small cell lung cancer. JAMA Oncol. 4, 739–742 (2018).
Qin, K., Hou, H., Liang, Y. & Zhang, X. Prognostic value of TP53 concurrent mutations for EGFR- TKIs and ALK-TKIs based targeted therapy in advanced non-small cell lung cancer: a meta-analysis. BMC Cancer 20, 328 (2020).
Suh, J. H. et al. Current approaches to the management of brain metastases. Nat. Rev. Clin. Oncol. 17, 279–299 (2020).
Ma, S. et al. The prognostic value of longitudinal circulating tumor DNA profiling during osimertinib treatment. Transl. Lung Cancer Res 10, 326–339 (2021).
Iuchi, T. et al. Frequency of brain metastases in non-small-cell lung cancer, and their association with epidermal growth factor receptor mutations. Int J. Clin. Oncol. 20, 674–679 (2015).
Schuler, M. et al. First-line afatinib versus chemotherapy in patients with non-small cell lung cancer and common epidermal growth factor receptor gene mutations and brain metastases. J. Thorac. Oncol. 11, 380–390 (2016).
Riely, G. J. et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin. Cancer Res. 12, 839–844 (2006).
Truini, A. et al. The EGFR exon 19 mutant L747-A750>P exhibits distinct sensitivity to tyrosine kinase inhibitors in lung Adenocarcinoma. Clin. Cancer Res. 25, 6382–6391 (2019).
Peng, X. et al. Clinical impact of uncommon epidermal growth factor receptor exon 19 insertion-deletion variants on epidermal growth factor receptor-tyrosine kinase inhibitor efficacy in non-small-cell lung cancer. Eur. J. Cancer 141, 199–208 (2020).
Maheswaran, S. et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 359, 366–377 (2008).
Marchetti, A. et al. Early prediction of response to tyrosine kinase inhibitors by quantification of EGFR mutations in plasma of NSCLC patients. J. Thorac. Oncol. 10, 1437–1443 (2015).
Wang, Z. et al. Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first-line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): a phase 2, single-arm, multicentre clinical trial. Lancet Respir. Med. 6, 681–690 (2018).
Song, Y. et al. Circulating tumor DNA clearance predicts prognosis across treatment regimen in a large real-world longitudinally monitored advanced non-small cell lung cancer cohort. Transl. Lung Cancer Res. 9, 269–279 (2020).
Duan, J. et al. Refined stratification based on baseline concomitant mutations and longitudinal circulating tumor DNA monitoring in advanced EGFR-mutant lung adenocarcinoma under gefitinib treatment. J. Thorac. Oncol. 15, 1857–1870 (2020).
Sequist, L. V. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3, 75ra26 (2011).
Ho, G. F. et al. Real-world experience of first-line afatinib in patients with EGFR-mutant advanced NSCLC: a multicenter observational study. BMC Cancer 19, 896 (2019).
Yang, J. C. et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 16, 830–838 (2015).
Yang, J. C. et al. Afatinib for the treatment of NSCLC harboring uncommon EGFR mutations: a database of 693 cases. J. Thorac. Oncol. 15, 803–815 (2020).
Schwartz, L. H. et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur. J. Cancer 62, 132–137 (2016).
Mao, X. et al. Capture-based targeted ultradeep sequencing in paired tissue and plasma samples demonstrates differential subclonal ctDNA-releasing capability in advanced lung cancer. J. Thorac. Oncol. 12, 663–672 (2017).
Li, Y. S. et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Ann. Oncol. 29, 945–952 (2018).
Acknowledgements
The authors thank all the patients who participated in this study and their families. We also thank the investigators, study coordinators, operation staff, and the whole project team who worked on this study. We would also like to thank the Department of Biostatistics, School of Public Health, Nanjing Medical University for assisting analysis of the clinical research data. We also thank the staff of Burning Rock Biotech, particularly Drs. Analyn Lizaso, Jing Lin, Jianxing Xiang, and Songan Chen for analyzing the gene biomarker data and for their active support. This work was sponsored by Hangzhou Zhongmei Huadong Pharmaceutical Co. Ltd. and supported by a grant from the Key R&D Program of Zhejiang Province (2020C03027). Hangzhou Zhongmei Huadong reviewed the data from the study and the final manuscript before submission, but the authors retained editorial control. The funding agencies had no role in the study design, data collection, analysis, interpretation, manuscript writing, and decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
P.W., Y.L., D.L., J.Z., and K.W. were responsible for study conception and design. L.Y. performed statistical analysis. All the authors contributed to clinical data collection, data analysis, project supervision, manuscript writing, editing, and final approval of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by relevant institutional review board of the participating institutions. All the patients provided written informed consent.
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, P., Li, Y., Lv, D. et al. Mefatinib as first-line treatment of patients with advanced EGFR-mutant non-small-cell lung cancer: a phase Ib/II efficacy and biomarker study. Sig Transduct Target Ther 6, 374 (2021). https://doi.org/10.1038/s41392-021-00773-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00773-3
This article is cited by
-
Development and validation of a new LC–MS/MS method for the determination of mefatinib in human plasma and its first application in pharmacokinetic studies
Journal of Analytical Science and Technology (2022)
