
Abstract
Background
This phase I, open-label, dose-escalation study evaluated the safety, pharmacokinetics and pharmacodynamics of combination therapy with the HDM2 inhibitor SAR405838 and the MEK1/2 inhibitor pimasertib administered orally once daily (QD) or twice daily (BID) in locally advanced or metastatic solid tumours (NCT01985191).
Methods
Patients with locally advanced or metastatic solid tumours with documented wild-type TP53 and RAS or RAF mutations were enroled. A 3 + 3 dose-escalation design was employed. The primary objective was to assess maximum tolerated dose (MTD).
Results
Twenty-six patients were treated with SAR405838 200 or 300 mg QD plus pimasertib 60 mg QD or 45 mg BID. The MTD was SAR405838 200 mg QD plus pimasertib 45 mg BID. The most common dose-limiting toxicity was thrombocytopenia. The most frequently occurring treatment-related adverse events were diarrhoea (81%), increased blood creatine phosphokinase (77%), nausea (62%) and vomiting (62%). No significant drug–drug interactions were observed. The biomarkers MIC-1 and pERK were, respectively, upregulated and downregulated in response to study treatment. In 24 efficacy-evaluable patients, one patient (4%) had a partial response and 63% had stable disease.
Conclusions
The safety profile of SAR405838 and pimasertib combined was consistent with the safety profiles of both drugs. Preliminary antitumour activity was observed.
Similar content being viewed by others


Introduction
The tumour suppressor p53 has a pivotal role in preventing tumourigenesis through the induction of cell-cycle arrest and apoptosis.1 TP53 is the most frequently mutated gene in human cancer; however, some tumours still harbour wild-type TP53.2 In these cases, wild-type TP53 function is usually inhibited by the negative regulator mouse double minute 2 (MDM2, HDM2 in humans). HDM2 binds to the transactivation domain of p53 and acts as an E3 ubiquitin ligase, leading to degradation in the proteasome. Overexpression of HDM2 has been reported in various tumour types, and small-molecule inhibitors of HDM2 have demonstrated antitumour activity in preclinical studies.3,4
SAR405838 is an oral, selective spirooxindole derivative antagonist of HDM2.4 SAR405838 treatment results in p53 pathway activation, leading to p53-dependent cell-cycle arrest and apoptosis in preclinical models.4 SAR405838 monotherapy was investigated in a phase I dose-escalation study in patients with solid tumours, including a maximum tolerated dose (MTD) expansion cohort of patients with de-differentiated liposarcoma.5 The main dose-limiting toxicity (DLT) observed was thrombocytopenia; the MTD and recommended phase II dose of SAR405838 was 300 mg once daily (QD).5 In the phase I study, it was also shown that TP53 mutations appeared in circulating cell-free DNA (cfDNA) from patients being treated over time with SAR405838. Furthermore, TP53 mutation burden increased over time and correlated with change in tumour size, suggesting emergence of resistance to HDM2 inhibition.6
MEK is a key component of the mitogen-activated protein kinase (MAPK) signalling pathway, which is integral to the proliferation and survival of cancer cells.7 Activation of the GTPase RAS leads to a phosphorylation cascade via the kinases RAF, MEK and ERK that results in the activation of oncogenic gene expression. Activating mutations in the upstream components B-RAF or RAS (KRAS, NRAS and H-RAS) are the most frequent cause of upregulation of the MAPK signalling pathway.8 However, targeting RAS directly has been unsuccessful to date. Therefore, strategies for inhibiting the MAPK pathway have focused on inhibiting B-RAF and MEK (MEK1 and MEK2 isoforms). MEK inhibitors have demonstrated encouraging activity in preclinical studies and preliminary clinical activity in solid tumours exhibiting RAS pathway activation.9
Pimasertib is an oral, selective, small-molecule inhibitor of MEK1/2. In a phase I dose-escalation trial, DLTs included skin rash, acneiform dermatitis, ocular events and stomatitis.10 The MTD for pimasertib monotherapy using either a QD or a twice daily (BID) regimen is 90 mg and 60 mg, respectively; the recommended phase II dose of pimasertib monotherapy was determined at 60 mg BID. Pimasertib was investigated in phase I/II clinical trials in a number of tumour types, and has demonstrated preliminary clinical activity as monotherapy and in combination with other agents.11,12
Together, the p53 and MAPK pathways are the most frequently mutated tumour suppressor and oncogene pathways. Preclinical studies have provided rationale to test the combination of SAR405838 and pimasertib in tumours with wild-type TP53 and MAPK pathway activation. In preclinical RAS pathway-activated, TP53 wild-type xenograft melanoma models (UACC62), a therapeutic benefit was observed for the SAR405838 and pimasertib combination over the activity of either single-agent; durable tumour regression was observed with the combination.13
This phase I, dose-escalation study evaluated the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of SAR405838 combined with pimasertib administered QD or BID in patients with locally advanced or metastatic solid tumours.
Materials and methods
Study design
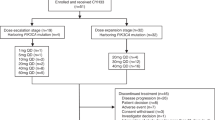
This was a phase I, open-label, dose-escalation, safety, PK and PD study of SAR405838 administered QD and combined with pimasertib administered either QD or BID in 21-day cycles in adult patients with advanced solid tumours (NCT01985191). Both study medications were administered orally using a gelatine capsule formulation. Patients fasted for 2 h prior to and 1 h after each dose. Each dose of SAR405838, except at Cycle 1 Day 1 (C1D1), was to be taken immediately after pimasertib administration, preferably in the morning of each day. Treatment could have continued until precluded by toxicity, incompliance, progression or death.
The primary endpoint was MTD, as assessed by DLT, of SAR405838 and pimasertib combination therapy in patients with locally advanced or metastatic solid tumours. Secondary endpoints included safety, PK, PD and tumour response, as well as determination of the impact of study combination regimen on the genetic status of TP53/RAS when compared with baseline.
Patient population
Patients eligible for inclusion were ≥ 18 years of age, with a histologically or cytologically confirmed solid tumour with documented wild-type TP53 and RAS/RAF mutations for which no further effective standard treatment was available. Eligible patients had locally advanced or metastatic disease with at least one measurable lesion defined by Response Evaluation Criteria in Solid Tumours (RECIST) Version 1.1,14 an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–1, life expectancy ≥ 12 weeks and sufficient bone marrow function.
Dose escalation and dose-limiting toxicities
A 3 + 3 design was used for dose escalation. The starting dose was SAR405838 200 mg QD/pimasertib 45 mg BID. Cohorts of three to six patients were enroled sequentially in ascending dose levels (DLs) per the protocol and decisions of the Study Committee (Investigators and Sponsor) based on the occurrence of DLTs within the first two cycles. Patients should have taken at least 80% of each study medication in order to be evaluable for DLT, unless precluded by the occurrence of a DLT. After confirmation of safety at the first DL1 of SAR405838 200 mg QD/pimasertib 45 mg BID, dose escalation was pursued independently and concomitantly according to the schedule of pimasertib. Using the pimasertib QD schedule, dose escalation was to sequentially proceed to DL2a (SAR405838 200 mg QD/pimasertib 60 mg QD) then DL3a (SAR405838 300 mg QD/pimasertib 60 mg QD). Using the pimasertib BID schedule, dose escalation was to sequentially proceed to DL2b (SAR405838 300 mg QD/pimasertib 45 mg BID) then DL3b (SAR405838 300 mg QD/pimasertib 60 mg BID). If one of three patients experienced a DLT in the first two cycles, the cohort was expanded to six patients for confirmation. If a DLT was observed in at least two out of a maximum of six patients at a DL, this was considered the maximum administered dose. The MTD was the highest DL where at most one patient of the cohort experienced a DLT.
A DLT was defined as any of the following drug-related adverse events (AEs) occurring during the first two cycles of treatment (Days 1–42): an AE that in the opinion of the safety committee was of potential clinical significance such that further dose escalation would expose patients to unacceptable risk; any grade ≥ 3 non-haematological toxicity (excluding Grade 3 fatigue persistent for < 7 days, Grade 3 vomiting or diarrhoea if controlled within 2 days with adequate therapy, asymptomatic Grade 3 creatinine phosphokinase (CPK) elevation, Grade 3 aspartate aminotransferase/alanine aminotransferase elevations < 7 days in duration, Grade 3/4 alkaline phosphatase (ALP) elevations in the context of bone metastasis, or Grade 3 hypertension that can be controlled within a week with oral antihypertensives); any Grade ≥ 3 thrombocytopenia; any Grade 4 neutropenia or febrile neutropenia; any Grade 4 anaemia; retinal vein occlusion; left-ventricular ejection fraction (LVEF) decrease > 20% from baseline or a decrease > 10% if baseline ejection fraction is 50%; Hy’s law; any treatment delay > 2 weeks owing to drug-related adverse effects; any severe or life-threatening complication or abnormality not defined in National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) that is attributable to the therapy; and any toxicities resulting in an inability to complete at least 80% of planned trial medication doses during the first two cycles.
Safety assessments
Safety was assessed by the evaluation of AEs, DLTs, changes in vital signs, ECOG PS, physical examinations,12-lead electrocardiograms, determination of LVEF, ophthalmological examinations and clinical laboratory tests (including haematology, coagulation, blood chemistry and urinalysis). AEs were graded according to NCI-CTCAE version 4.03.15
Pharmacokinetic assessments
Blood samples were collected on Days 1 (for pimasertib only), 2, 3, 8 and 15 of Cycle 1, on Days 1 and 2 of Cycle 2, and on Day 1 of Cycles 3 and 4, to determine the whole-blood and plasma concentrations of SAR405838 and pimasertib, respectively. Calculation of PK parameters included maximum concentration (Cmax), time to reach maximum concentration (tmax), time corresponding to the last concentration above the lowest limit of quantification (tlast), area under the concentration-versus-time curve (AUC) from time 0 to time t (AUC0–t; 12 h for BID or 24 h for QD), AUC from time 0 to tlast (AUClast), AUC extrapolated to infinity, AUC over the dosing interval (AUCtau) and terminal half-life (t1/2z).
Pharmacodynamic assessments
Blood samples for peripheral PD biomarkers analyses for both SAR405838 and pimasertib, including macrophage inhibitory cytokine-1 (MIC-1) levels in plasma and phosphorylated extracellular signal-regulated kinases (pERK) levels in peripheral blood mononuclear cells (PBMCs), were collected on Days 1, 2, 3, 8 and 15 of Cycle 1, on Days 1 and 2 of Cycle 2, and Day 1 of Cycles 3 and 4. MIC-1 protein concentrations were measured in plasma samples using an analytically validated enzyme-linked immunosorbent assay (Quantikine® Human GDF-15 immunoassay). pERK levels were determined by evaluating median value changes in response to ex vivo stimulation with or without phorbol myristate acetate (PMA) over time in whole-blood samples (CD 45+ and lymphocyte populations) by flow cytometry (BD FACSCanto™ II instrument). Stimulated pERK levels were calculated as percentage (PMA-dimethylsulphoxide [DMSO])/DMSO).
Molecular profiling assessments
Plasma preparation and cfDNA isolation from plasma
Plasma was prepared at clinical sites within 15–30 minutes from blood draw using double centrifugation as previously described.6 Blood samples were processed first by centrifugation at 1600 ( + 150) g for 10 minutes. The supernatant was transferred to a fresh 2 mL tube and was centrifuged again at 3000 ( + 150) g for 10 minutes. The resulting supernatant was transferred into a 3.5 mL polypropylene tube and stored at − 80 °C until cfDNA isolation. This process typically yielded ~ 1.2 mL of plasma for DNA isolation. For cfDNA isolation, we used the QIAamp® Circulating Nucleic Acid Kit (QIAGEN, Catalogue # 55114) using the QIAvac 24 Plus (QIAGEN, Catalogue # 19413) according to the manufacturer’s recommended procedures.
Targeted sequencing library preparation and mutation analysis
In order to monitor tumour genetic status using liquid biopsies, we developed a targeted deep-sequencing assay for mutation detection based on a hybrid-capture target enrichment strategy.6 Mutation analysis was done as previously described.6
Assessment of tumour genetic status of RAS/RAF and TP53 at baseline was performed at each site on either archival tissues (20 including 10 diagnostic specimen) or freshly biopsied tissues during screening (six patients).
Efficacy assessments
Radiological tumour assessments were made at least every two cycles or less frequently, if indicated. Tumour response was investigator assessed using RECIST 1.1. A partial response (PR) or complete response must have been confirmed on a second examination performed at least 4 weeks apart in order to be documented as a confirmed response.
Results
Patient population
Twenty-six patients with locally advanced or metastatic tumours were treated (first patient enroled: 03 December 2013; last patient completed: 15 February 2016): DL1 SAR405838 200 mg QD/pimasertib 45 mg BID (n = 7); DL2a SAR405838 200 mg QD/pimasertib 60 mg QD (n = 4); DL2b SAR405838 300 mg QD/pimasertib 45 mg BID (n = 7); and DL3a SAR405838 300 mg QD/pimasertib 60 mg QD (n = 8).
Patient baseline characteristics are shown in Table 1. Median age was 59.5 years (range 45–79 years). Patients had advanced cancer diagnoses, most commonly colorectal (42%) or lung (31%). Median number of prior anticancer therapies was 3 (range 1–7). Twenty-one of the 26 (81%) patients discontinued the study owing to disease progression and five (19%) discontinued owing to AEs.
Dose escalation and dose-limiting toxicities
Three of 12 DLT-evaluable patients treated with the BID-pimasertib regimen experienced a DLT: one of six evaluable patients experienced two DLTs at DL1 (Grade 2 pustular rash and Grade 2 thrombocytopenia that led to dose interruptions and < 80% of dose completion) and two of six evaluable patients experienced one DLT at DL2b (one patient with Grade 2 thrombocytopenia, for which the SAR405838 dose was interrupted and not resumed, and one patient with Grade 4 increased lipase). DL3b was not tested.
One of eight DLT-evaluable patients treated with the QD pimasertib regimen experienced a DLT. No DLT occurred in three DLT-evaluable patients treated at DL2a, and one of five DLT-evaluable patients experienced a DLT at DL3a (Grade 3 thrombocytopenia). At that same DL, an AE meeting DLT definition (Grade 2 thrombocytopenia) occurred in Cycle 4, post-DLT evaluation period, which led to discontinuation of study medication in an additional patient. A sixth patient was not recruited to complete the cohort as DL3a was considered not tolerated.
The MTD was SAR405838 200 mg QD plus pimasertib 45 mg BID (highest total daily dose of pimasertib).
Safety
Mean duration of treatment was 21 weeks (seven cycles per patient) for SAR405838 and pimasertib. The most frequently occurring AEs regardless of causality were diarrhoea (81%), increased blood CPK (77%), vomiting (73%), nausea (69%) and fatigue (58%; Supplementary Table 1). The most frequently occurring Grade ≥ 3 AEs regardless of causality were pulmonary embolism, fatigue and thrombocytopenia (15% each; Supplementary Table 1). The most frequently occurring treatment-related AEs were diarrhoea (81%), increased blood CPK (77%), nausea (62%) and vomiting (62%; Table 2). The most frequently occurring Grade ≥ 3 treatment-related AEs were increased blood CPK (12%), fatigue (12%) and thrombocytopenia (12%; Table 2).
AEs of interest included increased blood CPK (77%; Grade ≥ 3 12%), decreased ejection fraction (38%; Grade ≥ 3 4%), retinal detachment (31%; no Grade ≥ 3), macular detachment (27%; no Grade ≥ 3), thrombocytopenia (19%; Grade ≥ 3 15%) and increased troponin T (19%; no Grade ≥ 3). The most common Grade ≥ 3 haematological laboratory abnormality was lymphocyte count decreased (19%); the most common Grade ≥ 3 biochemistry laboratory abnormality was increased ALP (15%).
Fourteen patients (54%) had a serious AE (SAE), the most frequently occurring of which were disease progression (12%), constipation (8%) and accidental overdose (8%). Four patients (15%) had a treatment-related SAE, including macular detachment, pneumonitis, diarrhoea, nausea, vomiting and accidental overdose (4% each). Five patients (19%) had an AE leading to permanent treatment discontinuation, including rash pustular, dyspnoea, nausea, vomiting, increased blood CPK and ECG T-wave inversion (4% for each), and fatigue (8%). In addition, 17 patients had SAR405838 dose modification, reduction or omission, and 18 patients had pimasertib dose modification, reduction or omission. Four patients died during the study, with three deaths occurring in the post-treatment period. All deaths were owing to disease progression with no treatment-related deaths reported.
Pharmacokinetics
Table 3 summarises SAR405838 and pimasertib PK parameters. Figure 1 shows SAR405838 and pimasertib concentration–time profiles. PK of SAR405838 and pimasertib when administered in combination was similar to the PK of SAR405838 or pimasertib when administered as monotherapy. Owing to respective drug variability, only a substantial drug–drug interaction may have been evidenced.

Mean plasma concentration–time profiles for a SAR405838 and b pimasertib. BID twice daily, C cycle, D day, QD once daily
The dose increase (50%) between DL1/DL2a (SAR405838 200 mg) and DL2b/DL3a (SAR405838 300 mg) did not result in SAR405838 exposure increase. The dose increase (33%) between DL1/DL2b (pimasertib 45 mg BID) and DL2a/DL3a (pimasertib 60 mg BID) resulted in pimasertib exposure increase.
Pharmacodynamics
Inhibition of pERK was evaluated in PMA-stimulated PBMCs; ≥ 80% pERK inhibition was observed at C1D1, Cycle 1 Day 2 (C1D2) and Cycle 2 Day 1 (C2D1), at or around the tmax of pimasertib, at most DLs, and was maintained for 4 h (Fig. 2). Pimasertib dosing at 45 mg and 60 mg induced similar inhibition, which was not affected by SAR405838 administration.

Inhibition of pERK in PMA‑stimulated PBMCs at C1D1 (n = 26). BID twice daily, C cycle, D day, PBMC peripheral blood mononuclear cells, pERK phosphorylated extracellular signal-regulated kinases, PMA phorbol myristate acetate, QD once daily, SD stable disease
Induction of MIC-1, a non-tumour-specific soluble protein regulated by p53, was evaluated. MIC-1 elevation (mean 3.5+ /− 0.8-fold vs baseline) was observed in all patients at C1D2 and C2D1, peaking at 6 h on both days. SAR405838 dosing at 200 or 300 mg induced a similar increase, which was not affected by pimasertib administration (Supplementary Fig. 1). A patient with confirmed PR (endometrial tumour) had the greatest increase in MIC-1 (8.8‑fold) in the DL1 cohort.
Molecular profiling
The mutation status of TP53, RAS family genes and BRAF in cfDNA derived from plasma of 25 patients (one sample missing) was used to correlate with that from tumour tissue at screening. There was a partial concordance between tumour tissue (data provided by the clinical sites) and plasma cfDNA data collected at screening. Sixteen of 25 plasma samples had detectable level of KRAS (n = 14), BRAF (n = 1) or NRAS (n = 1) mutations, which were 100% concordant to reported tumour DNA genotype. Three of 25 plasma samples had detectable mutations in TP53, with a frequency ranging from 0.35% to 18%.
Mutation status was also analysed to look at the emergence of mutations after study treatment. Samples from 13 patients were collected. De novo TP53 mutations were only seen in two patients after study treatment, and this occurred after Cycle 3. Of the two patients with de novo TP53 mutations, one had best response of SD and progressive disease at Cycle 6, the other had a PR at Cycle 2 and progressive disease at Cycle 12.
Efficacy
In 24 efficacy-evaluable patients, the best overall response was PR for one patient (4%) with endometrial adenocarcinoma at SAR405838 200 mg QD/pimasertib 45 mg BID. Stable disease (SD) was the best overall response observed for 15 patients (63%), including patients with colorectal (seven patients), lung (five patients), pancreatic (one patient), intrahepatic bile duct (one patient) cancer and skin melanoma (one patient). Four patients had prolonged SD of > 6 months (three patients with colorectal cancer and one patient with intrahepatic bile duct cancer). Eight patients (33%) had progressive disease as best response. Best percentage change in target lesion diameters is shown in Fig. 3; changes in target lesion diameters were variable and were generally not dose dependent; however, tumour shrinkage was mostly observed in the pimasertib BID-based regimens.

Maximum percentage change from baseline in the sum of target lesion diameters. Dotted lines represent threshold required for partial response (− 30%) and progressive disease (+ 20%). * Pimasertib BID. BID twice daily
Discussion
This phase I, dose-escalation study evaluated the safety, PK and PD of combination therapy with SAR405838 and pimasertib (QD and BID) in advanced solid tumours. The MTD was SAR405838 200 mg QD plus pimasertib 45 mg BID; the main DLT was thrombocytopenia. The safety observations of thrombocytopenia are consistent with other drugs in the HDM2 antagonist class,16,17,18,19,20 and the mechanism of action.21
At the MTD, significant dose interruptions and reductions occurred after Cycle 2 owing to late toxicities and poor tolerance. Single-agent MTDs of SAR405838 and pimasertib could not be administered in combination, owing to overlapping toxicity. The most common treatment-related AEs were diarrhoea and blood CPK increase. Compared with pimasertib BID regimen, the QD regimen induced less diarrhoea, vomiting, electrolytes imbalance, skin reaction, ocular events, CPK increase and drop in LVEF. In addition, AEs meeting DLT definition occurring late, and responsible for dose delay and reduction, occurred in patients receiving the BID-pimasertib schedule.
No significant drug–drug interactions were observed. Considering respective variability of SAR405838 and pimasertib, the PK profiles were generally consistent with previous data for each drug alone. Only a substantial drug–drug interaction may have been evidenced. PD effect of both agents was demonstrated at all doses tested. The PD biomarkers MIC-1 and pERK were, respectively, upregulated and downregulated in response to study treatment.
In a phase I monotherapy study in solid tumours, emergence of TP53 mutations in patients being treated with SAR405838 was shown.6 In this combination study of SAR405838 + pimasertib, emergence of TP53 mutations during treatment was only seen in 2 of 13 patients tested, suggesting that combination with pimasertib may affect the emergence of TP53 mutations and thus resistance to HDM2 antagonist. There was only 64% concordance for KRAS mutation presence between tumour tissue and plasma samples where KRAS mutations were reported in the tumour but not in the plasma samples. There were also some discrepancies for TP53 mutations where three mutations were detected in plasma and not in the three patients’ tumour samples. This could be explained by the difference in date of collection for both samples or difference in assay sensitivity. For KRAS mutation discrepancies, tissue heterogeneity where a rare mutated clone would have been detected in a specific region of the tumour but not found in the plasma sample could also be a relevant explanation.
The best overall response was PR for one patient with an endometrial tumour. For the majority of patients (63%) the best overall response was SD. Prolonged SD ( > 6 months) was observed in three patients with colorectal cancer and one patient with intrahepatic bile duct cancer. The data suggest that pimasertib BID may have increased MAPK pathway inhibition and tumour shrinkage compared with the QD regimen.
In summary, this phase I dose-escalation study evaluated combination therapy with SAR405838 and pimasertib in locally advanced or metastatic solid tumours. SAR405838 and pimasertib could not be administered at the single-agent MTDs when combined. However, preliminary antitumour activity was observed, suggesting potential benefit of restoring p53 activity while inhibiting the MAPK pathway in TP53 wild-type and MAPK-mutated malignancies.
References
Levine, A. J. & Oren, M. The first 30 years ofp53: growing ever more complex. Nat. Rev. Cancer 9, 749–758 (2005).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 (2010).
Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004).
Wang, S. et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 74, 1–11 (2004).
de Jonge, M. et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur. J. Cancer 76, 144–151 (2017).
Jung, J. et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat. Commun. 7, 12609 (2016).
Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22 (2003).
Sebolt-Leopold, J. S. & Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 4, 937–947 (2004).
Duffy, A. & Kummar, S. Targeting mitogen-activated protein kinase kinase (MEK) in solid tumors. Target Oncol. 4, 267–273 (2009).
Awada, A. et al. Safety and recommended Phase II dose (RP2D) of the selective oral MEK1/2 inhibitor pimasertib (MSC1936369B/AS703026): results of a Phase I trial. Eur. J. Cancer 48, 185–186 (2012).
Ravandi, F. et al. Clinical, pharmacokinetic and pharmacodynamic data for the MEK1/2 inhibitor pimasertib in patients with advanced hematologic malignancies. Blood Cancer J. 5, e375 (2015).
Macarulla, T. et al. Phase I study of FOLFIRI plus pimasertib as second-line treatment for KRAS-mutated metastatic colorectal cancer. Br. J. Cancer 112, 1874–1881 (2015).
de Weger, V. A. et al. Phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumors. Eur. J. Cancer 51, S55 (2015).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. 2010. Available at: http://evs.nci.nih.gov/ftp1/CTCAE/About.html (accessed May 2018).
Iancu-Rubin, C. et al. Activation of p53 by the MDM2 inhibitor RG7112 impairs thrombopoiesis. Exp. Hematol. 42, 137–145 (2014).
Hyman, D. et al. Dose- and regimen-finding phase I study of NVP-HDM201 in patients (pts) with TP53 wild-type (wt) advanced tumors. Eur. J. Cancer 69, S128–S129 (2016).
Langenberg, M. H. et al. A phase I study of the MDM2 inhibitor AMG 232 in patients with advanced p53 wild type (p53WT) solid tumors or multiple myeloma. Eur. J. Cancer 69, S34 (2016).
Ray-Coquard, I. et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 13, 1133–1140 (2012).
Wagner A. J. et al. A phase I trial of the human double minute 2 (HDM2) inhibitor MK-8242 in patients (pts) with advanced solid tumors. J. Clin. Oncol. 3533, 1304–1311 (2015).
Mahfoudhi, E. et al. P53 activation inhibits all types of hematopoietic progenitors and all stages of megakaryopoiesis. Oncotarget 7, 31980–31992 (2016).
Acknowledgements
We received editorial support from Simone Blagg of MediTech Media, funded by Sanofi. We thank all patients who participated in the study, and all study team members and staff who worked on the study. We thank Bin Wu for her contribution to the study as a clinical biomarker lead, and Wei Zheng for his contribution to the biostatistical analyses. In memorium, we acknowledge Joonil Jung, who performed the molecular profiling analyses. This study was funded by Sanofi.
Author information
Authors and Affiliations
Contributions
VAdW, JHMS, BD, KT, KH, GT, SG, SM and ED designed the study. VAdW, MdJ, MHGL, JHMS, ML, AV and ED enroled and treated patients, and collected the data. All authors analysed and interpreted the data. All authors critically revised draft versions, and approved the final manuscript.
Corresponding author
Ethics declarations
Data availability
Data are available on request. Biomarker data were posted to EU registry in February 2017; all remaining samples were destroyed in January 2017. Raw PK data were archived electronically; all remaining samples were destroyed.
Competing interests
ML has received an ISS grant from Sanofi. KH, BD and SM are employees of Sanofi. GT is an employee of Sanofi and holds stock in Sanofi. KT is a former contractor with Sanofi. SG is an employee of EMD Serono. VDW, MDJ, MGHL, JS, AV and ED have no conflicts to declare.
Ethics approval and consent to participate
This clinical trial adhered to the principles outlined in the Helsinki declaration and was conducted in compliance with all applicable international and national laws and regulations. The protocol was approved by all relevant Institutional Review Boards/Independent Ethics 400 Committees. All patients provided written, informed consent.
Note
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International licence (CC BY 4.0).
Additional information
Ethics approval and consent to participate: This clinical trial adhered to the principles outlined in the Helsinki declaration and was conducted in compliance with all applicable international and national laws and regulations. The protocol was approved by all relevant Institutional Review Boards/Independent Ethics Committees. All patients provided written, informed consent.
Electronic supplementary material
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Weger, V.A., de Jonge, M., Langenberg, M.H.G. et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br J Cancer 120, 286–293 (2019). https://doi.org/10.1038/s41416-018-0355-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-018-0355-8
This article is cited by
-
Differentially expressed platelet activation-related genes in dogs with stage B2 myxomatous mitral valve disease
BMC Veterinary Research (2023)
-
Targeting p53 pathways: mechanisms, structures, and advances in therapy
Signal Transduction and Targeted Therapy (2023)
-
Targeting MDM2 for the development of a new cancer therapy: progress and challenges
Medicinal Chemistry Research (2023)
-
Targeting p53–MDM2 interaction by small-molecule inhibitors: learning from MDM2 inhibitors in clinical trials
Journal of Hematology & Oncology (2022)
-
Oncogenic KRAS blockade therapy: renewed enthusiasm and persistent challenges
Molecular Cancer (2021)
