
Abstracts
Bone mainly functions as a supportive framework for the whole body and is the major regulator of calcium homeostasis and hematopoietic function. Recently, an increasing number of studies have characterized the significance of bone as an endocrine organ, suggesting that bone-derived factors regulate local bone metabolism and metabolic functions. In addition, these factors can regulate global energy homeostasis by altering insulin sensitivity, feeding behavior, and adipocyte commitment. These findings may provide a new pathological mechanism for related metabolic diseases or be used in the diagnosis, treatment, and prevention of metabolic diseases such as osteoporosis, obesity, and diabetes mellitus. In this review, we summarize the regulatory effect of bone and bone-derived factors on energy metabolism and discuss directions for future research.
Similar content being viewed by others

Introduction
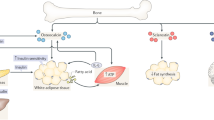
The skeleton constitutes up to ~15% of the total human body weight and mainly consists of bone matrix and osteoblasts, osteoclasts, osteocytes and chondrocytes1 (Fig. 1). As the fundamental framework of the skeletal system, the bone matrix includes organic matter and inorganic matter. Among them, organic matter contains type I collagen secreted by osteoblasts and a variety of noncollagenous proteins. Inorganic matter, also known as bone mineral, is mainly composed of calcium, phosphorus, magnesium, etc. It serves as an important calcium and phosphorus reservoir in the body.2 Osteoblasts can promote mineralization and bone formation by synthesizing osteoids and secreting matrix vesicles. Additionally, osteoclasts can secrete organic acids and proteases to dissolve and absorb bone matrix. Osteocytes, which are the most numerous cells in bone tissue, play an essential role in the regeneration and maintenance of bone matrix. Chondrocytes are the main component of cartilage, and endochondral ossification serves as an important process of bone formation. Bone has hematopoietic function, which is mainly achieved by hematopoietic cells in bone marrow. It is worth mentioning that not only hematopoietic cells but also adipocytes, fibroblasts, and bone marrow mesenchymal stem cells (BMSCs) participate in the regulation of hematopoiesis.3,4 However, the role of the skeleton in the pathogenesis of metabolic diseases is poorly understood.

The cells that make up bone mainly include osteocytes, osteoblasts, osteoclasts, and chondrocytes in bone tissue, as well as bone marrow mesenchymal cells and bone marrow adipocytes in the bone marrow cavity. Different cells can produce different endocrine factors, which can enter the blood circulation to regulate multiple organs in the body
Bone is an endocrine organ
The role of an endocrine organ is to regulate distant functions through the secretion of a peptide or steroid hormone. Several breakthroughs in bone science in the past few years have elucidated the endocrine role of the skeleton. FGF23 and osteocalcin, which function in a classic endocrine manner, are novel hormones produced by bone cells that control energy balance and mineral homeostasis.
Body homeostasis depends on a dynamic balance of energy metabolism. Once the balance is disrupted, it will lead to the risk of metabolic diseases. Various metabolic organs and tissues, such as the liver, islets, fat, muscle, and skeleton, are involved in energy metabolism. Metabolomic studies have determined the alteration of metabolic pathways during the pathological progression of osteoporosis, which provides strong evidence for the metabolic role of bone in endocrinology. Meanwhile, bone tissue is affected by other circulating hormones, such as adiponectin, leptin, and insulin. Reciprocally, bone-derived hormones exert an effect on energy metabolism throughout the body in return.5,6,7 Recent studies proved that bone cells, including osteoblasts, osteoclasts, BMSCs, and adipocytes, have respective endocrine functions. They can synthesize and secrete a variety of bioactive substances, such as proteins, polypeptides, cytokines, inflammatory factors, adipokines, and exosomes. These bioactive substances regulate bone remodeling by paracrine secretion from bone tissue itself. Additionally, they can be released into the circulation and function by targeting distal organs, thereby affecting the energy metabolism of the whole body.8,9,10,11,12
Additional studies have determined that osteocalcin (OCN), which is secreted by osteoblasts, is the first osteoprotein to regulate energy metabolism. It was initially found to promote the proliferation of pancreatic β-cells and insulin secretion,13 and later, its other functions in energy regulation were found, including its effects on intestinal epithelial cells, adipocytes, and hepatocytes.14,15,16
In this review, we summarized the bone-derived factors that regulate body energy homeostasis discovered in recent years and offered insights into the mechanisms of how these factors function in the interaction between bone metabolism and energy metabolism (Fig. 1). Concomitantly, this paper also introduces possible directions and challenges in current research, which would be helpful for further investigations.
Factors secreted by bone cells
Bone tissue harbors several types of specialized bone cells (osteocytes, osteoblasts, osteoclasts, and chondrocytes) that can secrete factors regulating energy metabolism throughout the body (Table 1). Interestingly, some of those factors, such as Lcn2 and sclerostin, are only produced by one type of cell, while others, such as osteopontin (OPN), can be secreted by various types of cells.
Osteocalcin
Osteocalcin (OCN) is a straight-chain polypeptide composed of 46–50 amino acid residues secreted by mature osteoblasts.17 After posttranslational modification, the 3 vitamin-K-dependent glutamic acid residues of OCN carboxylate form a γ-carboxy glutamic acid (gla) residue, which is also known as bone glutamic acid protein (gla protein) or γ-carboxylated glutamic acid protein.18 Due to the high affinity to mineral ions, the gla residue promotes the storage of carboxylated osteocalcin in bone. Once released from the bone matrix, carboxylated osteocalcin can regulate bone resorption and remodeling by promoting the activity of osteoclasts. However, the bone phenotype varies among different studies; thus, the mechanisms and functions of carboxylated osteocalcin in bone formation/resorption are still controversial.19,20,21,22 Interestingly, researchers found that OCN knockout mice have higher blood sugar and increased visceral fat.13,23 The embryonic stem cell phosphatase (ESP) gene encodes tyrosine phosphatase (OST PTP), which is only expressed in osteoblasts and Sertoli cells of the testis. Lee et al. found that in an ESP gene knockout mouse model, the concentration of circulating OCN was elevated, which was accompanied by an improvement in insulin sensitivity.17 These results suggest that OCN can be secreted or released into peripheral circulation in the form of uncarboxylated osteocalcin (unOCN)24,25 and promotes glucose uptake, participates in insulin signal transduction, and thus regulates energy metabolism in the whole body26,27,28 (Fig. 2a).

Metabolic regulation of factors derived from cells in bone tissue. a OCN can be decarboxylated in osteoblasts, transformed into unOCN, and secreted into the blood. It can primarily bind to the GPRC6A receptor on the cell membrane and activate downstream PI3K/mTOR/Akt signals to affect gene expression. OPG, as a competitive ligand of RANK and TRAIL receptors, can combine with RANK and TRAIL to reduce the distribution of RANK/RANKL and TRAILR/TRAIL on the cell membrane and further affect gene expression. FGF23 can regulate energy metabolism by forming a feedback regulation loop with 1,25(OH)2D3 and PTH. BMP is mainly derived from osteocytes. Different kinds of BMPs can combine with type I and type II serine/threonine kinase receptors on the cell membrane and can interfere with important downstream pathways, such as MAPK, PI3K/Akt, Wnt, hedgehog, and notch, through Smad protein-dependent or Smad-independent pathways, playing a role in regulating metabolism. b The BMP family influences the key steps of fat production and preadipocyte differentiation into WAT and BAT and regulates the function of adipocytes
A large number of in vivo and in vitro studies have proven the role of unOCN in the regulation of glucose metabolism. Studies have confirmed that unOCN directly inhibits the expression of the srebp1c and ChREBP genes in islet β-cells, thereby promoting β-cell proliferation.29,30 Moreover, unOCN activates the PLC/PKC/Ras/MEK pathway and inhibits kV channels to increase intracellular calcium levels, which plays a vital role in the exocytosis of islet cells.31 UnOCN can also promote insulin secretion by regulating other hormones. In intestinal epithelial cells, unOCN has been shown to increase the expression of the insulin stimulating protein glucagon-like peptide-1 (GLP-1) gene.15,32,33 In nonislet tissues, unOCN can also increase glucose utilization and improve cell sensitivity to insulin. It has been proven that unOCN can alleviate endoplasmic reticulum stress by activating the PI3K/AKT/NF-κB signaling pathway, thus improving insulin resistance of adipocytes, myocytes, vascular endothelial cells, etc.34,35,36,37,38 Interestingly, when NF-κB is activated, unOCN can interfere with NF-κB to prevent further inflammatory response expansion. Both a decrease in the proinflammatory cytokines tumor necrosis factor-α (TNF-α) and IL-6 and an increase in the secretion of the anti-inflammatory cytokine IL-10 slowed systemic inflammation and insulin resistance in mice. At the same time, enhanced expression of slc2a4/GLUT4 was also observed in these mice. As a downstream factor of the NF-κB pathway, slc2a4/GLUT4 mediates insulin-stimulated glucose uptake, which increases glucose utilization.26,27,28,39 In addition to the significant regulatory role of unOCN in systemic insulin signaling, insulin may, in turn, bind to the insulin receptor (IR) on osteoblasts. Thus, it activates insulin signaling in osteoblasts to promote bone remodeling by accelerating the decarboxylation of OCN and increasing circulating unOCN, which indicates a feedforward regulation loop between OCN and insulin.40,41
In addition to the regulatory role of insulin metabolism, unOCN can also affect lipid metabolism. unOCN activates the small GTPase Rap1 by expressing cAMP-response element binding protein (CREB) and peroxisome proliferator-activated receptor γ (PPARγ). Consequently, this effect results in adiponectin upregulation, a protein that controls glucose homeostasis and fatty acid oxidation.16,39,42 Another metabolic function of unOCN is thermogenesis, which can be detected in brown adipocytes. Upregulation of the T cell factor 7 (TCF7) and PR domain containing 16 (PRDM16) genes and uncoupling protein 1 (UCP1) facilitates the outcome.43,44
It is worth noting that unOCN can activate nuclear factor erythroid-2-related factor 2 (Nrf2) and inhibit c-Jun N-terminal kinase (JNK). Among them, Nrf2 is a key regulatory molecule that alleviates lipid peroxidation and oxidative stress. Disorders of Nrf2 trigger liver fibrosis and nonalcoholic fatty liver disease (NAFLD). Moreover, inhibition of the downstream factor JNK, which mediates insulin resistance, slows the occurrence of NAFLD.13,14,45 This provides evidence that unOCN can regulate liver metabolism through these two factors (Fig. 2a).
Current studies have indicated that unOCN may be related to the G protein-coupled, family C, group 6, member A (GPRC6A) receptor. GPRC6A receptors are widely distributed and can be activated by various ligands,46,47 such as hexapeptide (OCN-6a-c), derived from the C-terminus of OCN. OCN-6a-c acts as a direct ligand of the GPRC6A receptor by binding to GPRC6A.29,48 Many experiments have proven that deletion of the GPRC6A gene can eliminate the effect of unOCN on β-cells and adipocytes. This was evidenced by GPRC6A β-cell-cko mice, which exhibited lower circulating insulin levels and impaired glucose tolerance.29 Further studies have shown that unOCN activates ERK and cAMP second messenger pathways in GPRC6A-expressing cells in a dose-dependent manner but not in GPRC6A knockout cells.49 These results suggest that unOCN regulates energy metabolism by binding to the GPRC6A receptor and activating a series of downstream signal transduction pathways, such as ERK, cAMP, PI3K/Akt/mTOR, and AMPK.48,50,51,52,53
However, the regulation of unOCN on energy metabolism is still controversial. Recently, Cassandra reported that they did not find an effect of unOCN on blood glucose and insulin levels in OCN-KO mice.54 One of the possible reasons is that OCN impacts the internal environment to affect the regulation of some critical signaling pathways. For example, the PI3K pathway is affected by inflammation. In addition, exercise can change the state of the body and adjust the effect of OCN on related tissues. When studying the impact of OCN on skeletal muscle, it was found that the experimental result was closely associated with insulin concentration and muscle movement. Carboxylation of OCN is vitamin-K-dependent. This finding suggests that vitamin K may be an essential regulator affecting osteocalcin function in the human body, especially during bone formation.17,55 Another possible explanation is the differences in race and age between the studied subjects.56,57 Similarly, dose-dependent activation of GPRC6A induced by unOCN indicates that the concentration of unOCN affects the experimental results. However, these conclusions are conjectures because cross-sectional studies are not enough to explain the causal relationship between osteocalcin and energy metabolism in the human body. Energy homeostasis in the internal environment requires multiple, complex series of factors, and further research is needed in this field.
Osteoprotegerin
Osteoprotegerin (OPG) is a member of the TNF receptor superfamily expressed in bone, lung, kidney, the cardiovascular system, etc.58,59 It has been determined that OPG/receptor activator of NF-κB (RANK)/RANK ligand (RANKL) is an indispensable signaling network for maintaining and regulating bone homeostasis. In bone tissue, OPG inhibits the function of osteoclasts by combining with RANKL and then prevents excessive bone absorption.60,61,62
This regulatory pathway also plays a key role in energy metabolism. For instance, in islet cells, RANK/RANKL is a signal that inhibits the proliferation of β-cells. The phosphorylation of CREB and glycogen synthetase kinase-3 was suppressed by RANK. Then, it can be detected by OPG-treated cells, which hints that OPG can promote the proliferation of β-cells by restraining the RANK/RANKL pathway.63,64 Interestingly, epidemiological studies have found an apparent increase in OPG serum concentrations in patients with metabolic syndrome, accompanied by an increase in inflammatory marker C-reactive protein and insulin resistance,65 as well as a parallel increase in visceral fat.66,67 Similarly, OPG, soluble RANKL, insulin resistance markers, and C-reactive protein were higher in the prediabetic group than in the control group,68 and higher OPG levels were also found in obese adolescents.69,70
In addition to the OPG/RANK/RANKL signaling system, OPG has another ligand called tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). OPG, as the “fifth receptor” of TRAIL, can competitively inhibit TRAIL and its death receptors, trail-r1 and-r2, thus inhibiting cell apoptosis. Scientists discovered the expression of TRAIL and its receptors in human pancreatic β-cells.71 The activation of TRAIL signaling can kill normal pancreatic β-cells, which may contribute to the pathogenesis of type 1 diabetes.72 Some studies have certified that the OPG/TRAIL ratio is significantly decreased in the vascular wall of diabetic rats,73 suggesting that OPG may regulate energy metabolism through the pathway. In addition, inhibiting the activation of IL-1β signaling by p38 MAPK is a new mechanism by which OPG affects pancreatic β-cells. IL-1β-induced β-cell death requires sustained p38 MAPK activation, which is abolished by OPG.74,75,76 Apart from this, a manifest upregulation of osteocalcin expression and insulin sensitivity can be detected in OPG knockout mice. Therefore, OPG may also regulate bone resorption and glucose metabolism by affecting the secretion of OCN77 (Fig. 2a).
Some studies have shown that the level of serum OPG is associated with metabolic diseases, such as NAFLD.78,79,80 However, the relationship between the OPG or OPG/RANK/RANKL signaling system and metabolic disorders is still unclear. It was found that the increased OPG level in type 2 diabetes patients is not due to the onset of diabetes but the progression of disease.81 However, OPG and RANKL levels are not related to the risk of type 2 diabetes.82 Although osteoblasts secrete large amounts of OPG, the extensive expression of OPG in other organs makes it hard to determine whether bone-derived OPG affects β-cells directly. However, there are still some interesting findings on the OPG/RANK/RANKL signaling system. In addition to osteoblasts, BMSCs and chondrocytes, bone marrow adipocytes can also secrete RANKL.83,84 Experiments have shown that in the absence of parathyroid hormone 1 receptor (PTH1R), the expression of RANKL in marrow adipose tissue (MAT) and the content of RANKL in serum were evidently increased. In contrast, it remained unchanged in the thymus and spleen, as well as other tissues that produce RANKL. This result suggests that bone-derived RANKL can be secreted into the circulation to influence distant endocrine organs.84
Osteopontin
Osteopontin (OPN) is a secretory matrix cell protein. In 1985, Heingard et al. first isolated and identified three subtypes of human OPN from salivary protein of bovine bone matrix,85 including full-length subtype OPN a, OPN b lacking exon 5 and OPN c lacking exon 4.86 OPN is expressed in many cells, including osteoblasts, osteoclasts, chondrocytes, and BMSCs.87 It binds to several extracellular receptors, such as integrins (αvβ1, αvβ3, αvβ5, αvβ6, α4β1, α5β1, α8β1, and α9β1) and CD44.88,89
In bone tissues, OPN can be secreted by BMSCs and serve as an autocrine cytokine to regulate bone migration, adhesion, and resorption.90,91 BMSCs of OPN-null mice are prone to differentiate into adipocytes and exhibit higher body fat content.92 In other tissues and organs, the secretion of OPN is mostly related to inflammation. In adipocytes and hepatocytes, OPN primarily enhances inflammatory responses and interrupts glucose homeostasis in cells, which may affect the metabolism of phosphatidylcholine and cholesterol, aggravating the occurrence of nonalcoholic cirrhosis of the liver.93,94,95,96,97,98 However, some other investigations demonstrated that OPN can protect β-cells by reducing the generation of iNOS and preserving Ca2+ homeostasis.99,100,101 Existing research in the human population has confirmed the correlation between OPN levels and metabolic diseases.102,103,104,105,106 Furthermore, Marciano et al. found that the OPN-encoding gene SPP1 is a susceptibility gene for type 1 diabetes mellitus and regulates the autoimmunity process.107,108,109
Until very recently, studies have demonstrated that OPN secreted by adipocytes, hepatocytes, and macrophages regulates energy metabolism. However, whether bone-derived OPN can perform the same job remains elusive. OPN can regulate the osteogenic or lipogenic differentiation of BMSCs, and a higher body fat content can be detected in OPN global knockout mice. However, research on BMSC-specific deletion of OPN has not yet been reported.92 There are only some clinical studies about the effect of bone-derived OPN on metabolism. It is well accepted that a high-fat diet can induce chronic inflammation, while exercise can reduce inflammation. Previous studies showed that the content of OPN increased in mice fed a high-fat diet, and adipose tissue is the principal reservoir of circulating OPN. However, You et al. found that serum OPN in obese teenagers decreased significantly after exercising, while the body fat rate did not change significantly.110 These results suggest that tissues other than adipose tissue may contribute more to serum OPN. Since bone tissue is a vital source of OPN and exercise can ameliorate bone metabolism, it is reasonable to consider that dynamic bone metabolism can affect serum OPN levels. Therefore, the mechanism of bone-derived OPN involving global energy metabolism is worthy of deep study.
Bone morphogenetic protein
Bone morphogenetic protein (BMP) was first isolated from bovine bone. It can promote osteogenesis and chondrogenesis after implantation into bone tissue or extraosseous tissue of mice.111,112 It is a subfamily of the TGF-β ligand family.113 To date, more than 20 kinds of BMPs have been successfully isolated.114 According to their structure, BMP family members can be further divided into several subgroups, including BMP-2/-4, BMP-5/-6/-7/-8, BMP-9/-10, and BMP-12/-13/-14.115 BMPs and their receptors are widely distributed in the whole body and exert an essential regulatory role.116,117 The most critical regulatory pathway of BMP signaling depends on the phosphorylation of Smad proteins.118,119,120,121 In addition, BMP signaling also interferes with multiple signaling pathways, including MAPK/PI3K/Akt, Wnt, hedgehog, and notch, and participates in the regulation of various cytokines, such as the IL, INF-γ, and TNF-α. Due to the wide distribution and multiple functions of BMPs, disorder of BMPs may lead to developmental defects or diseases122,123,124 (Fig. 2a).
Bone-derived BMP can be used as an autocrine or paracrine factor to regulate bone itself. For example, in hBMSCs, BMP-2 can promote the expression of inhibitor of differentiation (ID) and runt-related transcription factor 2 (Runx2), thus enabling the development of osteoblasts.125,126 BMP also has a profound effect on whole-body metabolism. BMPs can regulate the formation, differentiation, maturation, and biological function of adipocytes127,128,129,130,131,132,133,134,135,136,137,138,139,140 (Fig. 2b). Meanwhile, BMPs are involved in insulin secretion of islet β-cells and glucose utilization in different types of cells.141,142,143,144 Related research in humans has investigated the relationship between circulating BMPs and energy metabolism. This finding demonstrates a positive correlation between fat content and circulating BMP-4 levels in obese individuals.145
However, whether the source of circulating BMPs detected in the experiment is bone tissue has not been completely determined. Although osteocytes and osteoblasts are the main sources of BMPs, bone is not the only tissue that secretes BMPs. BMPs are widely expressed in various tissues during the development of embryos.115 However, certain specific expressions are manifested when the individual matures. For instance, BMP-3/4/9/10 are located in the lung,146,147 BMP-3/7 can be found in nerve cells,148,149 and the liver is the primary organ producing BMP-9.150 Furthermore, the expression level of BMP-3b in adipocytes is equivalent to that in bone cells.131 Therefore, more research is needed to determine whether bone-derived BMPs can be secreted into the circulation and affect energy metabolism in all development stages of the organism.
Fibroblast growth factors
Fibroblast growth factors (FGFs) regulate multiple processes of growth and development of organisms.151,152 Among them, FGF19, FGF21, and FGF23 are called endocrine fibroblast growth factors (eFGFs) because their functions are closely related to metabolic regulation.153 eFGFs originate from metabolically active tissue. For example, FGF19 is mainly secreted by the colon,154 FGF21 is widely distributed in the liver and pancreas,155,156 and FGF23 is a bone-derived protein that is mostly secreted by osteocytes and osteoblasts.157 Genetically, it has been proven that serum FGF23 cannot be detected in mice with conditional deletion of the FGF23 gene in osteoblasts and osteocytes.158 The FGF23 gene has a high affinity for the FGFR/Klotho coreceptor complex, which is mainly located in the kidney and parathyroid gland.159,160 This gene was proven to be closely related to autosomal dominant hypophosphatemic rickets161 because it can affect the balance of vitamin D production and the balance between calcium and phosphorus in proximal renal tubules,12 which leads to metabolic disorders of bone. Later, it was proven that FGF23 could act directly on bone tissue as an autocrine or paracrine factor. Osteocytes, osteoblasts, and osteoclasts express the FGFR/Klotho coreceptor. Specific knockout of Klotho in osteocytes leads to osteogenic enhancement and an increase in bone mass.162 In parallel, related studies have shown that FGF23 can upregulate early growth response genes (EGR) 1 and 2 in osteoblasts and the RANKL/OPG ratio on the osteoclast surface by binding to the coreceptor. Regardless, the activation of the FGFR/Klotho coreceptor may also account for bone remodeling.163,164
FGF23 can also act on other tissues to regulate energy metabolism. The most familiar regulatory mechanism is the FGFR/Klotho coreceptor complex. Klotho can bind to membrane receptors and inhibit the phosphorylation of IRs and their intracellular signals, including insulin receptor substrate (IRS) 1 and 2.165 Therefore, increased insulin sensitivity and glucose tolerance were observed in FGF23−/− and Klotho−/− mice. The second mechanism is the complex feedback loop between FGF23, vitamin D, and PTH. PTH can promote the expression of FGF23 by increasing the activity of 1,25(OH)2D3. However, it can also enhance the secretion of FGF23 by acting on PTH1R of osteocytes and activating the cAMP and Wnt pathways.163 Conversely, the increased concentration of FGF23 suppresses the expression of PTH and 1,25(OH)2D3, thus forming a negative feedback loop.166,167 The specific role of this regulatory loop in energy metabolism has not yet been clearly studied. However, either PTH or 1,25(OH)2D3 is the factor regulating energy metabolism, and the imbalance of these factors will inevitably affect the metabolic state. For instance, a study showed that FGF23-null mice had higher insulin sensitivity and lower blood glucose levels, but double mutation of FGF23 and vitamin D receptor (VDR) exacerbated this situation.166 Considering the close interrelationship, it is concluded that FGF23 may also regulate energy metabolism through vitamin D (Fig. 2a).
Another mechanism may be related to systemic inflammatory reactions and obesity. FGF23 can reinforce the expression of inflammatory factors in mouse serum and hepatocytes and aggravate liver injury caused by inflammation. Accordingly, the high level of inflammatory factors in the circulation can induce the secretion of FGF23 in bone cells and contribute to a positive feedback cycle between inflammatory factors and FGF23.168,169 In hepatocytes, abnormally elevated FGF23 can activate the PLCγ/calcineurin/NFAT signaling pathway and promote the release of more inflammatory factors. However, the physiological concentration of FGF23 may not lead to such changes.168 In addition, a study of the elderly Caucasian population showed that serum FGF23 levels were positively correlated with BMI, waist circumference, waist-to-hip ratio, blood lipids, and fat quality.170 Because chronic inflammation is one of the causes of obesity, the relationship between FGF23 and inflammatory factors may be a potential mechanism for obesity and lipid metabolism (Fig. 2a).
Another explanation for the energy metabolism regulation function of FGF23 is that FGF23 and FGF19/21 are homologous in structure, and FGF19/21 is involved in maintaining bile acid homeostasis and regulating systemic insulin sensitivity.171,172,173 Therefore, FGF23 may have a similar effect.
Lcn2
Lcn2, a protein previously thought to be secreted exclusively by adipose tissue and related to obesity, has recently been found to be expressed in osteoblasts. The expression level in osteoblasts is at least 10 times higher than that in white adipose tissue (WAT) or other organs.174,175 In addition to acting as an autocrine regulator of adipocytes, Lcn2 also binds to various cell membrane surface receptors, including 1-microglobulin, glycodelin, retinol-binding protein, alpha-1-acid glycoprotein, beta-lactoglobulin, etc.176,177
Several lines of evidence have shown that Lcn2 exerts a beneficial effect on the energy metabolism of mice and humans. Its role in the central nervous system has been a significant discovery in recent years. The results showed that the Lcn2 concentration increased threefold in mice fed after fasting, suggesting that Lcn2 can regulate appetite. Lcn2 deficiency is characterized by increased food intake in mice, leading to insulin resistance and obesity. Melanocortin 4 receptor (MC4R), the receptor of Lcn2, controls appetite, body weight, and energy balance in the body.178,179 Physically, Lcn2 can penetrate the blood–brain barrier and bind to the MC4R of the PVH nucleus in the hypothalamus.174
In the periphery, Lcn2 has also been found to act on metabolism-related tissues. In vitro culture of islet cells showed that Lcn2 could directly act on islet cells and insulin secretion.174 In addition, some metabolic effects of Lcn2 have been discovered in adipose tissue. Although there is no evidence supporting that Lcn2 directly binds to the receptor on adipocytes, Lcn2 is a crucial mediator of retinoic acid that mediates the expression of the UCP1 gene and activation of thermogenesis in BAT. Therefore, Lcn2-deficient mice could suffer from decreased circulating retinoic acid levels and have hindered thermogenesis.180,181,182,183
However, some experimental results have proven the negative effect of Lcn2. GLUT1 and GLUT4 protein levels and glucose uptake in human adipocytes decreased significantly after treatment with rLcn2.184 Moreover, Lcn2 knockout mice showed markedly lower fasting blood glucose and higher glucose tolerance than wild-type mice.185 Its effect on glucose utilization is probably due to the upregulation of IL-6 gene expression and the reduction in PPARγ and adiponectin.184 Further studies in humans also showed that the level of Lcn2 in patients with type 2 diabetes mellitus was significantly higher than that in normal individuals, and its expression was positively correlated with the levels of inflammatory markers such as C-reactive protein, IL-6 and TNF-α.186
In general, Lcn2 influences energy metabolism via a complex regulatory network. The complex signaling pathways of Lcn2 and diverse research methods, for example, in vivo versus in vitro experiments and mouse genetic backgrounds, may explain the discrepancy in results from those studies. Another possibility is that Lcn2 secreted by different tissues may have various functions. For example, genome-wide Lcn2 knockout mice showed increased glucose tolerance and unchanged insulin sensitivity,185 while osteoblast-specific knockout mice showed decreased glucose tolerance and impaired insulin sensitivity.174 Additionally, the different mechanisms and target organs of Lcn2 may account for the different experimental results.
Sclerostin
Sclerostin (Scl) is a glycoprotein encoded by the SOST gene that is secreted by mature osteocytes.187 Several studies have shown that Scl is widely expressed in nearly all tissues and organs, with high expression in bone, especially in osteocytes.188,189 Scl can suppress osteoblast and osteoclast activity, which stabilizes the strength and toughness of bone under normal physiological conditions.187,190 The lack of Scl will lead to overhardening of bone, and its overexpression will inhibit the formation of bone.191,192,193 The Wnt signaling pathway has emerged as a key regulator of osteogenesis. Scl binds to lipoprotein receptor-related protein 4/5/6 in osteoblasts and osteocytes, a critical coreceptor of the Wnt signaling pathway, and then initiates subsequent signaling cascades.191,194,195,196
Metabolic diseases such as diabetes, obesity, and osteoporosis are usually accompanied by impaired bone formation and low bone mass caused by elevated Scl levels.197,198 During this state of metabolic disorder, Scl can not only act on bone tissue but also serve as an endocrine factor that functions in distant organs. Clinical studies by Daniele and Yu et al. found that Scl was associated with fasting insulin levels and insulin resistance in patients with type 2 diabetes.199,200 In addition, some other studies in mice with a SOST gene knockout background suggested a decrease in fat content and an increase in insulin sensitivity,201 while an increase in Scl levels could promote the formation of beige adipose tissue.202 These results indicate that the increase in Scl may not be due to the development of metabolic diseases but may be one of the pathological factors promoting the occurrence of these conditions. However, the specific mechanisms, including genetic drivers behind these findings, are still obscure. The potential role of Scl in whole-body energy metabolism is to combine with Wnt/β-Catenin, which is extensively expressed in the body.
Neuropeptide Y
Neuropeptide Y (NPY), one of the most abundant neuropeptides in the brain, is a peptide composed of 36 amino acids.203,204 In the central nervous system, NPY was initially found to be a robust appetite-stimulating neuropeptide released by agouti-related protein neurons to regulate appetite and energy balance.205,206,207,208,209 With a greater understanding of this peptide, it was gradually proven that NPY also had central regulatory effects on circadian rhythm, the cardiovascular system, stress, and anxiety.205,210 There are five kinds of NPY receptors (Y1, Y2, Y4, Y5, and Y6) in mammals, which are widely distributed in the central nervous system.211,212 An in-depth study of the NPY receptor found its expression in peripheral tissues, including adipose tissue, pancreas, and bone,213,214,215 and the peripheral effect of NPY has received considerable attention. For example, in the pancreas, the activation of the NPY receptor can reduce β-cell apoptosis and hyperglycemia.216 The role of NPY in adipose tissue is to promote adipocyte proliferation and adipogenesis.214 This suggests that in addition to its secretion in the brain, NPY secreted by peripheral tissues also has profound regulatory functions in the endocrine system.217,218
In bone, NPY is secreted by osteoblasts and osteocytes, where it acts on bone tissues through autocrine and paracrine functions.219 NPY is generally considered to have no direct effect on bone formation but rescues bone loss in high-fat diet-fed mice.220 However, another in vitro study showed that NPY might have proliferative and antiapoptotic effects on BMSCs.221 Apart from being secreted from bone, NPY and its receptor are both expressed in many peripheral tissues and play a role in regulating metabolism. A recent study showed that mice lacking NPY had increased fat cells.222 However, due to the limited research relating to site-specific knockout of NPY, the specific source of NPY has not been clearly studied.218 A study on NPY receptors found that specific knockout of the Y1 receptor in the early osteoblast lineage of mice can lead to increased fasting blood glucose levels and decreased glucose tolerance, which were caused by a decrease in both islet cells and insulin secretion.223 Since there is no evidence of NPY binding directly to islet cells, it is possible that NPY might influence the function of osteoblasts, resulting in changes in the metabolism of distant organs. However, the specific mechanism is still unclear.
PTHrP
Parathyroid hormone-related protein (PTHrP) can be produced by immature chondrocytes and feeds back to promote proliferation and suppress differentiation of chondrocytes. PTHrP can bind to either hypertrophic chondrocytes or hypertrophic mast cells, slowing their proliferation or differentiation.224,225,226 In addition, mice with osteoblast-specific targeted disruption of PTHrP showed decreased expression of PTHrP in bone, indicating that osteoblasts are one of the sources of PTHrP protein.227 A robust increase in PTHrP levels can be detected in blood circulation after enhancing the expression of PTHrP in osteoblasts, which indicates that bone-derived PTHrP can be released into the circulation.228 This provides a basis for its function of regulating global metabolism.
It was found that the increase in PTHrP gene expression in mouse osteoblasts could be secreted into the blood circulation, improve heat production and glucose tolerance, and decrease fasting blood glucose levels. However, the glucose level in mice was not related to the secretion of insulin and osteocalcin but to lipid metabolism. Further studies have revealed that PTHrP derived from osteoblasts could stimulate the secretion of adiponectin in WAT through the PKA/CAMP and Akt/Fox signaling pathways. As a result, “browning” of WAT and fatty acid oxidation were activated, and the process further reduced glucose production.228 Whether bone-derived PTHrP can directly affect β-cells remains to be further studied. Nevertheless, a recent study only found that PTHrP and its receptor are expressed in β-cells, and the overexpression of PTHrP increased the expression of the G1/S cell cycle activator CDK2 and cyclin E, thus promoting the proliferation of islet cells.229 The mechanisms by which PTHrP regulates energy metabolism in individuals warrant further exploration. Since PTHrP has been proven to be a safe drug for osteoporosis treatment,230 it may have great prospects in treating obesity and diabetes.
Factors secreted by BMSCs
BMSCs are multipotent stem cells with the potential to differentiate into many different types of cells. Moreover, BMSCs are a crucial part of the bone marrow microenvironment that regulates hematopoiesis. Metabolic disorders such as obesity and diabetes mellitus are prone to inducing adipogenic rather than osteogenic differentiation of BMSCs, which contributes to osteoporosis and bone fracture in patients.231 BMSCs have been used in cell transplantation therapy to treat metabolic diseases. In animal experiments and clinical trials, BMSC transplantation promoted the proliferation of islet cells and reduced insulin resistance.232,233 On the other hand, BMSCs can secrete some active substances and affect bone metabolism and energy metabolism throughout the body.234,235,236
Exosomes
Exosomes are extracellular vesicles (EVs), and researchers have focused on structural engineering approaches in recent years to drive regenerative concepts. The diameter of exosomes is between 40 nm and 150 nm, and they contain proteins, RNA, cytokines, etc. Exosomes are released by almost all cell types, including stem cells.237 Exosomes can enter the blood circulation where they mediate communication between cells, and they have a broad regulatory effect on the body.238,239,240,241
In bone tissue, exosomes are secreted by almost all kinds of cells.242,243,244,245 The components of vesicles are rich in variety, including NF-κB, RANKL, ephrinA2, semaphorin 4D, miR-146a, and miR-214-3p. These exosomes mediate the regulation of different kinds of cells and their functions in bone. For example, osteoclasts can release exosomes containing RANK to suppress osteoclastogenesis.246 Conversely, exosomes containing miR-214 are secreted by osteoblasts and inhibit their function.244,247,248 Recently, new studies have found that the expression of RNA in exosomes secreted by BMSCs is similar to that of exosomes produced by stem cell-derived adipocytes.249 This suggests that the two kinds of exosomes may share similar abilities to regulate whole-body energy metabolism.
Furthermore, Su et al.250 confirmed that BMSCs can exert an indispensable function in the pancreas, liver, and other metabolism-related organs through exosomes. They found that exosomes containing miR-29b-3p secreted by BMSCs increased significantly with age. In parallel, an in vitro study showed that these exosomes can induce aging-related insulin resistance and inhibit insulin signaling activation in 3T3-L1 adipocytes, C2C12 cardiomyocytes, and primary cultured hepatocytes. It is well known that the SIRT1 gene benefits insulin resistance and diabetic status. Further studies showed that the SIRT1 gene serves as the downstream target of miR-29b-3p regulating insulin sensitivity, and miR-2 9b-3p can directly conjugate to the SIRT1 gene and suppress its expression.250 In addition, because exosomes contain immunoregulatory factors, they also play a role in repairing liver fibrosis, including reducing collagen accumulation, enhancing liver function, inhibiting inflammation, and promoting liver cell regeneration.251 Cytologically, exosomes contain various bioactive substances, which make them attractive potential therapeutic sources. An in vivo experiment demonstrated that the injection of BMSC-derived exosomes into a diabetic mouse model could improve glucose tolerance by suppressing TGF-β/Smad3 signaling via exosomal miRNA, suggesting an important role of exosomes in the treatment of metabolic diseases.252 In another study, a mouse model of liver fibrosis was treated with exosomes derived from human BMSCs. The regeneration of hepatocytes was restored, and the expression of PPARγ, Wnt3a, Wnt10b, β-Catenin, WISP1, cyclin D1, α-SMA, and collagen I was inhibited.251 Furthermore, mitigation of age-related insulin intolerance was observed by injecting miR-29b-3p inhibitor into the bone marrow cavity of aged mice. Exosome contents can also be used as targets for the treatment of diseases250 (Fig. 3).

Exosomes and inflammatory factors are mainly secreted by BMSCs. After forming vesicles containing various active substances, exosomes are released into the extracellular matrix. The inflammatory factors released by BMSCs can be divided into proinflammatory factors and anti-inflammatory factors. Exosomes and inflammatory factors can regulate distant target organs, thus leading to an inflammatory response, obesity, insulin resistance, etc
Inflammatory cytokines
Chronic low-grade inflammation is associated with the pathogenesis of metabolic syndrome.253 It is characterized by abnormal cytokine production, increased acute-phase reactants and other mediators, and activation of inflammatory signaling pathway networks.254,255 For example, the activation of inflammation in cells of various organs and tissues can activate inflammatory signaling pathways, including NF-κB and JNK, which interfere with IR signal transduction and lead to insulin resistance.256 Inflammatory factors can be divided into proinflammatory factors and anti-inflammatory factors.257 Bone can regulate energy metabolism by secreting various inflammatory factors.
IL-6, TGF-α, and IL-1β are common proinflammatory factors that can be expressed by adipocytes.258 Inflammatory factors secreted by bone can regulate its growth.259 It has been determined that IL-6 secreted by osteoblasts can regulate bone resorption.260 However, BMSCs seem to be more capable of producing inflammatory factors than osteoblasts.253,261 Many inflammatory factors, such as IL-6, macrophage inflammatory protein-1α (MIP-1α), granulocyte colony stimulating factor and granulocyte macrophage colony stimulating factor, were demonstrated to be secreted by BMSCs.261 The higher levels of the chemokines CXCL1 and CXCL2 are involved in osteoclastogenesis and are associated with bone marrow adipocytes.253,262 Additionally, microarray analysis showed that inflammatory genes such as IL-6 and TNF-α were highly expressed by bone marrow adipocytes.263 However, serum levels of these bone-derived proinflammatory factors are usually low, and few studies target systemic effects. Researchers only found that human bone marrow adipocytes can secrete trace amounts of IL-1β and TNF-α but large quantities of IL-6 during an in vitro study. This implies that bone marrow adipocytes may participate in systemic fat metabolism and the inflammatory response.
IL-10 is a type II cytokine with anti-inflammatory properties.264 The anti-inflammatory effect requires its binding with the receptor complexes IL-10Rα and IL-10Rβ, which trigger the activation of signal transducer and activator of transcription 3 (STAT3).265,266 IL-10 is also expressed by bone marrow cells, and it can be attached to the receptor IL-10Rα on adipocytes. Compared to bone marrow cell-specific IL-10 knockout mice, global IL-10 gene knockout mice have greater WAT content, higher blood sugar levels, and lower glucose tolerance. We can rationally infer that bone marrow-derived IL-10 is a crucial factor affecting the browning of fat and insulin sensitivity.267 However, the role of BMSC-derived inflammatory factors in energy metabolism remains largely uncertain, which provides a potential field for exploration (Fig. 3).268
Factor secreted by MAT
Bone MAT accounts for more than 10% of the total human adipose tissue mass.269,270 Adipose tissue in bone marrow can be divided into two main categories. One category is regulated MAT (rMAT) in red bone marrow, which is mainly distributed in areas with activated hematopoietic function. The other is constitutive MAT (cMAT) in the yellow bone marrow, which is concentrated in the distal skeletal region. cMAT contains more unsaturated lipids than rMAT and has a similar structure to WAT.271,272 The structural difference between cMAT and rMAT leads to distinct functions. It has been found that MAT in vertebrae of mammals has BAT-like thermogenic characteristics, whereas it often exhibits a WAT-like phenotype in the tibia.273 Although studies on the formation and transformation of these two kinds of bone marrow adipocytes and their respective endocrine functions are still insufficient, MAT can indeed secrete various regulatory factors to exert its function.270 Among them, RANKL from MAT was recently found to be related to energy metabolism, which has been described previously in our review. This suggests that the role of MAT in bone and systemic energy metabolism cannot be underestimated.
Adiponectin
Adiponectin is abundant in serum.274 It was initially thought to be secreted by WAT,275 but later, a large amount of adiponectin expression was also observed in MAT. In fact, the expression of adiponectin in MAT was higher than that in WAT.276 Cawthorn et al. confirmed that under calorie restriction, the increased level of serum adiponectin is directly related to the volume of MAT but not WAT.269 These findings illustrate that MAT might offer alternative supplementary energy for patients with insufficient WAT.
Adiponectin produces a remarkable effect in regulating adipose metabolism, insulin secretion, and other metabolic pathways. The regulatory effect of adiponectin is achieved by acting on receptors 1 and 2 (AdipoR1 and AdipoR2), which are expressed in various tissues and organs, including skeletal muscle, liver, and pancreas.277,278,279,280,281,282 Adiponectin can activate PPARα, AMPK, and p38 MAPK by interacting with AdipoR,279,283 thereby regulating glycolipid metabolism. Additionally, adiponectin can even act on the central nervous system by crossing the blood–brain barrier. The increase in adiponectin in the ventricle can lead to weight loss and an increase in energy consumption, which may be mediated by overexpression of corticotropin-releasing hormone in the hypothalamus.284
The effect of adiponectin secreted by MAT on systemic metabolism has not been completely determined. The function of adiponectin secreted by MAT has only been proven in the metabolism of skeletal muscle cells. It can stimulate Ca2+ influx and liver kinase B1 activation, contributing to enhanced AMPK activity, PPARγ coactivator-1α expression, and mitochondrial biosynthesis.269 It is worth noting that adiponectin is highly expressed in MAT and can be secreted into the circulation. Therefore, adiponectin secreted by MAT may impact the metabolism of distal organs. In addition, insulin can inhibit the gene expression of adiponectin in human MAT, suggesting that MAT may be affected in hyperinsulinemia.285
The transcription and translation of adiponectin found in human osteoblasts was low.286 Mature osteoblasts collected from the tibia and femur were quantitatively analyzed by real-time polymerase chain reaction. The results show that it accounts for only 3% of the adiponectin level in human subcutaneous adipose tissue.286 Therefore, the role of adiponectin derived from bone tissue cells in energy metabolism is still under debate. (Fig. 4).

MAT can secrete RANKL, the ligand of RANK. RANKL regulates metabolism by binding with RANK on the cell membrane. In addition, MAT can also secrete the adipokines leptin and adiponectin. Leptin and adiponectin combine with ADIPOR and OBR on the cell membrane of peripheral tissue, regulating cellular sugar metabolism. They also act on the central nervous system, leading to reduced food intake and energy consumption
Leptin
Leptin, which is encoded by the obesity gene (Ob) and secreted primarily by WAT, is directly related to obesity. Mutations of the leptin gene lead to obesity.287 Most tissues express leptin receptor (ObR), among which the concentration of leptin receptor in the hypothalamic arcuate nucleus, lung, liver, spleen, kidney, and adrenal gland is relatively high.288 Leptin conjugates to ObR, thus triggering cascade reactions of the STAT, PI3K, and MAPK signaling pathways.289 For example, leptin can act directly on islet cells and inhibit insulin secretion.290,291,292 It can also regulate adipocytes and alter cell sensitivity to insulin293 or work on the feeding center of the brain, thereby inhibiting food intake and reducing fat content. At present, the regulatory mechanism of leptin on energy metabolism has been clearly studied.
It was recently discovered that MAT also expresses leptin.294 Rosiglitazone treatment can promote browning of adipose tissue in bone marrow and increase leptin expression.295 An in vitro study illustrated that the gene expression of leptin could be significantly inhibited by proinflammatory cytokines, hematopoietic cytokines, IL-1β, IL-6, TNF-α, and IFN-γ in human bone marrow adipocytes.296,297 Therefore, in the state of systemic inflammation, such as obesity, inflammatory factors can affect the endocrine function of MAT, leading to further deterioration of the inflammatory response. Additionally, leptin expressed in bone MAT can directly act on leptin receptors in bone and affect bone growth by activating FGF23 and regulating the secretion of osteocalcin.298 However, there is no clear evidence that leptin from MAT can regulate global metabolism (Fig. 4).
Circulating endocrine factors affect both bone and energy metabolism
There is a complex relationship between bone and energy metabolism. The main action of circulating endocrine factors is not limited to regulating energy metabolism but affects bone metabolism and remodeling.
Insulin
Insulin is an important hormone regulating energy metabolism in the human body, and its receptor exists in almost all cells. Insulin promotes glycogen, fat, and protein synthesis by binding to IRs and ultimately lowers blood sugar.299 In the skeleton, insulin can activate insulin signaling pathways in osteoblasts and osteoclasts by binding to IR and IRS.300 In osteoblasts, it can inhibit Twist2, a Runx2 inhibitor, and thus promote the differentiation of osteoblasts required for normal bone formation.301 Fulzele et al. demonstrated in mice that when osteoblasts lack IR, osteoporosis can easily develop after birth.302 In osteoclasts, insulin can upregulate the expression of RANK, increase the binding of RANK and RANKL, and ultimately contribute to the production of osteoclasts.303 In addition, IRS-1 deficiency inhibits the proliferation of chondrocytes through the PI3K/Akt pathway, which is an adverse factor for bone healing.304 Clinical observations have shown that excessive insulin seems to reduce bone turnover and increase bone mineral density. However, it may also lead to bone fragility by increasing cortical osteoporosis or other bone structural defects.305
The regulation of bone by insulin can affect whole-body energy metabolism. Insulin can promote osteoblast differentiation and glucose uptake by increasing the expression of Glut4 on the cell membrane. The absence of this pathway will lead to peripheral insulin resistance, which is mainly manifested by an increase in systemic insulin levels and a decrease in insulin sensitivity. However, no significant phenotypes were found in skeletal muscle cells and adipocytes. Therefore, the decrease in insulin absorption by bone may lead to systemic insulin resistance, which proves the influence of insulin and bone on whole-body energy metabolism from another aspect.306 On the other hand, insulin resistance in osteoblasts leads to decreased circulating osteocalcin levels, reducing insulin secretion and insulin sensitivity in skeletal muscle, thus leading to systemic glucose intolerance.307,308
Vitamin D
Vitamin D is a steroid hormone that plays a vital role in maintaining bone metabolism and calcium homeostasis.309 As an active form of vitamin D, 1,25(OH)2D3 can promote calcium absorption310 and regulate calcium reabsorption311 through conjugation to the VDR. On the other hand, when the serum calcium level declines, VDR in osteoblasts can enhance bone resorption, thereby mobilizing bone calcium into the blood to maintain serum calcium homeostasis.312 In recent years, it has been gradually found that vitamin D has other physiological effects. The relationship between vitamin D and metabolic diseases such as obesity, diabetes, and NAFLD is a trend in research. Adipocytes are the main storage sites of vitamin D and express active vitamin D and VDR.313,314 Meanwhile, vitamin D can regulate adipose formation from multiple perspectives, and its effect depends on the level of vitamin D, the type of adipocytes and the differentiation level.315,316,317,318 It was also found that patients with NAFLD were more likely to suffer from decreased bone mineral density and vitamin D deficiency. Vitamin D supplementation can reduce the risk of elevated blood glucose and insulin resistance.319,320 This suggests that bone may be a bridge for vitamin D to regulate energy metabolism.
Parathyroid hormone
Parathyroid hormone (PTH) can directly affect bone and kidney to regulate calcium and phosphorus metabolism. One of the key mechanisms of PTH in regulating calcium homeostasis is stimulating bone remodeling. It not only promotes bone resorption and mobilizes bone calcium into blood but also promotes osteogenesis and ameliorates osteoporosis. PTH1R is expressed on the surface of osteoblasts and many bone cells.84 The catabolic function of PTH is mainly achieved indirectly by acting on osteoclasts through the OPG-RANKL-RANK pathway.321 In contrast, the anabolic effect is conducted directly by working on PTH1R in osteoblasts and osteoclasts, further promoting the differentiation of precursor cells322,323 and inhibiting the apoptosis of mature cells and the expression of Scl.324,325,326 PTH can also enhance aerobic glycolysis in osteoblasts by promoting the transduction of the insulin-like growth factor signaling pathway, thereby enhancing the synthesis function of bone in mice.327
In addition to regulating systemic calcium and phosphorus metabolism, PTH also has some effects on energy metabolism, which may be related to the level of vitamin D. It was found that the level of blood sugar increased significantly after parathyroidectomy and decreased after PTH administration, while the secretion of insulin remained unchanged.328 Kimura et al. also demonstrated that PTH could reduce the blood sugar level in obese type 2 diabetic rats without changing the serum insulin level.329 However, PTH can affect insulin sensitivity. One piece of evidence is that the insulin sensitivity index is negatively correlated with plasma PTH levels.330 In addition, excessive PTH leads to disorders in lipid metabolism. PTH induces lipolysis of adipocytes by activating the cAMP-PKA pathway, leading to increased serum cholesterol and triglyceride concentrations in mice.331,332,333
Estrogen
In addition to reproductive function, estrogen also regulates bone metabolism and energy metabolism. Lack of estrogen increases the risk of metabolic syndromes such as obesity and type 2 diabetes mellitus.334,335 The main estrogen receptors are ERα and ERβ,336 which are distributed in multiple tissues of the body. In the central nervous system, activation of ERs in the ventromedial hypothalamus and arcuate nucleus can control dietary intake.337,338,339 In the periphery, estrogen can act on ERs to increase insulin sensitivity in adipose tissue, skeletal muscle, and liver.340,341,342 For example, estrogen can inhibit adipogenesis341 and reduce the expression of lipoprotein lipase, an important regulator of lipoprotein metabolism.340 Moreover, an experiment showed that ER agonist therapy increased the expression of GLUT4 and glucose uptake in rat skeletal muscle.342 The role of estrogen in bone has also been affirmed. Postmenopausal estrogen deficiency can lead to osteoporosis, which is due to estrogen inducing osteoclast apoptosis through ERs and protecting bone through the inflammatory mediators RANKL and Scl.343,344,345,346
Prospects
It has been known for a long time that skeletal health is closely related to the overall metabolism of the individual. For example, people with diabetes have a greater risk of developing osteoporosis than normal individuals,347,348 and a low body mass index causes bone loss.349,350 The presence of insulin, adiponectin and leptin-related receptors in bone proves that the energy metabolism of the whole body has a direct impact on bone.307,351,352 However, bone tissue’s constant turnover suggests active energy generation and consumption in osteogenesis and bone resorption, which will inevitably affect whole-body energy metabolism. Later, through the discovery of osteocalcin, the first bone-derived factor that regulates energy metabolism, researchers found that bone could also regulate energy metabolism throughout the body. Some findings showed that bone-derived factors, such as Lcn2 secreted by osteoblasts and adiponectin and leptin secreted by MAT, could cross the blood–brain barrier and act on the central nervous system, which is one of the discoveries in recent years. Further study on bone-derived factors and their interactions will help to understand the fundamentals of the central nervous system for energy balance. Bone-derived exosomes are also a new research area in recent years and have been found to regulate bone itself and other tissues.245 However, due to the universality of exosomes and the diversity of their contents, more functions of bone-derived exosomes need to be explored.
The effect of bone-derived factors on the regulation of energy metabolism-related organs is complicated. This may be due to the different distributions and proportions of receptors on the surfaces of other cells so that these factors have more than one regulatory pathway. Moreover, sometimes the results of experiments on animals and cell cultures in vitro may be different from those obtained from human subjects, indicating that the human body has a more complex internal environment, which impacts the results. Bone-derived factors may be affected by a variety of factors in the processes involved in secretion, circulation and function, including genetic background diversity, age and health status of the human body. For example, decarboxylation of OCN is vitamin K dependent, so the vitamin K concentration in the human body will affect the physiological function of osteocalcin.
One of the key points in studying the regulation of bone-derived factors on energy metabolism is to confirm whether the change in the levels of those factors is the cause but not the result of altered energy metabolism. Because of the close relationship between bone metabolism and whole-body energy metabolism, the levels of factors secreted by bone are likely to be determinants of energy homeostasis and metabolism. Second, it is of interest to determine whether those factors that play a crucial role in other tissues and organs are from bone. This could be easily determined for some factors, such as osteocalcin and FGF23, which are mostly secreted by bone cells. For other factors expressed in multiple tissues and organs, most of them need to be assessed through genetic engineering techniques by using animal models (Table 2). However, at present, not all bone-derived factors may possess the necessary gene-specific knockout animal models applicable to energy metabolism regulation experiments, such as BMP and OPN. Another reason is that although some autocrine factors in bone cannot be secreted into the blood circulation to act on other organs, they can indirectly affect systemic metabolism by regulating bone metabolism, which should not be ignored.
In view of the complex regulation of whole-body energy metabolism by bone, the clinical application of bone-derived factors will become one of the future research directions. For example, studying the role of bone-derived factors in aging, which is a severe problem facing society, may explain some diseases. In the process of human aging, bone metabolism and whole-body metabolism are continually changing. Aging may lead to metabolic disorders and affect bone metabolism. Macroscopically, the aging of bone is characterized by osteoporosis and a decrease in organic and inorganic components. At the same time, its significance is marked by a decrease in bone cells and the transformation of BMSCs into MAT and osteoblasts.353,354 Bone-derived factors from these cells are implicated in changes such as a decrease in glucose tolerance, the occurrence of insulin resistance, and the enhancement of the inflammatory response and may be caused by an imbalance in the levels of bone-derived factors, which leads to the occurrence of metabolic diseases. Bone is an important motor organ, so exercise can have a significant impact on bone metabolism.355 Not only bone but also exercise has been found to change the human body’s metabolic state and delay aging.356,357 One of the possible mechanisms is by affecting bone metabolism and the secretion of bone-derived factors. Therefore, exercise therapy may be used to treat or prevent metabolic diseases, especially those related to aging, in the future (Fig. 5).

The network relationship between bone and whole-body energy metabolism. Aging is one of the main causes of bone and whole-body metabolism disorder, and exercise training can alleviate it. Changes in bone metabolism will affect multiple organs and tissues, including the brain (a potential research direction in the future), and lead to metabolic diseases. Metabolic diseases, as well as changes in circulating endocrine factors, can in turn affect bone. On the other hand, bone-derived factors can also be used as autocrine factors to regulate their own metabolic state
The next question is whether we can use bone-derived factors as a marker for the diagnosis or as a new target for the treatment of metabolic diseases and their related complications. This conjecture has been verified by some research. For example, OPG has been found to be a potential marker for the diagnosis of diabetes in postmenopausal women,358 and experiments have explored whether regulating the proportion of OPG-RANKL-RANK signaling can interfere with the process of cardiovascular complications noted in patients with diabetes.359 More experiments are needed to further apply bone-derived factors to the treatment of clinical diseases.
References
Sommerfeldt, D. W. & Rubin, C. T. Biology of bone and how it orchestrates the form and function of the skeleton. Eur. Spine J. 10, S86–S95 (2001).
Bronner, F. Extracellular and intracellular regulation of calcium homeostasis. Sci. World J. 1, 919–925 (2001).
Askmyr, M., Quach, J. & Purton, L. E. Effects of the bone marrow microenvironment on hematopoietic malignancy. Bone 48, 115–120 (2011).
Abarrategi, A. et al. Modeling the human bone marrow niche in mice: from host bone marrow engraftment to bioengineering approaches. J. Exp. Med. 215, 729–743 (2018).
Kajimura, D. et al. Adiponectin regulates bone mass via opposite central and peripheral mechanisms through FoxO1. Cell Metab. 17, 901–915 (2013).
Karsenty, G. Convergence between bone and energy homeostases: leptin regulation of bone mass. Cell Metab. 4, 341–348 (2006).
Lecka-Czernik, B. Diabetes, bone and glucose-lowering agents: basic biology. Diabetologia 60, 1163–1169 (2017).
Andrukhova, O., Streicher, C., Zeitz, U. & Erben, R. G. Fgf23 and parathyroid hormone signaling interact in kidney and bone. Mol. Cell Endocrinol. 436, 224–239 (2016).
Cai, X., Xing, J., Long, C. L., Peng, Q. & Humphrey, M. B. DOK3 modulates bone remodeling by negatively regulating osteoclastogenesis and positively regulating osteoblastogenesis. J. Bone Min. Res. 32, 2207–2218 (2017).
Kalbasi Anaraki, P. et al. Urokinase receptor mediates osteoclastogenesis via M-CSF release from osteoblasts and the c-Fms/PI3K/Akt/NF-κB pathway in osteoclasts. J. Bone Min. Res. 30, 379–388 (2015).
Matsuoka, K., Park, K. A., Ito, M., Ikeda, K. & Takeshita, S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J. Bone Min. Res. 29, 1522–1530 (2014).
Shimada, T. et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Min. Res. 19, 429–435 (2004).
Gupte, A. A. et al. Osteocalcin protects against nonalcoholic steatohepatitis in a mouse model of metabolic syndrome. Endocrinology 155, 4697–4705 (2014).
Du, J. et al. Osteocalcin improves nonalcoholic fatty liver disease in mice through activation of Nrf2 and inhibition of JNK. Endocrine 53, 701–709 (2016).
Mizokami, A. et al. Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PLoS ONE 8, e57375 (2013).
Otani, T. et al. Signaling pathway for adiponectin expression in adipocytes by osteocalcin. Cell Signal 27, 532–544 (2015).
Booth, S. L., Centi, A., Smith, S. R. & Gundberg, C. The role of osteocalcin in human glucose metabolism: marker or mediator? Nat. Rev. Endocrinol. 9, 43–55 (2013).
Hauschka, P. V., Lian, J. B., Cole, D. E. & Gundberg, C. M. Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol. Rev. 69, 990–1047 (1989).
Ducy, P. et al. Increased bone formation in osteocalcin-deficient mice. Nature 382, 448–452 (1996).
Ishida, M. & Amano, S. Osteocalcin fragment in bone matrix enhances osteoclast maturation at a late stage of osteoclast differentiation. J. Bone Min. Metab. 22, 415–429 (2004).
Nikel, O., Poundarik, A. A., Bailey, S. & Vashishth, D. Structural role of osteocalcin and osteopontin in energy dissipation in bone. J. Biomech. 80, 45–52 (2018).
Hu, C. M. et al. High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metab. 29, 1334–1349.e10 (2019).
Lee, N. K. et al. Endocrine regulation of energy metabolism by the skeleton. Cell 130, 456–469 (2007).
Malashkevich, V. N., Almo, S. C. & Dowd, T. L. X-ray crystal structure of bovine 3 Glu-osteocalcin. Biochemistry 52, 8387–8392 (2013).
Rached, M.-T. et al. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J. Clin. Investig. 120, 357–368 (2010).
Guedes, J. A. C., Esteves, J. V., Morais, M. R., Zorn, T. M. & Furuya, D. T. Osteocalcin improves insulin resistance and inflammation in obese mice: Participation of white adipose tissue and bone. Bone 115, 68–82 (2018).
Levinger, I. et al. The effects of muscle contraction and recombinant osteocalcin on insulin sensitivity ex vivo. Osteoporos. Int. 27, 653–663 (2016).
Lin, X. et al. Recombinant uncarboxylated osteocalcin per se enhances mouse skeletal muscle glucose uptake in both extensor digitorum longus and soleus muscles. Front Endocrinol. 8, 330 (2017).
Pi, M. et al. Evidence for osteocalcin binding and activation of GPRC6A in β-cells. Endocrinology 157, 1866–1880 (2016).
Sanchez-Gurmaches, J. et al. Brown Fat AKT2 is a cold-induced kinase that stimulates ChREBP-mediated de novo lipogenesis to optimize fuel storage and thermogenesis. Cell Metab. 27, 195–209.e6 (2018).
Gao, J. et al. The PLC/PKC/Ras/MEK/Kv channel pathway is involved in uncarboxylated osteocalcin-regulated insulin secretion in rats. Peptides 86, 72–79 (2016).
Drucker, D. J. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 27, 740–756 (2018).
Mizokami, A. et al. Oral administration of osteocalcin improves glucose utilization by stimulating glucagon-like peptide-1 secretion. Bone 69, 68–79 (2014).
Zhou, B. et al. Osteocalcin reverses endoplasmic reticulum stress and improves impaired insulin sensitivity secondary to diet-induced obesity through nuclear factor-κB signaling pathway. Endocrinology 154, 1055–1068 (2013).
Guo, Q. et al. Undercarboxylated osteocalcin reverts insulin resistance induced by endoplasmic reticulum stress in human umbilical vein endothelial cells. Sci. Rep. 7, 46 (2017).
Jung, C. H. et al. The preventive effect of uncarboxylated osteocalcin against free fatty acid-induced endothelial apoptosis through the activation of phosphatidylinositol 3-kinase/Akt signaling pathway. Metabolism 62, 1250–1257 (2013).
Kalucka, J. et al. Quiescent endothelial cells upregulate fatty acid β-oxidation for vasculoprotection via redox homeostasis. Cell Metab. 28, 881–894.e13 (2018).
Li, X., Sun, X. & Carmeliet, P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 30, 414–433 (2019).
Hill, H. S. et al. Carboxylated and uncarboxylated forms of osteocalcin directly modulate the glucose transport system and inflammation in adipocytes. Horm. Metab. Res. 46, 341–347 (2014).
Clemens, T. L. & Karsenty, G. The osteoblast: an insulin target cell controlling glucose homeostasis. J. Bone Min. Res. 26, 677–680 (2011).
De Toni, L. et al. Osteocalcin, a bone-derived hormone with important andrological implications. Andrology 5, 664–670 (2017).
Otani, T. et al. Osteocalcin triggers Fas/FasL-mediated necroptosis in adipocytes via activation of p300. Cell Death Dis. 9, 1194 (2018).
Li, Q. et al. T Cell factor 7 (TCF7)/TCF1 feedback controls osteocalcin signaling in brown adipocytes independent of the Wnt/β-catenin pathway. Mol. Cell Biol. 38, e00562–17 (2018).
Mottillo, E. P., Ramseyer, V. D. & Granneman, J. G. SERCA2b cycles its way to UCP1-independent thermogenesis in beige fat. Cell Metab. 27, 7–9 (2018).
Deppermann, C. et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J. Exp. Med. 217, e20190723 (2020).
Wellendorph, P. & Bräuner-Osborne, H. Molecular cloning, expression, and sequence analysis of GPRC6A, a novel family C G-protein-coupled receptor. Gene 335, 37–46 (2004).
Ackerman, S. D. et al. GPR56/ADGRG1 regulates development and maintenance of peripheral myelin. J. Exp. Med. 215, 941–961 (2018).
Pi, M., Wu, Y. & Quarles, L. D. GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J. Bone Min. Res. 26, 1680–1683 (2011).
Fu, A., Eberhard, C. E. & Screaton, R. A. Role of AMPK in pancreatic beta cell function. Mol. Cell Endocrinol. 366, 127–134 (2013).
Ardestani, A., Lupse, B., Kido, Y., Leibowitz, G. & Maedler, K. mTORC1 Signaling: A Double-Edged Sword in Diabetic β Cells. Cell Metab. 27, 314–331 (2018).
Karmaus, P. W. F. et al. Critical roles of mTORC1 signaling and metabolic reprogramming for M-CSF-mediated myelopoiesis. J. Exp. Med. 214, 2629–2647 (2017).
Lin, S. C. & Hardie, D. G. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 27, 299–313 (2018).
Pi, M., Nishimoto, S. K. & Quarles, L. D. GPRC6A: Jack of all metabolism (or master of none). Mol. Metab. 6, 185–193 (2016).
Diegel, C. R. et al. An osteocalcin-deficient mouse strain without endocrine abnormalities. PLoS Genet. 16, e1008361 (2020).
Choi, H. J. et al. Vitamin K2 supplementation improves insulin sensitivity via osteocalcin metabolism: a placebo-controlled trial. Diabetes Care 34, e147 (2011).
Pollock, N. K. et al. Lower uncarboxylated osteocalcin concentrations in children with prediabetes is associated with beta-cell function. J. Clin. Endocrinol. Metab. 96, E1092–E1099 (2011).
Choudhury, A. B., Sarkar, P. D., Sakalley, D. K. & Petkar, S. B. Role of adiponectin in mediating the association of osteocalcin with insulin resistance and type 2 diabetes: a cross sectional study in pre- and post-menopausal women. Arch. Physiol. Biochem. 120, 73–79 (2014).
Martin, T. J. & Sims, N. A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 16, 131–139 (2015).
Simonet, W. S. et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89, 309–319 (1997).
Boyce, B. F. & Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther. 9, S1 (2007).
Nakashima, T. et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 (2011).
Chen, C. et al. MiR-503 regulates osteoclastogenesis via targeting RANK. J. Bone Min. Res. 29, 338–347 (2014).
Kondegowda, N. G. et al. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-κB ligand pathway. Cell Metab. 22, 77–85 (2015).
Sacco, F. et al. Phosphoproteomics reveals the GSK3-PDX1 axis as a key pathogenic signaling node in diabetic islets. Cell Metab. 29, 1422–1432.e3 (2019).
Musialik, K., Szulińska, M., Hen, K., Skrypnik, D. & Bogdański, P. The relation between osteoprotegerin, inflammatory processes, and atherosclerosis in patients with metabolic syndrome. Eur. Rev. Med Pharm. Sci. 21, 4379–4385 (2017).
Monseu, M. et al. Osteoprotegerin levels are associated with liver fat and liver markers in dysmetabolic adults. Diabetes Metab. 42, 364–367 (2016).
Cappel, D. A. et al. Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining TCA cycle and redox metabolism in liver. Cell Metab. 29, 1291–1305.e8 (2019).
Bilgir, O. et al. Relationship between insulin resistance, hs-CRP, and body fat and serum osteoprotegerin/RANKL in prediabetic patients. Minerva Endocrinol. 43, 19–26 (2018).
Suliburska, J. et al. The association of insulin resistance with serum osteoprotegerin in obese adolescents. J. Physiol. Biochem 69, 847–853 (2013).
Stekovic, S. et al. Alternate day fasting improves physiological and molecular markers of aging in healthy, non-obese humans. Cell Metab. 30, 462–476.e6 (2019).
Ou, D. et al. TNF-related apoptosis-inducing ligand death pathway-mediated human beta-cell destruction. Diabetologia 45, 1678–1688 (2002).
Chamoux, E., Houde, N., L’Eriger, K. & Roux, S. Osteoprotegerin decreases human osteoclast apoptosis by inhibiting the TRAIL pathway. J. Cell Physiol. 216, 536–542 (2008).
Vaccarezza, M., Bortul, R., Fadda, R. & Zweyer, M. Increased OPG expression and impaired OPG/TRAIL ratio in the aorta of diabetic rats. Med. Chem. 3, 387–391 (2007).
Knudsen, J. G. & Rorsman, P. β cell dysfunction in type 2 diabetes: drained of energy? Cell Metab. 29, 1–2 (2019).
Schrader, J. et al. Cytokine-induced osteoprotegerin expression protects pancreatic beta cells through p38 mitogen-activated protein kinase signalling against cell death. Diabetologia 50, 1243–1247 (2007).
Taylor, R. et al. Remission of human type 2 diabetes requires decrease in liver and pancreas fat content but is dependent upon capacity for β cell recovery. Cell Metab. 28, 547–556.e3 (2018).
Lacombe, J., Karsenty, G. & Ferron, M. In vivo analysis of the contribution of bone resorption to the control of glucose metabolism in mice. Mol. Metab. 2, 498–504 (2013).
Niu, Y. et al. Plasma osteoprotegerin levels are inversely associated with nonalcoholic fatty liver disease in patients with type 2 diabetes: a case-control study in China. Metabolism 65, 475–481 (2016).
Samuel, V. T. & Shulman, G. I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 27, 22–41 (2018).
Ayaz, T. et al. The relation between carotid intima media thickness and serum osteoprotegerin levels in nonalcoholic fatty liver disease. Metab. Syndr. Relat. Disord. 12, 283–289 (2014).
D’Amelio, P., Isaia, G., Fau, -, Isaia, G. C. & Isaia, G. C. The osteoprotegerin/RANK/RANKL system: a bone key to vascular disease. Expert Rev. Cardiovasc Ther. 4, 801–811 (2006).
Kiechl, S. et al. Blockade of receptor activator of nuclear factor-κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat. Med. 19, 358–363 (2013).
Lacey, D. L. et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176 (1998).
Fan, Y. et al. Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab. 25, 661–672 (2017).
Franzén, A. & Heinegård, D. Isolation and characterization of two sialoproteins present only in bone calcified matrix. Biochem. J. 232, 715–724 (1985).
Gimba, E. R. & Tilli, T. M. Human osteopontin splicing isoforms: known roles, potential clinical applications and activated signaling pathways. Cancer Lett. 331, 11–17 (2013).
Luukkonen, J. et al. Osteoclasts secrete osteopontin into resorption lacunae during bone resorption. Histochem. Cell Biol. 151, 475–487 (2019).
Oldberg, A., Franzén, A. & Heinegård, D. Cloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence. Proc. Natl Acad. Sci. USA 83, 8819–8823 (1986).
Wang, K. X. & Denhardt, D. T. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 19, 333–345 (2008).
Ge, Q. et al. Osteopontin regulates macrophage activation and osteoclast formation in hypertensive patients with vascular calcification. Sci. Rep. 7, 40253 (2017).
Ishijima, M. et al. Enhancement of osteoclastic bone resorption and suppression of osteoblastic bone formation in response to reduced mechanical stress do not occur in the absence of osteopontin. J. Exp. Med. 193, 399–404 (2001).
Chen, Q. et al. An osteopontin-integrin interaction plays a critical role in directing adipogenesis and osteogenesis by mesenchymal. Stem Cells 32, 327–337 (2014).
Chapman, J. et al. Osteopontin is required for the early onset of high fat diet-induced insulin resistance in mice. PLoS ONE 5, e13959 (2010).
Inoue, H. et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat. Med. 10, 168–174 (2004).
Kiefer, F. W. et al. Neutralization of osteopontin inhibits obesity-induced inflammation and insulin resistance. Diabetes 59, 935–946 (2010).
Kon, S. et al. Syndecan-4 protects against osteopontin-mediated acute hepatic injury by masking functional domains of osteopontin. J. Exp. Med. 205, 25–33 (2008).
Nuñez-Garcia, M. et al. Osteopontin regulates the cross-talk between phosphatidylcholine and cholesterol metabolism in mouse liver. J. Lipid Res. 58, 1903–1915 (2017).
Zeyda, M. et al. Osteopontin is an activator of human adipose tissue macrophages and directly affects adipocyte function. Endocrinology 152, 2219–2227 (2011).
Arafat, H. A. et al. Osteopontin protects the islets and beta-cells from interleukin-1 beta-mediated cytotoxicity through negative feedback regulation of nitric oxide. Endocrinology 148, 575–584 (2007).
Ma, D. & Leulier, F. A new transkingdom dimension to NO signaling. Cell Metab. 29, 513–515 (2019).
Wendt, A. et al. Osteopontin affects insulin vesicle localization and Ca2+ homeostasis in pancreatic beta cells from female mice. PLoS ONE 12, e0170498 (2017).
Ahmad, R. et al. Interaction of osteopontin with IL-18 in obese individuals: implications for insulin resistance. PLoS ONE 8, e63944 (2013).
Barchetta, I. et al. Increased circulating osteopontin levels in adult patients with type 1 diabetes mellitus and association with dysmetabolic profile. Eur. J. Endocrinol. 174, 187–192 (2016).
Carbone, F. et al. Serum levels of osteopontin predict diabetes remission after bariatric surgery. Diabetes Metab. 45, 356–362 (2019).
Kiefer, F. W. et al. Osteopontin expression in human and murine obesity: extensive local up-regulation in adipose tissue but minimal systemic alterations. Endocrinology 149, 1350–1357 (2008).
Talat, M. A. et al. The role of osteopontin in the pathogenesis and complications of type 1 diabetes mellitus in children. J. Clin. Res. Pediatr. Endocrinol. 8, 399–404 (2016).
Lee, H. et al. Beta cell dedifferentiation induced by IRE1α deletion prevents type 1 diabetes. Cell Metab. 31, 822–836.e5 (2020).
Marciano, R. et al. Association of alleles at polymorphic sites in the Osteopontin encoding gene in young type 1 diabetic patients. Clin. Immunol. 131, 84–91 (2009).
Warshauer, J. T., Bluestone, J. A. & Anderson, M. S. New frontiers in the treatment of type 1 diabetes. Cell Metab. 31, 46–61 (2020).
You, J. S. et al. Serum osteopontin concentration is decreased by exercise-induced fat loss but is not correlated with body fat percentage in obese humans. Mol. Med. Rep. 8, 579–584 (2013).
Israel, D. I. et al. Heterodimeric bone morphogenetic proteins show enhanced activity in vitro and in vivo. Growth Factors 13, 291–300 (1996).
Urist, M. R. Bone: formation by autoinduction. Science 150, 893–899 (1965).
Celeste, A. J. et al. Identification of transforming growth factor beta family members present in bone-inductive protein purified from bovine bone. Proc. Natl Acad. Sci. USA 87, 9843–9847 (1990).
Brazil, D. P., Church, R. H., Surae, S., Godson, C. & Martin, F. BMP signalling: agony and antagony in the family. Trends Cell Biol. 25, 249–264 (2015).
Chen, D., Zhao, M. & Mundy, G. R. Bone morphogenetic proteins. Growth Factors 22, 233–241 (2004).
Nohno, T. et al. Identification of a human type II receptor for bone morphogenetic protein-4 that forms differential heteromeric complexes with bone morphogenetic protein type I receptors. J. Biol. Chem. 270, 22522–22526 (1995).
ten Dijke, P., Miyazono, K. & Heldin, C. H. Signaling via hetero-oligomeric complexes of type I and type II serine/threonine kinase receptors. Curr. Opin. Cell Biol. 8, 139–145 (1996).
Ebara, S. & Nakayama, K. Mechanism for the action of bone morphogenetic proteins and regulation of their activity. Spine 27, S10–S15 (2002).
Heldin, C. H., Miyazono, K. & ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 (1997).
Wang, P. et al. Combined inhibition of DYRK1A, SMAD, and trithorax pathways synergizes to induce robust replication in adult human beta cells. Cell Metab. 29, 638–652.e5 (2019).
Lu, Q. et al. GDF11 inhibits bone formation by activating Smad2/3 in bone marrow mesenchymal stem cells. Calcif. Tissue Int. 99, 500–509 (2016).
Guo, X. & Wang, X.-F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 19, 71–88 (2009).
Lowery, J. W. & Rosen, V. The BMP pathway and its inhibitors in the skeleton. Physiol. Rev. 98, 2431–2452 (2018).
Yang, M. et al. MiR-497∼195 cluster regulates angiogenesis during coupling with osteogenesis by maintaining endothelial Notch and HIF-1α activity. Nat. Commun. 8, 16003 (2017).
Maeda, Y., Tsuji, K., Nifuji, A. & Noda, M. Inhibitory helix-loop-helix transcription factors Id1/Id3 promote bone formation in vivo. J. Cell Biochem. 93, 337–344 (2004).
Peng, Y. et al. Inhibitor of DNA binding/differentiation helix-loop-helix proteins mediate bone morphogenetic protein-induced osteoblast differentiation of mesenchymal stem cells. J. Biol. Chem. 279, 32941–32949 (2004).
Elsen, M. et al. BMP4 and BMP7 induce the white-to-brown transition of primary human adipose stem cells. Am. J. Physiol. Cell Physiol. 306, C431–C440 (2014).
Fabbiano, S. et al. Caloric restriction leads to browning of white adipose tissue through type 2 immune signaling. Cell Metab. 24, 434–446 (2016).
Gustafson, B. et al. BMP4 and BMP antagonists regulate human white and beige adipogenesis. Diabetes 64, 1670–1681 (2015).
Hata, K. et al. Differential roles of Smad1 and p38 kinase in regulation of peroxisome proliferator-activating receptor gamma during bone morphogenetic protein 2-induced adipogenesis. Mol. Biol. Cell 14, 545–555 (2003).
Hino, J. et al. Overexpression of bone morphogenetic protein-3b (BMP-3b) in adipose tissues protects against high-fat diet-induced obesity. Int J. Obes. 41, 483–488 (2017).
Hoffmann, J. M. et al. BMP4 gene therapy in mature mice reduces BAT activation but protects from obesity by browning subcutaneous adipose tissue. Cell Rep. 20, 1038–1049 (2017).
Huang, H. et al. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl Acad. Sci. USA 106, 12670–12675 (2009).
Kim, S., Choe, S. & Lee, D. K. BMP-9 enhances fibroblast growth factor 21 expression and suppresses obesity. Biochim. Biophys. Acta 1862, 1237–1246 (2016).
Tseng, Y. H. et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454, 1000–1004 (2008).
Wang, E. A., Israel, D. I., Kelly, S. & Luxenberg, D. P. Bone morphogenetic protein-2 causes commitment and differentiation in C3H10T1/2 and 3T3 cells. Growth Factors 9, 57–71 (1993).
Whittle, A. J. et al. BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell 149, 871–885 (2012).
Wang, Q. A. et al. Reversible de-differentiation of mature white adipocytes into preadipocyte-like precursors during lactation. Cell Metab. 28, 282–288.e3 (2018).
Li, C. J. et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J. Clin. Investig. 125, 1509–1522 (2015).
Zhang, R. et al. The role of microRNAs in adipocyte differentiation. Front. Med. 7, 223–230 (2013).
Chattopadhyay, T., Singh, R. R., Gupta, S. & Surolia, A. Bone morphogenetic protein-7 (BMP-7) augments insulin sensitivity in mice with type II diabetes mellitus by potentiating PI3K/AKT pathway. Biofactors 43, 195–209 (2017).
Luo, Y. et al. Decreased circulating BMP-9 levels in patients with Type 2 diabetes is a signature of insulin resistance. Clin. Sci. 131, 239–246 (2017).
Schreiber, I. et al. BMPs as new insulin sensitizers: enhanced glucose uptake in mature 3T3-L1 adipocytes via PPARγ and GLUT4 upregulation. Sci. Rep. 7, 17192 (2017).
Yang, M. et al. Role of bone morphogenetic protein-9 in the regulation of glucose and lipid metabolism. FASEB J. 33, 10077–10088 (2019).
Wang, X. et al. New association of bone morphogenetic protein 4 concentrations with fat distribution in obesity and Exenatide intervention on it. Lipids Health Dis. 16, 70 (2017).
Hodgson, J. et al. Characterization of GDF2 mutations and levels of BMP9 and BMP10 in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 201, 575–585 (2020).
Vukicevic, S., Helder, M. N. & Luyten, F. P. Developing human lung and kidney are major sites for synthesis of bone morphogenetic protein-3 (osteogenin). J. Histochem. Cytochem. 42, 869–875 (1994).
Kokubu, N., Tsujii, M., Akeda, K., Iino, T. & Sudo, A. BMP-7/Smad expression in dedifferentiated Schwann cells during axonal regeneration and upregulation of endogenous BMP-7 following administration of PTH (1–34). J. Orthop. Surg. 26, 2309499018812953 (2018).
Yamashita, K., Mikawa, S. & Sato, K. BMP3 expression in the adult rat CNS. Brain Res. 1643, 35–50 (2016).
Desroches-Castan, A. et al. Differential consequences of Bmp9 deletion on sinusoidal endothelial cell differentiation and liver fibrosis in 129/Ola and C57BL/6 mice. Cells 8, 1079 (2019).
Beenken, A. & Mohammadi, M. The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 8, 235–253 (2009).
Ornitz, D. M. & Marie, P. J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 29, 1463–1486 (2015).
Luo, Y., Ye, S., Li, X. & Lu, W. Emerging structure-function paradigm of endocrine FGFs in metabolic diseases. Trends Pharm. Sci. 40, 142–153 (2019).
Peng, M. et al. Developments in the study of gastrointestinal microbiome disorders affected by FGF19 in the occurrence and development of colorectal neoplasms. J. Cell Physiol. 235, 4060–4069 (2020).
Geller, S. et al. Tanycytes regulate lipid homeostasis by sensing free fatty acids and signaling to key hypothalamic neuronal populations via FGF21 secretion. Cell Metab. 30, 833–844.e7 (2019).
Nishimura, T., Nakatake, Y., Konishi, M. & Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta. 1492, 203–206 (2000).
Erben, R. G. Pleiotropic actions of FGF23. Toxicol. Pathol. 45, 904–910 (2017).
Clinkenbeard, E. L. et al. Conditional deletion of murine Fgf23: interruption of the normal skeletal responses to phosphate challenge and rescue of genetic hypophosphatemia. J. Bone Miner. Res. 31, 1247–1257 (2016).
Hu, M. C. et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 24, 3438–3450 (2010).
Strewler, G. J. Untangling klotho’s role in calcium homeostasis. Cell Metab. 6, 93–95 (2007).
Angelin, B., Larsson, T. E. & Rudling, M. Circulating fibroblast growth factors as metabolic regulators-a critical appraisal. Cell Metab. 16, 693–705 (2012).
Komaba, H. et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. 92, 599–611 (2017).
Rhee, Y. et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 49, 636–643 (2011).
Kaludjerovic, J. et al. Klotho expression in long bones regulates FGF23 production during renal failure. FASEB J. 31, 2050–2064 (2017).
Kurosu, H. et al. Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833 (2005).
Hesse, M., Fröhlich, L. F., Zeitz, U., Lanske, B. & Erben, R. G. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. 26, 75–84 (2007).
López, I. et al. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int. 80, 475–482 (2011).
Singh, S. et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 90, 985–996 (2016).
Ito, N. et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol. Cell. Endocrinol. 399, 208–218 (2015).
Mirza, M. A. I. et al. Circulating fibroblast growth factor-23 is associated with fat mass and dyslipidemia in two independent cohorts of elderly individuals. Arterioscler Thromb. Vasc. Biol. 31, 219–227 (2011).
Aljohani, A. et al. Hepatic stearoyl CoA desaturase 1 deficiency increases glucose uptake in adipose tissue partially through the PGC-1α-FGF21 axis in mice. J. Biol. Chem. 294, 19475–19485 (2019).
Chen, Y., Lu, J., Nemati, R., Plank, L. D. & Murphy, R. Acute changes of bile acids and FGF19 after sleeve gastrectomy and Roux-en-Y gastric bypass. Obes. Surg. 29, 3605–3621 (2019).
Lan, T. et al. FGF19, FGF21, and an FGFR1/β-Klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab. 26, 709–718.e3 (2017).
Mosialou, I. et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543, 385–390 (2017).
Zhang, J. et al. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol. Endocrinol. 22, 1416–1426 (2008).
Flower, D. R. Beyond the superfamily: the lipocalin receptors. Biochim Biophys. Acta 1482, 327–336 (2000).
Jha, M. K. et al. Diverse functional roles of lipocalin-2 in the central nervous system. Neurosci. Biobehav Rev. 49, 135–156 (2015).
Adriaenssens, A. E. et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. 30, 987–996.e6 (2019).
Liu, H. et al. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J. Neurosci. 23, 7143–7154 (2003).
Guo, H. et al. Lipocalin 2, a regulator of retinoid homeostasis and retinoid-mediated thermogenic activation in adipose tissue. J. Biol. Chem. 291, 11216–11229 (2016).
Guo, H. et al. Lipocalin-2 deficiency impairs thermogenesis and potentiates diet-induced insulin resistance in mice. Diabetes 59, 1376–1385 (2010).
Yu, B. et al. PGC-1α controls skeletal stem cell fate and bone-fat balance in osteoporosis and skeletal aging by inducing TAZ. Cell Stem Cell 23, 193–209.e5 (2018).
Zhang, Y. et al. Lipocalin 2 regulates brown fat activation via a nonadrenergic activation mechanism. J. Biol. Chem. 289, 22063–22077 (2014).
Kamble, P. G. et al. Lipocalin 2 produces insulin resistance and can be upregulated by glucocorticoids in human adipose tissue. Mol. Cell Endocrinol. 427, 124–132 (2016).
Capulli, M. et al. A complex role for lipocalin 2 in bone metabolism: global ablation in mice induces osteopenia caused by an altered energy metabolism. J. Bone Min. Res. 33, 1141–1153 (2018).
Wang, W. et al. Elevated serum lipocalin 2 levels are associated with indexes of both glucose and bone metabolism in type 2 diabetes mellitus. Endokrynol. Pol. 69, 276–282 (2018).
van Bezooijen, R. L. et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 199, 805–814 (2004).
Collette, N. M. et al. Sost and its paralog Sostdc1 coordinate digit number in a Gli3-dependent manner. Dev. Biol. 383, 90–105 (2013).
Winkler, D. G. et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 22, 6267–6276 (2003).
Li, C. et al. Lipoprotein receptor-related protein 6 is required for parathyroid hormone-induced Sost suppression. Ann. N. Y Acad. Sci. 1364, 62–73 (2016).
Bullock, W. A. et al. Lrp4 mediates bone homeostasis and mechanotransduction through interaction with sclerostin in vivo. iScience 20, 205–215 (2019).
Liu, W. et al. Osteocyte TSC1 promotes sclerostin secretion to restrain osteogenesis in mice. Open Biol. 9, 180262 (2019).
Stolina, M. et al. Temporal changes in systemic and local expression of bone turnover markers during six months of sclerostin antibody administration to ovariectomized rats. Bone 67, 305–313 (2014).
Fijalkowski, I. et al. A novel domain-specific mutation in a sclerosteosis patient suggests a role of LRP4 as an anchor for sclerostin in human bone. J. Bone Min. Res. 31, 874–881 (2016).
Haynes, K. R. et al. Treatment of a mouse model of ankylosing spondylitis with exogenous sclerostin has no effect on disease progression. BMC Musculoskelet. Disord. 16, 368 (2015).
Koide, M. et al. Bone formation is coupled to resorption via suppression of sclerostin expression by osteoclasts. J. Bone Min. Res. 32, 2074–2086 (2017).
Faienza, M. F. et al. High sclerostin and dickkopf-1 (DKK-1) serum levels in children and adolescents with type 1 diabetes mellitus. J. Clin. Endocrinol. Metab. 102, 1174–1181 (2017).
Hie, M., Iitsuka, N., Otsuka, T. & Tsukamoto, I. Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activation. Int J. Mol. Med. 28, 455–462 (2011).
Daniele, G. et al. Sclerostin and insulin resistance in prediabetes: evidence of a cross talk between bone and glucose metabolism. Diabetes Care 38, 1509–1517 (2015).
Yu, O. H. Y. et al. The association between sclerostin and incident type 2 diabetes risk: a cohort study. Clin. Endocrinol. 86, 520–525 (2017).
Kim, S. P. et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc. Natl Acad. Sci. USA 114, E11238–E11247 (2017).
Fulzele, K. et al. Osteocyte-secreted Wnt signaling inhibitor sclerostin contributes to beige adipogenesis in peripheral fat depots. J. Bone Min. Res. 32, 373–384 (2017).
Hofmann, S., Bellmann-Sickert, K. & Beck-Sickinger, A. G. Chemical modification of neuropeptide Y for human Y1 receptor targeting in health and disease. Biol. Chem. 400, 299–311 (2019).
Kawakami, Y. Neuropeptide Y. Nihon. Rinsho. 63, S421–S424 (2005).
Cedernaes, J. et al. Transcriptional basis for rhythmic control of hunger and metabolism within the AgRP neuron. Cell Metab. 29, 1078–1091.e5 (2019).
Ip, C. K. et al. Amygdala NPY circuits promote the development of accelerated obesity under chronic stress conditions. Cell Metab. 30, 111–128.e6 (2019).
Krashes, M. J. et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507, 238–242 (2014).
Chen, Z. Temporal control of appetite by AgRP Clocks. Cell Metab. 29, 1022–1023 (2019).
Makhmutova, M., Rodriguez-Diaz, R. & Caicedo, A. A nervous breakdown that may stop autoimmune diabetes. Cell Metab. 31, 215–216 (2020).
Lee, D. Y. et al. Neuropeptide Y mitigates ER stress-induced neuronal cell death by activating the PI3K-XBP1 pathway. Eur. J. Cell Biol. 97, 339–348 (2018).
Fetissov, S. O., Kopp, J. & Hökfelt, T. Distribution of NPY receptors in the hypothalamus. Neuropeptides 38, 175–188 (2004).
Huang, L. et al. Actions of NPY, and its Y1 and Y2 receptors on pulsatile growth hormone secretion during the fed and fasted state. J. Neurosci. 34, 16309–16319 (2014).
Park, M. H. et al. Neuropeptide Y induces hematopoietic stem/progenitor cell mobilization by regulating matrix metalloproteinase-9 activity through Y1 receptor in osteoblasts. Stem Cells 34, 2145–2156 (2016).
Shin, M. K. et al. Elevated pentraxin 3 in obese adipose tissue promotes adipogenic differentiation by activating neuropeptide Y signaling. Front. Immunol. 9, 1790 (2018).
Yang, K., Guan, H., Arany, E., Hill, D. J. & Cao, X. Neuropeptide Y is produced in visceral adipose tissue and promotes proliferation of adipocyte precursor cells via the Y1 receptor. FASEB J. 22, 2452–2464 (2008).
Franklin, Z. J. et al. Islet neuropeptide Y receptors are functionally conserved and novel targets for the preservation of beta-cell mass. Diabetes Obes. Metab. 20, 599–609 (2018).
Khan, D., Vasu, S., Moffett, R. C., Irwin, N. & Flatt, P. R. Influence of neuropeptide Y and pancreatic polypeptide on islet function and beta-cell survival. Biochim. Biophys. Acta Gen. Subj. 1861, 749–758 (2017).
Loh, K., Herzog, H. & Shi, Y. C. Regulation of energy homeostasis by the NPY system. Trends Endocrinol. Metab. 26, 125–135 (2015).
Igwe, J. C. et al. Neuropeptide Y is expressed by osteocytes and can inhibit osteoblastic activity. J. Cell Biochem. 108, 621–630 (2009).
Wee, N. K. Y. et al. Diet-induced obesity suppresses cortical bone accrual by a neuropeptide Y-dependent mechanism. Int J. Obes. 42, 1925–1938 (2018).
Wu, J. et al. Neuropeptide Y enhances proliferation and prevents apoptosis in rat bone marrow stromal cells in association with activation of the Wnt/β-catenin pathway in vitro. Stem Cell Res 21, 74–84 (2017).
Wee, N. K. Y. et al. Skeletal phenotype of the neuropeptide Y knockout mouse. Neuropeptides 73, 78–88 (2019).
Lee, N. J. et al. NPY signalling in early osteoblasts controls glucose homeostasis. Mol. Metab. 4, 164–174 (2015).
Kronenberg, H. M. PTHrP and skeletal development. Ann. N. Y Acad. Sci. 1068, 1–13 (2006).
Wysolmerski, J. J. Parathyroid hormone-related protein: an update. J. Clin. Endocrinol. Metab. 97, 2947–2956 (2012).
Dudeck, J. et al. Mast cells acquire MHCII from dendritic cells during skin inflammation. J. Exp. Med. 214, 3791–3811 (2017).
Miao, D. et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J. Clin. Investig. 115, 2402–2411 (2005).
Zhang, X., Cheng, Q., Wang, Y., Leung, P. S. & Mak, K. K. Hedgehog signaling in bone regulates whole-body energy metabolism through a bone-adipose endocrine relay mediated by PTHrP and adiponectin. Cell Death Differ. 24, 225–237 (2017).
Guthalu Kondegowda, N. et al. Parathyroid hormone-related protein enhances human ß-cell proliferation and function with associated induction of cyclin-dependent kinase 2 and cyclin E expression. Diabetes 59, 3131–3138 (2010).
Horwitz, M. J. et al. Parathyroid hormone-related protein for the treatment of postmenopausal osteoporosis: defining the maximal tolerable dose. J. Clin. Endocrinol. Metab. 95, 1279–1287 (2010).
Bukowska, J. et al. Bone marrow adipocyte developmental origin and biology. Curr. Osteoporos. Rep. 16, 312–319 (2018).
Bhansali, S. et al. Effect of mesenchymal stem cells transplantation on glycaemic profile & their localization in streptozotocin induced diabetic Wistar rats. Indian J. Med. Res. 142, 63–71 (2015).
Bhansali, S. et al. Efficacy of autologous bone marrow-derived mesenchymal stem cell and mononuclear cell transplantation in type 2 diabetes mellitus: a randomized, placebo-controlled comparative study. Stem Cells Dev. 26, 471–481 (2017).
Rydén, M. et al. Transplanted bone marrow-derived cells contribute to human adipogenesis. Cell Metab. 22, 408–417 (2015).
Goldberg, E. L. & Dixit, V. D. Bone marrow: an immunometabolic refuge during energy depletion. Cell Metab. 30, 621–623 (2019).
Zhang, J. et al. Metabolism in pluripotent stem cells and early mammalian development. Cell Metab. 27, 332–338 (2018).
Liu, J. et al. Bone-derived exosomes. Curr. Opin. Pharm. 34, 64–69 (2017).
Kita, S., Maeda, N. & Shimomura, I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J. Clin. Investig. 129, 4041–4049 (2019).
Raposo, G. & Stoorvogel, W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383 (2013).
Guay, C. et al. Lymphocyte-derived exosomal micrornas promote pancreatic β cell death and may contribute to type 1 diabetes development. Cell Metab. 29, 348–361.e6 (2019).
Whitham, M. et al. Extracellular vesicles provide a means for tissue crosstalk during exercise. Cell Metab. 27, 237–251.e4 (2018).
Deng, L. et al. Osteoblast-derived microvesicles: a novel mechanism for communication between osteoblasts and osteoclasts. Bone 79, 37–42 (2015).
Lyu, H., Xiao, Y., Guo, Q., Huang, Y. & Luo, X. The role of bone-derived exosomes in regulating skeletal metabolism and extraosseous diseases. Front. Cell Dev. Biol. 8, 89 (2020).
Sun, W. et al. Osteoclast-derived microRNA-containing exosomes selectively inhibit osteoblast activity. Cell Discov. 2, 16015 (2016).
Yeo, R. W. Y. et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 65, 336–341 (2013).
Huynh, N. et al. Characterization of regulatory extracellular vesicles from osteoclasts. J. Dent. Res 95, 673–679 (2016).
Li, D. et al. Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat. Commun. 7, 10872 (2016).
Mori, M. A., Ludwig, R. G., Garcia-Martin, R., Brandão, B. B. & Kahn, C. R. Extracellular miRNAs: from biomarkers to mediators of physiology and disease. Cell Metab. 30, 656–673 (2019).
Baglio, S. R. et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res Ther. 6, 127 (2015).
Su, T. et al. Bone marrow mesenchymal stem cells-derived exosomal MiR-29b-3p regulates aging-associated insulin resistance. ACS Nano 13, 2450–2462 (2019).
Rong, X. et al. Human bone marrow mesenchymal stem cells-derived exosomes alleviate liver fibrosis through the Wnt/β-catenin pathway. Stem Cell Res. Ther. 10, 98 (2019).
AbuBakr, N., Haggag, T., Sabry, D. & Salem, Z. A. Functional and histological evaluation of bone marrow stem cell-derived exosomes therapy on the submandibular salivary gland of diabetic Albino rats through TGFβ/ Smad3 signaling pathway. Heliyon 6, e03789 (2020).
Hotamisligil, G. S. Inflammation and metabolic disorders. Nature 444, 860–867 (2006).
Malle, E. K. et al. Nuclear factor κB-inducing kinase activation as a mechanism of pancreatic β cell failure in obesity. J. Exp. Med. 212, 1239–1254 (2015).
Lee, A. & Dixit, V. D. Energy sparing orexigenic inflammation of obesity. Cell Metab. 26, 10–12 (2017).
Saltiel, A. R. & Olefsky, J. M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 127, 1–4 (2017).
Laharrague, P. et al. Inflammatory/haematopoietic cytokine production by human bone marrow adipocytes. Eur. Cytokine Netw. 11, 634–639 (2000).
Sanchez-Lopez, E. et al. Choline uptake and metabolism modulate macrophage IL-1β and IL-18 production. Cell Metab. 29, 1350–1362.e7 (2019).
Romas, E. et al. The role of gp130-mediated signals in osteoclast development: regulation of interleukin 11 production by osteoblasts and distribution of its receptor in bone marrow cultures. J. Exp. Med. 183, 2581–2591 (1996).
Ishimi, Y. et al. IL-6 is produced by osteoblasts and induces bone resorption. J. Immunol. 145, 3297–3303 (1990).
Hardaway, A. L., Herroon, M. K., Rajagurubandara, E. & Podgorski, I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin. Exp. Metastasis 32, 353–368 (2015).
Cereijo, R. et al. CXCL14, a brown adipokine that mediates brown-fat-to-macrophage communication in thermogenic adaptation. Cell Metab. 28, 750–763.e6 (2018).
Saraiva, M. & O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 10, 170–181 (2010).
Saraiva, M., Vieira, P. & O’Garra, A. Biology and therapeutic potential of interleukin-10. J. Exp. Med. 217, e20190418 (2020).
Li, P. et al. Hematopoietic-derived galectin-3 causes cellular and systemic insulin resistance. Cell 167, 973–984.e12 (2016).
Lyons, J. J. et al. ERBIN deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J. Exp. Med. 214, 669–680 (2017).
Rajbhandari, P. et al. IL-10 signaling remodels adipose chromatin architecture to limit thermogenesis and energy expenditure. Cell 172, 218–233.e17 (2018).
Corre, J. et al. Human subcutaneous adipose cells support complete differentiation but not self-renewal of hematopoietic progenitors. J. Cell Physiol. 208, 282–288 (2006).
Cawthorn, W. P. et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. 20, 368–375 (2014).
Rendina-Ruedy, E. & Rosen, C. J. Lipids in the bone marrow: an evolving perspective. Cell Metab. 31, 219–231 (2020).
Kricun, M. E. Red-yellow marrow conversion: its effect on the location of some solitary bone lesions. Skelet. Radio. 14, 10–19 (1985).
Tavassoli, M. Marrow adipose cells. Histochemical identification of labile and stable components. Arch. Pathol. Lab Med 100, 16–18 (1976).
Nishio, M. et al. Production of functional classical brown adipocytes from human pluripotent stem cells using specific hemopoietin cocktail without gene transfer. Cell Metab. 16, 394–406 (2012).
Wang, Z. V. & Scherer, P. E. Adiponectin, the past two decades. J. Mol. Cell Biol. 8, 93–100 (2016).
Scherer, P. E., Williams, S., Fogliano, M., Baldini, G. & Lodish, H. F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 270, 26746–26749 (1995).
Scheller, E. L., Burr, A. A., MacDougald, O. A. & Cawthorn, W. P. Inside out: bone marrow adipose tissue as a source of circulating adiponectin. Adipocyte 5, 251–269 (2016).
Gil-Campos, M., Cañete, R. R. & Gil, A. Adiponectin, the missing link in insulin resistance and obesity. Clin. Nutr. 23, 963–974 (2004).
Yamauchi, T. & Kadowaki, T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 17, 185–196 (2013).
Yamauchi, T. et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423, 762–769 (2003).
Yamauchi, T. et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 7, 941–946 (2001).
Stanford, K. I. et al. 12,13-diHOME: an exercise-induced lipokine that increases skeletal muscle fatty acid uptake. Cell Metab. 27, 1111–1120.e3 (2018).
Manieri, E. et al. Adiponectin accounts for gender differences in hepatocellular carcinoma incidence. J. Exp. Med. 216, 1108–1119 (2019).
González, A., Hall, M. N., Lin, S. C. & Hardie, D. G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 31, 472–492 (2020).
Qi, Y. et al. Adiponectin acts in the brain to decrease body weight. Nat. Med. 10, 524–529 (2004).
Uchihashi, K. et al. Organotypic culture of human bone marrow adipose tissue. Pathol. Int. 60, 259–267 (2010).
Berner, H. S. et al. Adiponectin and its receptors are expressed in bone-forming cells. Bone 35, 842–849 (2004).
Dalamaga, M. et al. Leptin at the intersection of neuroendocrinology and metabolism: current evidence and therapeutic perspectives. Cell Metab. 18, 29–42 (2013).
Hoggard, N. et al. Localization of leptin receptor mRNA splice variants in murine peripheral tissues by RT-PCR and in situ hybridization. Biochem. Biophys. Res. Commun. 232, 383–387 (1997).
Harris, R. B. S. Direct and indirect effects of leptin on adipocyte metabolism. Biochim. Biophys. Acta. 1842, 414–423 (2014).
Shimomura, I., Hammer, R. E., Ikemoto, S., Brown, M. S. & Goldstein, J. L. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 401, 73–76 (1999).
Alfa, R. W. et al. Suppression of insulin production and secretion by a decretin hormone. Cell Metab. 27, 479 (2018).
Zhao, S. et al. Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab. 30, 706–719.e6 (2019).
Müller, G., Ertl, J., Gerl, M. & Preibisch, G. Leptin impairs metabolic actions of insulin in isolated rat adipocytes. J. Biol. Chem. 272, 10585–10593 (1997).
Laharrague, P. et al. High expression of leptin by human bone marrow adipocytes in primary culture. FASEB J. 12, 747–752 (1998).
Krings, A. et al. Bone marrow fat has brown adipose tissue characteristics, which are attenuated with aging and diabetes. Bone 50, 546–552 (2012).
Laharrague, P. et al. Regulation by cytokines of leptin expression in human bone marrow adipocytes. Horm. Metab. Res. 32, 381–385 (2000).
Münzberg, H. & Heymsfield, S. B. New insights into the regulation of leptin gene expression. Cell Metab. 29, 1013–1014 (2019).
Upadhyay, J., Farr, O. M. & Mantzoros, C. S. The role of leptin in regulating bone metabolism. Metabolism 64, 105–113 (2015).
Haeusler, R. A., McGraw, T. E. & Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 19, 31–44 (2018).
Pramojanee, S. N., Phimphilai, M., Chattipakorn, N. & Chattipakorn, S. C. Possible roles of insulin signaling in osteoblasts. Endocr. Res. 39, 144–151 (2014).
Fulzele, K. et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142, 309–319 (2010).
Fulzele, K. et al. Disruption of the insulin-like growth factor type 1 receptor in osteoblasts enhances insulin signaling and action. J. Biol. Chem. 282, 25649–25658 (2007).
Oh, J. H. & Lee, N. K. Up-regulation of RANK expression via ERK1/2 by insulin contributes to the enhancement of osteoclast differentiation. Mol. Cells 40, 371–377 (2017).
Shimoaka, T. et al. Impairment of bone healing by insulin receptor substrate-1 deficiency. J. Biol. Chem. 279, 15314–15322 (2004).
Conte, C., Epstein, S. & Napoli, N. Insulin resistance and bone: a biological partnership. Acta Diabetol. 55, 305–314 (2018).
Li, Z. et al. Glucose transporter-4 facilitates insulin-stimulated glucose uptake in osteoblasts. Endocrinology 157, 4094–4103 (2016).
Ferron, M. et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142, 296–308 (2010).
Mera, P. et al. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab. 25, 218 (2017).
Christakos, S., Dhawan, P., Verstuyf, A., Verlinden, L. & Carmeliet, G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol. Rev. 96, 365–408 (2016).
Hochberg, Z., Tiosano, D. & Even, L. Calcium therapy for calcitriol-resistant rickets. J. Pediatr. 121, 803–808 (1992).
Erben, R. G. et al. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol. Endocrinol. 16, 1524–1537 (2002).
Nakamichi, Y. et al. VDR in osteoblast-lineage cells primarily mediates vitamin D treatment-induced increase in bone mass by suppressing bone resorption. J. Bone Min. Res. 32, 1297–1308 (2017).
Matthews, D. G., D’Angelo, J., Drelich, J. & Welsh, J. Adipose-specific Vdr deletion alters body fat and enhances mammary epithelial density. J. Steroid Biochem. Mol. Biol. 164, 299–308 (2016).
Rosenstreich, S. J., Rich, C. & Volwiler, W. Deposition in and release of vitamin D3 from body fat: evidence for a storage site in the rat. J. Clin. Investig. 50, 679–687 (1971).
Abbas, M. A. Physiological functions of Vitamin D in adipose tissue. J. Steroid Biochem Mol. Biol. 165, 369–381 (2017).
Blumberg, J. M. et al. Complex role of the vitamin D receptor and its ligand in adipogenesis in 3T3-L1 cells. J. Biol. Chem. 281, 11205–11213 (2006).
Ricciardi, C. J. et al. 1,25-Dihydroxyvitamin D3/vitamin D receptor suppresses brown adipocyte differentiation and mitochondrial respiration. Eur. J. Nutr. 54, 1001–1012 (2015).
Sun, X. & Zemel, M. B. Role of uncoupling protein 2 (UCP2) expression and 1alpha, 25-dihydroxyvitamin D3 in modulating adipocyte apoptosis. FASEB J. 18, 1430–1432 (2004).
Eshraghian, A. Bone metabolism in non-alcoholic fatty liver disease: vitamin D status and bone mineral density. Minerva Endocrinol. 42, 164–172 (2017).
Pittas, A. G., Harris, S. S., Stark, P. C. & Dawson-Hughes, B. The effects of calcium and vitamin D supplementation on blood glucose and markers of inflammation in nondiabetic adults. Diabetes Care 30, 980–986 (2007).
Liu, S. et al. Bovine parathyroid hormone enhances osteoclast bone resorption by modulating V-ATPase through PTH1R. Int J. Mol. Med. 37, 284–292 (2016).
Guo, J. et al. Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab. 11, 161–171 (2010).
Vrahnas, C. et al. Anabolic action of parathyroid hormone (PTH) does not compromise bone matrix mineral composition or maturation. Bone 93, 146–154 (2016).
Jilka, R. L. et al. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Investig. 104, 439–446 (1999).
Keller, H. & Kneissel, M. SOST is a target gene for PTH in bone. Bone 37, 148–158 (2005).
Boucher, D. et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 215, 827–840 (2018).
Esen, E., Lee, S.-Y., Wice, B. M. & Long, F. PTH promotes bone anabolism by stimulating aerobic glycolysis via IGF signaling. J. Bone Min. Res. 30, 1959–1968 (2015).
Yamaguchi, M. Effect of parathyroid hormone on the increase in serum glucose and insulin levels after a glucose load to thyroparathyroidectomized rats. Endocrinol. Jpn 26, 353–358 (1979).
Kimura, S., Sasase, T., Ohta, T., Sato, E. & Matsushita, M. Parathyroid hormone (1-34) improves bone mineral density and glucose metabolism in Spontaneously Diabetic Torii-Lepr(fa) rats. J. Vet. Med. Sci. 74, 103–105 (2012).
Chiu, K. C. et al. Insulin sensitivity is inversely correlated with plasma intact parathyroid hormone level. Metabolism 49, 1501–1505 (2000).
Heuck, C. C. & Ritz, E. Does parathyroid hormone play a role in lipid metabolism? Contrib. Nephrol. 20, 118–128 (1980).
Lacour, B., Basile, C., Drüeke, T. & Funck-Brentano, J. L. Parathyroid function and lipid metabolism in the rat. Min. Electrolyte Metab. 7, 157–165 (1982).
Larsson, S., Jones, H. A., Göransson, O., Degerman, E. & Holm, C. Parathyroid hormone induces adipocyte lipolysis via PKA-mediated phosphorylation of hormone-sensitive lipase. Cell Signal 28, 204–213 (2016).
LeBlanc, M. E. et al. Secretogranin III as a disease-associated ligand for antiangiogenic therapy of diabetic retinopathy. J. Exp. Med. 214, 1029–1047 (2017).
Mauvais-Jarvis, F. Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol. Metab. 22, 24–33 (2011).
Jia, M., Dahlman-Wright, K. & Gustafsson, J.-Å. Estrogen receptor alpha and beta in health and disease. Best. Pr. Res Clin. Endocrinol. Metab. 29, 557–568 (2015).
Brown, L. M., Gent, L., Davis, K. & Clegg, D. J. Metabolic impact of sex hormones on obesity. Brain Res. 1350, 77–85 (2010).
Voisin, D. L., Simonian, S. X. & Herbison, A. E. Identification of estrogen receptor-containing neurons projecting to the rat supraoptic nucleus. Neuroscience 78, 215–228 (1997).
Xu, Y. et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 29, 1232 (2019).
Eckel, R. H. Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases. N. Engl. J. Med. 320, 1060–1068 (1989).
Cooke, P. S. & Naaz, A. Role of estrogens in adipocyte development and function. Exp. Biol. Med. 229, 1127–1135 (2004).
Gorres, B. K., Bomhoff, G. L., Morris, J. K. & Geiger, P. C. In vivo stimulation of oestrogen receptor α increases insulin-stimulated skeletal muscle glucose uptake. J. Physiol. 589, 2041–2054 (2011).
Hayashi, M. et al. Autoregulation of osteocyte Sema3A orchestrates estrogen action and counteracts bone aging. Cell Metab. 29, 627–637.e5 (2019).
Kondoh, S. et al. Estrogen receptor α in osteocytes regulates trabecular bone formation in female mice. Bone 60, 68–77 (2014).
Novack, D. V. Estrogen and bone: osteoclasts take center stage. Cell Metab. 6, 254–256 (2007).
Streicher, C. et al. Estrogen regulates bone turnover by targeting RANKL expression in bone lining cells. Sci. Rep. 7, 6460 (2017).
Janghorbani, M., Van Dam, R. M., Willett, W. C. & Hu, F. B. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am. J. Epidemiol. 166, 495–505 (2007).
Schwartz, A. V. et al. Older women with diabetes have an increased risk of fracture: a prospective study. J. Clin. Endocrinol. Metab. 86, 32–38 (2001).
Evans, A. L., Paggiosi, M. A., Eastell, R. & Walsh, J. S. Bone density, microstructure and strength in obese and normal weight men and women in younger and older adulthood. J. Bone Min. Res 30, 920–928 (2015).
Sornay-Rendu, E., Boutroy, S., Vilayphiou, N., Claustrat, B. & Chapurlat, R. D. In obese postmenopausal women, bone microarchitecture and strength are not commensurate to greater body weight: the Os des Femmes de Lyon (OFELY) study. J. Bone Min. Res 28, 1679–1687 (2013).
Driessler, F. & Baldock, P. A. Hypothalamic regulation of bone. J. Mol. Endocrinol. 45, 175–181 (2010).
Lin, Y. Y. et al. Adiponectin receptor 1 regulates bone formation and osteoblast differentiation by GSK-3β/β-catenin signaling in mice. Bone 64, 147–154 (2014).
Boskey, A. L. & Coleman, R. Aging and bone. J. Dent. Res 89, 1333–1348 (2010).
Grandl, G. & Wolfrum, C. Adipocytes at the core of bone function. Cell Stem Cell 20, 739–740 (2017).
Benedetti, M. G., Furlini, G., Zati, A. & Letizia Mauro, G. The effectiveness of physical exercise on bone density in osteoporotic patients. Biomed. Res. Int. 2018, 4840531 (2018).
Contrepois, K. et al. Molecular choreography of acute exercise. Cell 181, 1112–1130.e16 (2020).
Horowitz, A. M. et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 369, 167–173 (2020).
Duan, P., Yang, M., Wei, M., Liu, J. & Tu, P. Serum osteoprotegerin is a potential biomarker of insulin resistance in chinese postmenopausal women with prediabetes and type 2 diabetes. Int J. Endocrinol. 2017, 8724869 (2017).
Ndip, A., Wilkinson, F. L., Jude, E. B., Boulton, A. J. M. & Alexander, M. Y. RANKL-OPG and RAGE modulation in vascular calcification and diabetes: novel targets for therapy. Diabetologia 57, 2251–2260 (2014).
Cypess, A. M., Haft, C. R., Laughlin, M. R. & Hu, H. H. Brown fat in humans: consensus points and experimental guidelines. Cell Metab. 20, 408–415 (2014).
Yao, Q. et al. Wnt/β-catenin signaling in osteoblasts regulates global energy metabolism. Bone 97, 175–183 (2017).
Wang, P. et al. Osthole promotes bone fracture healing through activation of BMP signaling in chondrocytes. Int J. Biol. Sci. 13, 996–1007 (2017).
Yee, C. S. et al. Conditional deletion of Sost in MSC-derived lineages identifies specific cell-type contributions to bone mass and B-cell development. J. Bone Min. Res 33, 1748–1759 (2018).
Acknowledgements
We would like to thank all the researchers in the Endocrinology Research Center of Xiangya Hospital of Central South University for their generous help with our manuscript. There was no significant financial support for this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, R., Guo, Q., Xiao, Y. et al. Endocrine role of bone in the regulation of energy metabolism. Bone Res 9, 25 (2021). https://doi.org/10.1038/s41413-021-00142-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-021-00142-4
This article is cited by
-
Skeletal interoception in osteoarthritis
Bone Research (2024)
-
Osteoprotegerin deficiency aggravates methionine–choline-deficient diet-induced nonalcoholic steatohepatitis in mice
Scientific Reports (2023)
-
Reduced bone formation and increased bone resorption drive bone loss in Eimeria infected broilers
Scientific Reports (2023)
-
Association of bisphosphonates with diabetes risk and glycemic control: a meta-analysis
Osteoporosis International (2023)
-
Rapid trabecular bone growth in puberty associated with stiffer arteries in adulthood – longitudinal study on healthy young males
Archives of Osteoporosis (2023)
