
Abstract
Smoldering multiple myeloma (SMM) precedes multiple myeloma (MM). The risk of progression of SMM patients is not uniform, thus different progression-risk models have been developed, although they are mainly based on clinical parameters. Recently, genomic predictors of progression have been defined for untreated SMM. However, the usefulness of such markers in the context of clinical trials evaluating upfront treatment in high-risk SMM (HR SMM) has not been explored yet, precluding the identification of baseline genomic alterations leading to drug resistance. For this reason, we carried out next-generation sequencing and fluorescent in-situ hybridization studies on 57 HR and ultra-high risk (UHR) SMM patients treated in the phase II GEM-CESAR clinical trial (NCT02415413). DIS3, FAM46C, and FGFR3 mutations, as well as t(4;14) and 1q alterations, were enriched in HR SMM. TRAF3 mutations were specifically associated with UHR SMM but identified cases with improved outcomes. Importantly, novel potential predictors of treatment resistance were identified: NRAS mutations and the co-occurrence of t(4;14) plus FGFR3 mutations were associated with an increased risk of biological progression. In conclusion, we have carried out for the first time a molecular characterization of HR SMM patients treated with an intensive regimen, identifying genomic predictors of poor outcomes in this setting.
Similar content being viewed by others


Introduction
Smoldering multiple myeloma (SMM) is an asymptomatic precursor of multiple myeloma (MM), generally characterized by a higher rate of progression than monoclonal gammopathy of unknown significance (MGUS) [1]. Traditionally, the distinction between the three entities was based on the amount of serum and/or urine monoclonal component, bone marrow (BM) tumor plasma cell (PC) infiltration, and the presence of end-organ damage [2]. Clinically, SMM patients have a heterogeneous behavior and thus they can be stratified based on time to progression: patients in the low-risk group (~10% of all SMM patients) have similar outcomes than MGUS (~1% probability of progression per year), while high-risk patients (~30%) progress in a short term (50% probability of progression within 2 years). The rest of the patients (~60%) belong to an intermediate-risk group [1, 3]. In 2014, the International Myeloma Working Group (IMWG) updated the definition of MM [4], re-classifying ultra-high risk SMM patients (80% probability of progression within 2 years) as patients with active myeloma that should be treated accordingly. Many prognostic factors based on tumor burden, imaging or genomic markers have been described, and therefore several models for risk assessment have been designed for these patients. In 2020, aiming to have a consensus and easy-to-use model, a new scoring system, known as the ‘20/2/20’, was introduced by the IMWG [5], ensuring homogeneous risk evaluation in SMM hereinafter. Nevertheless, not all the prognostic models consider the underlying genomic architecture that could be a determinant for disease progression.
The mutational landscape of MM is highly complex, and includes primary translocations enhancing the expression of CCND1, FGFR3/MMSET, and MAF paralogues [t(11;14), t(4;14), t(14;16) and t(14;20), respectively], hyperdiploidy of odd chromosomes, copy number variations (CNV), secondary translocations, as well as single nucleotide variants (SNV) and short insertions/deletions (indels) [6]. In the last years, next-generation sequencing (NGS) strategies have made possible to gain insight into the genomics of MM precursor conditions and their role in progression, depicting the time-dependent acquisition of genetic aberrations through the evolution of the disease [7]. In MGUS and SMM, the presence of complex structural events, mutations affecting known driver genes, and mutational signatures identify patients with stable versus progressive disease profiles [8]. In particular, the molecular makeup of SMM pinpoints genetic predictors of progression [9, 10].
The significant clinical benefit of upfront treatment with lenalidomide alone or in combination with dexamethasone for high-risk SMM patients has been proven in two independent phase III clinical trials (ClinicalTrials.gov accession numbers NCT01169337 and NCT00480363) [11, 12] and encouraged further investigation of intensive regimens trying to cure myeloma, as in the GEM-CESAR or ASCENT trials [13, 14]. Therefore, those regimens may alter the usefulness of previously mentioned genomic aberrations for predicting resistance and progression, urging to explore novel biomarkers in this setting of intensive treatment at asymptomatic stages.
Here, we have combined an NGS capture panel and fluorescence in-situ hybridization (FISH) at baseline to detect SNV, indels, and structural alterations in 57 patients later treated in the GEM-CESAR trial in order to analyze their genomic profile, and to identify potential risk biomarkers of progression in the context of asymptomatic disease under intensive treatment.
Methods
Patients
Ninety [90] patients were recruited in the phase II GEM-CESAR trial (ClinicalTrials.gov NCT02415413) [13] conducted by the Spanish myeloma group. Patients with newly diagnosed high-risk SMM (HR SMM) were treated with a combination of carfilzomib, lenalidomide, and dexamethasone (KRd) as induction, followed by high-dose melphalan and autologous transplantation, consolidation with KRd and limited-duration maintenance with Rd.
The clinical trial was approved by the Ethics Committee of the University Hospital of Salamanca in accordance with the Spanish law and the Declaration of Helsinki principles. Written informed consent for biological studies was obtained from every patient prior to their inclusion.
Risk strata were defined at diagnosis based on BM infiltration by PC and the serum monoclonal component (Mayo criteria) [1]. If only >10% PC were present, immunoparesis and malignant PC infiltration in the BM (Spanish criteria) were considered [3]. The clinical trial was planned before the updated diagnostic criteria were published in 2014 [4]. Therefore, ultrahigh-risk SMM (UHR SMM) patients, currently considered patients with overt MM, were also recruited. The identification of such patients was based on the presence of at least one of the following biomarkers: serum free-light chain ratio (sFLCr) > 100, >1 focal lesion by magnetic resonance, and ≥ 60% BM PC.
Sample collection
CD138 + BM PC were isolated by autoMACS (Miltenyi Biotec, Auburn, CA, USA). Genomic DNA was extracted using the Qiagen’s AllPrep DNA/RNA kit (Qiagen, ThermoFisher Scientific, Waltham, MA, USA) and quantified using the Qubit 4.0 fluorometer and the dsDNA Broad Range kit (ThermoFisher Scientific).
Next-generation sequencing panel
A custom panel was designed in collaboration with SOPHIA GENETICS (Boston, MA, USA). This panel covers 145 Kb from 666 target regions, allowing the detection of SNV and indels located in 38 genes (complete coding regions or hotspots) previously reported in the literature as potentially relevant for disease initiation, progression or treatment resistance in MM: ACTG1, ATM, BIRC2, BRAF, CCND1, CDKN1B, CRBN, CYLD, DIS3, DUSP2, EGR1, FAM46C, FAT3, FGFR3, HIST1H1E, HUWE1, IRF4, KLHL6, KRAS, LTB, MAF, MAX, NF1, NFKB2, NRAS, PRDM1, PRKD2, PTPN11, RASA2, RB1, ROBO1, SP140, TP53, TRAF2, TRAF3, UBR5, ZFHX4 and ZNF292 [15,16,17,18,19].
Library preparation and targeted capture were performed according to the instructions provided by SOPHIA GENETICS, starting with 200 ng of genomic DNA. Enzymatic fragmentation and fragment size selection conditions were optimized to yield a library with an insert size of 300–700 bp. Multiplexed sequencing was carried out in a MiSeq platform (Illumina, San Diego, CA, USA) using v3 cartridges at 2 × 300 bp sequencing read length.
Pipeline for next-generation sequencing analysis
The SOPHIA-DDM-v5.10.11.1 platform was used for the preliminary analysis of FASTQ files obtained from sequencing. Briefly, this software automatically aligns FASTQ files to the human reference genome GRCh37/hg19 and then calls, classifies and filters variants. The platform performs an automated pre-classification of all variants of every sample based on the American College of Medical Genetics and Genomics’ (ACMG) criteria [20], and taking into account data available from multiple sources: frequencies in the population (gnomAD, ExAC, G1000 and ESP5400), in silico scores (SIFT, MutationTaster and PolyPhen-2), disease-specific data (ClinVar, OMIM, COSMIC), splicing predictors (dbscSNV), protein domains (InterPro), loss of function (ExAC pLI) and repetitive regions (RepeatMasker). Additional filters were applied manually to identify relevant mutations: only codifying variants, read depth ≥ 300x, variant allele frequency (VAF) ≥ 1%, and frequency in any ethnic population <1%. A visual confirmatory analysis of potentially significant variants was also performed using the Integrative Genomics Viewer, IGV [21].
FISH studies
FISH probes were used as previously described [22] for the routine analysis of the following translocations and CNA in CD138 + PC: t(4;14), t(14;16), t(11;14), 17p deletions (del17p), 1q gain or amplification (+1q), and 1p deletions. Moreover, structural aberrations in the 8q24 locus (gain/amplification and Ig translocations) were also evaluated. High-risk features as per the IMWG criteria included t(4;14), t(14;16) and 17p deletions [23]. VAF of SNV and indels were corrected based on the most clonal aberration detected by FISH, or the mean percentage in cases with more than one clonal variant (usually an IGH translocation & +1q). For cases without clonal structural variants detected by FISH, raw VAF were used.
Statistical analysis
Data were analyzed using the SPSS 26.0 software (IBM, Armonk, NY, USA) and the Maftools package for R [24]. Fisher and Mann-Whitney tests were used for discrete or continuous variables, respectively. Biochemical progression-free survival (bPFS) was defined as the time from inclusion to the last follow-up visit or the first of any of the following events: biochemical progression defined by biochemical relapse or progressive disease as per the IMWG criteria, MRD reappearance confirmed at least 2 months apart, or death. Progression-free survival (PFS) was defined as the time from inclusion to the last follow-up visit, clinical progression to overt MM or death by any cause. Time to progression (TTP) was defined as the time from inclusion to the last follow-up visit or clinical progression. Finally, overall survival (OS) was defined as the time from inclusion to the last follow-up visit or patient’s decease by any cause. The Kaplan–Meier method and the log-rank test were used to plot and compare bPFS, PFS, TTP and OS curves. The Cox regression model was used to perform univariate and multivariate analyses. For comparison purposes, previously published series were used as references for SMM and symptomatic MM [9, 10, 19]. Altered genes were grouped together based on signaling pathways as follows: mitogen-activated protein kinase, MAPK (BRAF, FGFR3, KRAS, NF1, NRAS, PTPN11, PRKD2, RASA2); nuclear factor kappa beta, NF-κB (LTB, CYLD, NFKB2, TRAF2, TRAF3); regulation of cell cycle (CCND1, CDKN1B); B-cell development (IRF4, PRDM1); RNA and protein processing (DIS3, FAM46C) and DNA damage repair (TP53, ATM). Multihit mutations were annotated when different SNV and/or indels (meeting all the previously mentioned requirements and passing filters to be considered relevant) were observed within the same gene. Clonality and subclonality of genomic alterations were defined as >80% or ≤80% by FISH and >40% or ≤40% by NGS, respectively. The CoMMpass data set was used as a validation cohort. All reported P values were obtained by a two-sided exact method, at the conventional 5% significance level (p < 0.05), correcting for multiple comparisons when needed. Confidence intervals (CI) for mean and median values were calculated at the standard 95% level.
Results
Patient characteristics
From the 90 patients included in the trial, 30 were initially excluded from NGS studies due to insufficient BM PC to perform CD138 positive selection or due to insufficient genomic DNA obtained after cell selection. Other 3 patients were excluded due to low total read counts obtained after sequencing. A total of 57 patients (63%) were successfully characterized using our custom NGS panel.
Clinical variables of the 57 patients are described and compared with the entire cohort of 90 patients in Table S1. Overall, the median age at diagnosis was similar (59 years) with identical distribution of cytogenetic risk categories. Other biochemical parameters were also comparable. According to the 2014 IMWG definition of smoldering and symptomatic disease, 44 patients met the current criteria to be considered HR SMM at diagnosis, while the remaining 13 cases, originally classified as UHR SMM, had active disease at baseline following the current criteria. Median follow-up after inclusion in the trial (cutoff date: October 31st, 2022) was 64.5 months (range: 23.7‒82). Six patients died; only 2 of them had previously experienced clinical progression (Figure S1). Biochemical relapses, including biochemical progression, MRD conversion from negative to positive at any time, and relapse from CR, occurred in 5, 4, and 11 patients, respectively. Projected 60-month bPFS, PFS, and OS were 66.2, 89.3, and 91.1%, respectively.
In addition to the clinics and the aforementioned three criteria used for their current identification, UHR SMM patients were associated with Bence-Jones (BJ) disease (3/4 BJ cases in this series were UHR, p = 0.023), but other parameters were similarly distributed between HR and UHR SMM cases.
Mutational profile of high-risk and ultrahigh-risk SMM patients
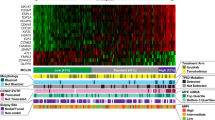
Overall, 96 SNV and indels were identified after filtering. A visual summary of SNV and indels is shown in Fig. 1. The median number of alterations per patient was 1 (range: 0‒9), with 11/57 patients (19.3%) harboring no alterations in the studied genes. Moreover, VAF from SNV and indels indicated that most alterations (69%) were subclonal (Fig. 2).

Both genomic and clinical data were integrated for each patient, represented by individual columns. Single nucleotide variants and indels are listed per gene, grouped based on the corresponding molecular pathway. Genes that do not belong to specific pathways were included in the category “other pathways”. Multihit mutations (i.e. several mutations in the same gene) are represented with an asterisk. The 8 structural variants evaluated by FISH were also incorporated as an additional molecular group in light green (positive), grey (negative) or white (not tested). On the top, the tumor mutation burden combining NGS and FISH is plotted, distinguishing between molecular pathways. On the lower side of the figure, diagnosis, MRD dynamics, biochemical progression, clinical progression and death events are color-coded. On the right side of the figure, the global percentage of each genomic event and the corresponding absolute number of altered patients are represented. bPFS biochemical progression-free survival, HR SMM high-risk smoldering myeloma, MAPK mitogen-activated protein kinase, MRD minimal residual disease, NF-κB nuclear factor-κB, TMB tumor mutation burden (total number of events per patient), transl translocation, UHR SMM ultrahigh risk smoldering myeloma.

A Represented with blue boxplots, Variant allele frequencies of single-nucleotide variants or indels, corrected based on FISH results. Note that local copy numbers were not evaluated and therefore not used to calculate cancer clonal fractions. B Represented with red boxplots, the proportion of altered cells as detected by FISH. Each event is represented by a blue/red dot. Those genes altered only once in our cohort were excluded. Clonality and subclonality of genomic alterations were defined as >80% or ≤80% by FISH, and as >40% or ≤40% by NGS, respectively. del: deletion; FISH: fluorescent in-situ hybridization.
First, we compared HR and UHR SMM patients (Table S2). TRAF3 mutations were exclusive of UHR SMM (3/13 vs 0/44, p = 0.01), while other mutations were equally distributed. Based on this result, further evaluations were performed, including all 57 patients. The mutational profile of our patients was enriched in alterations involving well-known drivers in MM, such as KRAS (17.5%), NRAS (12.3%), DIS3 (15.8%) or FAM46C (7%) (Table 1). When genes were grouped together based on their corresponding signaling pathways, MAPK gene mutations were the most abundant (52.6%), followed by those belonging to RNA and protein processing (22.8%), NF-κB (15.8%), DNA repair (10.5%), B-cell development (5.3%) and cell cycle (1.3%) pathways (Table 1). In 7 patients, we identified more than one SNV and/or indel in the same gene (multihit mutations), which were generally restricted to functionally relevant domains of the coding protein (Fig. 1, Table 2).
In addition, 56 chromosomal aberrations were detected by FISH (Fig. 1, Table 1). Combining NGS and FISH, only 3 patients had no genomic alterations detected. High-risk cytogenetic aberrations [t(4;14), t(14;16), and/or del17p] were identified in 13 cases, with two patients harboring both t(4;14) and del17p. Gains/amplifications of 1q and 1p deletions were detected in 51.9% and 3.8% of cases, respectively. Out of the 33 patients that could be tested for t(11;14), 4 (12.1%) were positive. Cytogenetic alterations involving 8q24 locus were evaluated in 40 patients and included the detection of 5 cases with 8q24 gains (12.5%) and 2 cases with 8q24 translocations (5%) involving the IGH locus. Looking at FISH aberrations shown in Fig. 2, t(11;14) and t(4;14) had high median clonal percentages, as expected for initiating events in myelomagenesis. Clonal proportions of +1q were also higher as compared to other structural events. Again, no significant differences between cytogenetic profiles of HR and UHR SMM patients were identified.
In terms of concurrent alterations (Fig. 3), FGFR3 mutations were frequently accompanied by t(4;14): out of 11 t(4;14) positive patients, 8 had FGFR3 mutations, and 5 of these consisted on amino acid substitutions to Cysteine residues in one of the three extracellular subdomains (Table S3). KRAS mutations and 1q alterations were mutually exclusive. NRAS mutations often co-occurred with t(11;14). 8q24 gains were associated with t(4;14) and DIS3 mutations. Finally, DUSP2 mutations specifically co-occurred with 8q24 gains and FGFR3 mutations. No double-hit events involving TP53 were identified in our cohort. Only one patient had a CCND1 mutation, with a concurrent t(11;14) translocation. In the same line, the only patient that showed a MAF mutation was the only t(14;16) positive one.

Colors blue and red are used to depict positive or negative associations, respectively. P-values were adjusted for significant associations at the levels of 0.05 (black circle) and 0.1 (white circle).
The mutational profile of high-risk SMM patients is intermediate between SMM and MM
Comparing our cohort of HR/UHR SMM with previously published series of SMM and symptomatic MM, we could observe that the burden of genomic aberrations consistently increased across the three entities, with HR/UHR SMM representing an intermediate link between low/intermediate SMM and MM (Table 1). Compared to previous observations in general populations of SMM, HR/UHR SMM patients showed an enrichment in DIS3, FAM46C, PRKD2, NF1, ZNF292 and ACTG1 mutations, as well as more +1q and t(4;14). Compared with a large MM cohort, our patients only showed a significantly lower incidence of 1p deletions. Only FGFR3 mutations were significantly enriched in HR/UHR SMM compared to the other two entities (p = 0.004 and p < 0.0001 compared with SMM, p = 0.001 compared to MM).
Comparing cell signaling pathways (Table 1), we found a higher proportion of mutations in the MAPK (52.6%), RNA processing (22.8%) and NF-κB (15.8%) pathways in our cohort, compared to those reported by Boyle and colleagues (24%, 3.7% and 4.9%, respectively). Conversely, in the other two cohorts, the proportions of each altered pathway were similar, probably indicating that patients described by Boyle et al. were mostly low-intermediate risk SMM.
Molecular and clinical associations in high-risk SMM
Evaluating specific subgroups of patients, we identified correlations between clinical and biological features. As mentioned above, the 13 UHR SMM patients were associated with BJ disease (p = 0.015) and, importantly, TRAF3 mutations were exclusive of this group, specifically of those with a sFLCr > 100. In the CoMMpass series, we checked the association between TRAF3 mutations and a sFCLr > 100 at diagnosis: 47/409 cases with a high sFLCr also had TRAF3 mutations, compared to only 21/483 cases with a lower sFLCr (11.5% vs 4.3%, p < 0.0001). Moreover, in our UHR SMM patients the presence of multihit mutations was enriched (30.7% of 13 UHR vs 6.8% of 44 HR patients, p = 0.041). No other mutation was specific to any clinical characteristic of our patients, although those with no somatic mutations detected with our panel most often had ≤ 20% BM PC infiltration, compared to mutated patients (8/11 vs 14/46, p = 0.015). Five out of the seven patients with multihit mutations had sustained positive minimal residual disease (MRD) assessed by next-generation flow cytometry (median sensitivity 2·10−6) or converted from undetectable to detectable MRD at any time point (p = 0.21). In cases with high-risk cytogenetics, compared to those with a standard-risk profile, the mean number of point mutations per patient was higher [2.62 (95% CI: 1.08‒4.15) vs 1.40 (95% CI: 1.03‒1.78); p = 0.021], as it was the mean serum M-protein levels [3.47 g/dL (95% CI: 2.26‒4.71) vs 2.34 g/dL (95% CI: 1.95‒2.73); p = 0.017].
Genomic predictors of treatment resistance and progression in high-risk SMM patients
For survival analyses, HR and UHR SMM were independently evaluated. First, we tested whether previously identified risk factors of progression would be still significant in the context of HR SMM patients receiving an intensive regimen. Neither t(4;14) alone, combined MAPK mutations, DNA repair pathway gene mutations nor structural variants affecting the 8q24 locus could effectively discriminate outcomes in the Kaplan‒Meier plots (Figure S2A-D). In contrast, we discovered new potential prognostic factors for these patients: the combination of t(4;14) plus FGFR3 mutation, as well as NRAS mutations, were both significantly associated with lower bPFS rates in univariate analyses; conversely, patients with KRAS mutations showed a trend towards longer bPFS (Fig. 4A-C).

Biochemical progression-free survival curves of the 44 high-risk SMM cases were plotted based on the presence (red) or absence (black) of different alterations at baseline. The 13 ultrahigh-risk patients were not considered here. (A) KRAS mutations; (B) NRAS mutations; (C) concurrent FGFR3 mutation and t(4;14). The number of patients for each category is shown in brackets. bPFS biochemical progression-free survival SV structural variant, WT wild type.
Among the 13 cases with UHR MM, only those with multihit mutations (Figure S3) showed a trend towards shorter bPFS rates (median: 20.10 months vs not reached, p = 0.053). TRAF3 mutations did not reach statistical significance, probably given the short size of the UHR MM subset, but in the MMRF’s CoMMpass cohort their presence predicted significantly improved outcomes as compared to TRAF3 wild-type patients (Fig. 5A-C).

A Survival plot of the 13 ultrahigh-risk patients in our cohort showed that patients with TRAF3 mutations may have improved outcomes, although this did not reach significance in our series. However, the prognostic significance of TRAF3 mutations was later explored in the CoMMpass series for confirmation. B Globally, TRAF3 wild-type patients in the CoMMpass cohort had a significantly worse PFS compared to TRAF3 mutated patients. C From the CoMMpass series, patients that had a sFLCr > 100 at diagnosis and experienced disease progression at any time (N = 181) were selected. In this high-risk subpopulation, TRAF3 wild-type patients also showed dismal prognosis with a significantly shorter TTP. CI Confidence interval, bPFS biochemical progression-free survival, HR hazard ratio, PFS progression-free survival, TTP time to progression, WT wild type.
Due to the low number of UHR cases, a multivariate Cox regression analysis was carried out only for the 44 HR SMM patients, including those variables with statistically significant impact in univariate analyses: NRAS mutations, combined t(4;14) plus FGFR3 mutations, the age at inclusion in the trial, and a high-risk cytogenetic status. Three variables retained its independent prognostic impact on bPFS: the combined presence of t(4;14) and FGFR3 mutations (HR: 6.58, 95% CI: 1.84‒23.47, p = 0.004), the age at inclusion (HR: 1.16, 95% CI: 1.04‒1.30, p = 0.006) and NRAS mutations (HR: 5.39, 95% CI: 1.54‒18.80, p = 0.008).
Discussion
In this study, we used NGS and FISH to analyze DNA from CD138 + BM PC obtained at diagnosis from 44 HR SMM and 13 UHR SMM patients. Unlike previous reports, these patients were treated with a combination of carfilzomib, lenalidomide and dexamethasone, followed by transplantation, consolidation, and maintenance, enabling us to determine the mutational features of this specific group of patients at baseline and their relationship with clinical outcomes in the context of an intensive treatment regimen.
Here and for the first time, we have shown how the intensive treatment of HR SMM patients may abrogate the negative clinical impact previously associated with certain genetic features. Thus, MAPK pathway somatic mutations (NRAS/KRAS/BRAF mutations), structural alterations in 8q24 (gains or translocations) and DNA repair pathway aberrations (TP53 and ATM mutations, as well as 17p deletions), in contrast to previous findings by Bustoros et al. [9], did not predict early biochemical or clinical progressions. In our cohort, only NRAS mutations, not KRAS, and the co-occurrence of t(4;14) and FGFR3 mutations were associated with a higher incidence of biochemical progressions in the multivariate survival analysis. If this preliminary observation is validated in larger cohorts with longer follow-up that could account for PFS or OS, these markers could be routinely used to identify high-risk disease at baseline for which conventional therapy would not be effective enough, perhaps indicating that targeted therapies could be added to improve outcomes.
The double-hit alteration of chromosome 4 in patients with t(4;14) plus FGFR3 mutation has been reported before [25, 26]. In addition, 4 of the FGFR3 mutations reported here are already classified as activating in MM as well as in other hematological malignancies, solid tumors and skeletal disorders. Notably, Stong et al. have recently described that FGFR3 mutations are exclusive of t(4;14) positive, newly-diagnosed MM patients, although the combination did not impact survival [27]. On the contrary, an additive negative prognostic value of FGFR3 mutations to t(4;14) patients was reported in an MMRF CoMMpass analysis [28]. The specific enrichment of the double-hit alteration in HR/UHR patients was, to the best of our knowledge, not described to date. Clonal fractions detected by NGS and FISH suggest that SNV are always preceded by the translocation, although this needs further confirmation. Surprisingly, 5 patients positive for t(4;14) harbored FGFR3 missense mutations changing the wild-type amino acid to Cysteine, and 4 of them progressed. Since mutant Cysteine residues in the extracellular domain lead to ligand-independent dimerization of FGFR3 on the cell surface [29], we believe this type of SNV may result in the constitutive activation of the receptor in MM, contributing to cell proliferation and survival. If replicated in functional assays, precision medicine using FGFR3 inhibitors for these patients could be explored as a potential treatment option [30].
The molecular characterization of UHR patients represents a novel finding. In our series, these patients seem to be associated with light-chain disease, enrichment of TRAF3 alterations, and multihit mutations. The role of TRAF3 mutations in MM has not been fully elucidated; while several publications indicate that they are a marker of aggressive disease and resistance to proteasome inhibitors [31], data from both the CoMMpass cohort and ours suggest that combined therapy, including proteasome inhibitors may ameliorate outcomes in these patients, in line with some preclinical studies [32]. Concerning multihit mutations in our cohort, our data suggest that the presence of multiple SNV targeting the same gene in the same patient may be a consequence of a more complex and unstable genomic landscape, perhaps with a higher baseline subclonal diversity [33], that may contribute to survival and a higher chance of treatment resistance (4 cases were always MRD positive and 1 converted from MRD negative to positive before starting maintenance). However, additional experiments (transcriptomics, functional assays, single-cell studies) would be mandatory to confirm this hypothesis.
In general terms, mutational frequencies in our cohort were closer to those of MM patients [16, 18, 19], but the allele frequencies of the corresponding point mutations were lower in HR/UHR SMM patients, most likely evidencing these are late events that remain at the subclonal level even in aggressive forms of asymptomatic disease. Chromosomal alterations considered as founder [t(11;14), t(4;14)] or early genetic events (+1q) were frequent and showed high clonal fractions in most cases, while secondary structural aberrations (del17p, 8q24 alterations or 1p deletions) were more rarely seen and showed reduced clonal fractions, supporting the notion that they tend to appear later on the evolution of the disease and mostly contribute to tumor progression [6, 9, 10, 17]. Certain aberrations were specifically enriched in HR SMM compared to the general SMM population [FGFR3, DIS3, FAM46C or PRKD2 mutations; t(4;14) and +1q], some of them already identified as genomic markers of disease aggressiveness [18, 34, 35]. On the contrary, biallelic TP53 alterations or 1p deletions were absent or infrequent in our series, and they seem to happen at later stages. However, in our cohort, the overall frequency of TP53 alterations (mutations or deletions: 8.9%) was similar to that observed in newly diagnosed MM patients, while 1p deletions were clearly underrepresented [19, 36, 37].
There are several limitations to note in our study. First, the lack of whole-exome or whole-genome sequencing data prevented the evaluation of large genomic regions and hampered the calculation of cancer clonal fractions, including corrections for local copy numbers. In addition, the availability of CD138+ cells also limited the number of probes used in FISH studies. Also, the panel design did not include either a set of single-nucleotide polymorphisms to track CNA or corrections based on matched sequencing of non-tumor cells. For these reasons, most genomic aberrations (hyperdiploidy, many CNA, complex structural changes, etc.) and mutational signatures have been missed, making our conclusions incomplete. Second, the number of patients analyzed is limited, but our results may pave the way for larger biological studies in phase III clinical trials treating a higher number of HR SMM patients to evaluate their complete genomic architecture, interactions between different alterations, and their prognostic value. Third, UHR SMM cases were recruited because the trial was designed before the updated diagnostic criteria were published in 2014. In addition, we evaluated reference series for SMM and MM that used heterogeneous methodologies for genomic analyses. Overall, this makes difficult to compare our results with other series. Finally, the current follow-up is insufficient to evaluate PFS and OS of SMM patients, in light of the high response rates and improved outcomes achieved with intensive regimens that most probably will translate into decades of survival for many patients [13, 14, 38].
In summary, our findings show how most genetic alterations have already been acquired at early stages of the disease, with a similar makeup of HR SMM compared to symptomatic MM. HR SMM is nonetheless enriched in specific alterations that could partially explain its inherent aggressiveness. In addition, the presence of t(4;14) plus FGFR3 mutations, or NRAS mutations, could be used to predict resistance, disease progression, and to discriminate which patients are suitable for intensive treatment strategies or targeted therapies.
Data availability
Raw data obtained from the NGS panel is freely available to any researcher wishing to use them for non-commercial purposes, without breaching participant confidentiality, in the European Nucleotide Archive repository (https://www.ebi.ac.uk) under Accession Project Number PRJEB72353.
References
Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, et al. Clinical course and prognosis of smoldering (Asymptomatic) multiple myeloma. N Engl J Med. 2007;356:2582–90.
International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–57.
Pérez-Persona E, Vidriales MB, Mateo G, García-Sanz R, Mateos MV, de Coca AG, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110:2586–92.
Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538–48.
Mateos MV, Kumar S, Dimopoulos MA, González-Calle V, Kastritis E, Hajek R, et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 2020;10:102.
Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14:100–13.
Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun. 2019;10:3835.
Oben B, Froyen G, Maclachlan KH, Leongamornlert D, Abascal F, Zheng-Lin B, et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat Commun. 2021;12:1861.
Bustoros M, Sklavenitis-Pistofidis R, Park J, Redd R, Zhitomirsky B, Dunford AJ, et al. Genomic profiling of smoldering multiple myeloma identifies patients at a high risk of disease progression. J Clin Oncol. 2020;38:2380–9.
Boyle EM, Deshpande S, Tytarenko R, Ashby C, Wang Y, Bauer MA, et al. The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat Commun. 2021;12:293.
Lonial S, Jacobus S, Fonseca R, Weiss M, Kumar S, Orlowski RZ, et al. Randomized trial of lenalidomide versus observation in smoldering multiple myeloma. J Clin Oncol. 2020;38:1126–37.
Mateos MV, Hernández MT, Giraldo P, de la Rubia J, de Arriba F, Corral LL, et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016;17:1127–36.
Mateos MV, Martinez Lopez J, Rodríguez-Otero P, Gonzalez-Calle V, Gonzalez MS, Oriol A, et al. Curative Strategy (GEM-CESAR) for High-Risk Smoldering Myeloma (SMM): Carfilzomib, Lenalidomide and Dexamethasone (KRd) As Induction Followed By HDT-ASCT, Consolidation with Krd and Maintenance with Rd. Blood. 2021;138:1829–1829.
Kumar SK, Alsina M, Laplant B, Badros AZ, Abdallah AO, Abonour R, et al. Fixed Duration Therapy with Daratumumab, Carfilzomib, Lenalidomide and Dexamethasone for High Risk Smoldering Multiple Myeloma-Results of the Ascent Trial. Blood. 2022;140:1830–2.
Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell. 2014;25:91–101.
Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997.
Bolli N, Biancon G, Moarii M, Gimondi S, Li Y, de Philippis C, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32:2604–16.
Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587–97.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Lopez-Corral, Mateos L, Corchete LA MV, Sarasquete ME, de la Rubia J, de Arriba F, et al. Genomic analysis of high-risk smoldering multiple myeloma. Haematologica. 2012;97:1439–43.
on behalf of the International Myeloma Working Group, Chng WJ, Dispenzieri A, Chim CS, Fonseca R, Goldschmidt H, et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia. 2014 ;28:269–77.
Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–56.
Chesi M, Nardini E, Brents LA, Schröck E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–4.
Kalff A, Spencer A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: prognostic implications and current clinical strategies. Blood Cancer J. 2012;2:e89–e89.
Stong N, Ortiz-Estévez M, Towfic F, Samur M, Agarwal A, Corre J, et al. The location of the t(4;14) translocation breakpoint within the NSD2 gene identifies a subset of patients with high-risk NDMM. Blood. 2023;141:1574–83.
Benard B, Christofferson A, Legendre C, Aldrich J, Nasser S, Yesil J, et al. FGFR3 Mutations Are an Adverse Prognostic Factor in Patients with t(4;14)(p16;q32) Multiple Myeloma: An Mmrf Commpass Analysis. Blood. 2017;130:3027–3027.
Qing J, Du X, Chen Y, Chan P, Li H, Wu P, et al. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J Clin Invest. 2009;119:1216–29.
Facchinetti F, Hollebecque A, Bahleda R, Loriot Y, Olaussen KA, Massard C, et al. Facts and New Hopes on Selective FGFR Inhibitors in Solid Tumors. Clin Cancer Res. 2020;26:764–74.
Neja SA. The roles of TRAF3 mutation in the oncogenic progression and drug response of multiple myeloma. Genome Instab Dis. 2020;1:278–85.
Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous Mutations Activate the Noncanonical NF-κB Pathway in Multiple Myeloma. Cancer Cell. 2007;12:131–44.
Melchor L, Brioli A, Wardell CP, Murison A, Potter NE, Kaiser MF, et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia. 2014;28:1705–15.
Schmidt TM, Fonseca R, Usmani SZ. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J. 2021;11:83.
Boyle EM, Ashby C, Tytarenko RG, Deshpande S, Wang H, Wang Y, et al. BRAF and DIS3 Mutations Associate with Adverse Outcome in a Long-term Follow-up of Patients with Multiple Myeloma. Clin Cancer Res. 2020;26:2422–32.
Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol. 2015;33:3911–20.
Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33:159–70.
Kazandjian D, Hill E, Dew A, Morrison C, Roswarski J, Korde N, et al. Carfilzomib, Lenalidomide, and Dexamethasone Followed by Lenalidomide Maintenance for Prevention of Symptomatic Multiple Myeloma in Patients With High-risk Smoldering Myeloma: A Phase 2 Nonrandomized Controlled Trial. JAMA Oncol. 2021;7:1678.
Acknowledgements
This work was partially supported by Amgen, the Asociación Española Contra el Cáncer (AECC, PROYE20047GUTI), the Instituto de Salud Carlos III (ISCIII, PI21/01917 and PMP21/00015), the Centro de Investigación Biomédica en Red (CIBERONC CB16/12/00233, CB16/12/00284, CB16/12/00369 and CB16/12/00489), the Cancer Research UK (C355/A26819) and FC AECC and AIRC under the Accelerator Award Program, and “Una manera de hacer Europa” (Innocampus; CEI-2010-1-0010). CJ was supported by the ISCIII (CD19/00030), AM-H by the European Social Fund through the IBSAL, the ISCIII (FI19/00320), the International Myeloma Society and the Spanish Society of Hematology (co-funded by Janssen). All Spanish funding is co-sponsored by the European Union FEDER program. MVM is the president of the Spanish Society of Hematology. JJL is the president of the GEM-PETHEMA group.
Author information
Authors and Affiliations
Consortia
Contributions
JML, MJC and RGS conceived and designed the study. NCG, VGC, PRO, AO, LR, AA, FE, JDLR, AIT, FDA, MTH, JLJ, EMO, NP, JJL, JB, JFSM, MVM, JML, and RGS recruited patients to the clinical trial. AMH, IV, IC, JMRR, BA, CJ, MF, MJL, NCG, MFG, NP, and BP produced and analyzed molecular, cytogenetics and flow cytometry results. AMH, IV, IC, JMRR, BA, and CJ reviewed the data and performed the statistical analyses. AMH, CJ, RGS, and MVM wrote the first draft of the manuscript. All the authors reviewed the final manuscript and agreed on its publication.
Corresponding author
Ethics declarations
Competing interests
The authors report personal fees from, or have consulted or served in an advisory role for Janssen (AMH, MVM, PRO, VGC, AO, LR, AA, JDLR, FDA, BP, EMO, JJL, JFSM, JML, MJC, RGS), BMS-Celgene (MVM, PRO, VGC, AO, LR, AA, JDLR, FDA, BP, EMO, JJL, JFSM, JML, MJC, RGS), Amgen (MVM, PRO, VGC, AO, LR, AA, JDLR, FDA, BP, EMO, JJL, JFSM, MJC), Takeda (MVM, AO, LR, AA, JDLR, PRO, BP, EMO, JJL, JFSM, RGS), Sanofi (MVM, AA, PRO, JDLR, FDA, BP, EMO, JJL, JFSM), GSK (MVM, PRO, AA, JDLR, FDA, EMO, BP, JFSM), Oncopeptides (MVM, AA, PRO, EMO), Regeneron (AA, EMO, MVM, PRO, JFSM), MSD (EMO, JFSM), AbbVie (EMO, PRO, MVM, JFSM), Pfizer (AA, EMO, PRO, JDLR, MVM), Roche (MVM, BP, JFSM), Karyopharm (JDLR, EMO, JFSM), Secura-Bio (EMO, JFSM), Novartis (JFSM, JML, MJC), Adaptive Biotechnologies (BP, MVM), Beckton-Dickinson (BP), Menarini-Stemline (EMO), Creative BioLabs (BP), H3 Biomedicine (PRO), Invivoscribe (AMH), Sea-Gen (MVM), Blue-Bird bio (MVM), Hospira (RGS), Pharmacyclics (RGS), Gilead (PRO, RGS) and Incyte (RGS).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Medina-Herrera, A., Vazquez, I., Cuenca, I. et al. The genomic profiling of high-risk smoldering myeloma patients treated with an intensive strategy unveils potential markers of resistance and progression. Blood Cancer J. 14, 74 (2024). https://doi.org/10.1038/s41408-024-01053-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-024-01053-3
