
Abstract
Myelodysplastic syndromes (MDS) are a heterogeneous group of hematopoietic stem cell disorders characterized by ineffective hematopoiesis with abnormal blood cell development (dysplasia) leading to cytopenias and an increased risk for progression to acute myeloid leukemia (AML). Patients with MDS can generally be classified as lower- (LR-MDS) or higher-risk (HR-MDS). As treatment goals for patients with LR-MDS and those with HR-MDS differ significantly, appropriate diagnosis, classification, and follow-up are critical for correct disease management. In this review, we focus on the diagnosis, prognosis, and treatment options, as well as the prediction of the disease course and monitoring of treatment response in patients with LR-MDS. We discuss how next-generation sequencing, increasing knowledge on mechanisms of MDS pathogenesis, and novel therapies may change the current treatment landscape in LR-MDS and why structured assessments of responses, toxicities, and patient-reported outcomes should be incorporated into routine clinical practice.
Similar content being viewed by others


Introduction
Myelodysplastic syndromes (MDS) comprise a heterogeneous group of hematopoietic stem and progenitor cell disorders characterized by ineffective hematopoiesis leading to dysplasia, cytopenias, and an increased risk of evolution to acute myeloid leukemia (AML) [1, 2]. MDS occurs in all age groups but mainly affects the elderly, with a median age of onset above 70 years [3, 4]. The majority of MDS diagnoses are “lower-risk” diseases (LR-MDS), indicating a relatively lower risk of death or progression to AML in the immediate period after diagnosis [5, 6]. However, the presence of anemia and complications related to cytopenias, transfusions, and inflammation can negatively affect comorbid conditions, potentially reducing the quality of life (QoL) and increasing the mortality of these patients relative to the general population [7, 8]. Molecular sub-characterization of MDS has emerged following the discovery of recurrent somatic driver mutations [9], though understanding of the mechanisms involved in clonal evolution and its impact on disease phenotype remains incomplete [10]. Together with the emergence of effective therapies for LR-MDS targeting disease-associated pathways and processes, e.g., involving transforming growth factor beta (TGF-β) signaling, DNA methylation, and other epigenetic targets, our understanding of the LR-MDS pathogenesis also advances [11].
Here we review the diagnosis, prognosis, and treatment—including treatment response monitoring—of patients with LR-MDS. We discuss emerging therapies and why structured assessments of responses, toxicities, and patient-reported outcomes (PROs) should be incorporated into guidelines and recommendations for daily clinical practice to improve clinical outcomes.
Diagnosis
Rapid and accurate diagnosis of MDS remains critical, and two new classification systems were recently proposed [12, 13]. The initial MDS assessment should provide data regarding disease prognostication and should inform about appropriate treatment choices. MDS diagnosis is based on the quantitative and qualitative assessment of a peripheral blood smear; bone marrow cytology and histology; cytogenetic and mutational analyses; and flow cytometry immunophenotyping [14]. Patients with suspected MDS should undergo a detailed medical history check on exposure to genotoxic agents (e.g., chemotherapy, therapeutic radiation, or organic solvents [e.g., benzene]). We also recommend assessing family history for potential signs of germline predisposition and constitutional stigmata (e.g., findings suggestive of telomere disease) and testing of germline tissue obtained through fibroblast cultures, when required. In addition, comprehensive molecular testing may identify patients with later onset germline mutations such as DDX41, some mutations in telomere disease, or RUNX1. This information should then be integrated with laboratory analyses (e.g., blood counts, peripheral blood smear, bone marrow aspirate/biopsy, cytogenetics, including a full karyotype, flow cytometry immunophenotyping, and mutational analysis) to exclude other conditions [15,16,17,18]. The International Working Group (IWG) for flow cytometry in MDS (IMDS Flow) of the MDS European LeukemiaNet (ELN) published guidelines for multi-parameter flow cytometry immunophenotyping in MDS outlining markers of particular interest [18,19,20,21] (Table S1). Finally, anemia symptoms, fatigue, bleeding, infections, and inflammation should be carefully assessed and checked during treatment. Numerous MDS diagnostic guidelines are available from several consortia, including the MDS ELN, European Society for Medical Oncology (ESMO), National Comprehensive Cancer Network® (NCCN®), and MDS-Right group [6, 15, 16, 22].
Until recently, the 2017 4th revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia provided guidelines for the diagnosis and subclassification of MDS (Table 1) [23] using an integrated approach based on clinical, hematologic, morphologic, genetic, flow cytometric, and molecular findings. A complete karyotype remains important in the diagnosis and prognosis of MDS; when conventional cytogenetics testing fails, fluorescence in situ hybridization (FISH) probes, for instance, of chromosomes 5, 7, 8, 17, and 20, can be useful in the prognostication and for monitoring disease response after treatment [24]. Subsequent data has underscored the importance of specific mutations in disease presentation and prognosis. Importantly, WHO classification may correlate with disease risk, but typically other prognostic tools are used for risk assessment (e.g., International Prognostic Scoring System [IPSS], Revised IPSS [IPSS-R], or molecular IPSS [IPSS-M]). For instance, some data suggest that SF3B1-mutant MDS, characterized by ring sideroblasts (RS), ineffective erythropoiesis, and an indolent clinical course, should be recognized as a distinct nosologic entity (Table 1) [23, 25]. This growing understanding of the molecular pathogenesis of disease has also identified overlapping features between MDS and other clonal marrow processes. This is particularly challenging in the era of molecular diagnostics, where clonal abnormalities may exist in the absence of MDS-defining dysplasia or cytogenetic criteria. Therefore, immunophenotypic or molecular alterations indicative of clonality have been recently introduced into the minimal diagnostic criteria for MDS in situations where morphological findings are insufficient (Fig. 1; Table S2) [23, 26]. These additions aim to support clinicians in finding the precise diagnosis for cases with inconclusive morphological and cytogenetic alterations [23, 26]. In patients with clonal hematopoiesis as well as cytopenias, distinguishing between clonally driven cytopenias and secondary causes of low blood counts remains challenging [26]. It is particularly important for patients with suspected LR-MDS, to distinguish MDS-related cytopenias from other causes of cytopenias presenting on top of background clonal hematopoiesis, including aplastic anemia, paroxysmal nocturnal hemoglobinuria, nutritional deficiencies, autoimmune disorders, and infections [9]. For instance, RS formation, which may masquerade as MDS, can also be associated with copper deficiency and alcohol dependency [27, 28]. Another condition that may be associated with MDS is the VEXAS syndrome, characterized by fever, inflammation, and vacuoles in hematopoietic cells and related to a mutation in the UBA1 gene [29].

Abbreviations: BM bone marrow, CCUS clonal cytopenias of uncertain significance, CHIP clonal hematopoiesis of indeterminate potential, ICUS idiopathic cytopenias of uncertain significance, IDUS idiopathic dysplasia of unknown significance, MDS myelodysplastic syndromes.
Recently, two updated classifications were published: the 5th edition of the WHO classification and the International Consensus Classification (ICC) of myeloid neoplasms and acute leukemias (Table 1) [12, 13]. There are minor differences between the classifications, which include the nomenclature of some MDS subgroups, minor variations in diagnostic thresholds, and several new diagnostic entities (Table 2). The ICC 2022 proposes categorizing MDS with single lineage dysplasia (MDS-SLD) and MDS with multilineage dysplasia (MDS-MLD), per the WHO 2017 revision, as MDS, not otherwise specified (MDS, NOS) with SLD or with MLD [13]. It also introduced a new MDS/AML category, defined as a cytopenic myeloid neoplasm with 10–19% blasts in peripheral blood or bone marrow, allowing patients to qualify for both MDS and AML clinical trials. The WHO 2022 revision replaced the term myelodysplastic syndromes with myelodysplastic neoplasms (still abbreviated as MDS) and regrouped MDS entities as MDS with defined genetic abnormalities and morphologically defined MDS [12]. It also categorizes MDS-SLD and MDS-MLD into a new category (MDS with low blasts; MDS-LB) and recognizes hypoplastic MDS (MDS-h) with <25% cellularity as a distinct entity. Both classifications replaced the MDS-RS category with the MDS with SF3B1 category [12, 13], however, the WHO 2022 classification also permits the use of the term MDS with low blasts and RS, if wildtype SF3B1 and ≥15% RS are present [12]. Both classifications include MDS-TP53 as a separate entity, recognizing the generally poor outcomes in this molecular subset, while MDS-del(5q) remains the same. Notably, the MDS unspecified category from the WHO 2017 revision no longer exists in either 2022 classification system, as all subtypes now fit into one of the categories [12, 13]. The impact of the differences between the WHO and ICC 2022 classifications on clinical practice is not yet clear; for instance, the MDS-del(5q) and MDS with SF3B1 entities remain identical, and the ICC 2022 MDS/AML category overlaps with the WHO 2022 MDS with increased blasts (MDS-IB) entity and is similar to the MDS with excess blasts (MDS-EB) category from the previous WHO edition. In general, prognostic models such as the IPSS, IPSS-R, or IPSS-M continue to guide clinical decision-making.
Relevance of next-generation sequencing (NGS) and discrimination of pre-MDS conditions
Identification of somatic gene mutations and establishment of comprehensive mutational profiles of MDS samples using next-generation sequencing (NGS) plays a growing role in the diagnosis, prognosis, treatment selection, and monitoring of MDS [11, 30]. Importantly, relevant mutations affecting processes such as DNA methylation, pre-mRNA splicing, chromatin modification, transcription, and cell signaling may inform the development of new therapies. Clonal heterogeneity and its progressive evolution characterize many myeloid malignancies [31,32,33]. The presence of a clonal population at a median variant allelic fraction (VAF) of ~10%, can be identified in ~10% of adults aged >70 years with otherwise normal blood counts, and in up to 30% of those aged >80 years [34]; a phenomenon termed clonal hematopoiesis of indeterminate potential (CHIP) (Table S2) [16, 35]. These patients have a higher risk of subsequent hematologic malignancy and reduced overall survival (OS) compared with individuals without detectable mutations and a higher risk for adverse cardiovascular events and other degenerative-inflammatory age-associated disorders [34,35,36,37]. Clonal cytopenia of undetermined significance (CCUS; cytopenias with clonal mutation, but not meeting MDS diagnostic criteria), idiopathic cytopenia of uncertain significance (ICUS; cytopenias without a clonal mutation detected), and idiopathic dysplasia of unknown significance (IDUS; bone marrow dysplasia without a clonal mutation) have been described as “pre-MDS” conditions (Table S2) [16, 25, 35]. The risk of progression in patients with these forms of pre-MDS varies and is lower in those without identified evidence of clonal expansion, although ongoing prospective studies (e.g., SEARCH consortium) may better define this risk [16, 25, 35, 38].
Risk stratification
To assess disease severity and treatment eligibility, patients with MDS are generally stratified by both disease- and patient-based risk. The most common risk-scoring systems are the IPSS and IPSS-R [5, 39]. The IPSS scoring system classifies patients into four risk categories: Low, Intermediate-1, Intermediate-2, and High, based on the number of cell lineages affected by cytopenias, blast percentages, and cytogenetic alterations [39]. The IPSS-R scoring system places greater emphasis on the impact of cytogenetic risk and bone marrow blast percentage and defines five risk categories: Very low, Low, Intermediate, High, and Very high (Table 3) [5]. The IPSS score is still considered for patient allocation to treatment, as most clinical trials for current MDS treatments have relied on IPSS classification [17]. Currently, patients are stratified into having either LR-MDS (IPSS-R categories: Very low-, Low-, or Intermediate-risk with a score of ≤3.5 points), with treatment focused on improving symptomatic cytopenias, or HR-MDS (Intermediate-risk category with a score of >3.5 points, and High-, or Very high-risk categories), with treatment focused on prolonging survival and delaying AML progression [40]. Likewise, patient-specific characteristics (i.e., patient age, presence of comorbidities, performance status, and frailty [reduced physical fitness]) have prognostic relevance in evaluating treatment-related mortality and hence treatment selection, including allogeneic hematopoietic stem cell transplantation (HSCT) [8, 41].
Recently, the IPSS-M scoring system was introduced [42]. This model includes similar clinical, morphological, and cytogenetic parameters as IPSS-R, with additional genetic parameters (16 main effect genes and 15 residual genes) to classify patients into six risk categories: Very low, Low, Moderate low, Moderate high, High, and Very high. It outlines the recommended gene selection, sequencing, and analysis that allowed the identification of mutations present in 31 genes that, together with cytogenetic parameters, improve prognostic discrimination of patients compared with the IPSS-R model [42]. In practice, we assess these mutations to a level of 1–5% VAF, with consideration of larger NGS panels that may also assess other relevant mutations, such as DDX41. Importantly, some mutations (TP53multihit, FLT3, KMT2A [MLLPTD]) provide additional adverse prognostic risk, while others may suggest a more favorable disease course (SF3B1), though the outcome may be modulated by co-mutation patterns (Table 3) [42]. Although molecular features are increasingly involved in prognosis, it is important to consider how these can be intertwined with morphology; for instance, how the favorable association with SF3B1 mutations may not add independent prognostic value after accounting for RS—like in the case of the WHO 2017 categories of refractory anemia with RS (RARS) or refractory cytopenia with multilineage dysplasia and RS (RCMD-RS) [43, 44]. Of note, the IPSS-M model includes patients with therapy-related MDS (t-MDS), which arises following cytotoxic chemotherapy and radiation treatment of a neoplastic or non-neoplastic disorder, or both [42]. Patients with t-MDS have previously been categorized within the WHO classification system as having a type of therapy-related myeloid neoplasm, alongside patients with therapy-related AML (t-AML) and t-MDS/myeloproliferative neoplasms (MPN), and historically were considered to have universally poor outcomes. However, the IPSS-M was able to stratify them into different risk groups, suggesting that molecular drivers of the disease may improve risk assessment more than clinical history alone [42].
The general approach to the management of patients with LR-MDS
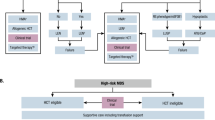
Following an appropriate MDS diagnosis and risk stratification, treatment is tailored toward the individual patient [10]. Most patients with LR-MDS will live with malignant hematopoiesis for many years, therefore, treatment goals focus on the improvement of disease-related symptoms and QoL. This is usually related to the management of cytopenias, most commonly anemia, and managing sequelae of disease and therapy (e.g., iron overload) [6, 15, 16, 22]. We, therefore, develop a disease management plan and treatment sequence with this in mind. Continuous development of diagnostics, therapies, and improving knowledge of MDS pathogenesis contribute to the evolving MDS management recommendations (e.g., ESMO [16, 45], ELN [15], NCCN® [22], and the MDS Europe platform [46]). Figure 2 outlines LR-MDS management recommendations based on ESMO guidelines [16, 45]. A recent study proposed 29 guideline-based indicators, defined as measurable elements in the areas of diagnosis, therapy, and care provider infrastructure, for the assessment of the quality of care, which is currently undergoing validation [17]. Nonetheless, such efforts underscore the importance of including patient-centered outcomes in MDS management.

Bold text indicates first-line therapy. aESMO 2014 [45]: RBC <2 per month or sEPO <500 U/L. bESMO 2014 [45]: if age <60–65 years and favorable features (including hypoplastic BM, blasts <5%, normal karyotype, HLA-DR15-positivity, younger age [<60 years], lower risk according to IPSS [10, 95]) for response to ATG; ESMO 2021 [16]: if age <65–70 years and favorable features for response to ATG. Abbreviations: ATG anti-thymocyte globulin, BM bone marrow, EPO erythropoietin, ESMO European Society for Medical Oncology, G-CSF granulocyte colony-stimulating factor, IPSS International Prognostic Scoring System, MDS myelodysplastic syndromes, MDS-RS myelodysplastic syndrome with ring sideroblasts, sEPO serum erythropoietin, TPO-RA thrombopoietin-receptor agonist.
Currently approved treatments for LR-MDS
The most common complication of LR-MDS is progressive anemia, which eventually leads to a requirement for regular red blood cell (RBC) transfusions [6, 47]. We administer erythropoiesis-stimulating agents (ESAs), which increase RBC production in the bone marrow, as first-line therapy for patients with LR-MDS and symptomatic anemia. There is, however, a significant variation in response quality (30–60%) and duration (1–2 years) with ESA use. Furthermore, as ESAs are not curative, eventually, patients will stop responding to therapy. Patients with low RBC transfusion requirement and serum erythropoietin (sEPO) below 200–500 mU/mL may be more likely to respond to ESAs, whereas those with high RBC transfusion requirement or high sEPO >500 mU/mL have a lower chance (<10%) of achieving a response [15, 16, 45]. A recent analysis of the EUMDS Registry study showed that patients with LR-MDS who received ESAs at the onset of anemia, but before starting RBC transfusion therapy, had improved survival, therefore supporting the consideration of early ESA treatment and further prospective validation of optimal ESA timing [48].
For some MDS subgroups, we consider other therapies in the frontline setting. The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) suggest that lenalidomide may be utilized as first- or second-line therapy for patients with MDS-del(5q) (typically ±1 other cytogenetic abnormality, excluding those involving chromosome 7) and transfusion dependency, or as second-line treatment after ESA failure. The NCCN Guidelines® also allow for a first-line trial of ESAs if desired [22], while ELN guidelines suggest a trial with growth factors before initiating lenalidomide [15]. Post hoc data show no differences in QoL with lenalidomide [49], and recent real-world studies in MDS-del(5q) have demonstrated long-term responses (from 21 to 32 months) and alleviation of anemia [50, 51]; nevertheless, further studies are needed to fully understand the impact of lenalidomide on QoL and to validate observed responses [49].
Generally, therapies such as lenalidomide have been reserved for transfusion-dependent patients; however, there are ongoing investigations exploring the possible benefits of starting treatments prior to transfusion dependence. An interim analysis of the phase 3 European Sintra-REV trial comparing lenalidomide to placebo in patients with non-transfusion-dependent del(5q) LR-MDS [52] showed that the patients receiving lenalidomide had a significantly longer time to transfusion dependence compared with patients receiving placebo (76 vs. 26 months; P = 0.021) [52]. However, a comparison to ESA would have been more in line with the current European guidelines.
For patients with non-del(5q) LR-MDS, there is less consensus on therapy options after the disease progresses while on ESA treatment or for those patients who are unlikely to respond to ESAs.
Responses to ESAs in combination with granulocyte colony-stimulating factors have been reported in specific subgroups, revealing an option for patients with insufficient ESA response, with the understanding that efficacy may be limited [53]. Additionally, two phase 3 trials have shown synergistic activity of epoetin alfa combined with lenalidomide compared with lenalidomide alone in patients without del(5q) who were not eligible for or were refractory to ESA [54, 55].
Another option for patients with MDS, particularly with RS—generally associated with mutations in SF3B1—who experience disease progression with ESA treatment, is luspatercept, which targets pathways associated with TGF-β signaling and enhances late-stage erythroid maturation [16]. ESMO 2021 and NCCN Guidelines® incorporate the use of luspatercept recognizing that patients with RS or SF3B1 mutations appear more likely to respond to this treatment [14, 16, 22]. The U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) have approved luspatercept for the treatment of patients following ESA therapy, particularly those with MDS with RS [16], as well as those with MDS/MPN with RS and thrombocytosis [56]. QoL was similar between patients receiving luspatercept and those receiving a placebo, despite a reduction in RBC transfusions, suggesting further work is needed to understand the impact of luspatercept on the patient experience [57]. The efficacy and safety of luspatercept is currently being compared to epoetin alfa in the ongoing phase 3 COMMANDS trial (NCT03682536), in RBC transfusion-dependent, ESA-naïve patients with LR-MDS with or without RS [58], and real-world experiences of MDS treatment with luspatercept are emerging. One study, which retrospectively evaluated luspatercept in MDS-RS patients in routine clinical practice, found limited value in securing durable anemia responses [59], while a single institution case series demonstrated potential clinical benefit in patients with LR-MDS with RS and SF3B1 mutation [60]. It needs to be noted however, that reported adverse events of bone pain and arthralgia warranted dose reduction or treatment suspension in some cases [60]. In other studies, thromboembolic events, and high blood pressure in patients with MDS and β-thalassemia treated with luspatercept have also been reported [61]. Further understanding of the long-term impact of luspatercept on patients with MDS, including the cost-effectiveness of this agent, remains to be addressed.
Patients’ dependence on regular RBC transfusions may lead to progressive iron overload, which can eventually affect multiple organs (i.e., liver, heart, and endocrine organs) and is known to reduce survival in hereditary transfusion-dependent anemias [62,63,64]. Event-free survival (EFS), iron overload, and safety of iron chelation therapy (ICT) with deferasirox were evaluated in patients with IPSS Low- and Intermediate-1 risk MDS in the randomized TELESTO trial [65]. The group receiving ICT showed superior EFS compared with the placebo; however, due to reduced patient enrollment (210 instead of 630), was insufficiently powered to answer the question of whether there was a survival benefit to ICT in MDS [65]. Multiple studies indicate an impact on other clinical endpoints, including cardiac and hepatic, marrow failure, and infections [47]. Guidelines recommend considering ICT for adults with serum ferritin levels >1000 µg/L, receiving >15–75 RBC units, and candidates for allogeneic HSCT, recognizing that preference should be given to minimizing iron overload by improving transfusion requirements with MDS medications where possible [15, 16, 47, 62, 63]. Potential side effects of current ICT must also be considered, including renal insufficiency and gastrointestinal disturbances (deferasirox), injection site reactions, ophthalmologic/ototoxicity (deferoxamine), and a risk of agranulocytosis with deferiprone (not approved by health authorities for this patient group in many jurisdictions) [47].
In patients with LR-MDS with anemia and other severe cytopenias, the selection of second-line therapies varies according to the mutation profile, specific cytopenias present, and blast counts. The hypomethylating agents (HMAs), azacitidine or decitabine, may be considered, but are often reserved for later lines of therapy unless another indication is present (e.g., excess blasts or evolution to higher-risk features) [66]. Limited efficacy and suboptimal trial design (i.e., poor patient selection, underdosing of one treatment arm in a trial comparing two HMAs) are important caveats to interpretation of data on HMAs in LR-MDS [67].
Highlighting the significance of a comprehensive MDS evaluation, to the identification of patients with MDS-h, a disease entity that may have some overlapping features with aplastic anemia [15], remains important. Indeed, it is now proposed as a distinct MDS subgroup in the WHO 2022 classification [12]. In these patients with LR-MDS, refractory cytopenia, and hypoplastic bone marrow (<25% cellularity), we consider immune-suppressive treatment with anti-thymocyte globulin (ATG), cyclosporine with or without thrombopoietin-receptor agonist (TPO-RA), analogous to the treatment of aplastic anemia [15, 68]. In patients with symptomatic thrombocytopenia, we may consider TPO-RAs, azacitidine, or androgens (Fig. 2). For patients with symptomatic neutropenia, treatments may include HMA or growth factor support at times of infections (Fig. 2).
Other factors guiding treatment selection may include age, patient-based risk factors, treatment goals, RBC transfusion dependence, lack or loss of response to first-line treatment, fibrosis, and somatic mutations [16]. Importantly, as none of these chemotherapeutic approaches are curative, patient participation in a clinical trial should be considered at any stage of treatment. Finally, allogenic HSCT may be considered for select patients with LR-MDS, particularly if they are young, failed multiple lines of therapy or treatment with HMAs, or if they present with higher-risk molecular features [69]. Notably, there are ongoing efforts to understand whether the new prognostic models (e.g., IPSS-M) can effectively risk stratifying patients with LR-MDS with high-risk features in order to recommend the most beneficial treatment options, including an allogeneic HSCT [70].
Assessment of response to treatment of LR-MDS
Historically, responses most relevant to LR-MDS included durable achievement of hematologic improvement (HI) or RBC-transfusion independence (RBC-TI), e.g., lasting ≥8 weeks. More recently, it is also recognized that response expectations may vary according to disease burden at the time of treatment initiation. For instance, in patients with high RBC transfusion burden (a receipt of ≥8 RBC U/16 weeks in ≥2 episodes), a 50% decrease in transfusions may be clinically meaningful, while for patients with lower RBC requirement at baseline (receipt of 3–7 RBC U/16 weeks in ≥2 episodes), achieving RBC-TI and improving baseline hemoglobin levels may be more meaningful. These considerations have led to a proposal for revisions to the IWG criteria for response assessment in LR-MDS, specifically pertaining to anemia and RBC transfusion needs [71]. These include defining a pre-treatment screening period of 16 weeks, dividing patients into three transfusion burden categories (non-transfused, low, and high transfusion burden), and an observation period of ≥16 weeks from treatment initiation for response assessment [71]. Improvement of QoL is relevant for patients with LR-MDS, and several PRO instruments focusing on QoL have been applied to patients with MDS [72]; however, there are limitations to the application of PROs, such as when they are administered and temporal events around their assessment (e.g., prior to or following transfusions). Additionally, the choice of instrument, frequency, and how this information should be applied to patient management, remains controversial [17]. Therefore, prospective assessment of standardized PROs in daily clinical care, including novel metric trackers (e.g., wearables), is urgently needed. The inclusion of PRO endpoints should be considered for future clinical trial design.
Emerging treatments for patients with LR-MDS
Numerous novel targets, which promise to change the LR-MDS treatment landscape, have recently been identified. Current studies in LR-MDS are outlined in Table 4, while specific therapeutics are outlined below.
Imetelstat
Imetelstat is a first-in-class competitive inhibitor of telomerase enzymatic activity. In the phase 2 part of the phase 2/3 IMerge study (NCT02598661), patients with LR-MDS refractory to, or ineligible for ESA treatment and with a high transfusion burden (≥4 RBC U/8 weeks), received intravenous imetelstat at a 7.5 mg/kg dose in a 2-h infusion every 4 weeks until disease progression. Overall, 37% of patients achieved the primary endpoint (RBC-TI for ≥8 weeks) [73]. The RBC-TI response was shown to be durable, with 42%, 32%, and 29% of patients achieving RBC-TI ≥ 8 weeks, ≥24 weeks, and ≥52 weeks, respectively [74]. The median and maximum RBC-TI durations were 20 months and 2.7 years, respectively [74]. A reduction in cytogenetic and mutational malignant clonal burden was observed in some patients, suggesting imetelstat’s disease-modifying activity [73], although further study is needed. The phase 3 part of the IMerge trial, comparing the efficacy of imetelstat versus placebo, has recently reached the recruitment target, and results are anticipated [75].
Roxadustat
Roxadustat is a hypoxia-inducible factor prolyl hydroxylase inhibitor approved in China for the treatment of anemia in patients with chronic kidney disease [76]. In a phase 3 study (NCT03263091), patients with non-del(5q) LR-MDS with <5% bone marrow blasts and low RBC transfusion burden (1–4 RBC U/8 weeks) received roxadustat (1.5, 2.0, or 2.5 mg/kg) orally three times weekly [77]. Roxadustat treatment resulted in RBC-TI lasting ≥56 consecutive days during the first 28 weeks of treatment in 37.5% of patients, while 54.2% achieved a ≥50% reduction in RBC transfusions [77]. At 1-year follow-up, the proportion of patients achieving RBC-TI ≥56 consecutive days remained at 37.5%, while the proportion of patients achieving a ≥50% reduction in RBC transfusions increased to 58.3% [77]. Subgroup analyses suggested that fewer patients with RS achieved RBC-TI for ≥56 consecutive days (23% vs. 55%), while baseline EPO had little effect on response (≤200 IU/L: 39%; 200–400 IU/L: 33%), although the sample size was small [77]. Notably, the FDA did not approve roxadustat for the treatment of anemia due to chronic kidney disease over concerns of increased risk of thrombotic and cardiovascular events [78].
Spliceosome modulators
Dysplasia-defining splicing factor mutations (e.g., SF3B1, SRSF2, and U2AF1) are found in over half of MDS patients and, therefore, are an appealing therapeutic target. Moreover, they tend to be early mutational events, and are mutually exclusive, suggesting that MDS cells do not tolerate multiple alterations in critical splicing factor proteins. H3B-8800, an orally available small molecule modulator of SF3B1, induced synthetic lethality in spliceosome-mutant cancer models [79]. It was tested in 84 patients with myeloid cancers (42 with HR-MDS or LR-MDS; 88% with spliceosome mutations of interest; NCT02841540) [80] and 14% of patients experienced reduced transfusion requirement (RBC or platelets), although marrow responses and changes in mutation burden were not seen [80]. Splicing modulators, or other targets essential to pre-mRNA splicing, such as protein arginine methyltransferase 5 (PRMT5) or ataxia telangiectasia and Rad3-related protein (ATR), are being actively investigated.
Oral HMAs
HMAs, or DNA methylation inhibitors (DNMTis), are used to treat patients with HR-MDS [14, 16, 22]. Important use limitations include the burden of treatment administration (subcutaneous) and local reactions, particularly in patients with LR-MDS for whom the burden of clinic visits relative to disease burden should be considered. However, oral administration of HMAs can allow for more flexible dosing and maintenance of patients’ autonomy. A phase 3 study of oral azacitidine (CC-486) versus placebo in patients with LR-MDS (NCT01566695) reported that 31% and 11% of patients, respectively, achieved the primary endpoint of RBC-TI ≥56 days [81]. Importantly, different formulations of azacitidine (oral vs. intravenous or subcutaneous) can have different pharmacokinetics, limiting them from being interchangeable, and different potential side effects, such as diarrhea with oral azacitidine (CC-586) and constipation with subcutaneous/intravenous azacitidine and its associated antiemetic regimens.
Combining oral cytidine deaminase inhibitors (e.g., cedazuridine) with oral DNMTi therapy allows for improved pharmacokinetics, similar to standard subcutaneous or intravenous DNMTi formulations. A combination of oral decitabine plus cedazuridine (ASTX727) was approved by the FDA for patients with IPSS Intermediate-1-, Intermediate-2-, and high-risk MDS, or chronic myelomonocytic leukemia, based on studies showing equivalence to intravenously administered decitabine (NCT02103478) [82]. A phase 1/2 study is currently evaluating the safety, pharmacodynamics, pharmacokinetics, and hematologic response to ASTX727 in patients with LR-MDS (NCT03502668). In phase 1 dose-escalation study in LR-MDS, a combination of oral azacitidine with cedazuridine (ASTX030) is also being assessed for equivalence with standard 7-day intravenous or subcutaneous azacitidine dosing (NCT04608110).
Immune-based therapies and inflammatory pathways in LR-MDS
Increasing evidence indicates that the pathogenesis and progression of MDS are influenced by immune mechanisms, suggesting that treatments that modulate the responses of innate and adaptive immunity by targeting immune checkpoints, tumor antigens (vaccines), and the inflammasome may be active [83, 84].
Allogeneic HSCT remains the only known curative approach for many myeloid malignancies, including MDS, thought in part related to a “graft-versus-leukemia” effect from immune mediator cells [85]. Novel approaches using immuno-oncology targets are being explored in MDS, either as monotherapy or in combination with azacitidine [86]. However, any immunotherapy approach will likely need to be more nuanced in MDS; for instance, inflammatory pathways have also been implicated in the progression and maintenance of clonal hematopoiesis and the disease context might be crucial for any therapeutics in this space [87, 88].
Tumor vaccines are promising with the hope of inducing an anti-tumor immune response in patients with LR-MDS. A pilot trial of the K562/GM-CSF (GVAX) vaccine in five patients with MDS (three with LR-MDS), reported a reduced transfusion requirement in one patient, and HI in another [89]. Further exploration of tumor vaccines, perhaps incorporating novel targets (e.g., mutation-specific moieties) or design (e.g., patient-specific mRNA vaccines) may lead to novel future treatments for MDS.
Finally, an increased understanding of MDS pathogenesis suggests a role of the Nod-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome activation, leading to cell death; NLRP3 inhibitors are in clinical development for LR-MDS treatment [84].
Conclusions and future directions
Management of patients with LR-MDS is increasingly nuanced, due to the heterogeneity of patient- and disease-based factors, and the expanding number of approved treatment options or combinations available. These patients will typically live with MDS for >3 years, and decisions around therapy depend on the burden of disease, symptomatic complications, mutational profile, and overall goals of therapy. The most common cytopenia in LR-MDS is anemia, but its degree and clinical impact on potential comorbid conditions vary. Several therapies are currently available for the treatment of patients with anemia due to MDS, and more are being evaluated, making an optimal selection of therapies and the sequence of interventions, more relevant to patient management. Increasingly, the use of NGS has refined prognostication and sometimes offers targeted therapeutic options. In the future, mutational profiles may be incorporated into risk stratification schemes and treatment algorithms, resulting in a more targeted treatment approach.
Notably, over the last 20 years, the number of clinical trials initiated for LR-MDS treatments has remained limited [90]. Furthermore, few agents are being developed specifically for MDS; many phase 1 trials investigate one drug for other cancers and may include MDS only as a subset of the study. Given the particularities around MDS management and response, such as the emergence of treatment-related cytopenias [91], exploration of novel therapeutics in MDS during early testing phases may be limited. To increase the number of potential treatment options and to maximize their chances for successful clinical development, factors, including the patient population characteristics, specific molecular targets and/or pathways involved in MDS pathology, and revision of relevant endpoints, need to be considered [90]. Improvement and standardization of molecular response criteria and PRO assessments will be fundamental for the development of new, effective, and tolerable therapies for LR-MDS. Although there are more potential therapies available than before, the progress remains slow. That said, there are reasons for optimism; our increasing understanding of MDS-associated molecular pathways, and a more refined understanding of clinically meaningful trial endpoints, suggest tangible ways to achieve improved clinical outcomes in LR-MDS patients in the near future.
Data availability
No datasets were generated or analyzed for this review paper.
References
Dao KT. Myelodysplastic syndromes: updates and nuances. Med Clin North Am. 2017;101:333–50.
Shastri A, Will B, Steidl U, Verma A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood. 2017;129:1586–94.
Sekeres MA, Schoonen WM, Kantarjian H, List A, Fryzek J, Paquette R, et al. Characteristics of US patients with myelodysplastic syndromes: results of six cross-sectional physician surveys. J Natl Cancer Inst. 2008;100:1542–51.
Bonadies N, Feller A, Rovo A, Ruefer A, Blum S, Gerber B, et al. Trends of classification, incidence, mortality, and survival of MDS patients in Switzerland between 2001 and 2012. Cancer Epidemiol. 2017;46:85–92.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
de Witte T, Malcovati L, Fenaux P, Bowen D, Symeonidis A, Mittelman M, et al. Novel dynamic outcome indicators and clinical endpoints in myelodysplastic syndrome; the European LeukemiaNet MDS Registry and MDS-RIGHT project perspective. Haematologica. 2020;105:2516–23.
Stauder R, Valent P, Theurl I. Anemia at older age: etiologies, clinical implications, and management. Blood. 2018;131:505–14.
Buckstein R, Wells RA, Zhu N, Leitch HA, Nevill TJ, Yee KW, et al. Patient-related factors independently impact overall survival in patients with myelodysplastic syndromes: an MDS-CAN prospective study. Br J Haematol. 2016;174:88–101.
Mufti GJ, McLornan DP, van de Loosdrecht AA, Germing U, Hasserjian RP. Diagnostic algorithm for lower-risk myelodysplastic syndromes. Leukemia. 2018;32:1679–96.
Chanias I, Stojkov K, Stehle GT, Daskalakis M, Simeunovic H, Njue LM, et al. Myelodysplastic syndrome in the postgenomic era and future perspectives for precision medicine. Cancers (Basel). 2021;13:3296.
Bonadies N, Bacher VU. What role can next-generation sequencing play in myelodysplastic syndrome care? Expert Rev Hematol. 2019;12:379–82.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36:1703–19.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of myeloid neoplasms and acute leukemia: integrating morphological, clinical, and genomic data. Blood. 2022;140:1200–28.
Chanias I, Bonadies N. Current standard of care in patients with myelodysplastic syndromes and future perspectives. Healthbook TIMES, Oncol Hematol. 2020;6:10–22.
Malcovati L, Hellström-Lindberg E, Bowen D, Adès L, Cermak J, Del Cañizo C, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122:2943–64.
Fenaux P, Haase D, Santini V, Sanz GF, Platzbecker U, Mey U, et al. Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32:142–56.
Stojkov K, Silzle T, Stussi G, Schwappach D, Bernhard J, Bowen D, et al. Guideline-based indicators for adult patients with myelodysplastic syndromes. Blood Adv. 2020;4:4029–44.
van de Loosdrecht AA, Kern W, Porwit A, Valent P, Kordasti S, Cremers E, et al. Clinical application of flow cytometry in patients with unexplained cytopenia and suspected myelodysplastic syndrome: a report of the European LeukemiaNet International MDS-Flow Cytometry Working Group. Cytometry B Clin Cytom. 2021; https://doi.org/10.1002/cyto.b.22044.
van der Velden VHJ, Preijers F, Johansson U, Westers TM, Dunlop A, Porwit A, et al. Flow cytometric analysis of myelodysplasia: pre-analytical and technical issues-recommendations from the European LeukemiaNet. Cytometry B Clin Cytom. 2021 Dec. https://doi.org/10.1002/cyto.b.22046. Online ahead of print.
Westers TM, Cremers EM, Oelschlaegel U, Johansson U, Bettelheim P, Matarraz S, et al. Immunophenotypic analysis of erythroid dysplasia in myelodysplastic syndromes. A report from the IMDSFlow working group. Haematologica. 2017;102:308–19.
Westers TM, Ireland R, Kern W, Alhan C, Balleisen JS, Bettelheim P, et al. Standardization of flow cytometry in myelodysplastic syndromes: a report from an international consortium and the European LeukemiaNet Working Group. Leukemia 2012;26:1730–41.
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Myelodysplastic syndromes. V1.2023. https://www.nccn.org/professionals/physician_gls/pdf/mds.pdf. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Myelodysplastic syndromes. V1.2023. © National Comprehensive Cancer Network, Inc. 2023. All rights reserved. Accessed November 21, 2022. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th ed. Lyon, France: IARC Press, 2017.
Killick SB, Ingram W, Culligan D, Enright H, Kell J, Payne EM, et al. British Society for Haematology guidelines for the management of adult myelodysplastic syndromes. Br J Haematol. 2021;194:267–81.
Malcovati L, Stevenson K, Papaemmanuil E, Neuberg D, Bejar R, Boultwood J, et al. SF3B1-mutant MDS as a distinct disease subtype: a proposal from the International Working Group for the Prognosis of MDS. Blood. 2020;136:157–70.
Valent P, Orazi A, Steensma DP, Ebert BL, Haase D, Malcovati L, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget. 2017;8:73483–73500.
D’Angelo G. Copper deficiency mimicking myelodysplastic syndrome. Blood Res. 2016;51:217–9.
Olcay L, Yetgin S Disorders mimicking myelodysplastic syndrome and difficulties in its diagnosis. In: (Ed.), Myelodysplastic Syndromes. IntechOpen: London, UK, 2016. https://doi.org/10.5772/64422
Templé M, Kosmider O. VEXAS Syndrome: a novelty in MDS landscape. Diagnostics (Basel). 2022;12:1590.
Swoboda DM, Gesiotto Q, Sallman DA. Novel therapies in myelodysplastic syndromes. Curr Opin Hematol. 2020;27:58–65.
Bonadies N, Rovó A, Porret N, Bacher U. When should we think of myelodysplasia or bone marrow failure in a thrombocytopenic patient? A practical approach to diagnosis. J Clin Med. 2021;10:1026.
Van Zeventer I, Salzbrunn JB, de Graaf AO, van der Reijden BA, Boezen HM, Vonk JM, et al. Prevalence, predictors, and outcomes of clonal hematopoiesis in individuals aged ≥80 years. Blood Adv. 2021;5:2115–22.
Galli A, Todisco G, Catamo E, Sala C, Elena C, Pozzi S, et al. Relationship between clone metrics and clinical outcome in clonal cytopenia. Blood. 2021;138:965–76.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–21.
Miller PG, Qiao D, Rojas-Quintero J, Honigberg MC, Sperling AS, Gibson CJ, et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood. 2022;139:357–68.
Osman AEWG. When are idiopathic and clonal cytopenias of unknown significance (ICUS or CCUS)? Hematol Am Soc Hematol Educ Program. 2021;2021:399–404.
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88.
Pfeilstöcker M, Tuechler H, Sanz G, Schanz J, Garcia-Manero G, Solé F, et al. Time-dependent changes in mortality and transformation risk in MDS. Blood. 2016;128:902–10.
Sorror ML, Storb RF, Sandmaier BM, Maziarz RT, Pulsipher MA, Maris MB, et al. Comorbidity-age index: a clinical measure of biologic age before allogenic hematopoietic cell transplant. J Clin Oncol. 2014;32:3249–56.
Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa J, Nannya Y et al. Molecular international prognosis scoring system for myelodysplastic syndromes. N Eng J Med Evid. 2022;1: https://doi.org/10.1056/evidoa2200008.
Tang Y, Miao M, Han S, Qi J, Wang H, Ruan C, et al. Prognostic value and clinical feature of SF3B1 mutations in myelodysplastic syndromes: a meta-analysis. Crit Rev Oncol Hematol. 2019;133:74–83.
Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood. 2012;119:569–72.
Fenaux P, Haase D, Sanz GF, Santini V, Buske C. ESMO Guidelines Working Group. Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii57–iii69.
MDS Europe. 2021. https://mds-europe.org/. Accessed May 2022.
Leitch H, Ezzat H, Merkeley H, Buckstein R, Zhu N, Nevill T, et al. MDS Iron Road: an internet-based algorithm for the diagnosis, workup and management of iron overload in MDS from the Canadian Consortium on MDS (CCMDS). Abstracts from the 16th International Congress on Myelodysplastic Syndromes; September 23–25, 2021; Toronto, ON, Canada. Abstract 75.
Garelius H, Smith A, Bagguley T, Taylor A, Fenaux P, Bowen D, et al. Erythropoietin stimulation agents significantly improves outcome in lower risk MDS. Abstract from the 2022 European Hematology Association (EHA) Annual Congress. HemaSphere. 2022;6(S3):S168.
Santini V, Almeida A, Giagounidis A, Platzbecker U, Buckstein R, Beach CL, et al. The effect of lenalidomide on health-related quality of life in patients with lower-risk non-del(5q) myelodysplastic syndromes: results from the MDS-005 study. Clin Lymphoma Myeloma Leuk. 2018;18:136–144.e7.
Gurnari C, Piciocchi A, Soddu S, Bonanni F, Scalzulli E, Niscola P, et al. Myelodysplastic syndromes with del(5q): a real-life study of determinants of long-term outcomes and response to lenalidomide. Blood Cancer J. 2022;12:132.
Singh A, Al-Kali A, Foran JM, Elliott MA, Begna K, Badar T, et al. Lenalidomide therapy for primary myelodysplastic syndromes with isolated del(5q): determinants of response and survival in a real-world setting. Am J Hematol. 2022;97:E377–E379.
López Cadenas F, Lumbreras E, Xicoy B, Sánchez J, Coll R, Slama B, et al. Phase 3 study of lenalidomide (LEN) vs placebo in non-transfusion dependent (TD) low risk del(5q) MDS patients—interim analysis of the European Sintra-REV trial. Abstract from the 2020 American Society of Hematology (ASH) Annual Meeting and Exposition. Blood. 2020;136:28–29.
Affentranger L, Bohlius J, Hallal M, Bonadies N. Efficacy of granulocyte colony stimulating factor in combination with erythropoiesis stimulating agents for treatment of anemia in patients with lower risk myelodysplastic syndromes: a systemic review. Crit Rev Oncol Hematol. 2019;136:37–47.
List AF, Sun Z, Verma A, Bennett JM, Komrokji RS, McGraw K, et al. Lenalidomide-epoietin alfa versus lenalidomide monotherapy in myelodysplastic syndromes refractory to recombinant erythropoietin. J Clin Oncol. 2021;39:1001–9.
Toma A, Kosmider O, Chevret S, Delaunay J, Stamatoullas A, Rose C, et al. Lenalidomide with or without erythropoietin in transfusion-dependent erythropoiesis-stimulating agent-refractory lower-risk MDS without 5q deletion. Leukemia. 2016;30:897–905.
Komrokji RS, Platzbecker U, Fenaux P, Garcia-Manero G, Mufti GJ, Santini V, et al. Efficacy and safety of luspatercept treatment in patients with myelodysplastic syndrome/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T): a retrospective analysis from the MEDALIST Study. Abstract from the 2020 American Society of Hematology (ASH) Annual Meeting and Exposition. Blood. 2020;136:13–15.
Oliva EN, Platzbecker U, Garcia-Manero G, Mufti GJ, Santini V, Sekeres MA, et al. Health-related quality of life outcomes in patients with myelodysplastic syndromes with ring sideroblasts treated with luspatercept in the MEDALIST phase 3 trial. J Clin Med. 2021;11:27.
Della Porta M, Platzbecker U, Santini V, Garcia-Manero G, Komrokji RS, Ito R, et al. The COMMANDS Trial: a phase 3 study of the efficacy and safety of luspatercept versus epoetin alfa for the treatment of anemia due to IPSS-R Very Low-, Low-, or Intermediate-Risk MDS in erythropoiesis stimulating agent-naive patients who require RBC transfusions. Abstract from the 2020 American Society of Hematology (ASH) Annual Meeting and Exposition. Blood. 2020;136:2198.
Farrukh F, Chetram D, Al-Kali A, Foran J, Patnaik M, Badar T, et al. Real-world experience with luspatercept and predictors of response in myelodysplastic syndromes with ring sideroblasts. Am J Hematol. 2022;97:E210–E214.
Khan S, Taveras Alam S, Torres Ramos R, Etumbani Mbue J, Apostplidou E, Rivero GA, et al. Luspatercept in low-risk myelodysplastic syndrome: a real-world single institution case series. Clin Hematol Int. 2022. https://doi.org/10.1007/s44228-022-00016-4.
Celgene Corporation. 2022. Reblozyl® (luspatercept). Summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/reblozyl-epar-product-information_en.pdf. Accessed August 2022.
Leitch HA. Improving clinical outcome in patients with myelodysplastic syndrome and iron overload using iron chelation therapy. Leuk Res. 2007;31(Suppl 3):S7–S9.
Leitch HA, Buckstein R, Zhu N, Nevill TJ, Yee KWL, Leber B, et al. Iron overload in myelodysplastic syndromes: evidence-based guidelines from the Canadian consortium on MDS. Leuk Res. 2018;74:21–41.
Gabutti V, Piga A. Results of long-term iron-chelation therapy. Acta Haematol. 1996;95:26–36.
Angelucci E, Li J, Greenberg P, Wu D, Hou M, Montano Figueroa EH, et al. Iron chelation in transfusion-dependent patients with low- to intermediate-1-risk myelodysplastic syndromes: a randomized trial. Ann Intern Med. 2020;172:513–22.
Komrokji R, Swern AS, Grinblatt D, Lyons RM, Tobiasson M, Silverman LR, et al. Azacitidine in lower-risk myelodysplastic syndromes: a meta-analysis of data from prospective studies. Oncologist. 2018;23:159–70.
Carraway HE. Saygin. Therapy for lower-risk MDS. Hematol Am Soc Hematol Educ Program. 2020;2020:426–33.
Passweg JR, Giagounidis AAN, Simcock M, Aul C, Dobbelstein C, Stadler M, et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care—SAKK 33/99. J Clin Oncol. 2011;29:303–9.
DeZern AE. Lower risk but high risk. Hematol Am Soc Hematol Educ Program. 2021;1:428–34.
de Witte T, Bowen D, Robin M, Malcovati L, Niederwieser D, Yakoub-Agha I, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood 2017;129:1753–62.
Platzbecker U, Fenaux P, Adès L, Giagounidis A, Santini V, van de Loosdrecht AA, et al. Proposals for revised IWG 2018 hematological response criteria in patients with MDS included in clinical trials. Blood. 2019;133:1020–30.
Trudeau JJ, He J, Rose E, Panter C, Randhawa S, Gater A. Content validity of patient-reported outcomes for use in lower-risk myelodysplastic syndromes. J Patient Rep. Outcomes 2020;4:69.
Steensma DP, Fenaux P, Van Eygen K, Raza A, Santini V, Germing U, et al. Imetelstat achieves meaningful and durable transfusion independence in high transfusion-burden patients with lower-risk myelodysplastic syndromes in a phase II study. J Clin Oncol. 2021;39:48–56.
Platzbecker U, Fenaux P, Steensma DP, Van Eygen K, Raza A, Germing U, et al. Treatment with imetelstat provides durable transfusion independence (TI) in heavily transfused non-del(5q) lower risk MDS (LR-MDS) relapsed/refractory (R/R) to erythropoiesis stimulating agents (ESAs). Abstract from the 2020 American Society of Hematology (ASH) Annual Meeting and Exposition. Blood 2020;136:658.
Platzbecker U, Fenaux P, Steensma DP, Van Eygen K, Raza A, Germing U, et al. IMerge: a phase 3 study to evaluate imetelstat in transfusion-dependent subjects with IPSS Low or Intermediate-1 risk myelodysplastic syndromes (MDS) that is relapsed/refractory to erythropoiesis-stimulating agent (ESA) treatment. Abstract from the 2020 American Society of Hematology (ASH) Annual Meeting and Exposition. Blood 2020;136:3113.
Dhillon S. Roxadustat: first global approval. Drugs 2019;79:563–72.
Henry DH, Glaspy J, Harrup R, Mittelman M, Zhou A, Carraway HE, et al. Roxadustat for the treatment of anemia in patients with lower-risk myelodysplastic syndrome: open-label, dose-selection, lead-in stage of a phase 3 study. Am J Hematol. 2022;97:174–84.
AstraZeneca. Status on FDA Advisory Committee vote on roxadustat in anaemia of chronic kidney disease [Press release]. 2021. https://www.astrazeneca.com/media-centre/press-releases/2021/status-on-us-fda-advisory-committee-for-roxadustat.html. Accessed October 7, 2022.
Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24:497–504.
Steensma DP, Wermke M, Klimek VM, Greenberg PL, Font P, Komrokji RS, et al. Results of a clinical trial of H3B-8800, a splicing modulator, in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) or chronic myelomonocytic leukemia (CMML). Abstract from the 2019 American Society of Hematology (ASH) Annual Meeting and Exposition. Blood 2019;134:673.
Garcia-Manero G, Santini V, Almeida A, Platzbecker U, Jonasova A, Silverman LR, et al. Phase III, randomized, placebo-controlled trial of CC-486 (oral azacitidine) in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2021;39:1426–36.
Garcia-Manero G, Griffiths EA, Steensma DP, Roboz GJ, Wells R, McCloskey J, et al. Oral cedazuridine/decitabine for MDS and CMML: a phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 2020;136:674–83.
Fozza C. Retuning the immune system in myelodysplastic syndromes: from immunomodulatory approaches to vaccination strategies and non-myeloablative hemopoietic cell transplant. Crit Rev Oncol Hematol. 2019;133:112–9.
Sallman DA, List A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood 2019;133:1039–48.
Sweeney C, Vyas P. The graft-versus-leukemia effect in AML. Front Oncol. 2019;9:1217.
Daver N, Boddu P, Garcia-Manero G, Yadav SS, Sharma P, Allison J, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia 2018;32:1094–105.
Young K, Eudy E, Bell R, Loberg M, Stearns T, Sharma D, et al. Cell Stem Cell. 2021;28:1473–82.e7. https://doi.org/10.1016/j.stem.2021.03.017. Epub 2021 Apr 12.
Andina N, Bonadies N, Allam R. Inflammasome activation in myeloid malignancies—friend or foe? Front Cell Dev Biol. 2022; https://doi.org/10.3389/fcell.2021.825611.
Robinson TM, Prince GT, Thoburn C, Warlick E, Ferguson A, Kasamon YL, et al. Pilot trial of K562/GM-CSF whole-cell vaccination in MDS patients. Leuk Lymphoma. 2018;59:2801–11.
Duetz C, Cucchi DGJ, Polak TB, Janssen JJWM, Ossenkoppele GJ, Estey EH, et al. The wider perspective: twenty years of clinical trials in myelodysplastic syndromes. Br J Haematol. 2022;196:329–35.
Sekeres MA, Maciejewski JP, Giagounidis AAN, Wride K, Knight R, Raza A, et al. Relationship of treatment-related cytopenias and response to lenalidomide in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2008;26:5943–9.
Drugs.com. Opdivo FDA approval history. 2021. https://www.drugs.com/history/opdivo.html. Accessed May 2022.
Garcia-Manero G, Sasaki K, Montalban-Bravo G, Daver NG, Jabbour EJ, Alvarado Y, et al. A phase II study of nivolumab or ipilimumab with or without azacitidine for patients with myelodysplastic syndrome (MDS). Abstract from the 2018 American Society of Hematology (ASH) Annual Meeting and Exposition. Blood 2018;132:465.
Drugs.com. Yervoy FDA approval history. 2020. https://www.drugs.com/history/yervoy.html. Accessed May 2022.
Sloand EM, Wu CO, Greenberg P, Young N, Barrett J. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol. 2008;26:2505–11.
Acknowledgements
The authors received writing and editorial assistance provided by Karolina Lech, Ph.D., of Excerpta Medica, funded by Bristol Myers Squibb.
Funding
This review was funded by Bristol Myers Squibb.
Author information
Authors and Affiliations
Contributions
Concept: All authors. Critical revision of the paper for important intellectual content: All authors.
Corresponding author
Ethics declarations
Competing interests
A.M.B. has received advisory board/research funding to institution from Acceleron Pharma, Agios, Alexion, Amgen, AstraZeneca, Celgene/BMS, Gilead, GSK, Janssen, Keros Therapeutics, Novartis, Taiho, and Takeda; and has received financial support for travel, consultancy honoraria, and research funding to institution from Pfizer. H.A.L. reports honoraria, advisory boards, and research funding from AbbVie, Alexion, AstraZeneca, BMS, Celgene, Janssen, Novartis, and Taiho. A.A.vdL. has received advisory board/research funding from Alexion, Amgen, Celgene/BMS, Novartis, Pfizer, and Roche. N.B. has received research funding to institution from Alexion, Astellas, Celgene/BMS, Novartis, Roche, Sandoz, Servier, and Takeda; financial support for travel from Amgen, Celgene/BMS, Gilead, Janssen, Novartis, and Roche; and consultancy honoraria from Janssen, Keros Therapeutics, and Novartis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brunner, A.M., Leitch, H.A., van de Loosdrecht, A.A. et al. Management of patients with lower-risk myelodysplastic syndromes. Blood Cancer J. 12, 166 (2022). https://doi.org/10.1038/s41408-022-00765-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-022-00765-8
