
Abstract
While the clinical impact of mutations in the ABL1 gene on response to therapy in chronic phase chronic myeloid leukemia (CP-CML) is well established, less is known about how other mutations affect prognosis. In a retrospective analysis, we identified 115 patients with CML (71 chronic, 15 accelerated and 29 blast phase) where targeted next-generation sequencing of genes recurrently mutated in myeloid leukemias was performed. ASXL1 was the most frequently mutated gene in the chronic (14%) and accelerated phase (40%) CML patients, whereas RUNX1 (20%) was the most common mutation in blast phase. Compared with wild-type ASXL1, CP-CML with mutant ASXL1 was associated with worse event-free survival (EFS) (median of 32.8 vs 88.3 months; P = 0.002) and failure-free survival (median of 13.8 vs 57.8 months; P = 0.04). In a multivariate analysis, ASXL1 mutation was the only independent risk factor associated with worse EFS in chronic phase CML with a hazard ratio of 4.25 (95% CI 1.59–11.35, P = 0.004). In conclusion, mutations in ASXL1 are associated with worse outcomes when detected in chronic phase CML.
Similar content being viewed by others

Introduction
Tyrosine kinase inhibitors (TKIs) have revolutionized the treatment of chronic myeloid leukemia (CML). However, despite success of this targeted therapy, resistance occurs with ABL1 kinase domain mutation as the best described mechanism [1]. Approximately 40% of TKI resistance is independent of BCR::ABL1 signaling [2], and could be mediated by chromosomal instability or possibly mutations in other genes recurrently mutated in myeloid malignancies [3,4,5].
Driver mutations in non-ABL1 genes, especially RUNX1, are commonly detected in blast phase CML, (BP-CML) [6,7,8,9,10], where there is a strong association between mutational profile and blast phenotype [8]. ASXL1, BCORL1, RUNX1, and TP53 mutations are associated with myeloid phenotype, whereas CDKN2A/B and IKZF1 mutations are more common in the lymphoid blast phase phenotype [8]. Despite the characterization of the mutational profile at blast-phase transformation, less is known about the frequency and clinical impact of non-ABL1 gene mutations in chronic phase CML (CP-CML). Mutations in ASXL1 have been detected in CP-CML, though their impact on clinical outcomes is not well-established [11,12,13,14]. These mutations could confer suboptimal response to TKIs, but it is unclear whether this would be due to higher co-occurrence with ABL1 kinase domain mutations [12, 13, 15], or independent epigenetic changes mediated by the polycomb repressive pathways [16]. In addition, other somatic mutations have been detected in CML Philadelphia-negative clones which persisted after optimal TKI response, suggesting a clonal event similar to clonal hematopoiesis of indeterminate potential (CHIP) [17, 18].
Given the unknown clinical impact of these mutations in CP-CML and the lack of clinical guidelines for testing and interpretation [19], we sought to describe the mutational profile of both chronic and advanced-phase CML and study the impact of mutations on TKI response and survival.
Methods
Patient selection and mutational analysis
We screened our databases for adult patients diagnosed with CML and identified those where mutational analysis was performed between 2017 and 2022. Targeted next-generation sequencing was done using a panel of 81 genes recurrently mutated in hematologic malignancies (Supplemental Table 1) [20]. To ensure the accuracy of ASXL1 p.G646fs mutation detection, we used a combination of sequencing chemistry, sequencing platform, internal VAF database, and a stringent VAF cut-off (>10%) to avoid sequencing artifacts across this homopolymer region [21]. ABL1 kinase domain mutations were identified using a previously described nested PCR-based cDNA sequencing assay, targeting codons 221 to 500, with an additional pyrosequencing step for T315I mutation detection [22]. Accelerated phase CML (AP-CML) and BP-CML were defined as per MD Anderson Cancer Center (MDACC) criteria [23]. A total of 115 evaluable patients were identified, among whom 71 had CP-CML (41 tested at diagnosis), 15 had AP-CML and 29 had BP-CML (Supplemental Fig. 1). This study was approved by the institutional review board and performed in accordance with the Declaration of Helsinki.
Response and outcome definitions
Response and survival outcomes were determined as previously described [24]. Major molecular response (MMR) was defined as a BCR::ABL1/ABL1 ratio of ≤0.1% on the international scale (IS). Molecular response with a 4-log reduction (MR4) and molecular response with a 4.5-log reduction (MR4.5) were defined as a BCR::ABL1/ABL1 ratio of ≤0.01% and ≤0.0032% (IS), respectively. Event-free survival (EFS) was measured from the start of treatment with first-line TKI to the date of any of the following events: loss of complete hematologic remission, loss of major cytogenetic response (MCyR), failure to achieve MCyR by 12 months, progression to accelerated or blast phase, or death from any cause. Failure-free survival (FFS) was measured similarly to EFS with the addition of treatment discontinuation due to resistance or intolerance as an event [25]. Overall survival (OS) was measured from the time of treatment with first-line TKI to time of death from any cause as an event.
Statistical analysis
Patient characteristics were summarized using median (range) for continuous and frequency (percentage) for categorical variables. Fisher’s exact test and Wilcoxon rank-sum test were used to assess differences in categorical and continuous variables, respectively. Survival probabilities were estimated by the Kaplan–Meier method and the log-rank test was used for comparisons. Univariate and multivariate analyses were used to assess the association between patient characteristics and survival outcomes.
Results
Baseline characteristics
Table 1 summarizes the baseline characteristics and treatments of patients with CP-CML by mutational status. The rate of ABL1 mutations was higher in patients with mutant ASXL1 compared to patients with no mutations, albeit not statistically significant (29% vs 11%, P = 0.3).
In BP-CML, the median bone marrow blast percentage at presentation was higher in the mutation group (45%) compared with the no mutation group (24%) albeit not statically significant (P = 0.07). The baseline characteristics for AP-CML and blast BP-CML are summarized in Supplemental Tables 2 and 3.
Mutational profile in CP-CML
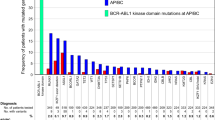
Among the 71 evaluable patients, 23 (32%) had at least one non-ABL1 mutation with 6 (8%) carrying two or more non-ABL1 mutations (Fig. 1A). The prevalence of mutations was lower in patients who were tested at diagnosis (10 out of 41) compared with patients who were tested at resistance or at loss of response (13 out of 30) (24% vs 43%; P = 0.1) The most common mutation was ASXL1 detected in 10 (14%) of all tested patients followed by DNMT3A in 5 (7%) and RUNX1 in 3 (4%) patients. Mutations in 12 other genes were detected at least once (Fig. 1A).

A Distribution of mutations in patients with CP-CML. B Frequency of various ASXL1 mutations identified. aa amino acids.
Seven different ASXL1 mutations were detected among 10 patients, with one patient carrying two concomitant ASXL1 mutations. These were either frameshift or nonsense mutations in exon 13 that resulted in a truncated ASXL1 protein. The most common mutation was G646fs, detected in five patients (Fig. 1B).
Response rates and time to response
Cytogenetic and molecular response rates as well as times to attaining these responses (TTR) are highlighted in Table 2 and Supplemental tables 4 and 5. One of the patients in the mutation group was lost to follow-up soon after treatment initiation and therefore was not included in response analyses. A trend of lower early response rates (33% vs 61%, P = 0.2) and longer time to MMR (17.5 vs 9.0 months, P = 0.2), MR4 (20.7 vs 16.2 months, P = 0.4) and MR4.5 (48.7 vs 23.0 months, P = 0.4) were observed in patients with mutant ASXL1, compared to wild-type counterpart, albeit not statistically significant (Supplemental Table 5).
Impact on survival
Patients who had at least one non-ABL1 mutation had worse FFS compared to those with no mutations (Median FFS 13.3 months vs 57.8 months; P = 0.02). There was no statistically significant difference in EFS or OS comparing the two groups (Supplemental Fig. 2). Compared with patients with no mutations, patients who had ASXL1 mutations had worse EFS (median of 32.8 months vs 88.3 months; P = 0.002) (Fig. 2A, Supplemental Fig. 3B) and worse FFS (median of 13.8 months vs 57.8 months; P = 0.04) (Fig. 2B). EFS and FFS of patients with mutations other than ASXL1 were not significantly different from those with no mutations. There was no significant difference in OS in our cohort based on the mutational profile of the patients (Fig. 2C), albeit a trend of worse OS in patients with mutated ASXL1 when considering only CML-related mortality (Supplemental Fig. 3C).

A Event-free survival (EFS). B Failure-free survival (FFS) and C overall survival (OS) comparing patients with mutations in ASXL1 vs mutations in other genes or no mutation. Univariate (UVA) and multivariate (MVA) analyses of factors predicting: D EFS, E FFS, and F OS. m months.
Among patients who were tested for mutations at diagnosis, those who had ASXL1 mutations had significantly worse EFS (median of 30.3 months vs not reached; P = 0.02) and FFS (median of 12.6 months vs not reached; P = 0.02) when compared to patients with no mutations (Supplemental figure 4). In patients tested at loss of response, there was still a trend of worse EFS in patients with ASXL1 mutation, albeit not statistically significant (Supplemental Fig. 5).
Multivariate analysis
In order to assess the impact of confounding variables on survival, we conducted univariate and multivariate analyses to predict the determinants of EFS, FFS, and OS. ASXL1 mutation was identified as an independent risk factor associated with worse EFS with a hazard ratio (HR) of 4.25 (95% CI 1.59–11.35, P = 0.004) (Fig. 2D). On the other hand, having any non-ABL1 mutation (HR of 2.33, 95% CI 1.16–4.68, P = 0.02) and treatment with imatinib (HR of 2.65, 95% CI 1.30–5.44, P = 0.008) were each independent risk factors associated with worse FFS (Fig. 2E).
Advanced phase CML
Fifteen patients with AP-CML and 29 patients with BP-CML had mutational analysis performed (Supplemental Tables 2 and 3). Among the 15 patients with AP-CML, 10 (67%) had at least one non-ABL1 mutation (Fig. 3A). The most common mutation was ASXL1 detected in 6 (40%) patients followed by RAS, SF3B1, and TET2 detected in 2 (13%) patients each. Mutations were also detected in 7 other non-ABL1 genes as highlighted in (Fig. 3A).

A Distribution of mutations in patients with AP-CML, and B BP-CML. C Association between non-ABL1 gene mutations and the blast phase phenotype.
ABL1 was the most common mutation in BP-CML, detected in 11 (38%) patients. Sixteen (56%) patients had at least one non-ABL1 mutation and 9 (31%) had two or more mutations (Fig. 3B). RUNX1 was the most frequently mutated gene in 6 (21%) patients, followed by WT1 in 4 (13%) and ASXL1 in 2 (7%) patients. PTEN mutation was detected in one patient who had lymphoid BP-CML and RAS mutation was detected in both lymphoid and myeloid phenotypes (one patient each). All the other gene mutations were associated with myeloid blast phenotype (Fig. 3C).
Among the 29 BP-CML patients, 26 were followed for a median of 18.1 months. Patients who had at least one non-ABL1 mutation had significantly worse EFS (1-year survival of 17% vs 61%, P = 0.007) (Supplemental figure 6). Multivariate analysis revealed that having at least one non-ABL1 mutation was an independent risk factor associated with worse EFS (HR = 5.42, 95% CI 1.2–23.8, P = 0.03) (Supplemental Table 6). Mutation status did not affect OS or FFS, whereas a myeloid phenotype was associated with worse OS (HR = 5.67, 95% CI 1.3–25.6, P = 0.02) (Supplemental Tables 7 and 8).
AML development
When screening our databases for patients for this study, we also identified patients who had history of CML and later developed de novo myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML), with low BCR::ABL1 levels (<1% IS). Interestingly, one of these patients with ASXL1, IDH2 and SRSF2 mutations identified in CP-CML, developed de novo AML 6 years after his initial CML diagnosis. Mutational analysis at the time of AML diagnosis revealed the same mutations.
Discussion
In this study we report on the mutational profile of CML patients who were tested in the clinical setting. Mutations in ASXL1 were the most common alteration in CP-CML, detected in 14% of all evaluable patients and in 12% of patients tested at diagnosis. These mutations were associated with significantly worse EFS and FFS.
The presence of non-ABL1 mutations and their potential adverse prognostic impact has been previously described [14]; however, to our knowledge, our study represents the first report on the impact of ASXL1 mutations on survival, adding to recently emerging evidence of suboptimal TKI response associated with these mutations [15]. There was no difference in OS when these mutations were detected, which could be due to the relatively short follow-up period (median follow-up of 42 months), or the excellent outcomes of CP-CML with the current standard of care. Mutations in RUNX1 were the most common non-ABL1 mutations among patients with BP-CML, mostly among those with a myeloid blast phenotype, an association reported in previous studies [8]. While none of the CP-CML patients in our cohort progressed to AP or BP, a small fraction with CP-CML developed de novo MDS or AML, including one patient who had the same mutational profile during his CML treatment. This could be explained by the presence of non-ABL1 mutations in Philadelphia-negative clones [17, 18]. Our findings could be biased by the fact that only 41 of 71 (58%) patients were tested for mutations at diagnosis, whereas 30 (42%) were tested only when clinicians suspect resistance or suboptimal response, therefore confounding our predicted associated risks with adverse outcomes. Prospective analysis with unbiased testing of these mutations could better determine their associated prognostic impact.
Although larger studies are needed, our findings support the role of broad mutational analysis in CP-CML, especially those with suboptimal response to therapy and absence of ABL1 mutations.
Data availability
Data generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Soverini S, Martinelli G, Rosti G, Bassi S, Amabile M, Poerio A, et al. ABL Mutations in Late Chronic Phase Chronic Myeloid Leukemia Patients With Up-Front Cytogenetic Resistance to Imatinib Are Associated With a Greater Likelihood of Progression to Blast Crisis and Shorter Survival: A Study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol. 2005;23:4100–9.
Wagle M, Eiring AM, Wongchenko M, Lu S, Guan Y, Wang Y, et al. A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia. 2016;30:1493–501.
Cortes JE, Talpaz M, Giles F, O’Brien S, Rios MB, Shan J, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood. 2003;101:3794–800.
Issa GC, Kantarjian HM, Gonzalez GN, Borthakur G, Tang G, Wierda W, et al. Clonal chromosomal abnormalities appearing in Philadelphia chromosome-negative metaphases during CML treatment. Blood. 2017;130:2084–91.
Alves R, Gonçalves AC, Rutella S, Almeida AM, De Las Rivas J, Trougakos IP, et al. Resistance to tyrosine kinase inhibitors in chronic myeloid leukemia-from molecular mechanisms to clinical relevance. Cancers. 2021;13:4820.
Adnan Awad S, Dufva O, Ianevski A, Ghimire B, Koski J, Maliniemi P, et al. RUNX1 mutations in blast-phase chronic myeloid leukemia associate with distinct phenotypes, transcriptional profiles, and drug responses. Leukemia. 2021;35:1087–99.
Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia. 2011;25:557–60.
Ochi Y, Yoshida K, Huang Y-J, Kuo M-C, Nannya Y, Sasaki K, et al. Clonal evolution and clinical implications of genetic abnormalities in blastic transformation of chronic myeloid leukaemia. Nat Commun. 2021;12:2833.
Branford S, Kim DDH, Apperley JF, Eide CA, Mustjoki S, Ong ST, et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia. 2019;33:1835–50.
Adnan-Awad S, Kankainen M, Mustjoki S. Mutational landscape of chronic myeloid leukemia: more than a single oncogene leukemia. Leuk Lymphoma. 2021;62:2064–78.
Fernandes A, Shanmuganathan N, Branford S. Genomic mechanisms influencing outcome in chronic myeloid leukemia. Cancers. 2022;14:620.
Branford S, Wang P, Yeung DT, Thomson D, Purins A, Wadham C, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. 2018;132:948–61.
Machnicki MM, Pepek M, Solarska I, Niesiobedzka-Krezel J, Seferynska I, Gora Tybor J, et al. ASXL1 mutations detectable at diagnosis may predict response to imatinib in patients with chronic myeloid leukemia. Blood. 2019;134:4148.
Mologni L, Piazza R, Khandelwal P, Pirola A, Gambacorti-Passerini C. Somatic mutations identified at diagnosis by exome sequencing can predict response to imatinib in chronic phase chronic myeloid leukemia (CML) patients. Am J Hematol. 2017;92:E623–E5.
Schönfeld L, Rinke J, Hinze A, Nagel SN, Schäfer V, Schenk T, et al. ASXL1 mutations predict inferior molecular response to nilotinib treatment in chronic myeloid leukemia. Leukemia. 2022;36:2242–2249.
Ko TK, Javed A, Lee KL, Pathiraja TN, Liu X, Malik S, et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood. 2020;135:2337–53.
Schmidt M, Rinke J, Schäfer V, Schnittger S, Kohlmann A, Obstfelder E, et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia. 2014;28:2292–9.
Kim T, Tyndel MS, Kim HJ, Ahn JS, Choi SH, Park HJ, et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood. 2017;129:38–47.
Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34:966–84.
Luthra R, Patel KP, Reddy NG, Haghshenas V, Routbort MJ, Harmon MA, et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014;99:465–73.
Montes-Moreno S, Routbort MJ, Lohman EJ, Barkoh BA, Kanagal-Shamanna R, Bueso-Ramos CE, et al. Clinical molecular testing for ASXL1 c.1934dupG p.Gly646fs mutation in hematologic neoplasms in the NGS era. PLoS ONE. 2018;13:e0204218.
Cortes J, Jabbour E, Kantarjian H, Yin CC, Shan J, O’Brien S, et al. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood. 2007;110:4005–11.
Cortes JE, Talpaz M, O’Brien S, Faderl S, Garcia-Manero G, Ferrajoli A, et al. Staging of chronic myeloid leukemia in the imatinib era. Cancer. 2006;106:1306–15.
Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol: Off J Am Soc Clin Oncol. 2009;27:6041–51.
O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004.
Acknowledgements
GCI received funding through the K12 Paul Calabresi Clinical Scholarship Award (NIH/NCI K12 CA088084).
Author information
Authors and Affiliations
Contributions
AB, HK, KS, and GCI designed the study. AB and GCI wrote the manuscript. AB, KS, and GCI collected and analyzed the data and performed the statistical analysis. HK, EJ, NJS, KP, and FR contributed to the analysis. All authors critically reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
HK received research grants from AbbVie, Amgen, Ascentage, BMS, Daiichi-Sankyo, Immunogen, Jazz, Novartis, Pfizer; and honoraria from AbbVie, Amgen, Aptitude Health, Ascentage, Astellas Health, Astra Zeneca, Ipsen, Pharmaceuticals, KAHR Medical Ltd, NOVA Research, Novartis, Pfizer, Precision Biosciences, Taiho Pharmaceutical Canada. EJ received research grants from Abbvie, adaptive biotechnologies, Amgen, Pfizer, and Takeda; and consultancy fees from Abbvie, adaptive biotechnologies, Amgen, BMS, Genentech, Incyte, Novartis, Pfizer, and Takeda. NJS reports consultancy for Takeda Oncology, Jazz Pharmaceuticals, NGMBio, AstraZeneca, and Amgen; research funding from Takeda Oncology and Astellas; honoraria from Amgen, and Novartis. FR reports honoraria from Astex, Taiho, Bristol Myers Squibb, Xencor, Agios, AstraZeneca, Novartis, AbbVie, Celgene, Jazz, Syros Pharmaceuticals, and Amgen; research funding from Astex, Taiho, Bristol Myers Squibb, Xencor, Agios, Prelude, AbbVie, Celgene, Jazz, Syros Pharmaceuticals, and Amgen; membership on the Board of Directors or advisory committees of Bristol Myers Squibb and Celgene; consultancy for Syros Pharmaceuticals KS received research funding and consultancy fees from Novartis; and is on the advisory board for Daiichi-Sankyo and Pfizer. GCI received research funding from Celgene, Kura Oncology, Syndax and Novartis; and consultancy fees from Novartis and Kura Oncology.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bidikian, A., Kantarjian, H., Jabbour, E. et al. Prognostic impact of ASXL1 mutations in chronic phase chronic myeloid leukemia. Blood Cancer J. 12, 144 (2022). https://doi.org/10.1038/s41408-022-00742-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-022-00742-1
This article is cited by
-
European LeukemiaNet laboratory recommendations for the diagnosis and management of chronic myeloid leukemia
Leukemia (2023)
-
Genetic landscape of chronic myeloid leukemia
International Journal of Hematology (2023)
-
Epigenetic regulation by ASXL1 in myeloid malignancies
International Journal of Hematology (2023)
