
Abstract
Patients with large granular lymphocytic leukemia (LGLL) frequently present with neutropenia. When present, anemia is usually accompanied by neutropenia and/or thrombocytopenia and isolated anemia is uncommon. We evaluated a cohort of 244 LGLL patients spanning 15 years and herein report the clinicopathologic features of 34 (14%) with isolated anemia. The patients with isolated anemia showed a significantly male predominance (p = 0.001), a lower level of hemoglobulin (p < 0.0001) and higher MCV (p = 0.017) and were less likely to have rheumatoid arthritis (p = 0.023) compared to the remaining 210 patients. Of the 34 LGLL patients with isolated anemia, 13 (38%) presented with pure red cell aplasia (PRCA), markedly decreased reticulocyte count and erythroid precursors, and more transfusion-dependence when compared to non-PRCA patients. There was no other significant clinicopathologic difference between PRCA and non-PRCA patients. 32 patients were followed for a median duration of 51 months (6–199). 24 patients were treated (11/11 PRCA and 13/21 non-PRCA patients, p < 0.02). The overall response rate to first-line therapy was 83% [8/11 (72.7%) for PRCA, 12/13 (92.3%) for non-PRCA], including 14 showing complete response and 6 showing partial response with a median response duration of 48 months (12–129). Half of non-PRCA patients who were observed experienced progressive anemia. During follow-up, no patients developed neutropenia; however, 5/27 (18.5%) patients developed thrombocytopenia. No significant difference in overall survival was noted between PRCA and non-PRCA patients. In summary, this study demonstrates the unique features of LGLL with isolated anemia and underscores the importance of recognizing LGLL as a potential cause of isolated anemia, which may benefit from disease-specific treatment. LGLL patients with PRCA were more likely to require treatment but demonstrated similar clinicopathologic features, therapeutic responses, and overall survival compared to isolated anemia without PRCA, suggesting PRCA and non-PRCA of T-LGLL belong to a common disease spectrum.
Similar content being viewed by others

Introduction
Large granular lymphocytes (LGL) are a morphologically distinct lymphoid subpopulation in the peripheral blood characterized by abundant azurophilic cytoplasmic granules. Large granular lymphocytic leukemia (LGLL) is a clonal hematopoietic disorder caused by an abnormal expansion of LGLs in the peripheral blood, bone marrow, and spleen [1]. The majority of LGLL cases (approximately 85%) arise from cytotoxic T lymphocytes (T-LGLL), while the remaining cases exhibit natural killer (NK) cell phenotype and are categorized under chronic lymphoproliferative disorder of NK cells (CLPD-NK) in the 2017 World Health Organization (WHO) classification of hematolymphoid neoplasms [1].
Although the pathophysiologic mechanism of LGLL is not well-understood, immunological stimuli, possibly from viruses or tumor antigens, have been implicated [2,3,4]. This theory has been supported by the high rate of association between LGLL and various hematologic malignancies, namely B-cell lymphoma, multiple myeloma, myeloid neoplasms, and autoimmune diseases such as rheumatoid arthritis [5,6,7,8,9,10].
The diagnosis of LGLL is established by the detection of persistent and clonal LGLL cells. LGLL cells can be identified by their specific morphology and immunophenotype in the peripheral blood or bone marrow, or detected by immunohistochemical stains (granzyme B, perforin, and/or TIA-1) in the bone marrow, highlighting the presence of intrasinusoidal cytotoxic lymphocyte infiltrates [11]. T-cell clonality can be established by monotypic TRBC1 expression or Vβ T-cell receptor (TCR) gene repertoire analysis by flow cytometry, or TCR gene arrangement by polymerase chain reaction (PCR) or next-generation sequencing (NGS) analysis [12, 13]. NK-cell clonality can be determined by the restriction of killer-cell immunoglobulin-like receptor (KIR) [14].
Performing a proper workup is the key to the diagnosis of LGLL, which can be straightforward when patients exhibit typical clinical or hematologic presentations such as repeated infections, unexplained neutropenia, or splenomegaly. However, the diagnosis of LGLL can be challenging or missed because of the lack of the typical clinical and hematologic presentations [15,16,17]. While anemia is common in LGLL, it is usually accompanied by neutropenia and/or thrombocytopenia. Although rare, LGLL associated with pure red cell aplasia (PRCA) has been previously reported [18,19,20], however, isolated anemia without PRCA has not been systematically investigated. In this report, we characterized the clinicopathologic features of LGLL presenting with isolated anemia in our practice, compared these features between patients with and without PRCA, and further evaluated the clinical outcomes of such patients.
Materials and methods
Patient selection
All patients who presented to Mayo Clinic, Rochester MN from 2005 to 2019 with a diagnosis of T-LGLL or CLPD-NK were included. We performed a retrospective medical record chart review for clinical and pathological data. This study was approved by the Mayo Clinic Institutional Review Board.
At least 3 of the following 4 criteria was used to diagnose T-LGLL or CLPD-NK: (1) a distinct T-cell population with coexpression of 1 or more natural killer (NK)-cell-associated antigens (CD16, CD56, or CD57) and decreased CD2, CD5, or CD7 expression; or a distinct NK-cell population with decreased CD2, CD7, CD16, or CD56; (2) a clonal T-cell population or a clonal NK-cell population; (3) intrasinusoidal cytotoxic T-cell or NK-cell infiltrates in bone marrow, spleen, or liver; and (4) persistence of the abnormal T-cell or NK-cell population or unexplained cytopenia for more than 6 months. Bone marrow biopsies were performed to evaluate other hematological malignancies and to support an LGLL diagnosis. Corresponding flow cytometry analyses were performed on peripheral blood and/or bone marrow specimens to characterize the LGLL population.
Isolated anemia was defined as: Unexplained hemoglobin (Hb) <13 g/dL for males or <12 g/dL for females, absolute neutrophil count ≥1.5 × 109/L, and platelet count ≥150 × 109/L. The diagnosis of T-LGLL associated PRCA was based on the following [21, 22]: (1) Markedly decreased to absent erythroid precursors in bone marrow biopsy (2) reticulocyte <0.5%; (3) Unremarkable granulopoiesis and megakaryopoiesis; and (4) No other known causes of PRCA. Patients with markedly decreased to absent erythroid precursors but borderline reticulocyte percentage (>0.5% but <1%) were also classified as PRCA.
Flow cytometry
T-cell and NK-cell flow cytometry was performed as previously described [13, 23, 24], on a FACSCanto II or FACSLyric flow cytometer (BD Biosciences, San Jose, CA), with antibodies to CD2, CD3, CD4, CD5, CD7, CD8, CD16, CD45, CD56, CD57, CD94, CD158a, CD158b, CD158e, NKG2A, TCR γ/δ (BD Biosciences, San Jose, CA), and TRBC1 (clone JOVI-1; Ancell Corp, Bayport, MN). The TCR Vβ repertoire kit (Beckman Coulter, Indianapolis, IN) was used to detect TCR Vβ clonality.
Molecular testing for TCR gene rearrangement
TCR gene rearrangement was analyzed using standard BIOMED-2 assay (Invivoscribe, San Diego, CA). Multiplex PCR for TCR gene rearrangement was performed for TCR Vβ and TCR Vγ, as described previously [25]. Standard electrophoresis visual evaluation criteria were applied for BIOMED-2 data analysis.
STAT3 gene mutation
STAT3 gene mutation by Sanger sequencing was performed on genomic DNA extracted from available peripheral blood samples, as previously described [23]. Three sets of primers were designed to cover the most commonly reported STAT3 mutations in the Src homology 2 (SH2), DNA-binding, and coiled-coil domains [26]. PCR was performed using HotStar Taq Master Mix (Qiagen, Germantown, MD). PCR products were subjected to bidirectional direct sequencing on an automatic DNA sequencer using the BigDye Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, MA).
Myeloid neoplasm-focused next-generation sequencing
NGS was performed on extracted DNA from bone marrow aspirate as previously described [27]. A targeted NGS includes 35 genes recurrently mutated in myeloid neoplasms: ASXL1, BCOR, BRAF, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, GATA1, GATA2, IDH1, IDH2, JAK2, KIT, KRAS, MPL, MYD88, NOTCH1, NPM1, NRAS, PHF6, PTPN11, RUNX1, SETBP1, SF3B1, SRSF2, TERT, TET2, TP53, U2AF1, WT1, and ZRSR2.
Patient follow-up
Treatment responses were assessed after a minimum of 16 weeks of therapy. Complete response (CR), partial response (PR), and no response were defined as previously described [28]. CR was defined as the attainment of a normal Hb. PR was defined as improvement in Hb by >1 g/dL without CR or a decrease in transfusion requirements by >50% for at least 4 months. No response was defined as a lack of CR/PR.
Statistical analysis
A χ2 test, Fisher’s exact test, two-tailed t-test, and Wilcoxon rank-sum/Mann-Whitney U test were used, as appropriate, for statistical evaluation of the results. “Log-rank (Mantel-Cox) test was performed using GraphPad Prism version 9.1.2 (226) for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com”. P < 0.05 was considered statistically significant.
Results
Prevalence of isolated anemia in LGLL patients and their clinical characteristics
A total of 244 LGLL patients were identified; 34 (14%) patients presented with isolated anemia at diagnosis. Among 213 identified T-LGLL patients, 31/213 (15%) presented with isolated anemia: 12/31 (39%) with PRCA, and 19/31 (61%) without PRCA. Of 31 diagnosed CLPD-NK patients, 3/31 (10%) presented with isolated anemia: 1/3 (33%) with PRCA and 2/3 (67%) without PRCA. No significant difference in the prevalence of isolated anemia was found between T-LGLL and CLPD-NK groups (p = 0.588).
Compared to 210 LGLL patients without isolated anemia, 34 LGLL patients with isolated anemia showed a significantly male predominance (p = 0.001) and fewer were associated with rheumatoid arthritis (p = 0.023). They also had much lower Hb levels (p < 0.0001), higher MCV (p = 0.017) and were more transfusion-dependent (p < 0.0001)(Table 1). Consistent with the selection criteria in this study, LGLL patients with isolated anemia had a higher absolute neutrophil count (ANC) and higher platelet count than those without isolated anemia (Table 1). There was no significant difference between LGLL patients with and without isolated anemia for age, other malignancies, history of bone marrow or solid organ transplantation, B symptoms, splenomegaly, lymphadenopathy, absolute lymphocyte count, or absolute LGLL count.
Among 34 LGLL patients with isolated anemia, 10 had a history of other hematological malignancies: 4 plasma cell neoplasms, 2 chronic lymphocytic leukemia (CLL), 1 each of nodal marginal zone lymphoma, diffuse large B-cell lymphoma NOS, acute myeloid leukemia, and classic Hodgkin lymphoma. At the time of LGLL diagnosis, 8 patients were in complete remission for the previous hematological malignancies, and 2 had a low level of involvement (5% of cellularity), 1 by plasma cell neoplasm and 1 by CLL.
LGLL patients with isolated anemia did not have known chemical/toxic/heavy alcohol exposure. An extensive workup was performed to rule out other potential causes of anemia. No deficiencies of iron, B12 and/or folate were detected in all 34 patients. 33 patients had at least one bone marrow biopsy that showed no evidence of myelodysplastic syndrome or other myeloid neoplasms by thorough morphologic and cytogenetic evaluation. An NGS myeloid neoplasm panel was performed on two available patients and revealed no pathogenic mutations. No definitive hemolysis was found based on absolute reticulocyte count, LDH, bilirubin, and haptoglobin levels. Six patients had chronic kidney disease; however, the stable and mildly decreased glomerular filtration rate (GFR) did not explain the worsening moderate to severe anemia (median Hb level was 8.5 g/dL, range 6.8–9.6 g/dL). In total, 4 of 6 patients with splenomegaly underwent splenectomy with no resolution of anemia. No parvoviral infection or thymoma was found in patients with PRCA.
Clinical and pathologic features of LGLL patients with isolated anemia presenting with and without PRCA
Of all 34 patients, 18 (53%) patients required transfusion, and patients with PRCA were more transfusion-dependent (100%) than patients without PRCA (24%) (p < 0.0001) (Table 1). Patients with PRCA also showed significantly lower MCV than non-PRCA patients (p = 0.035): PRCA patients usually presented with normocytic anemia while non-PRCA patients with macrocytic anemia. This finding could be due to the reticulocytopenia in PRCA patients and/or the transfusion effect of normal red blood cells on PRCA patients. There was no significant difference between LGLL patients with and without PRCA for gender, age, associated diseases, B symptoms, splenomegaly, lymphadenopathy, Hb, ANC, ALC, and platelet count (Table 1).
Among the 34 LGLL patients with isolated anemia, αβ T-LGLL was the most common type, followed by γδ T-LGLL and CLPD-NK, regardless of the presence or absence of PRCA (Table 2). As expected, patients with PRCA were found to have significantly lower bone marrow cellularity with median cellularity of 27.5% compared to 45% in patients without PRCA (p = 0.0007), mainly due to markedly reduced to absent erythroid precursors in PRCA patients (Table 2). The percentage of bone marrow involvement by LGLL cells revealed no significant difference between patients with and without PRCA (Table 2). STAT3 mutation status was available in 8 Caucasian patients, which showed mutations in 3 patients, including Y640F in 1 patient and N647I in 2 patients. STAT3 mutations were detected exclusively in LGLL patients without PRCA in this small cohort of patients. Similar to the 210 LGLL patients without isolated anemia (data not shown) and the previous reports [23, 24, 29], the 34 LGLL patients with isolated anemia had clonal T-cell or NK-cell populations and an intrasinusoidal distribution of cytotoxic T or NK-cells.
4 out of 31 patients had cytogenetic abnormalities: 1 PRCA and 1 non-PRCA patient had a loss of the Y chromosome, 1 PRCA patient had a loss of the Y chromosome and a trisomy 15 in a subset of cells tested. In adult males, the absence of a Y chromosome and trisomy 15 without any other abnormality in metaphases from bone marrow is likely age-related and not pathogenic [30, 31]. 1 non-PRCA patient showed del(1),der(3) in 2/20 metaphases, which completely disappeared after T-LGLL was treated, and appeared after T-LGLL relapsed. This result indicates del(1),der(3) represents the T-LGLL clone.
Response to therapy and clinical outcomes of LGLL patients with isolated anemia presenting with and without PRCA
32 patients were followed for a median duration of 51 months (range: 6–199), including 11 PRCA with a median follow-up of 80 months (range: 6–155) and 21 non-PRCA patients with a median follow-up of 45 months (range: 6–199). All 11 PRCA patients received treatment, while 13/21 (62%) of patients without PRCA received treatment, and the remaining 8 patients were observed. Cyclophosphamide, cyclosporin, or methotrexate was the first-line therapy for those patients. A minimum of 4 months of therapy was required before assessing response. 20 of 24 treated patients exhibited CR (n = 14) or PR (n = 6) to the first-line therapy with an overall response rate of 83% and a median response duration of 48 months (range: 12–129). Among 14 CR patients, 7 were in CR after cyclophosphamide therapy, 4 after cyclosporin therapy, and 3 after methotrexate therapy. Among 6 PR patients, 4 were in PR after methotrexate therapy, and 2 after cyclophosphamide therapy. The dosing and duration of treatment varied. In general, the dosing was as follows: cyclophosphamide 50 mg to 100 mg PO daily, cyclosporin 25 mg to 200 mg PO daily, methotrexate 7.5 mg to 20 mg PO weekly. The duration of treatment was between 4 and 136 months during follow-up. Cyclophosphamide was usually stopped after a median duration of 12 months of treatment, whereas methotrexate and cyclosporin were used until the last follow-up. Four patients did not respond to cyclophosphamide (n = 2) or methotrexate (n = 2) as first-line therapy; among the 3 patients who received subsequent lines of treatment, none responded. Steroid therapy was not used as first-line single-regimen therapy but was combined with other first-line agents in 9 (38%) patients (5 with cyclophosphamide, 3 with methotrexate, and 1 with cyclosporin). Four patients received prednisone 5–10 mg daily for 4–12 months; 5 patients started with prednisone 50–120 mg daily with a gradual taper over the ensuing months. Among 14 patients who achieved CR, 8 received steroids in addition to first-line therapy.
8/11 (72.7%) PRCA patients were in CR (n = 7) or PR (n = 1) after first-line treatment; 12/13 (92.3%) non-PRCA patients responded to first-line treatment with 7 in CR and 5 in PR. The median response duration was 45 months (range 21–118) for non-PRCA patients and 48 months (range 12–129) for PRCA patients, with no significant difference between the two groups (p = 0.989). The type of first-line therapy and overall responses to treatment in patients with and without PRCA were displayed in Table 3. The responses to different first-line therapy administrated to LGLL patients with and without PRCA are illustrated in Fig. 1.

X-axis represents the number of patients. Green, orange, and gray bars represent complete remission, partial remission, and no response, respectively.
Among the 14 CR patients, 6 (3 with PRCA, 3 without PRCA) had a relapse of disease after response to first-line therapy of cyclophosphamide (n = 3), cyclosporin (n = 2), or methotrexate (n = 1), after 1 to 10 years of CR. Three of them were treated subsequentially: one was treated again with cyclophosphamide and achieved CR; two were treated with second-line therapy (cyclosporin was the first-line, cyclophosphamide was the second-line therapy) and both patients achieved PR.
Among the 8 patients without PRCA who were under observation, 6 had follow-up (median:16 months; range: 13-199): 3 patients had progressive anemia (Hb level decreased by >1 g/dL), and 3 showed persistent anemia.
2 of 3 CLPD-NK patients (one with PRCA and the other without PRCA) were in complete remission when treated with methotrexate. The third patient was observed but no follow-up was available.
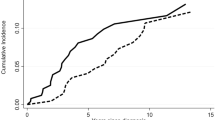
Although neutropenia is the most common presentation in LGLL patients, at the end of the follow-up period, no patients developed neutropenia, while 5/27 (18.5%) patients (4 with PRCA, 1 without PRCA) developed thrombocytopenia with a median platelet count at 108 × 109/L (range: 81–117). No significant difference in overall survival (OS) was noted between PRCA and non-PRCA patients (log-rank test p = 0.93) (Fig. 2).

Red line and black line represent patients with PRCA and patients without PRCA, respectively.
Discussion
Our study demonstrates the importance of recognizing LGLL as a potential cause of isolated anemia, including PRCA and non-PRCA. To the best of our knowledge this is the first report that addresses isolated anemia as a clinical presentation for LGLL patients. Because isolated anemia is a frequent cause for hematologic evaluation, including LGLL as one of the considerations is important for a proper evaluation, diagnosis, and treatment. The reliable clues that necessitate further LGLL workup in patients with isolated anemia include lymphocytosis, morphological LGL in the peripheral blood, and decreased normal NK-cell percentage/number in the peripheral blood as previously reported [29].
Anemia has been reported in 20–85% of LGLL patients, often in bicytopenic or pancytopenic patients [32, 33]. LGLL is rarely considered a cause when patients present with isolated anemia because this relationship has not been well established in the literature. In our cohort of 244 LGLL patients, isolated anemia was present in 34 (14%) patients. For these 34 patients, we excluded other causes as major contributors to the patients’ anemia through extensive workup and proposed LGLL was the underlying mechanism for their anemia. This is further supported by the fact that 20 of 24 treated patients in this cohort responded to the first-line therapy for LGLL (14 in CR and 6 in PR), and half of the observed patients experienced worsening anemia.
Most clinicopathologic features were similar among LGLL patients despite isolated anemia, and they were similar to those of LGLL patients reported by multiple groups [3, 32,33,34,35,36,37,38,39]. Interestingly, LGLL patients with isolated anemia were male predominant (30 of 34, 88%), which was significantly higher than the remaining 210 LGLL patients without isolated anemia (126/210, 60%). Low testosterone levels could cause mild anemia in older men [40, 41]. However, the vast majority of male patients in our study (28/30, 93%) had moderate to severe anemia (median Hb 9.1 g/dL, range 5.2–10.2), making this possibility unlikely. A sample bias could not be excluded, given the relatively small number of cases. Therefore, a larger cohort is required to confirm this finding. Rheumatoid arthritis was significantly less common in isolated anemia patients than those without isolated anemia. This could be related to the absence of neutropenia at diagnosis and during follow-up of LGLL, as neutrophil lysis plays an important role in the immunopathogenesis of rheumatoid arthritis [42].
PRCA is a rare hematologic syndrome characterized by anemia, reticulocytopenia, and severe erythroid hypoplasia of the bone marrow that could be caused by immune conditions, medications, viral infections, thymoma, LGLL, or idiopathy. Approximately 10–30% of PRCA patients have T-LGLL, and 10% of T-LGLL patients have PRCA [19] [43,44,45]. Among our cohort of 34 LGLL patients with isolated anemia, 38% of patients had PRCA and most did not. Although PRCA patients had markedly decreased reticulocyte count in the peripheral blood and erythroid precursors in the bone marrow and they are more transfusion-dependent, there was no other significant clinicopathologic difference between PRCA and non-PRCA patients.
The discovery of STAT3 mutations has shed light on the genetic basis of LGLL pathogenesis [26, 46]. Although T-LGLL patients with PRCA have been reported to preferentially harbor STAT3 mutations [44, 47, 48], none were found in the PRCA patients in this cohort. This is likely due to the small number of cases investigated.
More PRCA patients were given LGLL therapy than non-PRCA patients, likely because PRCA patients were more transfusion-dependent. Among the treated patients, both PRCA and non-PRCA groups exhibited higher therapeutic response rates (72.7% in PRCA and 92.3% in non-PRCA), compared to the reported overall response rate of 40–60% in typical LGLL patients [28] [32, 36, 49]. Our study confirmed the previous finding that LGLL patients with PRCA showed superior response to immunosuppressive therapy [19, 20]. In addition, this is the first study that demonstrates LGLL patients with non-PRCA related isolated anemia also having an excellent therapeutic response to LGLL treatment. Half of the observed non-PRCA patients experienced progressive anemia, which warrants LGLL treatment in the future. This cohort’s high therapeutic response rate (83%) further confirms that LGLL is the etiology for the anemia and emphasizes the importance of LGLL diagnosis in patients with isolated anemia who may benefit from LGLL treatment.
It is uncertain the mechanism of how LGLL causes cytopenia, but the direct cytotoxicity effect on hematopoietic cells is an appealing hypothesis. In patients with PRCA, both in vitro and in vivo data suggested that erythropoiesis at a level of differentiation between early and late erythroid progenitors was inhibited by large granular lymphocytes, which was mediated by direct cell-cell interaction [48, 50]. Immature erythroid elements were essentially absent in PRCA patients but present in non-PRCA patients, suggesting LGLL cells inhibited both immature and mature erythroid elements that might share similar cell surface antigens at successive stages of differentiation. Interestingly, no LGLL patients with isolated anemia developed neutropenia during a median follow-up of 51 months. It is consistent with the previous reports that neutropenia was uncommon in T-LGLL patients with PRCA [18]. The finding raises a possibility that LGLL sometimes might apply lineage-specific inhibition mechanisms, especially in this LGLL group with much lower incidence of rheumatoid arthritis. We recognize that anemia is associated with neutropenia in many LGLL patients, and they may occur at different time points in the course of LGLL [34, 36]. The relatively short median follow-up in this study could not be long enough for the development of neutropenia. Therefore, a longer follow-up may be informative. Nevertheless, it is crucial to recognize that LGLL could cause isolated anemia at initial presentation as these patients may not develop neutropenia until much later.
In conclusion, anemia can uncommonly be the sole cytopenia at the initial diagnosis of LGLL, with PRCA seen in one-third of these patients. Compared to PRCA patients, isolated anemia patients without PRCA demonstrate similar clinicopathologic features, similar therapeutic responses, and comparable overall survival, indicating they are the continuum of the same disorder. This study highlights the importance of recognizing LGLL as a potential cause of isolated anemia at the initial presentation which allows for beneficial LGLL-related treatment.
References
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375–90.
Lamy T, Moignet A, Loughran TP. LGL leukemia: from pathogenesis to treatment. Blood. 2017;129:1082–94.
Barila G, Calabretto G, Teramo A, Vicenzetto C, Gasparini VR, Semenzato G, et al. T cell large granular lymphocyte leukemia and chronic NK lymphocytosis. Best Pract Res Clin Haematol 2019;32:207–16.
Zambello R, Semenzato G. Large granular lymphocyte disorders: new etiopathogenetic clues as a rationale for innovative therapeutic approaches. Haematologica 2009;94:1341–5.
Viny AD, Maciejewski JP. High rate of both hematopoietic and solid tumors associated with large granular lymphocyte leukemia. Leuk Lymphoma. 2015;56:503–4.
Zhang R, Shah MV, Loughran TP Jr. The root of many evils: indolent large granular lymphocyte leukaemia and associated disorders. Hematol Oncol. 2010;28:105–17.
Durrani J, Awada H, Kishtagari A, Visconte V, Kerr C, Adema V, et al. Large granular lymphocytic leukemia coexists with myeloid clones and myelodysplastic syndrome. Leukemia 2020;34:957–62.
Cheng J, Talamo G, Malysz J, Ochmann M, Lamy T, Loughran TP Jr. Report of 6 cases of large granular lymphocytic leukemia and plasma cell dyscrasia. Clin Lymphoma Myeloma Leuk. 2014;14:e169–72.
Goyal T, Thakral B, Wang SA, Bueso-Ramos CE, Shi M, Jevremovic D, et al. T-cell large granular lymphocytic leukemia and coexisting B-cell lymphomas: a study from the bone marrow pathology group. Am J Clin Pathol. 2018;149:164–71.
Lesesve JF, Feugier P, Lamy T, Bene MC, Gregoire MJ, Lenormand B, et al. Association of B-chronic lymphocytic leukaemia and T-large granular lymphocyte leukaemia. Clin Lab Haematol. 2000;22:121–2.
Morice WG, Kurtin PJ, Tefferi A, Hanson CA. Distinct bone marrow findings in T-cell granular lymphocytic leukemia revealed by paraffin section immunoperoxidase stains for CD8, TIA-1, and granzyme B. Blood 2002;99:268–74.
Shi M, Jevremovic D, Otteson GE, Timm MM, Olteanu H, Horna P. Single antibody detection of T-cell receptor alphabeta clonality by flow cytometry rapidly identifies mature T-cell neoplasms and monotypic small CD8-positive subsets of uncertain significance. Cytom B Clin Cytom. 2020;98:99–107.
Horna P, Olteanu H, Jevremovic D, Otteson GE, Corley H, Ding W, et al. Single-antibody evaluation of T-cell receptor beta constant chain monotypia by flow cytometry facilitates the diagnosis of T-cell large granular lymphocytic leukemia. Am J Clin Pathol. 2021;156:139–48.
Morice WG, Jevremovic D, Olteanu H, Roden A, Nowakowski G, Kroft S, et al. Chronic lymphoproliferative disorder of natural killer cells: a distinct entity with subtypes correlating with normal natural killer cell subsets. Leukemia 2010;24:881–4.
Coffy S, Shi M. Giant intracytoplasmic inclusions in a T-cell large granular lymphocytic leukemia patient with acute severe anemia. Blood 2019;134:492.
Alfano G, Ferrari A, Fontana F, Damiano F, Solazzo A, Mori G, et al. Hemolytic anemia as presentation of T-cell large granular lymphocytic leukemia after kidney transplantation: a case report. Transplant Proc 2020;52:1617–8.
Bagacean C, Tempescul A, Patiu M, Fetica B, Bumbea H, Zdrenghea M. Atypical aleukemic presentation of large granular lymphocytic leukemia: a case report. Onco Targets Ther. 2017;10:31–4.
Go RS, Li CY, Tefferi A, Phyliky RL. Acquired pure red cell aplasia associated with lymphoproliferative disease of granular T lymphocytes. Blood 2001;98:483–5.
Lacy MQ, Kurtin PJ, Tefferi A. Pure red cell aplasia: association with large granular lymphocyte leukemia and the prognostic value of cytogenetic abnormalities. Blood 1996;87:3000–6.
Fujishima N, Sawada K, Hirokawa M, Oshimi K, Sugimoto K, Matsuda A, et al. Long-term responses and outcomes following immunosuppressive therapy in large granular lymphocyte leukemia-associated pure red cell aplasia: a Nationwide Cohort Study in Japan for the PRCA Collaborative Study Group. Haematologica 2008;93:1555–9.
Means RT Jr. Pure red cell aplasia. Hematol Am Soc Hematol Educ Program. 2016;2016:51–6.
Sawada K, Fujishima N, Hirokawa M. Acquired pure red cell aplasia: updated review of treatment. Br J Haematol. 2008;142:505–14.
Shi M, He R, Feldman AL, Viswanatha DS, Jevremovic D, Chen D, et al. STAT3 mutation and its clinical and histopathologic correlation in T-cell large granular lymphocytic leukemia. Hum Pathol. 2018;73:74–81.
Neff JL, Rangan A, Jevremovic D, Nguyen PL, Chiu A, Go RS, et al. Mixed-phenotype large granular lymphocytic leukemia: a rare subtype in the large granular lymphocytic leukemia spectrum. Hum Pathol. 2018;81:96–104.
Langerak AW, Groenen PJ, Brüggemann M, Beldjord K, Bellan C, Bonello L. et al. EuroClonality/BIOMED-2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia. 2012;26:2159–71.
Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng NG, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 2012;120:3048–57.
Ravindran A, He R, Ketterling RP, Jawad MD, Chen D, Oliveira JL, et al. The significance of genetic mutations and their prognostic impact on patients with incidental finding of isolated del(20q) in bone marrow without morphologic evidence of a myeloid neoplasm. Blood Cancer J. 2020;10:7.
Loughran TP Jr, Zickl L, Olson TL, Wang V, Zhang D, Rajala HL, et al. Immunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the Eastern Cooperative Oncology Group (E5998). Leukemia 2015;29:886–94.
Shi M, Neff JL, Jevremovic D, Morice WG. Decreased normal NK-cells is a characteristic of T-cell large granular lymphocytic leukemia and is strongly associated with cytopenia. Leuk Lymphoma. 2016;57:1230–3.
Hanson CA, Steensma DP, Hodnefield JM, Nguyen PL, Hoyer JD, Viswanatha DS, et al. Isolated trisomy 15: a clonal chromosome abnormality in bone marrow with doubtful hematologic significance. Am J Clin Pathol. 2008;129:478–85.
Wiktor AE, Van Dyke DL, Hodnefield JM, Eckel-Passow J, Hanson CA. The significance of isolated Y chromosome loss in bone marrow metaphase cells from males over age 50 years. Leuk Res. 2011;35:1297–300.
Bareau B, Rey J, Hamidou M, Donadieu J, Morcet J, Reman O, et al. Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica 2010;95:1534–41.
Zhu Y, Gao Q, Hu J, Liu X, Guan D, Zhang F. Clinical features and treatment outcomes in patients with T-cell large granular lymphocytic leukemia: a single-institution experience. Leuk Res. 2020;90:106299.
Braunstein Z, Mishra A, Staub A, Freud AG, Porcu P, Brammer JE. Clinical outcomes in T-cell large granular lymphocytic leukaemia: prognostic factors and treatment response. Br J Haematol. 2021;192:484–93.
Dong N, Castillo Tokumori F, Isenalumhe L, Zhang Y, Tandon A, Knepper TC, et al. Large granular lymphocytic leukemia - a retrospective study of 319 cases. Am J Hematol. 2021;96:772–80.
Sanikommu SR, Clemente MJ, Chomczynski P, Afable MG, Jerez A, Thota S, et al. Clinical features and treatment outcomes in large granular lymphocytic leukemia (LGLL). Leuk Lymphoma. 2018;59:416–22.
Thomas J, Haseman JK, Goodman JI, Ward JM, Loughran TP Jr, Spencer PJ. A review of large granular lymphocytic leukemia in Fischer 344 rats as an initial step toward evaluating the implication of the endpoint to human cancer risk assessment. Toxicol Sci. 2007;99:3–19.
Rivero A, Mozas P, Jimenez L, Lopez-Guerra M, Colomer D, Bataller A, et al. Clinicobiological characteristics and outcomes of patients with T-cell large granular lymphocytic leukemia and chronic lymphoproliferative disorder of natural killer cells from a single institution. Cancers. 2021;13:3900.
Awada H, Mahfouz RZ, Durrani J, Kishtagari A, Jagadeesh D, Lichtin AE, et al. Large granular lymphocytic leukaemia after solid organ and haematopoietic stem cell transplantation. Br J Haematol. 2020;189:318–22.
Ferrucci L, Maggio M, Bandinelli S, Basaria S, Lauretani F, Ble A, et al. Low testosterone levels and the risk of anemia in older men and women. Arch Intern Med. 2006;166:1380–8.
Fonseca R, Rajkumar SV, White WL, Tefferi A, Hoagland HC. Anemia after orchiectomy. Am J Hematol. 1998;59:230–3.
Gazitt T, Loughran TP Jr. Chronic neutropenia in LGL leukemia and rheumatoid arthritis. Hematol Am Soc Hematol Educ Program. 2017;2017:181–6.
Go RS, Lust JA, Phyliky RL. Aplastic anemia and pure red cell aplasia associated with large granular lymphocyte leukemia. Semin Hematol. 2003;40:196–200.
Kawakami T, Sekiguchi N, Kobayashi J, Imi T, Matsuda K, Yamane T, et al. Frequent STAT3 mutations in CD8(+) T cells from patients with pure red cell aplasia. Blood Adv. 2018;2:2704–12.
Balasubramanian SK, Sadaps M, Thota S, Aly M, Przychodzen BP, Hirsch CM, et al. Rational management approach to pure red cell aplasia. Haematologica 2018;103:221–30.
Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. New Engl J Med. 2012;366:1905–13.
Ishida F, Matsuda K, Sekiguchi N, Makishima H, Taira C, Momose K, et al. STAT3 gene mutations and their association with pure red cell aplasia in large granular lymphocyte leukemia. Cancer Sci. 2014;105:342–6.
Qiu ZY, Qin R, Tian GY, Wang Y, Zhang YQ. Pathophysiologic mechanisms and management of large granular lymphocytic leukemia associated pure red cell aplasia. Onco Targets Ther. 2019;12:8229–40.
Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood 2011;117:2764–74.
Abkowitz JL, Kadin ME, Powell JS, Adamson JW. Pure red cell aplasia: lymphocyte inhibition of erythropoiesis. Br J Haematol 1986;63:59–67.
Author information
Authors and Affiliations
Contributions
Y.S. and M.S. performed the analysis and drafted the manuscript. M.S., F.Z., and J.L.O. designed the study. M.S., F.Z., and J.Y. collected the data. All authors (J.L.O., J.Y., D.J., R.S.G., W.D., S.A.P., M.V.S., P.J.H., A.A., and W.G.M.) critically reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
S.A.P.: research funding has been provided to the institution from Pharmacyclics, MorphoSys, Janssen, AstraZeneca, TG Therapeutics, Bristol Myers Squibb, Merck, AbbVie, and Ascentage Pharma for clinical studies in which S.A.P. is a principal investigator. He has received honoraria for participation in consulting activities/advisory board meetings for Pharmacyclics, AstraZeneca, Genentech, GlaxoSmithKline, Adaptive Biotechnologies, and AbbVie (no personal compensation); and from Aptitude Health, Curio Science, and MedEd on the Go (with personal compensation). A.A.: received support to the institution from Novartis, Astex, Celgene/BMS, Onconova, Aprea, AstraZeneca. W.D.: research funding received from Merck, AbbVie, Astrazeneca, Bristol Myers Squibb, Octapharma, DTRM pharma directed to the institution where Dr. Ding is a principal investigator. She also has participated in advisory board meetings/steering committees for BeiGene, Octapharma, Alexion, AbbVie, and Merck without personal compensation. Other authors have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salama, Y., Zhao, F., Oliveira, J.L. et al. Isolated anemia in patients with large granular lymphocytic leukemia (LGLL). Blood Cancer J. 12, 30 (2022). https://doi.org/10.1038/s41408-022-00632-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-022-00632-6
