
Abstract
Multiple myeloma (MM) is a clonal plasma cell disorder that is characterized by a variety of genetic alterations. Recent studies have highlighted not only the importance of these genetic events but also epigenetic aberrations including DNA methylation, histone modifications, and non-coding RNAs in the biology of MM. Post-translational modifications of histone, such as methylation and acetylation, contribute to chromatin dynamics, and are modulated by histone modifying enzymes, and dysregulation of these enzymes is implicated in the pathogenesis of cancers, including MM. Histone modifiers also have non-histone substrates and enzymatically independent roles, which are also involved in tumorigenesis. Here we review and provide comprehensive insight into the biologic significance of histone methyl- and acetyl-modifiers in MM, and further provide an overview of the clinical applications of histone modifier inhibitors, especially histone deacetylase inhibitors. These findings underline the emerging roles of histone modifiers in the pathogenesis of MM, and further highlight the possibility of novel epigenetic therapies in MM.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is a plasma cell malignancy characterized by clonal evolution1. It arises from a premalignant stage known as monoclonal gammopathy of undetermined significance (MGUS), with subsequent multistep genetic alterations resulting in progression from MGUS to smoldering MM and later to symptomatic MM1. Recent studies have characterized these genetic events, including chromosomal translocation/deletion and somatic mutations2. In addition to genetic aberrations, accumulating evidence also indicates that epigenetic changes including DNA methylation, histone modifications, and non-coding RNAs also play crucial roles in the development of this disease. For example, global DNA hypomethylation with gene-specific DNA hypermethylation has been observed in MM3. MM-specific microRNA (miRNA) signatures have also been reported4. Furthermore, dysregulation of histone modifying enzymes, such as MM SET domain (MMSET) and lysine demethylase 6A (KDM6A), have shed light on aberrant histone modifications in MM5,6. The functional significance of other histone modifiers is also beginning to be elucidated.
In this review, we describe the biologic roles of histone modifying enzymes in MM, especially focusing on methylation and acetylation modifiers. Where relevant, we also discuss the clinical applications of histone modifier inhibitors, especially histone deacetylase (HDAC) inhibitors. For the reviews on the other epigenetics, including DNA methylation and miRNAs in MM, we respectfully refer readers to relevant reviews7,8,9.
Histone modifications
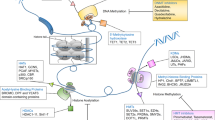
Since Allfrey et al.10 demonstrated that histones can be methylated and acetylated in 1964, at least 16 classes of histone modifications including methylation, acetylation, phosphorylation, and ubiquitination have been identified11,12. These modifications not only alter chromatin structure, but also contribute to the recruitment of protein complexes to the modification sites, which in turn affects DNA-templated processes, such as gene expression; dysregulation of these modifications can lead to diseases, including cancers11,13. Hence, these modifications are strictly controlled by histone modifying enzymes, the writers and erasers of these modifications (Fig. 1). Importantly, these histone modifiers also have non-histone targets and enzymatic activity-independent functions (Fig. 1). Here we will provide an overview of histone methylation and acetylation, as well as their modifiers.

a Histone modifiers modify the basic residues of histone tail, leading to alteration of chromatin structure and gene expression. b Histone modifiers also modify non-histone proteins, resulting in alteration of protein function. c Histone modifiers can alter chromatin structure and gene expression, as well as affect other protein function in an enzymatically independent manner. TF transcription factor, lncRNA long non-coding RNA
Histone methylation
Histone methylation can occur on the side chains of arginine, lysine, and histidine residues11,13. Arginines are monomethylated (me1) or symmetrically or asymmetrically dimethylated (me2s/me2a), whereas lysines are mono- (me1), di- (me2) or trimethylated (me3). Histidines have been shown to be monomethylated, although this methylation has not been well defined13. Various basic residues throughout the histone proteins are methylated14: among these methylation sites, histone H3 lysine4 (H3K4), H3K9, H3K27, H3K36, H3K79, and H4K20 have been well characterized. Not only the site of the methyl–lysine residue within a histone tail but also the status of methylation is linked to gene expression status. For example, H3K9me3 is associated with gene repression, while H3K9me1 is related to gene activation15. In general, H3K4me3, H3K36me3, and H3K79me3 are found near the transcriptional start site of active genes, whereas H3K9me3 and H3K27me3 are at silent promoters15. Histone methylation status is dynamically modified by histone methyltransferases (HMTs) and lysine demethylases (KDMs) in the context of specific biological processes.
HMTs
There are histone arginine methyltransferases and lysine methyltransferases. Arginine methyltransferases comprise nine members of the protein arginine methyltransferase (PRMT) family, which mediates three types of arginine methylation (me1/me2s/me2a)16. Lysine methyltransferases consist of a large family of proteins that possess the catalytic SET domain, except for one enzyme DOT1 like histone lysine methyltransferase (DOT1L)17. The SET domain-containing proteins are divided into six subfamilies based on sequence homology: the suppressor of variegation 39 (SUV39) family, the enhancer of zeste homolog (EZH) family, the SET domain-containing 1 (SET1) family, the SET2 family, the PR domain-containing (PRDM) family, and the SET and MYND domain-containing (SMYD) family. Lysine methyltransferases mediate mono-, di-, or trimethylation of lysine residues. Each methyltransferase has substrate specificity towards histone basic residues17. For example, the members of the SUV39 family methylate H3K9, while DOT1L catalyze H3K79 methylation. Importantly, HMTs also methylate various non-histone proteins, including p53 and vascular endothelial growth factor receptor 1 (VEGFR1), thereby altering their function and stability16,18.
KDMs
Histone arginine demethylases have not yet been identified. KDMs are divided into two families based on catalytic mechanism: the lysine-specific demethylase (LSD) family and the Jumonji C (JMJC) family13,19. The LSD family catalyzes demethylation of H3K4 or H3K9 by a flavin adenine dinucleotide-dependent amine oxidation reaction13,19. This family can remove mono- and dimethylation from lysine residues, but not trimethylation due to the requirement of a free electron pair at the methylated lysine residue for the demethylase activity19. The JMJC family is characterized by the conserved JMJC domain and is comprised of more than 30 proteins; however, not all family members possess enzymatic activity. This family members are further divided into subfamilies KDM2-8, based on structural similarity19. Each member has substrate specificity, and KDMs against H3K4, H3K9, H3K27, H3K36, and H4K20 have been identified. The JMJC demethylases catalyze demethylation of lysine residues by a dioxygenase reaction which requires iron and α-ketoglutarate13,19. Due to its distinct catalytic mechanism from the LSD family, the JMJC family can demethylate trimethylated lysine residues. As is the case in HMTs, KDMs also have non-histone targets19, as well as catalytically independent functions19.
Histone acetylation
Histones are acetylated on the side chains of lysine residues, which neutralizes lysine’s positive charge, leading to open chromatin structure by reducing interaction between histone and negatively charged DNA11,20. Thus, histone acetylation increases the accessibility of proteins, such as transcription factors, to promoters and enhancers, thereby mediating active gene expression. Acetylated histones also function as binding sites for numerous proteins with bromodomains, which often activates gene transcription20. In contrast, deacetylation of histone is associated with chromatin condensation and transcriptional repression20. Analogous to histone methylation, histone acetylation is reversibly controlled by two large families of enzymes: histone lysine acetyltransferases (KATs) and HDACs.
KATs
KATs comprise two major classes of enzymes: type A and type B KATs. Type A KATs are localized in the nucleus and involved in acetylation of histones in the context of chromatin11. Type A KATs include the Gcn5-related N-acetyltransferase (GNAT) family, the MYST (MOZ, Ybf2/Sas3, Sas2, Tip60) family, and the CREB binding protein (CBP)/p300 family. Type B KATs are engaged in the acetylation of newly synthesized free histones, but not nucleosomal histones11. Of note, KATs serve not only as histone acetyltransferases but also as acetyltransferases of other proteins. Similar to methylation, acetylation of non-histone proteins alters their protein functions21. KATs also function as molecular scaffolds to recruit protein complexes22.
HDACs
HDACs contain 18 enzymes, which are grouped into four classes (class I, II, III, and IV) based on structural homology11,20. Class I HDACs, which are homologous to yeast Rpd3, include HDAC1, 2, 3, and 8. Class II HDACs are closely related to yeast Hda1, and subdivided into two classes: class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6 and 10). Class IV HDAC consists of only one HDAC, HDAC11, whose catalytic domain shows similarity to those of both class I and class II, but does not possess enough homology to be placed in either class. Class I, II, and IV HDACs require a zinc metal ion for their enzymatic activity11. Class III HDACs (Sirtuin (SIRT)1–7) share homology with yeast Sir2, and have different enzymatic mechanism from other HDACs, which does not require a zinc metal ion, but has NAD+ dependence23. Similar to KATs, HDACs have numerous non-histone substrates, including oncoproteins and tumor suppressors, and affect their functions.
The roles of histone methylation modifiers in MM
MMSET
MMSET (also known as NSD2/WHSC1) is one of the HMTs containing the SET domain and catalyzes addition of H3K36me2, a methyl mark associated with active chromatin24,25. MMSET also functions as a transcriptional repressor by interacting with sin3a, HDAC1, HDAC2, and LSD126. MMSET is thought to be implicated in Wolf–Hirschhorn syndrome, a developmental disorder characterized by intellectual disability, craniofacial malformation, and heart and skeletal defects, since heterozygous deletion of MMSET is found in most patients with this syndrome27. MMSET is also involved in cancers. MMSET is overexpressed or somatically mutated in a variety of cancers, including MM28,29,30.
MMSET is the most extensively studied histone methyl modifier in MM because of its clinical relevance. The t(4;14)(p16;q32) is one of the most common translocations in MM, presenting in nearly 15% of patients, and is related to poor prognosis5,31. This translocation breakpoints on 4p16 are between genes encoding FGFR3 and MMSET, resulting in juxtaposition of both genes to the immunoglobulin heavy chain enhancer32,33. While MMSET is overexpressed in all t(4;14) MM samples, FGFR3 is expressed in only about 70% of them5,34. Moreover, the presence of t(4;14) is a predictor for poor prognosis regardless of FGFR3 expression, suggesting that activation of MMSET, not FGFR3, plays a critical role in the pathogenesis of this recurrent translocation5.
Recent studies have delineated the biological roles of MMSET in t(4;14) MM. Knockdown or knockout of MMSET induces cell cycle arrest and apoptosis, thereby reducing cell growth in t(4;14) MM cells24,25,35,36. Complementation of wild-type MMSET, but not enzymatically inactive mutant, restores cell growth in MMSET knockout cells24,25, indicating that MMSET stimulates MM cell growth depending on its methyltransferase activity. The underlying mechanisms of MMSET-mediated MM cell growth and survival are beginning to be deciphered (Fig. 2). Annunziata et al.37 have shown that MAF and its target genes are highly expressed not only in MM cells with MAF translocation, but also in those with MMSET translocation. They have further demonstrated that MMSET upregulates MAF transcription through activation of mitogen-activated protein kinase (MAPK) pathway, and that ectopic expression of MAF rescues t(4;14) MM cells from toxicity of MMSET depletion or MEK (MAPK kinase) inhibition, identifying MAF oncogene as a downstream effector of MMSET37. MMSET has also been shown to increase c-MYC expression by repressing expression of miR-126*, which targets 3’-untranslated region of c-MYC and inhibits its translation38. Overexpression of MMSET affects binding and distribution of another histone methyltransferase EZH2 across the genome, resulting in decreased global H3K27me3 level and increased H3K27me3 level at the specific loci39. Importantly, the miR-126* locus is one of these specific loci, and treatment with EZH2 inhibitor restores miR-126* expression, resulting in reduction of c-MYC expression and cell proliferation in MMSET-overexpressing cells39. Interferon regulatory factor 4 (IRF4) is a crucial survival factor in MM cells40. In t(4;14) MM cells, MMSET directly activates IRF4 expression through binding to the IRF4 promoter41. Overexpression of MMSET also confers chemotherapy resistance in MM cells. MMSET is involved in the DNA damage response42,43. Shah et al.44 have shown that DNA repair after treatment with DNA-damaging agents is facilitated in MMSET-high MM cells compared to MMSET-low MM cells, suggesting a reason underlying the poor prognosis of t(4;14) MM.

The t(4;14)(p16;q32) results in juxtaposition of MMSET to the immunoglobulin heavy chain (IgH) 5’ enhancer, leading to aberrant MMSET expression. MMSET confers MM cell growth and survival via (1) MAF upregulation through MAPK pathway, (2) c-MYC activation through inhibition of miR-126* by EZH2, and (3) direct IRF4 induction. MMSET also contributes to chemotherapy resistance by facilitating DNA damage repair
EZH2
Polycomb repressive complex 2 (PRC2) maintains the silent state of target genes such as HOX genes through trimethylation of H3K27, contributing to development and differentiation as well as tumorigenesis45,46. EZH2 is a core component of PRC2 that confers the HMT activity45,46. Overexpression of EZH2 is associated with progression in prostate and breast cancer45,46. Somatic activating mutations of EZH2 have also been found in diffuse large B-cell lymphoma and follicular lymphoma, whereas inactivating mutations have been reported in myeloid neoplasms, suggesting context-dependent roles of EZH2 in tumorigenesis45,46.
EZH2 is supposed to function as an oncogene in MM. Although EZH2 mutation has not been reported in MM, overexpression of EZH2 is linked to disease progression and poor prognosis47,48,49. Moreover, EZH2 is not expressed in normal bone marrow plasma cells50. The following studies further suggest involvement of EZH2 in MM pathogenesis. EZH2 is induced by interleukin 6 (IL-6) in IL-6-dependent MM cell lines, and expressed constitutively in IL-6-independent MM cell lines51. Furthermore, knockdown of EZH2 impairs MM cell growth, whereas ectopic expression of EZH2 induces IL-6 independence in IL-6-dependent MM cell lines51. c-Rel may be involved in constitutive EZH2 expression, since EZH2 expression is sustained by c-Rel in MM.1S, an IL-6-independent MM cell line52. Consistent with the notion that EZH2 is overexpressed in MM, H3K27me3 level is elevated at polycomb target genes in MM, and expression of these targets is decreased in MM compared to normal plasma cells53,54. This expression signature is more prominent in advanced stages of MM53,54. Recent studies have revealed the molecular mechanisms of EZH2-mediated MM cell growth and survival55,56. EZH2 interacts with the long non-coding RNA MALAT1, and EZH2 and MALAT1 cooperatively downregulate an anti-MM miRNA miR-29b by increasing H3K27me3 mark at its promoter55. Inhibition of EZH2 or MALAT1 induces miR-29b expression, thereby downregulating major miR-29b pro-survival targets such as SP1, CDK6, and MCL-1 and reducing MM cell growth55. Conversely, miR-29b inhibitor abrogates cell growth inhibition induced by EZH2 inhibitor, indicating that suppression of miR-29b mediates EZH2-driven MM cell growth and survival55. EZH2 and MALAT1 complex also promotes MM cell survival through epigenetic repression of KEAP1 expression56. Suppression of KEAP1 activates NRF1 and NRF2 expression, and then reduces endoplasmic reticulum (ER) stress-induced apoptosis by induction of proteasome gene expression56.
Recently, several EZH2-specific inhibitors have been developed, and pharmacologic inhibition of EZH2 by these inhibitors (E7438, UNC1999, GSK126, and EPZ005687) has been shown to exert anti-MM effects48,54,57,58,59,60. EZH2 inhibitors reduce global H3K27me3 level and trigger apoptosis in MM cells48,54,57,58,59,60. E7438 activates expression of epithelial tumor suppressor genes such as CDH1, EMP1, and EPHB2, although the role of these genes in MM remains unclear57. UNC1999 induces expression of miR-125a and miR-320c, resulting in the reduction of their targets IRF4, XBP1, and PRDM159. UNC1999 also induces NR4A1, which in turn suppresses expression of MYC60. GSK126 induces the intrinsic mitochondrial apoptosis pathway, as well as decreases MM stem-like cells by blocking Wnt/β-catenin pathway58. GSK126 also rescues osteoblast precursors from MM-induced suppression of osteoblast differentiation, suggesting that EZH2 inhibition may also be an effective treatment of lytic bone lesions in MM61. EPZ005687 upregulates cell cycle control genes, resulting in cell cycle arrest48.
EZH2 inhibition may or may not promote drug resistance in MM. Kikuchi et al.62 have shown that chemotherapy agents increase global H3K27me3 level, inducing MM cell death; conversely, MM cell adhesion to bone marrow stromal cells leads to EZH2 inactivation via phosphorylation of EZH2, counteracting drug-induced H3K27 hypermethylation and MM cell death. Moreover, knockdown of EZH2, as well as pharmacologic inhibition of EZH2, reduce the toxicity of chemotherapy agents in MM cells, confirming that EZH2 inactivation is the underlying mechanism of cell adhesion-mediated drug resistance62. On the other hand, Rastgoo et al.49 have shown that EZH2 is elevated via downregulation of miR-138 that is a suppressor of EZH2 in drug-resistant MM cell lines, and ectopic expression of EZH2 in parental cell lines induces drug resistance of these cell lines. Mechanistically, EZH2 directly suppresses RBPMS expression, which in turn activates MYC and BCL2 expression, conferring drug resistance in MM cells49. Conversely, restoration of RBPMS by forced expression of miR-138 restores the drug sensitivity in MM cells49.
PRMT5
PRMT5 (also known as JBP1/SKB1) is a type II arginine methyltransferase which catalyzes mono- and symmetric dimethylation of arginine. PRMT5 regulates various cellular processes via methylation of histone and other substrate proteins, including p53 and E2F1, and deregulation of this enzyme is implicated in cancers16.
PRMT5 is overexpressed in MM, and higher PRMT5 expression is associated with poor clinical outcome63. Indeed, PRMT5 confers MM cell growth. PRMT5 interacts with E3 ubiquitin ligase TRIM21, and this interaction prevents TRIM21-mediated degradation of IKKβ, thereby activating nuclear factor (NF)-κB signaling63. Notably, PRMT5 inhibitor (EPZ015666) decreases MM cell growth by blocking NF-κB activation, suggesting a potential clinical application of this drug63.
KDM3A
KDM3A (also known as JMJD1A/JHDM2A) is a member of the JMJC demethylases, which catalyze removal of H3K9me1 and H3K9me264. KDM3A is implicated in many biological processes including spermatogenesis, systemic metabolism, and sex determination65,66,67. In addition, oncogenic functions of KDM3A have been reported in cancers68,69.
We have recently shown the biological significance of KDM3A in MM. KDM3A is highly expressed in MM, and required for MM cell survival70. Mechanistically, KDM3A sustains expression of KLF2 and a MM master transcription factor IRF440 via H3K9 demethylation at their promoters70. KLF2 is a transcription factor that maintains homeostasis of B cells and plasma cells71. We further showed that KLF2 also plays a crucial role in MM cell survival70. Importantly, KLF2 directly stimulates IRF4 expression and IRF4 reciprocally activates KLF2 expression, forming a positive autoregulatory circuit downstream of KDM3A70. The interaction of MM cells with bone marrow microenvironment plays an essential role in MM cell survival72, and KDM3A also regulates MM cell adhesion and homing to the bone marrow, further promoting MM cell growth and survival70. A recent report has shown KDM3A functions in hypoxic conditions73. KDM3A is induced via hypoxia-inducible factor-1α under hypoxia in MM cells, and hypoxia-induced KDM3A stimulates expression of glycolytic genes through upregulation of MALAT1, conferring anti-apoptotic properties in MM cells73.
KDM6A
KDM6A (also known as UTX) is a JMJC demethylase which removes H3K27me2 and me3, methyl marks correlated with genomic silencing74,75. KDM6A regulates HOX genes transcription and contributes to development and differentiation, in concert with the H3K4me3 histone methyltransferase MLL2/374,75. Recent studies have shown that KDM6A is implicated in cancers such as acute lymphoblastic leukemia, chronic myelomonocytic leukemia, and bladder cancer76.
KDM6A is also implicated in MM pathogenesis since 10% MM samples have inactivating mutations in KDM6A6. Interestingly, samples with KDM6A mutations do not have t(4;14) translocation, implying potential mutual exclusion of KDM6A mutations and MMSET activation6. Recent studies using whole-exome sequencing have also revealed KDM6A mutations in MM patient samples at a frequency as low as 1–3%77,78. Of note, KDM6A mutations are associated with shorter survival in MM77. KDM6A is supposed to be a tumor suppressor in MM, given its inactivating mutations. Indeed, loss of KDM6A has recently been shown to promote MM cell proliferation via aberrant gene repression79. Interestingly, significant overlap was found between genes repressed by KDM6A loss and PRC2 target genes, and EZH2 inhibitors induce cell death by reactivating these repressed genes in KDM6A-null MM cells79.
KDM6B
KDM6B (also known as JMJD3) is another H3K27 demethylase which is closely related to KDM6A75,80. KDM6B is engaged in inflammatory response, stress-induced senescence, development, and differentiation75,80,81. KDM6B is also involved in the pathogenesis of cancers in a context-dependent manner76.
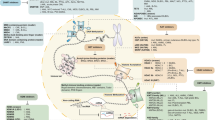
KDM6B acts as a mediator of MM cell survival in a demethylase activity-independent manner (Fig. 3)82. KDM6B expression in MM is induced by bone marrow stromal cells; conversely, this response is abrogated by IKKβ inhibitor MLN120B, indicating that KDM6B is activated via NF-κB signaling, a crucial survival pathway in MM cells82. The small hairpin RNA-mediated knockdown and CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-mediated knockout experiments revealed that KDM6B is necessary for MM cell survival82. Ectopic expression of KDM6B partially rescues MM cells from MLN120B-induced cytotoxicity, suggesting that KDM6B is one of the downstream effectors in the NF-κB pathway in MM82. Of note, KDM6B is recruited to the transcriptional start sites of MAPK signaling pathway-related genes such as ELK1 and FOS, and upregulates these gene expressions without altering H3K27 methylation level82. Overexpression of enzymatically dead KDM6B induces expression of MAPK pathway-related genes, indicating its catalytically independent function82. These data have revealed a novel function of KDM6B that links NF-κB and MAPK signaling pathways in MM.

KDM6B is upregulated by NF-κB, in part due to bone marrow stromal cells (BMSCs). KDM6B is in turn recruited to the loci of MAPK pathway-related genes, and activates expression of these genes in a catalytically independent manner. X represents an unknown scaffold for KDM6B
The roles of histone acetylation modifiers in MM
CBP/p300
CBP and its homolog p300 form CBP/p300 family, which is recently renamed as lysine acetyltransferase 3 (KAT3) family83. CBP/p300 acetylates H3K18 and H3K27, activating gene expression84. CBP/p300 also has multiple interaction domains and serves as a molecular scaffold to recruit transcriptional complexes to transcriptional machinery22. These proteins also have a variety of non-histone substrates, including TP53, CREB, E2F, and MYB22. CBP/p300 is required for normal development, and dysregulation of its functions is implicated in hematologic malignancies including acute myeloid leukemia and T-cell acute lymphoblastic leukemia22.
A recent study has shown that targeting the bromodomain of CBP/p300 is a promising therapeutic strategy in MM85. CBP/p300 bromodomain-specific inhibitors were identified, and their biological activity was evaluated using a panel of cell lines. Interestingly, most sensitive cell lines are derived from MM. Mechanistic analyses have revealed that CBP/p300 directly activates IRF4 expression through H3K18 and H3K27 acetylation at its super-enhancer and transcription start site; therefore, inhibition of CBP/p300 induces MM cell death via the reduction of IRF4 expression85.
HDAC1/HDAC3
Aberrant expression or recruitment to specific loci of HDACs has been identified in various types of cancers, and HDAC inhibitors have been shown to have anti-tumor activities, especially in hematologic cancers such as cutaneous T-cell lymphoma and myelodysplastic syndromes86. HDAC inhibitors also impair MM cell growth and survival. Preclinical studies have demonstrated that HDAC inhibitors trigger apoptosis, as well as induce cell cycle arrest, in MM cells87,88,89. Given that most HDAC inhibitors do not target class IIa enzymes at pharmacologically relevant concentrations90, class I or class IIb HDACs are implicated in MM pathogenesis. Among the class I HDACs, we have recently shown that HDAC3 is especially important in MM cell survival. Although both HDAC1 and HDAC3 knockdown inhibit MM cell growth, the growth inhibitory effect of HDAC3 knockdown is more significant than HDAC1 knockdown, in contrast to HDAC2 knockdown with minimal effect on MM cell growth91. Consistent with these findings, HDAC1, 2, and 3 inhibitor MS-275 is more toxic to MM cells than HDAC1 and 2 inhibitor Merck6091. Furthermore, HDAC3-specific inhibitor BG45 shows a potent cytotoxic effect in MM cells91. Notably, knockdown or pharmacologic inhibition of HDAC3 increases acetylation level of c-MYC and DNMT1 protein92. Acetylation of c-MYC and DNMT1 has been shown to promote turnover of these proteins through proteasome-dependent degradation93,94. Indeed, increased acetylation of c-MYC and DNMT1 after HDAC3 knockdown or inhibition enhances turnover and reduction of those proteins92. HDAC3 inhibition also reduces phosphorylation level of signal transducer and activator of transcription 3 (STAT3), and yet the underlying mechanism of this reduction remains elusive91. These data suggest that HDAC3 sustains MM cell survival via stabilization of oncoproteins c-MYC and DNMT1, as well as STAT3 activation (Fig. 4). Class I HDACs have also been shown to be the key molecules in bortezomib-induced cytotoxicity in MM95. Bortezomib decreases expression of class I HDACs via degradation of Sp1 protein, the transcriptional activator of class I HDAC genes95. Conversely, ectopic expression of HDAC1 induces bortezomib resistance in MM cells, suggesting that class I HDACs, especially HDAC1 downregulation, mediates bortezomib-induced MM cell death95. Importantly, patients with higher expression of class I HDACs have significantly shorter progression-free survival96.

HDAC3 deacetylates c-MYC and DNMT1 protein, thereby stabilizing these proteins. HDAC3 also activates STAT3 via an unknown mechanism, conferring MM cell survival. HDAC3 inhibitor increases acetylation level of c-MYC and DNMT1, resulting in reduction of these proteins. HDAC3 inhibitor also decreases phosphorylated STAT3
HDAC4
Among the class IIa HDACs, HDAC4 is essential for MM cell growth and survival97,98,99. Knockdown of HDAC4 inhibits MM cell growth via inducing apoptosis and autophagy97,98,99, whereas knockdown of HDAC5, 7, and 9 does not affect MM cell growth97. HDAC4 acts as a transcriptional corepressor by interacting with numerous transcription factors, including MEF2 and Runx2, during development and differentiation process100. Interestingly, HDAC4 forms a complex with the alternative NF-κB (RelB-p52) in MM cells, and represses pro-apoptotic genes Bim and BMF via deacetylation of H3 at their promoter regions, conferring MM cell survival99. This study further demonstrated that disruption of the RelB-HDAC4 complex using HDAC4-mimetic polypeptide induces Bim and MM cell death99. HDAC4 has also been shown to downregulate a tumor suppressor miRNA miR-29b by sustaining repressive chromatin at its promoter98. miR-29b is known to be an ep-miRNA which targets epigenetic regulators including DNA methyltransferases101. Importantly, HDAC4 is also a direct target of miR-29b, forming an epigenetic negative feedback loop in MM cells98. Introduction of miR-29b mimics reduced HDAC4 and increased the acetylation of both histone H4 and α-tubulin, indicating that miR-29b is involved in the acetylome in MM cells98. HDAC4 also counteracts ER stress response in MM: knockdown of HDAC4 under ER stress enhances ATF4 and CHOP induction, augmenting apoptosis in MM cells97. This result provides the rationale for combination therapy with class IIa HDAC inhibitors and proteasome inhibitors. Indeed, a class IIa HDAC inhibitor TM269 in combination with carfilzomib shows a strong synergistic cytotoxic effect in MM cell lines and patient MM cells. However, TM269 is not a clinical drug, and further studies are required for the development of clinical grade HDAC4 inhibitor97.
HDAC6
Because aggregation of unfolded/misfolded proteins are toxic to cells, unfolded/misfolded proteins are tightly monitored and processed in cells. Unfolded/misfolded proteins are usually polyubiquitinated and degraded by the proteasome. However, once unfolded/misfolded proteins are overloaded and aggregated, aggresomes are formed to degrade and remove these aggregates102. In the context of unfolded/misfolded protein processing, a microtubule-associated deacetylase HDAC6 plays a crucial role in aggresome formation103. HDAC6 bridges between polyubiquitinated proteins and the dynein motor complex, thereby recruiting polyubiquitinated proteins to aggresomes103. Indeed, depletion of HDAC6 results in failure of aggresome formation, and enhances induction of apoptosis in proteasome inhibitor-treated cells103. This is also the case in MM cells. HDAC6 inhibitor tubacin inhibits interaction of HDAC6 with dynein, and enhances bortezomib-induced cytotoxicity in MM cells104. Other HDAC6-specific inhibitors (ACY-1215, WT161) in combination with proteasome inhibitors also lead to synergistic accumulation of polyubiquitinated proteins and augment proteasome-induced cytotoxic effect in MM cells105,106,107. Indeed, a phase I/II trial of ACY-1215 in combination with bortezomib and dexamethasone for relapsed and refractory MM has shown promising results (described below). A recent study has suggested another HDAC6 function in MM: Imai et al.108 have shown that expression of PPP3CA, a catalytic subunit of calcineurin, is highly expressed in advanced MM patients’ samples, and that PPP3CA is indispensable for MM cell growth. HDAC6 is known to deacetylate HSP90 and maintain its chaperone function109. Consistent with this notion, treatment with ACY-1215 reduces the protein level of PPP3CA which is a HSP90 client protein, suggesting that HDAC6 maintains MM cell growth by preventing PPP3CA degradation108.
SIRT6
SIRT6 is a chromatin-associated deacetylase which deacetylates H3K9 and H3K56, as well as non-histone protein CtIP (C-terminal binding protein (CtBP) interacting protein)110,111,112. SIRT6 confers genomic stability by promoting DNA double-strand break repair, and prevents premature cellular senescence and aging via telomere maintenance110,111,112. SIRT6 also functions as a tumor suppressor by suppressing cancer metabolism113.
Consistent with the notion that SIRT6 contributes to genomic stability, it confers resistance to DNA damage agents in MM114. SIRT6 inactivates extracellular signal-regulated kinase (ERK)/p90RSK signaling, resulting in increased DNA repairs by Chk1114. Indeed, either depletion of SIRT6 or SIRT6-specific inhibitor OSS_128167 increases sensitivity to melphalan and doxorubicin in MM cells, and MEK1/2 inhibitors or RSK2 inhibitor abrogate sensitization to DNA-damaging agents in SIRT6-depleted cells114. A potential tumor suppressor function of SIRT6 in MM has also been shown, as knockdown of SIRT6 stimulates MM cell growth by activating MAPK pathway114.
Clinical studies of histone modifier inhibitors in MM
Among the inhibitors of histone methylation modifiers, the inhibitors of DOT1L, EZH2, and KDM1A have proceeded to phase I clinical trials for cancer therapy (https://clinicaltrials.gov). Relapsed and/or refractory MM patients were included in one of these trials, which studied the safety and clinical activity of EZH2 inhibitor GSK126 (GSK2816126), although this trial was terminated because of insufficient evidence of clinical activity (NCT02082977).
HDAC inhibitors are the most extensively investigated epigenetic drugs in clinical studies (Table 1). Among the non-selective HDAC inhibitors, romidepsin, vorinostat, and panobinostat have been well studied in MM. These HDAC inhibitors have shown a remarkable anti-MM effect in preclinical studies, and yet shown quite a modest clinical activity when used as single agents115,116,117. However, promising results have been obtained from trials in combination with other agents, especially bortezomib. In a phase I/II trial of the combination of romidepsin, bortezomib, and dexamethasone for relapsed or refractory MM, overall response rate was 60%118. Efficacy of vorinostat in combination with bortezomib has also been evaluated in VANTAGE trials119,120. In the phase III VANTAGE 088 trial, patients were randomly allocated to the vorinostat group (n = 315) or the placebo group (n = 320). Median progression-free survival was statistically longer in the vorinostat arm (7.63 months in the vorinostat group and 6.83 months in the placebo group; p = 0.0100), and yet this difference is not sufficient to be considered clinically relevant120. Different treatment schedules of vorinostat and bortezomib may further improve tolerability and efficacy of this combination. The combination of panobinostat, bortezomib plus dexamethasone has been studied in PANORAMA trials121,122. In the phase III PANORAMA 1 trials, patients were randomly assigned to panobinostat, bortezomib, and dexamethasone (n = 387), or to placebo, bortezomib, and dexamethasone (n = 381). Although the overall response rate did not differ between two groups (60.7% for the panobinostat group vs 54.6% for the placebo group; p = 0.09), the proportion of a complete or near complete response was significantly higher in the panobinostat group (27.6 vs 15.7%; p = 0.00006)122. Importantly, median progression-free survival was significantly longer in the panobinostat group than in the placebo group (11.99 months vs 8.08 months; p < 0.0001)122, resulting in the Food and Drug Administration (FDA) approval of panobinostat in combination with bortezomib and dexamethasone in relapsed or refractory MM. Combinations of panobinostat or vorinostat with the second-generation proteasome inhibitor carfilzomib and/or immunomodulatory drugs (IMiDs) have also been investigated in phase I/II studies, and have shown promising results123,124,125.
In the context of the use of non-selective HDAC inhibitors, a relatively high frequency of side effects, including diarrhea and thrombocytopenia, are observed, which limits dose and time of treatment, especially in combination therapy with other agents. Based on this background, selective HDAC inhibitors are under development to reduce adverse effects, keeping anti-tumor activity. Indeed, the first selective HDAC6 inhibitor ACY-1215 (ricolinostat) has been examined as a single agent or in combination with bortezomib and dexamethasone for relapsed or refractory MM in a phase I/II study126. Although single-agent ACY-1215 therapy resulted in no clinical responses similar to non-selective HDAC inhibitors, 37% of the overall response was observed in combination therapy with daily ACY-1215 at ≥160 mg126. Combination therapy at an ACY-1215 dose of 160 mg daily was well tolerated, with less severe adverse effects compared with published data on non-selective HDAC inhibitors, suggesting that selective inhibition of HDAC6 is promising in MM treatment126. ACY-1215 has also been examined in combination with lenalidomide and dexamethasone in relapsed or refractory MM127. In this multicenter phase Ib trial, ACY-1215 with lenalidomide and dexamethasone has been shown to be safe and well tolerated, with an overall response rate of 55%127. Further clinical studies with HDAC6 inhibitor are ongoing (NCT02189343, NCT01997840, NCT02400242).
Conclusions and perspectives
Accumulating studies have revealed the biological importance of histone modifiers in MM. Histone modifiers contribute to the pathogenesis of MM by mediating modifications not only of histone but also of non-histone proteins; as well as by catalytically independent functions. In this context, epigenetic drugs targeting histone modifiers are now being developed. Of note, an HDAC inhibitor panobinostat is already available for patients with refractory/relapsed MM. However, further studies are required to more comprehensively understand the roles of histone modifiers and develop related novel therapeutics in MM. The genome-wide locations of each of histone modifiers and histone modifications have to be defined. The methylome and acetylome analyses will identify novel non-histone substrates for histone modifiers. These basic studies will enable us to understand the more precise mechanisms whereby targeted therapies induce anti-MM activities, and also provide the rationale for combination therapies. While there is a need to develop selective inhibitors of each enzyme to reduce unfavorable adverse effects, this may be difficult due to structural homology within the catalytic domains between family proteins. Nevertheless, small-molecule inhibitors specific for EZH2 and HDAC6 have already been developed and proceeded to clinical trials, highlighting the potential of epigenetic therapies to improve patient outcome in MM.
Change history
06 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41408-021-00559-4
References
Palumbo, A. & Anderson, K. Multiple myeloma. N. Engl. J. Med. 364, 1046–1060 (2011).
Manier, S. et al. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 14, 100–113 (2017).
Walker, B. A. et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 117, 553–562 (2011).
Pichiorri, F. et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl Acad. Sci. USA 105, 12885–12890 (2008).
Keats, J. J. et al. In multiple myeloma, t(4;14)(p16; q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 101, 1520–1529 (2003).
van Haaften, G. et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 41, 521–523 (2009).
Rossi, M., Tagliaferri, P. & Tassone, P. MicroRNAs in multiple myeloma and related bone disease. Ann. Transl. Med. 3, 334 (2015).
Dimopoulos, K., Gimsing, P. & Gronbaek, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 4, e207 (2014).
Amodio, N., D’Aquila, P., Passarino, G., Tassone, P. & Bellizzi, D. Epigenetic modifications in multiple myeloma: recent advances on the role of DNA and histone methylation. Expert Opin. Ther. Targets 21, 91–101 (2017).
Allfrey, V. G., Faulkner, R. & Mirsky, A. E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl Acad. Sci. USA 51, 786–794 (1964).
Dawson, M. A. & Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 (2012).
Tan, M. et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 (2011).
Greer, E. L. & Shi, Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 (2012).
Young, N. L., Dimaggio, P. A. & Garcia, B. A. The significance, development and progress of high-throughput combinatorial histone code analysis. Cell. Mol. Life Sci. 67, 3983–4000 (2010).
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
Yang, Y. & Bedford, M. T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50 (2013).
Mozzetta, C., Boyarchuk, E., Pontis, J. & Ait-Si-Ali, S. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 16, 499–513 (2015).
Huang, J. & Berger, S. L. The emerging field of dynamic lysine methylation of non-histone proteins. Curr. Opin. Genet. Dev. 18, 152–158 (2008).
Kooistra, S. M. & Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311 (2012).
Haberland, M., Montgomery, R. L. & Olson, E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 (2009).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009).
Dutta, R., Tiu, B. & Sakamoto, K. M. CBP/p300 acetyltransferase activity in hematologic malignancies. Mol. Genet. Metab. 119, 37–43 (2016).
Longo, V. D. & Kennedy, B. K. Sirtuins in aging and age-related disease. Cell 126, 257–268 (2006).
Kuo, A. J. et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44, 609–620 (2011).
Martinez-Garcia, E. et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117, 211–220 (2011).
Marango, J. et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 111, 3145–3154 (2008).
Rutherford, E. L. & Lowery, L. A. Exploring the developmental mechanisms underlying Wolf-Hirschhorn Syndrome: evidence for defects in neural crest cell migration. Dev. Biol. 420, 1–10 (2016).
Oyer, J. A. et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 28, 198–201 (2014).
Hudlebusch, H. R. et al. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin. Cancer Res. 17, 2919–2933 (2011).
Keats, J. J. et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16; q32)-positive multiple myeloma patients. Blood 105, 4060–4069 (2005).
Avet-Loiseau, H. et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood 109, 3489–3495 (2007).
Chesi, M. et al. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 92, 3025–3034 (1998).
Stec, I. et al. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum. Mol. Genet. 7, 1071–1082 (1998).
Santra, M., Zhan, F., Tian, E., Barlogie, B. & Shaughnessy, J. Jr. A subset of multiple myeloma harboring the t(4;14)(p16; q32) translocation lacks FGFR3 expression but maintains an IGH/MMSET fusion transcript. Blood 101, 2374–2376 (2003).
Brito, J. L. et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 94, 78–86 (2009).
Lauring, J. et al. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 111, 856–864 (2008).
Annunziata, C. M. et al. A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression. Blood 117, 2396–2404 (2011).
Min, D. J. et al. MMSET stimulates myeloma cell growth through microRNA-mediated modulation of c-MYC. Leukemia 27, 686–694 (2013).
Popovic, R. et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 10, e1004566 (2014).
Shaffer, A. L. et al. IRF4 addiction in multiple myeloma. Nature 454, 226–231 (2008).
Xie, Z. et al. MMSET regulates expression of IRF4 in t(4;14) myeloma and its silencing potentiates the effect of bortezomib. Leukemia 29, 2347–2354 (2015).
Hajdu, I., Ciccia, A., Lewis, S. M. & Elledge, S. J. Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc. Natl Acad. Sci. USA 108, 13130–13134 (2011).
Pei, H. et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470, 124–128 (2011).
Shah, M. Y. et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 35, 5905–5915 (2016).
Margueron, R. & Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011).
Chase, A. & Cross, N. C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 17, 2613–2618 (2011).
Zhan, F. et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 99, 1745–1757 (2002).
Pawlyn, C. et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 7, e549 (2017).
Rastgoo, N., Pourabdollah, M., Abdi, J., Reece, D. & Chang, H. Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia e-pub ahead of print 24 April 2018; https://doi.org/10.1038/s41375-018-0140-y (2018).
Zhan, F. et al. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood 101, 1128–1140 (2003).
Croonquist, P. A. & Van Ness, B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene 24, 6269–6280 (2005).
Neo, W. H., Lim, J. F., Grumont, R., Gerondakis, S. & Su, I. H. c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J. Biol. Chem. 289, 31693–31707 (2014).
Kalushkova, A. et al. Polycomb target genes are silenced in multiple myeloma. PLoS ONE 5, e11483 (2010).
Agarwal, P. et al. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget 7, 6809–6823 (2016).
Stamato, M. A. et al. Inhibition of EZH2 triggers the tumor suppressive miR-29b network in multiple myeloma. Oncotarget 8, 106527–106537 (2017).
Amodio, N. et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia e-pub ahead of print 22 February 2018; https://doi.org/10.1038/s41375-018-0067-3 (2018).
Hernando, H. et al. EZH2 inhibition blocks multiple myeloma cell growth through upregulation of epithelial tumor suppressor genes. Mol. Cancer Ther. 15, 287–298 (2016).
Zeng, D., Liu, M. & Pan, J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget 8, 3396–3411 (2017).
Alzrigat, M. et al. EZH2 inhibition in multiple myeloma downregulates myeloma associated oncogenes and upregulates microRNAs with potential tumor suppressor functions. Oncotarget 8, 10213–10224 (2017).
Rizq, O. et al. Dual Inhibition of EZH2 and EZH1 sensitizes PRC2-dependent tumors to proteasome inhibition. Clin. Cancer Res. 23, 4817–4830 (2017).
Adamik, J. et al. EZH2 or HDAC1 inhibition reverses multiple myeloma-induced epigenetic suppression of osteoblast differentiation. Mol. Cancer Res. 15, 405–417 (2017).
Kikuchi, J. et al. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Invest. 125, 4375–4390 (2015).
Gulla, A. et al. Protein arginine methyltransferase 5 has prognostic relevance and is a druggable target in multiple myeloma. Leukemia 32, 996–1002 (2018).
Yamane, K. et al. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125, 483–495 (2006).
Okada, Y., Scott, G., Ray, M. K., Mishina, Y. & Zhang, Y. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 450, 119–123 (2007).
Tateishi, K., Okada, Y., Kallin, E. M. & Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 458, 757–761 (2009).
Kuroki, S. et al. Epigenetic regulation of mouse sex determination by the histone demethylase Jmjd1a. Science 341, 1106–1109 (2013).
Krieg, A. J. et al. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell Biol. 30, 344–353 (2010).
Ramadoss, S., Guo, G. & Wang, C. Y. Lysine demethylase KDM3A regulates breast cancer cell invasion and apoptosis by targeting histone and the non-histone protein p53. Oncogene 36, 47–59 (2017).
Ohguchi, H. et al. The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nat. Commun. 7, 10258 (2016).
Winkelmann, R. et al. B cell homeostasis and plasma cell homing controlled by Kruppel-like factor 2. Proc. Natl Acad. Sci. USA 108, 710–715 (2011).
Hideshima, T., Mitsiades, C., Tonon, G., Richardson, P. G. & Anderson, K. C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 7, 585–598 (2007).
Ikeda, S., Kitadate, A., Abe, F., Takahashi, N. & Tagawa, H. Hypoxia-inducible KDM3A addiction in multiple myeloma. Blood Adv. 2, 323–334 (2018).
Lee, M. G. et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450 (2007).
Agger, K. et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734 (2007).
Arcipowski, K. M., Martinez, C. A. & Ntziachristos, P. Histone demethylases in physiology and cancer: a tale of two enzymes, JMJD3 and UTX. Curr. Opin. Genet. Dev. 36, 59–67 (2016).
Pawlyn, C. et al. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. Clin. Cancer Res. 22, 5783–5794 (2016).
Bolli, N. et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 5, 2997 (2014).
Ezponda, T. et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. 21, 628–640 (2017).
De Santa, F. et al. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094 (2007).
Agger, K. et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176 (2009).
Ohguchi, H. et al. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia 31, 2661–2669 (2017).
Allis, C. D. et al. New nomenclature for chromatin-modifying enzymes. Cell 131, 633–636 (2007).
Jin, Q. et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262 (2011).
Conery, A. R. et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife 5, pii: e10483 (2016).
Bolden, J. E., Peart, M. J. & Johnstone, R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5, 769–784 (2006).
Catley, L. et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood 102, 2615–2622 (2003).
Mitsiades, N. et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood 101, 4055–4062 (2003).
Maiso, P. et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 66, 5781–5789 (2006).
Bradner, J. E. et al. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243 (2010).
Minami, J. et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 28, 680–689 (2014).
Harada, T. et al. HDAC3 regulates DNMT1 expression in multiple myeloma: therapeutic implications. Leukemia 31, 2670–2677 (2017).
Cheng, J. et al. Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat. Commun. 6, 7023 (2015).
Faiola, F. et al. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol. Cell Biol 25, 10220–10234 (2005).
Kikuchi, J. et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 116, 406–417 (2010).
Mithraprabhu, S., Kalff, A., Chow, A., Khong, T. & Spencer, A. Dysregulated class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 9, 1511–1520 (2014).
Kikuchi, S. et al. Class IIa HDAC inhibition enhances ER stress-mediated cell death in multiple myeloma. Leukemia 29, 1918–1927 (2015).
Amodio, N. et al. Therapeutic targeting of miR-29b/HDAC4 epigenetic loop in multiple myeloma. Mol. Cancer Ther. 15, 1364–1375 (2016).
Vallabhapurapu, S. D. et al. Transcriptional repression by the HDAC4-RelB-p52 complex regulates multiple myeloma survival and growth. Nat. Commun. 6, 8428 (2015).
Martin, M., Kettmann, R. & Dequiedt, F. Class IIa histone deacetylases: regulating the regulators. Oncogene 26, 5450–5467 (2007).
Amodio, N. et al. miR-29s: a family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget 6, 12837–12861 (2015).
Kopito, R. R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530 (2000).
Kawaguchi, Y. et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 (2003).
Hideshima, T. et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl Acad. Sci. USA 102, 8567–8572 (2005).
Santo, L. et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 119, 2579–2589 (2012).
Hideshima, T. et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl Acad. Sci. USA 113, 13162–13167 (2016).
Mishima, Y. et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 169, 423–434 (2015).
Imai, Y. et al. Histone deacetylase inhibitor panobinostat induces calcineurin degradation in multiple myeloma. JCI Insight 1, e85061 (2016).
Kovacs, J. J. et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607 (2005).
Michishita, E. et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496 (2008).
Kaidi, A., Weinert, B. T., Choudhary, C. & Jackson, S. P. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329, 1348–1353 (2010).
Toiber, D. et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 51, 454–468 (2013).
Sebastian, C. et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151, 1185–1199 (2012).
Cea, M. et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 127, 1138–1150 (2016).
Wolf, J. L. et al. Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk. Lymphoma 53, 1820–1823 (2012).
Richardson, P. et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk. Lymphoma 49, 502–507 (2008).
Niesvizky, R. et al. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer 117, 336–342 (2011).
Harrison, S. J. et al. A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood 118, 6274–6283 (2011).
Siegel, D. S. et al. VANTAGE 095: an international, multicenter, open-label study of vorinostat (MK-0683) in combination with bortezomib in patients with relapsed and refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 16, 329–334 e321 (2016).
Dimopoulos, M. et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 14, 1129–1140 (2013).
Richardson, P. G. et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 122, 2331–2337 (2013).
San-Miguel, J. F. et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 15, 1195–1206 (2014).
Berdeja, J. G. et al. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 100, 670–676 (2015).
Vesole, D. H. et al. Phase I study of carfilzomib, lenalidomide, vorinostat, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Br. J. Haematol. 171, 52–59 (2015).
Siegel, D. S. et al. Vorinostat in combination with lenalidomide and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood Cancer J. 4, e182 (2014).
Vogl, D. T. et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res. 23, 3307–3315 (2017).
Yee, A. J. et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial. Lancet Oncol. 17, 1569–1578 (2016).
Acknowledgements
The authors apologize to the researchers whose original work could not be cited due to space limitation. This study was supported by the National Institute of Health Grants SPORE P50-CA100707 (to H.O. and K.C.A.), R01-CA050947 (to K.C.A.), and R01-CA178264 (to T.H. and K.C.A.). K.C.A. is an American Cancer Society Clinical Research Professor.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
H.O. and T.H. declare that they have no conflict of interest. K.C.A. serves on advisory boards for Celgene, Millennium, and Gilead Sciences, and is a scientific founder of OncoPep and C4 Therapeutics.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ohguchi, H., Hideshima, T. & Anderson, K.C. The biological significance of histone modifiers in multiple myeloma: clinical applications. Blood Cancer Journal 8, 83 (2018). https://doi.org/10.1038/s41408-018-0119-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-018-0119-y
