
Abstract
Pyroptosis, an inflammatory form of lytic cell death, is a type of cell death mediated by the gasdermin (GSDM) protein family. Upon recognizing exogenous or endogenous signals, cells undergo inflammasome assembly, GSDM cleavage, the release of proinflammatory cytokines and other cellular contents, eventually leading to inflammatory cell death. In this review, we discuss the roles of the GSDM family for anti-cancer functions and various antitumor drugs that could activate the pyroptosis pathways.
Similar content being viewed by others
Introduction
The primary role of pyroptosis is defending the host from pathogens as part of the innate immunity system [1]. Pyroptosis has been regarded as caspase-1 dependent programmed cell death in response to certain pathogens. The identification of caspase-4/5/11 for detecting cytosolic lipopolysaccharide (LPS) broadened the spectrum of pyroptosis mediators (Fig. 1) [2, 3]. The recent discovery of the GSDM family’s pore formation activities led to the redefinition of the terms used to describe pyroptosis: gasdermin-mediated lytic programmed cell death often but not always requires the cleavage by inflammatory caspases [4,5,6]. Current chemotherapeutic agents are typically apoptosis inducers, while resistance to apoptosis is a common strategy of tumor cells to develop resistance. By triggering nonapoptotic cell death, inducing pyroptosis can be an appealing strategy to overcome chemotherapeutic drug resistance. Gasdermins are also linked to genetic diseases where their mechanism of function is gradually elucidated [7, 8].

GSDMD is common substrate for both caspase-1 and −11 while pro-IL-18 and pro-IL-1β are only catalyzed by caspase-1.
Gasdermin family
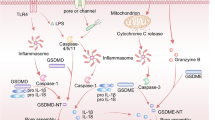
The GSDM protein family consists of six members, five of which are associated with various roles in inducing cell death via pore formation and pyroptosis. These five members are Gasdermin A (GSDMA), Gasdermin B (GSDMB), Gasdermin C (GSDMC), Gasdermin D (GSDMD), and Gasdermin E (GSDME). The other member, Pejvakin, also called DFNB59 (GSDMF), is related to autosomal recessive hearing loss and has a low sequence similarity to other GSDMs. Except for DFNB59, inactive GSDMs comprise an N-terminal effector domain and a C-terminal inhibitory domain connected by a flexible peptide linker. DFNB59 does not have a clear C-terminal inhibitory domain, suggesting that a different mechanism might regulate its activity. The C-terminal domain also binds to the N-terminal domain noncovalently, contributing to maintaining the N-terminus at its inactive structure. The activation of the N-terminus requires the cleavage of the linker, followed by N- and C- termini separation (Figs. 1, 2). The “freed” N-terminus then undergoes conformational changes for membrane insertion and oligomerization. Completing the oligomerization leads to the formation of transmembrane pores, through which cellular contents, such as cytokines and electrolytes, could leak out of the cells.

Steps 2–4 are based on the conclusions of reference (a). Step 1 is presented in both cartoon and structural views (b). PDB ID: 6N9O (before membrane insertion) and 6VFE (after membrane insertion).
Human GSDMA has no homologs, while there are three GSDMA homologs in mice (mGSDMA 1–3). Among the three homologs, mutations of mouse GSDMA3 are associated with diseases such as alopecia [9, 10], while polymorphisms in human GSDMA are associated with different autoimmune diseases like asthma [7]. In the meanwhile, GSDMA has been linked to gastric cancers when the gene transcription is silenced [11]. Upon removal of the inhibitory C-terminus, the genetically engineered N-terminus binds to inner leaflet lipids of the mammalian cell membrane, such as phosphatidylinositol (4,5)-bisphosphate [12]. Subsequently, GSDMA-N oligomerizes to form symmetrical pores. Disulfide bonds are also formed between the neighboring subunits to strengthen the pores. In cryo-EM structure, activated mouse GSDMA3-N forms pores of 18 nm using an average of 27 units [13]. The pores release cellular contents and permit the influx of extracellular molecules such as Ca2+. GSDMA3-N could also bind to lipids from bacterium and mitochondria, such as cardiolipin, which permeabilized these membranes from outside. Although engineered GSDMA can trigger pyroptosis by targeting inner leaflet lipids of mammalian cells and trigger bactericidal activity by targeting the outer membrane lipid of the bacterium, its physiological function is still not confirmed, and the protease that cleaves GSDMA remains to be discovered [14]. On the other hand, activatable GSDMA3 has been generated based on its pyroptotic abilities. A bioorthogonal system constructed using the compound phenylalanine trifluoroborate can enter cells, catalyze desilylation, and cleave the linker of a generated nanoparticle-GSDMA3 conjugate [15]. When used to treat implanted tumors in mice, the system enabled the controlled release of gasdermin proteins in specific tumor cells and resulted in substantial tumor suppression by inducing pyroptosis.
Like GSDMA, GSDMB is also associated with autoimmune diseases such as asthma and irritable bowel disease. When residues 1–275, the N-terminal domain of GSDMB were expressed in 293 T cells, ~80% of cells underwent pyroptosis, while the overexpression of the full-length (FL) or the C-terminus GSDMB did not cause cell death [12]. Consistently, Panganiban reported that the N-terminus of GSDMB proteins induced pyroptosis in the human bronchial epithelial cells, and the cleavage of GSDMB required the co-expression of caspase-1 [16]. The cleavage site of GSDMB was later determined to be at residue D236, without identifying the protease responsible for the cleavage. As such, the activation of GSDMB was proposed to be similar to that of GSDMD, where the C-terminus plays an inhibitory role while cleavage by caspase-1 dissociates the C-terminus and activates GSDMB for pyroptosis. Moreover, it was later discovered that CSDMB could be cleaved by apoptotic caspases −3/−6/−7, not by inflammatory caspase-1 [8]. In addition, the N-terminus of GSDMB binds to phosphoinositides and sulfatide of the membrane, although it was not shown if there was pore formation. Chen et al. showed that the overexpression of FL GSDMB promotes GSDMD cleavage and LPS-induced non-canonical pyroptosis, which was accompanied by increased LDH release [17]. In addition, FL GSDMB can directly bind to caspase-4 to enhance enzymatic activity. Zhou et al. discovered the more definitive role of GSDMB in cellular immunity where GSDMB of cancer cells was activated by granzyme A (GzmA) of nearby cytotoxic lymphocytes. During this process, GzmA cleaves GSDMB at Lys244 primarily and at Lys229 to a less extent, generating two products and liberating its pore-forming domain. The authors showed that cells with mutations at the two lysines were resistant to pyroptotic death. These findings collectively suggest that enforced expression of GSDMB may confer tumor clearance through the initiation of pyroptosis.
GSDMC is highly expressed in the GI tract, including the esophagus, stomach, colon, and spleen. It is also expressed in the skin and trachea. The physiological function of GSDMC is mostly unknown. This is in part because disease-related single nucleotide polymorphism or null mutation of GSDMC has not been reported. It is believed that GSDMC is related to oncogenesis and, under certain circumstances, it functions as an oncogene. The upregulation of GSDMC has been suggested to promote tumor proliferation in colorectal carcinogenesis [18]. Similarly, its upregulation is associated with the gaining metastasis of melanoma [19]. Interestingly, on the other hand, it was reported to be a tumor suppressor in some gastric cancers [11]. Similar to other GSDMs, its C-terminus plays an inhibitory role, albeit the enzyme to separate N- and C- termini has not been identified. When its N-terminus was overexpressed in a mammalian cell line 293 T cells, most cells underwent pyroptotic death [12], implying a functional potential akin to its other members in the GSDMs family.
GSDMD and GSDME are best studied for their functions when it comes to pore formation and pyroptosis. They play significant roles in defense against intracellular pathogens, conditions of inflammation, and tumors. GSDMD consists of an N-terminal domain and a C-terminal domain. The two domains are connected by a peptide linker, which could be cleaved by activated inflammatory caspases upon activation of the canonical or non-canonical pyroptotic pathway. In addition to the peptide linker, the C-terminus also interacts with the N-terminus using a large hydrophobic patch. Upon cleavage, the N-terminus dissociates from the C-terminus and becomes activated. Upon binding the inner leaflet of the cell membrane, it undergoes conformational changes to form a transmembrane β sheet and anchors itself to the membrane. It then oligomerizes and forms pores on the cell membrane to execute pyroptosis (Fig. 1). These pores will make the mammalian cells more permeable to water molecules and initiate pyroptosis. A recent pan cancer analysis by Qiu et al. showed that GSDMD-mediated pyroptosis is closely associated with the prognosis of adrenocortical carcinoma, renal clear cell carcinoma, lower grade glioma, hepatocellular carcinoma, cutaneous melanoma, and rectal adenocarcinoma [20], highlighting its role in antitumor immunity and arguing for developing future drugs for activating of GSDMD.
GSDME, also called DFNA5 (deafness, autosomal dominant 5), was originally associated with autosomal dominant nonsyndromic hearing loss [21] where GSDME was expressed as a truncated protein due to a mutation-induced skipping of exon 8 [22]. It was first identified to execute pyroptosis after cleavage by caspase-3 [23, 24], similar to the activation of GSDMD by caspase-1. Certain drugs, e.g., lobaplatin, cisplatin, and paclitaxel, induce pyroptosis by activating GSDME in cancers [25, 26]. GSDME has long been recognized to be a tumor suppressor in different cancer types, such as primary gastric cancers [27] and melanoma cells [28]. Recently, Ibrahim et al. analyzed hundreds patient samples and found that the putative GSDME promotors were hypermethylated in colorectal cancers [29], arguing for using GSDME methylation as a biomarker in colorectal cancers and activating GSDME as a regimen for tumor treatment. Until recently, GSDME has been shown to play its antitumor role directly by coordinating with immune cells and executing pyroptosis with coordination of killer lymphocytes, such as Natural Killer (NK) cells. NK cells release protease granzyme B (GzmB) and the pore-forming perforin to infiltrate target cancer cells, both of which, when coupled together, exhibit tumor-regression capabilities [5]. GzmB is secreted into the cancer cell from NK cell granules and directly cleaves GSDME after residue D270. Alternatively, GzmB indirectly induces cleavage by cutting and activating caspase-3 to cleave GSDME after D270, thereby forming pores on the cancer cell membrane. The cutting of GSDME by either GzmB or caspase 3 releases its functional pore-forming N-terminal fragment. The generated N-terminus translocates to and permeabilizes the cancer cell’s plasma membrane, triggering inflammatory cell death [5]. The mechanisms and antitumor effects of GSDME may extend to other GSDM proteins and be facilitated with a controlled release.
Canonical inflammasome pathway of pyroptosis
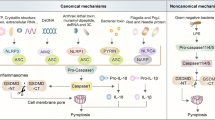
The pyroptotic pathways in which GSDMs are activated could be inflammasome-dependent or inflammasome-independent. Canonical inflammasome pathway is characterized by caspase-1 mediated cleavage of GSDMD, whereas the non-canonical inflammasome pathway is characterized by caspase-11 (in human caspase 4 and 5) mediated cleavage of GSDMD. The activation of other GSDMs requires the activity of other caspases, granzymes, and identified protease(s) without involving inflammasomes.
The inflammasome is a key player in the canonical pathway. Generally speaking, inflammasomes are composed of a sensor protein, an adapter protein (apoptosis-associated speck protein, ASC), and an enzyme (pro-caspase-1). In some cases, the sensor protein can directly recruit pro-caspase-1, such as NLRC4 inflammasome.
In the canonical pathway, the activation signals could come from various sources, including pathogen-associated molecular patterns (PAMPs) induced by invading pathogens or danger-associated molecular patterns (DAMPs) induced by endogenous cellular changes. There are multiple groups of cytosolic pattern recognition receptors (PRR) that can recognize these cytoplasmic signals, including absent in melanoma receptors (AIM2), Nod-like receptors (NLRs), nucleotide-binding domain, leucine-rich repeat-containing receptors (NLRs), and pyrin. Among these receptors, NLRs, AIM2, and pyrin can assemble into inflammasomes. Inflammasome activation leads to the activation of the canonical pathway, during which pro-caspase-1 is recruited and further cleaved into the active caspase-1. The active caspase-1 cleavages GSDMD into its N- and C- terminal fragments. The N-terminal fragments further oligomerize to form pores in the inner cell membrane. In the meantime, caspase-1 also processes inflammatory cytokines pro-IL-1β and pro-IL-18 into their corresponding active IL-1β and IL-18. Subsequently, cells undergo pyroptosis and lytic cell death.
There are several types of inflammasome sensors. NLRP3 inflammasomes are by far the most extensively studied [30,31,32]. It consists of an NLRP3 scaffold, ASC adapter, and pro-caspase-1. The NLRP3 scaffold further consists of an LRR domain, a NACHT domain, and a PYD domain. A diverse variety of PAMPs and DAMPs could activate NLRP3 [32], including bacteria such as Listeria and Staphylococcus species [33], fungi such as Candida [34] and Saccharomyces species [35], or viruses such as Sendai adenovirus [36] and influenza virus [37]. When the cells receive PAMP and DAMP signals, there will be upregulation and activation of the NLRP3 inflammasome. Upon NLRP3 activation, NLRP receptors oligomerize, leading to clustering of the pyrin domain, which recruits ASC complexes by pyrin-pyrin interaction. The caspase recruitment domain (CARD) of the ASC then recruits the CARD of pro-caspase-1. Clustering of pro-caspase-1 leads to the autocleavage and activation of caspase-1.
NLRP1 is the first identified sensor that forms an inflammasome [38]. Human NLRP1 comprises a nuclear binding domain, a pyrin domain, a leucine-rich repeat domain, a “function to find” domain (FIIND), and a C-terminal CARD domain. In mice, there are three NLRP1 homologs, NLRP1a, NLRP1b, and NLRP1c. The NLRP1b activation by lethal factor secreted by Bacillus Anthracis has been well established [38, 39]. There are two modes of activation of the NLRP1b in mice, direct activation and indirect activation. In direct activation, the N-terminal portion of the NLRP1b keeps the molecule in an inactive state. The lethal factor cleaves the N-terminus and thus activates NLRP1b. In the indirect activation, NLRP1b is inhibited by an unknown host factor X. Lethal factor cleaves host factor X and activates NLRP1b. NLRP1b is also known to be activated with Toxoplasma gondii infection in mice [40].
NLRC4 is a receptor that was identified to be capable of inducing inflammasome formation following Salmonella infection [41,42,43]. It has also been shown to be the main inflammasome activated upon Salmonella [44], Shigella [45], or Klebsiella pneumonia [46] infection. Bacterial flagellin and flagellin-associated secretion systems are the activators of NLRC4. Different from other sensors, NLRC4 senses the ligands via NLR family apoptosis inhibitory proteins (NAIPs) [42]. When an NAIP binds to its ligands such as flagellin or Type 3 secretion systems [42], NLRC4 oligomerizes to initiate downstream inflammasome assembly with ASC and pro-caspase-1 recruitment.
AIM2, a non-NRL family member, is another ligand sensor that binds to cytosolic double-strand DNA. AIM2 can recognize a wide range of dsDNA, including endogenous host, exogenous viral, or bacterial DNAs [47, 48]. Unlike the typical oligomerization process mediated by the central oligomerization domain of the inflammasomes, the oligomerization of the AIMs inflammasome is mediated by clustering of the multiple units to surround the ligand dsDNA [49]. Similar to NLRP1, The AIM2 sensor recruits ASC through pyrin-pyrin interaction, followed by CARD-CARD interaction between ASC and pro-caspase-1. Self-reactivation of pro-caspase-1 occurs further triggering the proinflammatory cytokine release.
Pyrin inflammasome has also been identified as an inflammasome sensor in recent years [50]. Its expression is mainly confined to cells involved in the innate immune system [51]. In humans, pyrin is composed of an N-terminal pyrin domain, a center B box domain, and a C-terminal B30.2 domain. Pyrin inflammasome is activated by covalent modification of some cytoskeletal proteins. TcdB, a virulence factor of Clostridium Difficile, which is shown to glycosylate and downregulate RhoA, can activate pyrin inflammasome [52]. Similar to the NLRP3 inflammasome assembly, via pyrin-pyrin interaction, ASC is recruited to the pyrin domain of pyrin receptor, thus, in turn, recruiting pro-caspase-1 via CARD-CARD interaction.
Non-canonical inflammasome pathway of pyroptosis
Non-canonical inflammasome pyroptosis, a pathway mediated by caspase-4/5/11, is central to a cell’s immunological response to gram-negative bacteria. The catalytic domain of caspase-1 and caspase-11 (also to −4/−5) are similar in primary sequence and structure (Fig. 3); however, their activation mechanism is different. The process involves lipopolysaccharides (LPS), an endotoxin found on the outer membrane of gram-negative bacteria, to be directly bound to these caspases, which then cleave GSDMD (Fig. 1). Extracellular LPS can center a host cell as part of the membrane of a gram-negative bacteria or be carried by outer membrane vesicles (OMVs). Intracellular LPS is sensed by caspase-4/5/11 directly or relayed by GBP1 to form non-canonical inflammasomes [53].

In the structural alignment, dimeric caspase-1 was shown while one of the symmetric monomer of caspase-11 was not shown. PDB ID: 1SC1 (capase-1) and 6KMV (caspase-11).
GBP1 constructs a GBP coat on cytosolic bacteria. When paired together, GBP1 and GBP4 instigate the recruitment of caspase-4 to the bacterial surface. GBP3 functions to initialize caspase activation. GBP1 uses positive charges to attract the negative charges of LPS for binding. The lipid A moiety of LPS binds to the CARD and oligomerizes caspase-4 (caspase-11 and caspase-5 were not reported and are likely to have the same mechanism). Caspase-4 then cleaves the GSDMD linker, liberating the GSDMD-N and GSDMD-C fragments and inducing pyroptosis [53].
It is important to note that in the non-canonical pathway, caspase-4/5/11 activation precedes caspase-1 activation, and both require the expression of interferon (IFN)-stimulated human macrophages infected with Salmonella Typhimurium. Such an overlap indicates crosstalk between the canonical and non-canonical pathways. This is further supported in that caspase-1 and caspase-4 undergo dual activation in the same cell. This crosstalk occurs at the inflammasome NLRP3, which mediates caspase-1 activation. Together, the two pathways trigger pyroptosis through the formation of GSDMD-N [54].
The implications of non-canonical pyroptosis can extend beyond defense against intracellular pathogens and into many inflammation-related conditions, including sepsis. Overactivated pyroptosis causes GSDMD-N to release an excess of cytokines, leading to sepsis or eventually septic shock. Correlation between the inhibition of pyroptosis and reduction in sepsis-related deaths suggests that research in drugs inhibiting the pyroptosis pathway could potentially lead to breakthroughs in sepsis treatment.
Inflammasome-independent pathways
In contrast to GSDMD activation, other GSDMs seem only to require the actions of respective proteases without the assembly of inflammasome complexes. Their primary functions are not defending pathogens. Instead, they seem to play important roles in tumor suppressive immunity. GSDMB in tumor cells could be activated by GzmA released from nearly CTLs or NK cells, resulting in tumor cell death through pyroptosis [4]. GSDME, upon chemotherapy, can often be activated by caspase-3 cleavage and switch cell death from apoptosis to pyroptosis [55]. Hou et al. reported that nuclear PD-L1 promotes the transcription of the GSDMC gene in hypoxia, where GSDMC is preferentially cleaved by caspase-8 in response to TNFalpha treatment, resulting in the generation of GSDMC-N, pores in the cell membrane and pyroptosis in cancer cells [6].
Cellular response to pore formation by GSDMD
The pore-forming protein GSDMD demonstrates an ability to mediate pyroptosis in cells by damaging the plasma membrane and disrupting electrolyte homeostasis, thereby inducing cell death. GSDMD is cleaved via one of two ways—with canonical inflammasomes mediated via the activation of caspase-1 or with non-canonical inflammasomes via caspase-4/5 in humans and caspase-11 in mice. The GSDMD cleavage generates a C-terminal and an N-terminal, which targets and translocates to the cell membrane. The N-terminal binds to phosphatidylinositides on the membrane and proceeds to construct arc-, slit-, and ring-shaped oligomers. These oligomers permeabilize the membrane to induce lysis and begin the process of pyroptotic cell death [56].
However, cell death is not always the end result, as the cell may attempt to survive by activating lysosomes, vesicles, and the cytoskeleton. The disruption to the cell membrane causes an imbalance in electrolyte concentration between the cytosol and the extracellular fluids. In a response known as the “cellular wound-healing response,” the cell triggers a rapid release of potassium ions and an influx of calcium and sodium ions to maintain homeostasis and repair the pores [1]. The influx of calcium ions signals for the cytoskeleton and vesicles to repair the GSDMD-induced damage. The cytoskeleton transports active vesicles towards the wound site via a network of microtubules using kinesin and myosin [57]. These vesicles proceed to fuse with one other, assembling a barrier around the lesion for repair [58]. Additionally, calcium regulates the exocytosis of lysosomes, specifically the lysosomal synaptotagmin isoform Syt VII. Syt VII prompts cells to reseal the wounded plasma membrane [59]. Cooperatively, these processes are potentially able to patch the damaged area and prevent pyroptosis.
These cellular mechanisms work together to reseal the GSDMD-induced injury in the plasma membrane. If these efforts are unsuccessful or the GSDMD pores overwhelm the cell, pyroptotic cell death commences.
Itis worth noting that rupturing the plasma membrane during various types of cell death was previously thought to be a passive process. An unregulated influx of water leads to the rupture of the plasma membrane. Plasma membrane rupture was recently reported to be an active process mediated by transmembrane ninjurin-1, which is triggered by cell death stimuli and oligomerizes to rupture the plasma membrane and releases inflammatory cellular contents [60]. Future studies are needed to detail its functional association with GSDMD.
Potential anti-cancer molecules that trigger pyroptosis
Now it has been widely accepted that pyroptosis pathways are involved in tumor suppression. In general, activating the pyroptosis pathways enhances tumor-suppressive immunity, whereas suppression of the pyroptosis pathways promotes tumor growth and metastasis. Some cancer cells have developed strategies to evade tumor suppression via pyroptosis pathways, which are usually achieved by epigenetic suppressions or genetic mutations. Here we review the current literature on the molecules that induce or enhance pyroptosis (Fig. 4 and Table 1). Interestingly, some of these molecules have been used for years to treat cancers, and their mechanisms have been solely attributed to inducing apoptosis, such as cisplatin.

The names of the pyroptosis-inducing drugs/compounds are in blue. Double arrow means multiple steps are involved, although these steps for TMPs and α-NTEA are not elucidated.
Compounds that activate pyroptosis
CARD-8, NLRP1/Caspase-1/GSDMD
DPP 8 and 9 (DPP8/9) are two serine proteases in host cells. Johnson et al. showed that DPP8/9 inhibitors induce pyroptosis in myeloid cells, including most of the available AML cell lines and AML samples from patients [61]. Importantly, cells of other lineages did not undergo pyroptosis. NLRP1 and the CARD-containing protein CARD8 are two related pattern recognition receptors (PRRs) that form canonical inflammasomes. Small molecule inhibitors of DPP8/9 activate both NLRP1 and CARD8 by protease-mediated destruction of their N terminal, thus releasing the active C terminus from autoinhibition. In mouse myeloid cells, the inhibitors inhibit AML progression by activating NLRP1b inflammasome, which recruits and activates procaspase-1 for pyroptosis; in human myeloid cells, the inhibitors induce the activation CARD8, which drives pyroptosis by recruiting procaspase-1 independently of NLRP1 [61, 62]. Consistently, DPP9 has been reported to inhibit CARD8-mediated pyroptosis in human T cells [63]. Interestingly, human DPP9 was recently reported to inhibit the NLRP1 inflammasome by binding to the NLRP1 receptor and regulating the inflammatory response in cells [64]. It is unclear if CARD8 and NLRP1 are differentially used among cell types, but DPP9 is the upstream inhibitor and could be a good target for myeloid or lymphoid origin cancers.
NLRP3/Caspase-1/GSDMD
FL118, a camptothecin analog that is well tolerated when taken orally, has anti-cancer activity against several chemoresistant cancers [65]. It has a better resistance profile when compared to other first-line drugs such as irinotecan or topotecan. Tang et al. had shown that colorectal cancer cells, SW480 and HT29, treated with FL118, display both an increase in GSDMD expression and LDH release rate, indicating FL118’s ability to induce pyroptosis in colorectal cancer cells. FL118 facilitates caspase-1 dependent pyroptosis by promoting IL-18 and IL-1β upregulation and the upregulation of upstream markers NLRP3 and ASC [66]. NLRP3 inflammasome activation initiates the release of downstream inflammatory factors, with the pyroptosis end products being detected by Western blot, ELISA, and immunofluorescence staining. In both mouse models and a lung metastasis model, FL118 effectively inhibits cancer cell proliferation, migration, and invasion. FL118 inhibits many proteins, including survivin, Mcl-1, XIAP, cIAP2, MdmX, ERCC1/6, while playing its antitumor activity [65]. Presumably, the inhibition of these proteins leads to the activation of NLRP3 by activating a factor upstream of NLRP3. However, the factor, which is the linkage of these proteins and NLRP3, remains elusive.
Anthocyanin is a pigment in fruits and vegetables. It has been known for its anti-inflammatory, bacteriostatic, anti-aging, and antitumor activities [67, 68]. It is a class of flavonoids that dissolves easily in water. Bai et al. reported that anthocyanin promotes pyroptosis cell death in oral squamous cell carcinoma (OSCC) cells [69]. The authors found increased levels of NLRP3, caspase-1, and IL-1β as studied using qRT-PCR, immunofluorescence, and Western blot. When caspase-1 inhibitors were used to pretreat the cells, the antitumor effects were antagonized, suggesting that pyroptosis through NRRP3 inflammasome was causing cell death. Anthocyanin-induced pyroptosis in OSCC was confirmed by the increased GSDMD level using Western blot and membrane localization using confocal microscopy. On the other hand, it has been widely reported that anthocyanin induces apoptosis, and as such, the two mechanisms may complement each other in anti-cancer therapies.
Caspase-1/GSDMD
Docosahexaenoic acid (DHA), the most abundant omega-3 fatty acid in CNS, modulates the inflammatory response and suppresses tumor cell proliferation [70]. Pizato et al. found that tumor cells stimulated by DHA exhibited significant pyroptotic characteristics and led to decreased cell viability [71]. Human breast cancer cells, MDA-MB-231, when treated with DHA, underwent pore formation in the plasma membrane [71]. DHA achieves this effect by inflammasome activation, particularly lysosomal damage, reactive oxygen species (ROS) formation, ASC, and caspase-1 activation. By activating caspase-1, DHA triggers GSDMD cleavage and N-terminal release. DHA also promotes high mobility group box 1 (HMGB1) translocation from the cell nucleus to the cytoplasm and causes IL-1β secretion [71]. These mechanisms together enhance pore formation, membrane damage, and eventually pyroptosis of breast cancer cells. Since DHA has been suggested to induce apoptosis in the same cell line [70], DHA seems to have dual effects on cancer cells. Unlike many other drugs that induce apoptosis and pyroptosis using caspase-3/GSDME dependent mechanism in pyroptosis, DHA induces apoptosis using caspase-8 pathway [72] while inducing pyroptosis using inflammasome/caspase-1/GSDMD canonical pathway. Hence, DHA represents a case of inducing programmed cell death through both canonical pyroptosis and apoptosis pathways.
Compounds that activate the non-canonical pathway
Caspase-4/GSDMD
α-NETA, originally designed to treat multiple sclerosis (MS) as a choline acetylcholine transferase inhibitor, also inhibited the binding between β-arrestin-2 to chemerin chemokine-like receptor 1 (CMKLR1) [73]. The administration of α-NETA improved the symptoms of experimental autoimmune encephalomyelitis (EAE) [73], an accepted mice model for MS. It has been known that the knockout of GSDMD alleviated EAE [74]. Qiao et al. reported that α-NETA induced pyroptosis in epithelial ovarian cancer (EOC) cells using caspase-4/GSDMD pathway [75]. Compared to untreated cells, the expression of both caspase-4 and GSDMD expression increased significantly in cells treated with α-NETA. At the same time, knockout of GSDMD or caspases-4 strongly reduced the cell killing effects of α-NETA. There were inhibitory effects of α-NETA on EOC in vivo [75], suggesting that α-NETA may serve as a potential drug candidate for ovarian cancers. Still, there are multiple questions to be answered to elucidate the role of α-NETA in pyroptosis further. First, Caspase-4 and GSDMD are often present in immune cells, and their functions in EOC cells are to be determined. In addition, it will be important to know if α-NETA is selective toward cancer cells expressing high levels of caspase-4 and GSDMD and the different expression levels of caspase-4 and GSDMD in various EOC subtypes.
Caspase-3/GSDME
Cisplatin and paclitaxel are two traditional agents used in lung cancer treatment that induce apoptosis via different mechanisms. Cisplatin and paclitaxel have been shown to activate caspase-3 and −7 to trigger apoptosis, where the ratio of Bax and Bcl-2 was increased in cancer cells [76]. Ouyang et al. showed that on top of inducing apoptosis, there are dying cells displaying characteristics of pyroptosis [25] with blebbing on the cell membrane and increasing levels of caspase-3 and GSDME-NT. It is suggested that cisplatin and paclitaxel induce pyroptosis through the GSDME pathway, whereby activated caspase-3 cleaves GSDME to generate active N terminal fragments for pore formation. Paradoxically, caspase-3 specific inhibitor (Ac-DEVD-CHO) suppressed the production of cisplatin- but not paclitaxel-induced GSDME cleavage, indicating that paclitaxel-induced GSDME activation might be independent of caspase-3. In addition, GSDME knockdown significantly inhibited pyroptosis induced by cisplatin but not by paclitaxel [25]. Cisplatin and paclitaxel have been used for years to treat different cancers. It will be important to understand if the therapeutic effects are from their differential activation of apoptosis and pyroptosis.
Recently, Wu et al. showed that the combination of BI2536, a PLK1 inhibitor, and cisplatin, an alkylating agent commonly used in cancer treatment, could trigger a strong pyroptosis response in esophageal squamous cell carcinoma (ESCC) tissue at a low dose. PLK 1 gene and protein are involved in DNA repair response. In addition, the authors showed that the pyroptosis in ESCC was caspase-3 and GSDME dependent [77]. Consistently, GSDME is significantly overexpressed in esophageal cancer tissues than adjacent normal tissues. Cisplatin, a DNA alkylating agent interfering cell cycle, and BI2536, an inhibitor of mitosis, could both impair DNA repairs. It was shown that BI2536 sensitizes ESCC cells to cisplatin treatment. ESCC cells treated with BI2536 and cisplatin showed typical pyroptosis cell morphology such as cell membrane “bubbling”; however, cells treated with BI2536 or cisplatin alone failed to show the morphology of pyroptosis. Also, ESCC cells treated with BI2536 and cisplatin showed increased GSDME accumulation in the cytoplasm adjacent to the cell membrane. These are promising data that could lead to a change in esophageal cancer treatment and the treatment of other types of cancers.
As a carcinogen, Arsenic Trioxide (As2O3) paradoxically has antitumor activity and has been used for centuries to treat chronic myeloid leukemia (CML) and other malignancies [78]. Currently, As2O3 has been approved by FDA for the treatment of acute promyelocytic leukemia (APL) [79], in which apoptosis of the tumor cells is induced [80]. As2O3 and other arsenical compounds have been reported to inhibit NLRP1, NLRP3, and NLRC4 inflammasomes in BMDMs [81], suggesting As2O3 interferes with canonical pathway pyroptosis. In hepatocellular carcinoma (HCC) cell lines Huh7 and Bel-7402, As2O3 and As2O3 nanoparticles (As2O3-NPs) consistently induce pyroptosis [82]. Notably, As2O3 treatment triggers GSDME cleavage and downstream pyroptosis in Huh7 cells, triggering GSDMD cleavage in Bel-7402 cells. As2O3-NP triggers considerably higher levels of caspase-3 cleavage in HCC cells. In a Huh7 xenograft mouse model, both free As2O3 and As2O3-NPs showed the ability to inhibit tumor growth, with As2O3-NPs showing more significant tumor inhibition effects. The difference in effects could be due to the local injection of nano delivery system utilized by As2O3-NPs, which increased local drug concentration [82]. DNA methyltransferase proteins are associated with hepatocarcinogenesis. These proteins are significantly less in As2O3 treated groups, indicating that As2O3-induced pyroptosis in cancer cells could be achieved by downregulation of DNA methyltransferase expression. Since As2O3-NPs triggered GSDMD-mediated pyroptosis in Bel-7402 cell lines and As2O3 inhibits inflammasomes in BMDMs, it is elusive what is responsible for the different cellular effects of As2O3-NPs and As2O3 in HCC cells and BMDMs, respectively.
Curcumin, a natural compound, has anti-cancer activity but with low bioavailability in vivo [83]. L61H10, a thiopyran derivative of curcumin, shows good in vitro and in vivo antitumor activity for lung cancers. In addition, Bcl-2 was downregulated while Bax was upregulated, suggesting the induction of apoptosis. Chen et al. has shown that L61H10 was also able to induce pyroptosis using caspase-3/GSDME pathway [84]. The author further showed that L61H10 could induce a switch from apoptosis to pyroptosis by regulating NF-κB, promoting the expression of anti-apoptotic genes. It was shown that L61H10 effectively inhibits tumor cells both in vivo and in vitro with no observed toxicity, making it a promising drug for lung cancer. In light of its promising in vitro and in vivo effects, it requires further study to elucidate the contributions of apoptosis and pyroptosis during L61H10 treatment.
Iron can trigger oxidative stress by elevating ROS, which induces different forms of programmed cell death, such as apoptosis, necroptosis, and ferroptosis [85]. Zhou et al. reported iron induced pyroptosis through caspase-3 and GSDME pathway in melanoma cells. The authors showed that iron, in synergy with carbonyl cyanide m-chlorophenyl hydrazine (CCCP), enhanced ROS signaling that is sensed by Tom20 [86]. During the process, CCCP, an uncoupler of mitochondrial oxidative phosphorylation, induced ROS generation and activated ROS-associated pathways, which were enhanced by the addition of iron. As a sensor for iron-activated ROS signaling, Tom20 becomes oxidized upon encountering ROS and oligomerizes on the outer membrane of the mitochondria. The oxidized and oligomerized Tom20 recruits Bax, a pore-forming apoptotic protein, to mitochondria. Bax-mediated pores permeabilize the mitochondria’s outer membrane to release cytochrome c into the cytosol, which indirectly activates caspase-3. Interestingly, the activated caspase-3 barely induces apoptosis in the iron/CCCP treated melanoma cells. Instead, it cleaves GSDME to start pyroptosis. A few drugs are known to moderately increase ROS and are used to treat myeloma cells A375 [86]. In the absence of iron, none of these drugs triggers pyroptosis. The role of iron and ROS-induced pyroptosis was further verified using melanoma-bearing mice, where tumor burden could be reduced by co-administration of an ROS-inducing drug and iron. Melanoma is the most aggressive skin cancer and is very resistant to apoptosis by chemotherapy and radiotherapy [87]. The research provided an exciting alternative mechanism to treat melanoma, where co-administration of ROS-inducing agents will induce pyroptotic cell death. In this process, activating Tom20 signaling is a key step for triggering pyroptosis, but it is unclear if Tom20 is the coordinator of apoptosis and pyroptosis.
Eukaryotic elongation factor-2 kinase (eEF2K) phosphorylates and inhibits eEF2 at Thr56 under stress conditions, which negatively regulates the elongation stage of protein synthesis. It plays an important role in modulating the crosstalks between autophagy and apoptosis in cancer cells under various stress conditions, such as insufficient nutrition [88]. Under cellular stress, eEF2K promotes cancer cell survival by mediating autophagy while inhibiting apoptosis. Interestingly, crosstalk between autophagy and pyroptosis recently reported autophagy inhibited pyroptotic death of Legionella-infected macrophages [89]. Recently, Yu et al. showed that doxorubicin treatment triggered pyroptosis in melanoma cell lines, SK-MEL-5, SK-MEL-28, and A-375, where GSDME is highly expressed. In the meantime, autophagy was also activated by doxorubicin, and the inhibition of autophagy further augmented pyroptosis induced by doxorubicin [90]. Consistent with this, MCF-7 cells, which express a low level of GSDME, did not undergo pyroptosis upon doxorubicin treatment. The authors further discovered that doxorubicin activated eEF2K in the melanoma cells, which promoted autophagy and negatively regulated pyroptosis [90]. As such, eEF2K bridges the crosstalk between autophagy and pyroptosis in myeloma cells and modulates the chemotherapeutic effects of doxorubicin. Since eEF2K regulates translations under cell stress, such as doxorubicin treatment, it will be important to elucidate the interplay among the key mediators of autophagy and pyroptosis.
Pyridoxine, vitamin B6, plays multiple roles in cellular metabolism [91]. Yang et al. discovered that THP-1 and U937 cells, 2 AML cell lines, treated with pyridoxine trigger programmed cell death. Surprisingly, it triggered apoptosis in the U937 cell line and pyroptosis in the THP-1 cell line [92]. While caspase-3 is activated in both cells, THP-1 cells undergo GSDME-dependent pyroptosis. In addition, inhibition of caspase-3 could switch apoptosis/pyroptosis to MLKL-dependent necroptosis, accompanied by a strong inflammatory response. When tested in vivo, pyridoxine significantly reduced tumor burden and degree of tumor invasion. Pyridoxine is an orally available vitamin with low toxicity, making the research exciting for AML treatment. Paradoxically, pyridoxine level has been suggested to be a tumor marker in a few cancer types [91], suggesting it might play different roles in different cell types. THP-1 cell line is a typical cell line for pyroptosis induction. It is unclear if the induction of pyroptosis by pyridoxine can be extended to other AML cell lines.
Lobaplatin, an isomeric mixture of platinum complexes, attacks tumors using a mechanism similar to cisplatin [93]. Yu et al. showed that lobaplatin exhibits pyroptotic effects in colon cancer cells [26]. HT-29 and HCT116 cells treated with lobaplatin developed swelling and pore formation in the plasma membrane, resulting in cell death. This effect is initiated by caspase-3 activation, which leads to the release of the N-terminal fragment of GSDME and induction of pyroptosis. Lobaplatin-treated HT-29 and HCT116 cells exhibit an elevation in ROS levels. ROS signals to protein kinase JNK, which responds with an increase in phosphorylation in HT-29 and HCT116 cells, suggesting that JNK functions as an upstream regulator of GSDME-dependent pyroptosis. JNK then induces transportation of Bax to the mitochondria, triggering caspase-3 activation and later pyroptosis of colon cancer cells. Interestingly, knocking out GSDME did not affect lobaplatin’s inhibitory effects for the growths of HT-29 and HCT116 cells, while lobaplatin-mediated cell death switched from pyroptosis to apoptosis [26]. One apparent difference between apoptosis and pyroptosis that may affect the drug effects is the release of inflammatory cytokines. Future studies investigating the role of these cytokines in tumor treatment will help elucidate the benefit of pyroptosis over apoptosis. It will be essential to elucidate the detailed mechanistic studies of various cytokines in individual cell death pathways.
Miltirone, an active compound from herbal medicine Salvia miltiorrhiza Bunge, has been shown to have antileukemic activity by triggering ROS production [94]. Zhang et al. demonstrated the ability of militrone to reduce the viability of hepatocellular carcinoma cells and induce pyroptosis in HCC cells [95]. Miltirone dose- and time-dependently inhibits HCC cell growth and viability by inducing typical pyroptotic morphological changes, including swelling and bubble formation on the cell surface, eventually leading to cell rupture. GSDME, whose activation depends on caspase-3 cleavage, is a central mediator during militrone pyroptosis. After the treatment of militrone, ROS was induced to activate Bax, which induces the activation of caspase-3. Previously, militrone was suggested to use ROS to inhibit MEK/ERK pathway to induce apoptosis for its antitumor activity [94]. The authors showed that the inhibition of the MEK/ERK pathway could also induce pyroptosis. In the meantime, militrone could play a ROS-independent role in cells by inhibiting the PI3K-Akt pathway [96]. It will be interesting to know which pathway plays a major role in miltirone’s activity.
Drug induced activation of pyroptosis could also be seen in normal tissues, such as in epithelial cells, which raises the concern for the side effects during tumor treatment. Cisplatin, for example, could also trigger pyroptosis in various normal cells [55].
MiR-497/PELPI/GSDMD
Metformin is a class drug in the treatment of type 2 diabetes with few side effects. Proline, glutamic acid, and leucine-rich protein 1 (PELP1) is a protein of multiple subunits, playing an important role in cancer development and metastasis. Wang et al. searched the database and found an increase in both the mRNA and protein levels in different cell lines of ESCC. PELP1 was further correlated with recurrence and poor diagnosis of ESCC. When metformin was tested for its antitumor effects, it reduced the tumor size significantly in mice, which was associated with the decrease in PELP1 level by upregulation of MiR-497 [97]. Large solid tumors acquired chemotherapeutic resistance by evading apoptosis. Wang et al. searched for the antitumor effects by investigating the mRNA level of death effectors, and GSDMD was found to be significantly upregulated. Importantly, GSDMD-N was significantly increased, suggesting pyroptosis was responsible for cell death. As such, metformin seemed to induce pyroptosis using a MiR-497/PELP1/GSDMD pathway [97]. However, it was unclear which proteases cleaved GSDMD during the process. Thus it was not certain which pathway led to GSDMD cleavage/activation. In addition, there are many molecules involved in the effects of metformin, and further elucidation of the roles of these molecules is critical to establishing metformin’s therapeutic potential. Considering that metformin is a widely used drug and its safety has been well evaluated, the research has great potential for subsequent drug development.
Pyroptosis inhibitors
Inflammasome-driven infectious illnesses demonstrate the potential to be treated and controlled by pyroptosis inhibitors through mechanisms that prevent caspase activation. HIV infection-induced cell death is mediated by caspase-1 through IL-1β secretion and CD4 T-cell depletion. Therapy that blocks caspase-1 cleavage thereby would inhibit HIV-induced CD4 T-cell death and further blocks the development of chronic inflammation [98]. Similarly, sepsis is triggered by uncontrolled pyroptosis in response to infection. Targeting GSDMD or caspase-11 on the non-canonical pathway and reducing cytokine levels alleviates system-wide inflammation response and decreases the risk of septic shock and death [99]. Diabetes and associated severe complications are likewise propagated by greater levels of IL-1β secretion, NLRP3 activation, and caspase-1 interaction [100]. Such inflammatory conditions propelled by gasdermin-mediated pyroptosis may be alleviated using pyroptosis inhibitors, including disulfiram or dimethyl fumarate (DMF), which orchestrate blockage of the inflammasome pathway.
Disulfiram dose-dependently inhibits canonical human and non-canonical mouse inflammasome pyroptosis pathways to prevent cell death. Although it has previously been used in treating chronic alcoholism, disulfiram has since been identified as a potential treatment for cancer and other inflammatory illnesses exacerbated by pyroptosis. Disulfiram prevents liposome leakage in cleaved GSDMD but notably did not have this effect in cleaved GSDMA3, suggesting GSDMD to be the primary operation mechanism. Additionally, disulfiram works to block further pore formation on the GSDMD membrane surface but does not act to repair pre-existing pores. This is achieved by targeting Cys191 in human GSDMD to undergo covalent modification, thereby inhibiting caspase activity. Disulfiram obstructs IL-1β secretion primarily through pore formation prevention but not GSDMD cleavage, proteolytic processes, or prior upstream events. This is further substantiated by disulfiram’s ability to suppress the effects of LPS-induced sepsis in mice. Mice treated with disulfiram after LPS administration and the inflammatory cascade initiation displayed significant delays in death. Cu(II) may improve this effect, as shown in combination treatment of disulfiram and Cu(II), which exhibited a moderate increase in survival rates. Disulfiram’s mechanisms pose a contrast with other Cys-reactive GSDMD inhibitors. Cys-reactive necroptotic inhibitor NSA blocks GSDMD-mediated pyroptosis, while Bay 11–7082 inhibits the NLRP3 inflammasome. This is facilitated via liposome leakage control, though to a lesser degree than disulfiram. Moreover, unlike disulfiram, these two inhibitors primarily target caspase activity. These findings collectively indicate disulfiram to be the most effective and only direct inhibitor of GSDMD-mediated pyroptosis by directly inhibiting pore formation [101].
Humphries et al. showed that DMF, independent of NRD2 or GAPDH, can reduce GSDMD-N formation, impair pore formation, and ultimately block cell death. DMF targets and inhibits LPS-Nig-induced LDH and IL-1β secretion through GSDMD. This effect is displayed in mice that, administration of a treatment dose of DMF, could counteract a lethal dose of LPS and resist LPS shock, thereby increasing survival rates. DMF then reacts with GSDMD to generate 2-(succinyl)-cysteine at Cys191 in human GSDMD and Cys192 in mice GSDMD. Cys52, Cys77, and Cys192 undergo a combination of 2-monomethyl and 2-dimethyl GSDMD succination by exogenous or endogenous fumarate. This reduces GSDMD interacting with and binding to caspase-1. Through these mechanisms, DMF is able to limit GSDMD and GSDME pyroptotic cell death considerably. DMF reduced T helper, CD4+, and CD8+ T cell levels and myeloid cell infiltration in muroid tests. This effect reveals the potential of DMF and similar analogs in treating inflammatory conditions including multiple sclerosis, familial Mediterranean fever, experimental autoimmune encephalitis, or demyelination [102].
Some natural products have been shown to reduce inflammation by inhibiting NLPR3 inflammasome [103,104,105]. Interestingly, scutellarin, a flavone that can induce apoptosis, regulates protein kinase A (PKA) signaling to suppress caspase-11 activation and pyroptosis in macrophages [106]. Scutellarin treated mouse macrophages displayed increased phosphorylation of caspase 11, which reduces its proteolytic activity toward GSDMD. These phosphorylation sites were attributed to the action of PKA, as either PKA inhibitor, H89, or adenylyl cyclase (AC) inhibitor, MDL12330A, could promote pyroptosis.
PANoptosis
Panoptosis, an integration of pyroptosis, apoptosis, and necroptosis, has recently emerged as a critical mechanism to fight pathogens and cancer development [107,108,109]. PANoptosis is a process of inflammatory programmed cell death that includes essential characteristics of pyroptosis, apoptosis, and/or necroptosis [110]. The process, which cannot be explained by one of the three programmed cell death pathways, is mediated by the PANoptosome complex [110] (Fig. 5).

The ZBP1 PANoptosome is simultaneously triggering all three pathways.
Although pyroptosis, apoptosis, and necroptosis were previously considered independent processes, recent data suggests that significant interactions and regulations exist among the three pathways during some cellular events related to inflammation [111, 112]. A well studied example is the discovery of Z-DNA Binding Protein 1 (ZBP1) [113], a central regulator for the three pathways after detecting influenza A virus infection in microphages. During the infection, ZBP1 activates caspase 1/3/8 and (Receptor Interacting Serine/Threonine Kinase 3) RIPK3 [113, 114], the hallmark proteins in pyroptosis, apoptosis, and necroptosis. ZBP1 deficiency effectively prevents against cells death caused by influenza A virus infection, while deletion of essential proteins of a single pathway does not rescue the cell death [113, 114]. In light of its role in cell death, PANoptosis attracts significant attention for tumor treatment. It has been established the synergy of coadministration of TNFalpha and IFNgamma could trigger PANoptosis to inhibit tumor growth [107]. In addition, many of the drugs in Table 1 that could activate more than one programmed cell death may promote PANoptosis.
Conclusions
Pyroptosis, which is a form of programmed cell death, has been extensively studied in recent years. Compounds that activate the pyroptosis pathway have been shown to be effective in cancer treatment. In addition to the executor role for direct cell killing, gasdermins can indirectly play a tumor suppressor role and mediate the release of tumor antigens for immunogenic cell death (ICD) of tumor cells. Meanwhile, inhibitors of the pyroptosis pathways can suppress the inflammatory response during sepsis and other immune-related diseases. More studies are needed in several aspects of cancer treatment in light of the promising anti-cancer role of pyroptosis. e.g., it is crucial to elucidate more detailed mechanisms for the activation of gasdermins in tumor cells, which will direct the development of agonists or activators; it will be of diagnostic advantage to develop a non-invasive method, such as using ultrasensitive IR detection, to evaluate pyroptosis before/during/after treatment; it will significantly reduce the side effects to suppress tumors without inducing pyroptosis.
References
Lieberman J, Wu H, Kagan JC. Gasdermin D activity in inflammation and host defense. Sci Immunol. 2019;4:eaav1447.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368:eaaz7548.
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–20.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–75.
Yu J, Kang MJ, Kim BJ, Kwon JW, Song YH, Choi WA, et al. Polymorphisms in GSDMA and GSDMB are associated with asthma susceptibility, atopy and BHR. Pediatr Pulmonol. 2011;46:701–8.
Chao KL, Kulakova L, Herzberg O. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci USA. 2017;114:E1128–E37.
Tanaka S, Mizushina Y, Kato Y, Tamura M, Shiroishi T. Functional conservation of Gsdma cluster genes specifically duplicated in the mouse genome. G3 (Bethesda). 2013;3:1843–50.
Ruge F, Glavini A, Gallimore AM, Richards HE, Thomas CP, O’Donnell VB, et al. Delineating immune-mediated mechanisms underlying hair follicle destruction in the mouse mutant defolliculated. J Invest Dermatol. 2011;131:572–9.
Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer. 2009;48:261–71.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6.
Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature. 2018;557:62–7.
Zou J, Zheng Y, Huang Y, Tang D, Kang R, Chen R. The versatile gasdermin family: their function and roles in diseases. Front Immunol. 2021;12:751533.
Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579:421–6.
Panganiban RA, Sun M, Dahlin A, Park HR, Kan M, Himes BE, et al. A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. J Allergy Clin Immunol. 2018;142:1469–78.
Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, et al. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J Mol Cell Biol. 2019;11:496–508.
Miguchi M, Hinoi T, Shimomura M, Adachi T, Saito Y, Niitsu H, et al. Gasdermin C is upregulated by inactivation of transforming growth factor beta receptor type II in the presence of mutated Apc, promoting colorectal cancer proliferation. PLoS One. 2016;11:e0166422.
Watabe K, Ito A, Asada H, Endo Y, Kobayashi T, Nakamoto K, et al. Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Jpn J Cancer Res. 2001;92:140–51.
Qiu S, Hu Y, Dong S. Pan-cancer analysis reveals the expression, genetic alteration and prognosis of pyroptosis key gene GSDMD. Int Immunopharmacol. 2021;101:108270.
Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20:194–7.
Cheng J, Han DY, Dai P, Sun HJ, Tao R, Sun Q, et al. A novel DFNA5 mutation, IVS8+4 A>G, in the splice donor site of intron 8 causes late-onset non-syndromic hearing loss in a Chinese family. Clin Genet. 2007;72:471–7.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
Xu WF, Zhang Q, Ding CJ, Sun HY, Che Y, Huang H, et al. Gasdermin E-derived caspase-3 inhibitors effectively protect mice from acute hepatic failure. Acta Pharmacol Sin. 2021;42:68–76.
Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24:312–25.
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193.
Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2007;98:88–95.
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689.
Ibrahim J, Op de Beeck K, Fransen E, Croes L, Beyens M, Suls A, et al. Methylation analysis of Gasdermin E shows great promise as a biomarker for colorectal cancer. Cancer Med. 2019;8:2133–45.
Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:688.
Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32.
Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe. 2009;5:487–97.
Kumar H, Kumagai Y, Tsuchida T, Koenig PA, Satoh T, Guo Z, et al. Involvement of the NLRP3 inflammasome in innate and humoral adaptive immune responses to fungal beta-glucan. J Immunol. 2009;183:8061–7.
Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–7.
Thomas PG, Dash P, Aldridge JR Jr., Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–75.
Chavarria-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol Rev. 2015;265:22–34.
Taabazuing CY, Griswold AR, Bachovchin DA. The NLRP1 and CARD8 inflammasomes. Immunol Rev. 2020;297:13–25.
Wang Y, Cirelli KM, Barros PDC, Sangare LO, Butty V, Hassan MA, et al. Three toxoplasma gondii dense granule proteins are required for induction of Lewis rat macrophage pyroptosis. mBio. 2019;10:e02388–18.
Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46:1140–6.
Vance RE. The NAIP/NLRC4 inflammasomes. Curr Opin Immunol. 2015;32:84–9.
Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE. Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol Cell. 2014;54:17–29.
Qu Y, Misaghi S, Newton K, Maltzman A, Izrael-Tomasevic A, Arnott D, et al. NLRP3 recruitment by NLRC4 during Salmonella infection. J Exp Med. 2016;213:877–85.
Mitchell PS, Roncaioli JL, Turcotte EA, Goers L, Chavez RA, Lee AY, et al. NAIP-NLRC4-deficient mice are susceptible to shigellosis. Elife. 2020;9:e59022.
Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S. NLRC4 inflammasome-mediated production of IL-1beta modulates mucosal immunity in the lung against gram-negative bacterial infection. J Immunol. 2012;188:5623–35.
Lugrin J, Martinon F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol Rev. 2018;281:99–114.
Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur J Immunol. 2016;46:269–80.
Zahid A, Ismail H, Li B, Jin T. Molecular and structural basis of DNA sensors in antiviral innate immunity. Front Immunol. 2020;11:613039.
Loeven NA, Medici NP, Bliska JB. The pyrin inflammasome in host-microbe interactions. Curr Opin Microbiol. 2020;54:77–86.
Heilig R, Broz P. Function and mechanism of the pyrin inflammasome. Eur J Immunol. 2018;48:230–8.
Saavedra PHV, Huang L, Ghazavi F, Kourula S, Vanden Berghe T, Takahashi N, et al. Apoptosis of intestinal epithelial cells restricts Clostridium difficile infection in a model of pseudomembranous colitis. Nat Commun. 2018;9:4846.
Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, et al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun. 2020;11:3276.
Fisch D, Clough B, Domart MC, Encheva V, Bando H, Snijders AP, et al. Human GBP1 differentially targets salmonella and toxoplasma to license recognition of microbial ligands and caspase-mediated death. Cell Rep. 2020;32:108008.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Muller DJ. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. 2018;37:e98321.
Abreu-Blanco MT, Watts JJ, Verboon JM, Parkhurst SM. Cytoskeleton responses in wound repair. Cell Mol Life Sci. 2012;69:2469–83.
McNeil PL, Terasaki M. Coping with the inevitable: how cells repair a torn surface membrane. Nat Cell Biol. 2001;3:E124–9.
Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell. 2001;106:157–69.
Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591:131–6.
Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med. 2018;24:1151–6.
Johnson DC, Okondo MC, Orth EL, Rao SD, Huang HC, Ball DP, et al. DPP8/9 inhibitors activate the CARD8 inflammasome in resting lymphocytes. Cell Death Dis. 2020;11:628.
Linder A, Bauernfried S, Cheng Y, Albanese M, Jung C, Keppler OT, et al. CARD8 inflammasome activation triggers pyroptosis in human T cells. EMBO J. 2020;39:e105071.
Zhong FL, Robinson K, Teo DET, Tan KY, Lim C, Harapas CR, et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J Biol Chem. 2018;293:18864–78.
Li F, Aljahdali I, Ling X. Cancer therapeutics using survivin BIRC5 as a target: what can we do after over two decades of study? J Exp Clin Cancer Res. 2019;38:368.
Tang Z, Ji L, Han M, Xie J, Zhong F, Zhang X, et al. Pyroptosis is involved in the inhibitory effect of FL118 on growth and metastasis in colorectal cancer. Life Sci. 2020;257:118065.
He J, Giusti MM. Anthocyanins: natural colorants with health-promoting properties. Annu Rev Food Sci Technol. 2010;1:163–87.
Lin BW, Gong CC, Song HF, Cui YY. Effects of anthocyanins on the prevention and treatment of cancer. Br J Pharmacol. 2017;174:1226–43.
Yue E, Tuguzbaeva G, Chen X, Qin Y, Li A, Sun X, et al. Anthocyanin is involved in the activation of pyroptosis in oral squamous cell carcinoma. Phytomedicine. 2019;56:286–94.
Blanckaert V, Ulmann L, Mimouni V, Antol J, Brancquart L, Chenais B. Docosahexaenoic acid intake decreases proliferation, increases apoptosis and decreases the invasive potential of the human breast carcinoma cell line MDA-MB-231. Int J Oncol. 2010;36:737–42.
Pizato N, Luzete BC, Kiffer L, Correa LH, de Oliveira Santos I, Assumpcao JAF, et al. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci Rep. 2018;8:1952.
Kang KS, Wang P, Yamabe N, Fukui M, Jay T, Zhu BT. Docosahexaenoic acid induces apoptosis in MCF-7 cells in vitro and in vivo via reactive oxygen species formation and caspase 8 activation. PLoS One. 2010;5:e10296.
Graham KL, Zhang JV, Lewen S, Burke TM, Dang T, Zoudilova M, et al. A novel CMKLR1 small molecule antagonist suppresses CNS autoimmune inflammatory disease. PLoS One. 2014;9:e112925.
Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, et al. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J Exp Med. 2019;216:2562–81.
Qiao L, Wu X, Zhang J, Liu L, Sui X, Zhang R, et al. alpha-NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. FASEB J. 2019;33:12760–7.
Eliopoulos AG, Kerr DJ, Herod J, Hodgkins L, Krajewski S, Reed JC, et al. The control of apoptosis and drug resistance in ovarian cancer: influence of p53 and Bcl-2. Oncogene. 1995;11:1217–28.
Wu M, Wang Y, Yang D, Gong Y, Rao F, Liu R, et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine. 2019;41:244–55.
Kwong YL, Todd D. Delicious poison: arsenic trioxide for the treatment of leukemia. Blood. 1997;89:3487–8.
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–60.
Kinjo K, Kizaki M, Muto A, Fukuchi Y, Umezawa A, Yamato K, et al. Arsenic trioxide (As2O3)-induced apoptosis and differentiation in retinoic acid-resistant acute promyelocytic leukemia model in hGM-CSF-producing transgenic SCID mice. Leukemia. 2000;14:431–8.
Maier NK, Crown D, Liu J, Leppla SH, Moayeri M. Arsenic trioxide and other arsenical compounds inhibit the NLRP1, NLRP3, and NAIP5/NLRC4 inflammasomes. J Immunol. 2014;192:763–70.
Hu J, Dong Y, Ding L, Dong Y, Wu Z, Wang W, et al. Local delivery of arsenic trioxide nanoparticles for hepatocellular carcinoma treatment. Signal Transduct Target Ther. 2019;4:28.
Zhao C, Liu Z, Liang G. Promising curcumin-based drug design: mono-carbonyl analogues of curcumin (MACs). Curr Pharm Des. 2013;19:2114–35.
Chen L, Weng B, Li H, Wang H, Li Q, Wei X, et al. A thiopyran derivative with low murine toxicity with therapeutic potential on lung cancer acting through a NF-kappaB mediated apoptosis-to-pyroptosis switch. Apoptosis. 2019;24:74–82.
Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41:274–86.
Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171–85.
Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–51.
Ashour AA, Abdel-Aziz AA, Mansour AM, Alpay SN, Huo L, Ozpolat B. Targeting elongation factor-2 kinase (eEF-2K) induces apoptosis in human pancreatic cancer cells. Apoptosis. 2014;19:241–58.
Byrne BG, Dubuisson JF, Joshi AD, Persson JJ, Swanson MS. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. mBio. 2013;4:e00620–12.
Yu P, Wang HY, Tian M, Li AX, Chen XS, Wang XL, et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol Sin. 2019;40:1237–44.
Galluzzi L, Vacchelli E, Michels J, Garcia P, Kepp O, Senovilla L, et al. Effects of vitamin B6 metabolism on oncogenesis, tumor progression and therapeutic responses. Oncogene. 2013;32:4995–5004.
Yang W, Liu S, Li Y, Wang Y, Deng Y, Sun W, et al. Pyridoxine induces monocyte-macrophages death as specific treatment of acute myeloid leukemia. Cancer Lett. 2020;492:96–105.
McKeage MJ. Lobaplatin: a new antitumour platinum drug. Expert Opin Investig Drugs. 2001;10:119–28.
Zhou L, Jiang L, Xu M, Liu Q, Gao N, Li P, et al. Miltirone exhibits antileukemic activity by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction pathways. Sci Rep. 2016;6:20585.
Zhang X, Zhang P, An L, Sun N, Peng L, Tang W, et al. Miltirone induces cell death in hepatocellular carcinoma cell through GSDME-dependent pyroptosis. Acta Pharm Sin B. 2020;10:1397–413.
Song W, Ma YY, Miao S, Yang RP, Zhu Y, Shu D, et al. Pharmacological actions of miltirone in the modulation of platelet function. Acta Pharmacol Sin. 2019;40:199–207.
Wang L, Li K, Lin X, Yao Z, Wang S, Xiong X, et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 2019;450:22–31.
Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–14.
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21.
Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, et al. NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol. 2017;8:519.
Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21:736–45.
Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369:1633–7.
Wang Z, Xu G, Gao Y, Zhan X, Qin N, Fu S, et al. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm Sin B. 2019;9:734–44.
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187:61–70.
He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018;9:2550.
Ye J, Zeng B, Zhong M, Li H, Xu L, Shu J, et al. Scutellarin inhibits caspase-11 activation and pyroptosis in macrophages via regulating PKA signaling. Acta Pharm Sin B. 2021;11:112–26.
Malireddi RKS, Karki R, Sundaram B, Kancharana B, Lee S, Samir P, et al. Inflammatory cell death, PANoptosis, mediated by cytokines in diverse cancer lineages inhibits tumor growth. Immunohorizons. 2021;5:568–80.
Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021;37:109858.
Jiang M, Qi L, Li L, Wu Y, Song D, Li Y. Caspase-8: a key protein of cross-talk signal way in “PANoptosis” in cancer. Int J Cancer. 2021;149:1408–20.
Wang Y, Kanneganti TD. From pyroptosis, apoptosis and necroptosis to PANoptosis: a mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J. 2021;19:4641–57.
Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med. 2018;215:1023–34.
Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med. 2020;217:e.20191644.
Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:aag2045.
Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell. 2020;181:674–87.
Acknowledgements
This publication was made possible by the Arkansas INBRE program (P20 GM103429) and grants from National Heart, Lung and Blood Institute (HL153876, USA) and National Eye Institute (EY030621, USA).
Author information
Authors and Affiliations
Contributions
All authors participated in writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Yang, F., Bettadapura, S.N., Smeltzer, M.S. et al. Pyroptosis and pyroptosis-inducing cancer drugs. Acta Pharmacol Sin 43, 2462–2473 (2022). https://doi.org/10.1038/s41401-022-00887-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00887-6
Keywords
This article is cited by
-
Nanoparticle delivery of si-Notch1 modulates metabolic reprogramming to affect 5-FU resistance and cell pyroptosis in colorectal cancer
Cancer Nanotechnology (2024)
-
Hydralazine loaded nanodroplets combined with ultrasound-targeted microbubble destruction to induce pyroptosis for tumor treatment
Journal of Nanobiotechnology (2024)
-
The Role of Pyroptosis and Autophagy in the Nervous System
Molecular Neurobiology (2024)
-
New prospects of cancer therapy based on pyroptosis and pyroptosis inducers
Apoptosis (2024)
-
Research Progress of Caspase in Endometriosis
Reproductive Sciences (2024)
