
Abstract
Nonalcoholic steatohepatitis (NASH) is a progressive form of nonalcoholic fatty liver disease (NAFLD), characterized with hepatocellular steatosis, ballooning, lobular inflammation, fibrotic progression, and insulin resistance. NASH may progress to cirrhosis and hepatocellular carcinoma (HCC), which are the major indications for liver transplantation and the causes for mortality. Thus far, there are no approved pharmacotherapeutics for the treatment of NASH. Given the complexity of NASH pathogenesis at multifaceted aspects, such as lipotoxicity, inflammation, insulin resistance, mitochondrial dysfunction and fibrotic progression, pharmacotherapeutics under investigation target different key pathogenic pathways to gain either the resolution of steatohepatitis or regression of fibrosis, ideally both. Varieties of pharmacologic candidates have been tested in clinical trials and have generated some positive results. On the other hand, recent failure or termination of a few phase II and III trials is disappointing in this field. In face to growing challenges in pharmaceutical development, this review intends to summarize the latest data of new medications which have completed phase II or III trials, and discuss the rationale and preliminary results of several combinatory options. It is anticipated that with improved understanding of NASH pathogenesis and critical endpoints, efficient pharmacotherapeutics will be available for the treatment of NASH with an acceptable safety profile.
Similar content being viewed by others
Introduction
NAFLD is one of the most common chronic liver diseases worldwide and its prevalence in general population is about 25% [1]. It was reported that the prevalence of NAFLD has increased from 15% to 29.2% in somewhere in China [2, 3]. It refers to a series of progressive disorders with fat accumulation in the liver, and ranges from nonalcoholic fatty liver (NAFL), nonalcoholic steatohepatitis (NASH), which may progresses to fibrosis, further to cirrhosis or hepatocellular carcinoma (HCC). At an early stage of NAFL, it could be recovered through diet control and exercise. NASH is an active stage with hepatocellular injury and inflammatory responses, and requires medical intervention to reduce the risk of cirrhosis and HCC. Growing evidence demonstrates when over 10% weight loss is achieved by lifestyle changes, it actually improves histological features including NASH resolution and fibrosis regression in a large proportion of patients with NASH [4]. However, this extent of weight loss is challenging for most patients to achieve and maintain for a long time, and body weight rebound may occur thereafter. Bariatric surgery often leads to durable weight reduction, at the same time liver fat content is reduced, and hepatic fibrosis is improved [5]; nevertheless this procedure is not suggested for NASH patients as a practical guideline due to the surgical risk. So far, no pharmacotherapeutic candidates have been approved by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA) or Center for Drug Evaluation of China for the treatment of NASH, and therefore there is an urgent need to move pharmacologic candidates from pipelines to clinical trials.
As a liver complication of a spectrum of metabolic disorders, such as obese, metabolic syndrome, hyperlipidemia or type 2 diabetes (T2DM), NASH varies in manifestations, course progression, and treatment responses dramatically among different individuals. This heterogeneity dictates the complexity of its pathogenesis and multiple layers of metabolic, genetic and epigenetic abnormalities [6]. Thereby, no etiologic interventions are available; instead, intermediate pathways and molecular targets in lipid metabolism, lipotoxicity, hepatocellular injury and death, inflammatory or immunoresponses, fibrotic progression have been selected for pharmacologic intervention. These targets are critical receptors, such as farnesoid X receptor (FXR), peroxisome proliferator-activated receptor (PPAR); thyroid hormone receptor-β (THRβ), fibroblast growth factor (FGF) and glucagon-like peptide-1 (GLP1), anti-inflammation pathway (chemokine receptor type-2/5); rate-limiting enzymes involved in de novo fatty acid synthesis (acetyl-CoA carboxylase (ACC), stearoyl CoA desaturase-1 (SCD1) or fatty acid synthase); or molecules involved in hepatic fibrosis (galectin-3), and many others. Molecular targets and potential agents in pharmaceutical developing pipelines are extensively summarized in recent reviews [7,8,9]. The present overview intends to cover pharmacologic mechanisms and new results of these agents in randomized phase II and III trials focusing on efficacy, adverse effects, and possible limitations in the interpretation of trial results.
NASH pathophysiology and clinical treatment endpoints
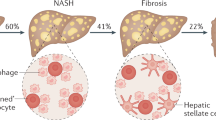
NASH pathophysiology appears to be complicated because of the interdependence and cross-talks between the liver and other organs (particularly the gut, kidneys and adipose tissue). The key event starts with accumulation of lipid droplets with lipotoxicity in hepatocytes [10]. Excessive nutrition causes expansion of adipose tissue as well as ectopic fat accumulation. Transformation of anti-inflammatory to pro-inflammatory macrophages leads to cytokine secretion and inflammation in visceral adipose tissue, which promotes insulin resistance and metabolic disorders. Insulin resistance drives the increased hepatic conversion of carbohydrates into fatty acids through de novo lipogenesis (DNL) and accelerates lipolysis of adipose tissue, resulting in elevated non-esterified fatty acids levels in the blood, which are taken up by the liver in a concentration-dependent manner [11, 12]. Abnormal accumulation of lipids in the liver overwhelms its metabolic capacity, following the formation of lipotoxic lipids that contribute to oxidative stress, inflammasome activation, hepatocellular damage, and cell death through necrotic, apoptotic or pyroptotic pathways (often described as necroptosis in histopathology) [13]. Subsequently, due to the damaging signals from injured hepatocytes and/or stimulation of inflammatory responses, hepatic stellate cells (HSCs) are activated leading to fibrogenesis [14, 15]. During the course of disease progression, gut microbiota may also play an important role and it appears that dysbiosis and gut microbial components exacerbate the initiation and progression of NASH [16, 17]. A schematic illustration is depicted to reflect the key pathophysiologic events in NASH as well as effects of other tissues or organs (Fig. 1).

The major pathological changes in NASH include steatohepatitis (representing steatosis, lipotoxicity, necroptosis, inflammatory responses), insulin resistance and fibrotic progression. These pathophysiologic alterations are the intervention focuses. Genetic alterations, such as single-nucleotide polymorphism (SNP) in PNPLA3, TM6SF2 or MBOAT7 enhance the predisposition and risk for NAFLD. The excessive fatty accumulation is an overall result of influx of lipolysis products, de novo lipogenesis and insufficient out-ward transportation through VLDL, LDL, or HDL, etc. Lipotoxicity originates from excessive fatty acids, and leads to oxidant stress and endoplasmic reticulum (ER) stress, further to cell death through necroptosis or pyroptosis. Hepatocellular injury and death elicit inflammatory and immunogenic responses, as well as fibrotic initiation and progression. Steatotic hepatocytes with oxidant stress or ER stress often accompany with insulin resistance, which could be the part of systemically disordered metabolism, and may exacerbate hepatic metabolism in various arrays. A variety of chemokines, adipokines or lipolysis products from extrahepatic tissues, such as adipose tissue, gut, and neuroendocrine system affect metabolic status and energy homeostasis in the liver, and may overwhelm inflammatory, immunogenic or fibrotic responses in a steatotic liver. Moreover, gut dysbiosis and toxic products worsen the disordered metabolism, necroptosis, insulin resistance and fibrotic progression in the liver. The overall consequence of these pathologic changes drives the liver to enter into a loop of lipid metabolic disturbance → steatotic necroptosis → insulin resistance → fibrotic progression → worsened liver and systemic metabolic abnormalities. DNL de novo lipogenesis, DAMPs damage-associated molecular patterns, HCC hepatocellular carcinoma, HDL high density lipoprotein, HSC hepatic stellate cells, LDL low density lipoprotein, VLDL very low density lipoprotein, PAMPs pathogen-associated molecular patterns, PNPLA3 patatin-like phospholipase domain-containing protein-3, TM6SF2 transmembrane 6 superfamily member-2, MBOAT7 membrane bound O-acyltransferase domain-containing-7.
The gold standard for a diagnosis of NASH requires liver biopsy and histological confirmation of fat accumulation, hepatocellular ballooning, inflammatory infiltration, and fibrosis [18]. NAFLD activity score (NAS) was developed to determine histopathologic features, including hepatocellular steatosis, ballooning and lobular inflammation in a semi-quantitative manner [18]. A NAS ≥ 4 correlates highly with a diagnosis of NASH [19]. Clinical investigations demonstrate that various degree of fibrosis is typical in NASH and that hepatic fibrosis at an advanced stage (defined histologically as fibrosis stage F2 or higher) is in at least a quarter of patients at diagnosis [20]. Severity of hepatic fibrosis is on a scale ranging from F0 to F4 as follows: no fibrosis [F0], portal fibrosis without septa [F1], portal fibrosis with few septa [F2], bridging septa between central and portal veins [F3], and cirrhosis [F4] [10, 21]. Notably, severity of hepatic fibrosis is the only histologic measure that independently predicts long-term liver-related complications, needs for liver transplantation, and liver-related death in patients with NASH [10]. Accumulating evidence exists demonstrating that the severity of fibrotic stages was positively correlated with frequency of long-term comorbidity and the risk for all-cause and liver-related mortality in NASH patients; and that survival duration of patients with F2 and F3 was shortened by 10 and 20 years in comparison to those without fibrosis [22, 23]. Patients with cirrhosis have a higher incidence of HCC which was amplified by the presence of T2DM [24]. Thus, fibrosis regression is commonly used as a primary or secondary endpoint in the treatment of NASH in phase III trials, and recruited NASH subjects are those with F2–F3 or F4 stage of fibrosis [25, 26] as indicated in Table 1. Due to the high co-linearity between NASH and fibrosis severity, there exists a strong correlation between fibrosis regression and NASH resolution [22]. Patients diagnosed as NASH without cirrhosis have a higher risk for HCC than other etiologies of liver disease [27, 28]. Therefore, NASH resolution is a major focus in phase I and II trials, and remains one of the primary endpoints in phase III studies [29, 30]. Since the majority of patients with NASH will ultimately die as a result of complications of T2DM and cardio-metabolic illnesses, clinical trials of novel therapeutic candidates also evaluate effects on metabolic improvements, a composite of long-term outcomes and all-cause mortality up to several years [31]. On-going, terminated or completed phase III trials of NASH pharmacotherapeutics are summarized in Table 1 for an overview of molecular targets, number of subjects recruited, duration of trials and subject selection categories.
Disappointing and encouraging results of NASH phase II and III trials
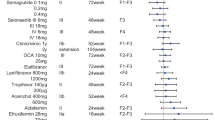
As indicated in Table 1, the only phase III trial completed during 2020 is obeticholic acid (OCA), other three (cenicriviroc, elafibranor, selonsertib) trials were terminated because the failure to reach endpoints during the mid-term assessment, which brought some disappointments in pharmaceutical industry, medical professionals and patients. However, promising candidates are moving quickly from phase II to phase III trials, as shown in Fig. 2, and an increasing number of phase III trials, such as resmetirom, lanifibranor, aramchol and semaglutide, etc. are on-going with different subject profiles and arms. Hopefully, anticipated results are encouraging. For better understanding of therapeutic potential and safety concern, their targets and results of phase II trials are briefly discussed.

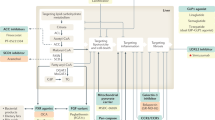
Based on the pathogenesis of NASH initiation and fibrotic progression as illustrated in Fig. 1, most pharmacologic interventions are focused on the fatty acid synthetic pathway, lipotoxicity and metabolic stress, inflammation and apoptosis, as well as fibrosis although overlapping in pharmacologic actions exists. Under each particular target, such as ACC, SCD1, or CCR2/5, etc. there exist a couple of pharmacotherapeutic candidates, which are either in phase II or III trials. Emerging candidates are added to the list, and evolving on the daily base. The status of trials is color-coded as phage II on-going, phage III on-going, completed or failure. ⊥ indicates inhibitory action, dashed arrows imply improving effects. Solid arrows represent entering or moving to next steps. FASN fatty acid synthase, LOXL2 Lysyl oxidase-like molecule 2.
Of note, key issues in NASH pathophysiology include accumulation of fat droplets, lipotoxicity and fibrotic progression as well as systemically metabolic abnormalities [10]. Pharmacotherapeutic agents are developed to target these pathways, such as triglyceride synthesis, insulin sensitivity and energy consumption to reduce hepatic lipid accumulation, or indirectly regulating metabolic homeostasis by modulating bile acid production or reducing energy intake. Presumably, reduction of fatty accumulation, attenuation of lipotoxicity, improvement of insulin sensitivity and systemic improvement of metabolic status may achieve NASH resolution, and subsequently halt or reverse fibrotic progression, the two major endpoints in the phase III trials, and either single or both benefits are heavily focused in clinical data evaluation [32].
Glucagon-like peptide-1 (GLP1) receptor agonists
GLP1 is an endogenous hormone, secreted by intestinal endocrine L-cells in response to luminal stimuli. It enhances the production and release of insulin, indirectly inhibits glucagon secretion, slows gastric emptying and reduces food intake by modulators acting through G protein-coupled GLP1 receptor (GLP1R) [33]. As key modulators of GLP1 function in glucose homeostasis, multiple GLP1R agonists (exenatide, liraglutide, dulaglutide, and semaglutide) are approved for the treatment of T2DM [12]. A study has revealed that hepatic fibroblast growth factor-21 (FGF21) is involved in mediating functions of liraglutide to lower body weight and improve hepatic lipid homeostasis in mice [34]. Separate randomized phase II trials of liraglutide and semaglutide for NASH treatment have been completed with satisfying results [26, 35].
A randomized, double-blind, phase II trial of liraglutide vs. placebo was conducted in 52 patients with biopsy-proven NASH, and has demonstrated higher resolution of NASH (39% vs. 9%, P = 0.02) and less fibrotic progression (9% vs. 36%, P = 0.04) than placebo at 48 weeks [35]. Due to a small sample size, extensive and longer-term studies are needed to evaluate the efficacy of liraglutide. In a phase II trial, 320 patients with biopsy-confirmed NASH and fibrosis of stage F1–F3 were randomly assigned to daily subcutaneous injection of semaglutide at 0.1, 0.2, or 0.4 mg or placebo for 72 weeks. The results from patients with fibrosis F2–F3 stages documented that NASH was resolved without worsening of fibrosis in 59% of subjects receiving semaglutide at 0.4 mg compared to 17% in the placebo group (P < 0.001) [26]. Although there was no difference in the secondary endpoint (improvement in at least one fibrosis stage without worsening of NASH) compared to placebo, fewer participants experienced worsening of fibrosis stage in the semaglutide 0.4 mg dose group vs. the placebo group (5% vs. 19%). Gastrointestinal discomforts were the most common adverse events. The percentage of patients with nausea was higher in the semaglutide 0.4 mg group than in the placebo (42% vs. 11%) group [26, 36]. However, from NASH, obesity and T2DM trials, the improvement in NASH with semaglutide may stem from significant weight loss; and it remains to be investigated regarding whether semaglutide has direct and independent effects to ameliorate NASH in human. The safety of semaglutide has been investigated to be the same as it was used for T2DM treatment. If the benefits are confirmed in its phase III trials, the approval of semaglutide for NASH treatment may become possible.
Specific inhibitors against enzymes in the fatty acid synthesis pathway
A key feature of NASH is the aberrant regulation of lipids within hepatocytes (e.g., increased DNL or impaired fatty acid β-oxidation). The regulation of DNL plays a central role in controlling fatty acid synthesis and catabolism. Since ACC is a rate-limiting enzyme of converting acetyl-coenzyme A (acetyl-CoA) to malonyl-CoA in fatty acid synthesis, inhibition of ACC1 and ACC2 isoforms would reduce DNL; and ACC inhibitors, such as firsocostat (GS-0976), were selected as potential pharmacotherapeutic agents for NASH treatment [37]. One hundred and six patients with NASH were randomly assigned (2:2:1) to groups given firsocostat (20 mg or 5 mg), or placebo daily. At week 12, 47.8% of patients given firsocostat at 20 mg achieved a ≥30% relative fat decrease in the liver from the baseline as indicated by magnetic resonance imaging proton density fat fraction (MRI-PDFF) compared to 15.4% in the placebo group (P = 0.004). However, clinically asymptomatic hypertriglyceridemia (>500 mg/dL) in patients receiving firsocostat treatment was observed in 14% of patients given firsocostat at 20 mg, 18% of them at 5 mg, and none of them receiving placebo [38]. Therefore, its safety needs to be evaluated in long-term trials. Firsocostat has been tested for combinatory trials with FXR agonists (Hepatology 2020; 72: 901A-902A).
SCD1 controls a rate-limiting step in mono-unsaturated fatty acid synthesis. Aramchol is a conjugate of cholic and arachidic acid that was shown to inhibit SCD1 and fatty acid synthesis in in vitro and animal experiments [39]. Sixty patients with biopsy-confirmed NAFLD (6 with NASH) were randomly assigned to receive aramchol (100 or 300 mg) or placebo once daily for 3 months. Liver fat content was decreased by 12.6% in those receiving aramchol at 300 mg/day, but increased by 6.4% in the placebo group. Although significant reduction in liver fat content in those receiving aramchol was seen, there were no significant improvements in serum levels of alanine aminotransferase (ALT) and adiponectin [40]. A phase IIb study is currently evaluating the effects of higher doses (400 and 600 mg/day) in patients with biopsy-proven NASH (NAS ≥ 4). The primary endpoint is absolute change in liver fat content measured by PDFF at week 52. The absolute changes from baseline of mean liver fat were achieved as follows: −3.18% in aramchol 600 mg group, −3.41% in aramchol at 400 mg vs. −0.09% in the placebo group. And 16.7% of population in higher dose group (7.5% in lower dose group, 5% in placebo) met the second endpoint, NASH resolution without worsening of fibrosis. There was a significant reduction in serum ALT and aspartate aminotransferase (AST) from the baseline in both aramchol groups compared to placebo (NCT02279524). This agent is currently being evaluated in a phase III ARMOR trial.
FXR agonists targeting bile acid metabolism pathway
Farnesoid X receptor (FXR), a nuclear receptor for bile acids, is widely expressed in the intestine, liver and kidneys, and is responsible for the homeostasis of cholesterol metabolism [41]. It induces expression of a typical nuclear receptor small heterodimer partner that may repress CYP7A1 gene transcription [41]. CYP7A1 is a rate-limiting enzyme for cholesterol conversion into bile acids, and accounts for ~75% of bile acid production under a physiological condition [42]. A study with GSK2324, a potent and specific synthetic FXR agonist, demonstrated that hepatic FXR controls the GSK2324-dependent decreases in lipogenic genes; whereas intestinal FXR largely controls changes in lipid absorption [43]. Obeticholic acid (OCA) is the most extensively studied FXR agonist for NASH treatment. Patients in the global REGENERATE phase III study were confirmed with NASH by liver biopsy and randomized in 1:1:1 ratio as follows: OCA at 10, 25 mg daily or placebo. A total of 931 patients with stage F2–F3 fibrosis were enrolled in a primary assessment at 18 months and fibrosis improvement endpoint was achieved by 12% of patients in the placebo group, 18% in the OCA 10 mg group (P = 0.045), and 23% in the 25 mg group (P = 0.0002); whereas, the NASH resolution endpoint was not met. The most common adverse event was pruritus which occurred more frequently in the 25 mg group than the placebo (51% vs. 19%). Furthermore, elevation in LDL-C level was observed in the OCA treatment group, which may be associated with cardiovascular adverse events or pose a potential atherosclerotic risk [25]. Because the benefit of OCA based on histopathologic endpoint remains uncertain and it does not sufficiently outweigh the potential risks, the FDA has not approved it for NASH treatment yet (https://ir.interceptpharma.com). The REGENERATE study continues to determine its long-term benefits and safety. Other FXR agonists of different chemotypes, both steroidal and non-steroidal, including EDP-305, Tropifexor, Cilofexor and MET-409, have been demonstrated that pruritus is a common manifestation of FXR agonists associated with dose elevation [44,45,46,47]. Thus, identifying the optimal dose to alleviate adverse effects (hyperlipidemia and pruritus) while maintaining sufficient efficacy is the current objective remaining to be achieved. For instance, MET-409 administration strikingly lowered liver fat content as measured by MRI-PDFF. However there are warning signs with a transient ALT elevation in MET-409 administration group [45]. A phase IIa trial of another FXR agonist, TERN-101, has been completed and the data have not been released yet. FXR agonists are in combinatory trials with cenicriviroc (CVC), atorvastatin (AVT), semaglutide or firsocostat for better efficacy and an acceptable safety profile. Attention has been paid to the fact that excessive activation of intestinal FXR triggers a steady increase of fibroblast growth factor-19 (FGF19), which is a key driver of hepatocyte proliferation and may contribute to HCC progression in cirrhotic patients [48, 49]. Future studies are needed to ensure the safety of FXR agonists in these aspects.
Analogues of FGF19
Human FGF19 is a gastrointestinal hormone that regulates bile acid synthesis, glucose metabolism and hepatic fatty acid oxidation, and is known to be a target of FXR [50]. Aldafermin (NGM282) is an engineered non-tumorigenic analogue of FGF19, which retains the ability to potently suppress CYP7A1 but does not elicit activation of STAT3, a signaling pathway critical for FGF19-mediated hepatic carcinogenesis. No liver tumors were observed in preclinical animal models after exposure to aldafermin for 24 weeks; in contrast, mice exhibited high tumor penetrance following AAV–FGF19 delivery [51]. In a 24-week phase II trial, patients with NASH and fibrosis receiving aldafermin 1 mg once daily by subcutaneous injection achieved significant improvements in liver fat content, liver transaminases and novel markers of fibrosis. In this trial, 38% of patients in the aldafermin group attained fibrotic regression without worsening of NASH vs. 18% in the placebo group (P = 0.10); whilst 24% of patients in the aldafermin group gained NASH resolution without worsening of fibrosis vs. 9% in the placebo group (P = 0.20). And there were few adverse events, such as diarrhea and nausea with aldafermin treatment. Aldafermin was generally well-tolerated with an overall safety profile similar to placebo [50]. However, it was found that aldafermin reduced cholesterol catabolism and produced a significant increase in serum LDL-C levels [50]. In considering the increase in cardiovascular risk associated with NASH, it is important to explore the concomitant use of lipid-lowering therapies, such as statins, to support chronic administration of aldafermin [52]. A phase IIb study for 24 weeks evaluating aldafermin vs. placebo with histologic improvement as the primary endpoint was conducted in patients of NASH with F2/3 fibrosis. Although the study achieved NASH resolution (at a 3 mg dose) with statistical significance, it did not meet primary endpoint of fibrosis regression by ≥1 stage without worsening of NASH vs. placebo. In addition, a phase IIb study in patients with biopsy-confirmed NASH with F4 fibrosis and compensated cirrhosis is ongoing (https://ir.ngmbio.com).
Agents for improving insulin sensitivity
MSDC-0602K is a novel insulin sensitizer designed to preferentially target the mitochondrial pyruvate carrier while minimizing direct binding to the transcriptional factor PPARγ, aiming at improving disordered lipid metabolism, inflammation, and insulin resistance as well as fibrosis [53]. Patients with biopsy-confirmed NASH and fibrosis (F1–F3) were randomized to daily oral placebo, or 1 of 3 MSDC-0602K doses in a 52-week study. MSDC-0602K did not exhibit statistically significant effects on the primary endpoint or secondary endpoints including NAS improvement without worsening fibrosis, NASH resolution, and fibrosis reduction at 12 months. MSDC-0602K treatment was associated with better glucose metabolism, reduced insulin levels, and improvement in hepatic injury parameters [54].
Agonists for peroxisome proliferation-associated receptor (PPAR) subtypes
PPARs act as ligand-activated transcription factors to regulate metabolic and energy homeostasis, which are comprised of PPAR-α, PPAR-γ, and PPAR-β/δ subtypes. Each PPAR has distinct tissue distribution and physiological function [55]. PPARα has relatively high expression in the liver, and its activation reduces hepatic triglyceride levels [55]. Activation of PPARγ prevents the increased flux of free fatty acids and adipokines from the adipose tissue to other organs, especially to the liver, while activation of PPARβ/δ promotes fatty acid metabolism and suppresses macrophage-mediated inflammation [56]. Lanifibranor (IVA337) is a pan-PPAR agonist, and a phase IIb trial for NASH treatment has been completed [57]. In this study, 247 patients with NASH confirmed by biopsy were randomly assigned into placebo or lanifibranor-treatment groups at 1200 or 800 mg daily doses. The steatosis activity fibrosis (SAF) score is considered as a clinically useful histopathologic measure [58] and SAF activity scoring A, which combines a score for lobular inflammation and ballooning, is used to assess lanifibranor efficacy. The primary endpoint is SAF activity scoring A decrease of at least two points without worsening of fibrosis. At 24th week, 49% of patients treated with lanifibranor 1200 mg daily and 41% of patients in 800 mg group met the primary endpoint, whereas only 27% in placebo group. Both lanifibranor dose groups met resolution of NASH and fibrosis regression, i.e., lanifibranor 1200 mg (31.3%) and 800 mg (20.5%) vs. placebo (7.4%). Moreover, serum ALT and AST levels were reduced significantly in the treatment groups compared to baseline. Regarding adverse events, lanifibranor was associated with diarrhea and weight gain in about 10% of patients (NCT03008070).
Saroglitazar, a PPAR agonist with predominant PPARα and moderate PPARγ activity, was designed to reduce untoward side effects related to a PPARγ agonist, pioglitazone (PGZ), (e.g., weight gain). In a phase II study, 106 patients with NAFLD/NASH with ALT ≥ 50 U/L at baseline and BMI ≥ 25 kg/m2 were randomized in a 1:1:1:1 ratio to receive Saroglitazar at 1, 2, or 4 mg or placebo for 16 weeks. The reduction from baseline in ALT at week 16 was −25.5%, −27.7% and −45.8% with Saroglitazar at 1, 2, and 4 mg, respectively, vs. 3.4% in placebo (P < 0.001 for all). Saroglitazar at 4 mg improved homeostatic model assessment-insulin resistance, and reduced toxic lipid species and fat content in the liver. In the Saroglitazar 4 mg group, 40.7% of patients had a reduction of at least 30% in liver fat content when compared to only 8.0% of patients in the placebo group (P = 0.01) as assessed by MRI-PDFF [59]. Saroglitazar was used for management of diabetic dyslipidemia and hypertriglyceridemia in those with T2DM uncontrolled by a statin alone and was approved for non-cirrhotic NASH treatment in 2020 in India by Drug Controller General of India.
Elafibranor is a PPAR-α/δ dual agonist and showed some favorable effects in a phase II trial [60], which was not confirmed in the RESOLVE-IT phase III trial. The phase III trial has been discontinued in 2020, because interim results of the trial at week 72 did not meet the predefined primary endpoint with a response rate of 19.2% in the elafibranor group compared to 14.7% in the placebo group. The results didn’t show improvement in fibrosis or metabolic parameters compared to placebo (https://ir.genfit.com/). Investigators are carefully reviewing the outcomes and the responder percentage of early phase trials to determine whether the effect of the drug is adequate to support the continuation of the next phase trial.
Agonists for thyroid hormone receptor-β (THRβ)
THRβ is highly expressed in hepatocytes and is responsible for regulating the level of systemic lipid and the metabolic pathways in the liver [61]. The incidence of clinical and subclinical hypothyroidism is higher in patients with NASH than age-matched controls [62]. Resmetirom (MGL-3196) is an oral THRβ selective agonist and a phase IIb study enrolled 125 patients and randomly assigned in 2:1 ratio to receive resmetirom at 80 mg or matching placebo. Liver biopsy assessment was performed in 108 patients at 36th week. Resmetirom therapy resulted in a significant reduction in absolute and relative liver fat content from the baseline compared to placebo. At week 12, the change in median relative fat content from baseline was −36.3% in resmetirom vs. −9.6% in placebo groups (P < 0.0001), and the proportion of patients with a ≥ 30% relative fat reduction was greater (60% vs. 18%) than the placebo group. Furthermore, 27% of patients in the resmetirom group attained NASH resolution compared to 6% in the placebo group at week 36. The most common adverse events were diarrhea and nausea, which were generally well-tolerated [63]. In addition, a separate study suggests that resmetirom treatment led to an improvement in health-related quality of life in NASH patients, particularly in those with more than 30% relative fat reduction at week 12 [64]. From these two studies, it appears that targeting THRβ may have positive effects in NASH resolution without fibrosis improvement, which needs to be confirmed in the on-going MAESTRO phase III trial.
Agents in suppressing apoptosis and inflammation
Emricasan is a pan-caspase inhibitor that attenuates hepatocellular apoptosis and inflammatory responses. In a phase II study, 318 patients with NASH fibrosis were randomized in 1:1:1 ratio into groups of treatment with emricasan (5 or 50 mg) or matching placebo. Emricasan treatment didn’t improve liver histology and may have worsened fibrosis and ballooning at week 72 [65]. On the other hand, emricasan has been shown to decrease portal pressure and improve synthetic function in mice with carbon tetrachloride-induced cirrhosis. Based on this animal study, 217 individuals with decompensated NASH cirrhosis was randomly assigned in 1:1:1 ratio into emricasan (5 or 25 mg) or placebo. The primary endpoint comprised all-cause mortality and a new decompensation event. Although emricasan was safe, it was ineffective for the treatment of decompensated NASH cirrhosis from the released results of the trial [66].
CCR2/5 antagonist, Cenicriviroc (CVC)
During hepatic injury, C-C chemokine receptors type 2 (CCR2) and 5 (CCR5) and C-C chemokine ligands 2 and 5 (CCL2 and CCL5) worsen liver fibrosis through activation of inflammatory signaling and immune cell infiltration [67]. Cenicriviroc (CVC) is a CCR2 and CCR5 dual antagonist under evaluation for treating liver fibrosis in adults with NASH [67]. Improvement of fibrosis has been observed in the phase II trial for NASH treatment [68]. The phase III trial was conducted to confirm the efficacy and safety of cenicriviroc for the treatment of hepatic fibrosis in adult subjects with NASH. However, it was terminated early due to lack of efficacy based on the results of the phase III AURORA trial (NCT03028740).
Apoptosis signal-regulating kinase-1 (ASK1) inhibitor, Selonsertib
Selonsertib is an apoptosis inhibitor. The phase III trial was discontinued as it did not reach the primary endpoint (NCT03053063) [69]. As a few phase II and III trials failed to reach primary and secondary endpoints in the past two years, disappointing data reflect a number of issues in clinical evaluation of pharmacological candidates targeting early stages of necroptosis and inflammatory responses, and prompt the combination of selonertib with other agents, such as FXR agonists, as discussed below.
Analogues of fibroblast growth factor-21 (FGF21)
FGF21 is a novel endocrine messenger that activates a cell membrane co-receptor complex of β-klotho and one of its cognate FGF receptors (FGFRs), FGFR1c, FGFR2c or FGFR3c. Circulating FGF21 is liver-derived, and is also expressed in a number of other tissues, such as pancreas, muscle and adipose [70]. It regulates individual organ metabolism and systematic energy homeostasis [71]. FGF21-induced inhibition of lipolysis in adipose tissue is mediated by FGF receptor-1c (FGFR1c), and direct suppression of DNL in the liver is mediated by FGFR2c or FGFR3c [72, 73]. Efruxifermin is a fusion protein of human IgG1 Fc domain linked to a modified human FGF21 (Fc-FGF21) with balanced in vitro agonist potency at the three cognate FGFRs. A phase IIa trial aimed to test the safety and efficacy of weekly subcutaneous administration for 16 weeks in 80 patients with NASH stratified by hepatic fat content and fibrosis stage. One primary endpoint, absolute change from baseline in hepatic fat content assessed by MRI-PDFF at week 12 was met. Absolute changes of hepatic fat content from baseline were −12.3%, −13.4% and −14.1% in the 28-, 50- and 70 mg groups, respectively, vs. 0.3% in the placebo group. The reduction in hepatic fat content was accompanied by rapid and marked decreases in markers of liver stress and injury. All patients in efruxifermin-treated groups at week 12 achieved ≥30% relative reduction of hepatic fat content, and only two patients in the placebo group achieved [73]. Forty-eight percent of efruxifermin-treated patients had NASH resolution without worsening of fibrosis; whilst markers of collagen synthesis and fibrogenesis were also reduced at week 20. Moreover, 55% (22/40) had a fibrosis improvement ≥1 stage [73]. Because only two patients receiving placebo were evaluated by biopsy, it is impossible to compare the treatment group with placebo. The most frequent efruxifermin treatment-associated adverse events were gastrointestinal, mainly mild and transient in nature [73]. Although an investigation indicates that FGF21 promotes bone loss [74, 75], no clinically significant safety findings were identified based on markers of bone turnover, and bone mineral density was unchanged from baseline to week 16 in efruxifermin-treated group [73]. Longer-duration studies are warranted to further evaluate the safety and efficiency of efruxifermin in NASH patients.
Pegbelfermin (BMS-986036), a PEGylated human FGF21 analogue, has previously been shown to improve metabolism and liver fibrosis in obese patients with T2DM. In a phase IIa study, patients with biopsy-confirmed NASH received PEG-belfermin at 10 mg or 20 mg once weekly or placebo by subcutaneous injections. In both PEG-belfermin-treated groups, patients achieved an absolute reduction in hepatic fat content (−6.8% in 10 mg group, −5.2% in 20 mg group vs. −1.3% in placebo, P < 0.01) as measured by MRI-PDFF. More than half of the patients achieved at least 30% relative reduction in hepatic fat content with PEG-belfermin [76]. Since both efruxifermin and PEG-belfermin are FGF21 mimetics, it is worried that they might elicit immunogenic responses in some patients. Anti-FGF21 antibodies were detectable in most patients who completed the trials while the antibody titers were generally low, indicating FGF21 mimetics did not elicit clinically meaningful immunogenicity [73, 76]. Future studies with PEG-belfermin in NASH should be conducted with more convincing evidence, such as histological endpoints.
Berberine ursodeoxycholate
HTD1801 (berberine ursodeoxycholate) is an ionic salt combining berberine with ursodeoxycholic acid, which is dissociated into two parts before it is absorbed within the gastrointestinal tract. Berberine has been shown to have a hypoglycemic effect, and its underlying mechanisms on NASH improvement have been gradually revealed [77, 78]. It is known that ursodeoxycholate is a hepatoprotective agent, and has a cholesterol-lowering effect. Given the benefits of berberine and ursodeoxycholate, HTD1801 is assumed to be beneficial for NAFLD. A prospective phase II trial with two doses of HTD1801 for NASH patients with type 2 diabetes has been completed; and the findings of the trial have been released recently (NCT03656744). Patients in the treatment groups met the primary endpoint; and those in the high dose group had significantly more reduction in liver fat content than the placebo group as measured by MRI-PDFF at 18 weeks (absolute decrease −4.8% vs. −2.0%). In addition, HTD1801 improved serum glycemic and ALT levels, and decreased body weight; whilst it was relatively well-tolerated. Gastrointestinal side effects were the most frequently reported adverse events, and about 12% patients discontinued the drug during the study [79]. Future studies are under the way to explore liver histopathologic improvement with HTD1801.
Combinatory treatment
The pathophysiology of NASH is complex and represents as “multiple-hits” [80], which underscores that various organs, i.e., the liver, adipose tissue, endocrine pancreas, immune system, and gut are involved in the progression. Excessive and sustained energy input over the output of the body brings overwhelming stress on hepatocytes, leading to hepatocellular damage, inflammation, and fibrosis [14]. As discussed above, various drug candidates target a single pathway in the paradigm of NASH pathogenesis with multiple-hits. On the other hands, the endpoints of clinical trials are assessed by histopathologic evidence of NASH resolution and fibrosis regression, the two late events in the disease progression [19]. Unsurprisingly, the efficacy of a few trials is limited and they were disappointedly terminated in the mid-term evaluation. To increase efficacy and reduce side effects, combination of therapeutic agents for different targets is an alternative solution [81]. Although the safety has been assessed in monotherapy trials, it needs to be re-evaluated in a combinatory regimen with hope that combinatory trial may achieve synergistic effects while adverse effects are acceptable.
An ACC inhibitor has been shown to inhibit DNL and improve hepatic steatosis; however, it elevates plasma triglycerides as a result of adaptive upregulation of a lipogenic transcription factor, sterol regulatory element-binding protein 1 (SREBP-1) in the liver [82]. On the contrary, diacylglycerol acyltransferase 2 (DGAT2) inhibitor has been shown to downregulate SREBP-1 [83]. In a phase IIa trial, an ACC inhibitor (PF-05221304) was co-administered with a DGAT2 inhibitor (PF-06865571) for the treatment of NAFLD. After 6 weeks, co-administration of these two agents has decreased liver fat content from baseline; and placebo-adjusted least squares mean (90% CI) was −44.5%, which was similar to PF-05221304 monotherapy, but higher than PF-06865571 alone. Although these two agents produced a transient increase in serum ALT and AST levels, the enzymes declined toward baseline after 4 weeks. Significantly, the combination of PF-05221304 with PF-06865571 normalized the ACC inhibitor-induced increase in serum triglyceride levels in NAFLD patients, demonstrating the advantages of the combinatory regimen over monotherapy [84].
In a phase IIb trial, 392 patients with bridging fibrosis or compensated cirrhosis (F3–F4) were randomized to receive placebo, Selonsertib at 18 mg (ASK1 inhibitor), cilofexor at 30 mg (FXR agonist), or firsocostat at 20 mg (ACC inhibitor), alone or in two-drug combination, once daily for 48 weeks. The combination did not meet the primary endpoint, i.e., ≥1-stage improvement in fibrosis without worsening of NASH. The highest response rate (21%) was observed in the combination of cilofexor with firsocostat (vs. placebo 11%, P = 0.17). Moreover, the treatment combining cilofexor with firsocostat led to significant improvements in noninvasive biomarkers, including hepatic steatosis by MRI-PDFF, AST, ALT and liver stiffness compared to placebo or monotherapy [81]. These findings suggest synergistic effects of firsocostat with cilofexor may be seen in the endpoints of improving fibrosis, inflammation, and steatosis when a comprehensive design of clinical trial is successfully conducted [81].
Another study is ongoing to test the combination of cilofexor with firsocostat plus semaglutide in NASH patients with F2–F3 fibrosis for 16 weeks. The rates of adverse events were similar between different groups. Greater improvements were observed in serum ALT, AST and liver steatosis in combinations of these three agents than semaglutide monotherapy (J Hepatol 2021; 75: S561). Many other combinations are ongoing in different clinical trials, such as an FXR agonist tropifexor combined with a sodium-glucose cotransporter (SGLT) 1/2 inhibitor licogliflozin; cenicriviroc with tropifexor; tropifexor with licogliflozin; OCA with AVT, and so on [55]. On-going combinatory phase II trials are summarized in Table 2, and the primary endpoints of most trials are to evaluate adverse events, as their benefits have been tested in monotherapy trials. From Table 2, it is obvious that a large portion of designs of combinatory trials integrates metabolic modulation with intervention of NASH plus fibrosis, and consider systemic impacts and metabolic control as a whole in addition to molecular intervention at specific pathways in NASH progression. It is anticipating that encouraging findings from these clinical investigations will be available in the near future.
Future prospective and conclusions
Liver biopsy is still the key standard of NASH diagnosis. FDA and EMA guidance indicates that for clinical approval of new drugs in the treatment of NASH, trials should include patients who have biopsy-proven NASH with stage 2 fibrosis or higher. Pharmacological candidates for NASH are required to demonstrate clinical benefits in improving liver-related outcomes, which may take several years to be proven due to slow progression of the disease. To expedite drug development, liver histological improvements have been accepted as surrogates for clinical outcomes. Despite several drugs are in phase III trials, histological improvement of NASH has not exceeded 50% and NASH resolution rates are remarkably lower in treatment groups. The underlying explanations may be attributed to significant heterogeneity in etiology, variation in pathologic progression, as well as insufficient understanding of this disorder. The dilemma of pathogenic complexity and multiple targets of pharmacologic intervention increase difficulties in achieving impressing efficacy similar to anti-HCV direct active agents for the viral eradication. Moreover, lack of organ or tissue specificity of pharmacotherapeutic agents in development will hardly avoid systemic or organ-specific adverse effects. As a reminder, NAFLD is a liver complication of a variety of metabolic disorders, such as obese, metabolic syndrome, hyperlipidemia or T2DM with significant metabolic abnormalities. Based on this, an international panel proposed a new nomenclature, metabolism-associated fatty liver disease (MAFLD) instead of NAFLD to better reflect its complex pathophysiology and therapeutic emphasis. The new nomenclature helps understand the pathophysiology and enhance etiologic awareness [85]; however, the regulatory authorities still use NAFLD or NASH as an approvable indication in clinical trials, and the change of the nomenclature needs a wide acceptance in authoritative professional societies.
Since pathological evaluation of NASH resolution and fibrotic regression is the fundamental evidence of efficacy assessment, any discrepancy in the use of pathological criteria between individual pathologists results in inter-personal errors in NASH diagnosis in addition to sampling difference in performing liver biopsy. Thus, liver biopsy sections are required to be evaluated by at least two independent pathologists with the same criteria. Inevitably, variations in semi-quantitative scoring between histopathologists may generate the disagreement or misinterpretation in efficacy assessments. For example, MSDC-0602K did not meet the clinical endpoints. In subsequently analyzing the results of the study, researchers organized re-reading of baseline biopsies by the same pathologist using the same digital images, 24.5% of the patients were found to not meet the study’s entry criteria upon re-reading, and 12.6% were deemed to not have NASH [54]. Alternatively, more objective approaches are under development, for example, second harmonic generation/two-photon excitation fluorescence imaging technology as an automated tool was used to assess histological features of NASH [86]. There is an urgent need to develop an artificial intelligent (AI) deep learning system to facilitate the histopathologic assessment to reduce subjectivity, inter-personal discrepancy, and diagnostic errors [87].
Liver biopsy is an invasive procedure, may cause liver bleeding, and possesses risks of fatality. Thus, emerging noninvasive imaging modalities with high sensitivity and specificity have been used for assessment of steatosis and fibrosis, including MRI-PDFF [88] and magnetic resonance elastography [89]. However, inflammation components of NASH are still lacking biomarkers, and new surrogates are emerging, such as ceramides, CK-18, tripalmitin for lipotoxicity; fragments of propeptide of type III procollagen (ProC3), tissue inhibitor of metalloproteinase and procollagen N-terminal peptide for fibrosis; and FGF21 and adiponectin for insulin sensitivity [90, 91]. Although several animal models of NASH are available for preclinical assessment, there are obvious differences in metabolic pathways, pathologic progression and therapeutic efficacy between animals and humans; hence, findings from animal models may not always predict outcome of clinical investigations [92, 93]. It is hoping that humanized liver organoids be popularized to help understand NASH pathogenesis for new drug development [94].
In the present review the efficacy and safety profiles of pharmacotherapeutic candidates currently in phase II and III trials have been summarized for recent advancements in the treatment of NASH. As a chronically progressive disorder, NASH demands a long-term intervention; therefore, the therapeutic benefits and safety profiles are equally critical for any treatment options. As a liver complication of systemic and metabolic disorders, not only does any intervention aim to attain improvement in liver pathology, but also to systemically correct metabolic abnormalities and reduce extrahepatic complications. Hence, to fulfill these objectives, a sequential and combinatory strategy is considered to be in place for evidence-based practice in a precise manner. In waiting for promising pharmacotherapeutics available from on-going clinical trials, keeping healthy lifestyle, and sufficient exercises are still the corner stones for a group of metabolic disorders, including NASH.
References
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84.
Fan JG, Kim SU, Wong VW. New trends on obesity and NAFLD in Asia. J Hepatol. 2017;67:862–73.
Zhou J, Zhou F, Wang W, Zhang XJ, Ji YX, Zhang P, et al. Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology. 2020;71:1851–64.
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015;149:367–78.
Lassailly G, Caiazzo R, Ntandja-Wandji LC, Gnemmi V, Baud G, Verkindt H, et al. Bariatric surgery provides long-term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology 2020;159:1290–301.e5.
Carlsson B, Linden D, Brolen G, Liljeblad M, Bjursell M, Romeo S, et al. Review article: the emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2020;51:1305–20.
Wang XK, Peng ZG. Targeting liver sinusoidal endothelial cells: an attractive therapeutic strategy to control inflammation in nonalcoholic fatty liver disease. Front Pharmacol. 2021;12:655557.
Sun D, Yang X, Wu B, Zhang XJ, Li H, She ZG. Therapeutic potential of G protein-coupled receptors against nonalcoholic steatohepatitis. Hepatology 2021;74:2831–38.
Fianchi F, Liguori A, Gasbarrini A, Grieco A, Miele L. Nonalcoholic fatty liver disease (NAFLD) as model of gut-liver axis interaction: from pathophysiology to potential target of treatment for personalized therapy. Int J Mol Sci. 2021;22:6485.
Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med. 2017;377:2063–72.
Korenblat KM, Fabbrini E, Mohammed BS, Klein S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology. 2008;134:1369–75.
Ferguson D, Finck BN. Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus. Nat Rev Endocrinol. 2021;17:484–95.
Liu XJ, Duan NN, Liu C, Niu C, Liu XP, Wu J. Characterization of a murine nonalcoholic steatohepatitis model induced by high fat high calorie diet plus fructose and glucose in drinking water. Lab Investig. 2018;98:1184–99.
Powell EE, Wong VWS, Rinella M. Non-alcoholic fatty liver disease. Lancet. 2021;397:2212–24.
Liu X-J, Xie L, Du K, Liu C, Zhang N-P, Gu C-J, et al. Succinate-GPR-91 receptor signalling is responsible for nonalcoholic steatohepatitis-associated fibrosis: Effects of DHA supplementation. Liver Int. 2020;40:830–43.
Lang S, Schnabl B. Microbiota and fatty liver disease - the known, the unknown, and the future. Cell Host Microbe. 2020;28:233–44.
Liu C, Wang YL, Yang YY, Zhang NP, Niu C, Shen XZ, et al. Novel approaches to intervene gut microbiota in the treatment of chronic liver diseases. FASEB J. 2021;35:e21871.
Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M, et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw Open. 2019;2:e1912565.
Vuppalanchi R, Noureddin M, Alkhouri N, Sanyal AJ. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2021;18:373–92.
Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149:389–97.
Angulo P, Machado MV, Diehl AM. Fibrosis in nonalcoholic fatty liver disease: mechanisms and clinical implications. Semin Liver Dis. 2015;35:132–45.
Hagström H, Nasr P, Ekstedt M, Hammar U, Stål P, Hultcrantz R, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol. 2017;67:1265–73.
Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology. 2017;65:1557–65.
Simon TG, Roelstraete B, Sharma R, Khalili H, Hagstrom H, Ludvigsson JF. Cancer risk in patients with biopsy-confirmed nonalcoholic fatty liver disease: a population-based cohort study. Hepatology. 2021;74:2410–23.
Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019;394:2184–96.
Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113–24.
Ioannou GN. Epidemiology and risk-stratification of NAFLD-associated HCC. J Hepatol. 2021;75:1476–84.
Stine JG, Wentworth BJ, Zimmet A, Rinella ME, Loomba R, Caldwell SH, et al. Systematic review with meta-analysis: risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment Pharmacol Ther. 2018;48:696–703.
Lassailly G, Caiazzo R, Pattou F, Mathurin P. Perspectives on treatment for nonalcoholic steatohepatitis. Gastroenterology. 2016;150:1835–48.
Ratziu V. A critical review of endpoints for non-cirrhotic NASH therapeutic trials. J Hepatol. 2018;68:353–61.
Rowe IA, Wai-Sun Wong V, Loomba R. Treatment candidacy for pharmacologic therapies for NASH. Clin Gastroenterol Hepatol. 2021. https://doi.org/10.1016/j.cgh.2021.03.005.
Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–22.
Qin W, Ying W, Hamaker B, Zhang G. Slow digestion-oriented dietary strategy to sustain the secretion of GLP-1 for improved glucose homeostasis. Compr Rev Food Sci Food Saf. 2021;20:5173–96.
Liu DH, Pang J, Shao WJ, Gu JQ, Zeng Y, He HH, et al. Hepatic fibroblast growth factor 21 is involved in mediating functions of liraglutide in mice with dietary challenge. Hepatology. 2021;74:2154–69.
Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679–90.
Dichtel LE. The glucagon-like peptide-1 receptor agonist, semaglutide, for the treatment of nonalcoholic steatohepatitis. Hepatology. 2021;74:2290–2.
Harriman G, Greenwood J, Bhat S, Huang X, Wang R. Paul D, et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci USA. 2016;113:E1796–805.
Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology. 2018;155:1463–73.e6.
Leikin-Frenkel A, Gonen A, Shaish A, Goldiner I, Leikin-Gobbi D, Konikoff FM, et al. Fatty acid bile acid conjugate inhibits hepatic stearoyl coenzyme A desaturase and is non-atherogenic. Arch Med Res. 2010;41:397–404.
Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12:2085–91.e1.
Fiorucci S, Biagioli M, Sepe V, Zampella A, Distrutti E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin Investig Drugs. 2020;29:623–32.
Sun L, Pang Y, Wang X, Wu Q, Liu H, Liu B, et al. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharmacol Sin B. 2019;9:702–10.
Clifford BL, Sedgeman LR, Williams KJ, Morand P, Cheng A, Jarrett KE, et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33:1671–84.e4.
Kremoser C. FXR agonists for NASH: how are they different and what difference do they make? J Hepatol. 2021;75:12–5.
Harrison SA, Bashir MR, Lee KJ, Shim-Lopez J, Lee J, Wagner B, et al. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J Hepatol. 2021;75:25–33.
Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020;72:58–71.
Ratziu V, Rinella ME, Neuschwander-Tetri BA, Lawitz E, Denham D, Kayali Z, et al. EDP-305 in patients with NASH: A phase II double-blind placebo-controlled dose-ranging study. J Hepatol. 2021. https://doi.org/10.1016/j.jhep.2021.10.018.
Kong B, Sun R, Huang M, Chow MD, Zhong XB, Xie W, et al. Fibroblast growth factor 15-dependent and bile acid-independent promotion of liver regeneration in mice. Hepatology. 2018;68:1961–76.
Miura S, Mitsuhashi N, Shimizu H, Kimura F, Yoshidome H, Otsuka M, et al. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. 2012;12:56.
Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and safety of Aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology. 2021;160:219–31.e1.
Zhou M, Wang X, Phung V, Lindhout DA, Mondal K, Hsu JY, et al. Separating tumorigenicity from bile acid regulatory activity for endocrine hormone FGF19. Cancer Res. 2014;74:3306–16.
Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2018;391:1174–85.
Colca JR, McDonald WG, Adams WJ. MSDC-0602K, a metabolic modulator directed at the core pathology of non-alcoholic steatohepatitis. Expert Opin Investig Drugs. 2018;27:631–6.
Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J Hepatol. 2020;72:613–26.
Boeckmans J, Natale A, Rombaut M, Buyl K, Rogiers V, De Kock J, et al. Anti-NASH drug development hitches a lift on PPAR agonism. Cells. 2019;9:37.
Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol Ther. 2017;179:142–57.
Boubia B, Poupardin O, Barth M, Binet J, Peralba P, Mounier L, et al. Design, synthesis, and evaluation of a novel series of indole sulfonamide peroxisome proliferator activated receptor (PPAR) alpha/gamma/delta triple activators: discovery of lanifibranor, a new antifibrotic clinical candidate. J Med Chem. 2018;61:2246–65.
Adams LA. End-points for drug treatment in NASH. Hepatol Int. 2019;13:253–8.
Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR-alpha/gamma agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology. 2021;74:1809–24.
Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–59.
Kannt A, Wohlfart P, Madsen AN, Veidal SS, Feigh M, Schmoll D. Activation of thyroid hormone receptor-beta improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br J Pharmacol. 2021;178:2412–23.
Kim D, Kim W, Joo SK, Bae JM, Kim JH, Ahmed A. Subclinical hypothyroidism and low-normal thyroid function are associated with nonalcoholic steatohepatitis and fibrosis. Clin Gastroenterol Hepatol. 2018;16:123–31.e1.
Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394:2012–24.
Younossi ZM, Stepanova M, Taub RA, Barbone JM, Harrison SA. Hepatic fat reduction due to resmetirom in patients with nonalcoholic steatohepatitis is associated with improvement of quality of life. Clin Gastroenterol Hepatol. 2021. https://doi.org/10.1016/j.cgh.2021.07.039.
Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol. 2020;72:816–27.
Frenette C, Kayali Z, Mena E, Mantry PS, Lucas KJ, Neff G, et al. Emricasan to prevent new decompensation in patients with NASH-related decompensated cirrhosis. J Hepatol. 2021;74:274–82.
Friedman S, Sanyal A, Goodman Z, Lefebvre E, Gottwald M, Fischer L, et al. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp Clin Trials. 2016;47:356–65.
Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. 2020;72:892–905.
Rinella ME, Noureddin M. STELLAR 3 and STELLAR 4: lessons from the fall of Icarus. J Hepatol. 2020;73:9–11.
Talukdar S, Kharitonenkov A. FGF19 and FGF21: In NASH we trust. Mol Metab. 2020;46:101152.
Henriksson E, Andersen B. FGF19 and FGF21 for the treatment of NASH-two sides of the same coin? differential and overlapping effects of FGF19 and FGF21 from mice to human. Front Endocrinol (Lausanne). 2020;11:601349.
Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–9.
Harrison SA, Ruane PJ, Freilich BL, Neff G, Patil R, Behling CA, et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat Med. 2021;27:1262–71.
Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor gamma. Proc Natl Acad Sci USA. 2012;109:3143–8.
Wang X, Wei W, Krzeszinski JY, Wang Y, Wan Y. A liver-bone endocrine relay by IGFBP1 promotes osteoclastogenesis and mediates FGF21-induced bone resorption. Cell Metab. 2015;22:811–24.
Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019;392:2705–17.
Sun R, Kong B, Yang N, Cao B, Feng D, Yu X, et al. The hypoglycemic effect of Berberine and Berberrubine involves modulation of intestinal farnesoid X receptor signaling pathway and inhibition of hepatic gluconeogenesis. Drug Metab Dispos. 2021;49:276–86.
Mai W, Xu Y, Xu J, Zhao D, Ye L, Yu G, et al. Berberine inhibits Nod-like receptor family pyrin domain containing 3 inflammasome activation and pyroptosis in nonalcoholic steatohepatitis via the ROS/TXNIP axis. Front Pharmacol. 2020;11:185.
Harrison SA, Gunn N, Neff GW, Kohli A, Liu L, Flyer A, et al. A phase 2, proof of concept, randomised controlled trial of berberine ursodeoxycholate in patients with presumed non-alcoholic steatohepatitis and type 2 diabetes. Nat Commun. 2021;12:5503.
Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038–48.
Loomba R, Noureddin M, Kowdley KV, Kohli A, Sheikh A, Neff G, et al. Combination therapies including Cilofexor and Firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73:625–43.
Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 2017;26:394–406.e6.
Yu XX, Murray SF, Pandey SK, Booten SL, Bao D, Song XZ, et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology. 2005;42:362–71.
Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S, et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat Med. 2021;27:1836–48.
Tilg H, Effenberger M. From NAFLD to MAFLD: when pathophysiology succeeds. Nat Rev Gastroenterol Hepatol. 2020;17:387–8.
Liu F, Goh GB, Tiniakos D, Wee A, Leow WQ, Zhao JM, et al. qFIBS: an automated technique for quantitative evaluation of fibrosis, inflammation, ballooning, and steatosis in patients with nonalcoholic steatohepatitis. Hepatology. 2020;71:1953–66.
Wainberg M, Merico D, Delong A, Frey BJ. Deep learning in biomedicine. Nat Biotechnol. 2018;36:829–38.
Noureddin M, Lam J, Peterson MR, Middleton M, Hamilton G, Le TA, et al. Utility of magnetic resonance imaging versus histology for quantifying changes in liver fat in nonalcoholic fatty liver disease trials. Hepatology. 2013;58:1930–40.
Loomba R, Wolfson T, Ang B, Hooker J, Behling C, Peterson M, et al. Magnetic resonance elastography predicts advanced fibrosis in patients with nonalcoholic fatty liver disease: a prospective study. Hepatology. 2014;60:1920–8.
Marques V, Afonso MB, Bierig N, Duarte-Ramos F, Santos-Laso A, Jimenez-Aguero R, et al. Adiponectin, leptin, and IGF-1 are useful diagnostic and stratification biomarkers of NAFLD. Front Med (Lausanne). 2021;8:683250.
Pirola CJ, Sookoian S. Multiomics biomarkers for the prediction of nonalcoholic fatty liver disease severity. World J Gastroenterol. 2018;24:1601–15.
Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology. 2016;151:513–25.e0.
McLaren DG, Han S, Murphy BA, Wilsie L, Stout SJ, Zhou H, et al. DGAT2 inhibition alters aspects of triglyceride metabolism in rodents but not in non-human primates. Cell Metab. 2018;27:1236–48.e6.
McCarron S, Bathon B, Conlon DM, Abbey D, Rader DJ, Gawronski K, et al. Functional characterization of organoids derived from irreversibly damaged liver of patients with NASH. Hepatology. 2021;74:1825–44.
Acknowledgements
This article is supported by the National Key R&D Program of China (#2016YFE0107400); the National Natural Science Foundation of China (NSFC# 81572356, 81871997, 82170624).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Yang, Yy., Xie, L., Zhang, Np. et al. Updates on novel pharmacotherapeutics for the treatment of nonalcoholic steatohepatitis. Acta Pharmacol Sin 43, 1180–1190 (2022). https://doi.org/10.1038/s41401-022-00860-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00860-3
Keywords
This article is cited by
-
An effective MASH drug is good, but biotech can make it better
Nature Biotechnology (2024)
-
A revisit of drugs and potential therapeutic targets against non-alcoholic fatty liver disease: learning from clinical trials
Journal of Endocrinological Investigation (2023)
-
All about NASH: disease biology, targets, and opportunities on the road to NASH drugs
Acta Pharmacologica Sinica (2022)
-
Data mining of key genes expression in hepatocellular carcinoma: novel potential biomarkers of diagnosis prognosis or progression
Clinical & Experimental Metastasis (2022)
