
Abstract
Acute liver failure (ALF) is a fatal clinical syndrome with no special drug. Recent evidence shows that modulation of macrophage to inhibit inflammation may be a promising strategy for ALF treatment. In this study we investigated the potential therapeutic effects of melittin, a major peptide component of bee venom both in mice model of ALF and in LPS-stimulated macrophages in vitro, and elucidated the underlying mechanisms. ALF was induced in mice by intraperitoneal injection of d-galactosamine/LPS. Then the mice were treated with melittin (2, 4, and 8 mg/kg, ip). We showed that melittin treatment markedly improved mortality, attenuated severe symptoms and signs, and alleviated hepatic inflammation in d-galactosamine/LPS-induced ALF mice with the optimal dose being 4 mg/kg. In addition, melittin within the effective doses did not cause significant in vivo toxicity. In LPS-stimulated RAW264.7 macrophages, melittin (0.7 μM) exerted anti-oxidation and anti-inflammation effects. We showed that LPS stimulation promoted aerobic glycolysis of macrophages through increasing glycolytic rate, upregulated the levels of Warburg effect-related enzymes and metabolites including lactate, LDHA, LDH, and GLUT-1, and activated Akt/mTOR/PKM2/HIF-1α signaling. Melittin treatment suppressed M2 isoform of pyruvate kinase (PKM2), thus disrupted the Warburg effect to alleviate inflammation. Molecular docking analysis confirmed that melittin targeted PKM2. In LPS-stimulated RAW264.7 macrophages, knockdown of PKM2 caused similar anti-inflammation effects as melittin did. In d-galactosamine/LPS-induced ALF mice, melittin treatment markedly decreased the expression levels of PKM2 and HIF-1α in liver. This work demonstrates that melittin inhibits macrophage activation-mediated inflammation via inhibition of aerobic glycolysis by targeting PKM2, which highlights a novel strategy of using melittin for ALF treatment.
Similar content being viewed by others


Introduction
Acute liver failure (ALF) is a fatal clinical syndrome characterized by rapid progression and massive hepatocyte death and has a high mortality rate [1]. ALF can develop over a span of <26 weeks in patients without preexisting liver disease [2]. Although the most viable treatment is emergency liver transplantation, it is critically limited due to organ rejection, a lack of donors and high cost. Unfortunately, to date, there are no drugs available to alter the course of ALF [3]. Only 14% of patients with ALF recover through existing medical therapies [4]. Pharmacologists and clinicians hope to explore novel strategies for treating ALF.
Based on the pathophysiology of ALF, severe dysregulation of the systemic inflammatory response rather than the primary etiology is responsible for most mortality [5]. Systemic inflammation plays a central role in the process of liver injury and determines the clinical course and outcome [6,7,8]. As hepatic macrophages, both Kupffer cells and monocyte-derived macrophages predominantly contribute to the regulation of the hepatic inflammatory response [9, 10]. In the early stages of ALF, there are a large number of hepatic macrophages derived from the proliferation of resident Kupffer cells and the recruitment from the circulating pool of monocytes [7]. This expansion triggers the macrophages toward an ‘M1-like’ activation state (categorized as a proinflammatory phenotype), further causing the propagation and perpetuation of inflammation through the overproduction of proinflammatory cytokines and mediators, which reflect the severity of liver insult, the development of extrahepatic organ dysfunction and the risk of death [11, 12]. Thus, targeting macrophages to treat ALF may be a promising therapeutic strategy [13].
ALF results in severe hypoxia in the liver [14], driving excessive proliferation and inflammation induced by macrophage activation toward aerobic glycolysis (known as the Warburg effect or metabolic reprogramming) [15, 16]. Under hypoxic conditions, pyruvate cannot be oxidized further because of a lack of oxygen. Instead, pyruvate is reduced to form high levels of lactate. To support proliferation and the increased biosynthetic demands, rapidly growing macrophages undergo profound metabolic changes to adapt to these new physiological requirements, exhibiting the features of aerobic glycolysis, which increases both glycolytic rates and lactate release [17]. Consequently, the Warburg metabolism caused by macrophages is implicated in hepatic inflammation. Inhibiting aerobic glycolysis in overactive M1 macrophages has become a therapeutic strategy for inflammatory attenuation and tissue repair [16]. When the oxygen supply is limited, aerobic glycolysis enables the conversion of glucose to pyruvate with the involvement of the M2 isoform of pyruvate kinase (PKM2) [18], a protein kinase that functions in the final and rate-limiting reaction step of the glycolytic pathway [19]. PKM2 is a critical determinant of metabolic reprogramming in macrophages via hypoxia-inducible factor-1α (HIF-1α) [20]. Following macrophage proliferation, PKM2 is highly expressed through activation of the mTOR signaling pathway, further stabilizing HIF-1α and regulating HIF-1α target inflammatory genes, as well as genes encoding the glycolytic machinery [21]. The critical determinant of macrophage activation and inflammatory promotion is PKM2, which is an auspicious target for interfering with aerobic glycolysis in inflammation [22, 23].
Melittin, a major peptide component of bee venom in traditional Chinese medicine, is a water-soluble, linear, cationic, hemolytic, and amphipathic peptide weighing 2840 Da and consisting of 26 amino acids (Fig. 1a) [24]. It is recognized that this natural product has pharmacological properties against a variety of diseases, such as cancer [24], atopic dermatitis [25], and bacterial infection [26]. Although melittin has been found to protect against inflammation and apoptosis in ALF [27], the potential mechanisms remain unclear. Furthermore, there have been no reports on melittin-mediated inhibition of aerobic glycolysis in ALF.

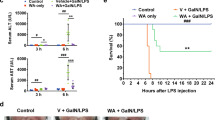
a Amino acid sequence of melittin. b Schematic of melittin treatments in the mouse model of ALF. c After intraperitoneal administration of various doses of melittin, the survival rate was analyzed, n = 8 mice per group. d, e Serum markers, including AST and ALT, were measured. f Macroscopic analysis of the liver; hematoxylin-eosin and TUNEL staining of liver sections. *P < 0.05, **P < 0.01, compared with d-GalN/LPS.
In this study, we showed that melittin treatment increased the survival rate and attenuated inflammation in mice with d-galactosamine/lipopolysaccharide (d-GalN/LPS)-induced ALF. Further experiments revealed that melittin decreased M1 macrophage activation-based inflammation through the inhibition of aerobic glycolysis by targeting PKM2. The present findings indicate that melittin functions as a novel agent for ALF treatment.
Materials and methods
Animals and ALF models
Male C57BL/6 mice (18–22 g, 6–8 weeks old) were purchased from the Laboratory Animal Research Center of Central South University. All mice were allowed to acclimate for at least 1 week under constant temperature (23 ± 2 °C) and humidity (60% ± 5%) with a 12-h light-dark cycle. The mice were supplied with standard laboratory chow and water ad libitum and housed six per cage in plastic cages. ALF model mice were intraperitoneally challenged with 700 mg/kg d-GalN (Sigma-Aldrich, Darmstadt, Germany) and 100 μg/kg LPS (Sigma-Aldrich, Darmstadt, Germany) and then treated with PBS or melittin by intraperitoneal (i.p.) injection. We treated the different groups for 6 h and collected organ tissues and blood samples. We performed dynamic observational studies using additional mice that were treated simultaneously. As shown in Fig. 1b, we measured the survival rates of the mice as the primary outcome. Additionally, the mice were anaesthetized with 0.3% pentobarbital sodium (80 mg/kg) for the collection of organ tissues and blood samples. Protocols for the animal experiments were approved by the Medical Ethics Committee of Xiangya Hospital of Central South University. We performed the animal experiments according to the guidelines for the care and use of laboratory animals established by Central South University.
Hematology and serum analysis
A fully automatic hematology analyzer was used to measure white blood cells (WBCs), red blood cells (RBCs), hemoglobin (HGB), platelets (PLTs), hematocrit (HCT), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), and mean corpuscular hemoglobin concentration (MCHC; Ac. T 5diff AL, Beckman Coulter, CA, USA) in the Clinical Laboratory of Xiangya Hospital, Central South University.
The biochemical markers, including serum alanine transaminase (ALT), aspartate aminotransferase (AST), total bilirubin (TB), urea, albumin (ALB), total protein (TP), total bile acid (TBA), and uric acid (UA), were measured by a fully automatic biochemical analyzer (AU680, Beckman Coulter, CA, USA) in the Clinical Laboratory of Xiangya Hospital, Central South University.
Histology and TUNEL assay
Organ tissues were fixed in 4% paraformaldehyde, paraffin-embedded and cut into 4-μm-thick sections. Hematoxylin and eosin (H&E, Beijing Solarbio Science & Technology Co., Ltd, Beijing, China) or terminal dUTP nick end-labeling (TUNEL) kit (Roche, IN, USA) was used to stain the sections. The levels of tissue damage and immune complexes were assessed using light microscopy.
Cell culture and MTT assay
RAW264.7 murine macrophages were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (HyClone, Utah, USA) and 1% antibiotic-antimycotic solution (NCM Biotech, Suzhou, China) at 37 °C and 5% CO2. The cells were seeded in 96-well plates at a cell density of 5 × 103/well and incubated until the cells reached 70% confluence. Then, 100 ng/ml LPS was added to each well, followed by treatment with melittin (0.35, 0.70, 1.40, and 2.80 μM). After incubation for 24 h, the cell viability was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays according to the manufacturer’s instructions (Beyotime Biotechnology, Shanghai, China). Control or LPS-treated cells received PBS.
Determination of the cellular oxygen consumption rate and extracellular acidification rate
We measured the cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of RAW264.7 macrophages using an XFe96 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA, USA). OCR and ECAR were examined using a Seahorse XF cell mito stress test kit and Seahorse XF glycolysis stress test kit, respectively. The experiments were performed according to the manufacturer’s protocols. Briefly, RAW264.7 cells were seeded into Seahorse XFe96 cell culture microplates at a cell density of 5 × 103/well. There were three basal measurements: for OCR analysis, oligomycin, the reversible oxidative phosphorylation inhibitor FCCP (p-trifluoromethoxy carbonyl cyanide phenylhydrazone), and the mitochondrial complex I inhibitor rotenone plus the mitochondrial complex III inhibitor antimycin A (Rote/AA) were sequentially injected into the well plates at the indicated time points; for ECAR analysis, glucose, the oxidative phosphorylation inhibitor oligomycin, and the glycolytic inhibitor 2-DG were sequentially injected. The data were analyzed by Seahorse XFe Wave software (Agilent Technologies, Santa Clara, USA).
ELISA
The levels of TNF-α and IL-1β in mouse serum and cell supernatants were measured by ELISA kits (Cusabio, Wuhan, China) according to the manufacturer’s instructions. We measured the absorbance values at 450 nm using a microplate reader (MB-530, Heales, Shenzhen, China).
Evaluation of biochemical markers of energy metabolism and oxidative status
The levels of biochemical markers (energy metabolism and oxidative status) in tissue or cell lysates, including lactate, lactate dehydrogenase (LDH), acetyl-CoA, superoxide dismutase (SOD), malondialdehyde (MDA), catalase (CAT), and glutathione (GSH), was measured according to the manufacturer’s recommendations (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Immunofluorescence analysis
For dual immunofluorescence staining, tissue sections or RAW264.7 cells were fixed in 4% paraformaldehyde and then permeabilized with 0.3% Triton X-100. After being blocked with 5% bovine serum albumin (BSA), the tissue sections or cells were immunostained with the following primary antibodies: IL-1β (ab9722, 1:100, Abcam), TNF-α (ab66579, 1:200, Abcam), PKM2 (ab150377, 1:4000, Abcam), and CD68 (ab125212, 1:250, Abcam). After being incubated at 4 °C overnight, the cells were further incubated with the corresponding Alexa Fluor 594- or Alexa Fluor 488-conjugated secondary antibodies (1:1000, Proteintech) for 1 h at room temperature. Finally, the nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma). We acquired images using an immunofluorescence microscope (Axio Imager M2, Zeiss, Germany).
Western blotting
Mouse liver homogenates or RAW264.7 cell lysates were prepared. The protein concentrations were determined by the BCA method. The proteins were combined with SDS loading buffer and separated by 10% SDS polyacrylamide gel electrophoresis (PAGE). After the proteins were transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), the membranes were blocked, incubated overnight at 4 °C with an appropriate primary antibody, and then incubated for 2 h at room temperature with an appropriate secondary antibody. The primary antibodies were as follows: IL-1β (ab9722, 1:100, Abcam), TNF-α (ab66579, 1:500, Abcam), GLUT-1 (ab652, 1:1000, Abcam), LDHA (ab101562, 1:4000, Abcam), PKM2 (ab150377, 1:4000, Abcam), HIF-1α (ab113642, 1:1000, Abcam), p-Akt (#4060, 1:2000, CST), Akt (#9272, 1:1000, CST), p-mTOR (ab109268, 1:4000, Abcam), mTOR (ab32028, 1:2000, Abcam), and β-actin (60008-1-Ig, 1:5000, Proteintech). Protein brands were visualized using ECL Plus reagent (Beyotime, Shanghai, China). Each experiment was replicated at least three times, and the acquired images were quantified by ImageJ software.
RT-PCR
Total RNA from liver tissues or RAW264.7 cells was extracted using TRIzol reagent (Invitrogen, USA). Then, reverse transcription into cDNA was performed using a cDNA reverse transcription kit (CWBIO, Beijing, China). Quantitative RT-PCR with fluorescent SYBR GREEN I (CWBIO, Beijing, China) was performed to determine the gene expression of TNF-α and IL-1β. The relative mRNA expression was evaluated by the 2−ΔΔCt method. β-actin was used as the internal control. The primer sequences are shown in Table 1.
Molecular docking analysis
Molecular docking was used to investigate the binding mode between melittin and the PKM2 protein with the ZDOCK server (http://zdock.umassmed.edu/). The three-dimensional (3D) structures of melittin (PDB ID: 2MLT) and PKM2 (PDB ID: 1T5A) were both downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). For docking analysis, the default parameters were used as described in the ZDOCK server. The top ranked pose as judged by the docking score was subjected to visual analysis using PyMOL 1.7.6 software (http://www.pymol.org/).
Lentivirus packaging and shRNA-mediated PKM2 knockdown
A lentivirus-mediated shRNA against PKM2 was purchased from Cyagen (Guangzhou, China). Mouse PKM2 shRNA #1 (5′-CCCGCAACACTGGCATCATTT-3′), mouse PKM2 shRNA #2 (5′-ATCATTGCCGTGACTCGAAAT-3′), mouse PKM2 shRNA #3 (5′-GACATGGTGTTTGCATCTTTC-3′), and negative control shRNA (5′-CCTAAGGTTAAGTCGCCCTCG-3′) were transfected into RAW264.7 cells using the third generation lentivirus shRNA knockdown vector pLVshRNA-EGFP(2A)Puro (Cyagen, Guangzhou, China). According to the protein expression results, RAW264.7 cells expressing PKM2 shRNA #2 were selected for further studies (Supporting Information Fig. S1).
Statistical analysis
The results are presented as the mean ± standard deviation (SD). Statistical differences between groups were determined by one-way analysis of variance (ANOVA) for multiple comparisons, followed by Tukey’s post hoc test using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). A P value < 0.05 was considered statistically significant.
Results
Melittin prevents d-GalN/LPS-induced ALF in mice
As a classic model that mimics the clinical manifestations of ALF, the well-characterized d-GalN/LPS mouse model was used in the present study (Fig. 1b) [28, 29]. To investigate the effects on mortality, the survival rates were examined. PBS-treated model mice all died within 15 h, whereas melittin-treated (2, 4, and 8 mg/kg) mice had improved survival (Fig. 1c). Among these doses, 4 mg/kg melittin had a stunning impact and prevented the lethal outcome in 75% of GalN/LPS-induced mice (Fig. 1c). To observe the inhibition of liver injury, serum indexes were assessed and histological analysis was performed. Compared with those of the control, d-GalN/LPS markedly increased serum levels of AST (Fig. 1d), ALT (Fig. 1e), and TB (Supporting Information Fig. S2). Melittin (2, 4, and 8 mg/kg) treatment markedly improved hepatic functions by reducing the serum levels of TB, ALT, and AST. Additionally, melittin improved the gross liver appearance following d-GalN/LPS administration with an absence of typical signs of hemorrhage (Fig. 1f). H&E staining showed substantial necrosis, damaged architecture, and massive hemorrhage and inflammatory cell infiltration in the d-GalN/LPS group (Fig. 1f). These histological changes were ameliorated by melittin treatment (Fig. 1f and Supporting Information Fig. S6f), and the score was based on the area of acinar damage, the structural integrity of hepatic lobules, and the red blood cell count according to Liver Pathology by Demos Medical Publishing. Decreased hepatocyte death was observed as determined by reduced numbers of TUNEL-positive cells after melittin treatment (Fig. 1f). Therefore, these results indicated that melittin reduced mortality and attenuated liver injury in GalN/LPS-induced mice. Intriguingly, the optimal dose of melittin to achieve the best effects was 4 mg/kg, which was selected for subsequent experiments.
Melittin reduces hepatic inflammation in vivo
To investigate the anti-inflammatory effects of melittin, we examined the expression of inflammatory indexes. As the proinflammatory response culminates in increased expression of TNF-α and IL-1β [22, 30], we used these two cytokines as inflammatory markers. The inflammatory responses were monitored by fluorescence microscopy and showed that d-GalN/LPS caused excessive expression of TNF-α and IL-1β in liver tissue (Fig. 2a, d and Supporting Information Fig. S6a, b). In contrast, melittin markedly reduced TNF-α and IL-1β production. Additionally, melittin alleviated the expression of these two proinflammatory cytokines at both the transcription and serum levels (Fig. 2b, c, e, f).

a, d Fluorescence microscopy analysis of proinflammatory factors, including TNF-α and IL-1β, in liver tissues. b, e The levels of TNF-α and IL-1β in serum were measured by ELISA. c, f The mRNA levels of TNF-α and IL-1β were analyzed by RT-PCR. n = 6 mice per group. *P < 0.05, **P < 0.01, compared with d-GalN/LPS.
Melittin induces no obvious in vivo toxicity in mice treated with the effective doses
Next, we investigated the in vivo toxicity of melittin within the effective doses (1, 2, and 4 mg/kg). The hematological, blood biochemical, and histological analyses were performed on day 14 after melittin injection. As shown in Fig. 3 and Supporting Information Fig. S2, melittin did not cause meaningful changes in body weight (Fig. 3a). In addition, compared with those of the control group, melittin treatment induced no statistically significant differences in hematological parameters (Fig. 3b–e and Supporting Information Fig. S3a–d), blood biochemical markers (Fig. 3f–i and Supporting Information Fig. S3e–h), gross pathology or histopathology (Fig. 3j and Supporting Information Fig. S3i). These results demonstrate that melittin leads to no toxicity in mice treated with the effective doses. The LD50 of mice that were intraperitoneally injected with melittin was 7.4 mg/kg (PubChem Database. Melittin, CID = 16129627). The hematological and blood biochemical parameters of the high dose (8 mg/kg) group were higher than those of the control group and the effective dose groups (1, 2, and 4 mg/kg), and the histological analyses showed that the hepatic lobules of mice were disorganized (Supporting Information Fig. S11).

The in vivo toxicity was evaluated in healthy C57BL/6 mice. a–e Mean body weights of the model mice treated with melittin. Hematological parameters, including WBCs, RBCs, HGB, and PLTs, were measured. f–i Blood biochemical parameters, including ALT, AST, UREA, and total bilirubin, were measured. j Representative images of hematoxylin-eosin-stained liver, kidney, heart, lung, and cortex tissues under a ×40 microscope. n = 5 mice per group. a–i Group comparisons had no statistical significance.
Melittin exerts antioxidative effects in LPS-induced macrophages
LPS-induced injury has been commonly used as a model to trigger the immune response, liver dysfunction, and liver failure [31]. Thus, we used LPS-induced RAW264.7 macrophages to estimate the in vitro inflammatory response and macrophage activation. To test whether melittin exerted cytotoxicity in RAW264.7 macrophages, we measured cell viability using the MTT assay. The results suggested that at ranges of 0.35–0.70 μM, melittin was not cytotoxic to RAW264.7 macrophages, whereas 1.40 μM melittin started to induce cellular toxicity (Fig. 4a). To ensure that the effects of melittin treatment were not due to its cytotoxicity, a dose of 0.70 μM was selected for subsequent in vitro experiments. Moreover, d-GalN/LPS dramatically reduced SOD, (Supporting Information Fig. S4a), CAT (Supporting Information Fig. S4c) and GSH (Supporting Information Fig. S4d) activities, as well as increased MDA (Supporting Information Fig. S4b). Treatment with melittin reversed these effects, indicating the enhancement of antioxidant capacity.

a Cell viability of RAW264.7 cells treated with melittin was determined by the MTT assay. b, c The oxygen consumption rate and extracellular acidification rate were measured. d–f The expression levels of acetyl-CoA, lactate, and LDH were measured. g Immunoblot analysis of GLUT-1 and LDHA protein levels. h Cytosolic total and phosphorylated Akt and mTOR, as well as PKM2 and HIF-1α expression, were determined by Western blotting, and representative images are shown. i, j Typical fluorescence microscopy images of PKM2, TNF-α, and IL-1β are shown. k Immunoblot analysis of the protein levels of TNF-α and IL-1β were performed. l, m The levels of TNF-α and IL-1β in the cell supernatant were measured by ELISA. n, o The mRNA levels of TNF-α and IL-1β in cell lines were analyzed by RT-PCR. The nuclei were stained with DAPI (blue). The white arrows indicate the nuclear translocation of PKM2. *P < 0.05, **P < 0.01, compared with LPS.
Melittin suppresses the Warburg effect in LPS-activated macrophages
Because of melittin-mediated anti-inflammatory prevention of the Warburg effect in activated macrophages, we investigated the expression profiles of factors involved in aerobic glycolysis and inflammation. Measurements of the OCR and ECAR indicated that LPS decreased the OCR, which is an indicator of mitochondrial respiration, and increased the ECAR, which reflects overall glycolytic flux (Fig. 4b, c and Supporting Information Fig. S8). In contrast, 0.70 μM melittin notably reversed these effects in LPS-induced macrophages, demonstrating that melittin dampened glycolysis following macrophage activation. Additionally, the ELISA and Western blot results showed that LPS led to reduced acetyl-CoA expression and increased levels of LDH, lactate, glucose transporter 1 (GLUT-1), and lactate dehydrogenase A (LDHA) compared with those of the control (Fig. 4d–g and Supporting Information Fig. S7a, b). Melittin treatment rescued these effects. Notably, melittin inhibited aerobic glycolysis in LPS-induced macrophages.
Since the Akt/mTOR pathway mainly contributes to aerobic glycolysis and related inflammation, we next explored the potential mechanisms regulated by melittin. As shown in Fig. 4h and Supporting Information Fig. S7c, d, the expression levels of PKM2 and HIF-1α were markedly increased by activation of the Akt/mTOR signaling pathway after LPS administration. Melittin reduced the enrichment of PKM2 and HIF-1α by attenuating the activation of Akt/mTOR. In particular, the immunofluorescence imaging results suggested that excessive PKM2 was translocated into the nucleus in LPS-stimulated macrophages, and melittin obviously prevented this translocation (Fig. 4i). Further analysis by ELISA, immunofluorescence, cellular RT-PCR, and Western blotting showed that melittin strikingly reduced TNF-α and IL-1β levels (Fig. 4j–o and Supporting Information Fig. S7e, f). These results indicated that melittin suppressed aerobic glycolysis-mediated inflammatory responses.
Melittin disrupts the Warburg effect by targeting PKM2 to alleviate inflammation in LPS-treated macrophages
Given the essential role of PKM2 in the Warburg effect in ALF, we next investigated the effect of melittin-mediated PKM2 inhibition on inflammation. We used molecular docking analysis to explore the binding mode between melittin and PKM2 (Fig. 5a–c and Supporting Information Fig. S5). Figure 5b and Supporting Information Fig. S5b show that melittin binds to gaps in chains B, C, and D of PKM2. As shown in Fig. 5c, the detailed analysis revealed that a hydrophobic interaction was observed between residues Ile-2, Val-5, Leu-6, Val-8, and Leu-9 of melittin and residues C/Ile-40, C/Ala-42, C/Cys-423, C/Cys-424, C/Phe-502, C/Phe-503, and C/Val-508 of PKM2. Additionally, another hydrophobic interaction was observed between the residues Leu-13, Pro-14, Leu-16, Ile-17, Trp-19, and Ile-20 of melittin and residues C/Leu-18, C/Met-22, D/Leu-401, and D/Ile-404 of PKM2. Moreover, residue Trp-19 of melittin formed a cation-π interaction with residue C/Arg-32 of PKM2. In addition, residue Arg-24 of melittin formed electrostatic interactions with residues B/Asp-24 and C/Asp-24 of PKM2. Importantly, four hydrogen bond interactions were shown between Ile-2 of melittin and C/Thr-41 (bond length: 3.3 Å) and C/Gly-501 (bond length: 2.4 Å) of PKM2, Gly-3 of melittin and C/Gly-501 (bond length: 3.5 Å) of PKM2, and Arg-24 of melittin and C/Asp-24 (bond length: 3.0 Å) of PKM2, which describes the main binding affinity between melittin and PKM2. Therefore, the molecular docking information illustrated that PKM2 could be a potential druggable target of melittin.

a Interactions between PKM2 and melittin were examined by MD simulation. PKM2 and melittin adjusted their orientations and approach to each other and formed a complex. b Detailed view of the interactions between PKM2 and melittin. The interface is represented with a solid surface. The melittin is represented with cartoon and stick models and is colored rose red. c Detailed view of the interactions between PKM2 and melittin. PKM2 is represented with sticks colored cyan, green, and blue. Melittin is represented with rose red sticks. The hydrogen bonds are shown as yellow dotted lines. d Immunoblot analysis of TNF-α and IL-1β expression after lentiviral transfection-mediated PKM2 knockdown (PKM2 shRNA) in macrophages. e, f The levels of TNF-α and IL-1β in the cell supernatant after PKM2 shRNA transfection were measured by ELISA. g Protein expression levels of GLUT-1, LDHA, and HIF-1α after PKM2 shRNA transfection were determined by Western blotting, and representative images are shown. h, i Lactate and LDH assays of cells transfected with PKM2 shRNA. *P < 0.05, **P < 0.01 compared with LPS. #P < 0.05, ##P < 0.01 compared with control.
We next transfected RAW264.7 cells with shRNA against PKM2 or the negative control (NC; Fig. 5d–i and Supporting Information Fig. S9d, e). PKM2 knockdown in LPS-mediated macrophages led to reduced levels of lactate and LDH (ELISA tests, Fig. 5h, i), as well as the expression of GLUT-1, LDHA, and HIF-1α (Western blotting, Fig. 5g and Supporting Information Fig. S9a–c), compared with those of the LPS-induced group and NC shRNA-transfected group. In addition, compared with PKM2 silencing alone, cotreatment with melittin and PKM2 shRNA slightly decreased these aerobic glycolysis-related factors in LPS-mediated macrophages, which indicates that PKM2 inhibition by melittin mainly contributes to the disruption of the Warburg effect in LPS-activated macrophages.
Afterwards, we determined whether the suppression of the Warburg effect by melittin targeting of PKM2 induced anti-inflammatory effects. The Western blot and ELISA results indicated that melittin reduced the secretion of TNF-α and IL-1β but no more effective results were observed after cotreatment with melittin and PKM2 shRNA (Fig. 5d–f). Thus, melittin-mediated PKM2 inhibition is mainly responsible for its anti-inflammatory effects.
Melittin represses the Warburg effect-associated signaling cascade to attenuate inflammation in a mouse model
As far as we know, PKM2/HIF-1α activation caused by the Akt/mTOR signaling pathway functions as a pivot in the Warburg effect [32, 33]. We further investigated the effects of melittin on Warburg effect-induced inflammation in mice with d-GalN/LPS-induced ALF (Fig. 6). After the mice underwent d-GalN/LPS administration, the expression of PKM2 and HIF-1α was upregulated in hepatic tissues, followed by activation of the Akt/mTOR signaling pathway (Fig. 6a and Supporting Information Fig. S10a–d). Melittin treatment exerted marked inhibitory effects by blocking the Akt/mTOR/PKM2/HIF-1α signaling cascade (Fig. 6a). Subsequently, we observed marked reductions in TNF-α and IL-1β in the liver tissue compared with those of the model group (Fig. 6b and Supporting Information Fig. S10e, f).

a Expression levels of p-Akt, Akt, p-mTOR, mTOR, PKM2, and HIF-1α in liver tissues were determined by Western blotting. b Immunoblot analysis of TNF-α and IL-1β protein levels was performed. c Working model showing that melittin ameliorates inflammatory responses via inhibition of the PKM2-mediated Warburg effect in a mouse model of ALF.
Discussion
ALF is characterized by profound activation of innate immune responses, with macrophages being the main orchestrator during disease development. Exploring novel therapeutic agents targeting the function of macrophages offers the opportunity to improve hepatic repair in ALF. In this study, melittin prevented liver injury, as shown by decreased mortality, decreased hepatic function, the restoration of tissue architecture, and the attenuation of inflammation. Further experiments on the underlying mechanisms revealed that melittin notably inhibited the PKM2-mediated Warburg effect to alleviate inflammation (Fig. 6c). Thus, the present report highlights a novel use of melittin in ALF treatment.
Melittin is the main bioactive compound in bee venom, accounting for 40%–50% of its total dry weight. Inhibition of inflammation through TNF-α/IFN-γ-stimulated nuclear factor-kappa B (NF-κB) phosphorylation has been described, but other pathways have not been elucidated [34]. Studies have shown that melittin inhibited the PgLPS-induced expression of TLR-4 and proinflammatory cytokines, including IL-1β, IL-6, and IFN-γ, and blocked the NF-κB signaling pathway and the expression of Akt and ERK1/2. Akt and ERK1/2 are involved in cellular proliferation, survival, differentiation, and inflammation. The activation of Akt and ERK1/2 increases the production of proinflammatory factors (such as IL-1β and TNF-α) [35, 36]. A previous study showed a protective effect of melittin on inflammation and apoptosis by inhibiting the transcription factor NF-κB [27]. However, deeply exploring the underlying mechanisms is urgent. In this study, we found that 4 mg/kg melittin optimally improved the survival rate, restored abnormal liver function, ameliorated histological injuries, and attenuated hepatic inflammation in ALF mice. These data support the therapeutic role of melittin in the treatment of ALF. However, its clinical translation of melittin faces several challenges, including nonspecific cytotoxicity and hemolytic activity [37]. Therefore, we conducted histopathological analysis, hemanalysis, and biochemistry to evaluate the side effects and systemic toxicity of different doses of melittin (1, 2, 4, and 8 mg/kg). The assessments revealed that 4 mg/kg melittin induced no obvious damage to normal tissues (heart, liver, lung, kidney, small intestine, and brain). Additionally, there was no loss of body weight in any group during the examination period. Furthermore, we observed no significant differences regarding the values of RBC, HGB, WBC, MCH, MCHC, ALT, AST, and urea nitrogen between the treatment and control groups. These results demonstrate that doses equal to or <4 mg/kg melittin tended to be safe without systemic toxicity. The hematological and blood biochemical parameters of the high dose (8 mg/kg) group were higher than those of the control group and the effective dose groups (1, 2, and 4 mg/kg), and the histological analyses showed damage in the mice, indicating that 8 mg/kg melittin caused significant toxicological damage to the mice. Based on these data, 4 mg/kg melittin was selected for subsequent in vivo and in vitro studies.
Inflammation is recognized as determining the clinical course and outcome of ALF, which is characterized by accumulation and activation of macrophages [6, 7]. During ALF, rapidly proliferating macrophages promote aerobic glycolysis (the Warburg effect), which becomes a distinctive feature of all rapidly proliferating cells, including macrophages [15]. The present work indicates that an in vitro model promotes aerobic glycolysis in macrophages by increasing the glycolytic rate, upregulating Warburg effect-associated enzymes and metabolites, including lactate, LDHA, LDH, and GLUT-1, and lowering acetyl-CoA, which is associated with the tricarboxylic acid cycle. Furthermore, PKM2 and HIF-1α expression via Akt/mTOR highlights the key signaling in the regulation of the Warburg effect. In this study, LPS stimulation not only caused oxidative stress and inflammatory responses but also led to the activation of Akt/mTOR/PKM2/HIF-1α, triggering the nuclear translocation of PKM2 [19]. Interestingly, melittin reduced oxidative stress and suppressed aerobic glycolysis, thus attenuating inflammation of activated macrophages. The underlying molecular mechanism is associated with the inhibition of the Akt/mTOR/PKM2/HIF-1α pathway, as well as the inhibition of PKM2 nuclear translocation. Additionally, the data from the in vivo study also confirmed that melittin alleviated inflammatory responses by inhibiting the Akt/mTOR/PKM2/HIF-1α signaling pathway.
The ability of melittin to alleviate inflammation by regulating aerobic glycolysis in macrophages provides a novel insight into ALF treatment. To develop a clinical treatment, we next focused on potential drug targets for melittin. As far as we know, as the crucial regulator of the Warburg effect, PKM2 represents a potential therapeutic target for inflammatory diseases [38]. PKM2 not only controls aerobic glycolysis but also regulates inflammatory responses in immune cells, including macrophages, which was validated by the results of the PKM2 knockdown experiment. In this study, we showed that PKM2 was a critical determinant of macrophage activation and promotion of inflammation. Molecular docking analysis revealed that melittin could bind to PKM2 through cation-π interactions and hydrogen bond interactions. Furthermore, the binding of melittin to PKM2 exerts inhibitory effects on aerobic glycolysis and downstream inflammatory responses.
In summary, this work demonstrated that melittin treatment reduced mortality, attenuated liver injury, and improved hepatic inflammation in a mouse model of ALF. Moreover, melittin inhibited macrophage activation-mediated inflammation through the inhibition of aerobic glycolysis by targeting PKM2. The present findings highlight a novel strategy for the use of melittin in ALF treatment.
References
Flamm SL, Yang YX, Singh S, Falck-Ytter YT, AGA Institute Clinical Guidelines Committee. American Gastroenterological Association Institute Guidelines for the diagnosis and management of acute liver failure. Gastroenterology. 2017;152:644–7.
Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013;369:2525–34.
Ferriero R, Nusco E, De Cegli R, Carissimo A, Manco G, Brunetti-Pierri N. Pyruvate dehydrogenase complex and lactate dehydrogenase are targets for therapy of acute. J Hepatol. 2018;S0168-8278:30210–1.
O’Grady J. Timing and benefit of liver transplantation in acute liver failure. J Hepatol. 2014;60:663–70.
Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734–9.
Xia XM, Fu JL, Song XF, Shi Q, Su CY, Song EQ, et al. Neohesperidin dihydrochalcone down-regulates MyD88-dependent and -independent signaling by inhibiting endotoxin-induced trafficking of TLR4 to lipid rafts. Free Radic Biol Med. 2015;89:522–32.
Puengel T, Tacke F. Repair macrophages in acute liver failure. Gut. 2018;67:202–3.
Zhou RR, Zhao SS, Zou MX, Zhang P, Zhang BX, Dai XH, et al. HMGB1 cytoplasmic translocation in patients with acute liver failure. BMC Gastroenterol. 2011;11:21.
Ehud Z, Shany SG, Metsada PC, Eli B, Oren S, Zamir H, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. 2014;193:344–53.
You Q, Holt M, Yin H, Li G, Hu CJ, Ju C. Role of hepatic resident and in filtrating macrophages in liver repair after acute injury. Biochem Pharmacol. 2013;86:836–43.
Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol. 2015;62:S112–20.
Possamai LA, Thursz MR, Wendon JA, Antoniades CG. Modulation of monocyte/macrophage function: a therapeutic strategy in the treatment of acute liver failure. J Hepatol. 2014;61:439–45.
Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66:1300–12.
Tak E, Jung DH, Kim SH, Park GC, Jun DY, Lee J, et al. Protective role of hypoxia-inducible factor-1α-dependent CD39 and CD73 in fulminant acute liver failure. Toxicol Appl Pharmacol. 2017;314:72–81.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:513–9.
Meiser J, Krämer L, Sapcariu SC, Battello N, Ghelfi J, D’Herouel AF, et al. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J Biol Chem. 2016;291:3932–46.
Yang LC, Xie M, Yang MH, Yu Y, Zhu S, Hou W, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436.
Zhang ZX, Deng WJ, Kang R, Xie M, Billiar T, Wang HC, et al. Plumbagin protects mice from lethal sepsis by modulating immunometabolism upstream of PKM2. Mol Med. 2016;22:162–72.
Corcoran SE, O’Neill LA. HIF-1α and metabolic reprogramming in inflammation. J Clin Invest. 2016;126:3699–707.
Luo WB, Hu HX, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–44.
Palsson-McDermott EM, O’Neill LA. The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays. 2013;35:965–73.
Liu N, Parry S, Xiao Y, Zhou S, Liu Q. Molecular targets of the Warburg effect and inflammatory cytokines in the pathogenesis of pulmonary artery hypertension. Clin Chim Acta. 2017;466:98–104.
Rady I, Siddiqui IA, Rady M, Mukhtar H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett. 2017;402:16–31.
An HJ, Kim JY, Kim WH, Gwon MG, Gu HM, Jeon MJ, et al. Therapeutic effects of bee venom and its major component, melittin, on atopic dermatitis in vivo and in vitro. Br J Pharmacol. 2018;175:4310–24.
Al-Ani I, Zimmermann S, Reichling J, Wink M. Pharmacological synergism of bee venom and melittin with antibiotics and plant secondary metabolites against multi-drug resistant microbial pathogens. Phytomedicine. 2015;22:245–55.
Park JH, Kim KH, Lee WR, Han SM, Park KK. Protective effect of melittin on inflammation and apoptosis in acute liver failure. Apoptosis. 2012;17:61–9.
Farghali H, Kgalalelo Kemelo M, Wojnarová L, Kutinová Canová N. In vitro and in vivo experimental hepatotoxic models in liver research: applications to the assessment of potential hepatoprotective drugs. Physiol Res. 2016;65:S417–25.
Nakama T, Hirono S, Moriuchi A, Hasuike S, Nagata K, Hori T, et al. Etoposide prevents apoptosis in mouse liver with D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure resulting in reduction of lethality. Hepatology. 2001;33:1441–50.
Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–52.
Reddy DB, Reddanna P. Chebulagic acid (CA) attenuates LPS-induced inflammation by suppressing NF-kappaB and MAPK activation in RAW 264.7 macrophages. Biochem Biophys Res Commun. 2009;381:112–7.
Wang HJ, Hsieh YJ, Cheng WC, Lin CP, Lin YS, Yang SF, et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α-mediated glucose metabolism. Proc Natl Acad Sci U S A. 2014;111:279–84.
Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108:4129–34.
Kim WH, An HJ, Kim JY, Gwon MG, Gu H, Jeon M, et al. Beneficial effects of melittin on ovalbumin-induced atopic dermatitis in mouse. Sci Rep. 2017;7:17679.
Park SY, Park DJ, Kim YH, Kim YH, Choi YW, Lee SJ, et al. Schisandra chinensis alpha-iso-cubebenol induces heme oxygenase-1 expression through PI3K/Akt and Nrf2 signaling and has anti-inflammatory activity in Porphyromonas gingivalis lipopolysaccharide-stimulated macrophages. Int Immunopharmacol. 2011;11:1907–15.
Moon DO, Park SY, Lee KJ, Heo MS, Kim KC, Kim MO, et al. Bee venom and melittin reduce proinflammatory mediators in lipopolysaccharide-stimulated BV2 microglia. Int Immunopharmacol. 2007;7:1092–101.
Jia HR, Zhu YX, Xu KF, Wu FG. Turning toxicants into safe therapeutic drugs: cytolytic peptide-photosensitizer assemblies for optimized in vivo delivery of melittin. Adv Healthc Mater. 2018;7:e1800380.
Xie M, Yu Y, Kang R, Zhu S, Yang LC, Zeng L, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280.
Acknowledgements
This work was supported by the project funded by China Postdoctoral Science Foundation (no’s. 2016M600639 and 2017T100614), the National Natural Science Foundation of China (no’s. 81673719, 81970550, 81700561 and 81873574), and Natural Science Foundation of Hunan Province (no’s. 2019JJ30042 and 11JJ6072).
Author information
Authors and Affiliations
Contributions
NL performed animal experiments and wrote the manuscript; DZ, SYP, and PCZ performed the cell experiments; XWH and TL performed the molecular docking analysis; ZBH and YH wrote part of the manuscript; XGF designed the experiments and edited the manuscript; YW and XGF designed the experiments, analyzed the data, XGF and SYP revised all figures, and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Fan, Xg., Pei, Sy., Zhou, D. et al. Melittin ameliorates inflammation in mouse acute liver failure via inhibition of PKM2-mediated Warburg effect. Acta Pharmacol Sin 42, 1256–1266 (2021). https://doi.org/10.1038/s41401-020-00516-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00516-0
Keywords
This article is cited by
-
Neuroprotective Effects of Melittin Against Cerebral Ischemia and Inflammatory Injury via Upregulation of MCPIP1 to Suppress NF-κB Activation In Vivo and In Vitro
Neurochemical Research (2024)
-
CB2R agonist GW405833 alleviates acute liver failure in mice via inhibiting HIF-1α-mediated reprogramming of glycometabolism and macrophage proliferation
Acta Pharmacologica Sinica (2023)
-
The role of Glut-1 and H+/K+-ATPase expression in hyperplasia of mice laryngeal epithelium induced by pepsin
European Archives of Oto-Rhino-Laryngology (2022)
-
Pyruvate kinase M2 in chronic inflammations: a potpourri of crucial protein–protein interactions
Cell Biology and Toxicology (2021)
