
Abstract
Fulminant hepatitis (FH) is an incurable clinical syndrome where novel therapeutics are warranted. Withaferin A (WA), isolated from herb Withania Somnifera, is a hepatoprotective agent. Whether and how WA improves D-galactosamine (GalN)/lipopolysaccharide (LPS)-induced FH is unknown. This study was to evaluate the hepatoprotective role and mechanism of WA in GalN/LPS-induced FH. To determine the preventive and therapeutic effects of WA, wild-type mice were dosed with WA 0.5 h before or 2 h after GalN treatment, followed by LPS 30 min later, and then killed 6 h after LPS treatment. To explore the mechanism of the protective effect, the macrophage scavenger clodronate, autophagy inhibitor 3-methyladenine, or gene knockout mouse lines NLR family pyrin domain containing 3 (Nlrp3)-null, nuclear factor-erythroid 2-related factor 2 (Nrf2)-null, liver-specific AMP-activated protein kinase (Ampk)a1 knockout (Ampka1ΔHep) and liver-specific inhibitor of KB kinase β (Ikkb) knockout (IkkbΔHep) mice were subjected to GalN/LPS-induced FH. In wild-type mice, WA potently prevented GalN/LPS-induced FH and inhibited hepatic NLRP3 inflammasome activation, and upregulated NRF2 and autophagy signaling. Studies with Nrf2-null, Ampka1ΔHep, and IkkbΔHep mice demonstrated that the hepatoprotective effect was independent of NRF2, hepatic AMPKα1, and IκκB. Similarly, 3-methyladenine cotreatment failed to abolish the hepatoprotective effect of WA. The hepatoprotective effect of WA against GalN/LPS-induced FH was abolished after macrophage depletion, and partially reduced in Nlrp3-null mice. Consistently, WA alleviated LPS-induced inflammation partially dependent on the presence of NLRP3 in primary macrophage in vitro. Notably, WA potently and therapeutically attenuated GalN/LPS-induced hepatotoxicity. In conclusion, WA improves GalN/LPS-induced hepatotoxicity by targeting macrophage partially dependent on NLRP3 antagonism, while largely independent of NRF2 signaling, autophagy induction, and hepatic AMPKα1 and IκκB. These results support the concept of treating FH by pharmacologically targeting macrophage and suggest that WA has the potential to be repurposed for clinically treating FH as an immunoregulator.
Similar content being viewed by others


Introduction
Fulminant hepatitis (FH) is a global life-threatening clinical syndrome with no curable drugs and measures available other than liver transplantation, and thus novel drug discovery is imperative1,2. In the clinic, lipopolysaccharide (LPS) released from intestinal bacterial stimulates macrophage to release tumor necrosis factor (TNF)-α3,4,5. While LPS alone only induces systematic inflammation and LPS pretreatment can even ameliorate hepatotoxicity3,6,7, LPS-induced hepatotoxicity is usually enhanced when livers are pre-exposed to various other hepatic insults that are frequently involved with humans in the clinic, such as viruses, toxins and alcohol1,2,8. These hepatic insults could deplete hepatic glutathione levels9,10 and/or inhibit the protein and RNA transcription, both of which could sensitize hepatocytes to TNF-α-induced apoptosis11,12,13. To mimic the clinical FH, the D-galactosamine (GalN) and LPS-induced acute liver injury model has been established in mice7,14,15. GalN/LPS induces FH by GalN-induced inhibition of protein and RNA synthesis that sensitizes hepatocytes to TNF-α-induced apoptosis16, followed by TNF-α release through LPS-stimulated macrophage14. Therefore, FH progresses through two processes, sensitizing livers by GalN, and LPS activation of macrophage to release TNF-α, which together contribute to extensive TNF-α-induced hepatocyte apoptosis17,18.
Many targets in the liver and macrophage, which could contribute to hepatocellular stress including oxidative stress, inflammation and hepatocyte death, mediate GalN/LPS-induced FH4,19,20,21,22,23,24,25. Nuclear factor-erythroid 2-related factor 2 (NRF2) activation by various drugs protects against hepatic oxidative stress26,27 and the related liver injury28,29,30,31. Proinflammatory cytokines released from macrophage usually induce hepatic inflammation, and TNF-α among these cytokines, could induce liver apoptosis in GalN-pre-sensitized hepatocytes9,10,16. Protective autophagy, which could be regulated by AMP-activated protein kinase (AMPK) activation, protects against GalN/LPS-induced liver injury by alleviating apoptosis22,32. On the other hand, macrophage activation-induced inflammation also mediates the progression of hepatotoxicity33,34, among which NLRP3 inflammasome activation is one most well-documented mechanisms for activating inflammation30,35. Accordingly, strategies were proposed to target macrophage/NLRP3 activation for treating liver disease36,37.
Traditional herbs have been extensively investigated for treating liver diseases both preclinically and clinically37,38,39,40. Withaferin A (WA), a natural steroidal lactone isolated from the traditional herb Withania Somnifera, possesses various pharmacological activities, such as antitumor, anti-inflammation41, and also acts as a leptin sensitizer42. WA potently improves acetaminophen-induced hepatotoxicity43,44 and diet-induced nonalcoholic steatohepatitis45 in rodents. However, whether and how WA improves GalN/LPS-induced FH has not been explored. Mechanically, WA is an NRF2 inducer and WA NRF2-dependently protects against acetaminophen-induced hepatotoxicity44. WA also inhibits inflammation by directly inhibiting IκκB activity46,47 or NLRP3 inflammasome activation in vitro in immune cells48,49, and WA was suggested to AMPKα-dependently protect against cardiovascular disease50. Thus, WA probably protects against FH by targeting the macrophage and/or hepatocyte stress via activating NRF2, AMPKα, autophagy, inhibiting IκκB activity or antagonizing the NLRP3 inflammasome activation. In this study, the effects and mechanisms of WA in GalN/LPS-induced FH were investigated, particularly concentrating on the regulation of NLRP3, NRF2, AMPKα, and autophagy signaling.
Results
WA attenuated GalN/LPS-induced hepatotoxicity and the inflammatory response
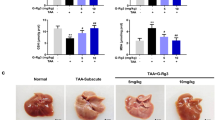
To test the preventive effect of WA (structure shown in Fig. 1a) in GalN/LPS-induced FH, mice were pretreated as schemed (Fig. 1b). Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were slightly increased at 3 h and markedly increased at 6 h, both of which were decreased by WA (Fig. 1c). WA also sharply attenuated the GalN/LPS-induced histological liver damage, consistent with the normalized gross liver appearance (Fig. 1d). Furthermore, WA significantly improved the survival rate of GalN/LPS-treated mice (Fig. 1e). While FH is accompanied by cytokine release and hepatic inflammation, WA significantly reduced the GalN/LPS-induced increase of serum IL-1β, IL-6, and TNF-α (Fig. 2a–c). WA significantly attenuated the GalN/LPS-induced increase of hepatic Il1b and Il6 mRNAs, with a tendency to decrease Tnfa mRNA (Fig. 2d–f). Thus, WA potently protects against GalN/LPS-induced hepatotoxicity and inflammation.

a The chemical structure of WA. b Dosing scheme of animal experiments. c Serum ALT and AST levels at 3 h and 6 h after GalN/LSP treatment. d Representative liver images and liver histological H&E staining, scale bar 50 μm. e Survival curve (n = 10). Data are presented as means ± SD (n = 5 unless otherwise indicated). Control, mice dosed with control vehicle and saline; WA only, mice dosed with WA and saline; V + GalN/LPS, mice dosed with control vehicle (V), and GalN/LPS; WA + GalN/LPS, mice dosed with WA and GalN/LPS. One-way ANOVA was used for statistical analyses. #p < 0.05, ##p < 0.01, and ###p < 0.005 versus Control group; *p < 0.05, **p < 0.01, and ***p < 0.005 versus V + GalN/LPS group.

Serum IL-1β levels (a), Serum IL-6 levels (b), and Serum TNF-α levels (c). mRNA levels of hepatic Il1b (d), Il6 (e), and Tnfa (f). Data are presented as means ± SD (n = 5 unless otherwise indicated). Groups were same as described in Fig. 1 legend. One-way ANOVA was used for statistical analyses. ###p < 0.005 versus Control group; **p < 0.01 and ***p < 0.005 versus V+GalN/LPS group.
WA suppressed hepatic apoptosis in vivo, but not TNF-α-induced hepatocyte death in vitro
Typical apoptosis markers, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, poly-(ADP-ribose) polymerase (PARP1) and caspase 3 (CASP3) cleavage, were next examined to determine whether WA rescued GalN/LPS-induced hepatocyte apoptosis in vivo. WA was found to significantly decrease GalN/LPS-induced positive TUNEL staining (Fig. 3a, b) and attenuate the increase of cleaved CASP3 and cleaved PARP1 (Fig. 3c, d). To test whether WA has a direct anti-apoptosis effect, the ACTD/TNF-α and GalN/TNF-α-induced apoptosis models were employed in primary mouse hepatocytes. As expected, either ACTD or GalN synergized with TNF-α to induce a decrease in cell viability. However, WA pretreatment at 0.01–0.5 μM had no significant effect in reducing ACTD/TNF-α or GalN/TNF-α-induced cell death, while ZVAD efficiently prevented cell death in both models as a positive control (Supplementary Fig. S1). The doses of WA were chosen based on a previous publication43 and no toxicity of WA was found at 0.01–0.5 μM (Supplementary Fig. S1). Thus, WA alleviates GalN/LPS-induced apoptotic hepatocyte death in vivo, but shows no direct effect against TNF-α-induced hepatocyte apoptosis in vitro.

a Representative TUNEL staining of liver sections. b Statistical analysis of TUNEL staining (n = 3). c Western blot analysis of CASP3, cleaved PARP1 and cleaved CASP3 expression. d quantitation analyses of cleaved PARP1, CASP3 and cleaved CASP3 (n = 3). e Hepatic GSH levels. f Hepatic GSSG levels. g Hepatic GSH/GSSG ratio. Data are presented as means ± SD (n = 5 unless otherwise indicated). Groups were the same as described in Fig. 1 legend. One-way ANOVA or unpaired two-tailed t test was used for statistical analyses. #p < 0.05, ##p < 0.01, ###p < 0.005 versus Control group; *p < 0.05, **p < 0.01, ***p < 0.005 versus V + GalN/LPS group.
WA alleviated hepatic oxidative stress and rescued the expression of NRF2 target genes in GalN/LPS-treated mice
The effect of WA on oxidative damage was explored. Hepatic glutathione (GSH) levels were significantly depleted by ~50% 6 h after GalN/LPS administration and were recovered to levels comparable with that of control mice by WA treatment (Fig. 3e). While the hepatic levels of oxidized glutathione (GSSG) remained unchanged between groups (Fig. 3f), the GSH/GSSG ratio was significantly decreased and rescued by WA (Fig. 3g), indicative of normalized oxidative stress. Further analyses for how WA affected the expression of NRF2 target gene mRNAs was next performed. In the absence of GalN/LPS treatment, WA significantly upregulated the mRNA levels of several NRF2 target genes in WT mice, and not in Nrf2−/− mice (Supplementary Fig. S3a), suggesting an NRF2-dependent effect. GalN/LPS was found to induce NRF2 protein levels when compared with the control group, consistent with a previous study showing a similar role for LPS in inducing NRF2 protein expression although decreasing Nrf2 mRNA expression20. After GalN/LPS administration, WA treatment was found to further increase hepatic NRF2 protein (Supplementary Fig. S2a), and to significantly increase the mRNA levels of Nrf2 target genes in both WT mice (Supplementary Fig. S2b) and Nrf2−/− mice (Supplementary Fig. S3b). Given that NRF2 target gene mRNAs were sharply decreased by GalN/LPS treatment (Supplementary Fig. S2b), WA likely upregulates the mRNA levels of NRF2 target genes in GalN/LPS-treated Nrf2−/− mice as a result of its hepatoprotective effect.
NRF2 was not required for the hepatoprotective effect of WA
WA is known to induce NRF2 signaling and NRF2-dependently alleviates acetaminophen-induced liver injury43,44. To further determine if WA protects against FH depending on NRF2, Nrf2−/− mice were employed. The results revealed that WA sharply decreased serum ALT levels in GalN/LPS-treated Nrf2−/− mice (Supplementary Fig. S2c), and this hepatoprotective effect was confirmed by hematoxylin and eosin (H&E) staining (Supplementary Fig. S2d). Analysis of hepatic Nrf2 mRNA levels validated the Nrf2 knockout efficiency of Nrf2−/− mice (Supplementary Fig. S3a). These data demonstrate that the presence of NRF2 does not influence the hepatoprotective effect of WA in treating GalN/LPS-induced liver injury, at least under the present experiment conditions.
WA induced hepatic autophagy in GalN/LPS-treated mice
Previously, another herbal NRF2 activator, licochalcone A, potently alleviated GalN/LPS-induced liver injury in Nrf2−/− mice by inducing autophagy31. Thus, whether WA alleviated the FH via induced autophagy similar to licochalcone A was examined. WA significantly rescued the GalN/LPS-induced decrease of autophagy related 3 (ATG3), microtubule-associated protein light chain 3A/B (LC3II), the ratio of LC3II/LC3I (Fig. 4a, b), indicating that WA upregulated hepatic autophagy signaling in GalN/LPS-treated mice.

a Western blot analysis for ATG3, LC3I, and LC3II (n = 3). b Quantitation analysis of relative protein levels (n = 3). c Experimental time scheme. d Effect of 3-MA in the expression of ATG3, LC3I, and LC3II. e Serum ALT and AST levels in control saline-pretreated mice treated with or without WA. f Serum ALT levels and AST levels in 3-MA-pretreated mice treated with or without WA. g Representative liver histological H&E staining of control saline-pretreated and 3-MA-pretreated mice treated with or without WA, scale bar 50 μm. Data are presented as means ± SD (n = 5 unless otherwise indicated). One-way ANOVA or unpaired two-tailed t test was used for statistical analyses. ##p < 0.01, ###p < 0.005 versus Control; *p < 0.05, **p < 0.01 and ***p < 0.005 versus V + GalN/LPS.
Autophagy, hepatocyte AMPKα, or IκκB were not required for the protective effect of WA
3-MA, as a widely-used autophagy inhibitor51, was then applied to block WA-induced autophagy. Mice were treated as designed based on previous publications (Fig. 4c)20,22,31,52,53. 3-MA at the tested dose, significantly inhibited the expression of ATG3 and decreased the LC3II/LC3I ratio, two typical autophagy markers (Fig. 4d), indicative of the efficient autophagy inhibition by 3-MA. In 3-MA-pretreated mice, WA significantly decreased the GalN/LPS-induced increase of the serum ALT and AST levels to a similar extent with control vehicle-pretreated mice (Fig. 4e, f), consistent with the H&E staining (Fig. 4g). Therefore, 3-MA does not abolish the hepatoprotective effect of WA.
WA was found to protect against myocardial ischemia/reperfusion injury via AMPKα activation50, while hepatic AMPKα1 activation protects against GalN/LPS-induced liver injury22,39. Whether WA protected against GalN/LPS-induced FH by AMPKα activation was investigated. WA showed no significant effect on expression of AMPKα and p-AMPKα proteins (Supplementary Fig. S4a, b). Ampka1ΔHep mice were next employed and analyses of serum ALT levels revealed that the hepatoprotective effect of WA in Ampka1ΔHep mice was as potent as in the Ampka1fl/fl littermates (Supplementary Fig. S4c), supporting an AMPKα1-independent hepatoprotective effect of WA. Western blot analyses confirmed that the AMPKα1 expression was efficiently depleted in livers of Ampka1ΔHep mice (Supplementary Fig. S4d, e). While WA directly targets IκκB to produce its anti-inflammatory effects46,47, WA still showed a potent effect in alleviating GalN/LPS-induced liver injury in IkkbΔHep mice by analyses of serum ALT levels (Supplementary Fig. S4f) and H&E staining (Supplementary Fig. S4g). Thus, the hepatoprotective effect of WA is independent of hepatocyte AMPKα1, hepatocyte IκκB and hepatic autophagy signaling.
Macrophages depletion abrogated the hepatoprotective effect of WA
Given that WA still elicited a potent effect in Nrf2−/− mice, autophagy-inhibited WT mice, and Ampka1ΔHep and IkkbΔHep mice, the hepatoprotective effect of WA was then suspected to not directly target hepatocytes, but target macrophage, another key determinant of hepatotoxicity. To answer this question, clodronate liposomes were used to deplete macrophage, and mice were treated as schemed (Fig. 5a). In control liposome-injected mice, WA alleviated the GalN/LPS-induced increase of serum ALT and AST levels (Fig. 5b), while upon clodronate injection, WA failed to significantly decrease serum ALT and AST levels (Fig. 5c). Liver F4/80 mRNA was markedly decreased in clodronate-treated mice compared with control liposome treatment (Fig. 5d), indicative of efficient macrophage depletion by clodronate. Further analyses of liver histology and hepatic proinflammatory cytokines mRNA expression confirmed that the hepatoprotective effect of WA was largely compromised in clodronate-injected mice compared with that in the control liposome-injected mice (Fig. 5e–g). Thus, WA targets macrophage to protect against GalN/LPS-induced FH.

a Experimental time scheme. b Serum ALT and AST levels in control liposomes-pretreated mice. c Serum ALT and AST levels clodronate liposomes-pretreated mice. d Hepatic F4/80 mRNA levels of control or clodronate alone-treated mice. e Hepatic mRNA levels of proinflammatory cytokines including Tnfa, Il6, Il1b, and Ccl2 in control liposomes-pretreated mice. f Hepatic mRNA levels of inflammation pathway including Tnfa, Il6, Il1b, and Ccl2 in clodronate liposomes-pretreated mice. g Representative images of liver H&E staining, scale bar 50 µm. Data are presented as means ± SD, n = 5. Groups were the same as described in Fig. 1 legend. One-way ANOVA was used for statistical analyses. #p < 0.05, ##p < 0.01, and ###p < 0.005 versus Control group; ***p < 0.005 versus V + GalN/LPS group.
WA prevented GalN/LPS-induced FH partially by inhibiting activation of the NLRP3 inflammasome
NLRP3 inflammasome activation is a well-documented pathway for inflammation induction in macrophage in vitro and in various diseases models in rodents30,35,54,55, while WA directly antagonizes NLRP3 inflammasome activation in vitro48,49. Thus, the question arose whether WA attenuated GalN/LPS-induced FH by inhibiting NLRP3 inflammasome activation. The time course of NLRP3 inflammasome-mediated inflammation signaling activation was examined, and western blot analyses demonstrated that interleukin 1β (IL-1β) was significantly induced as early as 1 h, which peaked 3 h after LPS dosing, and the protein levels of both NLRP3 and ASC were significantly upregulated and peaked 6 h post LPS dosing (Supplementary Fig. S5a, b). The time-course change of NLRP3 inflammasome signaling correlated well with the time-course change of serum ALT and AST levels, both of which were also sharply upregulated 6 h post LPS dosing, while only showing a slight increase 3 h post LPS dosing (Fig. 1c). Given that all tested proteins involved in NLRP3 inflammasome signaling were consistently upregulated 6 h post LPS dosing, how WA affected the hepatic NLRP3 inflammasome signaling activation in GalN/LPS-dosed mice was next investigated. WA significantly attenuated the GalN/LPS-induced increase of the proteins including ASC, cleaved-CASP1 and IL-1β in liver, all of which play a key role in NLRP3 inflammasome activation (Fig. 6a, b). To examine whether the inhibitory effect of WA in hepatic NLRP3 inflammasome activation is a cause or result of its hepatoprotective effect, Nlrp3−/− mice were employed. Analyses of serum ALT and AST levels showed that Nlrp3 deficiency did not abrogate, but significantly reduced the hepatoprotective effect of WA by ~20% percent (Fig. 6c), which were further supported by histology analyses (Fig. 6d). Thus, WA alleviated GalN/LPS-induced FH partially dependent on NLRP3.

a Representative images of western blot analyses for CASP1, ASC and IL-1β in WT livers (n = 3). b Quantitation for CASP1, ASC and IL-1β (n = 3). c Serum ALT and AST levels in GalN/LPS-treated WT or Nlrp3−/− mice treated with or without WA. d Representative hepatic H&E staining of V or WA-treated WT or Nlrp3−/− mice subjected to GalN/LPS challenge, scale bar 50 µm. Effect of WA in mRNA levels of Tnfa (e), Il6 (f), Il1b (g), and Ccl2 (h), in LPS-treated WT and Nlrp3−/− macrophage. One-way ANOVA or two-tailed t test was used for statistical analyses. Data were presented as mean ± SD (n = 5 unless otherwise indicated). For the animal study, #p < 0.05 versus Control; *p < 0.05 or ***p < 0.005 versus V+GalN/LPS. For the macrophage culture study, #p < 0.05, ##p < 0.01, or ###p < 0.005 versus WT Control or Nlrp3−/− Control, respectively; *p < 0.05, **p < 0.01, or ***p < 0.005 versus WT LPS or Nlrp3−/− LPS, respectively. &&&p < 0.005 represents comparison between WT LPS and Nlrp3−/− LPS group.
To test whether the direct anti-inflammatory role of WA depends on NLRP3, primary macrophages were isolated from WT mice and matched Nlrp3−/− mice. The knockout efficacy of Nlrp3 was validated (Supplementary Fig. S5c). In WT macrophage, WA, at 0.2 µM and 0.5 µM, dose-dependently attenuated LPS-induced Tnfa, Il6, Il1b mRNAs (Fig. 5e–g) and Nlrp3 mRNA (Supplementary Fig. S5d). The inhibitory effects of WA in LPS-induced upregulation of Tnfa, Il6, Il1b, were lost in Nlrp3−/− macrophage when WA was used at the lower dose of 0.2 µM, while this effect was still found with WA at the higher dose of 0.5 µM in Nlrp3−/− macrophage (Fig. 6e–g), suggesting that WA has both NLRP3-dependent and NLRP3-independent anti-inflammatory effects. Consistent with the possibility that WA has an NLRP3-independent anti-inflammatory effect, LPS-induced upregulation of chemokine (C-C motif) ligand 2 (Ccl2) mRNA, which does not act directly downstream of NLRP3, was dose-dependently decreased by WA at 0.2 µM and 0.5 µM in both WT macrophage and Nlrp3−/− macrophage (Fig. 6h). Notably, LPS-induced upregulation of Tnfa, Il6, Il1b and Ccl2 mRNAs were all markedly decreased in Nlrp3−/− macrophage compared with WT macrophage (Fig. 6e–h), indicating a key role for NLRP3 in mediating LPS-induced inflammation. WA at 0.2 µM and 0.5 µM was nontoxic to both WT and Nlrp3−/− macrophage (Supplementary Fig. S5e, f). These data suggest that WA produces a strong anti-inflammatory effect in macrophages, which is only partially dependent on NLRP3.
WA therapeutically improved GalN/LPS-induced hepatotoxicity
To further explore the therapeutic potential of WA in GalN/LPS-induced hepatotoxicity, a therapeutic dosing scheme was designed to treat WT mice (Fig. 7a). Notably, WA was found to markedly alleviate the GalN/LPS-induced increase of serum ALT and AST levels as well as histological damage even when WA was dosed 2 h after GalN dosing compared with control vehicle (Fig. 7b, c). These data support the view that WA has a potent therapeutic effect in GalN/LPS-induced liver injury in mice.

a Experimental time scheme. b Serum ALT and AST levels in GalN/LPS-treated mice followed by vehicle (V) or WA treatment. c Representative hepatic H&E staining of V or WA-treated WT mice subjected to GalN/LPS challenge, scale bar 50 µm. d graphic abstract for major findings and proposed mechanism. Unpaired two-tailed Student’s t test was used for statistical analyses. GalN/LPS + V, mice treated with GalN/LPS followed by vehicle treatment; GalN/LPS + WA, mice treated with GalN/LPS followed by WA treatment. Data were presented as mean ± SD (n = 5). **p < 0.01 versus GalN/LPS + V group.
Discussion
FH, also known as acute liver failure, could lead to lethality with liver transplantation as the only curable treatment1,2. Thus, pharmacotherapeutic discoveries are needed to treat this disease. WA was reported as a hepatoprotective agent in acetaminophen-induced hepatotoxicity mainly due to hepatocyte necrosis43,44, and diet-induced nonalcoholic steatohepatitis45 that is characterized by hepatic steatosis, inflammation and fibrosis, while little is known about its role and mechanism in alleviating GalN/LPS-induced FH that is associated with apoptotic hepatocyte death and inflammation.
In the present study, hepatocellular stress alleviation that could be mediated by rescuing expression of hepatic NRF2 target genes, hepatic IκκB inhibition, and protective autophagy induction via AMPKα signaling were first excluded as contributors to the hepatoprotective effects of WA by using Nrf2−/− mice, IkkbΔHep mice, Ampka1ΔHep mice and 3-MA-injected WT mice under the conditions used in this study. The hepatoprotective properties of WA is likely due to effects on macrophage rather than by directly protecting hepatocytes. The protective effects of WA was sharply deminished by macrophage depletion, supporting the view that WA targets the macrophage to treat experimental FH. Further studies demonstrated that the protective effect of WA against the GalN/LPS-induced FH was partially dependent on NLRP3 inflammasome antagonism in vivo and has a direct anti-inflammatory influence partially dependent on the presence of NLRP3 in macrophage in vitro, indicating that other targets, beyond NLRP3, mediate the anti-inflammatory effect of WA. WA inhibits inflammation by direct hyperphosphorylation of IκκB to inhibit the IκκB activity46,47. While the possibility that hepatocyte IκκB plays a role in the hepatoprotective effect of WA on GalN/LPS-induced FH was excluded by using IkkbΔHep mice, it is still possible that WA targets the IκκB present in macrophage to produce its anti-inflammatory effects. Thus, molecular targets present in macrophage other than NLRP3, such as IκκB, that mediate the anti-inflammatory effect of WA deserve further study.
Previously, WA was found to alleviate acetaminophen-induced liver injury dependent on NRF2 induction44. In contrast, the hepatoprotective effect of WA was currently found to be independent of NRF2 in the GalN/LPS model as revealed by using Nrf2−/− mice. This may be due to the different pathological hepatotoxicity mechanisms for these two liver injury models, where acetaminophen mainly induces hepatocyte necrosis56 and GalN/LPS mainly initiates hepatocyte apoptosis15. More importantly, the roles of NRF2 deficiency in these two models are different. Nrf2−/− mice were protected from GalN/LPS-induced FH31, but showed an enhanced hepatotoxicity in acetaminophen-treated mice57. Consistent with these findings, another herbal NRF2 activator, licochalcone A, also showed a potent hepatoprotective effect in GalN/LPS-treated Nrf2−/− mice31. This same study demonstrated that the autophagy inhibitor 3-MA did not abolish the hepatoprotective effect of licochalcone A31. Similarly, the current study found that 3-MA did not abolish the hepatoprotective effect of WA. These results suggest that the NRF2 presence and autophagy induction play only minor roles in mediating the hepatoprotective effects of these herbs at the employed experiment conditions. Thus, WA targets macrophage instead of directly protecting hepatocyte stress pathways to alleviate GalN/LPS-induced FH, at least under the present experiment conditions.
Evidence in the current study reveal that the macrophage-NLRP3 axis at least partially determines the hepatoprotective effect of WA. Indeed, previous studies also support the concept of targeting macrophage to alleviate liver injury23,33,34,36,37,58,59,60 and that pharmacotherapies alleviate GalN/LPS-induced liver injury via inhibiting NLRP3 inflammasome activation30,61. Both macrophage and NLRP3 have pleotropic effects in maintaining systemic homeostasis. For example, macrophage are divided into two polarized types, the M2-like macrophage that are hepatoprotective and the M1-like macrophage that stimulate inflammation62. NLRP3 is physiologically beneficial in maintaining metabolic and immune homeostasis but could be detrimental upon overactivation63,64. In the current study, the hepatoprotective effect of WA on GalN/LPS-induced FH depends on the presence of macrophage and partially dependent on NLRP3, which may be explained by the potent inhibitory effects of WA on the adverse events caused by overactivation of the macrophage-NLRP3 axis in the GalN/LPS model. However, no statistically significant difference in sensitivity to GalN/LPS-induced FH was found between Nlrp3-/- mice and the background strain-matched WT mice, as well as between macrophage-depleted mice and vehicle-treated control mice, which could result from a pooled effect of both the diminished beneficial effects and harmful effects upon loss of macrophage or the Nlrp3 gene, or due to the potent toxic effects of GalN/LPS that overwhelm the potential influence of macrophage and NLRP3 on the disease phenotype. These results suggest that macrophage could have different effects between modulating the onset of the disease and WA suppression of the disease. In line with this point, inflammasome-deficient mice have increased severity to induced nonalcoholic steatohepatitis65, and NLRP3 inflammasome overactivation is required for fibrosis development and thus antagonism of NLRP3 inflammasome activation reduces nonalcoholic fatty liver disease66,67.
In summary, the current study demonstrates a novel role for WA in both preventing and therapeutically alleviating GalN/LPS-induced FH via targeting the macrophage that is partially dependent on NLRP3 (Fig. 7d). WA, although having effects of inducing hepatic NRF2 and autophagy signaling, the dependence of its hepatoprotective effect in these two signaling pathways is minor under the experimental conditions used in this study. These results demonstrate that WA targets macrophages to treat GalN/LPS-induced FH, which further support the possibility of targeting hepatic macrophage to treat liver disease36 and support the potential of WA to be repurposed as a hepatoprotective agent against the clinical FH, particularly featured with TNF-α-related apoptosis.
Methods and Materials
Reagents
WA was purchased from ChromaDex (Irvine, CA, USA). ACTD, GalN, LPS, 3-methyladenine (3-MA), dimethyl sulfoxide, N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone (ZVAD) and other reagents, if not otherwise indicated, were purchased from Sigma Aldrich (St. Louis, MO, USA). Recombinant mouse TNF-α was from Peprotech (Rocky Hill, NJ, USA). Cell-counting kit 8 was from Dojindo Molecular Technologies Inc (Rockville, MD, USA). ALT and AST kits were from Catachem (Oxford, CT, USA). Clodronate liposomes (neutral) were obtained from FormuMax (Sunnyvale, CA, USA). Antibodies against CASP3, cleaved PARP1, LC3I/II, ATG3, ASC, IL-1β, AMPKα, p-AMPKα, NRF2 and β-actin (ACTB) were from Cell Signaling Technology (Danvers, MA, USA). CASP1 antibody was from Santa Cruz Biotechnology (Dallas, Texas, USA). Details for the antibodies are described in Supplementary Table S1.
Animal Experiments
Wild-type (WT) mice, Nrf2−/−, Nlrp3−/−, or Ampka1fl/fl mice on a C57BL/6J background were purchased from the Jackson Laboratory, while IkkbΔHep mice on the C57BL/6J background were described previously68,69. All mice were housed in the National Cancer Institute animal facility that was a pathogen-free environment controlled for temperature, light (25 °C, 12-h light/dark cycle) and humidity (45–65%). Albumin-Cre mice as described previously69 were bred with Ampka1fl/fl mice to generate Ampka1ΔHep mice. Age and body weight-matched 6- to 8-week-old males were randomized into groups (at least N = 5) and treated with 700 mg/kg of GalN and 50 µg/kg of LPS or other reagents as described in detail in the Supplementary materials. The National Cancer Institute Animal Care and Use Committee approved all animal experiments conducted in this study.
Cell culture
Primary hepatocytes, isolated as described previously70, were pretreated with WA at 0, 0.01, 0.1, and 0.5 μM or 10 μM ZVAD for 30 min, followed by 5 mM of GalN or 0.3 μM ACTD for 30 min, and then 25 ng/mL of recombinant mouse TNF-α for additional 13 h. Cell counting kit-8 reagent was used to measure cell viability at 13 h after TNF-α treatment. Dimethyl sulfoxide (0.1%) was used as control vehicle. Primary peritoneal macrophage were isolated as described previously with minor modifications54. The macrophage were pretreated with WA at 0, 0.2, and 0.5 µM for 2 h, followed by 100 ng/mL of LPS treatment for additional 18 h. Other details can be found in Supplementary Materials.
Blinding, randomization, statistical analysis, and reproducibility of experiments
The investigators were blind to the experiment treatments. The experiments were repeated at least twice. The animal experiments were randomized. Statistical analyses were determined by two-tailed unpaired student’s t test between two groups or by one-way ANOVA followed by Dunnett’s multiple comparisons test among multiple groups for independent samples using GraphPad Prism 7.0 (San Diego, CA, USA). To predetermine sample sizes, power analysis was performed using StatMate version 2.0 (GraphPad Software). All data were presented as mean ± SD. P values < 0.05 were considered statistically significant.
References
Bernal, W. & Wendon, J. Acute liver failure. N. Engl. J. Med. 369, 2525–2534 (2013).
William Bernal, G. A., Dhawan, Anil & Wendon, Julia Acute liver failure. Lancet 376, 190–201 (2010).
Opal, S. M. & van der Poll, T. Endothelial barrier dysfunction in septic shock. J. Intern. Med. 277, 277–293 (2015).
Wu, Z., Han, M., Chen, T., Yan, W. & Ning, Q. Acute liver failure: mechanisms of immune-mediated liver injury. Liver Int. 30, 782–794 (2010).
Ding, W. X. & Yin, X. M. Dissection of the multiple mechanisms of TNF-α-induced apoptosis in liver injury. J. Cell Mol. Med. 8, 445–454 (2004).
Zhang, S. N. et al. Pretreatment of lipopolysaccharide (LPS) ameliorates D-GalN/LPS induced acute liver failure through TLR4 signaling pathway. Int. J. Clin. Exp. Pathol. 7, 6626–6634 (2014).
Endo, Y. et al. Enhancement by galactosamine of lipopolysaccharide(LPS)-induced tumour necrosis factor production and lethality: its suppression by LPS pretreatment. Br. J. Pharm. 128, 5–12 (1999).
Schiodt, F. V. et al. Viral hepatitis-related acute liver failure. Am. J. Gastroenterol. 98, 448–453 (2003).
Nagai, H., Matsumaru, K., Feng, G. & Kaplowitz, N. Reduced glutathione depletion causes necrosis and sensitization to tumor necrosis factor-α-induced apoptosis in cultured mouse hepatocytes. Hepatology 36, 55–64 (2002).
Matsumaru, K., Ji, C. & Kaplowitz, N. Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology 37, 1425–1434 (2003).
Su, F. & Schneider, R. J. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor α. Proc. Natl Acad. Sci. USA 94, 8744–8749 (1997).
Janssen, H. L., Higuchi, H., Abdulkarim, A. & Gores, G. J. Hepatitis B virus enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) cytotoxicity by increasing TRAIL-R1/death receptor 4 expression. J. Hepatol. 39, 414–420 (2003).
Kim, W. H. et al. Hepatitis B virus X protein sensitizes primary mouse hepatocytes to ethanol- and TNF-α-induced apoptosis by a caspase-3-dependent mechanism. Cell Mol. Immunol. 2, 40–48 (2005).
Nowak, M. et al. LPS-induced liver injury in D-galactosamine-sensitized mice requires secreted TNF-α and the TNF-p55 receptor. Am. J. Physiol. Regul. Integr. Comp. Physiol. 278, R1202–R1209 (2000).
Leist, M. et al. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am. J. Pathol. 146, 1220–1234 (1995).
Leist, M. et al. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-α requires transcriptional arrest. J. Immunol. 153, 1778–1788 (1994).
Wen, Z. et al. Circulating histones exacerbate inflammation in mice with acute liver failure. J. Cell Biochem. 114, 2384–2391 (2013).
Seki, E. & Brenner, D. A. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48, 322–335 (2008).
Amir, M. et al. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 20, 878–887 (2013).
Jiao, M. et al. Peroxisome proliferator-activated receptor α activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis. 5, e1397 (2014).
Liu, L. M. et al. A role of cell apoptosis in lipopolysaccharide (LPS)-induced nonlethal liver injury in D-galactosamine (D-GalN)-sensitized rats. Dig. Dis. Sci. 53, 1316–1324 (2008).
Liu, Y. M. et al. AMPK activation ameliorates D-GalN/LPS-induced acute liver failure by upregulating Foxo3A to induce autophagy. Exp. Cell Res. 358, 335–342 (2017).
Ilyas, G. et al. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1 β. J. Hepatol. 64, 118–127 (2016).
Ilyas, U., Katare, D. P., Aeri, V. & Naseef, P. P. A review on hepatoprotective and immunomodulatory herbal plants. Pharmacogn. Rev. 10, 66–70 (2016).
Gehrke, N. et al. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J. Hepatol. 68, 986–995 (2018).
Ahmed, S. M., Luo, L., Namani, A., Wang, X. J. & Tang, X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys. Acta Mol. Basis Dis. 1863, 585–597 (2017).
Morito, N. et al. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene 22, 9275–9281 (2003).
Xie, Y. L. et al. Curcumin attenuates lipopolysaccharide/d-galactosamine-induced acute liver injury by activating Nrf2 nuclear translocation and inhibiting NF-kB activation. Biomed. Pharmacother. 91, 70–77 (2017).
Hashem, R. M., Rashd, L. A., Hashem, K. S. & Soliman, H. M. Cerium oxide nanoparticles alleviate oxidative stress and decreases Nrf-2/HO-1 in D-GALN/LPS induced hepatotoxicity. Biomed. Pharmacother. 73, 80–86 (2015).
Liu, X. et al. Biochanin A protects lipopolysaccharide/D-galactosamine-induced acute liver injury in mice by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Int. Immunopharmacol. 38, 324–331 (2016).
Lv, H. et al. Nrf2 signaling and autophagy are complementary in protecting lipopolysaccharide/d-galactosamine-induced acute liver injury by licochalcone A. Cell Death Dis. 10, 313 (2019).
Li, J. et al. AMP-activated protein kinase agonist N6-(3-hydroxyphenyl)adenosine protects against fulminant hepatitis by suppressing inflammation and apoptosis. Cell Death Dis. 9, 37 (2018).
Ito, S. et al. GADD34 suppresses lipopolysaccharide-induced sepsis and tissue injury through the regulation of macrophage activation. Cell Death Dis. 7(2016).
Wang, T. et al. PER1 prevents excessive innate immune response during endotoxin-induced liver injury through regulation of macrophage recruitment in mice. Cell Death Dis. 7(2016).
Seo, M. J., Hong, J. M., Kim, S. J. & Lee, S. M. Genipin protects D-galactosamine and lipopolysaccharide-induced hepatic injury through suppression of the necroptosis-mediated inflammasome signaling. Eur. J. Pharm. 812, 128–137 (2017).
Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 66, 1300–1312 (2017).
Yan, T. et al. Herbal drug discovery for the treatment of nonalcoholic fatty liver disease. Acta Pharm. Sin. B 10, 3–18 (2020).
Sun, R. et al. Orally administered berberine modulates hepatic lipid metabolism by altering microbial bile acid metabolism and the intestinal FXR signaling pathway. Mol. Pharm. 91, 110–122 (2017).
Yan, T. et al. Glycyrrhizin alleviates nonalcoholic steatohepatitis via modulating bile acids and meta-inflammation. Drug Metab. Dispos. 46, 1310–1319 (2018).
Yan, T. et al. Glycyrrhizin protects against acetaminophen-induced acute liver Injury via alleviating tumor necrosis factor α-mediated apoptosis. Drug Metab. Dispos. 44, 720–731 (2016).
Vanden Berghe, W., Sabbe, L., Kaileh, M., Haegeman, G. & Heyninck, K. Molecular insight in the multifunctional activities of Withaferin A. Biochem Pharm. 84, 1282–1291 (2012).
Lee, J. et al. Withaferin A is a leptin sensitizer with strong antidiabetic properties in mice. Nat. Med. 22, 1023–1032 (2016).
Jadeja, R. N., Urrunaga, N. H., Dash, S., Khurana, S. & Saxena, N. K. Withaferin-A reduces acetaminophen-induced liver injury in mice. Biochem. Pharm. 97, 122–132 (2015).
Palliyaguru, D. L. et al. Withaferin A induces Nrf2-dependent protection against liver injury: Role of Keap1-independent mechanisms. Free Radic. Biol. Med. 101, 116–128 (2016).
Patel, D. P. et al. Withaferin A improves nonalcoholic steatohepatitis in mice. J. Pharm. Exp. Ther. 371, 360–374 (2019).
Heyninck, K., Lahtela-Kakkonen, M., Van der Veken, P., Haegeman, G. & Vanden Berghe, W. Withaferin A inhibits NF-kappaB activation by targeting cysteine 179 in IKKβ. Biochem. Pharm. 91, 501–509 (2014).
Kaileh, M. et al. Withaferin a strongly elicits IkappaB kinase β hyperphosphorylation concomitant with potent inhibition of its kinase activity. J. Biol. Chem. 282, 4253–4264 (2007).
Kim, J. E. et al. Withaferin A inhibits helicobacter pylori-induced production of IL-1β in dendritic cells by regulating NF-kappaB and NLRP3 inflammasome activation. Immune Netw. 15, 269–277 (2015).
Dubey, S. et al. Withaferin A associated differential regulation of inflammatory cytokines. Front. Immunol. 9, 195 (2018).
Guo, R. et al. Withaferin A prevents myocardial ischemia/reperfusion injury by upregulating AMP-activated protein kinase-dependent B-cell lymphoma 2 signaling. Circ. J. 83, 1726–1736 (2019).
Hou, H. et al. Inhibitors of phosphatidylinositol 3′-kinases promote mitotic cell death in HeLa cells. PLoS ONE 7, e35665 (2012).
Zhao, J. et al. Celastrol ameliorates experimental colitis in IL-10 deficient mice via the up-regulation of autophagy. Int. Immunopharmacol. 26, 221–228 (2015).
Ren, F. et al. Inhibition of glycogen synthase kinase 3β promotes autophagy to protect mice from acute liver failure mediated by peroxisome proliferator-activated receptor α. Cell Death Dis. 7, e2151 (2016).
Hao, H. et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 25, 856–867 (2017).
Mangan, M. S. J. et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 17, 588–606 (2018).
Boyer, T. D. & Rouff, S. L. Acetaminophen-induced hepatic necrosis and renal failure. JAMA 218, 440–441 (1971).
Enomoto, A. et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 59, 169–177 (2001).
Zimmermann, H. W., Trautwein, C. & Tacke, F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front. Physiol. 3, 56 (2012).
Sica, A., Invernizzi, P. & Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 59, 2034–2042 (2014).
Dong, X., Liu, J., Xu, Y. & Cao, H. Role of macrophages in experimental liver injury and repair in mice. Exp. Ther. Med. 17, 3835–3847 (2019).
Pan, C. W. et al. Mangiferin alleviates lipopolysaccharide and D-galactosamine-induced acute liver injury by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Eur. J. Pharm. 770, 85–91 (2016).
Tacke, F. & Zimmermann, H. W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 60, 1090–1096 (2014).
Coll, R. C., O’Neill, L. & Schroder, K. Questions and controversies in innate immune research: what is the physiological role of NLRP3? Cell Death Discov. 2, 16019 (2016).
Zaki, M. H., Lamkanfi, M. & Kanneganti, T. D. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 32, 171–179 (2011).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Wree, A. et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 92, 1069–1082 (2014).
Mridha, A. R. et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 66, 1037–1046 (2017).
Maeda, S. et al. IKKβ is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFα. Immunity 19, 725–737 (2003).
Kim, I. et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 48, 2664–2672 (2007).
Seglen, P. O. Preparation of isolated rat liver cells. Methods Cell Biol. 13, 29–83 (1976).
Acknowledgements
This project was supported by the National Cancer Institute Intramural Research Program, Center of Cancer Research, National Institutes of Health. We thank Linda G. Byrd for preparation of the animal protocols, John Buckley for animal experiments, Yuhong Luo, Lulu Sun and Tomoki Yagai for expert advice. Y.X. was supported by the Chinese Scholarship Council (201806065070) and P.W. was supported by the Natural Science Foundation of Shanghai (18ZR1436500).
Author information
Authors and Affiliations
Contributions
T.Y, F.J.G. conceived and designed the project; Y.X., P.W., T.Y., N.Y. performed the experiments; Y.X., P.W., T.Y. organized the figures and performed data analysis; T.Y., Y.X. wrote the manuscript; F.J.G., T.Y. supervised this project and edited the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. M. Fimia
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xia, Y., Wang, P., Yan, N. et al. Withaferin A alleviates fulminant hepatitis by targeting macrophage and NLRP3. Cell Death Dis 12, 174 (2021). https://doi.org/10.1038/s41419-020-03243-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03243-w
