
Abstract
Circulating or tissue-related biomarkers are of clinical value for risk stratification in patients with abdominal aortic aneurysms. Relaxin-2 (RL2) has been linked to the presence and size of arterial aneurysms, and to the extent of atherosclerosis in human subjects. Here, we assessed the expression levels of RL2 in aneurysmal (AA, n = 16) and atherosclerotic (ATH, n = 22) arteries, and established the correlation between RL2 levels and the presence/size of AA and the clinical severity of atherosclerosis. The expression levels of metalloproteinases (MMPs) and endothelial nitric oxide synthetase (eNOS) were also detected for correlations with different phenotypes of atherosclerosis and AA. Temporal artery biopsy specimens (n = 6) and abdominal aortic tissues harvested from accident victims during autopsy (n = 10) were used as controls. Quantitative tissue biomarker analysis revealed that tissue-specific RL2 was increased in patients with larger or symptomatic AA compared to subjects with atherosclerotic disease and healthy controls. In situ RL2 levels were proportional to the size and the severity of aneurysmatic disease, and were substantially elevated in patients with symptomatic aneurysm of any diameter or asymptomatic aneurysm of a diameter >350% of that of the normal artery. In contrast, tissue RL2 was inversely associated with the clinical severity of atherosclerotic lesions. Correlation between RL2 and MMP2 was different between ATH1 and ATH2, depending on atherosclerosis grade. Overall, tissue RL2 is differentially associated with discrete phenotypes of arterial disease and might exert multipotent biological effects on vascular wall integrity and remodeling in human subjects.
Similar content being viewed by others

Introduction
Abdominal aortic aneurysms (AAAs) represent the most widely studied lesions among arterial aneurysms (AAs). Although the prevalence and incidence rates of AAAs have decreased during the past two decades [1], early detection is crucial for timely and successful management because the morbidity [2] and mortality [3,4,5,6] are high in patients with a ruptured AAA (rAAA) who manage to reach the hospital alive in order to undergo emergency open or endovascular repair. Biomarkers potentially related to aneurysm formation and expansion have been investigated and reviewed twice [7, 8] in order to augment timely diagnoses, but currently, there is no clinical applicability for any of the studied biomarkers due to either no association or a weak association with the natural history of aneurysms. Our group has recently discovered a positive correlation between the serum levels of the novel biomarker relaxin-2 (RL2) and the presence/size of AAs in human subjects [9]. RL2, a nonglycosylated peptide with a structural and post-translational processing resemblance to insulin and insulin-like growth factors (ILGF), was initially identified as a reproductive hormone implicated in vasoregulation during pregnancy [10]. Ever since the discovery [11] of its receptor, known today as relaxin family peptide receptor-1 (RFXP1), research on RL2 has expanded on various tissues and systems, including cardiovascular tissues such as arteries, veins, and the atrial and ventricular myocardium [12,13,14].
We hypothesized [9] that serum RL2 is related to the presence and size of an AA based on the knowledge that RL2 upregulates the synthesis of matrix metalloproteinase (MMP)-2 and MMP9 [15,16,17], which are MMPs with a stronger association with AAs [18, 19]. Meanwhile, existing knowledge suggests that RL2 upregulates the synthesis of nitric oxide (NO) [12, 20,21,22]. Reduction in endothelium-derived NO is, along with oxidative stress, the common pathway of action of all major risk factors leading to endothelial dysfunction and atherosclerosis [23,24,25,26,27], and our group demonstrated recently [9] that serum RL2 is inversely correlated with the severity of atherosclerotic disease.
In the present study, we aimed to verify and expand the findings of our preliminary study for serum RL2 [9] by investigating the levels of tissue-specific RL2 in aneurysmal (AA) and atherosclerotic (ATH) arteries and establishing a correlation of tissue-specific RL2 levels with the presence/size of AA and the clinical severity of atherosclerosis. Comparisons were made between the AA and ATH artery groups and the control artery group, including temporal artery biopsy specimens (TAB) and abdominal aortic tissues harvested from accident victims during autopsy (AG). Moreover, we carried out tissue-specific measurements of MMP2, MMP9 and endothelial nitric oxide synthetase (eNOS) in the aforementioned groups of arterial specimens to investigate further correlations with different phenotypes of atherosclerosis and AA.
Materials and methods
Study population
In this study, a total of 53 subjects were enrolled. Our interventional groups consisted of 21 patients who had open surgery for different forms of atherosclerotic disease and 16 patients who had open surgery for an AA. These 37 patients underwent surgery at Laiko Hospital in Athens, Greece and along with 6 patients who underwent TAB in the same center were the same subjects originally recruited in our preliminary study [9]. Ten accident victims who underwent a postmortem autopsy (AG) in the Department of Anatomy, Medical School of Athens, Kapodistrian University, were further enrolled to serve as controls. The study was conducted in accordance with the Declaration of Helsinki. Apart from the accident victims, all subjects provided informed consent for the use of their clinical and laboratory results for scientific purposes. Data collection and processing remained anonymous.
Phenotypes per group
Study groups and subcategorization along with baseline characteristics have been previously described [9]. In brief, our study population was allocated to the following groups:
AA group
The AA group included patients (69.73 ± 8.69, mean ± SD; age; male sex) with AAAs (n = 13), thoracic aortic aneurysms (TAA) (n = 1), internal iliac aneurysms (n = 1), or popliteal aneurysms (n = 1). Further subcategorization with respect to the size and clinical presentation of aneurysmatic disease was implemented as described previously [9].
-
(1)
Aneurysm group 1 (AA1) (n = 5; male sex): patients with an asymptomatic aneurysm of a diameter 250%–300% of that of the normal artery.
-
(2)
Aneurysm group 2 (AA2) (n = 4, male sex): patients with an asymptomatic aneurysm of a diameter 300%–350% of that of normal artery.
-
(3)
Aneurysm group 3 (AA3) (n = 7; male sex): patients with either a symptomatic aneurysm of any diameter or an asymptomatic aneurysm of a diameter >350% of that of normal artery.
ATH group
The ATH group included a total of 22 patients (69.52 ± 14.14, mean ± SD; age; male/female sex): 12 patients with extracranial carotid artery disease and 10 patients with peripheral arterial disease (PAD). Subsequently, we further categorized patients according to the clinical severity of underlying atherosclerosis into three groups:
-
(1)
Atherosclerosis group 1 (ATH1, n = 10; male/female sex): patients with moderate functional consequences [asymptomatic internal carotid stenosis >70% or claudication (Rutherford stage 3)].
-
(2)
Atherosclerosis group 2 (ATH2, n = 6; male/female sex): patients with severe functional consequences [transient ischemic attack or rest leg pain (Rutherford stage 4)].
-
(3)
Atherosclerosis group 3 (ATH3, n = 5; male/female sex): patients with permanent disability [stroke or tissue loss (Rutherford stage 5–6)].
Details about diagnostic modalities and diagnostic approaches in classifying the severity of atherosclerosis can be found elsewhere [9].
TAB and AG control groups
The TAB group (n = 6) (69.67 ± 11.31, means ± SD; age; male/female sex) had a normal pathology report of temporal arteritis and had no history or clinical signs of AA, an extracranial carotid artery, or PAD. The AG group (n = 10) (43.5 ± 3.4, means ± SD; age; male/female sex) included accident victims who were autopsied within 24 h postmortem without AA found during autopsy and without a history of atherosclerotic disease. As stated in our previous report [9], the AA, ATH, and TAB study groups were well matched for age, gender, medications, medical comorbidities, smoking status, and renal and liver biochemistry. AG group subjects were substantially younger and did not have any medical comorbidities.
Tissue collection and preservation
Arterial specimens from all subjects were collected with an aseptic technique at room temperature. In the AA group, part of the aneurysmatic sac, consisting of all arterial layers, was collected during open repair of the aneurysm. In the ATH group, only the atherosclerotic plaque was collected during carotid endarterectomy or the lower extremity revascularization procedure. In the TA group, part of the temporal artery, consisting of all arterial layers, was collected during TAB. In the AG group, part of the infrarenal abdominal aorta, consisting of all arterial layers, was collected during autopsy. The samples were flushed with normal saline and immediately stabilized with a commercially available tissue reagent (Allprotect Tissue Reagent, QIAGEN GmbH, D-40724 Hilden, Germany). Stabilized tissues were transported within the time and temperature limits set by the manufacturer and stored at −20 °C until analysis was performed.
Analysis of atherosclerotic and aneurysm tissues
Immunohistochemical staining
Ten serial paraffin sections of 5 μm thickness along the arterial specimens were mounted on polylysine-coated microscope slides and allowed to dry overnight, pending immunohistological staining. Sections were deparaffinized in xylene and a series of graded ethanol and finally stained with the appropriate antibodies (a) rabbit anti-human Relaxin-2 (Meridian, Life Science, Inc., UK) (working concentration: 5 µg/mL), (b) rabbit anti-human MMP2 (MBL, USA) (working concentration: 15 µg/mL), (c) rabbit anti-human MMP9 (Thermo Fisher Scientific, USA) (working concentration: 10 µg/mL), (d) eNOS (Thermo Fisher Scientific, USA) (working concentration: 2 µg/mL). The Zytochem Plus Detection Kit (Germany) was used for the development as described by the manufacturer.
qRT-PCR
Total RNA was isolated from acquired samples using the Tri Reagent (Sigma, Saint Louis, MO, USA), according to the manufacturer’s protocol [28]. cDNA was synthesized by RT (M-MLV, reverse transcriptase; Sigma), and real-time quantitative polymerase chain reaction was performed by using SYBR Green (Invitrogen, Life Technologies, NY, USA), according to the manufacturer’s protocol. The following primers synthesized by Eurofins Genomic (Ebersberg Germany) were used: (a) RL2: Forward: 5′-AGAAATTGTGCCATCCTTCATC-3′, Reverse: 5′-AGGGTTAACTTCAGCTCCTGTG-3′; (b) MMP2: Forward: 5′-ATGACGATGAGCTATGGACCTT-3′, Reverse: 5′- CTGTTGTACTCCTTGCCATTGA-3′; (c) MMP9: Forward: 5′-ACTTTGACAGCGACAAGAAGTG-3′, Reverse: 5′-GGCACTGAGGAATGATCTAAGC-3′; (d) eNOS: Forward: 5′-CATCACCAGGAAGAAGACCTTT-3′, Reverse: 5′-ATACAGGATTGTCGCCTTCACT-3′; (e) β-actin: Forward: 5′-GATCAAGATCATTGCTCCTCCT-3′, Reverse: 5′-ATACTCCTGCTTGCTGATCCAC-3′.
The thermal cycling protocol was as follows: 10 min at 95 °C, followed by 40 cycles of 95 °C for 15 s and then 60 °C for 1 min. Negative PCR controls were run to verify the absence of genomic DNA contamination (no reverse transcription control). Fluorescence was recorded at regular intervals following the 60 °C annealing/extension segment of the PCR, and real-time data showing the relative fluorescence versus cycle number were analyzed. Because of the paucity of good internal PCR controls for tissue specimens, RL2, MMP2, MMP9, and eNOS expression (for consistency of measurement) was determined from a ΔCt value [expression = 2(−ΔΔCt)] where ΔCt was derived for each individual specimen and calculated by subtracting the mean Ct value for all targets measured from the individual Ct value of a given PCR target, as previously described. The results were then reported as the mean ± SEM for each peptide (RL2, MMP2, MMP9, and eNOS) measured in tissue.
Statistical analysis
Data are presented as the mean ± standard deviation (mean ± SD). Statistical significance between groups was calculated using Tukey's multiple comparison test. Correlation between measured parameters was assessed by Pearson analysis. Differences were considered significant when a two-tailed P < 0.05 was calculated. All statistical calculations were performed using GraphPad Prism, version 4.03 (GraphPad, Inc., CA, USA).
Results
Immunohistochemical tissue biomarkers-qualitative analysis
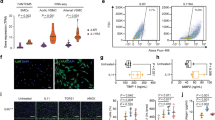
Tissue biomarkers are presented in Fig. 1. Our qualitative analysis showed that MMPs and eNOS were colocalized to residual elastic fiber fragments in aneurysmal tissue and in atherosclerotic plaques. Interestingly, we found that Relaxin-2 lined endothelial cells (ECs) in all groups; the tunica media was composed of alternate layers of elastic lamellae in both AA and ATH. Metalloproteinase staining was also found in endothelial cells of both AA and ATH. We also found thickening of the tunica media marked with disarrangement of smooth muscle cells (in all groups). In both the AA and ATH groups, we observed an accumulation of amorphous material and plasma membrane rupture, while in the TAB group, MMP staining showed a diffusion of the endothelial lining bound by tight junctions. Finally, eNOS staining showed features of endothelial cell vacuolation (all groups) and plasma membrane rupture in the AA group.

Representative immunopositive images from the 3 groups with AA and ATH; TAB showed a brown color for Relaxin 2, MMP2, MMP9 and eNOS. IEL inner elastic lamina, EC endothelial cells, TM tunica media.
Expression tissue mRNA levels of RL2, MMP2, MMP9, and eNOS in all groups
In all three ATH subgroups, the RL2 level was significantly higher than those of MMP2, MMP9, and eNOS (P < 0.05) (Fig. 2). Similarly, RL2 tissue levels were increased in comparison to MMP2, MMP9, and eNOS levels (P < 0.05). In TAB patients, RL2 tissue levels were increased compared to those of MMPs and eNOS (P < 0.05 for all). There was no significant difference between RL2 and MMPs and eNOS in the AG group. All data of the mRNA tissue levels are presented in Supplementary Table 1 for the AA and ATH groups and Supplementary Table 1 for the TAB and AG groups.

The green line indicates a significant difference between subgroups (ATH 1,2,3; AA 1,2,3) and groups (TAB, AG). # indicates a significant difference between TAB or AG and ATH; $ indicates a significant difference between TAB or AG and AA.
Differences in tissue RL2, MMPs, and eNOS mRNA levels across increasing severities of aneurysmatic and atherosclerotic disease
Across increasing severity of the ATH group, MMP2, MMP9, and eNOS expression decreased in patients with a more severe presentation of atherosclerotic disease (P < 0.05) (Fig. 2a). In contrast, within the aneurysmatic disease (subgroup AA1 to AA3), RL2, MMP2, and MMP9 as well as eNOS tissue levels increased (P < 0.05) (Fig. 2b).
Comparison of RL2, MMPs, and eNOS mRNA levels between all study groups
RL2, MMPs, and eNOS mRNA levels were substantially higher in the AA group than in the ATH, TAB, and AG groups (Supplemental graphs a, b, c and d). There were no significant differences between RL2, MMP2, and eNOS levels in the ATH and TAB groups. Only MMP9 was significantly higher in the ATH group than in the TAB group. The AG group had significantly lower levels of RL2, MMPs, and eNOS mRNA than the other groups had.
Comparison of RL2, MMPs, and eNOS mRNA levels between subcategorized study groups
In the ATH subgroups, further statistical analysis (Fig. 2a–d) revealed that RL2, MMPs, and eNOS mRNA levels were significantly higher in the ATH1 and ATH2 subgroups than in the TAB group, with the exception of RL2 mRNA levels in the ATH2 subgroup, which were not significantly different from TAB. mRNA levels were lower or not significantly different in patients with clinically severe atherosclerotic disease (ATH3 subgroup) compared to the TAB group. Finally, the AG group had significantly lower mRNA levels than all subgroups had.
Further statistical analysis revealed that only in arterial specimens from larger or symptomatic aneurysms (AA2 and AA3 groups) were the RL2, MMPs, and eNOS mRNA levels consistently higher than those in the ATH subgroups or in the TAB and AG groups. Specimens from smaller aneurysms (AA1 group) had significantly higher RL2, MMPs, and eNOS mRNA levels only compared to the levels in patients with clinically severe atherosclerotic disease (ATH3 subgroup) and to the levels in the AG group and generally had significantly lower or not significantly different levels compared to those in less clinically advanced atherosclerotic disease (ATH1 and ATH2 subgroups) and in the TAB group.
Correlation between RL2 and MMPs and eNOS in the AA and ATH groups
In patients with mild atherosclerosis (group ATH1), RL2 was positively correlated with eNOS and inversely correlated with MMP2, whereas it was positively associated with MMP2 and eNOS in moderate atherosclerotic disease (ATH2) (Table 1, upper right). In severe atherosclerosis (ATH3), no correlations were found between RL2 and MMP2, MMP9, and eNOS (Table 1, upper right). With respect to AAs, RL2 was positively associated with eNOS and inversely correlated with both MMP2 and MMP9 only in patients with mild dilatation (AA1 group). In contrast, no significant correlations of RL2 were found in patients with moderate and severe arterial aneurysmatic disease (AA2 and AA3) (Table 1, lower left). Furthermore, RL2 was positively correlated with MMP2, MMP9, and eNOS in the TAB group. In the AG group, RL2 did not correlate with either MMPs or eNOS (P > 0.05).
Discussion
The principal finding of this study is that tissue-specific RL2 is increased in patients with a larger or symptomatic AA in comparison to subjects with atherosclerotic disease and to healthy controls. Notably, in situ RL2 levels were proportionally increased according to the size and severity of aneurysmatic disease and were substantially elevated in patients with a symptomatic aneurysm of any diameter or an asymptomatic aneurysm of a diameter >350% of that of the normal artery in comparison to (a) patients with asymptomatic or smaller AAs, (b) subjects with atherosclerosis, and (c) control subjects. On the other hand, an inverse association of tissue RL2 with the clinical severity of atherosclerotic lesions was observed. These findings are generally consistent with our previously published data on serum RL2 [9], and to the best of our knowledge, this is the first report on the correlation of tissue-specific RL2 mRNA levels in human subjects with aneurysms and atherosclerosis. Further to our previous work, our current study indicated that tissue MMPs and eNOS fluctuate with exactly the same pattern as RL2 in various phenotypes of both aneurysmatic and atherosclerotic disease, and importantly, RL2 was not related to MMPs in moderate and severe aneurysmatic and atherosclerotic disease, suggesting an independent role of this multipotent molecule on vascular wall integrity and remodeling.
AAs are characterized by structural alterations of the vascular wall resulting, in part, from the degradation of collagen and elastin fibers. These changes are associated with inflammatory infiltrates [29] and excessive production of MMPs [30, 31], which are assumed to orchestrate the widespread matrix destruction. An association between AA and MMPs has been described in both AAA [32, 33] and PAD [34, 35]. MMP transcripts expressed physiologically at low levels increase rapidly in the presence of inflammation-associated vascular remodeling. Notably, increased expression of MMPs has been observed by other authors in human aneurysm tissue [31] and has been linked to extracellular matrix degradation and increased risk of rupture in response to distending intra-arterial pressure [36, 37]. Previous studies have shown a complementary role of MMP2 and MMP9 in aneurysmal dilatation. In brief, MMP2 primarily acts as a collagenase that initiates cleavage of the collagen triple helix, and subsequently, single chains are subject to degradation by MMP9 [38, 39]. In our study, we found that increased expression of both MMPs is related to a more severe phenotype of AA disease, even though previous studies have reported contradicting conclusions on the clinical utility of circulating plasma [40] and serum [41] levels of MMP2 and MMP9. In addition, we observed an inverse correlation of RL2 with MMPs in the early stages of AA. However, the association of MMPs with RL2 in advanced stages was not noted; this suggests that further mechanisms are involved.
Endothelial cells express eNOS to serve as an important source of NO, a potent vasodilator and inhibitor of inflammation, platelet aggregation, and smooth muscle cell proliferation [42]. Interestingly, eNOS gene polymorphisms have been associated with vascular diseases [43]; two recent meta-analyses have identified the T786C polymorphism of eNOS as a predictor for the development of intracranial aneurysms in the cerebral vascular system [44, 45], and various polymorphisms of eNOS have been linked to AAA [46,47,48]. Our findings of increased eNOS levels in larger aneurysms are in accordance with a recent animal study [49], in which increased eNOS activity reduced smooth muscle α-actin and upregulated MMPs during flow-induced intracranial aneurysm formation. However, experiments in mice have shown that eNOS deficiency results in an increased incidence of AAA formation [40], and Aoki et al. [50] showed that eNOS suppresses cerebral aneurysm formation by reducing hemodynamic stress to arterial walls. Currently, there is no clinical utility of eNOS as a biomarker for predicting the natural history of human aneurysms, which are a multifactorial disease, and the contradicting evidence suggests that eNOS may play a lesser role in aneurysm formation and progression than other factors.
Destabilization of plaques is implicated as a clinical manifestations of atherosclerotic disease [51, 52]. Among various proteinases, MMP2 and MMP9 have been shown to be the predominant ones secreted by T lymphocytes and macrophages [53, 54] across atherosclerotic plaque development [55]. MMP2 is constitutively expressed in all human vascular cells [56], but plaques express increased MMP2 compared to that in normal vessels, especially unstable ones [57]. Overall, MMP2 levels are increased in patients with atherosclerotic [58, 59] PAD, especially in combination with type II diabetes [60] or acute coronary syndromes [61, 62]. Additionally, circulating MMP9 levels are increased in patients with atherosclerosis [62], acute coronary syndromes [62, 63], and type II diabetes [64]. There is also substantial evidence for a link between heightened MMP9 and plaque vulnerability [65, 66] through facilitation of basement membrane breakdown, smooth muscle cell migration, plaque neovascularization, and inflammatory cell infiltration [67, 68].
Indeed, in our current study, increased levels of MMP2 and MMP9 were related to the severity of atherosclerotic disease, while the RL2 level was inversely associated with atherosclerotic burden. Our group has recently shown serum RL2 to be increased in the early clinical stages of atherosclerosis and decreased in more advanced stages of atherosclerotic disease [9], and it was hypothesized that RL2 upregulation represents a form of protective mechanism; however, it was unclear whether this early increase was actually beneficial. The inverse correlation of RL2 with MMP2 in mild atherosclerosis and the positive association with eNOS in moderate atherosclerosis is suggestive of a beneficial effect. Enhanced NO bioavailability through eNOS upregulation and decreased expression of H2S generating enzymes might increase plaque stability and integrity [69, 70]. It remains unclear why this protective mechanism is ameliorated in advanced stages of atherosclerosis, but our findings provide further evidence of potential therapeutic use of RL2 in atherosclerotic disease, as other investigators have recently demonstrated [71].
There is a good size of evidence that RL2 increases NO synthesis in both an acute and a delayed fashion, although the actual mechanism and whether this is related directly or indirectly to RFXP1 is not clear [72, 73]. The activity and expression of not only eNOS [74,75,76,77] but also inducible-NOS (iNOS) [74, 76, 78,79,80,81,82,83,84] and neuronal-NOS (nNOS) is increased by RL2. Due to the production of NO and activation of guanyl cyclase, NOS activation by RL2 leads to an increase in cyclic guanyl monophosphate (cGMP) [78, 79, 83, 85]. Based on activation of NOS and cGMP, NO production by RL2 can be further enhanced by an increase in eNOS activity by Akt (Protein Kinase B) phosphorylation [86], upregulation of iNOS activity following stimulation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κΒ)-controlled transcription [79, 81], and finally by increased activity of eNOS, following activation of the endothelin B (ETB) receptor by increased activity of MMP2 and MMP9 by RL2 [15]. Interestingly, inhibition of NF-κΒ-mediated transcription reduces the RL2-related increase in MMP9 expression and activity [86], suggesting a relationship between the effects of RL2 upon MMPs and NO [12]. Furthermore, RL2 can decrease the expression of tissue inhibitors of MMPs (TIMPs) [87,88,89], and recently, it was shown that the known pathways of RL2/RFXP1 signaling [including activation of phosphoinositide 3-kinase (PI3K), Akt, protein kinase C (PKC)-ζ, and extracellular signal-regulated protein kinases 1 and 2 (ERK1/2)] are connected with the increased expression of mRNA for MMP9 [4]. These insights could be useful for investigating the utility of RL2 in the clinical setting [12].
Certain limitations should be acknowledged in our study. We enrolled four relatively small groups of patients, which might have hampered the identification of associations. Furthermore, our study is limited by the fact that the AG group was not matched with other groups for age and comorbidities, while in the ATH group, subjects presented with heterogeneous arterial pathologies. These limitations warrant caution when interpreting the results and drawing conclusions. In our study, we included data from autopsy subjects, as Gupta et al. [90] have shown that under conditions similar to ours, autopsy specimens are reliable for conduction molecular estimations. Importantly, no definite conclusions can be made regarding the underlying mechanisms of RL2 in vascular diseases. Contemporary literature does not provide mechanistic data on possible RL2 effects on AAs. Consequently, the potential applicability of RL2 as a novel therapeutic target merits further investigation. A postulation may be advanced that RL2 represents a feedback protective mechanism to counteract an ongoing detrimental process.
In conclusion, tissue-specific RL2 is higher in patients with an AA, showing a positive relationship with the size of the aneurysmatic dilatation. Levels of RL2 are also inversely correlated with the severity of atherosclerotic disease. Future studies with larger cohorts are warranted to verify and expand our results with the ultimate aim to reveal possible etiopathogenetic backgrounds of atherosclerosis and aneurysm formation.
References
Sampson UK, Norman PE, Fowkes FG, Aboyans V, Song Y, Harrell FE Jr, et al. Estimation of global and regional incidence and prevalence of abdominal aortic aneurysms 1990 to 2010. Glob Heart. 2014;9:159–60.
Siracuse JJ, Krafcik BM, Farber A, Kalish JA, McChesney A, Rybin D, et al. Contemporary open repair of ruptured abdominal aortic aneurysms. J Vasc Surg. 2017;65:1023–8.
Desgranges P, Kobeiter H, Katsahian S, Bouffi M, Gouny P, Favre JP, et al. A French randomized controlled trial of endovascular versus open surgical repair of ruptured aorto-iliac aneurysms. Eur J Vasc Endovasc Surg. 2015;50:303–10.
Hinchliffe RJ, Bruijstens L, MacSweeney ST, Braithwaite BD. A randomised trial of endovascular and open surgery for ruptured abdominal aortic aneurysm-results of a pilot study and lessons learned for future studies. Eur J Vasc Endovasc Surg. 2006;32:506–13.
Powell JT, Sweeting MJ, Thompson MM, Ashleigh R, Bell R, et al. Endovascular or open repair strategy for ruptured abdominal aortic aneurysm: 30 day outcomes from IMPROVE randomized trial. BMJ. 2014;348:7661.
Reimerink JJ, Hoornweg LL, Vahl AC, Wisselink W, van den Broek TA, Legemate DA, et al., Amsterdam Acute Aneurysm Trial Collaborators. Endovascular repair versus open repair of ruptured abdominal aortic aneurysms: a multicenter randomized controlled trial. Ann Surg. 2013;258:248–56.
Moris D, Mantonakis E, Avgerinos E, Makris M, Bakoyiannis C, Pikoulis E, et al. Novel biomarkers of abdominal aortic aneurysm disease: identifying gaps and dispelling misperceptions. Biomed Res Int. 2014;2014:925840.
Urbonavicius S, Urbonaviciene G, Honore B, Henneberg EW, Vorum H, Lindholt JS. Potential circulating biomarkers for abdominal aortic aneurysm expansion and rupture-a systematic review. Eur J Vasc Endovasc Surg. 2008;36:273–80.
Papoutsis K, Kapelouzou A, Tsilimigras DI, Patelis N, Kouvelos G, Schizas D, et al. Associations between serum relaxin 2, aneurysm formation/size and severity of atherosclerosis: a preliminary prospective analysis. Acta Pharmacol Sin. 2018;39:1243–8.
Bani D. Relaxin: a pleiotropic hormone. Gen Pharmacol. 1997;28:13–22.
Hsu SY, Nakabayashi K, Nishi S, Kumagai J, Kudo M, Sherwood OD, et al. Activation of orphan receptors by the hormone relaxin. Science. 2002;295:671–4.
Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93:405–80.
Novak J, Parry LJ, Matthews JE, Kerchner LJ, Indovina K, Hanley-Yanez K, et al. Evidence for local relaxin ligand-receptor expression and function in arteries. FASEB J. 2006;20:2352–62.
Jelinic M, Leo CH, Post Uiterweer ED, Sandow SL, Gooi JH, Wlodek ME, et al. Localization of relaxin receptors in arteries and veins, and region-specific increases in compliance and bradykinin-mediated relaxation after in vivo serelaxin treatment. FASEB J. 2014;28:275–87.
Jeyabalan A, Novak J, Doty KD, Matthews J, Fisher MC, Kerchner LJ, et al. Vascular matrix metalloproteinase-9 mediates the inhibition of myogenic reactivity in small arteries isolated from rats after short term administration of relaxin. Endocrinology. 2007;148:189–97.
Kapila S, Wang W, Uston K. Matrix metalloproteinase induction by relaxin causes cartilage matrix degradation in target synovial joints. Ann NY Acad Sci. 2009;1160:322–8.
Maruo N, Nakabayashi K, Wakahashi S, Yata A, Maruo T. Effects of recombinant H2 relaxin on the expression of matrix metalloproteinases and tissue inhibitor metalloproteinase in cultured early placental extravillous trophoblasts. Endocrine. 2007;32:303–10.
McMillan WD, Tamarina NA, Cipollone M, Johnson DA, Parker MA, Pearce WH. Size matters: the relationship between MMP-9 expression and aortic diameter. Circulation. 1997;96:2228–82.
Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–32.
Feijóo-Bandín S, Aragón-Herrera A, Rodríguez-Penas D, Portolés M, Roselló-Lletí E, Rivera M, et al. Relaxin-2 in cardiometabolic diseases: mechanisms of action and future perspectives. Front Physiol. 2017;8:599. https://doi.org/10.3389/fphys.2017.00599.
Wilkinson TN, Speed TP, Tregear GW, Bathgate RA. Evolution of the relaxin-like peptide family: from neuropeptide to reproduction. Ann NY Acad Sci. 2005;1041:530–3.
Siddle K. Signalling by insulin and IGF receptors: supporting acts and new players. J Mol Endocrinol. 2011;47:R1–10.
Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation: contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–23.
Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91:2546–51.
Tesfamariam B, Cohen RA. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol. 1992;263:H321.
Tsao PS, Buitrago R, Chang H. Effects of diabetes on monocyte-endothelial interactions and endothelial superoxide production in fructose-induced insulin-resistant and hypertensive rats. Circulation. 1995;92:A2666.
Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032–7.
Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9.
Bobryshev YV, Lord RS, Parsson H. Immunophenotypic analysis of the aortic aneurysm wall suggests that vascular dendritic cells are involved in immune responses. Cardiovasc Surg. 1998;6:240–9.
Curci JA, Liao S, Huffman MD, Shapiro SD, Thompson RW. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. J Clin Invest. 1998;102:1900–10.
Davis V, Persidskaia R, Baca-Regen L, Itoh Y, Nagase H, Persidsky Y, et al. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 1998;18:1625–33.
Van Laake LW, Vainas T, Dammers R, Kitslaar PJ, Hoeks AP, Schurink GW. Systemic dilation diathesis in patients with abdominal aortic aneurysms: a role for matrix metalloproteinase-9? Eur J Vasc Endovasc Surg. 2005;29:371–7.
Mao D, VanVickle SJ, Curci JA, Thompson RW. Expression of matrix metalloproteinases and TIMPs in human abdominal aortic aneurysms. Ann Vasc Surg. 1999;13:236–7.
Zhou W, Chai H, Ding R, Lam HY. Distribution of inflammatory mediators in carotid and femoral plaques. J Am Coll Surg. 2010;211:92–8.
Abdul-Hussien H, Hanemaaijer R, KleemannR, Verhaaren BF, van Bockel JH, Lindeman JH. The pathophysiology of abdominal aortic aneurysm growth: corresponding and discordant inflammatory and proteolytic processes in abdominal aortic and popliteal artery aneurysms. J Vasc Surg. 2010;51:1479–87.
Nosoudi N, Nahar-Gohad P, Sinha A, Chowdhury A, Gerard P, Carsten CG, et al. Prevention of abdominal aortic aneurysm progression by targeted inhibition of matrix metalloproteinase activity with batimastat-loaded nanoparticles. Circ Res. 2015;117:80–9.
Petersen E, Wagberg F, Angquist KA. Proteolysis of the abdominal aortic aneurysm wall and the association with rupture. Eur J Vasc Endovasc Surg. 2002;23:153–7.
Aimes R, Quigley J. Matrix metalloproteinase-2 is an interstitial collagenase. J Biol Chem. 1995;270:5872–6.
Wielockx B, Lannoy K, Shapiro SD, Itoh T, Itohara S, Vandekerckhove J, et al. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat Med. 2001;7:1202–8.
Lindholt JS, Vammen S, Fasting H, Henneberg EW, Heickendorff L. The plasma level of matrix metalloproteinase 9 may predict the natural history of small abdominal aortic aneurysms. A preliminary study. Eur J Vasc Endovasc Surg. 2000;20:281–5.
Eugster T, Huber A, Obeid T, Schwegler I, Gurke L, Stierli P. Aminoterminal propeptide of type III procollagen and matrix metalloproteinases-2 and -9 failed to serve as serum markers for abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2005;29:378–82.
Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–54.
Wang XL, Wang J. Endothelial nitric oxide synthase gene sequence variations and vascular disease. Mol Genet Metab. 2000;70:241–51.
Paschoal EHA, Yamaki VN, Teixeira RKC, Paschoal Junior FM, Jong-A-Liem GS, et al. Relationship between endothelial nitric oxide synthase (eNOS) and natural history of intracranial aneurysms: meta-analysis. Neurosurg Rev. 2018;41:87–94.
Yang C, Qi ZY, Shao C, Xing WK, Wang Z. Association between three eNOS polymorphisms and intracranial aneurysms risk: a meta-analysis. Medicine (Baltimore). 2015;94:e452. https://doi.org/10.1097/MD.0000000000000452.
Kotani K, Shimomura T, Murakami F, Ikawa S, Kanaoka Y, Ohgi S, et al. Allele frequency of human endothelial nitric oxide synthase gene polymorphism in abdominal aortic aneurysm. Intern Med. 2000;39:537–9.
Fatini C, Sofi F, Sticchi E, Bolli P, Sestini I, Falciani M, et al. eNOS G894T polymorphism as a mild predisposing factor for abdominal aortic aneurysm. J Vasc Surg. 2005;42:415–9.
Atli FH, Manduz S, Katrancioglu N, Ozum U, Disli OM, Atahan E, et al. eNOS G894T polymorphism and abdominal aortic aneurysms. Angiology. 2010;61:125–30.
Liaw N, Dolan Fox JM, Siddiqui AH, Meng H, Kolega J. Endothelial nitric oxide synthase and superoxide mediate hemodynamic initiation of intracranial aneurysms. PLoS One. 2014;9:101721.
Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Miyamoto S. Complementary inhibition of cerebral aneurysm formation by eNOS and nNOS. Lab Invest. 2011;91:619–26.
Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–71.
Burke AP, Farb A, Malcom GT, Liang LH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336:1276–82.
Oviedo-Orta E, Bermudez-Fajardo A, Karanam S, Benbow U, Newby AC. Comparison of MMP-2 and MMP-9 secretion from T helper 0, 1 and 2 lymphocytes alone and in coculture with macrophages. Immunology. 2008;124:42–50.
Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483–93.
Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19.
Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–62.
Sluijter JP, Pulskens WP, Schoneveld AH, Velema E, Strijder CF, Moll F, et al. Matrix metalloproteinase 2 is associated with stable and matrix metalloproteinases 8 and 9 with vulnerable carotid atherosclerotic lesions: a study in human endarterectomy specimen pointing to a role for different extracellular matrix metalloproteinase inducer glycosylation forms. Stroke. 2006;37:235–9.
Muller A, Kramer SD, Meletta R, Beck K, Selivanova SV, Rancic Z, et al. Gene expression levels of matrix metalloproteinases in human atherosclerotic plaques and evaluation of radiolabeled inhibitors as imaging agents for plaque vulnerability. Nucl Med Biol. 2014;41:562–9.
Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–503.
Signorelli SS, Malaponte G, Libra M, Di Pino L, Celotta G, Bevelacqua V, et al. Plasma levels and zymographic activities of matrix metalloproteinases 2 and 9 in type II diabetics with peripheral arterial disease. Vasc Med. 2005;10:1–6.
Ghaderian SM, Akbarzadeh Najar R, Tabatabaei Panah AS. Genetic polymorphisms and plasma levels of matrix metalloproteinases and their relationships with developing acute myocardial infarction. Coron Artery Dis. 2010;21:330–5.
Ghaderian SM, Najar RA, Tabatabaei Panah AS, Rezaie G, Rezaei Farimani A, Beigi Harchegani A, et al. Matrix metalloproteinase: investigation from gene to protein as effective factor in myocardial infarction. J Thromb Thrombolysis. 2010;30:404–11.
Kai H, Ikeda H, Yasukawa H, Kai M, Seki Y, Kuwahara F, et al. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J Am Coll Cardiol. 1998;32:368–72.
Ju C, Ye M, Li F. Plasma brain natriuretic peptide, endothelin-1, and matrix metalloproteinase 9 expression and significance in type 2 diabetes mellitus patients with ischemic heart disease. Med Sci Monit. 2015;21:2094–9.
Jefferis BJ, Whincup P, Welsh P, Wannamethee G, Rumley A, Lennon L, et al. Prospective study of matrix metalloproteinase-9 and risk of myocardial infarction and stroke in older men and women. Atherosclerosis. 2010;208:557–63.
Tan C, Liu Y, Li W, Deng F, Liu X, Wang X, et al. Associations of matrix metalloproteinase-9 and monocyte chemoattractant protein-1 concentrations with carotid atherosclerosis, based on measurements of plaque and intima-media thickness. Atherosclerosis. 2014;232:199–203.
Johnson C, Galis ZS. Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol. 2004;24:54–60.
Papalambros E, Sigala F, Georgopoulos S, Panou N, Kavatzas N, Agapitos M, et al. Vascular endothelial growth factor and matrix metalloproteinase 9 expression in human carotid atherosclerotic plaques: relationship with plaque destabilization via neovascularization. Cerebrovasc Dis. 2004;18:160–5.
Ul Ain Q, Chung H, Chung JY, Choi JH, Kim YH. Amelioration of atherosclerotic inflammation and plaques via endothelial adrenoceptor-targeted eNOS gene delivery using redox-sensitive polymer bearing l-arginine. J Control Release. 2017;262:72–86.
Sigala F, Efentakis P, Karageorgiadi D, Filis K, Zampas P, Iliodromitis EK, et al. Reciprocal regulation of eNOS, H2S and CO-synthesizing enzymes in human atheroma: correlation with plaque stability and effects of simvastatin. Redox Biol. 2017;12:70–81.
Sonaglia F, Milia P, Caserio M, Bigazzi B, Bigazzi B, Ricotta S, et al. Efficacy and safety of oral porcine relaxin (pRLX) in adjunct to physical exercise in the treatment of peripheral arterial disease (PAD). Ital J Anat Embryol. 2013;118:84–91.
Conrad KP, Novak J. Emerging role of relaxin in renal and cardiovascular function. Am J Physiol Regul Integr Comp Physiol. 2004;287:R250–61.
Nistri S, Bani D. Relaxin receptors and nitric oxide synthases: search for the missing link. Reprod Biol Endocrinol. 2003;1:5.
Baccari MC, Bani D, Bigazzi M, Calamai F. Influence of relaxin on the neurally induced relaxant responses of the mouse gastric fundus. Biol Reprod. 2004;71:1325–9.
Baccari MC, Nistri S, Vannucchi MG, Calamai F, Bani D. Reversal by relaxin of altered ileal spontaneous contractions in dystrophic (mdx) mice through a nitric oxide-mediated mechanism. Am J Physiol Regul Integr Comp Physiol. 2007;293:R662–8.
Bani D, Baccari MC, Nistri S, Calamai F, Bigazzi M, Sacchi TB. Relaxin up-regulates the nitric oxide biosynthetic pathway in the mouse uterus: involvement in the inhibition ofmyometrial contractility. Endocrinology. 1999;140:4434–41.
Bani D, Baccari MC, Quattrone S, Nistri S, Calamai F, Bigazzi M, et al. Relaxin depresses small bowel motility through a nitric oxide-mediated mechanism: studies in mice. Biol Reprod. 2002;66:778–84.
Bani D, Bigazzi M, Masini E, Bani G, Sacchi TB. Relaxin depresses platelet aggregation: in vitro studies on isolated human and rabbit platelets. Lab Invest. 1995;73:709–16.
Bani D, Failli P, Bello MG, Thiemermann C, Bani Sacchi T, Bigazzi M, et al. Relaxin activates the L-arginine-nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension. 1998;31:1240–7.
Cardoso LC, Nascimento AR, Royer C, Porto CS, Lazari MF. Locally produced relaxin may affect testis and vas deferens function in rats. Reproduction. 2010;139:185–96.
Failli P, Nistri S, Quattrone S, Mazzetti L, Bigazzi M, Sacchi TB, et al. Relaxin up-regulates inducible nitric oxide synthase expression and nitric oxide generation in rat coronary endothelial cells. FASEB J. 2002;16:252–4.
Masini E, Nistri S, Vannacci A, Bani Sacchi T, Novelli A, Bani D. Relaxin inhibits the activation of human neutrophils: involvement of the nitric oxide pathway. Endocrinology. 2004;145:1106–12.
Masini E, Zagli G, Ndisang JF, Solazzo M, Mannaioni PF, Bani D. Protective effect of relaxin in cardiac anaphylaxis: involvement of the nitric oxide pathway. Br J Pharmacol. 2002;137:337–44.
Quattrone S, Chiappini L, Scapagnini G, Bigazzi B, Bani D. Relaxin potentiates the expression of inducible nitric oxide synthase by endothelial cells from human umbilical vein in in vitro culture. Mol Hum Reprod. 2004;10:325–30.
Bani D, Masini E, Bello MG, Bigazzi M, Sacchi TB. Relaxin activates the L-arginine-nitric oxide pathway in human breast cancer cells. Cancer Res. 1995;55:5272–5.
Ho TY, Yan W, Bagnell CA. Relaxin-induced matrix metalloproteinase-9 expression is associated with activation of the NF-B pathway in human THP-1 cells. J Leukoc Biol. 2007;81:1303–10.
Unemori EN, Amento EP. Relaxin modulates synthesis and secretion of procollagenase and collagen by human dermal fibroblasts. J Biol Chem. 1990;265:10681–5.
Bennett RG, Kharbanda KK, Tuma DJ. Inhibition of markers of hepatic stellate cell activation by the hormone relaxin. Biochem Pharmacol. 2003;66:867–74.
Williams EJ, Benyon RC, Trim N, Hadwin R, Grove BH, Arthur MJ, et al. Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut. 2001;49:577–83.
Gupta S, Halushka MK, Hilton GM, Arking DE. Postmortem cardiac tissue maintains gene expression profile even after late harvesting. BMC Genomics. 2012;13:26. https://doi.org/10.1186/1471-2164-13-26.
Author information
Authors and Affiliations
Contributions
KP wrote the paper and designed the study; AK performed the research; GG and NP analyzed the data; C. Kontogiannis, C. Kourek, KSM and DVC wrote the paper; and IK and SG designed the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Papoutsis, K., Kapelouzou, A., Georgiopoulos, G. et al. Tissue-specific relaxin-2 is differentially associated with the presence/size of an arterial aneurysm and the severity of atherosclerotic disease in humans. Acta Pharmacol Sin 41, 745–752 (2020). https://doi.org/10.1038/s41401-019-0350-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0350-5
Keywords
This article is cited by
-
Extracellular vesicles in vascular remodeling
Acta Pharmacologica Sinica (2022)

{kind=link}
{kind=link}
{kind=link}