
Abstract
Traumatic events may lead to post-traumatic stress disorder (PTSD), with higher prevalence in women. Adverse childhood experiences (ACE) increase PTSD risk in adulthood. Epigenetic mechanisms play important roles in PTSD pathogenesis and a mutation in the methyl-CpG binding protein 2 (MECP2) in mice provide susceptibility to PTSD-like alterations, with sex-dependent biological signatures. The present study examined whether the increased risk of PTSD associated with ACE exposure is accompanied by reduced MECP2 blood levels in humans, with an influence of sex. MECP2 mRNA levels were analyzed in the blood of 132 subjects (58 women). Participants were interviewed to assess PTSD symptomatology, and asked to retrospectively report ACE. Among trauma-exposed women, MECP2 downregulation was associated with the intensification of PTSD symptoms linked to ACE exposure. MECP2 expression emerges as a potential contributor to post-trauma pathophysiology fostering novel studies on the molecular mechanisms underlying its potential sex-dependent role in PTSD onset and progression.
Similar content being viewed by others

Introduction
Trauma exposure is a common experience worldwide, with 70% of people exposed to at least one traumatic event throughout their lives [1]. This may lead to the onset of post-traumatic stress disorder (PTSD), a chronic mental disorder characterized by severely debilitating and long-lasting symptoms, that is more prevalent in women than men [2]. PTSD symptoms can be grouped in four main categories [3]: (i) re-experiencing, defined as the appearance of intrusive thoughts, nightmares and flashbacks; (ii) avoidance of internal or external trauma reminders; (iii) hyperarousal, manifested as attentional threat bias, sleep problems and enhanced startle reactivity; (iv) negative alterations in cognition and mood, including patients’ inability to recall important aspects of the traumatic event and persistent negative emotional state. Only an average of 5.6% of traumatized individuals in the world develop a chronic PTSD symptomatology [4]. Nevertheless, the high socio-economic costs and the burden that PTSD symptomatology represents for the affected individuals urge the identification of the risk factors involved in disease development in traumatized people [5].
In recent years the neurobiological bases of PTSD have been deeply explored [6], and a growing body of evidence has underscored the contribution of epigenetic mechanisms to PTSD pathogenesis and symptom presentation in the aftermath of trauma exposure [7]. Among the multiple epigenetic signatures, altered DNA methylation has been especially linked to traumatic stress consequences [8]. Both candidate gene and epigenome-wide association studies have in fact identified PTSD-related alterations in methylated DNA (mDNA) at multiple genetic loci involved in stress, inflammation and neurotransmission pathways [9,10,11]. On these grounds previous works also found variations in the enzymes responsible for DNA methylation in association with risk for PTSD [12]. In spite of this evidence, the potential role in PTSD pathophysiology of mDNA reader binding proteins, such as the X-linked methyl-CpG binding protein 2 (MECP2), has not yet been addressed. MECP2 in particular serves as a scaffold protein for the recruitment of chromatin remodeling complexes [13,14,15] and DNA methyltransferases [16, 17] on methylated DNA loci, thus representing an excellent candidate for mediating post-trauma epigenetic rearrangements. Moreover, the activity and expression of MECP2 in the rodent brain, where MECP2 is known to regulate learning and memory processes [18], is very responsive to environmental challenges such as exposure to early life stress as well as to stressors in adulthood [19,20,21]. In this line, previous studies searching for genetic markers of PTSD risk described an altered expression of multiple targets of MECP2 in subjects who developed the disorder in the aftermath of a traumatic experience [22,23,24,25]. Consistently, MECP2 is known to control the transcription of stress response-regulating genes [26, 27], and to tune immune function and cytokine production [28], whose alterations have been described in patients with PTSD [22, 29,30,31,32,33]. These characteristics make MECP2 a promising mediator of the lasting epigenetic adjustments taking place following stress or trauma exposure that could direct towards vulnerability or resilience to PTSD [34]. Based on this, we recently addressed the potential involvement of altered MECP2 functionality in the onset of PTSD-like pathophysiology in transgenic mice carrying a hypofunctional form of MECP2. We demonstrated that MECP2-mutated mice display an increased propensity to develop enduring neurobehavioral alterations, comparable to those observed in patients with PTSD, when exposed to intense, acute stressors [35].
Notably, male and female carriers of the MECP2 hypomorphic mutation, while being both behaviorally sensitive to stressors, exhibited an opposite modulation of stress markers at the molecular level [36]. This finding opens up the intriguing possibility that the MECP2 protein may be involved in the regulation of sex-dependent differences in vulnerability to PTSD. In this line, it is noteworthy that MECP2 has been proposed to participate in the sexual differentiation of the developing rodent brain [37, 38], suggesting that it might play a role in setting the basis for the existing sex bias in vulnerability to PTSD [39, 40].
Interestingly, a sex-specific modulation of MECP2 expression has previously been described in rodents following stress exposure early in life [41, 42]. Furthermore, in a non-clinical population sample we recently demonstrated that reduced levels of MECP2 are linked to an increased risk of psychopathology following childhood adversities selectively in women [43]. Given that early-life adversities, vulnerability to psychopathology and female sex are factors known to intensify the impact of exposure to traumatic experiences in adulthood, eventually increasing the risk of PTSD onset [44, 45], these findings confirm the need of further exploring the role of MECP2 in the pathogenesis of PTSD.
Based on this body of evidence, we hypothesized that MECP2 levels might be altered within a traumatized population, with its downregulation possibly representing a risk factor for developing PTSD. As exposure to stressful experiences at critical developmental periods, such as childhood, dramatically increases vulnerability to the pathological outcomes of subsequent trauma exposure, we explored the possibility that adverse childhood experiences (ACE) strengthen the association between reduced MECP2 and an increased PTSD risk. We also reasoned that this association might be more marked in women, in line with the existing sex bias in vulnerability to PTSD. To test our assumptions we evaluated MECP2 mRNA levels in the blood of 132 male and female participants who were trauma-exposed with or without ensuing symptoms of PTSD in adulthood (PTSD and trauma controls, TC) or non-exposed throughout their lifetime (non-traumatized controls, NTC), and assessed whether MECP2 expression varied as a function of sex, ACE and trauma exposure. Focusing on traumatized individuals, we tested the possibility that the increased severity of PTSD symptoms associated with exposure to ACE was accompanied by reduced MECP2 blood levels, with an influence of sex.
Materials and methods
Study participants
Study participants were civilians recruited between 2010 and 2018 to take part in multiple independent studies on the psychobiological alterations characterizing people suffering from PTSD (see Supplementary Methods for further information). A total of 132 subjects (58 women, mean age 41.72 ± 13.91 years) were included in the present study; among them, 85 subjects reported traumatic experiences, of whom 37 received a diagnosis of PTSD (see Table 1 for detailed information on the study sample). PTSD diagnosis and possible comorbidities were evaluated by the Structured Clinical Interviews for DSM-IV [46] I and II (SCID) [47, 48] (see Supplementary Methods). Participants were excluded in case of clinically significant traumatic experiences before 18 years of age, comorbid psychotic symptoms, borderline personality disorder, alcohol/drug dependence or abuse, and cardiovascular or neurological disorders. The study conformed to the Code of Ethics of the World Medical Association (Declaration of Helsinki, 6th revision, 2008) and was approved by the Ethics Committee of the Medical Faculty Mannheim, Heidelberg University. All participants gave written informed consent.
Psychometric measures
Posttraumatic Diagnostic Scale
Traumatic experiences were assessed by the means of the German version of the Posttraumatic Diagnostic Scale (PDS) [49, 50], a self-report instrument aimed at assessing the severity of post-traumatic stress symptoms. The first part of the questionnaire consists of a short checklist of potentially traumatizing events. Among the experienced events, respondents are required to indicate the one that has troubled them the most in the past month (index trauma). Participants experiencing the index trauma before 18 years of age were excluded, since clinically significant childhood trauma experiences are expected to have differential impacts on PTSD pathophysiology [22]. The subjects reporting an index trauma were then required to rate, on a 4-point scale (0 - never to 3 - daily), 17 items representing the frequency of the occurrence of cardinal PTSD symptoms in the last 30 days. Finally, respondents rated the degree of impairment caused by symptoms across different areas of life functioning. The symptom severity score was obtained by adding up the responses to selected items and ranges from 0 to 51.
Clinician-Administered PTSD Scale
PTSD symptomatology was assessed by the means of the Clinician-Administered PTSD Scale interview (CAPS) [51, 52], a 30-item structured interview that corresponds to the DSM-IV criteria for PTSD [46]. Frequency and severity of each item are rated on a 5-point Likert scale ranging from 0, never/not affected to 4, every day/extremely affected. Three subscales measuring re-experiencing, avoidance and arousal symptom clusters were then calculated as the mean frequency and severity values of the relative items (5 for re-experiencing and arousal and 7 for avoidance symptoms). The total CAPS score was calculated as the overall sum of the ratings, ranging from 0 to 68.
Adverse childhood experiences
Participants completed the Childhood Trauma Questionnaire [53] in order to retrospectively evaluate the severity of ACE (<18 years of age), including emotional, physical abuse/neglect and sexual abuse. This is a psychometrically validated self-report inventory composed of 28 items each rated on a 5-point Likert scale (1, never true – 5, very often true). The total score ranges from 25 to 125. We exploited the classification of the total score into severity quartiles (none/minimal, low to moderate, moderate to severe, severe to extreme) contained in the manual (Bernstein & Fink, 1998; MacDonald et al., 2016) to include ACE as a discrete independent factor in tests of analysis of variance (ANOVA). By merging quartiles 2-4 we obtained a dichotomous variable: 1, none/minimal (total score ≤ 36) - for individuals who did not report ACE, and 2, low to extreme (total score > 36) - for individuals who recalled ACE of different intensities.
Trier Inventory for Chronic Stress
The load of current chronic stress was assessed by the means of the Trier Inventory for Chronic Stress (TICS) [54], a self-assessment instrument composed of 57 items evaluating 9 chronic stressors: work and social overload, pressure to perform, work discontent, excessive demand at work, lack of social recognition, social tensions or isolation and chronic worrying. Each item is rated on a 5-point Likert scale indicating how often the subject had experienced a certain situation within the last 3 months (0, never – 4, very often).
Center for Epidemiological Studies Depression Scale and State-Trait Anxiety Inventory
See Supplementary Methods.
MECP2 expression
Whole blood was collected in PAXgene Blood RNA Tubes (PreAnalytiX, Hombrechtikon Switzerland) and stored until analysis at −80 °C [43, 55]. A PAXgene Blood miRNA Kit (Qiagen, Hilden, Germany) was used to extract total RNA, following the manufacturer´s instructions. RNA concentration and sample purity were assessed with a NanoDrop 1000 Spectralphotometer (Thermo Scientific, Waltham, MA, USA), and RNA integrity was determined with the Agilent 2100 Bioanalyzer System (Agilent Technologies, Santa Clara, CA, USA). The cDNA was synthesized by a reversed transcription reaction using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA). Quantitative PCR was performed on the QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems by Life Technologies, Carlsbad, CA, USA) by using TaqMan Fast Advanced Mastermix (Applied Biosystems), and the MECP2 TaqMan Gene Expression Assay Hs00172845_m1 (Applied Biosystems). The Actin Beta ACTB TaqMan Gene Expression Assay Hs01060665_g1 (Applied Biosystems) was used as an internal standard. Results were calculated with the QuantStudio Real-Time PCR Software v1.3 (Applied Biosystems by Thermo Fisher Scientific). Analyses were carried out in triplicates. All data were normalized to the endogenous reference gene ACTB. For statistical analyses, the relative expression with respect to participants not reporting traumas or ACE (controls) was calculated by the Delta-Delta threshold cycles (∆∆Ct) method, and converted to the relative expression ratio (2-∆∆Ct), separately for men and women [56].
Statistical analyses
All statistical analyses were conducted using SPSS 20.0 and AMOS 20.0 (IBM Statistics, Armonk, NY, USA).
A logarithmic transformation was performed to reduce skewedness and kurtosis of non-normally distributed variables (see Supplementary Methods and Table S1). Outliers, defined as observations lying three standard deviations outside from the mean, were excluded (2 observations, in total).
A three-way ANOVA was performed to evaluate the relative role of sex (men & women), ACE (none/minimal & low to extreme) and trauma exposure (non-traumatized & traumatized in adulthood) in the modulation of peripheral MECP2 expression. Normality and homoscedasticity of residuals were assessed by the means of Shapiro Wilk, Levene and Breush Pagan tests. Post hoc comparisons were performed by Tukey’s test.
Structural Equation Modeling (SEM) with maximum likelihood estimation was used to test the hypothesis that reduced MECP2 expression is associated with the increased risk of developing PTSD following an index trauma, as the result of previous exposure to ACE. Exclusion criteria for the model were: failure to converge after 240 iterations, the presence of squared multiple correlation values greater than 1 (R2 > 1) and poor fit, estimated via the following goodness-of-fit (GOF) measures: the χ2statistic (with a good fit indicated by χ2/degrees of freedom (df) < 3), the root mean square error of approximation (RMSEA, with a good fit indicated by an index < 0.08) and the comparative fit index (CFI, with a good fit indicated by an index > 0.95) [57]. To establish mediation, indirect paths were tested for significance using a Bias-Corrected (BC) Bootstrapping method (95% confidence intervals; 2000 resamples) [58]. At least 10 observations per measured variable were included [59]. We checked whether the final model predicted equally well PTSD symptoms while using different scales established in the literature (CAPS and PDS). To examine whether the final model was specific for ACE, it was retested with a measure of current perceived chronic stress replacing the ACE score.
To dissect the moderating role of sex, the model was separately re-specified on male and female subsamples. For each of the analyses the alpha level was set to 0.05 [60, 61].
Results
MECP2 expression reflects exposure to adversities in childhood and traumas in adulthood as a function of sex
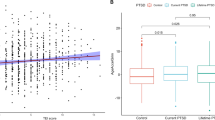
To evaluate whether exposure to traumatic experiences in adulthood or a history of ACE are associated with an altered peripheral expression of MECP2, and to test the moderating effect of sex, we exploited a three-way ANOVA model. The levels of blood MECP2 mRNA were significantly higher in men compared to women (sex: F1,124 = 37.89, p < 0.001, ηp2 = 0.23). This difference was driven by a sex-dependent effect of trauma exposure, which was associated with an increase of MECP2 expression selectively in men (p = 0.009 for non-traumatized vs traumatized men after post hoc comparisons on an almost significant sex*trauma interaction: F1,124 = 3.79, p = 0.054, ηp2 = 0.03; Fig. 1A) without affecting women. Conversely, peripheral MECP2 expression was significantly decreased in participants reporting ACE (ACE: F1,124 = 7.46, p = 0.007, ηp2 = 0.06), especially among women (p = 0.006 for women with none/minimal vs low to extreme ACE after post hoc comparisons on sex*ACE interaction: F1,124 = 7.21, p = 0.008, ηp2 = 0.06; Fig. 1B). The three-way interaction between sex, ACE and index trauma was not significant (sex*ACE*trauma: F1,124 = 2.32, p = 0.130, ηp2 = 0.02).

A Blood mRNA levels of methyl-CpG binding protein 2 (MECP2) are increased in traumatized men, compared to non-traumatized men and women. B MECP2 is downregulated in the blood of women exposed to adverse childhood experiences (ACE) compared to non-stressed women and men. MECP2 levels were normalized to total actin beta (ACTB) contents and expressed as a proportion of those of non-traumatized participants, not exposed to ACE (ctrl), separately for men and women. Statistical significance was calculated by the means of three-way ANOVA, and Tukey’s post hoc tests. Symbols: **p < 0.01; ***p < 0.001. Data are mean ± standard error of the mean.
Reduced MECP2 expression accompanies the increase in the severity of PTSD symptoms associated with ACE exposure in traumatized participants
Given that ACE are known to increase the risk of PTSD onset in the aftermath of traumatic events, using structural equation modeling we tested the hypothesis that the increase in the severity of PTSD symptoms (total CAPS score) associated with ACE exposure in individuals reporting an index trauma is accompanied by reduced MECP2 levels (Fig. 2). Overall PTSD symptomatology was represented as a single latent factor in the present model, based on the a priori assumption that the three PTSD symptom subscales may be all associated within the same latent construct [51, 62].

Among traumatized participants, reduced methyl-CpG binding protein 2 (MECP2) expression is directly associated with higher adverse childhood experiences (ACE) scores (R2 = 5.6%; β = −0.24, p = 0.037), which in turn predict increased post-traumatic stress disorder (PTSD) symptom severity (R2 = 7.4%; β = 0.27, p = 0.015). PTSD symptom severity is measured by means of the clinician-administered PTSD scale (CAPS). Symbols: Plain arrows - directed arcs, positive path coefficients (p < 0.05); dashed arrows - directed arcs, negative path coefficients (p < 0.05); black numbers - standardized coefficients; black underlined numbers – proportion of total variation explained by the model (R2); r – residual variances (errors).
The model fit was satisfactory (Tables 1, 2), suggesting that the hypothesized path (Fig. 2) describes the data well, thus allowing further interpretations of the results to be obtained. MECP2 expression was inversely proportional to ACE severity (R2 = 5.6%), implying that decreased MECP2 levels were associated with a stressful childhood (β = −0.24, p = 0.037). As expected, higher ACE scores were also linked to increased PTSD symptom severity (R2 = 7.2%) among traumatized individuals (β = 0.27, p = 0.022). Overall, MECP2 expression turned out to be indirectly associated with PTSD symptoms, with ACE mediating the association between lower levels of MECP2 and higher total CAPS scores (β = −0.06, p = 0.037). This suggests that MECP2 downregulation accompanies the increased PTSD vulnerability emerging from a history of ACE (see Table 3 and Supplementary Table S2 for further details on direct and indirect effects in the hypothesized model).
Importantly, the present results were further confirmed after retesting the hypothesized path on the prediction of PTSD symptom severity measured through a distinct psychometric scale (total PDS score, R2 = 19.2%) (PTSD symptom scale substitution model, see Tables 2, 2i, Table 3 and Supplementary Table S3 for detailed statistical results, and Supplementary Fig. S1).
The increase in PTSD symptomatology associated with current stress load is not paralleled by changes in MECP2 expression
To examine the specificity of the observed effects for stress experienced during childhood, we assessed whether the hypothesized path was still valid when replacing the ACE score with a measure of current chronic stress load (Supplementary Fig. S1).
The model had acceptable fit indices (Table 2, 2ii), and explained a relatively high proportion of total PTSD symptom variation (R2 = 26%), which was due to the highly significant association between chronic stress load and PTSD symptoms (β = 0.51, p < 0.001) (see Supplementary Table S4 for further information on direct and indirect effects). Indeed, MECP2 was not significantly associated with chronic stress and failed to have significant indirect effects on PTSD symptomatology in the present model (see Table 3), suggesting that the association between MECP2 expression and PTSD symptomatology is indirectly mediated specifically by stressors experienced during childhood.
The link between MECP2 downregulation and the increase in the severity of PTSD symptoms associated with ACE exposure is particularly relevant in women
In order to dissect the effects of sex, we explored the validity of the selected path on two different subsamples, composed of men or women only (Supplementary Fig. S2). The GOF indices for both subsamples were acceptable (see Table 2, 2iii and 2iv). In terms of R2 the model explained up to 27.8% of PTSD symptom variance in the female subsample, but failed to significantly explain PTSD symptom variance in the male subsample.
Importantly, in both samples, MECP2 expression failed to be directly associated with the severity of ACE, which, conversely, significantly predicted PTSD symptoms selectively in women (β = 0.53, p < 0.001). Of note, the total indirect effect of MECP2 expression on PTSD symptoms was significant in the female (β = −0.14, p = 0.033), but not in the male subsample (see Table 3 and Supplementary Table S5 for further details).
Discussion
The present findings provide evidence of an association between the epigenetic factor MECP2 and symptom severity in traumatized individuals diagnosed with PTSD, a mental illness with severe impact on quality of life and high cost to the health care system [5]. This association appears to occur especially among women, who are typically most affected by PTSD, and is mediated by the quality of early life experiences. These findings suggest that MECP2 may represent a key sex-dependent player in PTSD pathogenesis, and point to MECP2 expression as a putative marker of vulnerability to stress and trauma–related disorders. Further studies dissecting the underlying mechanisms may unravel targetable pathways for sex-specific preventive interventions.
Previous evidence from the preclinical setting support the existence of a tight link between MECP2 and early life events, and point to MECP2 as a key transducer of perinatal experiences into lasting epigenetic signatures, which ultimately modulate an individual’s ability to cope with future challenges [19, 21, 41, 42, 63,64,65,66]. We recently demonstrated the translatability of this framework to the human species, by evidencing an association between MECP2 levels and subclinical symptoms of anxiety and depression after ACE [43]. The present findings substantiate and transfer this link to a clinical framework by showing that the connection between the severity of PTSD symptoms and MECP2 levels in traumatized individuals is significantly influenced by exposure to ACE. Indeed, MECP2 downregulation was related to reporting more ACE and the associated onset of severe PTSD symptoms following exposure to an index trauma in adulthood. Conversely, MECP2 levels had no connection with the exacerbation of PTSD symptoms linked to ongoing chronic stressors in traumatized participants. An intriguing hypothesis explaining the specific influence of ACE on the link between MECP2 and PTSD vulnerability concerns the possibility that MECP2 downregulation may blur the participants’ recall of childhood experiences, without affecting current stress perception, which is in line with the key role exerted by MECP2 in cognition and memory processes [18]. However, MECP2 downregulation might also be an immediate consequence of the experience of more ACE. Further studies are certainly needed to ultimately delineate the precise nature of the relationship between MECP2 and early life events.
An intriguing aspect of the present results is that exposure to stressors of varying intensity in different periods of life is accompanied by sex-specific patterns of MECP2 expression. In fact, women reporting ACE display reduced levels of MECP2, while men exposed to traumas in adulthood show MECP2 overexpression. These results are in line with the evidence describing MECP2 as a critical environmental sensor [19, 67] and suggest that peripheral MECP2 expression may “quantify” lifetime stress exposure, possibly representing a sex-specific biomarker of vulnerability [68]. Our results in fact point to females as the sex most affected by reduced levels of MECP2 and the associated negative outcomes of early life challenges. Although the small sample size imposes a cautious interpretation of the data, present results are in line with our previous findings of a sex-dependent association of MECP2 with vulnerability to psychopathology in healthy individuals exposed to ACE [43]. Consistently, females were described to be more vulnerable than males to the detrimental and lasting consequences of ACE [69, 70]. It is thus conceivable that MECP2 may take part in sex-dependent biological mechanisms that make females more vulnerable than males to stress-related disorders [71]. In this line, other factors that lie within the MECP2 network have been associated with PTSD in a sex-specific manner across rodents and people (e.g. FKBP5, HDAC4) [72, 73] Furthermore, it is important to underline that developmental sex differences in MECP2 expression and its contribution to the emergence of sex dimorphisms have been previously acknowledged [37, 38]. The fact that these dimorphisms interest brain regions implicated in PTSD [72] further strengthen our hypothesis of an involvement of MECP2 in the establishment of sex differences in vulnerability to traumas. Gaining further insight into the mechanisms involved in the sex-specific regulation of MECP2 expression after exposure to stressful experiences will be of great help for the identification of vulnerability or pathogenic pathways to be targeted with the aim of increasing resilience.
The present results should be considered in light of some limitations. Indeed, participants were mainly of European ancestry, thus limiting the possibility to extend our findings to multiple ethnicities. This is important, given that the genetic and epigenetic underpinnings of PTSD have previously been demonstrated to differ among ethnic groups [74]. Moreover, although several studies point to a major involvement of altered DNA methylation processing as a fundamental mechanism providing vulnerability to traumas [7, 12, 40], within the present study we cannot draw conclusions about the functionality of the methylation machinery in patients with PTSD. Further studies are needed to explore the expression of other enzymes involved in DNA methylation and assess MECP2 protein levels, which would be of interest to unveil a functional role for MECP2 and the methylation machinery in the periphery. In this context, it is relevant to clarify that, while analyzing human blood samples allowed us to provide novel evidence of an association between MECP2 levels and PTSD symptoms, gene expression in blood does not necessarily reflect the molecular processes that may take place within the brain. Although there is evidence that peripheral epigenetic responses might, in some instances, reflect brain-related states [11, 39, 75], the present findings need to be reinforced by animal studies addressing brain MECP2 levels.
Beyond these considerations, the evidence of an existing link, in the clinical setting, between MECP2 and the negative outcome of ACE looks promising in the search for vulnerability markers. Indeed ACE represent a risk factor common to multiple mental disorders, including depression and schizophrenia [76, 77], with whom PTSD shares a substantial proportion of genetic variance [10, 78, 79]. In this light, a better understanding of the role of MECP2 in the pathophysiology of mental disorders may benefit from a research focusing on pathological traits, rather than on strict diagnostic categories [80].
Collectively, the present study suggests that MECP2 downregulation may represent a step in the pathogenic process leading to PTSD onset in patients, especially women, exposed to childhood adversities. Studies focusing on dissecting the mechanisms involved in the regulation of MECP2 expression could shed new light on the biological pathways underlying the sex and gender bias in trauma vulnerability and could provide a more detailed mechanistic understanding of the pathophysiology of the disorder, hopefully leading to more effective, individualized interventions.
Data availability
Ethical restrictions to protect participant confidentiality prevent us from making anonymised study data publicly available. Readers seeking access to the study data and materials should contact the corresponding author based on a formal collaboration agreement. This formal collaboration agreement indicates that data will be shared with other researchers who agree to work with the authors, and for the sole purpose of verifying the claims in the paper. The data and materials will be released to requestors after approval of this formal collaboration agreement by the local Ethics Committee of the Medical Faculty Mannheim.
References
Kessler RC, Aguilar-Gaxiola S, Alonso J, Benjet C, Bromet EJ, Cardoso G, et al. Trauma and PTSD in the WHO World Mental Health Surveys. Eur J Psychotraumatol 2017;8:1353383. https://doi.org/10.1080/20008198.2017.1353383.
Ramikie T, Ressler K. Mechanisms of Sex Differences in Fear and Posttraumatic Stress Disorder. Biol Psychiatry 2018;83:876–85. https://doi.org/10.1016/j.biopsych.2017.11.016.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed.). 2013. https://doi.org/10.1176/appi.books.9780890425596.744053.
Koenen KC, Ratanatharathorn A, Ng L, McLaughlin KA, Bromet EJ, Stein DJ, et al. Posttraumatic stress disorder in the World Mental Health Surveys. Psychol Med 2017;47:2260–74. https://doi.org/10.1017/S0033291717000708.
Watson P. PTSD as a Public Mental Health Priority. Curr Psychiatry Rep. 2019;21:61. https://doi.org/10.1007/s11920-019-1032-1.
Ressler K. Molecular Signatures of Stress and Posttraumatic Stress Disorder: An Overview. Biol Psychiatry 2018;83:792–4. https://doi.org/10.1016/j.biopsych.2018.03.007.
Howie H, Rijal C, Ressler K. A review of epigenetic contributions to post-traumatic stress disorder. Dialogues Clin Neurosci. 2019;21:417–28. https://doi.org/10.31887/DCNS.2019.21.4/kressler.
Smith A, Ratanatharathorn A, Maihofer A, Naviaux R, Aiello A, Amstadter A, et al. Epigenome-wide meta-analysis of PTSD across 10 military and civilian cohorts identifies methylation changes in AHRR. Nat Commun 2020;11:5965. https://doi.org/10.1038/s41467-020-19615-x.
Zannas AS, Provençal N, Binder EB. Epigenetics of posttraumatic stress disorder: current evidence, challenges, and future directions. Biol Psychiatry 2015;78:327–35. https://doi.org/10.1016/j.biopsych.2015.04.003.
Daskalakis N, Rijal C, King C, Huckins L, Ressle KJ. Recent Genetics and Epigenetics Approaches to PTSD. Curr Psychiatry Rep. 2018;20:30. https://doi.org/10.1007/s11920-018-0898-7.
Logue M, Miller M, Wolf E, Huber B, Morrison F, Zhou Z, et al. An epigenome-wide association study of posttraumatic stress disorder in US veterans implicates several new DNA methylation loci. Clin Epigenet 2020;12:46. https://doi.org/10.1186/s13148-020-0820-0.
Sipahi L, Wildman D, Aiello A, Koenen K, Galea S, Abbas A, et al. Longitudinal epigenetic variation of DNA methyltransferase genes is associated with vulnerability to post-traumatic stress disorder. Psychol Med 2014;44:3165–79. https://doi.org/10.1017/S0033291714000968.
Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci 2013;16:898–902. https://doi.org/10.1038/nn.3434.
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–9. https://doi.org/10.1038/30764.
Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 2003;278:4035–40. https://doi.org/10.1074/jbc.M210256200.
Rajavelu A, Lungu C, Emperle M, Dukatz M, Bröhm A, Broche J, et al. Chromatin-dependent allosteric regulation of DNMT3A activity by MeCP2. Jurkowska RZ, Jeltsch A. Nucleic Acids Res 2018;46:9044–56. https://doi.org/10.1093/nar/gky715.
Kimura H, Shiota K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem 2003;278:4806–12. https://doi.org/10.1074/jbc.M209923200.
Robinson H, Pozzo-Miller L. The role of MeCP2 in learning and memory. Learn Mem 2019;26:343–50. https://doi.org/10.1101/lm.048876.118.
Zimmermann CA, Hoffmann A, Raabe F, Spengler D. Role of Mecp2 in experience-dependent epigenetic programming. Genes (Basel) 2015;6:60–86. https://doi.org/10.3390/genes6010060.
Finsterwald C, Steinmetz AB, Travaglia A, Alberini CM. From Memory Impairment to Posttraumatic Stress Disorder-Like Phenotypes: The Critical Role of an Unpredictable Second Traumatic Experience. J Neurosci 2015;35:15903–15. https://doi.org/10.1523/JNEUROSCI.0771-15.2015.
Singh-Taylor A, Molet J, Jiang S, Korosi A, Bolton J, Noam Y, et al. NRSF-dependent epigenetic mechanisms contribute to programming of stress-sensitive neurons by neonatal experience, promoting resilience. Mol Psychiatry 2018;23:648–57. https://doi.org/10.1038/mp.2016.240.
Mehta D, Klengel T, Conneely K, Smith A, Altmann A, Pace T, et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci USA. 2013;110:8302–7. https://doi.org/10.1073/pnas.1217750110.
Sarapas C, Cai G, Bierer L, Golier J, Galea S, Ising M, et al. Genetic markers for PTSD risk and resilience among survivors of the World Trade Center attacks. Dis Markers 2011;30:101–10. https://doi.org/10.3233/DMA-2011-0764.
Yehuda R, Cai G, Golier J, Sarapas C, Galea S, Ising M, et al. Gene expression patterns associated with posttraumatic stress disorder following exposure to the World Trade Center attacks. Biol Psychiatry 2009;66:708–11. https://doi.org/10.1016/j.biopsych.2009.02.034.
Dell’Osso L, Carmassi C, Del Debbio A, Catena Dell’Osso M, Bianchi C, da Pozzo E, et al. Brain-derived neurotrophic factor plasma levels in patients suffering from post-traumatic stress disorder. Prog Neuropsychopharmacol Biol Psychiatry 2009;33:899–902. https://doi.org/10.1016/j.pnpbp.2009.04.018.
McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2006;103:18267–72. https://doi.org/10.1073/pnas.0608702103.
Nuber UA, Kriaucionis S, Roloff TC, Guy J, Selfridge J, Steinhoff C, et al. Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet 2005;14:2247–56. https://doi.org/10.1093/hmg/ddi229.
Zalosnik M, Fabio M, Bertoldi M, Castañares C, Degano A. MeCP2 deficiency exacerbates the neuroinflammatory setting and autoreactive response during an autoimmune challenge. Sci Rep. 2021;11:10997. https://doi.org/10.1038/s41598-021-90517-8.
Baker D, West S, Nicholson W, Ekhator N, Kasckow J, Hill K, et al. Serial CSF Corticotropin-Releasing Hormone Levels and Adrenocortical Activity in Combat Veterans With Posttraumatic Stress Disorder. Am J Psychiatry 1999;156:585–8. https://doi.org/10.1176/ajp.156.4.585.
van Zuiden M, Geuze E, Willemen HL, Vermetten E, Maas M, Amarouchi K, et al. Glucocorticoid receptor pathway components predict posttraumatic stress disorder symptom development: a prospective study. Biol Psychiatry 2012;71:309–16. https://doi.org/10.1016/j.biopsych.2011.10.026.
Liberzon I, Ressler K. Neurobiology of PTSD: from brain to mind. New York, USA, Oxford University Press, 2016. https://doi.org/10.1093/med/9780190215422.001.0001.
Wang Z, Caughron B, Young M. Posttraumatic stress disorder: An immunological disorder? Front Psychiatry 2017;8:222. https://doi.org/10.3389/fpsyt.2017.00222.
Morrison F, Miller M, Logue M, Assef M, Wolf E. DNA Methylation correlates of PTSD: Recent findings and technical challenges. Prog Neuropsychopharmacol Biol Psychiatry 2019;90:223–34. https://doi.org/10.1016/j.pnpbp.2018.11.011.
Dudley K, Xiang L, Kobor M, Kippin T, Bredy T. Epigenetic mechanisms mediating vulnerability and resilience to psychiatric disorders. Neurosci Biobehav Rev 2011;35:1544–51. https://doi.org/10.1016/j.neubiorev.2010.12.016.
Cosentino L, Vigli D, Medici V, Flor H, Lucarelli M, Fuso A, et al. Methyl-CpG binding protein 2 functional alterations provide vulnerability to develop behavioral and molecular features of post-traumatic stress disorder in male mice. Neuropharmacology 2019;160:107664. https://doi.org/10.1016/j.neuropharm.2019.06.003.
Cosentino L, Bellia F, Pavoncello N, Vigli D, D’Addario C, De Filippis B. Methyl-CpG binding protein 2 dysfunction provides stress vulnerability with sex- and zygosity-dependent outcomes. Eur J Neurosci 2022;55:2766–76. https://doi.org/10.1111/ejn.15165.
Kurian J, Forbes-Lorman R, Auger A. Sex Difference in Mecp2 Expression During a Critical Period of Rat Brain Development. Epigenetics 2007;2:173–8. https://doi.org/10.4161/epi.2.3.4841.
Forbes-Lorman RM, Rautio JJ, Kurian JR, Auger AP, Auger CJ. Neonatal MeCP2 is important for the organization of sex differences in vasopressin expression. Epigenetics 2012;7:230–8. https://doi.org/10.4161/epi.7.3.19265.
Uddin M, Sipahi L, Li J, Koenen K. Sex differences in DNA methylation may contribute to risk of PTSD and depression: A review of existing evidence. Depress Anxiety 2013;30:1151–60. https://doi.org/10.1002/da.22167.
Rusiecki J, Uddin M, Alexander M, Moore L. Post-traumatic Stress Disorder and DNA Methylation. In: Martin C, Preedy V, Patel V (eds). Comprehensive Guide to Post-Traumatic Stress Disorder. Springer, Cham, 2015. https://doi.org/10.1007/978-3-319-08613-2_98-1.
Sobolewski M, Varma G, Adams B, Anderson DW, Schneider JS, Cory-Slechta DA. Developmental lead exposure and prenatal stress result in sex-specific reprograming of adult stress physiology and epigenetic profiles in brain. Toxicol Sci 2018;163:478–89. https://doi.org/10.1093/toxsci/kfy046.
Blaze J, Roth T. Exposure to caregiver maltreatment alters expression levels of epigenetic regulators in the medial prefrontal cortex. Int J Dev Neurosci 2013;31:804–10. https://doi.org/10.1016/j.ijdevneu.2013.10.001.
Cosentino L, Zidda F, Dukal H, Witt S, De Filippis B, Flor H. Low levels of Methyl-CpG binding protein 2 are accompanied by an increased vulnerability to the negative outcomes of stress exposure during childhood in healthy women. Transl Psychiatry 2022;12:506. https://doi.org/10.1038/s41398-022-02259-4.
Koenen KC, Moffitt TE, Poulton R, Martin J, Caspi A. Early childhood factors associated with the development of post-traumatic stress disorder: Results from a longitudinal birth cohort. Psychol Med 2007. https://doi.org/10.1017/S0033291706009019.
Shalev A, Liberzon I, Marmar C. Post-traumatic stress disorder. N. Engl J Med 2017;376:2459–69. https://doi.org/10.1056/NEJMra1612499.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) (Vol. 1). 2000. https://doi.org/10.1176/appi.books.9780890423349.
Fydrich T, Renneberg B, Schmitz B, Wittchen H. Strukturiertes Klinisches Interview für DSM-IV Achse II: Persönlichkeitsstörungen (SKID-II) [Structured clinical interview for DSM-IV, Axis II: Personality disorders]. Göttingen: Hogrefe, 1997.
Wittchen HU, Wunderlich U, Gruschwitz S, Zaudig M. Strukturiertes klinisches Interview für DSM-IV, Achse I: Psychische Störungen (SKID-I) [Structured clinical interview for DSM-IV, Axis I: Mental disorders]. Göttingen: Hogrefe, 1997.
Ehlers A, Steil R, Winter H, Foa EB. Deutschsprachige Ubersetzung der Posttraumatic Diagnostic Scale von Foa (1995) [German translation of the Posttraumatic Diagnostic Scale by Foa]. Unpublished manuscript, Department of Psychiatry, Warneford Hospital, Oxford, England, 1997.
Foa EB, Cashman L, Jaycox L, Perry K. The validation of a self-report measure of posttraumatic stress disorder: the Posttraumatic Diagnostic Scale. Psychol Assess 1997;9:445–51. https://doi.org/10.1037/1040-3590.9.4.445.
Schnyder U, Moergeli H. German version of Clinician-Administered PTSD Scale. J Trauma Stress 2002;15:487–92. https://doi.org/10.1023/A:1020922023090.
Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, et al. The development of a clinician-administered PTSD scale. J Trauma Stress 1995;8:75–90. https://doi.org/10.1007/BF02105408.
Bernstein D, Fink L. Childhood Trauma Questionnaire: a retrospective self-report (manual). San Antonio, Psychological Corporation, 1998.
Schultz P, Schlotz W. Trierer Inventar zur Erfassung von chronischem Stress (TICS): Skalenkonstruktion, teststatistische Überprüfung und Validierung der Skala Arbeitsüberlastung [The Trier Inventory for the Assessment of Chronic Stress (TICS)]. Diagnostica 1999;45:8–19. https://doi.org/10.1026//0012-1924.45.1.8.
Witt S, Dukal H, Hohmeyer C, Radosavljevic-Bjelic S, Schendel D, Frank J, et al. Biobank of Psychiatric Diseases Mannheim – BioPsy. Open J Bioresour 2016. https://doi.org/10.5334/ojb.18.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001;25:402–8. https://doi.org/10.1006/meth.2001.1262.
Schreiber JB, Stage FK, King J, Nora A, Barlow EA. Reporting structural equation modeling and confirmatory factor analysis results: A review. J Educ Res 2006;99:323–38. https://doi.org/10.3200/JOER.99.6.323-338.
Preacher KJ, Hayes AF. Asymptotic and resampling strategies for assessing and comparing indirect effects in multiple mediator models. Behav Res Methods 2008;40:879–91. https://doi.org/10.3758/brm.40.3.879.
Nunnally J. Psychometric theory. New York, USA, McGraw-Hill, 1967.
Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry 2009;14:681–95. https://doi.org/10.1038/mp.2008.143.
Sainani KL. The Problem of Multiple Testing. PM&R. 2009;1:1098–1103. https://doi.org/10.1016/j.pmrj.2009.10.004.
Müller-Engelmann M, Schnyder U, Dittmann C, Priebe K, Bohus M, Thome J, et al. Psychometric Properties and Factor Structure of the German Version of the Clinician-Administered PTSD Scale for DSM-5. Assessment 2020;27:1128–38. https://doi.org/10.1177/1073191118774840.
Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci 2009;12:1559–66. https://doi.org/10.1038/nn.2436.
Eid A, Bihaqi S, Renehan W, Zawia N. Developmental lead exposure and lifespan alterations in epigenetic regulators and their correspondence to biomarkers of Alzheimer’s disease. Alzheimers Dement (Amst) 2016;15:123–31. https://doi.org/10.1016/j.dadm.2016.02.002.
Glendining K, Fisher L, Jasoni C. Maternal high fat diet alters offspring epigenetic regulators, amygdala glutamatergic profile and anxiety. Psychoneuroendocrinology 2018;96:132–41. https://doi.org/10.1016/j.psyneuen.2018.06.015.
Schneider J, Kidd S, Anderson D. Influence of developmental lead exposure on expression of DNA methyltransferases and methyl cytosine-binding proteins in hippocampus. Toxicol Lett 2013;217:75–81. https://doi.org/10.1016/j.toxlet.2012.12.004.
Picard N, Fagiolini M. MeCP2: an epigenetic regulator of critical periods. Curr Opin Neurobiol 2019;59:95–101. https://doi.org/10.1016/j.conb.2019.04.004.
Lebow MA, Schroeder M, Tsoory M, Holzman-Karniel D, Mehta D, Ben-Dor S, et al. Glucocorticoid-induced leucine zipper “quantifies” stressors and increases male susceptibility to PTSD. Transl Psychiatry 2019;9:178. https://doi.org/10.1038/s41398-019-0509-3.
Monteiro S, Matos A, Oliveira S. The moderating effect of gender: Traumatic experiences and depression in adolescence. Procedia - Soc Behav Sci 2015;165:251–9. https://doi.org/10.1016/j.sbspro.2014.12.629.
Cecil H, Matson S. Psychological functioning and family discord among African-American adolescent females with and without a history of childhood sexual abuse. Child Abus Negl 2001;27:973–88. https://doi.org/10.1037/0033-2909.132.6.959.
Tolin D, Foa E. Sex differences in trauma and posttraumatic stress disorder: A quantitative review of 25 years of research. Psychol Bull 2006;132:959–92. https://doi.org/10.1037/0033-2909.132.6.959.
Ponomareva O, Ressler K. Genomic factors underlying sex differences in trauma-related disorders. Neurobiol Stress 2021;14:100330. https://doi.org/10.1016/j.ynstr.2021.100330.
Maddox S, Kilaru V, Shin J, Jovanovic T, Almli L, Dias B, et al. Estrogen-dependent association of HDAC4 with fear in female mice and women with PTSD. Mol Psychiatry 2018;23:658–65. https://doi.org/10.1038/mp.2016.250.
Nievergelt C, Maihofer A, Klengel T, Atkinson E, Chen C, Choi K, et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat Commun 2019;10:4558. https://doi.org/10.1038/s41467-019-12576-w.
Sanfeliu A, Hokamp K, Gill M, Tropea D. Transcriptomic Analysis of Mecp2 Mutant Mice Reveals Differentially Expressed Genes and Altered Mechanisms in Both Blood and Brain. Front Psychiatry 2019;10:278. https://doi.org/10.3389/fpsyt.2019.00278.
Ausió J. MeCP2 and the enigmatic organization of brain chromatin. Implications for depression and cocaine addiction. Clin Epigenetics 2016;8:58. https://doi.org/10.1186/s13148-016-0214-5.
Popovic D, Schmitt A, Kaurani L, Senner F, Papiol S, Malchow B, et al. Childhood trauma in Schizophrenia: Current findings and research perspectives. Front Neurosci 2019;13:274. https://doi.org/10.3389/fnins.2019.00274.
Duncan L, Ratanatharathorn A, Aiello A, Almli L, Amstadter A, Ashley-Koch A, et al. Largest GWAS of PTSD (N = 20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry 2018;23:666–73. https://doi.org/10.1038/mp.2017.77.
Misganaw B, Guffanti G, Lori A, Abu-Amara D, Flory JD, Hammamieh R, et al. Polygenic risk associated with post-traumatic stress disorder onset and severity. Transl Psychiatry 2019;9:165. https://doi.org/10.1038/s41398-019-0497-3.
Cuthbert B. Research Domain Criteria: Toward future psychiatric nosologies. Dialogues Clin Neurosci 2015;17:89–97. https://doi.org/10.31887/DCNS.2015.17.1/bcuthbert.
Acknowledgements
The authors would like to thank Frauke Nees, PhD for help with data analysis. Results presented in this manuscript are part of LC’s doctoral dissertation.
Funding
The study was supported by funding of the Deutsche Forschungsgemeinschaft to HF (SFB636/C1), of the Italian Ministry of Health to BDF (#GR-2018-12366210) and by Open Access funding of the Istituto Superiore di Sanità.
Author information
Authors and Affiliations
Contributions
Conceptualization: LC, BDF, HF. Methodology: LC, SS, FZ, HD, SHW. Software: LC, FZ, SS. Validation: SHW, BDF, HF. Formal analysis: LC, HD. Investigation: FZ, SS, HD. Resources: SHW, BDF, HF. Data curation: LC, FZ, SS, HD. Writing – original draft: LC. Writing – Review & Editing: all authors. Visualization: LC, BDF. Supervision: BDF, HF. Project administration: BDF, HF. Funding acquisition: BDF, HF.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cosentino, L., Witt, S.H., Dukal, H. et al. Methyl-CpG binding protein 2 expression is associated with symptom severity in patients with PTSD in a sex-dependent manner. Transl Psychiatry 13, 249 (2023). https://doi.org/10.1038/s41398-023-02529-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-023-02529-9
