
Abstract
Methanotrophic microorganisms play a critical role in controlling the flux of methane from natural sediments into the atmosphere. Methanotrophs have been shown to couple the oxidation of methane to the reduction of diverse electron acceptors (e.g., oxygen, sulfate, nitrate, and metal oxides), either independently or in consortia with other microbial partners. Although several studies have reported the phenomenon of methane oxidation linked to selenate reduction, neither the microorganisms involved nor the underlying trophic interaction has been clearly identified. Here, we provide the first detailed evidence for interspecies electron transfer between bacterial populations in a bioreactor community where the reduction of selenate is linked to methane oxidation. Metagenomic and metaproteomic analyses of the community revealed a novel species of Methylocystis as the most abundant methanotroph, which actively expressed proteins for oxygen-dependent methane oxidation and fermentation pathways, but lacked the genetic potential for selenate reduction. Pseudoxanthomonas, Piscinibacter, and Rhodocyclaceae populations appeared to be responsible for the observed selenate reduction using proteins initially annotated as periplasmic nitrate reductases, with fermentation by-products released by the methanotrophs as electron donors. The ability for the annotated nitrate reductases to reduce selenate was confirmed by gene knockout studies in an isolate of Pseudoxanthomonas. Overall, this study provides novel insights into the metabolic flexibility of the aerobic methanotrophs that likely allows them to thrive across natural oxygen gradients, and highlights the potential role for similar microbial consortia in linking methane and other biogeochemical cycles in environments where oxygen is limited.
Similar content being viewed by others

Introduction
Methanotrophic microorganisms play a major role in the cycling of methane (CH4) in both oxic and anoxic environments [1, 2], with relevance to global carbon transformations and the Earth’s climate. Aerobic methanotrophs have been identified within several bacterial lineages of the Proteobacteria and Verrucomicrobia, and are widely distributed in nature [3]. The key enzyme possessed by these microorganisms is an oxygen-dependent methane monooxygenase (MMO), catalyzing the oxidation of methane to methanol [3]. Methanotrophs possessing an MMO are routinely observed in hypoxic environments, where they have been suggested to utilize alternate electron acceptors (e.g., nitrate), or fermentation pathways, to achieve redox balance for oxygen-limited growth [4,5,6,7].
In anoxic environments, the bacterium “Candidatus Methylomirabilis oxyfera” has been shown to couple MMO-mediated methane oxidation to the reduction of nitrite by generating its own oxygen from nitric oxide [8]. Several uncultured anaerobic methanotrophic (ANME) archaea oxidize methane, essentially through the reversal of the classical methanogenesis pathway, in consortia with sulfate-reducing bacteria (SRB) [9,10,11]. In addition, members of the Methanoperedenaceae (formerly ANME-2d), can also perform nitrate- and metal-oxide-dependent anaerobic oxidation of methane independent of a syntrophic partner [12,13,14,15,16], and have the genetic potential to utilize other terminal electron acceptors, including selenate [17].
Selenium (Se) compounds are abundant in the Earth’s crust and have been found in different aquatic ecosystems, such as agricultural drainage and wetland sediments [18, 19], which are known hot spots for methane cycling [20,21,22]. Selenium is mainly found in the form of selenate (SeO42−), selenite (SeO32−), and elemental selenium (Se0), with the former two being more mobile and toxic than the latter. Transformations between these states of Se can be mediated by both chemical and biological agents [23]. High concentrations of selenium in the environment may result in significant ecological damage, with only one order of magnitude separating essential and toxic levels for living organisms [23]. Anthropogenic activities associated with a variety of industries, including production of photoelectric devices, coal mining and combustion, and flue-gas desulfurization [18, 24], are important sources for selenium pollution, leading to a wider distribution in natural environments.
Although the phenomenon of methane oxidation linked to selenate reduction has been reported [25,26,27], neither the microorganisms involved nor the underlying trophic interaction has been resolved. To fill this knowledge gap, we established a bioreactor microbial consortium from wetland sediments that coupled methane oxidation to the reduction of selenate. Spectroscopy, isotope tracer experiments, metagenomics combined with metaproteomics, and gene knockout studies were applied to understand trophic interactions in the enriched microbial community. Based on the results, we propose a novel, synergistic pathway for methane-fueled selenate reduction with organic acids acting as interspecies electron shuttles.
Results and discussion
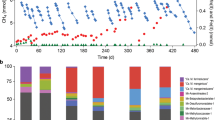
A wetland sediment sample was used to inoculate a membrane biofilm batch reactor system (MBBR; Fig. S1) fed with methane and selenate as the sole electron donor and acceptor, respectively, under hypoxic conditions. A stable selenate reduction rate of 9.4–11.7 μM/day was achieved after 1 month of operation (Fig. S2). Batch tests containing microbes from the established MBBR system confirmed the oxidation of supplemented 13C-labeled methane to 13CO2 during 40 days (Fig. 1a). In parallel, a total of ~550 μM selenate (added in three lots) was reduced to selenite (~340 μM) and elemental selenium (~210 μM; Fig. 1a). The formation of precipitated elemental selenium was shown by scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS) (Fig. 1b, c). These results demonstrated a stoichiometric balance for Se, with selenite and elemental selenium as the products of selenate reduction. However, selenate was not consumed in the absence of methane, indicating that its reduction was strictly dependent on the addition of methane or other carbon sources (Fig. S3).

a Concentrations of selenate (black squares), selenite (red circles), elemental selenium (gray diamonds), and 13CO2 (blue triangles) during oxidation of 13CH4 and selenate reduction. The symbols show the average of three replicates, and error bars indicate standard deviations. b SEM image of Se nanospheres produced during these incubations. Scale bar, 1 μm. c Se 3d XPS spectrum of the precipitates.
In order to understand the metabolic pathways responsible for the observed transformations, metagenomics and metaproteomics were applied to samples collected at the end of the batch incubations. Metagenomics enabled the recovery of 15 metagenome assembled genomes (MAGs), after quality filtration, accounting for ~90% of the sequenced reads (Table 1 and Fig. S4). Two MAGs were associated with canonical aerobic Type II methanotrophs, being classified with the Genome Taxonomy Database (GTDB) [28] as novel species within the genera Methylocystis (S2B18) and Methylosinus (S3B16), respectively. These MAGs encoded and expressed the complete aerobic methane oxidation pathway, including a particulate methane monooxygenase (pMMO), methanol dehydrogenase, the tetrahydromethanopterin (H4MPT)-linked C1 transfer pathway and a formate dehydrogenase (Table S1). Together these two methanotrophic species accounted for 14.5% of the total sequenced reads. None of the other populations possessed the annotated genetic potential for methane metabolism (Table 1). No archaea were detected in abundance in the present study (~0.3% of the reads). This is in contrast to a similar MBBR seeded from the system described here, where Archaea, predominantly the genus Methanosarcina spp., made up ~2.5% of the microbial community [27]. Methanosarcina is typically known as a methanogen but was suggested to oxidize methane in that system through a reversal of the methanogenic pathway like related ANME. Batch experiments with the biomass of that system using selective inhibitors of MMO (acetylene) and methyl-coenzyme M reductase (bromoethanesulphonate) suggested that both Methanosarcina and Methylocystis were important for the observed methane-linked selenate reduction, but the metabolic mechanism was not further investigated [27].
The ability of aerobic methanotrophs to thrive under hypoxic conditions is consistent with several previous pure culture studies and environmental surveys [5, 29,30,31,32]. Under hypoxic conditions, the limited available oxygen is known to be used for methane oxidation to methanol, whilst net electron balance is achieved through the reduction of nitrate or fermentation of methane-derived formaldehyde [4, 5, 7]. In our experiments, the fermentation of formaldehyde by the abundant methanotrophs was supported by the accumulation of micromolar organic acids, including formate, acetate, propionate, butyrate, and lactate, in the absence of selenate (Fig. S5 and Table S2). The ratios of isotopically labeled carbon could not be measured for individual organic compounds due to their very low concentrations. However, the 13C/(13C + 12C) ratio was found to increase dramatically for total dissolved carbon (i.e., inorganic plus organic) but not for total inorganic carbon (Fig. S6), in selenate-free incubations supplemented with 13CH4. These observations strongly support the idea that the methanotrophs are oxidizing methane and fermenting the resulting formaldehyde to organic compounds.
The dominance of aerobic methanotrophs in the system is interesting given that oxygen was not supplied to the batches and yet was consistently detected at low levels during the incubations (<250 nM). A supply of around 250 μM O2 would be required to account for the amount of methane oxidized (i.e., ~250 μM CH4, calculated from the produced 13CO2 in Fig. 1a) by the expressed pMMOs of Methylocystis (MAG ID in Table 1: S2B18) and Methylosinus (S3B16) in this study. To investigate the possibility that oxygen was produced as a metabolic intermediate, several 18O-labeling batch experiments were performed similar to those used to confirm the intra-aerobic pathway utilized by “Ca. M. oxyfera” [8]. No 18O2 was ever detected in the headspace when the biomass was incubated with either 18O-selenate or 18O-H2O in the presence of methane (Table S3), suggesting that an analogous intra-oxygenic pathway to “Ca. M. oxyfera” is unlikely. The methanotrophs may be able to scavenge any oxygen leaking into the vials [33], although substantial ingress was ruled out by parallel abiotic incubations (Fig. S7). Several previous studies have described similar observations, in which aerobic methane oxidation also preceded despite very low O2 concentrations [5, 30, 31]. For example, Kits, et al. [5] reported methane oxidation by the aerobic methanotroph Methylomonas when the O2 concentration was below 50 nM throughout the experiment. Further investigation of the potential for cryptic oxygen cycles in experimental and natural ecosystems is warranted.
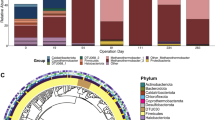
Unexpectedly, none of the MAGs generated in this study encoded a specific selenate reductase (e.g., Ser, Srd) [34, 35]. However, phylogenetic analyses of annotated members of the dimethyl sulfoxide (DMSO) reductase superfamily revealed that some of the MAGs possessed genes that clustered with periplasmic nitrate reductases known to reduce selenate [36, 37]. These enzymes were found in Pseudoxanthomonas sp. (S2B13), Piscinibacter sp. (S3B8), and unclassified Rhodocyclaceae (S3B23) (Figs. 2 and S8, Table S4). Selenate reduction was previously reported for members of the family Rhodocyclaceae [38], but not for the genera Pseudoxanthomonas or Piscinibacter.

Amino acid sequences recovered from the enrichment culture metagenomes (Table 1) are highlighted in yellow, blue, and gray. Putative selenate reductases are shown in red typeface. Periplasmic nitrate reductases indicated by asterisks have previously been shown to have selenate reductase activity. Red and green circles indicate 100% and >90% bootstrap support (1000 replicates), respectively. Full names and accession numbers of references are presented in Table S4.
To experimentally validate the proposed role of these periplasmic nitrate reductases in selenate reduction, gene-knockout experiments were performed with an isolate of Pseudoxanthomonas wuyuanensis [39] that possessed a napA gene similar to the enzymes above (i.e., percent identity = 74.6–86.6%, query coverage = 88.0–99.0%). Successful knockout of the napA gene in this isolate was confirmed by the absence of PCR amplification of DNA from the mutant with primers targeting the gene (Fig. 3a) and a relatively shorter product with primers targeting motifs flanking the gene (Fig. 3b). Oxygen respiration of wild and mutant P. wuyuanensis strains was similar (Fig. 3c), indicating that the napA-deletion did not harm the essential metabolism of this facultative anaerobic microorganism. Despite not possessing a canonical selenate reductase, the wild-type P. wuyuanensis was able to reduce selenate, with observed upregulation of the napA gene by 101.1 ± 8.0 fold when selenate was added to the media (Fig. 3d). Conversely, the napA-deletion mutant could not reduce selenate, providing strong evidence for the role of the annotated periplasmic nitrate reductase in selenate reduction in this microorganism and, based on the presence of very similar napA, in Pseudoxanthomonas sp. (S2B13), Rhodocyclaceae (S3B23) and Piscinibacter sp. (S3B8) in our study.

a PCR amplification of the napA gene, with a product size of 152 bp, showing a negative result for the mutant napA-knockout strain. b PCR amplification with primers targeting motifs flanking the napA gene showing amplified product of 3007 and 514 bp for the wild-type and napA-knockout mutant, respectively. The numbers in a, b correspond to bp. Electron acceptor profiles for the wild-type and mutant strains grown with c oxygen and d selenate. Error bars indicate standard deviations. d The transcriptomic activity of napA of the wild strain was determined at 0 and 44 h in triplicate for the culture grown on selenate.
The enzymes involved in the observed reduction of selenite by the MBBR community were less easily identified. The enzymes responsible for selenite reduction are generally less well studied and few specific reductases have been reported in the literature. The selenate-reducing Pseudoxanthomonas sp. (S2B13) and Piscinibacter sp. (S3B8) are potentially also responsible for the reduction of selenite to elemental selenium, with both possessing genes of the NIR/SIR reductase family, as some members of this family have shown the ability of selenite reduction (Fig. S9, Table S5) [40, 41]. No genes were identified in this study that had close homology with the few genes with reported selenite-reducing activity belonging to the fumarate reductase (FAD-binding superfamily) and NADH:flavin oxidoreductase (Old Yellow Enzyme family) (Figs. S10 and S11, Tables S6 and S7) [42,43,44]. It is worth noting that selenite reduction is often associated with detoxification, with several biomolecules also shown to have selenite-reducing activity, including glutathione, glutaredoxin, and siderophores [45].
As none of the putatively identified selenate-reducing populations (i.e., Pseudoxanthomonas, Rhodocyclaceae, and Piscinibacter) had the annotated-potential for methane oxidation, they likely relied on the methane-derived fermentation by-products released by methanotrophs (Figs. S5 and S6, Table S2). In support of this hypothesis, compared to the average rate of selenate reduction in the methane-fed incubation (i.e., 1.15 ± 0.04 μM-h−1, Fig. 4a), the rates were an order of magnitude higher for parallel incubations with formate (10.20 ± 0.37 μM-h−1, Fig. 4b), acetate (8.19 ± 0.64 μM-h−1, Fig. 4c), propionate (7.60 ± 0.95 μM-h−1, Fig. 4d), butyrate (7.79 ± 0.52 μM-h−1, Fig. 4e), and lactate (8.08 ± 0.80 μM-h−1; Fig. 4f), suggesting that these identified intermediates were preferred electron donors for selenate reduction by these bacteria.

The enrichment was incubated with methane (a), formate (b), acetate (c), propionate (d), butyrate (e), and lactate (f). Error bars indicate standard deviations of triplicate setups. The rates, as shown in each graph, are calculated by fitting the curves of which the confidence levels are 95%. Please note the different x-axis scales between (a) and (b–f).
Genes responsible for the activation of formate, acetate, propionate, and lactate were encoded in the MAGs representing all three of the putative selenate-reducing community members, as well as other heterotrophic populations. The expression of these genes was detected during methane-driven selenate reduction (Figs. 4, 5 and S12). The absence of canonical butyrate-activating genes suggests an alternate pathway for butyrate metabolism in these selenate-reducing populations, noting that the addition of this substrate to batches stimulated selenate reduction (Fig. 4e). The importance of fatty acids as interspecies electron shuttles was supported by the diffuse floc structure of the biomass and the presence of suspended methanotroph cells (Fig. S13). In contrast, direct interspecies electron transfer (DIET) would require microcolonies with much more compact physical association [46, 47]. Genes for multi-heme cytochromes or pili associated with DIET were also not annotated in the MAGs representing the methanotrophs and putative selenate reducers in this study, lending more supports to the involvement of interspecies electron shuttles.

The color of the symbols indicates the number of expressed unique peptides identified with proteomics. Empty symbols indicate genes missing in the corresponding MAGs. Pmo particulate methane monooxygenase, Medh PQQ-dependent methanol dehydrogenase, Fae formaldehyde-activating enzyme, Fdh NAD-dependent formate dehydrogenase, Gly serine hydroxymethyltransferase, AGXT serine-glyoxylate aminotransferase, Hpr hydroxypyruvate reductase, Gck glycerate kinase, ENO enolase, Pyk pyruvate kinase, Ppc phosphoenolpyruvate carboxylase, Madh malate dehydrogenase, Mtk malate-CoA ligase, Mcl malyl-CoA lyase, Fum fumarate hydratase, Sdh succinate dehydrogenase, CS citrate synthase, Ace pyruvate dehydrogenase, Pta phosphate acetyltransferase, Ack acetate kinase, Lld L-lactate dehydrogenase, Suc succinyl-CoA synthetase, Mut methylmalonyl-CoA mutase, Mce methylmalonyl-CoA epimerase, Mmd methylmalonyl-CoA decarboxylase, Pct propionyl-CoA transferase, Aca acetyl-CoA C-acetyltransferase, Hbd 3-hydroxybutyryl-CoA dehydrogenase, Ech enoyl-CoA hydratase, Ccr crotonyl-CoA reductase, Bct butyryl-CoA transferase, Nap periplasmic nitrate reductase, Nir copper-containing nitrite reductase, Pcs propionyl-CoA synthetase, Acs acetyl-CoA synthetase, Ldh lactate dehydrogenase.
To compare the contribution of the two populations of methanotrophs to fatty acid production, we measured the δ13C of each population with proteomics [48]. When incubated with unlabeled methane, Methylocystis and Methylosinus had quite different δ13C values of −14.3‰ and −43.9‰, respectively (Table 1). Being Type II methanotrophs, these bacteria use the serine cycle to assimilate approximately equal amounts of formaldehyde and CO2, both originating from methane oxidation [3, 49,50,51]. Because the CO2 in the external medium is heavier than methane, the lower δ13C value for Methylosinus is consistent with it completely oxidizing more of the supplied methane and assimilating the lighter methane-derived CO2 relative to Methylocystis, where the latter methanotroph is suggested to release more 13C-depleted intermediates and assimilate heavier CO2 from the culture medium. As such, Methylosinus appears to be more reliant on scavenging available oxygen and less on fermentation for redox balance. These results are consistent with the observation that the Methylocystis MAG possesses a more extensive suite of genes for fermentation than the Methylosinus MAG which would rely more on fermentation to acetate and formate (Fig. 5).
Among the putative selenate reducers, Pseudoxanthomonas sp. displayed a δ13C value of −31.8‰, suggesting its assimilation of isotopically depleted methane-derived intermediates. The Rhodocyclaceae displayed a much higher δ13C value of −10.1‰, indicating that this population incorporated less of the organics into its biomass and possibly fixed heavier carbon from CO2 via the Calvin Cycle annotated in the representative MAG (Fig. S14).
In conclusion, we report a novel trophic relationship linking methane oxidation to selenate reduction under hypoxic conditions. In our bioreactor enrichment community, a species of Methylocystis appeared to be primarily responsible for the oxidation and fermentation of methane-derived carbon to organic acids, which were in turn utilized by co-abundant heterotrophic populations as a source of carbon and electrons for selenate reduction. This novel synergistic process may link the global carbon and selenium cycles in a new way, supported by the widespread co-occurrence of the key bacterial genera in natural environments, at relative abundances of >1.0%, including the wetland sediment used to seed the MBBR (Fig. S15 and Dataset S1). The discovered trophic interaction could also potentially be applied for the treatment of wastewater or contaminated environmental sites, as the reduction product Se0 is insoluble and can be easily removed from the environment by filtration [52].
Although previous studies have suggested that methane-derived carbon released by methanotrophs supports the growth of a broad range of co-existing heterotrophic populations in the environment [53,54,55,56], specific trophic relationships have not been identified. Diffusible intermediates of methane oxidation—such as hydrogen, formate, acetate, methanol, and methanethiol—have been suggested to mediate the transfer of electrons from anaerobic archaeal methanotrophs to their SRB partners [57,58,59,60], but none of these have been confirmed experimentally. Subsequent studies have demonstrated that DIET is more probable for the ANME-SRB consortia [46, 47, 61]. Our study provides a clear example of community metabolism of methane and highlights the possibility that similar trophic relationships may link methane metabolism with a range of terminal electron acceptors in hypoxic environments. Our study highlights the metabolic flexibility of aerobic methanotrophs, likely contributing to their success along the entire oxygen gradient of natural systems.
Materials and methods
Description of the inoculum
In this study, a membrane biofilm batch reactor was set up to enrich for a microbial community coupling methane oxidation to the reduction of selenate (Fig. S1). The reactor had 32 ~10-cm long composite bubble-less hollow fibers with 280-μm outer diameter and 180-μm inner diameter, and a total membrane surface area of 2.8 × 10−3 m2 (Mitsubishi Rayon). Fibers were glued and sealed in Norprene tubing and supplied with CH4 at a pressure of 15 psig (1.97 atm). The MBBR had an overall volume of 1200 mL, and active volume of 1000 mL of liquid. A magnetic stirrer (HJ-1, Xinbao, Ltd., China) was used to mix the contents of reactor. The reactor was operated in batch mode at a temperature of 35 ± 1 °C and pH at 7.2 ± 0.2 throughout the experiment. Dissolved O2 in the medium was calculated to be <250 nM according to Henry’s Law, based on the measured O2 concentration in headspace being below the detection limit (i.e., 200 ppm) of gas chromatography (GC, Agilent 7890A, USA). The GC was configured with Molesieve 5A column and thermal conductivity detector, using Argon as the carrier gas. The MBBR was inoculated with a sediment sample (~5 g) from the wetland in Zhejiang University (30°17′53″ N and 120°05′10″ E, Hangzhou, China) and 1000 mL of anoxic mineral salt medium (Table S8). Selenate was introduced at a concentration of ~30 mg Se/L in the form of selenic acid solution and was replenished after being completely consumed.
Selenate reduction linked to methane oxidation in serum bottles
For batch experiments, the MBBR was gently shaken to detach the biofilm on Day 328. Approximately 20 mL of the suspended enriched culture from the MBBR was inoculated into 120-mL serum bottles containing 60 mL of anoxic medium. All the bottles were sparged with Argon for 25 min, and then tightly closed with butyl rubber stoppers, sealed with crimped aluminum caps. Stock solutions of selenate were added to the medium at a final concentration of 185 μM, serving as the sole electron acceptor, and 10 mL of 13C-labeled CH4 (99.5% 13CH4, purchased from Sigma-Aldrich) was introduced simultaneously as the sole electron donor. The electron equivalents of the supplied methane (8 mol e− per mol) was in excess of that required to completely reduce the supplied Se(VI) to Se(0). Selenate was re-introduced when its concentration reached below 10 μM. The O2 in the headspace was measured over time and the calculated dissolved oxygen was always below the detection limit of 250 nM. For the negative controls, only selenate or 13CH4 (at the same concentration) was added. Experimental batches were performed in triplicate and negative controls in duplicate.
Comparison of selenate reduction with different electron donors
To assess the ability of the enrichment community to utilize fermentation products, batch experiments were set-up in 120-ml bottles containing ~20 mL enriched culture from the MBBR (Day 433). For the methane-fed batches, enrichment cultures were amended with selenate at a final concentration of 120 μM and 10 mL of CH4 (0.45 mmol). For the organic-fed batches, enrichment cultures were amended with selenate at a final concentration of ~200 μM and formate of 22.5 mM (1.8 mmol), acetate of 5.62 mM (0.45 mmol), propionate of 3.22 mM (0.26 mmol), butyrate of 2.25 mM (0.18 mmol), and lactate of 3.75 mM (0.3 mmol). The concentration of the added electron donor was selected to give the same electron equivalents (based on: 8 mol e− per mol of methane; 2 mol e− per mol of formate; 8 mol e− per mol of acetate; 14 mol e− per mol of propionate; 20 mol e− per mol of butyrate; 12 mol e− per mol of lactate) and in excess of that required to fully reduce Se(VI) to Se(0). Batches were performed in triplicate.
Exploration of oxygen source
To assess the possible intrusion of air into batch vials, sterile anoxic medium was introduced in similar serum bottles and degassed with Argon as described above. After closing and sealing, oxygen gas was injected to give a final concentration of ~0.4% and 2.4% (v/v), respectively. These bottles were set up in triplicate and incubated with the same conditions as detailed for the batch experiments.
To investigate possible metabolic sources of oxygen, batch experiments were performed as detailed earlier with several combinations of 18O-enriched components: (1) abiotic setup with medium only; biotic setups with (2) CH4, Se18O42−, and H216O; (3) CH4, Se16O42−, and H218O; (4) CH4 and H218O; (5) Se18O42− and H216O. Incubations with 18O-water were used to cover other metabolites as potential sources of O2—reliant on 18O exchange with the added water—as applied previously to assess nitrate as the source of oxygen for “Ca. M. oxyfera” [8]. Incubations with 18O-enriched selenate were included to assess the possibility that O2 was derived from selenate, noting that there will be no 18O exchange between selenate and water under the batch incubation conditions [62].
18O-enriched selenate was synthesized through oxygen isotope exchange between 16O-selenate and 18O-enriched water following the reported protocol [62]. Briefly, 18O-enriched water (18O, 98%, Shanghai Research Institute of Chemical Industry) was incubated with selenate at pH of 1.00 and 80 °C for 100 h. Then the pH was adjusted to 6–7 by NaOH to quench the isotope exchange reaction. The mixed solution was evaporated and complemented using 16O-water five times to achieve the final 18O-enriched selenate stock solution. The ratio of 18O to total oxygen atoms is 3.23 ± 0.28% for the synthetic 18O-enriched selenate, compared to 0.20 ± 0.0015% for the commercial 16O-selenate.
Chemical analysis procedures
Liquid samples were filtered with a 0.22-μm membrane filter, and selenate, selenite, organic acids, and nitrate concentrations were measured using ion chromatography configured with an AS 11 column (Dionex ICS-1500, ThermoFisher Scientific). For selenate, selenite, and nitrate, the concentration of the eluent KOH was set at 20 mM. To better quantify organic acids (e.g., formate, acetate, propionate, butyrate, and lactate), the eluent concentration was set at 0.5 mM initially for 80 min, and then increased to 40 mM to wash the column. Precipitated elemental selenium was centrifuged, washed with phosphate buffer, and digested by 69% HNO3 at 100 °C for 10 min, after which the total soluble Se was measured using inductively coupled plasma-mass spectroscopy (ICP-MS, PerkinElmer NexION 300X). The quantity and isotopic composition of 13CO2 in the headspace was measured using GC/MS (GC 7890A coupled to MSD 5977B, Agilent). The 13DIC (Dissolved Inorganic Carbon) in the liquid was measured using isotope-ratio mass spectrometry (IR-MS, MAT 253, Thermo Scientific). Total 13CO2 was calculated as the sum of 13CO2 in the headspace and 13DIC in the liquid, and then normalized by the total liquid volume. The total carbon (i.e., inorganic and organic) in the liquid was measured using the same mass spectrometry as described above, by oxidizing the solution with a mixture of H2SO4 and KMnO4 to convert all the organic carbon to CO2. Please note that the oxidants H2SO4 and KMnO4 would not oxidize any CH4 present, excluding the contribution of supplied 13C–CH4 to any observed increase in the 13C/(12C + 13C) ratios. In support of this, Day 0 samples for total carbon measurements were taken after injecting CH4 and there was no difference in measured 13C/(12C + 13C) ratios between the 13C–CH4 and 12C–CH4 amended batches (Fig. S6). The 18O2 in the headspace was measured using IR-MS (Sercon 20–22) configured with Molesieve 5 A column.
The oxygen isotopic compositions of selenate were measured according to the previously described method [62]. Briefly, selenate was precipitated by reaction with excess BaCl2. Produced BaSeO4 was centrifuged, with the pellet dried and reacted with glassy carbon at 1300 °C in silver capsules to completely convert selenate-derived oxygen to CO. Released CO was loaded into IR-MS (MAT 253, Thermo Scientific) for measuring δ18O values. The δ18O value of CO is theoretically equal to that of selenate.
Electron spectroscopy analysis of the precipitates was performed with SEM (Hitachi) following the manufacturer’s instructions. An electron spectrometer (Thermo Electron) with Al Kλ radiation (1486.6 eV) at 150 W was used to analyze the valence state of Se in lyophilized samples.
DNA extraction and sequencing
The DNA was extracted from the wetland inoculum using the DNeasy PowerSoil Kit (QIAGEN, USA) according to the instruction manual, and then used as template for the PCR amplification of the 16S rRNA gene with 341F (5′-CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACNNGGGTATCTAAT-3′) primers. The resulting amplicons were sequenced on the MiSeq platform (Illumina). Community profiles from this sequencing data were generated with the SHAMAN platform [63].
DNA samples for metagenomic analyses were collected at the end of the batch experiment that was incubated with 13C-labeled CH4 and selenate (Fig. 1). We first centrifuged the suspensions in serum bottles at 6000 × g for 20 min at 4 °C, and then extracted DNA from the biomass pellet using the DNeasy PowerSoil Kit. Shotgun metagenomic sequencing (2 × 150 bp) was performed using the HiSeq X Ten (Illumina) for 4 replicate samples. The TruSeq DNA Nano Library Prep Kit was used for library preparation. For the first sample, about 70 million paired-end reads were generated; for the latter three samples, 201–216 million reads per sample were produced.
The quality of the raw data was confirmed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The partial primers and adapters were trimmed from the reads using the BBmap package (https://sourceforge.net/projects/bbmap/), and reads were filtered out which: (i) contained the spike-in PhiX sequences; (ii) were low-quality (Phred Quality Score <15) or short (<30 bp); (iii) had low complexity. All reads that passed quality control were assembled into contigs individually using metaSPAdes [64] and collectively using MEGAHIT v1.1.1 [65], yielding five assemblies (four individual samples and one co-assembly). Quality-controlled reads were mapped to assembled contigs (>500 bp) and annotated using MetaErg [66]. Metabat and CheckM were used to bin the contigs and select the best binning results, respectively [67, 68]. All the bins were combined and dereplicated using dRep to obtain the final high-quality draft genomes [69].
The bacterial genome tree was constructed based on the concatenated 120 bacterial-specific conserved marker genes using the Genome Taxonomy Database (GTDB v2.3.2) [28]. The toolkit GTDB-Tk was performed to identify, align, and concatenate marker genes in each genome, and further infer the tree topology and classify the MAGs [70]. The protein phylogenetic trees were constructed using MEGA 6 program with recovered sequences in the study and selected references from NCBI public database [71].
Gene knockout and physiology test
The Pseudoxanthomonas wuyuanensis isolate (CGMCC: 1.10978) was purchased from the Institute of Microbiology, Chinese Academy of Sciences. The whole-genome sequence of this isolated has previously been deposited in the NCBI database under the accession number of OCND00000000. The upstream and downstream fragments of napA were amplified using the primers Up-FsmaI (5′-TCCCCCGGGGCATCAGCCAAGCCGTGATT-3′) and Up-R (5′-TACGCATGACAACAACCCTCCAGTCTGCTCCGGTCGGTACG-3′), Down-F (5′-ACCGACCGGAGCAGACTGGAGGGTTGTTGTCATGCGTA-3′) and Down-KnpIR (5′-GGGGTACCGGCCTTCAGGCGATCCCATTC-3′), with the genomic DNA of the wild strain as the template. The generated fragments were recovered and ligated by PCR. The joint fragment was then digested with SmaI and KpnI, as well as the vector pTCX18Cm, and ligated to produce the pT-ΔnapA. The synthetic plasmid was transformed into P. wuyuanensis competent cells using electroporation. Transformed cells were plated on agar plates containing clindamycin and kanamycin for selection of the first homologous recombination. The mutants were further identified via sucrose counter-selection of the second homologous recombination [72].
The potential mutants were confirmed by PCR amplification using primers V17m (5′-TGGACVATGGGYTTYAAYC-3′) and napA4r (5′-ACYTCRCGHGCVGTRCCRCA-3′) [73], and Conf-F (5′-GGTGCATATCGCCAGTTTCGT-3′) and Conf-R (5′-GCTGCCTTCCTGGTCCACCG-3′). The former pair of primers target sites within the napA gene, while the latter target sites up- and downstream of the gene. The respective amplified products are 152 and 3007 bp for wild-type strain, and absent and 514 bp for the mutant strain. Both the wild-type strain and confirmed mutants were grown in trypticase soy broth medium to an OD600 of 0.3, and incubated with O2 and selenate in serum bottles using the above mineral salt medium amended with glucose as the carbon and electron source. RNA samples of wild-type strains amended with selenate were extracted, reverse-transcribed, and quantified using the primer V17m and napA4r at 0 and 44 h. The 2(-Delta Delta C(T)) method was used to calculate the relative expression of napA [74], using amplification of the 16S rRNA gene, with the primer 16SF (5′-GTGSTGCAYGGYTGTCGTCA-3′) and 16SR (5′-ACGTCRTCCMCACCTTCCTC-3′) [75], as a reference.
Protein extraction, identification, and quantification
For protein analyses, the biosample was sonicated on ice in lysis buffer (8 M urea, 1% Protease Inhibitor Cocktail). The remaining debris was removed by centrifugation at 12,000 g at 4 °C for 10 min. Finally, the supernatant was collected and the protein concentration was determined with BCA kit according to the manufacturer’s instructions. For digestion, the protein solution was reduced with 5 mM dithiothreitol for 30 min at 56 °C and alkylated with 11 mM iodoacetamide for 15 min in darkness. Trypsin was added at 1:50 trypsin-to-protein mass ratio for the first digestion overnight and 1:100 trypsin-to-protein mass ratio for a second 4 h-digestion. The tryptic peptides were dissolved in 0.1% formic acid (solvent A), directly loaded onto a custom-made reversed-phase analytical column (15-cm length, 75 μm i.d.). The gradient was comprised of an increase from 6 to 23% solvent B (0.1% formic acid in 98% acetonitrile) over 26 min, 23 to 35% in 8 min and climbing to 80% in 3 min then holding at 80% for the last 3 min, all at a constant flow rate of 400 nl/min on an EASY-nLC 1000 UPLC system.
The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Q Exactive Plus (Thermo) coupled online to the UPLC. The m/z scan range was 350–1800 for a full scan, and intact peptides were detected in the Orbitrap at a resolution of 70,000. The resulting MS/MS data were processed using Maxquant search engine (v.1.5.2.8) [76]. Tandem mass spectra were searched against the target proteomic database concatenated with a reverse decoy database. The target database was created by combining all protein sequences generated from the metagenomic data and those downloaded from the NCBI database of reference microorganisms (Table S9). The final database contained 396,698 protein sequences. FDR was used for protein identification, which was adjusted to <1%, and the minimum score for peptides was set at >40. The stable isotope fingerprints were performed by the Calis-p software package as previously described [48]. In brief, the Calis-p program first (1) extracted isotope peak intensities for identified peptides, (2) pre-filtered and cleaned isotope patterns on the basis of peptide ID confidence scores and peak intensity, (3) estimated peptide 13C/12C ratios by fitting the experimental spectra using Fast Fourier Transforms, (4) reported δ13C values for all peptides and calculated averages and standard errors for each taxon, and finally (5) corrected δ13C values using the known δ13C value of the reference material.
Fluorescence in situ hybridization
FISH was performed essentially as previously detailed [77]. Biomass was fixed with 4% paraformaldehyde (w/v) and stored at −20 °C. The 5′ and 3′ ends of the oligonucleotide FISH probe was labeled with the Alexa488 dye (Integrated DNA technologies, Singapore). The NON-EUB nonsense probe was used as a negative hybridization control [78]. Type II methanotrophs were identified with the Mα450 probe at a hybridization formamide concentration of 30% (v/v) [79]. Cells were stained with DAPI (1 ng/µl) for 15 min in the dark. Microscopy was performed with a Stellaris5 white light laser confocal microscope (Leica, Germany).
Global distribution of key genera
The almost full-length 16S rRNA gene sequences of representative Methylocystis (NR_044946.1) and Pseudoxanthomonas (OCND00000000.1) species were used to screen Short Read Archive (SRA) datasets by IMNGS [80]. To explore the natural distribution of these two genera, the minimum identity threshold was set at 95% with a minimum nucleotide size of 200 [81]. Samples were tagged when (1) their category description was associated with a natural environment (e.g., soil) and (2) they contained both Methylocystis and Pseudoxanthomonas at relative abundance of more than 1.0%. Geographic locations of tagged samples were extracted and plotted using R packages (ggplot2, sf, rnaturalearth, and rnaturalearthdata).
Data availability
All raw Illumina metagenomic sequence data were submitted to the Sequence Read Archive under accession numbers SRP136677, SRP136696, SRP136790, and SRP136859. The 16S rRNA gene sequences generated from the wetland inoculum were submitted under the accession number SRR14328346. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [82] partner repository with the dataset identifier PXD011889. Public release of the PRIDE projects will be requested as soon as a citable pre-print is online. The username and password of temporary reviewer account are: reviewer66716@ebi.ac.uk and Y5JAcO59, respectively.
References
Trotsenko YA, Murrell JC. Metabolic aspects of aerobic obligate methanotrophy. Adv Appl Microbiol. 2008;63:183–229.
Knittel K, Boetius A. Anaerobic oxidation of methane: progress with an unknown process. Annu Rev Microbiol. 2009;63:311–34.
Khmelenina VN, Murrell JC, Smith TJ, Trotsenko YA. Physiology and Biochemistry of the Aerobic Methanotrophs. In: Rojo F (editor) Aerobic Utilization of Hydrocarbons, Oils and Lipids. Handbook of Hydrocarbon and Lipid Microbiology. Springer; Cham; 2018;1–25. https://doi.org/10.1007/978-3-319-39782-5_4-1.
Kalyuzhnaya MG, Yang S, Rozova ON, Smalley NE, Clubb J, Lamb A, et al. Highly efficient methane biocatalysis revealed in a methanotrophic bacterium. Nat Commun. 2013;4:2785.
Kits KD, Klotz MG, Stein LY. Methane oxidation coupled to nitrate reduction under hypoxia by the Gammaproteobacterium Methylomonas denitrificans, sp. nov type strain FJG1. Environ Microbiol. 2015;17:3219–32.
Kits KD, Campbell DJ, Rosana AR, Stein LY. Diverse electron sources support denitrification under hypoxia in the obligate methanotroph Methylomicrobium album strain BG8. Front Microbiol. 2015;6:1072.
Gilman A, Fu Y, Hendershott M, Chu F, Puri AW, Smith AL, et al. Oxygen-limited metabolism in the methanotroph Methylomicrobium buryatense 5GB1C. PeerJ. 2017;5:5.
Ettwig KF, Butler MK, Le Paslier D, Pelletier E, Mangenot S, Kuypers MM, et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature. 2010;464:543–8.
Devol AH, Ahmed SI. Are high-rates of sulfate reduction associated with anaerobic oxidation of methane. Nature. 1981;291:407–8.
Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A, et al. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 2000;407:623–6.
Orphan VJ, House CH, Hinrichs KU, McKeegan KD, DeLong EF. Methane-consuming archaea revealed by directly coupled isotopic and phylogenetic analysis. Science. 2001;293:484–7.
Ettwig KF, Zhu BL, Speth D, Keltjens JT, Jetten MSM, Kartal B. Archaea catalyze iron-dependent anaerobic oxidation of methane. Proc Natl Acad Sci USA. 2016;113:12792–6.
Cai C, Leu AO, Xie GJ, Guo J, Feng Y, Zhao JX, et al. A methanotrophic archaeon couples anaerobic oxidation of methane to Fe(III) reduction. ISME J. 2018;12:1929–39.
Leu AO, Cai C, McIlroy SJ, Southam G, Orphan VJ, Yuan Z, et al. Anaerobic methane oxidation coupled to manganese reduction by members of the Methanoperedenaceae. ISME J. 2020;14:1030–41.
Haroon MF, Hu S, Shi Y, Imelfort M, Keller J, Hugenholtz P, et al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature. 2013;500:567–70.
Shi LD, Guo T, Lv PL, Niu ZF, Zhou YJ, Tang XJ, et al. Coupled anaerobic methane oxidation and reductive arsenic mobilization in wetland soils. Nat Geosci. 2020;13:799–805. -+
Leu AO, McIlroy SJ, Ye J, Parks DH, Orphan VJ, Tyson GW. Lateral gene transfer drives metabolic flexibility in the anaerobic methane-oxidizing archaeal family Methanoperedenaceae. Mbio 2020;11:11.
Lemly AD. Aquatic selenium pollution is a global environmental safety issue. Ecotox Environ Safe. 2004;59:44–56.
Simmons DBD, Wallschlager D. A critical review of the biogeochemistry and ecotoxicology of selenium in lotic and lentic environments. Environ Toxicol Chem. 2005;24:1331–43.
Reim A, Luke C, Krause S, Pratscher J, Frenzel P. One millimetre makes the difference: high-resolution analysis of methane-oxidizing bacteria and their specific activity at the oxic-anoxic interface in a flooded paddy soil. ISME J. 2012;6:2128–39.
Danilova OV, Suzina NE, Van De Kamp J, Svenning MM, Bodrossy L, Dedysh SN. A new cell morphotype among methane oxidizers: a spiral-shaped obligately microaerophilic methanotroph from northern low-oxygen environments. ISME J. 2016;10:2734–43.
Smith GJ, Angle JC, Solden LM, Borton MA, Morin TH, Daly RA, et al. Members of the genus methylobacter are inferred to account for the majority of aerobic methane oxidation in oxic soils from a freshwater wetland. Mbio 2018;9:9.
Fernández-Martínez A, Charlet L. Selenium environmental cycling and bioavailability: a structural chemist point of view. Rev Environ Sci Biotechnol. 2009;8:81–110.
Chung J, Nerenberg R, Rittmann BE. Bioreduction of selenate using a hydrogen-based membrane biofilm reactor. Environ Sci Technol. 2006;40:1664–71.
Lai CY, Wen LL, Shi LD, Zhao KK, Wang YQ, Yang X, et al. Selenate and nitrate bioreductions using methane as the electron donor in a membrane biofilm reactor. Environ Sci Technol. 2016;50:10179–86.
Luo JH, Chen H, Hu S, Cai C, Yuan Z, Guo J. Microbial selenate reduction driven by a denitrifying anaerobic methane oxidation biofilm. Environ Sci Technol. 2018;52:4006–12.
Shi LD, Lv PL, Wang M, Lai CY, Zhao HP. A mixed consortium of methanotrophic archaea and bacteria boosts methane-dependent selenate reduction. Sci Total Environ. 2020;732:139310.
Parks DH, Chuvochina M, Chaumeil PA, Rinke C, Mussig AJ, Hugenholtz P. A complete domain-to-species taxonomy for bacteria and archaea. Nat Biotechnol. 2020;38:1079–86.
Jakobs G, Labrenz M, Rehder G, Hietanen S, Kießlich K, Vogts A, et al. A bioreactor approach to investigate the linkage between methane oxidation and nitrate/nitrite reduction in the pelagic oxic-anoxic transition zone of the central baltic sea. Front Mar Sci. 2016;3:3.
Naqvi SWA, Lam P, Narvenkar G, Sarkar A, Naik H, Pratihary A, et al. Methane stimulates massive nitrogen loss from freshwater reservoirs in India. Nat Commun. 2018;9:9.
Martinez-Cruz K, Leewis MC, Herriott IC, Sepulveda-Jauregui A, Anthony KW, Thalasso F, et al. Anaerobic oxidation of methane by aerobic methanotrophs in sub-arctic lake sediments. Sci Total Environ. 2017;607:23–31.
Zheng Y, Wang H, Liu Y, Zhu B, Li J, Yang Y, et al. Methane-dependent mineral reduction by aerobic methanotrophs under hypoxia. Environ Sci Tech Lett. 2020;7:606–12.
Oswald K, Milucka J, Brand A, Hach P, Littmann S, Wehrli B, et al. Aerobic gammaproteobacterial methanotrophs mitigate methane emissions from oxic and anoxic lake waters. Limnol Oceanogr. 2016;61:S101–S118.
Schroder I, Rech S, Krafft T, Macy JM. Purification and characterization of the selenate reductase from Thauera selenatis. J Biol Chem. 1997;272:23765–8.
Ridley H, Watts CA, Richardson DJ, Butler CS. Resolution of distinct membrane-bound enzymes from Enterobacter cloacae SLD1a-1 that are responsible for selective reduction of nitrate and selenate oxyanions. Appl Environ Microbiol. 2006;72:5173–80.
Sabaty M, Avazeri C, Pignol D, Vermeglio A. Characterization of the reduction of selenate and tellurite by nitrate reductases. Appl Environ Microbiol 2001;67:5122–6.
Turner RJ, Weiner JH, Taylor DE. Selenium metabolism in Escherichia coli. Biometals 1998;11:223–7.
Hunter WJ. An Azospira oryzae (syn Dechlorosoma suillum) strain that reduces selenate and selenite to elemental red selenium. Curr Microbiol. 2007;54:376–81.
Li D, Pang HC, Sun LC, Fan JP, Li YY, Zhang JL. Pseudoxanthomonas wuyuanensis sp. nov., isolated from saline-alkali soil. Int J Syst Evol Microbiol. 2014;64:799–804.
Harrison G, Curle C, Laishley EJ. Purification and characterization of an inducible dissimilatory type sulfite reductase from Clostridium pasteurianum. Arch Microbiol. 1984;138:72–8.
Basaglia M, Toffanin A, Baldan E, Bottegal M, Shapleigh JP, Casella S. Selenite-reducing capacity of the copper-containing nitrite reductase of Rhizobium sullae. FEMS Microbiol Lett. 2007;269:124–30.
Li DB, Cheng YY, Wu C, Li WW, Li N, Yang ZC, et al. Selenite reduction by Shewanella oneidensis MR-1 is mediated by fumarate reductase in periplasm. Sci Rep. 2014;4:3735.
Song DG, Li XX, Cheng YZ, Xiao X, Lu ZQ, Wang YZ, et al. Aerobic biogenesis of selenium nanoparticles by Enterobacter cloacae Z0206 as a consequence of fumarate reductase mediated selenite reduction. Sci Rep. 2017;7:3239.
Hunter WJ. A rhizobium selenitireducens protein showing selenite reductase activity. Curr Microbiol. 2014;68:311–6.
Nancharaiah YV, Lens PNL. Selenium biomineralization for biotechnological applications. Trends Biotechnol. 2015;33:323–30.
McGlynn SE, Chadwick GL, Kempes CP, Orphan VJ. Single cell activity reveals direct electron transfer in methanotrophic consortia. Nature. 2015;526:531–U146.
Wegener G, Krukenberg V, Riedel D, Tegetmeyer HE, Boetius A. Intercellular wiring enables electron transfer between methanotrophic archaea and bacteria. Nature. 2015;526:587–U315.
Kleiner M, Dong X, Hinzke T, Wippler J, Thorson E, Mayer B, et al. Metaproteomics method to determine carbon sources and assimilation pathways of species in microbial communities. Proc Natl Acad Sci USA. 2018;115:E5576–84.
Anthony C. How half a century of research was required to understand bacterial growth on C1 and C2 compounds; the story of the serine cycle and the ethylmalonyl-CoA pathway. Sci Prog. 2011;94:109–37.
Salem AR, Hacking AJ, Quayle JR. Cleavage of malyl-coenzyme-A into acetyl-coenzyme-A and glyoxylate by Pseudomonas AM1 and other C1-unit utilizing bacteria. Biochem J. 1973;136:89–96.
Large PJ, Peel D, Quayle JR. Microbial growth on C1 compounds. 3. Distribution of radioactivity in metabolites of methanol-grown Pseudomonas AM1 after incubation with 14C-methanol and 14C-bicarbonate. Biochem J 1962;82:483–8.
Shi LD, Wang Z, Liu T, Wu M, Lai CY, Rittmann BE, et al. Making good use of methane to remove oxidized contaminants from wastewater. Water Res. 2021;197:117082.
Chistoserdova L, Kalyuzhnaya MG. Current trends in methylotrophy. Trends Microbiol. 2018;26:703–14.
Karwautz C, Kus G, Stockl M, Neu TR, Lueders T. Microbial megacities fueled by methane oxidation in a mineral spring cave. ISME J. 2018;12:87–100.
Hernandez ME, Beck DAC, Lidstrom ME, Chistoserdova L. Oxygen availability is a major factor in determining the composition of microbial communities involved in methane oxidation. PeerJ. 2015;3:3.
Oshkin IY, Beck DA, Lamb AE, Tchesnokova V, Benuska G, McTaggart TL, et al. Methane-fed microbial microcosms show differential community dynamics and pinpoint taxa involved in communal response. ISME J. 2015;9:1119–29.
Hoehler TM, Alperin MJ, Albert DB, Martens CS. Field and laboratory studies of methane oxidation in an anoxic marine sediment—evidence for a methanogen-sulfate reducer consortium. Glob Biogeochem Cycles. 1994;8:451–63.
Nauhaus K, Treude T, Boetius A, Kruger M. Environmental regulation of the anaerobic oxidation of methane: a comparison of ANME-I and ANME-II communities. Environ Microbiol. 2005;7:98–106.
Meulepas RJW, Jagersma CG, Khadem AF, Stams AJM, Lens PNL. Effect of methanogenic substrates on anaerobic oxidation of methane and sulfate reduction by an anaerobic methanotrophic enrichment. Appl Microbiol Biotechnol 2010;87:1499–506.
Sorensen KB, Finster K, Ramsing NB. Thermodynamic and kinetic requirements in anaerobic methane oxidizing consortia exclude hydrogen, acetate, and methanol as possible electron shuttles. Micro Ecol. 2001;42:1–10.
Krukenberg V, Riedel D, Gruber-Vodicka HR, Buttigieg PL, Tegetmeyer HE, Boetius A, et al. Gene expression and ultrastructure of meso- and thermophilic methanotrophic consortia. Environ Microbiol. 2018;20:1651–66.
Kaneko M, Poulson SR. Rate of oxygen isotope exchange between selenate and water. Environ Sci Technol. 2012;46:4539–45.
Volant S, Lechat P, Woringer P, Motreff L, Campagne P, Malabat C, et al. Shaman: a user-friendly website for metataxonomic analysis from raw reads to statistical analysis. BMC Bioinform. 2020;21:345.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. Metaspades: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34.
Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, et al. Megahit v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 2016;102:3–11.
Dong XL, Strous M. An integrated pipeline for annotation and visualization of metagenomic contigs. Front Genet. 2019;10:999.
Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. Metabat 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Olm MR, Brown CT, Brooks B, Banfield JF. Drep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–8.
Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2020;36:1925–7.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Huang WL, Wilks A. A rapid seamless method for gene knockout in Pseudomonas aeruginosa. BMC Microbiol. 2017;17:199.
Bru D, Sarr A, Philippot L. Relative abundances of proteobacterial membrane-bound and periplasmic nitrate reductases in selected environments. Appl Environ Microbiol 2007;73:5971–4.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(t)(-delta delta c) method. Methods. 2001;25:402–8.
Maeda H, Fujimoto C, Haruki Y, Maeda T, Kokeguchi S, Petelin M, et al. Quantitative real-time PCR using Taqman and SYBR green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunol Med Microbiol. 2003;39:81–6.
Tyanova S, Temu T, Cox J. The maxquant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11:2301–19.
Daims H, Stoecker K, Wagner M. “Fluorescence in situ hybridization for the detection of prokaryotes,”. In: Osborn AM, Smith cJ, editors. Molecular microbial ecology. New york: Taylor & francis; 2005. p. 213–39.
Wallner G, Amann R, Beisker W. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry. 1993;14:136–43.
Eller G, Stubner S, Frenzel P. Group-specific 16S rRNA targeted probes for the detection of Type I and Type II methanotrophs by fluorescence in situ hybridisation. FEMS Microbiol Lett. 2001;198:91–7.
Lagkouvardos I, Joseph D, Kapfhammer M, Giritli S, Horn M, Haller D, et al. Imngs: a comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci Rep. 2016;6:33721.
Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–45.
Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, et al. 2016 update of the pride database and its related tools. Nucleic Acids Res. 2016;44:D447–D456.
Acknowledgements
The authors greatly thank the “National Natural Science Foundation of China (Grant No. 51878596, 21577123)”, the “Natural Science Funds for Distinguished Young Scholar of Zhejiang Province (LR17B070001)”, and the “National Key Technology R&D Program (2018YFC1802203)” for their financial support. A.K. and M.S. are supported by NSERC, CFI, CFREF and the Government of Alberta. GWT and S.M are supported by Australian Research Council (ARC) Future Fellowships (FT170100070 and FT190100211, respectively). The authors also thank Dr. Erica M. Hartmann for her assistance in manuscript preparation.
Author information
Authors and Affiliations
Contributions
LDS designed the study, performed the experiments, collected and analyzed the data, evaluated and arranged the results, and drafted the manuscript; LPL, ZW, and CYL characterized the reduction product and collected the data; SJM performed FISH, analyzed the results, and contributed to the manuscript preparation; XLD helped to analyze the metagenomic data; AK performed proteomics and analyzed the data; GWT and MS helped to analyze and discuss the results, and contributed to the manuscript preparation; HPZ initiated and supervised the project, conceived the experiments, and wrote the manuscript; all authors contributed to revising the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Shi, LD., Lv, PL., McIlroy, S.J. et al. Methane-dependent selenate reduction by a bacterial consortium. ISME J 15, 3683–3692 (2021). https://doi.org/10.1038/s41396-021-01044-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-01044-3
This article is cited by
-
Methane-dependent complete denitrification by a single Methylomirabilis bacterium
Nature Microbiology (2024)
-
Annotation-free discovery of functional groups in microbial communities
Nature Ecology & Evolution (2023)
