
Abstract
Copper-containing membrane monooxygenases (CuMMOs) are encoded by xmoCAB(D) gene clusters and catalyze the oxidation of methane, ammonia, or some short-chain alkanes and alkenes. In a metagenome constructed from an oilsands tailings pond we detected an xmoCABD gene cluster with <59% derived protein sequence identity to genes from known bacteria. Stable isotope probing experiments combined with a specific xmoA qPCR assay demonstrated that the bacteria possessing these genes were incapable of methane assimilation, but did grow on ethane and propane. Single-cell amplified genomes (SAGs) from propane-enriched samples were screened with the specific PCR assay to identify bacteria possessing the target gene cluster. Multiple SAGs of Betaproteobacteria belonging to the genera Rhodoferax and Polaromonas possessed homologues of the metagenomic xmoCABD gene cluster. Unexpectedly, each of these two genera also possessed other xmoCABD paralogs, representing two additional lineages in phylogenetic analyses. Metabolic reconstructions from SAGs predicted that neither bacterium encoded enzymes with the potential to support catabolic methane or ammonia oxidation, but that both were capable of higher n-alkane degradation. The involvement of the encoded CuMMOs in alkane oxidation was further suggested by reverse transcription PCR analyses, which detected elevated transcription of the xmoA genes upon enrichment of water samples with propane as the sole energy source. Enrichments, isotope incorporation studies, genome reconstructions, and gene expression studies therefore all agreed that the unknown xmoCABD operons did not encode methane or ammonia monooxygenases, but rather n-alkane monooxygenases. This study broadens the known diversity of CuMMOs and identifies these enzymes in non-nitrifying Betaproteobacteria.
Similar content being viewed by others


Introduction
Enzymes in the copper-containing membrane monooxygenase family (CuMMOs) catalyze diverse reactions. Particularly important are CuMMOs that act as ammonia and methane monooxygenases, as these play important roles in global carbon and nitrogen cycles [1,2,3,4]. Nitrifying Bacteria and Archaea use ammonia monooxygenase (AMO) to catalyze the oxidation of ammonium to hydroxylamine, while methanotrophic bacteria use particulate methane monooxygenase (pMMO) to convert methane into methanol. Evidence for CuMMO-mediated metabolism of other compounds like short-chain alkanes and alkenes has emerged in recent years. CuMMOs have been reported to catalyse C2–C4 alkane oxidation in a number of actinobacterial strains including Mycobacterium chubuense NBB4 [5], Mycobacterium rhodesiae NBB3 [6] and Nocardiodes sp. CF8 [7]. Ethylene-assimilating Haliea spp. within the class Gammaproteobacteria have also been shown to possess CuMMOs, although the role of the enzymes in ethylene oxidation is not firmly established [8]. CuMMOs are also known to oxidise numerous other substrates via competitive co-oxidation, particularly hydrocarbons containing methyl and alkyl groups, but the converted substrates do not support growth [3].
CuMMOs are encoded in an operon of three to four genes in the usual order CAB(D). The D gene is sometimes encoded separate from the operon, or can be absent entirely [9]. Operons are by convention named amoCAB(D) (encoding AMO), pmoCAB(D) (encoding pMMO), or other names depending on the substrate-specificity. However, all are homologous and for simplicity we will refer to them collectively as xmoCAB(D) [10]. Functionally and taxonomically coherent groups of methane and ammonia oxidisers are distinguishable on the basis of xmoA phylogeny, making these genes excellent biomarkers to identify environmental populations [10,11,12]. Broad-spectrum PCR primers targeting these genes are therefore extensively used in ecological studies of methanotroph and nitrifier diversity. However, a common result of such studies is the detection of divergent sequences of unknown affiliation or function [12]. Genome and metagenome studies are also uncovering new operons encoding divergent CuMMOs. Notable examples include the three different xmoCAB operons reported in verrucomicrobial methanotrophs [13,14,15] as well as the “pxm-group” identified in some alphaproteobacterial and gammaproteobacterial methanotrophs [16,17,18]. Other divergent operons have been identified in sequenced genomes, including those of the gammaproteobacterium Solimonas aquatica DSM 25927 [19] and the betaproteobacterium Hydrogenophaga sp. T4 (Genbank accession number: AZSO00000000), but the functions of the encoded CuMMOs are unknown. Collectively, there is increasing evidence that the diversity of bacteria encoding CuMMO enzymes, and the diversity of substrates these enzymes act on, may be greater than currently appreciated.
Petroleum-impacted environments are good habitats to explore for new hydrocarbon degrading oxygenases. In the Athabascan oilsands of Alberta, Canada, surface oil extraction involves a combination of alkali-hot water treatment and addition of chemical diluents (naphtha). The extraction process generates fluid tailings that are stored in open ponds to allow for particle settling, surface water recycling and long-term pollutant containment [20]. These tailings ponds contain high (up to 10 mM) concentrations of ammonia/ammonium [21, 22], along with residual bitumen-derived and naphtha-derived hydrocarbons including C3–C14 alkanes and monoaromatics (benzene, toluene, ethylbenzene and xylene) [23, 24]. Some oilsands tailings ponds are strongly methanogenic, and emit methane along with traces of other C2–C10 volatile organic compounds [23,24,25,26]. Aerobic methanotrophs possessing pMMO are abundant in oxic surface waters of these ponds [22].
Given the wealth of known CuMMO substrates in these tailings ponds, the oxic surface waters may offer a unique environment in which to discover new CuMMOs. Numerous investigations of the microbial communities in oilsands tailings ponds have been undertaken [20, 27], including metagenomic analyses [27,28,29]. Through data mining of these metagenomes, a CuMMO-encoding operon highly divergent from any previously recognized operon was discovered. The objective of this study was to identify the bacteria encoding this sequence, and to gain insights into their metabolism.
Materials and methods
Sample sites and metagenomes
A metagenome (IMG Genome ID: 3300002856) of the surface oxic water of an active oilsands tailings pond near Fort McMurray, Alberta (West-In Pit or WIP) was generated on Illumina and Roche 454 platforms and assembled as described previously [29]. An unusual xmoCABD operon (draft_100068512-draft_100068515) was identified in this metagenome and is referred to in this manuscript as “WIPMG xmoCABD1.” The WIP pond was decommissioned in 2012 and repurposed as an End-Pit Lake (Base Mine Lake or BML) that no longer receives fresh tailings material [30]. Therefore, fresh samples could not be obtained from WIP for the present study, and were instead obtained from another active pond, Mildred Lake Settling Basin (MLSB). Until 2012 water was cycled between the two ponds and their microbial communities were similar [22, 29], so we expected that MLSB would be a suitable proxy for the pre-2012 WIP community. Samples were obtained from the surface 10-cm of MLSB at several times between 2015 and 2017. The pond locations, chemical compositions, and the sampling methods used have been described previously [22, 29].
Bacterial enrichments
Surface water samples (0–10 cm depth) of MLSB sampled in August 2015 were used for enrichments and stable isotope probing (SIP) experiments. Twenty-millilitre amounts were added to 100-ml serum bottles sealed with butyl rubber stoppers. Headspaces of triplicate capped bottles were augmented with 10% v/v methane, ethane or propane. Alternatively, ammonium chloride (20 mM) was added to enrich for nitrifying bacteria. The headspace of each bottle was supplemented with 5% v/v CO2 to support autotrophy or anapleurotic CO2 fixation. Bottles were incubated at 23 °C with shaking (180 rpm) for up to 6 weeks. Gaseous hydrocarbon consumption and CO2 production were determined using a Varian 450-gas chromatograph (Varian, Walnut Creek, CA) equipped with a thermal conductivity detector (detector T 150°) after separation in a 2 mm × 0.5 m Hayesep N column and a 2 mm × 1.2 m molecular sieve 16X column in series (column T 70°).
Detection and quantification of the WIPMG xmoA1 gene
Water samples were centrifuged for 10 min at 10 000 × g prior to DNA extraction using the FastDNA Spin Kit for Soil (MP Biomedical, Santa Ana, CA). Eluted DNA was stored at −80 °C. Specific PCR primers to target the WIPMG xmoA1 gene were designed using the “Probe Design” tool in ARB [31] on a curated database of xmoA genes from public domain genomes. Primers and PCR assay conditions are detailed in Supplementary Table S1. Primer specificity was verified using Primer Blast [32] with eight maximum allowed mismatches and the largest E-value of 105 against the NR database, including uncultured and environmental sample sequences. No unspecific hits were found. The PCR was optimized via temperature-gradient analysis, and reaction specificity verified via Sanger sequencing of selected PCR products, and melt curve analyses during qPCR. A PCR product was cloned into the vector pJET1.2 using the CloneJET PCR cloning kit (Thermo Fisher Scientific, Waltham, MA), transformed into E. coli, recovered via colony PCR and used to construct qPCR standards ranging from 102–108 gene copies per microliter [33]. qPCR was performed on a Qiagen RotorGene-Q (Qiagen, Toronto, ON) using SsoAdvanced Universal SYBR green supermix (Bio-Rad, Hercules, CA).
DNA-stable isotope probing (SIP)
One-liter bottles containing 150 mL of MLSB water were sealed using butyl rubber stoppers and the headspace supplemented with 10% v/v of isotopically light (12C) or heavy (99 mol% 13C, Sigma-Aldrich, Oakville, Canada) methane, ethane, propane, or no added alkane (duplicate bottles of each). Five percent (v/v) 12CO2 was also added to minimise cross-feeding of 13CO2. Bottles were incubated as described above and gas depletion measured via GC. Experiments were stopped after ten days when between 21 and 33% of the supplied alkanes had been consumed. Extracted DNA was separated via isopycnic ultracentrifugation in cesium chloride and divided into twelve fractions of ~0.4 mL each, as described previously [34]. The density of each fraction was measured using an AR200 refractometer (Reichert Technologies, Depew, NY). Recovered DNA was precipitated with polyethylene glycol and glycogen, washed with 70% ethanol, eluted, and quantified using the Quant-iT dsDNA HS assay kit (Invitrogen) [34]. Samples from the SIP density fractions, unamended controls, and the initial (t = 0) community were investigated via qPCR of xmoA1 genes, as well as via Illumina sequencing of 16S rRNA gene amplicons.
For amplicon sequencing multiple DNA density fractions of 1.72–1.74 g ml−1 were pooled to form a single representative ‘heavy DNA pool’. Fractions were selected if they contained much more DNA in 13C vs. 12C incubations. Two controls were used: unfractionated DNA from the initial community; and the heaviest PCR-amplifiable fractions (1.71–1.73 g ml−1) of the unamended samples. The latter control verified that designated “heavy” fractions were not simply GC-rich organisms. Amplification of the 341–785 region of 16S rRNA genes [35], and amplicon sequencing using an Illumina MiSeq (Illumina, San Diego, CA) was carried out as described previously [36]. Reads were paired, filtered to exclude samples with quality-scores below 19 and analyzed using QIIME [37] with parameter settings described previously [29] Taxonomic identities were assigned via BLAST comparison to the Silva database (v. 123) [38]. OTUs representing >1% of any relative read-set were validated through manual BLAST against the NCBI NR database.
Single-cell genomics
MLSB water sampled in September 2016 was enriched under 10% propane and 5% CO2 as described above. Propane consumption was monitored using an SRI-8610C gas chromatograph (SRI Instruments, Torrance, CA) containing a HayeSep-D column (column T 190 °C) coupled to a flame ionization detector (detector T 300 °C) using N2 as the carrier gas. When propane consumption slowed, bottle headspaces were flushed with air and reconstituted with propane and CO2. After 6 weeks total, 2-ml aliquots were removed and centrifuged at 300 × g for 2 min to remove inorganic particulate matter. The supernatant was transferred and cell biomass recovered via centrifugation at 6000 × g for 3 min. Cell pellets were washed three times in 50% strength PBS, then resuspended in 1 ml of 50 mM Tris-EDTA buffer (pH 8.0) containing 10% v/v glycerol. The prepared cells were then sorted into 384-well plates and single-cell amplified genomes (SAGs) prepared using methods described previously [39, 40]. SAGs were screened for 16S rRNA genes with standard protocols, and each well containing an identified 16S rRNA gene was then screened via the specific WIPMG xmoA1 PCR assay. Ten SAGs positive for both 16S rRNA and WIPMG xmoA1 were selected for complete genome sequencing on an Illumina NextSeq [40], followed by assembly and annotation using the standard operating procedure of the Joint Genome Institute’s microbial annotation pipeline [41]. Genome completeness and contamination for individual and combined SAGs were estimated using CheckM [42].
Comparative phylogenetics and DNA–DNA hybridizations
Phylogenetic analysis was performed on concatenated derived amino acid sequences of the three operonic xmoCAB genes. Sequences from publicly available genomes/metagenomes and sequences determined in this study were aligned via Clustal Omega [43] and the tree constructed using maximum likelihood employing the LG model in Seaview 4.4.12 [44].
In silico DNA–DNA hybridizations were performed using the online Genome-to-Genome Distance Calculator v. 2.1 [45]. Fasta nucleic acid sequence files containing all assembled scaffolds for a specific SAG were compared against other SAGs within the genus in a pairwise fashion. Values were calculated by determining the sum of all identities found in high-scoring segment pairs divided by the overall high-scoring segment pair length (Formula 2 in the programme) as recommended for incomplete, draft genomes [45].
Analysis of xmoA transcripts in propane-fed enrichment culture
To our knowledge, cloning and expression of CuMMO-encoding operons has previously shown to be lethal to expression hosts [46, 47] and has been achieved only once in any model organism, for the butane MMO of a Mycobacterium [6]. Therefore, a cloning approach was considered unlikely to succeed, and we instead applied RT-qPCR to address the function of the WIPMG xmoCABD1. A sealed 2-L Duran glass bottle containing 1.5 L of mineral salts M10 medium [48] was inoculated with 75 mL of MLSB water that had been pre-enriched with propane as described above. The reactor was fed with a flow-through of mixed gas (propane and air at a ratio of 1:12) at a flow rate of 2.6 mL min−1. The fed-batch reactor was maintained at 30 °C in the dark and the aqueous phase stirred at 250 rpm. Cell density reached OD600nm of 0.171 after 96 h of incubation, after which the propane was shut off and gas feed continued with air alone for another 24 h.
At intervals (0, 48, 96 and 120 h), 0.5-mL samples were taken, immediately treated with 1 mL RNAprotect bacteria reagent (Qiagen), and centrifuged at 5000 × g for 10 min for RNA extraction. Parallel samples for DNA extraction were prepared without the RNAprotect treatment. Cell pellets were stored at −80 °C until analyses. Genomic DNA and the total RNA were extracted using the DNeasy PowerSoil Kit (Qiagen) and RNeasy Mini Kit (Qiagen), respectively. Three separate samples were processed in parallel to ensure reproducibility. Before lysing the cells for the extraction of the total RNA, 1 μL of luciferase mRNA solution (Promega, Madison, WI) diluted to 1010 copies μL−1 was added to each RNA extraction vial to account for the RNA loss during the extraction and purification procedures [49]. The extracted RNA samples were treated with DNase I (Qiagen) and purified with RNeasy MinElute Cleanup Kit (Qiagen) as previously described [50]. The purified total RNA samples were reverse-transcribed using SuperScript III reverse transcriptase (Invitrogen).
The WIPMG xmoA1 genes in the DNA and cDNA samples were quantified via qPCR using the primer set TP2f + TP2r (Table S1). The primer set and assay were redesigned from the xmoA1 assay described earlier in order to universally target the entire cluster of related xmoA1 genes found in SAGs and metagenome sequences. Specific assays were also designed to target the xmoA2 gene from the Rhodoferax SAGs and the xmoA2 gene from the Polaromonas SAGs (Table S1). Primer specificity was verified as described earlier.
Results
Sequence discovery and phylogenetic analyses
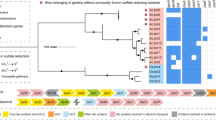
Analysis of a previously published metagenome [29] identified a scaffold of 4485 bp that encoded a four-gene cluster homologous to known CuMMO-encoding operons. Like most known pMMO- and AMO-encoding operons the genes were organized in the C-A-B order, with an additional xmoD gene. Phylogenetic analyses (Fig. 1) showed that the xmoCAB (designated as WIPMG xmoCAB1) is most closely related to an operon in Solimonas aquatica DSM 25927, a gammaproteobacterium isolated from a freshwater spring in Taiwan [51]. However, the individual CAB subunits share only 54–66% derived protein sequence identity with this strain (59% overall). Only one other gene was annotated on the genomic scaffold, a long-chain fatty acid transport protein showing a maximal protein sequence identity of 67% to a protein in the alkane-oxidizing betaproteobacterium, Thauera butanivorans [52].

Maximum-likelihood tree of concatenated derived amino acid sequences of CuMMO-encoding genes xmoC, xmoA and xmoB. The tree was constructed as described in the “Materials and Methods” section. Preferred substrates, if known, for the CuMMOs are indicated in brackets. Accession numbers for sequences are given in Table S2. For the Rhodoferax and Polaromonas SAGs, the specific genomes encoding the CuMMOs are found in Table S3. For genomes encoding multiple CuMMOs, numerical identifiers were assigned to unique sequences (e.g. xmoCAB1 or xmoCAB2). The scale bar represents substitutions per site. Branch support values are shown at each node and were determined based on 100 bootstrap replicates
Enrichment and stable isotope probing with potential CuMMO substrates
Given the high sequence divergence of the WIPMG xmoCABD1 operon relative to sequences of homologues from known methanotrophs and nitrifiers (Fig. 1, Tables S2 and S3), we sought to identify a possible ecological role for the organism(s) possessing it. A specific qPCR assay targeting the WIPMG xmoA1 gene was used to analyse tailings pond water samples enriched with methane, ammonium, ethane or propane. The number of WIPMG xmoA1 gene copies was low (1.28 × 104 ± 2.63 × 103 gene copies ml−1) at the onset of the experiment and stayed relatively constant over six weeks of incubation in controls and in methane or ammonium enrichments (Fig. 2). The only clearly stimulatory treatment was propane, where gene counts increased by over an order of magnitude, although ethane may have caused a small, transient increase (significant at week 4).

Abundance of WIPMG xmoA1 gene copies (per ml of water) during enrichment of oilsands tailings pond water under methane, ethane, propane, ammonium chloride or no added substrate. Error bars indicate ±1 SEM of triplicates
In order to verify the enrichment experiments over shorter incubation times that would limit disturbance of community structure, DNA-SIP was performed using isotopically light (12C) or heavy (13C) methane, ethane and propane. Rapid oxidation was observed using both the 12C and 13C alkanes, showing maximal potential oxidation rates of 117, 90 and 63 μmol L−1 d−1 for methane, ethane and propane, respectively (Fig. S1). In the density gradient-fractionated DNA from an unamended control sample, as well as in all enrichments using 12C substrates, the peak amount of total DNA and the peak number of WIPMG xmoA1 copies were detected in a DNA fraction of 1.69–1.70 g ml−1 (Fig. 3). This therefore represented the natural peak density of the DNA from the entire community, and from the organisms containing the xmoA1 gene.

Abundance of WIPMG xmoA1 gene copies (per ml) in density-fractionated DNA extracts from SIP enrichments. Samples were enriched using isotopically light (12C) or heavy (13C) methane, ethane and propane. Controls were unamended. The bar graph indicates the number of xmoA1 gene copies per density fraction. Error bars indicate ±1 SEM of two separate density gradients. The line graph indicates the relative DNA concentration per fraction, with the highest quantity detected in any fraction set to 1
In the 13C-methane-incubated samples the peak amount of DNA shifted to a density of 1.71–1.74 g ml−1 due to incorporation of the heavy-isotope by methanotrophs. However, there was no shift in the density fraction containing the highest WIPMG xmoA1 gene copy number after incubation with 13C methane. In both the 13C and 12C methane enrichments, peak WIPMG xmoA1 copies were observed in light (1.69 g ml−1) fractions, suggesting that the bacteria possessing these genes did not assimilate methane-derived carbon (Fig. 3a, b).
In contrast, the density fraction showing the maximum WIPMG xmoA1 copy numbers did shift after 13C-ethane and 13C-propane enrichment (Fig. 3d, f). In both cases, WIPMG xmoA1 gene copy numbers were highest at densities of 1.69–1.70 g ml−1 in the 12C enrichments but shifted to densities >1.71 g ml−1 after enrichment with 13C-labelled n-alkanes (Fig. 3c, e). This shift suggests that the bacteria encoding WIPMG xmoCAB1 were capable of assimilating carbon from ethane and propane. Peak WIPMG xmoA1 copy numbers were three orders of magnitude higher in the propane enrichment, suggesting that this substrate was preferred over ethane.
16S rRNA gene amplicons of DNA from control samples (i.e. the entire unenriched tailings pond DNA sample, as well as just the heaviest PCR-amplifiable DNA fraction) showed diverse communities, with 707–1098 OTUs detected. The heavy DNA fractions from the methane, ethane and propane enrichments were dominated by fewer OTUs (367–622). Gammaproteobacteria was the predominant class in the unamended sample as well as the methane enrichment (Fig. S2). The methane enrichment was dominated by a single OTU (Fig. S3) closely related to the methanotrophs Methyloparacoccus and Methylocaldum (97% nucleic acid sequence identity) agreeing with previous studies [22]. Betaproteobacteria were much more abundant in the ethane and propane enrichments, comprising 48 and 77% of the total read sets, respectively (Fig. S2). Relative abundances of OTUs within the genera Methyloversatilis, Hydrogenophaga, Pedomicrobium, Arenimonas, Acidovorax, Rhodoferax and Oxalicibacterium increased after propane enrichment (Fig. S3).
Screening single-cell genomes for xmoCAB genes
About 98% of the identified sorted cells from a propane enrichment belonged to the class Betaproteobacteria and six distinct genera were identified overall (Table S4). SIP experiments (Fig. S3) had already suggested that three of the genera sorted (Rhodoferax, Hydrogenophaga and Methyloversatilis) could assimilate propane. However, the most abundant bacterium sorted was a Polaromonas, which was not enriched in the propane SIP experiments. The water samples used for these two experiments were taken in different years, perhaps accounting for the difference. Industrial management parameters of the lake are irregular, with intermittent surface water draw-off and fresh tailings addition depending on industrial needs. A severe wildfire in 2016 also resulted in complete shutdown of industrial operations for several weeks prior to sampling. Chemical and biological properties of separate samples can therefore vary.
Aliquots of amplified genomic DNA from the sorted cells were screened using the WIPMG xmoA1-specific PCR assay. Bands of the expected size were observed in multiple SAGs identified as Rhodoferax and Polaromonas, so five SAGs of each genus were selected for genome sequencing (Table S5). Comparative analyses suggested that the 5 SAGs of each genus were nearly clonal. In silico DNA–DNA hybridizations [45] revealed that the nucleic acid sequences were >70% identical within each genus suggesting that each genus was represented by a single species in the sorted plates (Tables S6 and S7). The 16S rRNA gene sequences from the Polaromonas SAGs were identical and showed 98.0% nucleotide identity to Polaromonas naphthalenivorans CJ2, an aromatic hydrocarbon degrading bacterium [53]. For the Rhodoferax genomes, the full length 16S rRNA gene sequences were identical except for a single nucleotide mismatch in SAG-1 and closely matched (98.5%) Rhodoferax ferrireducens T118 [54]. Finished genomes for both P. naphthalenivorans CJ2 and R. ferrireducens T118 are available, but neither organism (nor any closely related genome-sequenced strain) possesses CuMMO-encoding genes.
Collectively, the Rhodoferax SAGs possessed two divergent xmoCABD operons (Figs. 1 and 4). One showed 99.9% nucleic acid sequence identity to the WIPMG xmoCAB1 genes found in the original oilsands tailings pond metagenome. However, the second operon (xmoCAB2) clustered in a distinct clade (Fig. 1). The Polaromonas SAGs also encoded two divergent operons. Again, one was homologous to the WIPMG xmoCAB1. The other formed a third new lineage not homologous to the second operon in the Rhodoferax SAGs (Fig. 1, Table S3). An orphan xmoC (e.g. Ga0215891_10812) was also identified in three of the five Polaromonas genomes, with flanking genes on the scaffolds confirming the absence of a complete operon.

Gene arrangements of xmoCABD in SAGs: a 17101 bp section of Rhodoferax SAG JGI 00BML02F20; b 8526 bp section of Rhodoferax SAG JGI 00BML02C18; c 9822 bp section of Polaromonas SAG JGI 00BML02G21; d 9887 bp segment of Polaromonas SAG JGI 00BML02L09. a, c The gene clusters corresponding to the WIPMG xmoCAB1 cluster in Fig. 1. A single sigma-70 promoter (indicated by an arrow) was predicted in front of each xmoCABD operon via Virtual Footprint
The four new xmo gene clusters and selected neighbouring genes are shown in Fig. 4. Each of the four clusters included xmoD, a gene occasionally part of an xmoCAB(D) operon in methanotrophs and nitrifiers, and occasionally present elsewhere in the genome [9]. Promoter prediction with Virtual Footprint [55] indicated that these genes are expressed as single xmoCABD operons under control of a sigma70 promoter in all four cases. Other genes located nearby include genes encoding predicted alcohol and aldehyde dehydrogenases, which may be involved in degrading the downstream products of the monooxygenase reaction (Fig. 4).
Metabolic potential
While the major goal of sequencing the SAGs was to identify the organisms containing WIPMG xmoCABD1 operons, they were also analysed to indicate any potential for ammonia, methane, or alkane oxidation. The CheckM genome completeness estimates for the combined Polaromonas SAGs was 96% and for the combined Rhodoferax SAGs was 82% (Table S5). Contamination was estimated to below (≤0.03%) or zero (Table S5). Therefore, the combined SAGs should give nearly-complete overviews of metabolic capacity, especially for the Polaromonas.
The SAGs encoded multiple oxygenases, including toluene, benzene, phenol and n-alkane monooxygenases, attesting to their adaptation to an oil-contaminated environment (Supplementary Table S8). Complete pathways were predicted in the Polaromonas for benzene, toluene, biphenyl and phenol oxidation via dioxygenase or two-monooxygenase reactions to methylcatechol or catechol, followed by meta-cleavage of catechol or methylcatechol to Acetyl-CoA. A Sox system for thiosulfate metabolism was also predicted. However, as the aim of this study was to characterize CuMMOs, we focused on the potential for oxidation of ammonia, methane or n-alkanes.
Genes encoding a complete Calvin Benson Bassham (CBB) cycle for autotrophic CO2 fixation, including the large subunit of ribulose bisphosphate carboxylase, were detected in the Polaromonas (Ga0215911_14316; Ga215901_1152) but not in the Rhodoferax. The Polaromonas may therefore be capable of 1-C fixation via the CBB cycle, a prerequisite for autotrophic nitrification. However, genes encoding for hydroxylamine dehydrogenase, an enzyme essential for ammonia oxidation [56], were not annotated in any SAG.
Pathways typical of proteobacterial methanotrophs were also mostly missing, although some genes encoding the catabolism of 1-C substrates (formate and possibly methanol) were predicted. Neither a ribulose monophosphate (RuMP) nor a serine cycle for fixation of 1-C intermediates of methane oxidation was complete in either organism. Genes encoding the key RuMP enzymes 3-hexulose-6-phosphate synthase and 6-phospho-3-hexuloisomerase were not found, nor were genes for the key serine cycle enzyme serine glyoxylate aminotransferase. A hydroxypyruvate reductase encoding gene was annotated, but was different from the form other methanotrophs use for the serine cycle (EC 1.1.1.81 instead of 1.1.1.29). There was no clear mxaFI or xoxF-encoded methanol dehydrogenase, which is common in methanotrophs [57, 58]. However, two other pyrrolquinoline quinone (PQQ)-binding alcohol dehydrogenases were encoded in both Rhodoferax and Polaromonas genomes: a homologue to the single subunit mdh2-type methanol dehydrogenase identified in the methylotroph Methyloversatilis universalis FAM5 [59] (e.g. Ga0215885_1254 – Rhodoferax; Ga0215901_10418– Polaromonas), and a second PQQ-binding alcohol dehydrogenase (e.g. Ga0215904_10692 – Rhodoferax; Ga0215901_1397– Polaromonas) located just downstream of CuMMO-encoding subunits (Fig. 4). Genes encoding PQQ synthesis were also identified in the Polaromonas (e.g Ga0215902_10817–108110). Formaldehyde dehydrogenase or a tetrahydromethanopterin-linked pathway to convert formaldehyde to formate was not encoded, although other aldehyde dehydrogenases were, via genes often adjacent to the xmoCABD operons (Fig. 4). The Polaromonas did encode multiple subunits of a formate dehydrogenase (e.g. Ga0215892_11102 to 11104).
On the other hand, complete pathways for propane oxidation were predicted in both Rhodoferax and Polaromonas genomes (Fig. 5). In addition to the two CuMMOs, both genera encoded multiple other alkane monooxygenases, including a group 3 (methane/alkane) soluble di-iron monooxygenase in the Polaromonas that is homologous (87% protein sequence identity) to the butane monooxygenase of Thauera butanivorans [60, 61] (Table S8). The oxidation of propane could be initiated by one or several of these enzymes, either terminally forming 1-propanol or sub-terminally forming 2-propanol [62, 63]. The terminal oxidation of 1-propanol could proceed via propionaldehyde and propionate [63], which could then be further oxidized by a number of described heterotrophic pathways [64]. In brief, both organisms can theoretically convert 1-propanol to propionate and then propionyl-CoA (Fig. 5). At this branch point, one possible degradation route includes oxidation via the citramalate cycle where propionyl-CoA is converted into succinyl-CoA [64, 65]. In the Rhodoferax, the genes encoding a propionyl-CoA carboxylase (e.g. Ga0215895_1164–1165) and a methylmalonyl-CoA mutase (e.g. Ga0215895_1161) were adjacent in the genome. A similar gene neighbourhood architecture was observed in the Polaromonas genomes. Neither organism had gene homologues encoding known methylmalonyl-CoA epimerases, which catalyze the conversion between 2R-methylmalonyl-CoA and 2S-methylmalonyl-CoA (Fig. 5). Multiple other epimerases were annotated in each genome, however, which may act as functional equivalents.

Possible pathways for terminal (a) or sub-terminal (b) oxidation of propane encoded in the Rhodoferax and Polaromonas SAGs. Both the citramalate (i) and methylcitrate (ii) pathways are shown in a. The propane monooxygenase could potentially include several enzymes, one of which may be a CuMMO
Another possible pathway includes the conversion of propionyl-CoA (plus oxaloacetate) to pyruvate (plus succinate) via the methylcitrate pathway (Fig. 5a). In Polaromonas, genes encoding 2-methylcitrate synthase, 2-methylcitrate dehydratase and 2-methylisocitrate lyase were adjacent in the genome (e.g. Ga0215912_10019–100111). Genes for 2-methylcitrate synthase could not be identified in the Rhodoferax genomes although a citrate synthase encoding gene (e.g. Ga0215903_11011) was located just downstream of annotated 2-methylcitrate dehydratase and 2-methylcitrate lyase encoding genes.
A number of possible sub-terminal oxidation pathways have been described [63, 66]. No known 2-propanol degradation pathways were encoded in the Rhodoferax SAGs (Fig. 5b), but in Polaromonas, three distinct NAD(P)-dependent alcohol dehydrogenase encoding genes (ADH) were adjacent to genes for acetone/cyclohexanone monooxygenase (e.g. Ga0215909_1235). In the actinomycete Gordonia sp. TY-5, where the pathway of 2-propanol oxidation via acetone was first described [67], the acetone monooxygenase-encoding genes were adjacent to genes for a methylacetate hydrolase (an esterase). A homologous methylacetate hydrolase was not encoded in the Polaromonas SAGs, but an esterase of unspecified activity was encoded just upstream of an acetone monooxygenase.
Transcription of xmoA in tailings pond water enriched with propane
The transcription of WIPMG xmoA1 was quantified in an enrichment culture grown in a batch reactor continuously fed with propane (Table 1). The number of xmoA1 genes increased from 8.0 × 104 ± 7.9 × 103 copies mL−1 immediately after inoculation (t = 0) to 3.9 × 106 ± 6.0 × 105 copies mL−1 at t = 96. Up-regulation of xmoA1 transcription during growth on propane was indicated by significantly higher transcript-to-gene ratios (p < 0.05) observed at t = 48 h and t = 96 h than at t = 120 h, 24 h after shutdown of propane supply (Table 1). Sequencing of the xmoA1 gene PCR-amplified from this enrichment culture verified that it fell phylogenetically into the WIPMG xmoA1 cluster. Although it did not match the gene from either of the SAGs perfectly, it was 93–94% identical in sequence and probably represented another very closely related strain.
The xmoA2 gene found in Polaromonas was not detectable with a specific PCR assay, however, the xmoA2 gene in the Rhodoferax SAGs was. Fewer Rhodoferax xmoA2 gene copies were detected compared with WIPMG xmoA1 genes, suggesting that fewer bacteria in the enrichment had a close homologue of this xmoA2 cluster (Table 1). However, like xmoA1, expression of the Rhodoferax xmoA2 was also dependent on the propane supply, although not as dramatically. Expression of xmoA2 decreased only fourfold (rather than 30 fold for the xmoA1) after removal of propane.
Discussion
Phylogenies of genes encoding CuMMOs can clearly delineate some functional and taxonomic groups of bacteria. Different ammonia oxidisers (in the phylum Thaumarchaeota, and the proteobacterial classes Gammaproteobacteria, and Betaproteobacteria), methane oxidisers (phyla Verrucomicrobia and NC10, proteobacterial classes Gammaproteobacteria, and Alphaproteobacteria) and butane oxidisers (phylum Actinobacteria) can all be reliably separated on the basis of phylogenetic clustering [10,11,12, 16]. This coherent phylogenetic structure has served as a useful backbone to establishing community structure-function relationships in numerous molecular ecology surveys investigating methane and ammonium oxidisers [10, 12]. However, phylogenetic clusters of CuMMO-encoding genes with unknown function and taxonomic affiliation are also found in genomes, metagenomes and environmental PCR amplicons produced with broad-specificity PCR primers. Here we investigated one such unknown xmoCABD operon identified in the metagenome of an oilsands tailings pond, in order to assign it to a probable taxon and function.
Single-cell genomics positively identified bacteria possessing this operon in our samples as members of the class Betaproteobacteria, in the family Comamonadaceae and the genera Rhodoferax and Polaromonas. Both genomes also encoded multiple predicted oxygenases for degradation of aromatic compounds and n-alkanes. Comamonadaceae are abundant in hydrocarbon-contaminated environments such as oilsands tailings, and specific members possess multiple aerobic and anaerobic petroleum-degrading pathways [20, 27, 29, 68]. However, none are known to oxidise methane or ammonia, and the presence of CuMMOs has not formally been described previously. The presence of CuMMO-encoding genes in Comamonadaceae, where many other sequenced genomes do not possess them, is unusual. Methanotrophs and ammonia oxidisers tend to occur in coherent taxonomic clusters where all members possess the genes [10,11,12, 16]. This raises the possibility that the genes have been acquired by the Comamonadaceae via recent gene transfer events. Unfortunately, the SAG data are not appropriate to address this issue more closely, and too few related genes have been discovered to develop a robust phylogenetic picture.
The use of a single-cell genomics approach was preferred to metagenomic binning of genomes because nearly all xmo operons differ significantly in their nucleotide compositional biases compared with their overall host genomes [10], which could potentially cause problems with compositional binning. The assignment of the xmo operons to the Rhodoferax and Polaromonas genomes was verified in multiple uncontaminated SAGs of each bacterium. Unexpectedly, the SAGs also revealed other divergent CuMMO-encoding operons in both bacteria. Collectively, the CuMMOs clustered into three distinct phylogenetic clades. In each clade the closest genes from identified, cultured bacteria are only distantly related to those from our SAGs, and formal studies into gene expression or enzyme function of the xmo genes in these bacteria have not been reported. The closest genome sequence to the WIPMG xmoCAB1 cluster showed only 59% protein sequence identity and is encoded by the gammaproteobacterium Solimonas aquatica NAA16T. Solimonas aquatica is a metabolically versatile bacterium [51] whose CuMMO-encoding genes were identified solely through genome sequencing as part of the Genomic Encyclopedia of Archaeal and Bacterial Type Strains, Phase-II sequencing initiative [19]. Characterization of gene expression and alkane metabolism in the type strain Solimonas aquatica NAA16T could be valuable to explore the diversity of CuMMOs, however the low sequence identities of the Solimonas xmo operon to those of the Polaromonas/Rhodoferax characterized in this study suggests that the functions of the encoded CuMMOs may not be the same.
The low numbers of WIPMG xmoA1 genes observed under ammonia enrichments (Fig. 2) suggested that neither the Rhodoferax nor the Polaromonas strains were capable of growth via nitrification. This was further supported by the lack of a hydroxylamine dehydrogenase encoding genes in any of the SAGs. Methane oxidation also seemed an unlikely function based on SIP and qPCR analyses after incubation under methane-containing atmospheres (Figs. 2 and 3). No enrichment of the xmoA or 16S rRNA genes of these bacteria was observed in the heavy fraction of the 13C-methane SIP experiments, verifying these bacteria did not assimilate methane. The genomes lacked many genes typical of methanotrophs, although a limited capacity to catabolise some 1-C compounds was indicated. Neither bacterium encoded methanol and formaldehyde oxidation pathways typical of methanotrophs and neither encoded pathways for assimilating methane-carbon via formaldehyde or formate. The Polaromonas encoded some capability for 1-C catabolism (formate and possibly methanol), and although methane assimilation was not encoded, the CBB cycle would provide an alternative path of C assimilation, as practiced by Verrucomicrobia methanotrophs [34]. Given this potential, and the known promiscuous nature of CuMMOs [3], methane cannot be completely discounted as a possible substrate for the new CuMMOs. It is possible that the Polaromonas/Rhodoferax obtain energy from the oxidation of methane, but are unable to assimilate it. However, a clear test of this hypothesis would require a pure culture. Our cultivation efforts to date have included plating and batch-culture dilution under propane-containing atmospheres, however, the target organisms have proven elusive, and are easily overgrown by other species. Successful isolation will likely require optimizing incubations conditions such as pO2.
Contrary to methane and ammonia oxidation, n-alkane metabolism in the target bacteria was supported by multiple lines of evidence: enrichment, SIP, genome analysis and gene expression. Genomic analyses suggested two plausible pathways for terminal propane oxidation in both Rhodoferax and Polaromonas (Fig. 4a) along with a pathway in Polaromonas for the oxidation of 2-propanol (Fig. 4b). These genome predictions, along with the strong assimilation of propane-derived C into the xmoA genes of Rhodoferax and Polaromonas demonstrated by SIP studies, show clearly that these bacteria are capable of alkanotrophy. They do not together prove that the CuMMOs are key enzymes in propane oxidation, since multiple other hydrocarbon monooxygenases were also identified in the genomes (Table S8). However, RT-qPCR studies indicated that WIPMG xmoA1 gene expression in an enrichment culture (as well as the expression of the xmoA2 gene detected in the Rhodoferax) was regulated depending on the availability of propane, strongly suggesting the involvement of these CuMMO enzymes in propane oxygenation.
CuMMO-enabled n-alkane (butane, ethane and propane) oxygenation has already been established in some Actinomycetes [5,6,7]. Although our bacteria showed a clear preference for growth on propane, ethane also supported a lower growth and C-assimilation rate, and could also have been a CuMMO substrate. A difference between the CuMMO-encoding genes in our Comamonadaceae vs. those of Actinobacteria is the presence of xmoD in the former. The product of xmoD was recently described as a critical component of some CuMMOs: a Cu-containing polypeptide that may facilitate assembly and stabilization of the CuMMO complex, or facilitate electron delivery to the active site [9]. The gene is present in all bacteria encoding AMO or pMMO for ammonia/methane oxidation, sometimes as part of the xmo operon and sometimes elsewhere in the genome. However, homologues are not found in the genomes of the actinobacteria Mycobacteria and Nocardiodes that encode n-alkane targeting CuMMOs [9]. This suggests that the Rhodoferax/Polaromonas CuMMOs identified in our study are functionally more similar to the better known proteobacterial pMMO/AMO enzymes than to the actinobacterial CuMMOs, a proposition supported by phylogenetic analysis, which places the actinobacterial CuMMOs well apart from all proteobacterial CuMMOs (Fig. 1).
This study has expanded the known diversity of xmoCAB(D) operons encoding CuMMOs and the taxonomic groups that encode this enzyme. Definitive functional roles for any of the encoded CuMMOs could only be inferred, and conclusive evidence will require further experimentation using laboratory cultures. However, multiple lines of cultivation-independent evidence suggest that these CuMMOs are probably involved primarily in n-alkane oxidation, rather than methane or ammonia oxidation.
References
Hakemian AS, Rosenzweig AC. The biochemistry of methane oxidation. Annu Rev Biochem. 2007;76:223–41.
Canfield DE, Glazer AN, Falkowski PG. The evolution and future of Earth’s nitrogen cycle. Science. 2010;330:192–6.
Semrau JD. Bioremediation via methanotrophy: overview of recent findings and suggestions for future research. Front Microbiol. 2011;2:209.
Bodelier PLE, Steenbergh AK. Interactions between methane and the nitrogen cycle in light of climate change. Curr Opin Environ Sustain. 2014;9-10:26–36.
Coleman NV, Yau S, Wilson NL, Nolan LM, Migocki MD, Ly MA, et al. Untangling the multiple monooxygenases of Mycobacterium chubuense strain NBB4, a versatile hydrocarbon degrader. Environ Microbiol Rep. 2011;3:297–307.
Coleman NV, Le NB, Ly MA, Ogawa HE, McCarl V, Wilson NL, et al. Hydrocarbon monooxygenase in Mycobacterium: recombinant expression of a member of the ammonia monooxygenase superfamily. ISME J. 2012;6:171–82.
Sayavedra-Soto LA, Hamamura N, Liu CW, Kimbrel JA, Chang JH, Arp DJ. The membrane-associated monooxygenase in the butane-oxidizing Gram-positive bacterium Nocardioides sp. strain CF8 is a novel member of the AMO/PMO family. Environ Microbiol Rep. 2011;3:390–6.
Suzuki T, Nakamura T, Fuse H. Isolation of two novel marine ethylene-assimilating bacteria, Haliea species ETY-M and ETY-NAG, containing particulate methane monooxygenase-like genes. Microbes Environ. 2012;27:54–60.
Fisher OS, Kenney GE, Ross MO, Ro SY, Lemma BE, Batelu S, et al. Characterization of a long overlooked copper protein from methane- and ammonia-oxidizing bacteria. Nat Commun. 2018;9:4276.
Khadka R, Clothier L, Wang L, Lim CK, Klotz MG, Dunfield PF. Evolutionary history of copper membrane monooxygenases. Front Microbiol. 2018;9:2493.
Purkhold U, Pommerening-Röser A, Juretschko S, Schmid MC, Koops HP, Wagner M. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl Environ Microbiol. 2000;66:5368–82.
Knief C. Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front Microbiol. 2015;6:1346.
Op den Camp HJM, Islam T, Stott MB, Harhangi HR, Hynes A, Schouten S, et al. Environmental, genomic and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ Microbiol Rep. 2009;1:293–306.
Khadem AF, van Teeseling MC, van Niftrik L, Jetten MSM, Op den Camp HJM, Pol A. Genomic and physiological analysis of carbon storage in the verrucomicrobial methanotroph “Ca. Methylacidiphilum fumariolicum” SolV. Front Microbiol. 2012;3:345.
Anvar SY, Frank J, Pol A, Schmitz A, Kraaijeveld K, den Dunnen JT, et al. The genomic landscape of the verrucomicrobial methanotroph Methacidiphilum fumariolicum SolV. BMC Genomics. 2014;15:914.
Tavormina PL, Orphan VJ, Kalyuzhnaya MG, Jetten MS, Klotz MG. A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ Microbiol Rep. 2011;3:91–100.
Vorobev A, Jagadevan S, Jain S, Anantharaman K, Dick GJ, Vuilleumier S, et al. Genomic and transcriptomic analyses of the facultative methantroph Methylocystis sp. Strain SB2 grown on methane or ethanol. Appl Environ Microbiol. 2014;80:3044–52.
Kits KD, Klotz MG, Stein LY. Methane oxidation coupled to nitrate reduction under hypoxia by the Gammaproteobacterium Methylomonas denitrificans, sp. nov. type strain FJG1. Environ Microbiol. 2015;17:3219–32.
Whitman WB, Woyke T, Klenk HP, Zhou Y, Lilburn TG, Beck BJ, et al. Genomic Encyclopedia of Bacterial and Archaeal Type Strains, Phase III: the genomes of soil and plant-associated and newly described type strains. Stand Genom Sci. 2015;10:26.
Foght JM, Gieg LM, Siddique T. The microbiology of oil sands tailings: past, present, future. FEMS Microbiol Ecol. 2017;93:fix034.
Allen EW. Process water treatment in Canada’s oil sands industry: I. Target pollutants and treatment objectives. J Environ Eng Sci. 2008;7:123–38.
Saidi-Mehrabad A, He Z, Tamas I, Sharp CE, Brady AL, Rochman FF, et al. Methanotrophic bacteria in oilsands tailings ponds of northern Alberta. ISME J. 2013;7:908–21.
Siddique T, Shahimin MFM, Zamir S, Semple K, Li C, Foght JM. Long-term incubation reveals methanogenic biodegradation of C5 and C6 iso-alkanes in oil sands tailings. Environ Sci Technol. 2015;49:14732–9.
Siddique T, Fedorak PM, Mackinnon MD, Foght JM. Metabolism of BTEX and naphtha compounds to methane in oil sands tailings. Environ Sci Technol. 2007;41:2350–6.
Simpson IJ, Blake NJ, Barletta B, Diskin GS, Fuelberg HE, Gorham K, et al. Characterization of trace gases measured over Alberta oil sands mining operations: 76 speciated C2–C10 volatile organic compounds (VOCs), CO2, CH4, CO, NO, NO2, NOy, O3 and SO2. Atmos Chem Phys. 2010;10:11931–54.
Small CC, Cho S, Hashisho Z, Ulrich AC. Emission from oil sands tailings ponds: Review of tailings pond parameters and emission estimates. J Pet Sci Eng. 2015;127:490–501.
An D, Caffrey SM, Soh J, Agrawal A, Brown D, Budwill K, et al. Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common. Environ Sci Technol. 2013;47:10708–17.
Aguilar M, Richardson E, Tan B, Walker G, Dunfield PF, Bass D, et al. Next-generation sequencing assessment of eukaryotic diversity in oil sands tailings ponds sediments and surface water. J Eukaryot Microbiol. 2016;63:732–43.
Rochman FF, Sheremet A, Tamas I, Saidi-Mehrabad A, Kim JJ, Dong X, et al. Benzene and naphthalene degrading bacterial communities in an oil sands tailings pond. Front Microbiol. 2017;8:1845.
Syncrude. “Mildred Lake extension project”. Public Disclosure Document (Fort McMurray: Syncrude Canada Ltd). 2012. http://www.syncrude.ca/assets/pdf/News-Room/MLX-Project-Public-Disclosure-Document.pdf.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012;13:134.
Sharp CE, Smirnova AV, Graham JM, Stott MB, Khadka R, Moore TR, et al. Distribution and diversity of Verrucomicrobia methanotrophs in geothermal and acidic environments. Environ Microbiol. 2014;16:1867–78.
Sharp CE, Stott MB, Dunfield PF. Detection of autotrophic verrucomicrobial methanotrophs in a geothermal environment using stable isotope probing. Front Microbiol. 2012;3:303.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1.
Ruhl IA, Grasby SE, Haupt ES, Dunfield PF. Analysis of microbial communities in natural halite springs reveals a strong but domain-dependent relationship of species diversity to osmotic stress. Environ Microbiol Rep. 2018;10:695–703.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Method. 2010;7:335–6.
Glockner FO, Yilmaz P, Quast C, Gerken J, Beccati A, Ciuprina A, et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J Biotechnol. 2017;261:169–76.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–7.
Rinke C, Lee J, Nath N, Goudeau D, Thompson B, Poulton N, et al. Obtaining genomes from uncultivated environmental microorganisms using FACS-based single-cell genomics. Nat Protoc. 2014;9:1038–48.
Huntemann M, Ivanova NN, Mavromatis K, Tripp HJ, Paez-Espino D, Palaniappan K, et al. The standard operating procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP v.4). Stand Genom Sci. 2015;10:86.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2014;25:1043–55.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–4.
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013;14:60.
Klotz MG, Alzerreca J, Norton JM. A gene encoding a membrane protein exists upstream of the amoA/amoB genes in ammonia oxidizing bacteria: a third member of the amo operon? FEMS Microbiol Lett. 1997;150:65–73.
Norton JM, Low JM, Klotz MG. The gene encoding ammonia monooxygenase subunit A exists in three nearly identical copies in Nitrosospirasp. NpAV. FEMS Microbiol Lett. 1996;139:181–8.
Heyer J, Galchenko VF, Dunfield PF. Molecular phylogeny of type II methane-oxidizing bacteria isolated from various environments. Microbiology. 2002;148:2831–46.
Johnson DR, Lee PKH, Holmes VF, Alvarez-Cohen L. An internal reference technique for accurately quantifying specific mRNAs by real-time PCR with application to the tceA reductive dehalogenase gene. Appl Environ Microbiol. 2005;71:3866–71.
Yoon S, Cruz-Garcia C, Sanford R, Ritalahti KM, Loffler FE. Denitrification versus respiratory ammonification: environmental controls of two competing dissimilatory NO3 −/NO2 − reduction pathways in Shewanella loihica strain PV-4. ISME J. 2015;9:1093–104.
Sheu SY, Cho NT, Arun AB, Chen WM. Proposal of Solimonas aquatica sp. nov., reclassification of Sinobacter flavus Zhou et al. 2008 as Solimonas flava comb. nov. and Singularimonas variicoloris Friedrich and Lipski 2008 as Solimonas variicoloris comb. nov. and emended descriptions of the genus Solimonas and its type species Solimonas soli. Int J Syst Evol Microbiol. 2011;61:2284–91.
Dubbels BL, Sayavedra-Soto LA, Bottomley PJ, Arp DJ. Thauera butanivorans sp. nov., a C2-C9 alkane-oxidizing bacterium previously referred to as ‘Pseudomonas butanovora’. Int J Syst Evol Microbiol. 2009;59:1576–8.
Yagi JM, Sims D, Brettin T, Bruce D, Madsen EL. The genome of Polaromonas naphthalenivorans strain CJ2, isolated from coal tar-contaminated sediment, reveals physiological and metabolic versatility and evolution through extensive horizontal gene transfer. Environ Microbiol. 2009;11:2253–70.
Finneran KT, Johnsen CV, Lovley DR. Rhodoferax ferrireducens sp. nov., a psychrotolerant, facultatively anaerobic bacterium that oxidizes acetate with the reduction of Fe(III). Int J Syst Evol Microbiol. 2003;53:669–73.
Münch R, Hiller K, Grote A, Scheer M, Klein J, Schobert M, et al. Virtual Footprint and PRODORIC: an integrative framework for regulon prediction in prokaryotes. Bioinformatics. 2005;15:4187–9.
Schmid MC, Hooper AB, Klotz MG, Woebken D, Lam P, Kuypers MM, et al. Environmental detection of octahaem cytochrome c hydroxylamine/hydrazine oxidoreductase genes of aerobic and anaerobic ammonium-oxidizing bacteria. Environ Microbiol. 2008;10:3140–9.
Lau E, Fisher MC, Steudler PA, Cavanaugh CM. The methanol dehydrogenase gene, mxaF, as a functional and phylogenetic marker for proteobacterial methanotrophs in natural environments. PloS ONE. 2013;8:e56993.
Chu F, Lidstrom ME. XoxF acts as the predominant methanol dehydrogenase in the type I methanotroph Methylomicrobium buryatense. J Bacteriol. 2016;198:1317–25.
Kalyuzhnaya MG, Hristova KR, Lidstrom ME, Chistoserdova L. Characterization of a novel methanol dehydrogenase in representatives of Burkholderiales: implications for environmental detection of methylotrophy and evidence for convergent evolution. J Bacteriol. 2008;190:3817–23.
Cooley RB, Dubbels BL, Sayavedra-Soto LA, Bottomley PJ, Arp DJ. Kinetic characterization of the soluble butane monooxygenase from Thauera butanivorans, formerly ‘Pseudomonas butanovora’. Microbiol. 2009;155:2086–96.
Holmes AJ. The diversity of soluble di-iron monooxygenases with bioremediation applications. In: Singh A, Kuhad R, Ward O, editors. Advances in applied bioremediation soil biology. Berlin: Springer; 2009.
Kotani T, Kawashima Y, Yurimoto H, Kato N, Sakai Y. Gene structure and regulation of alkane monooxygenases in propane-utilizing Mycobacterium sp. TY-6 and Pseudonocardia sp. TY-7. J Biosci Bioeng. 2006;102:184–92.
Ashraf W, Mihdhir A, Murrell JC. Bacterial oxidation of propane. FEMS Microbiol Lett. 1994;122:1–6.
Textor S, Wendisch VF, De Graaf AA, Müller U, Linder MI, Linder D, et al. Propionate oxidation in Escherichia coli: evidence for operation of a methylcitrate cycle in bacteria. Arch Microbiol. 1997;168:428–36.
Suvorova IA, Ravcheev DA, Gelfand MS. Regulation and evolution of malonate and propionate catabolism in proteobacteria. J Bacteriol. 2012;194:3234–40.
Hausinger RP. New insights into acetone metabolism. J Bacteriol. 2007;189:671–3.
Kotani T, Yurimoto H, Kato N, Sakai Y. Novel acetone metabolism in a propane-utilizing bacterium, Gordonia sp. strain TY-5. J Bacteriol. 2007;189:886–93.
Willems A. The family Comamonadaceae. In: Rosenberg E, editor. The prokaryotes—alphaproteobacteria and betaproteobacteria. Berlin: Springer; 2014. p. 777–851.
Acknowledgements
This work was made possible through the NSERC (Natural Sciences and Engineering Research Council of Canada) Collaborative Research and Development program (Grant number CRDPJ478071–14); an NSERC Discovery grant (2014–05067); and a grant from the National Research Foundation of Korea (2018K1A3A1A74065626). Initial metagenomic data were created with financial assistance from Genome Canada, Genome Alberta, Genome BC and the Government of Alberta (GC Grant 1203). SAG sequencing and analysis was supported by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility supported under Contract No. DE-AC02–05CH11231. We acknowledge the assistance of Syncrude Canada, Ltd in providing samples. AA Lopez-Jauregui was supported by a Mitacs Globalink internship award.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Rochman, F.F., Kwon, M., Khadka, R. et al. Novel copper-containing membrane monooxygenases (CuMMOs) encoded by alkane-utilizing Betaproteobacteria. ISME J 14, 714–726 (2020). https://doi.org/10.1038/s41396-019-0561-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0561-2
This article is cited by
-
Identification of active gaseous-alkane degraders at natural gas seeps
The ISME Journal (2022)
-
Nitrogen cycling and microbial cooperation in the terrestrial subsurface
The ISME Journal (2022)
-
Universal activity-based labeling method for ammonia- and alkane-oxidizing bacteria
The ISME Journal (2022)
-
Verrucomicrobial methanotrophs grow on diverse C3 compounds and use a homolog of particulate methane monooxygenase to oxidize acetone
The ISME Journal (2021)
