
Abstract
Various plant species establish intimate symbioses with bacteria within their aerial organs. The bacteria are contained within nodules or glands often present in distinctive patterns on the leaves in what is commonly referred to as leaf nodule symbiosis. We describe here a highly specific symbiosis between a wild yam species from Madagascar, Dioscorea sansibarensis and bacteria of the species Orrella dioscoreae. Using whole-genome sequencing of plastids and bacteria from wild-collected samples, we show phylogenetic patterns consistent with a dominant vertical mode of transmission of the symbionts. Unique so far among leaf nodule symbioses, the bacteria can be cultured and are amenable to comparative transcriptomics, revealing a potential role in complementing the host’s arsenal of secondary metabolites. We propose a recent establishment of a vertical mode of transmission in this symbiosis which, together with a large effective population size explains the cultivability and apparent lack of genome reductive evolution in O. dioscoreae. We leverage these unique features to reveal pathways and functions under positive selection in these specialized endophytes, highlighting the candidate mechanisms enabling a permanent association in the phyllosphere.
Similar content being viewed by others

Introduction
Microorganisms can establish a wide range of beneficial interactions with plants, often contributing to mineral uptake, nitrogen fixation, or plant defense. Most of the mutualistic associations with bacteria are facultative and have been widely studied at the root level [1, 2], with much less focus on the phyllosphere or endosphere despite recent findings that plants can shape phyllosphere microbial communities [3, 4]. Furthermore, the molecular mechanisms enabling the establishment of these interactions are not well characterized outside of a few and usually pathogenic model systems [5].
Leaf nodule symbioses represent some of the most intimate associations between plants and bacteria. Most leaf nodule symbioses are found in species of the Rubiaceae (Psychotria and Pavetta) and Primulaceae (Ardisia) families, and their symbionts are members of the Burkholderiaceae family of β-proteobacteria. The symbionts reside in dedicated structures called leaf glands or nodules, and are transmitted between generations via seeds [6]. The association is essential for both hosts and symbionts: Candidatus Burkholderia (Ca. Burkholderia) species cannot be cultured outside of their host and bacteria-free Psychotria kirkii and Ardisia crenata display severe growth defects [7, 8]. This co-dependence between host and symbiont is likely the result of co-evolution over several million years, compounded by small effective population sizes and genetic drift [6]. Typical of vertically transmitted symbiotic bacteria, Ca. Burkholderia leaf nodule symbionts show extensive signs of reductive genome evolution, with coding capacities ranging from 41.7 to 67.3% and an accumulation of pseudogenes and insertion sequences [9,10,11]. Despite extensive genome erosion, some symbionts have been shown to produce secondary metabolites, likely involved in the protection of the host from herbivory, such as the insecticidal kirkamide and the depsipeptide FR900359, as well as the herbicidal streptol-glucoside possibly involved in allelopathic interactions [11,12,13]. Due to genomic instability and evolved co-dependence, it is unclear whether secondary metabolism was present in the ancestor of leaf nodule Burkholderia or acquired as a secondary trait [6].
We recently described a leaf nodule symbiosis in the monocot species Dioscorea sansibarensis [14]. D. sansibarensis, or the Zanzibar yam, is a true yam native to Madagascar and tropical Africa [15]. This fast growing vine, like many yam species, reproduces asexually through aerial bulbils and underground tubers and is not known to produce viable seeds [16]. The leaf of D. sansibarensis displays a prominent acumen or ‘drip-tip’, harboring high titers (>109 cfu/g) of Orrella dioscoreae (O. dioscoreae), a newly described species of the Alcaligenaceae family [14]. Similar to leaf nodule symbioses in dicot species, the bacteria are hosted extracellularly in the leaf gland and do not invade the mesophyll or vasculature of the host and do not spread systemically [6]. In contrast to other leaf nodule symbioses, O. dioscoreae can be cultured [14].
The aim of this study was to (i) characterize the prevalence and mode of transmission of O. dioscoreae in wild populations of D. sansibarensis; (ii) propose hypotheses regarding the recruitment of functions in leaf nodule symbiosis, and (iii) leverage the unique tractability of the D. sansibarensis leaf symbiosis to uncover the characteristics of a strict endophytic lifestyle. We show that the association with O. dioscoreae is ubiquitous and highly specific in D. sansibarensis. Based on phylogenetic data of extant specimens, we propose that the symbiosis and vertical transmission evolved during the Pleistocene, offering a unique opportunity to document the early events shaping the evolution of a hereditary plant-microbe symbiosis. Finally, secondary metabolism seems to play a central role in the D. sansibarensis leaf nodule symbiosis, suggesting that the acquisition of novel metabolism is a pre-requisite for the evolution of symbiont capture in the phyllosphere.
Material and methods
Bacterial strains and growth conditions
All O. dioscoreae strains (Table S1) were grown at 28 °C on tryptic soy agar (TSA) medium or AB minimal medium [17] supplemented with 20 g/L sodium citrate and 0.5 g/L yeast extract unless otherwise indicated. Aerobic cultures were grown with vigorous shaking (200 rpm) in 500 mL Erlenmeyer flasks containing 100 ml of medium. Growth curves and additional details on media composition are given in supplementary information.
Sampling and identification of wild Dioscorea sansibarensis samples
Leaf nodule samples from wild Dioscorea sansibarensis plants were collected from 12 different sites in Madagascar during two field collections in November 2016 and May 2017, with research permit 158/16/MEEF/SG/DGF/DSAP/SCB.Re issued by the Ministry of Environment, Ecology and Forests of the Republic of Madagascar. At each sampling location, ~10 leaf nodules from distinct plants were harvested. Samples were immediately placed in sealed plastic sampling bags containing 5–10 g of silica gel (Carl Roth) for dehydration and shipping. The GPS coordinates of the sampling locations are given in Table S2. Appropriate measures were taken to comply with Nagoya protocol guidelines.
DNA extraction and PCR
Silica-dried samples were processed using a combination of bead-beating (Retsch MM400, Haan, Germany) and a Maxwell® 16 DNA Purification Kit (Promega, Madison, WI, USA). More details are given in supplementary information. PCR amplification and sequencing of the nrdA gene (coding for ribonucleoside-diphosphate reductase 1 subunit alpha and a common marker used for typing of Alcaligenaceae species) was used to confirm the presence of Orrella dioscoreae. PCR analysis and Sanger sequencing using primers specific for the chloroplastic markers matK, rbcL, and rpl32-trnL were used to confirm plant species against reference sequences obtained from a vouchered D. sansibarensis specimen from the live collection of the botanical garden of Ghent University (accession 19001189). All oligos used in this study are listed in Table S3.
Metagenome assembly and annotation
Sequencing reads were prepared for assembly by adapter trimming and read filtering using Trimmomatic [18], removing reads with phred scores below 30 and discarding non-paired reads. To assemble sequencing reads derived from Orrella dioscoreae, an approach based on Albertsen et al. [19] was used. In short, filtered reads were assembled using SPAdes v3.10.1 [20] in metagenome mode using kmer-lengths of 21, 33, 55, and 77. The read coverage and GC-content of the resulting contigs were calculated and plotted using the Matplotlib package in Python [21]. Taxonomic classification of the contigs was done using the Kraken software and overlaid on the plot [22]. Contigs consistent with O. dioscoreae were selected and re-assembled as previously described using SPAdes in careful mode, using kmer-lengths of 21, 33, 55, 77, 99, and 121 [11]. Assembly statistics of the resulting assemblies were generated using Quast v4.5 [23]. Contigs smaller than 500 nt, with low coverage (<1/3 of average coverage) or classified as eukaryotic were discarded from the final assembly. Annotation was performed with the RAST online service [24] with gene prediction enabled. Orthologs were computed using OrthoMCL v1.4 [25], using a Blastp e-value cut-off of 1.0 × 10−6, 50% identity over 50% query length, and an inflation factor of 1.5. EGGNOGmapper was used to assign GO, EggNOG, and COG category annotations to the proteins [26]. Analysis of putative secondary metabolite gene clusters, including NRPS adenylation domain substrate prediction were done using the AntiSMASH web server [27]. Analysis and phylogenetic clustering of NRPS condensation domains was done using the NaPDoS web server [28]. Genome comparisons were done using the NCBI blastn program and blast ring diagrams were drawn using the Circos v0.63 software [29]. Sequencing reads and genome assemblies were deposited in the European Nucleotide Archive under accession PRJEB30075.
Microbial diversity analysis of leaf nodules
To assess the diversity of bacteria within the leaf nodule, whole-genome shotgun (WGS) sequencing reads of each nodule were classified using Kraken [22], using a custom kraken database built from the ‘bacteria’ and ‘plastid’ components of the NCBI RefSeq database (downloaded Feb 2017). In addition, sequencing reads were analyzed using MetaPhlAn2 [30] for detection of microbial eukaryotes. In parallel to the analysis of field-collected D. sansibarensis nodules, strain-level diversity of O. dioscoreae within one plant was assessed by WGS of 20 nodules sampled from a single D. sansibarensis specimen kept in the greenhouse of the botanical garden of Ghent University. Total DNA was extracted, pooled in 4 pools of 5 samples and sequenced using shotgun methods as described above. The resulting WGS reads were trimmed and filtered as described above and mapped to the repeat-masked reference genome sequence. Reads mapping to multiple sites in the genome were discarded from the analysis. Polymorphic sites were detected using CLC genomics workbench v7.5, using a base quality filter of 25, with a minimum quality score of 20 in the neighborhood of 5 bases. Only polymorphisms supported by at least 5 reads were considered.
Metagenome project mining
To investigate the presence of O. dioscoreae in the environment, metagenomics data from the MG-RAST [31] database was screened. Project data of 71 metagenome projects (totaling 1677 metagenome samples) was acquired using the MG-RAST toolkit, and the rRNA sequences were extracted (totaling over 85 Gb of rRNA data). Blast [32] was used to search for sequences matching the 16S rRNA sequence of O. dioscoreae. Sequence hits with ≥98% identities were further analyzed using SILVA [33], and compared to the NCBI nr dna database to determine the most likely origin of the sequence. No sequence from any of these 1677 samples from environments as diverse as soil, plants, water bodies, feces, and anthropogenic environments could be confidently assigned to the genus Orrella above the 98% rRNA identity threshold.
Gnotobiotic culture of D. sansibarensis
Five D. sansibarensis bulbils were collected from the greenhouse of the botanical garden of Ghent University, thoroughly washed with tap water, surface sterilized with 70% ethanol and 1.4% sodium hypochloride for 5 min each and rinsed three times with sterile MilliQ water. Each bulbil was transferred to an autoclaved (121 °C for 15 min) microbox container (SacO2, Belgium) containing half-strength Murashige and Skoog (MS) medium and incubated for 2 months at 28 °C with a 16 h photoperiod and light intensity of 50 µmol m−2 s−1. After germination, leaf nodules were aseptically dissected and ground in sterile 0.4% NaCl. The macerate was streaked on TSA medium and incubated at 28 °C for 48 h. Identification and typing of isolates was done by colony PCR amplification and sequencing of the nrdA gene as described above.
RNA isolation and sequencing
Dioscorea sansibarensis were grown in the greenhouse at Ghent University at 25 °C with a 14 h-light/10 h-dark photoperiod cycle (~75 µmol m−2 s−1 at time of harvest). Previous typing of nodule isolates from our stock plant used for propagation revealed identical 16S and nrdA sequences as well as identical REP-(GTG)5-PCR and RAPD-PCR profiles to type strain LMG 29303T [14]. Nodules from three 3-month-old plants were dissected, at the middle of the light phase (2 p.m.) and middle of the dark phase (1 a.m.), using sterile scissors decontaminated with RNAase ZAP (Sigma Aldrich, St. Louis, MI, USA). Each pair of collected nodules (day/night), were collected from the same plant. In parallel, O. dioscoreae strain LMG 29303T was cultured in AB medium supplemented with 10 mM citrate and 0.05% yeast extract, in triplicate. Cells were harvested at mid-exponential phase (OD590nm = 0.2–0.3). RNA was isolated using the AurumTM Total RNA Mini Kit (BioRad, USA) according to manufacturer’s recommendations. Stranded cDNA libraries were constructed and sequenced at the Wellcome Trust Centre for Human Genetics (Oxford, UK). Analysis of the resulting sequencing reads was done using the DEseq2 software using the LMG 29303T reference genome [34]. RNA-sequencing reads are deposited in the European Nucleotide Archive under study accession number PRJEB30089. Additional details on library preparation and data analysis are listed in the supplementary information.
Phenotypic analysis
O. dioscoreae was grown on R2A agar (Oxoid) and grown overnight at 28 °C. Cells were suspended in the inoculation fluid IF-0 (Biolog, Hayward, CA) supplemented with dye A to a final turbidity of 85%T according to manufacturer’s recommendation. The suspension was then inoculated on Biolog plates PM1 and PM2A and incubated at 28 °C for 48 h under aerobic conditions. Development of color indicating substrate respiration was monitored at 24 h and 48 h.
Additional methods are described in the supplementary section.
Results
The D. sansibarensis/O. dioscoreae association is common in nature
We previously reported the isolation of O. dioscoreae from several D. sansibarensis specimens from European botanical gardens [14]. To investigate whether the association is also prevalent in nature, we isolated total DNA from 47 D. sansibarensis leaf acumens collected in 12 sites in the eastern Atsinanana and northern Diana regions of Madagascar (Fig. 1). We could detect the presence of O. dioscoreae DNA by PCR in all samples using primers specific to the nrdA gene sequence of O. dioscoreae LMG 29303T. Sequence analysis of the nrdA PCR products revealed very low diversity among samples, with a minimum of 97.6% identity to the type strain LMG 29303T. To investigate the prevalence of O. dioscoreae inside the leaf nodule, we generated shotgun metagenome sequences for 20 samples representative of the sites sampled and nrdA sequence types (Figure S1). O. dioscoreae was the only microbial species consistently found in all samples (Figure S2), comprising on average 95.65% of reads classified as bacterial (min 92%; max 98%). Furthermore, O. dioscoreae has never been isolated outside of D. sansibarensis leaf nodules and an exhaustive search for Orrella rRNA sequences in 1677 samples from 71 public metagenome projects failed to retrieve sequences above the 98% identity threshold (Supplementary information). A Blast search of the NCBI non-redundant nucleotide (nr/nt) and rRNA databases using the 16S sequence of O. dioscoreae LMG 29303T yielded four hits with >99% nucleotide identity. All corresponded to uncultured sequences found in the midgut of the cicada Meimuna mongolica [35]. Cicadas are phytophagous sap-sucking insects, and there is a distinct possibility that Orrella bacteria were accidentally ingested upon feeding. These results suggest that O. dioscoreae is limited to its unique niche in D. sansibarensis and that the association is prevalent and specific in nature.

Sample site locations of collected D. sansibarensis leaf nodules. Red markers represent collection sites in the Atsinanana region accessed in November 2016. Blue markers represent sites in the Diana region accessed in May 2017
Within-host population structure of O. dioscoreae
Populations of O. dioscoreae reach on average 3.4 × 108 symbiont cells within a single nodule [14] and have the potential to display high levels of genetic diversity due to de novo mutations or mixed infections. To investigate the diversity of O. dioscoreae in one host plant, we sequenced the contents of 20 nodules from a single host achieving an average coverage of the O. dioscoreae reference genome in excess of 600x. In total, we found 216 SNPs and 67 insertion-deletions. Of these, only 5 SNPs were represented in more than 10% of the reads mapped at the site. Intra-host diversity is thus low, with low frequencies of individual SNPs indicating that de novo mutational processes drive intra-host diversity rather than co-infection by multiple strains.
Dominant vertical mode of transmission of O. dioscoreae
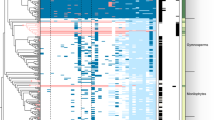
Low intra-host diversity in symbiont populations may be the result of strict controls on infection or a vertical mode of transmission accompanied by population bottlenecks [36, 37]. The lack of evidence for a reservoir also suggests that O. dioscoreae cannot easily be acquired from the environment. To test if O. dioscoreae is transmitted vertically, bulbils were surface-sterilized and germinated under gnotobiotic conditions. The bulbils all gave rise to plants colonized by bacteria with identical nrdA gene sequences to the parent (data not shown). Furthermore, O. dioscoreae could be isolated from macerated, surface-sterilized bulbils in high numbers (on average 2.2 × 105 ± 1.2 × 105 cfu/g or about 4.5 × 105 per bulbil), as well as from axillary buds from which bulbils emerge. Furthermore, co-phylogenetic analysis of chloroplast and symbiont genomes of wild-collected samples revealed broad patterns of co-speciation, with a clear biogeographical component. First, chloroplast whole-genome phylogenetic analysis resolved two distinct clusters according to sampling location in the Atsinanana and Diana regions (Fig. 2). Symbiont whole-genome phylogenies displayed partial congruence with the chloroplast phylogeny and a general conservation of the biogeographic signal (Fig. 2 and Figure S1 B). However, statistical analysis rejected strict co-speciation between host and symbionts (p > 0.9), while reconciliation analysis introduced 4 co-speciation events, 2 losses and 3 host-switching events (Fig. 2). These data are consistent with a dominant vertical mode of transmission with occasional horizontal or host-switching events.

Population structure of the D. sansibarensis/O. dioscoreae symbiosis. Co-phylogenetic analysis of D. sansibarensis (left) and O. dioscoreae (right). Host phylogeny was reconstructed based on a concatenated SNP-based alignment of whole chloroplast sequences including invariant sites (see supplementary information for details). Bootstrap values are shown. Nodes supported by <75% bootstrap values were collapsed. Highlighted in dark grey: samples collected in the Atsinanana region; highlighted in light grey: samples collected in the Diana region; Black: Samples collected in the botanical garden of Ghent University
Vertical transmission without genome reduction
Reductive genome evolution, a process by which the size and coding capacity of genomes tends to shrink over time, is a nearly universal phenomenon among vertically transmitted symbionts and obligate pathogens, including leaf nodule symbionts of Rubiaceae and Primulaceae [9,10,11, 38, 39]. The genome of O. dioscoreae LMG 29303T is of average size for the family Alcaligenaceae (Figure S3). Reduced genomes also commonly have lower %G+C compared to free-living relatives, but the average %G+C of 66.15% does not deviate significantly from the average of 65.34% calculated from genomes of neighboring Bordetella and Achromobacter genomes. Coding density is also high (90%), with only 40 predicted pseudogenes and 20 putative IS elements present in the genome of the type strain LMG 29303T. This lack of evidence for genome reduction could be explained by two, non-mutually exclusive reasons: (i), the Dioscorea leaf nodule symbiosis evolved only recently, leaving little time for the effects of genome erosion to accrue, or (ii), a large effective population size and efficient selection accounts for the maintenance of genomic structure. To obtain a measure of the efficiency of selection, we calculated genome-wide non-synonymous to synonymous substitutions rates (dN/dS) of O. dioscoreae. The genome-wide dN/dS of O. dioscoreae is low (dN/dS = 0.0597) and in the reported range for free-living bacteria [40]. Furthermore, core genes do not display significantly elevated dN/dS compared to free-living Alcaligenaceae species (Figure S4). These data indicate that O. dioscoreae experiences low levels of genetic drift, typical for bacteria with large effective populations but highly unusual for vertically transmitted symbionts [41, 42].
Recent capture of a vertically transmitted endophyte
A recent evolution of a vertical mode of transmission may also contribute to the lack of evidence for genome erosion. The ancestor of D. sansibarensis diverged from sister Malagasy yam species around 22.6 Mya [43]. However, the estimated time of divergence between D. sansibarensis and non-nodulated sister yam species is likely a gross overestimate of the age of the symbiosis because of incomplete sampling of extant and extinct species. Using chloroplastic whole-genome sequences and calibration data from Viruel et al. [43], we estimated that the specimens included in our study diverged during the Pleistocene and perhaps as recently as 20,000 years ago (95% confidence interval: 0.0201 to 3.1902 Mya; see Supplementary information for details). To provide another independent estimate of the divergence time of our samples, we calculated the mutation rate of O. dioscoreae in a single lineage sampled at 2 years interval. Using an estimated mutation rate of 2.06 × 10−7 substitutions/site/year, we inferred that all O. dioscoreae strains (including samples collected in continental Africa) diverged from a common ancestor about 124,000 years ago (supplementary information). This estimate falls within the confidence interval derived from the chloroplast whole-genome phylogenetic analysis and supports a recent emergence of the symbiosis.
Large complement of differentially regulated genes during symbiosis
To elucidate the function and the nature of the metabolic exchange between host and symbiont, we generated transcriptomic data of O. dioscoreae in the leaf gland of D. sansibarensis compared to O. dioscoreae LMG 29303T grown to exponential phase on minimal media supplemented with citrate and ammonia as carbon and nitrogen sources (Table S4). A total of 1639 out of 4363 (37.5%) genes were differentially expressed (p-value < 0.05; absolute log2 fold change ≥1.5) between growth in planta or on minimal medium, with a balanced proportion of upregulated (834) and downregulated (805) genes across all major functional categories (Figure S5). A majority of genes assigned to translation, ribosomal structure and biogenesis were upregulated in the leaf nodule, while most differentially expressed genes of inorganic ion transport and metabolism as well as lipid transport and metabolism were down-regulated, perhaps indicative of a low diversity of substrates available for growth. Of the 70 upregulated genes linked to ribosome structure and function, 17 were among the top 100 upregulated genes (>13-fold). This higher expression of ribosome components was concomitant with an upregulation of the translation elongation factors efp, tsf, tuf, and fusA, as well as more than 30% of genes involved in cell cycle control and chromosome partitioning (COG category D) and an upregulation of genes coding for subunits of the RNA polymerase (ODI_R0061, ODI_R0131-2). Increased synthesis of ribosomes and components of the translation, transcription, and replication machinery are indicative of faster growth in planta [44].
The leaf gland niche is characterized by micro-oxia, iron limitation, and a sessile lifestyle
Faster apparent growth suggests that O. dioscoreae is highly adapted to conditions within the leaf gland environment. Upregulation of a cytochrome d ubiquinol oxidase in the leaf gland (ODI_00363-4) is reminiscent of other Burkholderiales bacteria grown under micro-oxic conditions and may be a response to low oxygen concentration in the leaf gland [45]. Direct free oxygen measurements taken at ca. 1 mm depth beneath the leaf gland surface confirm conditions of micro-oxia, with dissolved O2 concentration of 35.7 ± 10.8 μM at 21 °C (data not shown). Most genes related to motility and chemotaxis (ODI_R2117-R2164) are downregulated, while genes coding for cellulose synthase (ODI_R2609-2610) and capsular polysaccharides (ODI_R0994-1001) are upregulated, all of which typical of a biofilm mode of growth [46]. Furthermore, upregulation of fur by more than 6-fold on average in the leaf nodule, together with upregulation of genes related to siderophore biosynthesis and uptake (ODI_R2471-78 and R2482) indicate that iron may be a factor limiting growth in the leaf gland [47,48,49,50].
Organic acids fuel the growth of O. dioscoreae in planta
The genome of O. dioscoreae does not encode enzymes for the degradation of complex carbohydrates, and O. dioscoreae cannot utilize monosaccharides in culture [14]. Upregulation of the genes of the TCA in the leaf nodule by an average of 3.9-fold and a lack of overall differential expression of the pentose phosphate pathway, glycolysis, or branched-chain amino acid degradation pathways (Table S4) suggest that growth inside the leaf nodule is fueled by short-chain amino acids or organic acids. To validate this interpretation, we tested growth of O. dioscoreae on 190 distinct carbon sources. Strain LMG 29303T could only utilize organic acids, predominantly substrates of the TCA (Table S5). In addition, growth was supported by l-proline and l-glutamate. Two putative NADP-dependent glutamate dehydrogenases were upregulated in the leaf gland (ODI_R4072, 3.6-fold and ODI_R2231, 6.3-fold), suggesting that deamination of glutamate to 2-oxo-glutarate could provide substrates for the TCA cycle. However, a putA homolog (ODI_R1770), encoding a bi-functional proline dehydrogenase and delta-1-pyrroline-5-carboxylate dehydrogenase was not differentially regulated. This suggests that glutamate, rather than proline, possibly serves as an energy source in the leaf nodule. Growth could also be supported by d-galactonic acid (supplementary information). The dgo operon (ODI_R1138-1141) of d-galactonate utilization was upregulated by an average of more than 7-fold in the leaf nodule. However, lack of conservation in other strains suggests that utilization of d-galactonate may not be essential for growth of O. dioscoreae in leaf nodule symbiosis (Fig. 3).

Conservation and expression of O. dioscoreae genes. Inner circles represent nucleotide conservation (% Blastn identity) of that genome with the type strain LMG29303T as histograms. A legend showing the order of the genomes used for comparison is shown at the bottom right. Blue and red rings correspond to samples collected in the Diana and Atsinanana regions, respectively. Outer rings show the log2 fold change of predicted genes in the leaf nodule compared to in culture as measured by RNA-Seq. Loci discussed in the text are indicated with gray rectangles
Simple metabolic needs of leaf nodule bacteria
Components of the GS/GOGAT pathway (ODI_R0289–ODI_R0288; OD_R2281), were only slightly upregulated (<3-fold) in the leaf nodule and a putative ammonium transporter (ODI_R2565) was not differentially regulated in planta compared to growth with ammonium as a nitrogen source. Neither the assimilatory nitrate reductase (ODI_R3120) or the nitrite reductase (ODI_R2365-2367) were differentially regulated. These observations indicate that nitrogen is taken up as ammonium or through deamination of amino-acids. Amino-acid biosynthetic pathways were either slightly upregulated or not differentially regulated in planta, except for the pathways for the biosynthesis of branched-chain amino acids (Table S4) which were significantly downregulated. Several putative branched-chain amino acid transporters were simultaneously upregulated, suggesting that valine, leucine, and/or isoleucine are abundant in the nodule. A metE homolog, coding for the cobalamin-independent methionine synthase (ODI_R2167), is upregulated by more than 150-fold on average, indicating that this pathway is preferred over the MetH pathway (ODI_R0578, not differentially regulated). LMG 29303T cultures grew in the presence of low concentrations of yeast extract, which contains small amounts of vitamins and cofactors. The vast majority of genes (64%) involved in these pathways were not differentially regulated in the nodule. Only genes involved in vitamin B6 biosynthesis showed moderately increased expression (4.5-fold) compared to axenic cultures and may reflect a higher demand for the pyridoxal phosphate coenzyme, for example needed for the transamination reactions required by increased demand for amino-acid biosynthesis.
Symbiont response to light cycle
The genome of O. dioscoreae does not encode photosystems or carbon assimilation pathways, and isolates are not pigmented, ruling out direct participation to photosynthesis [14]. However, the leaf acumen hosting the bacterial glands is composed of green tissue, raising the possibility that O. dioscoreae participates to or complements the metabolism of photosynthates. To test this, we generated transcriptome data of whole leaf nodules harvested in the middle of the light and dark phases. Only 8 O. dioscoreae genes were upregulated under light conditions (3.7-fold average), and 1 hypothetical gene was downregulated (3-fold). Six of the seven upregulated genes (ODI_R3721-3727) belonged to a putative operon conserved in all O. dioscoreae genomes. Aside from putative regulatory proteins and a putative ECF sigma factor, the operon encodes a short-chain dehydrogenase, a flavin-containing amine oxidase, a hypothetical protein, and a cyclopropane fatty acyl transferase (ODI_R3722-3725). Homologs of these, sharing on average 30–40% identity at the protein level, are induced by singlet oxygen in the purple bacterium Rhodobacter sphaeroides and may be involved in detoxifying reactive oxygen species (ROS) during photosynthesis [51]. Together, these data indicate that leaf nodule bacteria do not play a significant role in photosynthesis or the host’s carbon assimilation pathways, and reveal that detoxification of singlet oxygen, a by-product of photosynthesis, may be a significant challenge for leaf endophytes.
The leaf nodule is dedicated to secondary metabolite production and exchange
Thirty-eight of the fifty most upregulated genes in the leaf nodule belonged to only 2 gene clusters. The largest cluster, called smp for its likely role in secondary metabolite production comprises 23 genes upregulated by 165- to 720-fold in the nodule and is organized in 2 divergent operons, smp1 (ODI_R1487-1493) and smp2 (ODI_R1497-1509), separated by a regulatory region (supplementary information). Transcripts of the smp cluster have among the highest absolute abundance values in the leaf nodule, making up between 23.5 and 27.4% of all non-rRNA reads. The smp cluster is highly conserved in all O. dioscoreae genomes with average nucleotide identities ranging from 96.6 to 99.3% but is otherwise unique within the Alcaligenaceae. Despite this exclusivity, we could not detect any of the common signs associated with recent acquisition by horizontal gene transfer: the average %G+C of the smp cluster is only slightly higher than genome average (69.0% vs. 67.4%) and codon usage does not significantly deviate from the rest of the genome (Pearson chi-square test p-value > 0.25). Further, we could not detect evidence of mobile elements flanking the smp cluster and the phylogeny of individual smp genes tracked that of a core gene set (data not shown). Sequence analysis of the smp genes revealed a highly unusual arrangement of genes linked to non-ribosomal peptide synthesis (NRPS), putative NRPS tailoring enzymes and genes involved in acyl chain biosynthesis (Supplementary information). Iron-chelating activity on reporter medium was unchanged for a smpD null mutant, ruling out a possible role of smp in siderophore synthesis (supplementary information). A second gene cluster of 19 predicted genes linked to polyketide synthesis accounts for another 11 of the top 50 most differentially expressed genes (ODI_R2247-2265). Genes of the opk (for Orrella polyketide) cluster are also among the most highly expressed in the leaf nodule, making up between 8.8 and 9.3% of all mRNA reads mapped. As for the smp cluster, the opk cluster is unique to O. dioscoreae among Alcaligenaceae but displays partial similarity to uncharacterized gene clusters of Burkholderia glumae BGR1 and B. ubonensis MSMB818. All opk genes appear conserved in both Burkholderia strains, but the homologous Burkholderia gene clusters contain additional genes coding for an iron containing redox enzyme and an acyl-coA dehydrogenase. Sequence analysis of the opk genes reveal the presence of a set of genes linked to polyketide synthesis, a class of natural products with broad activities [52], as well as an ATP transporter highly upregulated in planta (Supplementary information).
Recognition of symbiotic partners and maintenance of the symbiosis
We previously reported that the genomes of Burkholderia leaf nodule symbionts of Psychotria and Ardisia did not encode functions found in other plant symbioses such as Type III or IV secretion, plant hormone metabolism or Nod factor synthesis [9,10,11]. How these symbionts avoid triggering plant defenses during symbiosis remains unknown. The genome of O. dioscoreae LMG 29303T encodes a type III secretion system (T3SS) (ODI_R0595-0615) and an ACC-deaminase (ODI_R1068), possibly involved in modulation of the ethylene defense pathway [53]. However, neither pathways were differentially regulated in planta. Furthermore, homologs of the T3SS are absent from the genomes of strains RAN3, ANT2, and ANT3 (Fig. 3). Similarly, two putative type IV secretion systems (ODI_R1296-1315 and ODI_R2705-2718) are downregulated in the leaf nodule and are not conserved in all genomes. T3SS and T4SS are thus unlikely to play major roles in the interaction with the host. Two distinct type VI secretion systems (T6SS), T6SS-1 and T6SS-2 (ODI_R3980-4005 and ODI_R0780-0812, respectively), were highly upregulated in planta and conserved in all O. dioscoreae genomes (Fig. 3). Both clusters contain all 13 core components (TssA–TssM) and display a similar organization but with overall low sequence identity, ruling out recent duplication. Four Rhs-VgrG effectors (three in T6SS-1, one in T6SS-2) are conserved in all strains, while one effector is only conserved in ANT2 and ANT3 genomes. In addition, a Rhs-core domain-containing gene was found only in the BER1 and BER2 genomes.
Genes under positive selection
In the absence of evidence for relaxed purifying selection affecting many vertically transmitted symbionts, adaptive mutations may still accumulate in distinct populations of O. dioscoreae [54]. We found 215 genes which showed evidence of positively selected sites in O. dioscoreae (Table S6). Among these, 2 genes of T6SS-1 seem to be under positive selective pressure. These code for a TssA homolog (ODI_R4005) and a TssM homolog (ODI_R3983), which have respectively been proposed to be part of the T6SS baseplate and the membrane complex [55, 56]. One of four conserved VgrG effector proteins in the O. dioscoreae genome (ODI_R0793) also contains multiple sites with dN/dS > 1. Gene ODI_R3363, coding for a catalase and highly expressed in the leaf nodule, contains 3 sites under positive selection. Catalase is an important enzyme mitigating damage that may arise from ROS such as H2O2. ROS is often produced by plants in response to pathogen infection, but also plays an important role in plant signaling. The sites under positive selection localize within a non-catalytic domain at the C-terminal of the predicted protein. A homologous domain in the KatA catalase of Helicobacter pylori has been found to mediate T-cell interaction [57] and facilitate evading the host’s defenses [58]. Two putative proteins containing a diguanylate cyclase GGDEF domain (ODI_R2651, ODI_R0990), as well as proteins involved in iron uptake and siderophore synthesis (ODI_R1761, ODI_R2287) and flagellar motility (ODI_R2135) also contained sites with elevated dN/dS. Homologs of these proteins have recently been found to elicit plant defenses [59]. Together, these suggest that environmental factors such as microbe-microbe competition, but also adaptation to host immunity may be significant factors shaping the evolution of O. dioscoreae. Intriguingly, five genes of the smp cluster (ODI_R1487, ODI_R1489, ODI_R1490, ODI_R1498, and ODI_R1507) display signs of positive selection, indicating that secondary metabolism may be diversifying in O. dioscoreae (Table S6).
Discussion
Recent evolution of a vertically transmitted symbiosis in plants
Heritable symbiosis is relatively common in the animal kingdom but affects only a handful of plants [39, 60]. We describe here a new heritable symbiosis between bacteria of the Alcaligenaceae family and D. sansibarensis, the first of its kind in monocots. These symbiotic bacteria are found within conspicuous galls or nodules on leaves and are transmitted vertically via bulbils. Vertical transmission of O. dioscoreae likely relies on the colonization of lateral buds which give rise to already colonized bulbils [61]. In support of this hypothesis, high titers of O. dioscoreae are found in apical and lateral buds as well as bulbils and are sufficient to ensure colonization of seedlings under gnotobiotic conditions. A bacterial population maintained in proximity of developing shoot meristems (~103–104 cfu, data not shown), may allow both the allocation of founder colonies to the developing leaf nodules and the transmission to the next generation (Fig. 4). We speculate that bacteria whose fate are either to fulfill the symbiotic function in the leaf nodule or to be transmitted to the next generation are drawn from this common pool. This unique mode of infection may have important consequences for the evolution of the symbiosis in plants. However, partial congruence between host and symbiont phylogenies suggests a mixed mode of transmission at population level, combining vertical transmission and occasional horizontal or host switching events [62]. Because even very rare horizontal transmission would completely degrade the correlation between host and symbiont phylogenetic signals [63], the vertical mode of transmission is probably largely dominant in the D. sansibarensis/O. dioscoreae symbiosis. Rare horizontal transmission may occur by host switching via insect vectors, since the only evidence for a possible reservoir of environmental O. dioscoreae is the gut of sap-feeding insects.

General overview of the O. dioscoreae lifecycle and functional predictions. D. sansibarensis harbors symbiotic bacteria (O. dioscoreae) which are contained within leaf nodules, bulbils, and apical or axillary buds. The apical bud is the site of post-embryonic growth and gives rise to new leaves or aerial bulbils used for propagation. Images center: microscopic images of O. dioscoreae in key plant tissues. Top center: O. dioscoreae in the apical bud labeled with FISH probe BETA42a specific for betaproteobacteria; L: young leaf; T: trichome; white arrows: bacteria (T. Acar, unpublished). Bottom center: Leaf nodule section triple stained with acridine red, chrysoidine and astra blue, which enhance contrast in sections of plant organs containing both primary and secondary cell walls. Bacteria are often found near trichomes, but never inside plant cells. G: bacterial gland; VT: vascular tissue. Right panel: pathways or functions presumed important for symbiosis and survival inside the leaf nodule. ROS, reactive oxygen species; LPS, lipopolysaccharide; EPS, exopolysaccharide, TCA, tricarboxylic acid cycle; T6SS, Type 6 secretion system; N, nitrogenous compounds such as ammonium or amino acids. LPS and EPS may play a role in host/symbiont recognition. T6SS may play a role in preserving the specificity of the association by excluding competitors. See main text for details
Vertical transmission is thought to be a particularly effective mechanism to enforce cooperation in symbiotic associations [64,65,66], but is not synonymous with mutualism [60, 67]. Transcriptomic analysis of leaf nodule contents show that O. dioscoreae relies on organic acids for growth in planta and scavenges iron by upregulating iron-acquisition pathways, while the pathways for biosynthesis of amino acids, as well as vitamins were expressed to levels comparable or slightly higher than growth on minimal medium. This is in direct contrast to plant pathogens, which typically show increased breadth of substrate utilization and suppression of siderophore synthesis during infection [68,69,70]. These data indicate that the metabolic needs of O. dioscoreae in the leaf nodule are relatively simple and avoid exploitation of costly host resources such as sugars, complex carbohydrates, and amino-acids. Further supporting a mutualistic rather than parasitic lifestyle, T3SS genes which are essential virulence factors for a majority of gram-negative plant pathogens [71], were not significantly expressed in the leaf nodule and were not conserved in all O. dioscoreae genomes.
We did not find evidence for roles of the bacterial symbiont in carbon assimilation, or other roles commonly associated with beneficial endophytic bacteria such as nitrogen fixation or hormone metabolism. Moderate expression of vitamin or amino-acid biosynthetic pathways, mostly consistent with growth on simple carbon and nitrogen sources, makes nutrient supply to the host unlikely. Instead, over 30% of transcripts in leaf nodule bacteria stemmed from only two gene clusters related to bacterial secondary metabolism. The role and nature of the secondary metabolites produced in the D. sansibarensis/O. dioscoreae symbiosis remains to be elucidated but appear to result from a very unusual combination of NRPS, fatty acid, and polyketide synthesis, possibly representing new molecules. We have previously shown that more than 10% of the proteome of Ca. Burkholderia kirkii, the obligate symbiont of Psychotria kirkii, was dedicated to the synthesis of cyclitol compounds with insecticidal and herbicidal properties [72, 73]. Similarly, the leaf nodule symbiont of Ardisia crenata synthesizes FR900359, a depsipeptide inhibitor of mammalian Gq proteins and potent insecticide [9, 13]. Taken together, these data suggest that leaf nodule symbionts were recruited independently in three plant families to complement the host’s secondary metabolism. Interestingly, the lack of evidence for gene conversion or horizontal transfer of the smp and opk clusters indicates that these genes were present in the last common ancestor of leaf nodule O. dioscoreae. Synthesis of secondary metabolites may thus be a pre-requisite for symbiont capture in the phyllosphere. Further characterization of the metabolites of the D. sansibarensis leaf nodule symbiosis will be essential to uncover the ecological role of this symbiosis and may provide new leads with biological activities of interest.
Vertical transmission of symbionts is a major evolutionary transition which enables the fixation of complex heritable traits in a lineage. However, this often comes at the cost of co-dependence between the partners, with the microsymbiont unable to replicate independently and in some extreme cases with the host unable to survive alone [60, 66, 74]. O. dioscoreae is among the very few vertically transmitted bacterial symbionts which can be cultured, along with symbionts of fungi [75], the Tsetse fly [76], and earthworms [77]. Unlike these examples, the genome of O. dioscoreae seem completely devoid of any of the hallmarks of reductive genome evolution. This unusual characteristic may be due to a recent evolution of the association and a large effective population. The strong purifying selection acting on the O. dioscoreae genome would suggest the latter, but large populations would also be expected to result in high intra-host symbiont diversity [36, 66, 78, 79]. Surprisingly, we found intra-host diversity to be low and consistent with accumulation of low frequency de novo mutations. Perhaps as another consequence of the unique infection mode directly coupled to the host’s post-embryonic growth, intra-host competition between genotypes may instead account for this low observed diversity [80]. Because host controls such as sanctions have never been documented in heritable symbioses [65, 81, 82], hypothetical mechanisms preventing the fixation of non-cooperating genotypes as a result of intense intra-host competition remain to be elucidated.
How leaf nodule symbionts avoid triggering innate plant immune defenses remains an open question. We show that T3SS and T4SS are not conserved in O. dioscoreae genomes, suggesting that these secretion systems do not play an active role in modulating the immune response of the host. The genomes of O. dioscoreae do not encode cell-wall degrading enzymes and the metabolism of organic acids rather than complex sugars may avoid the release of damage associated molecular patterns (DAMPs) [83]. Alternatively, EPS, upregulated in the leaf nodule, is known to be crucial for the establishment of successful symbiosis with legumes [84, 85] and may play an important role in protection against host defense [86]. Interestingly, we could also detect signatures of positive selection in natural populations of O. dioscoreae in various genes coding for products implicated in the elicitation of plant defenses including several GGDEF domain proteins and proteins involved in iron uptake and siderophore synthesis [59]. These potential M/PAMPs may have evolved to lower immune recognition and enable the bacteria to multiply within host tissue (e.g. in the shoot apical bud) without triggering defenses. Specific epitopes may also enable active recognition of beneficial symbionts, similar to what has been described in some invertebrate systems such as Hydra sp. or in the squid-Vibrio symbiosis [87, 88]. In this respect, patterns of positive selection on smp genes are of particular interest. Diversifying selection on genes of the secondary metabolism may reflect adaptation to biotic pressure in distinct environments, e.g. the presence of different herbivores, but may also reflect pressure by the host to indirectly gauge the contribution of the leaf nodule symbionts. These genes make attractive targets for mutagenesis or heterologous expression to further elucidate partner recognition in leaf nodule symbiosis.
As the lifecycle of O. dioscoreae is strictly extracellular and leaf nodules initially appear open to the outside environment [15], it is unclear how specificity is maintained in the leaf nodule. A complex interplay of immune modulation or recognition and filtering due to environmental parameters may enable the host to control access to the nodule. Oxidative stress, possibly as a result of singlet oxygen produced through photosynthesis, may guard against potential invading microorganisms [89]. In addition, we found two conserved O. dioscoreae T6SS to be upregulated in the leaf nodule. T6SS is a common mediator of antagonistic microbe–microbe interactions in many bacteria and may actively contribute to gatekeeping inside the leaf nodule [90]. Evidence of positive selection in genes of the T6SS, including diversification of putative effectors, could indicate ongoing adaptation of the symbiont to maintain its niche against biotic challenges. This phenomenon was recently observed in bee gut symbionts and was hypothesized to have a significant impact into shaping the evolution of the bee gut microbiome [91].
In conclusion, this study broadens our knowledge of plant–bacteria interactions in the phyllosphere and highlights features specific to heritable symbioses in plants. We demonstrate striking commonalities between the leaf nodule symbiosis in D. sansibarensis and those of Rubiaceae and Primulaceae, including a vertical mode of symbiont transmission and a seemingly central role of bacterial secondary metabolism. As O. dioscoreae can easily be cultured, and the host plant can easily be grown and propagated using standard methods, this binary symbiosis provides an attractive model system for the study of beneficial plant-bacteria interactions in the phyllosphere. However, O. dioscoreae cannot be easily removed from bulbils, even after prolonged treatment with antibiotics (data not shown). We are currently developing protocols for the generation and re-inoculation of aposymbiotic plants based on plant-tissue culture techniques.
References
Sheibani-Tezerji R, Rattei T, Sessitsch A, Trognitz F, Mitter B. Transcriptome profiling of the endophyte Burkholderia phytofirmans PsJN indicates sensing of the plant environment and drought stress. MBio. 2015;6:e00621–15.
Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH. Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol. 2013;11:789–99.
Bodenhausen N, Bortfeld-Miller M, Ackermann M, Vorholt JA. A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 2014;10:e1004283.
Horton MW, Bodenhausen N, Beilsmith K, Meng D, Muegge BD, Subramanian S, et al. Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat Commun. 2014;5:5320.
Yu X, Lund SP, Scott RA, Greenwald JW, Records AH, Nettleton D, et al. Transcriptional responses of Pseudomonas syringae to growth in epiphytic versus apoplastic leaf sites. Proc Natl Acad Sci USA. 2013;110:E425–34.
Pinto-Carbó M, Eberl L, Gademann K, Carlier A. Leaf nodule symbiosis: function and transmission of obligate bacterial endophytes. Curr Opin Plant Biol. 2018;44:23–31.
Lersten NR, Horner HT Jr. Development and structure of bacterial leaf nodules in Psychotria bacteriophila Val. (Rubiaceae). J Bacteriol. 1967;94:2027–36.
Miller IM. Bacterial leaf nodule symbiosis. Adv Bot Res. 1990;17:163–234.
Carlier A, Fehr L, Pinto-Carbó M, Schäberle T, Reher R, Dessein S, et al. The genome analysis of Candidatus Burkholderia crenata reveals that secondary metabolism may be a key function of the A rdisia crenata leaf nodule symbiosis. Environ Microbiol. 2016;18:2507–22.
Carlier AL, Eberl L. The eroded genome of a leaf symbiont: hypotheses about lifestyle and interactions with its plant host. Environ Microbiol. 2012;14:2757–69.
Pinto-Carbó M, Sieber S, Dessein S, Wicker T, Verstraete B, Gademann K, et al. Evidence of horizontal gene transfer between obligate leaf nodule symbionts. ISME J. 2016;10:2092–105.
Sieber S, Carlier A, Neuburger M, Grabenweger G, Eberl L, Gademann K. Isolation and total synthesis of kirkamide, an aminocyclitol from an obligate leaf nodule symbiont. Angew Chem Int Ed. 2015;54:7968–70.
Crüsemann M, Reher R, Schamari I, Brachmann AO, Ohbayashi T, Kuschak M, et al. Heterologous expression, biosynthetic studies, and ecological function of the selective Gq-signaling inhibitor FR900359. Angew Chem Int Ed. 2018;57:836–40.
Carlier A, Cnockaert M, Fehr L, Vandamme P, Eberl L. Draft genome and description of Orrella dioscoreae gen. nov. sp. nov., a new species of Alcaligenaceae isolated from leaf acumens of Dioscorea sansibarensis. Syst Appl Microbiol. 2017;40:11–21.
Miller IM, Reporter M. Bacterial leaf symbiosis in Dioscorea sansibarensis: morphology and ultrastructure of the acuminate leaf glands. Plant Cell Environ. 1987;10:413–24.
Burkill H. The useful plants of west tropical Africa. Volume 1, families A-D. Kew, England: Royal Botanic Gardens, Kew; 1985.
Clark JD, Maaloe O. DNA replication and the division cycle in Escherichia coli. J Mol Biol. 1967;23:99–112.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol. 2013;31:533–8.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–95.
Wood DE, Salzberg SL. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–5.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Li L, Stoeckert CJ, Roos DSC-P. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol. 2017;34:2115–22.
Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, et al. AntiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017;45:W36–W41.
Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR. The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012;7:e34064.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12:902–3.
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M. The metagenomics RAST server-a public resource for the automatic phylo- genetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Zhou W, Nan X, Zheng Z, Wei C, He H. Analysis of inter-individual bacterial variation in gut of Cicada Meimuna mongolica (Hemiptera: Cicadidae). J Insect Sci. 2015;15:131.
Wollenberg MS, Ruby EG. Population structure of Vibrio fischeri within the light organs of Euprymna scolopes squid from Two Oahu (Hawaii) populations. Appl Environ Microbiol. 2009;75:193–202.
Russell SL, Cavanaugh CM. Intrahost genetic diversity of bacterial symbionts exhibits evidence of mixed infections and recombinant haplotypes. Mol Biol Evol. 2017;34:2747–61.
Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–90.
Fisher RM, Henry LM, Cornwallis CK, Kiers ET, West SA. The evolution of host-symbiont dependence. Nat Commun. 2017;8:15973.
Kuo C-H, Moran NA, Ochman H. The consequences of genetic drift for bacterial genome complexity. Genome Res. 2009;19:1450–4.
Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304.
Mondo SJ, Salvioli A, Bonfante P, Morton JB, Pawlowska TE. Nondegenerative evolution in ancient heritable bacterial endosymbionts of fungi. Mol Biol Evol. 2016;33:2216–31.
Viruel J, Segarra-Moragues JG, Raz L, Forest F, Wilkin P, Sanmartín I, et al. Late Cretaceous-early Eocene origin of yams (Dioscorea, Dioscoreaceae) in the Laurasian Palaearctic and their subsequent Oligocene-Miocene diversification. J Biogeogr. 2016;43:750–62.
Bosdriesz E, Molenaar D, Teusink B, Bruggeman FJ. How fast-growing bacteria robustly tune their ribosome concentration to approximate growth-rate maximization. FEBS J. 2015;282:2029–44.
Pessi G, Braunwalder R, Grunau A, Omasits U, Ahrens CH, Eberl L. Response of Burkholderia cenocepacia H111 to Micro-Oxia. PLoS ONE 2013;8:e72939.
Flemming H-C, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol. 2016;14:563–75.
Mey AR, Craig SA, Payne SM. Characterization of Vibrio cholerae RyhB: the RyhB regulon and Role of ryhB in biofilm Formation. Infect Immun. 2005;73:5706–19.
Wu Y, Outten FW. IscR controls iron-dependent biofilm formation in Escherichia coli by regulating type I fimbria expression. J Bacteriol. 2009;191:1248–57.
Lin M-H, Shu J-C, Huang H-Y, Cheng Y-C. Involvement of iron in biofilm formation by Staphylococcus aureus. PLoS ONE 2012;7:e34388.
Banin E, Vasil ML, Greenberg EP. Iron and Pseudomonas aeruginosa biofilm formation. Proc Natl Acad Sci USA. 2005;102:11076–81.
Anthony JR, Warczak KL, Donohue TJ. A transcriptional response to singlet oxygen, a toxic byproduct of photosynthesis. Proc Natl Acad Sci USA. 2005;102:6502–7.
Helfrich EJN, Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep. 2016;33:231–316.
Glick BR. Modulation of plant ethylene levels by the bacterial enzyme ACC deaminase. FEMS Microbiol Lett. 2005;251:1–7.
Kjeldsen KU, Bataillon T, Pinel N, De Mita S, Lund MB, Panitz F, et al. Purifying selection and molecular adaptation in the genome of Verminephrobacter, the heritable symbiotic bacteria of earthworms. Genome Biol Evol. 2012;4:307–15.
Planamente S, Salih O, Manoli E, Albesa‐Jové D, Freemont PS, Filloux A. TssA forms a gp6‐like ring attached to the type VI secretion sheath. EMBO J. 2016;35:1613–27.
Santin YG, Cascales E. Domestication of a housekeeping transglycosylase for assembly of a Type VI secretion system. EMBO Rep. 2017;18:138–49.
Guy B, Krell T, Sanchez V, Kennel A, Manin C, Sodoyer R. Do Th1 or Th2 sequence motifs exist in proteins? Immunol Lett. 2005;96:261–75.
Richter C, Mukherjee O, Ermert D, Singh B, Su Y-C, Agarwal V, et al. Moonlighting of Helicobacter pylori catalase protects against complement-mediated killing by utilising the host molecule vitronectin. Sci Rep. 2016;6:24391.
McCann HC, Nahal H, Thakur S, Guttman DS. Identification of innate immunity elicitors using molecular signatures of natural selection. Proc Natl Acad Sci USA. 2012;109:4215–20.
Sachs JL, Skophammer RG, Regus JU. Evolutionary transitions in bacterial symbiosis. Proc Natl Acad Sci USA. 2011;108:10800–7.
Rao AN, Tan AS. Shoot apex and bulbil development in Dioscorea sansibarensis Pax. Bot J Linn Soc. 1976;72:285–98.
Bright M, Bulgheresi S. A complex journey: transmission of microbial symbionts. Nat Rev Microbiol. 2010;8:218–30.
Brandvain Y, Goodnight C, Wade MJ. Horizontal transmission rapidly erodes disequilibria between organelle and symbiont genomes. Genetics. 2011;189:397–404.
Douglas AE. Host benefit and the evolution of specialization in symbiosis. Heredity. 1998;81:599–603.
Douglas AE. Conflict, cheats and the persistence of symbioses. New Phytol. 2008;177:849–58.
Bennett GM, Moran NA. Heritable symbiosis: the advantages and perils of an evolutionary rabbit hole. Proc Natl Acad Sci USA. 2015;112:10169–76.
Bordenstein SR, Paraskevopoulos C, Dunning Hotopp JC, Sapountzis P, Lo N, Bandi C, et al. Parasitism and mutualism in Wolbachia: what the phylogenomic trees can and cannot say. Mol Biol Evol. 2009;26:231–41.
González-Mula A, Lang J, Grandclément C, Naquin D, Ahmar M, Soulère L, et al. Lifestyle of the biotroph Agrobacterium tumefaciens in the ecological niche constructed on its host plant. New Phytol. 2018;219:350–62.
Nobori T, Velásquez AC, Wu J, Kvitko BH, Kremer JM, Wang Y, et al. Transcriptome landscape of a bacterial pathogen under plant immunity. Proc Natl Acad Sci. 2018;115:E3055–E3064.
Puławska J, Kałużna M, Warabieda W, Mikiciński A. Comparative transcriptome analysis of a lowly virulent strain of Erwinia amylovora in shoots of two apple cultivars-susceptible and resistant to fire blight. BMC Genom. 2017;18:868.
Buttner D, He SY. Type III protein secretion in plant pathogenic bacteria. Plant Physiol. 2009;150:1656–64.
Carlier AL, Omasits U, Ahrens CH, Eberl L. Proteomics analysis of Psychotria leaf nodule symbiosis: improved genome annotation and metabolic predictions. Mol Plant Microbe Interact. 2013;26:1325–33.
Hsiao C-C, Sieber S, Georgiou A, Bailly A, Emmanouilidou D, Carlier A, et al. Synthesis and biological evaluation of the novel growth inhibitor streptol glucoside, isolated from an obligate plant symbiont. Chem A Eur J. 2018;25:1722–6.
De Mazancourt C, Loreau Mi, Dieckmann U. Understanding mutualism when there is adaptation to the partner. J Ecol. 2005;93:305–14.
Lackner G, Moebius N, Partida-Martinez LP, Boland S, Hertweck CC-P. Evolution of an endofungal Lifestyle: deductions from the Burkholderia rhizoxinica Genome. BMC Genomics. 2011;12:210.
Dale C, Maudlin I. Sodalis gen. nov. and Sodalis glossinidius sp. nov., a microaerophilic secondary endosymbiont of the tsetse fly Glossina morsitans morsitans. Int J Syst Bacteriol. 1999;49:267–75.
Lund MB, Kjeldsen KU, Schramm A. The earthworm-Verminephrobacter symbiosis: an emerging experimental system to study extracellular symbiosis. Front Microbiol. 2014;5:128.
Mira A, Moran NA. Estimating population size and transmission bottlenecks in maternally transmitted endosymbiotic bacteria. Microb Ecol. 2008;44:137–43.
Greiner S, Sobanski J, Bock R. Why are most organelle genomes transmitted maternally? Bioessays. 2015;37:80–94.
Frank SA. Host–symbiont conflict over the mixing of symbiotic lineages. Proc Biol Sci. 1996;263:339–44.
Kiers ET, Rousseau RA, West SA, Denison RF. Host sanctions and the legume-rhizobium mutualism. Nature. 2003;425:78–81.
Sachs JL, Russell JE, Lii YE, Black KC, Lopez G, Patil AS. Host control over infection and proliferation of a cheater symbiont. J Evol Biol. 2010;23:1919–27.
Pieterse CMJ, Zamioudis C, Berendsen RL, Weller DM, Van Wees SCM, Bakker PAHM. Induced systemic resistance by beneficial microbes. Annu Rev Phytopathol. 2014;52:347–75.
Fraysse N, Couderc F, Poinsot V. Surface polysaccharide involvement in establishing the rhizobium-legume symbiosis. Eur J Biochem. 2003;270:1365–80.
Okazaki S, Tittabutr P, Teulet A, Thouin J, Fardoux J, Chaintreuil C, et al. Rhizobium–legume symbiosis in the absence of Nod factors: two possible scenarios with or without the T3SS. ISME J. 2016;10:64–74.
D’Haeze W, Holsters M. Surface polysaccharides enable bacteria to evade plant immunity. Trends Microbiol. 2004;12:555–61.
Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. 2013;14:668–75.
Nyholm SV, Graf J. Knowing your friends: invertebrate innate immunity fosters beneficial bacterial symbioses. Nat Rev Microbiol. 2012;10:815–27.
Triantaphylidès C, Krischke M, Hoeberichts FA, Ksas B, Gresser G, Havaux M, et al. Singlet oxygen is the major reactive oxygen species involved in photooxidative damage to plants. Plant Physiol. 2008;148:960–8.
Jani AJ, Cotter PA. Type VI secretion: not just for pathogenesis anymore. Cell Host Microbe. 2010;8:2–6.
Steele MI, Kwong WK, Whiteley M, Moran NA. Diversification of type VI secretion system toxins reveals ancient antagonism among bee gut microbes. MBio. 2017;8:e01630–17.
Acknowledgements
We wish to thank Dr. Olivier Leroux (Ghent University) for his help with microscopy. This work was supported by the Flemish Fonds Wetenschappelijk Onderzoek under grant G017717N and the Special Research fund of Ghent University under grant BOF17/STA/024. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank the Oxford Genomics Centre at the Wellcome Centre for Human Genetics (funded by Wellcome Trust grant reference 203141/Z/16/Z) for the generation and initial processing of the sequencing data. Finally, We thank three anonymous reviewers for constructive comments which substantially improved the manuscript.
Author information
Authors and Affiliations
Contributions
A.C. designed the research, F.D.M. performed the molecular biology experiments, including isolation of DNA and RNA, and phenotyping experiments. B.D. performed the genome assemblies and annotation, metagenome mining and phylogenetic analyses. T.A. provided microscopy images and microbiological data. R.R., M.T.R., and V.J. identified and collected field samples. F.D.M, B.D., and A.C. analyzed and interpreted the data; F.D.M, B.D., T.A., and A.C. wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
De Meyer, F., Danneels, B., Acar, T. et al. Adaptations and evolution of a heritable leaf nodule symbiosis between Dioscorea sansibarensis and Orrella dioscoreae. ISME J 13, 1831–1844 (2019). https://doi.org/10.1038/s41396-019-0398-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0398-8
