
Abstract
The human gastrointestinal tract is populated with a diverse microbial community. The vast genetic and metabolic potential of the gut microbiome underpins its ubiquity in nearly every aspect of human biology, including health maintenance, development, aging, and disease. The advent of new sequencing technologies and culture-independent methods has allowed researchers to move beyond correlative studies toward mechanistic explorations to shed light on microbiome–host interactions. Evidence has unveiled the bidirectional communication between the gut microbiome and the central nervous system, referred to as the “microbiota–gut–brain axis”. The microbiota–gut–brain axis represents an important regulator of glial functions, making it an actionable target to ameliorate the development and progression of neurodegenerative diseases. In this review, we discuss the mechanisms of the microbiota–gut–brain axis in neurodegenerative diseases. As the gut microbiome provides essential cues to microglia, astrocytes, and oligodendrocytes, we examine the communications between gut microbiota and these glial cells during healthy states and neurodegenerative diseases. Subsequently, we discuss the mechanisms of the microbiota–gut–brain axis in neurodegenerative diseases using a metabolite-centric approach, while also examining the role of gut microbiota-related neurotransmitters and gut hormones. Next, we examine the potential of targeting the intestinal barrier, blood–brain barrier, meninges, and peripheral immune system to counteract glial dysfunction in neurodegeneration. Finally, we conclude by assessing the pre-clinical and clinical evidence of probiotics, prebiotics, and fecal microbiota transplantation in neurodegenerative diseases. A thorough comprehension of the microbiota–gut–brain axis will foster the development of effective therapeutic interventions for the management of neurodegenerative diseases.
Similar content being viewed by others
Introduction
Microbes have always been an essential part of human life. The co-evolution between the human host and microbes has established a mutualistic symbiosis in which the host provides a hospitable environment and nutrients for the microbiota, while the microbiota exerts substantial influence on the host during homeostasis and disease.1 The human gastrointestinal (GI) tract is populated with the most diverse microbial community in the human body, including bacteria, fungi, viruses, and archaea.2,3 Approximately 2000 bacterial species have been identified in the human gut, and it is estimated that the gut microbiota contains nearly 150 times more genes than the human genome.4,5 The vast genetic and metabolic potential of the gut microbiome underpins its ubiquity in nearly every aspect of human biology, including health maintenance, development, aging, and disease.6
The biological importance of the gut microbiome is evident from the early stages of life. The human gut microbiota develops after birth and contributes to the development of the immune system in newborns.7,8 Furthermore, microbial colonization in the GI tract of infants enables the production of essential amino acids and vitamins, which begins around 4 months of life.9 The gut microbiome gradually reaches an adult-like configuration by the age of 3–6 years old and remains stable throughout adulthood.10,11,12 Notable biological functions of the adult gut microbiome include regulation of nutrient harvest from the diet,13 regulation of immunity and auto-immunity,3,14 maintenance of intestinal barrier integrity,15,16 cholesterol metabolism,17,18,19 transformation of bile acids (BAs),20,21 production of antimicrobial peptides,22,23 and drug metabolism.24,25 Recent studies have revealed that the human gut microbiome is a major determinant of plasma metabolome, potentially playing a more dominant role than genetics.26,27,28 Notably, dysbiosis has been recognized as one of the 12 updated hallmarks of aging, further emphasizing the importance of the microbiome.29
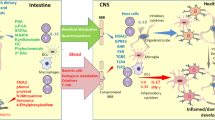
Accumulating evidence has unveiled the bidirectional communication between the gut microbiome and central nervous system (CNS), referred to as the “microbiota–gut–brain axis”.30,31 Although the gut and brain are anatomically separated, several pathways by which the gut microbiota communicates with the CNS have been proposed. These include modulation of the immune system, vagus nerve, enteric nervous system (ENS), neuroendocrine system, and circulatory system via the production of neuroactive substances, metabolites, and hormones (Fig. 1).31,32 Studies have shown that gut microbiota is capable of producing or stimulating the production of neurotransmitters, including serotonin,33,34,35 dopamine,36 and γ-aminobutyric acid (GABA).37 Earlier studies reporting correlations between gut microbiota and CNS functions have largely relied on simplified animal models, which are insufficient to elucidate the underlying mechanisms of action. Nevertheless, the development of new technologies and culture-independent methods has allowed researchers to move beyond correlative studies toward mechanistic exploration to shed light on microbiome–host interactions.31 Pre-clinical and human studies have demonstrated the intricate involvement of gut microbiota in the regulation of social behavior,38,39,40,41,42 depressive-like behavior,43,44,45,46,47 physical performance, and motivation.48,49,50

The microbiota–gut–brain axis. The bidirectional communication between the gut microbiome and the brain is mediated by the immune system, vagus nerve, enteric nervous system, neuroendocrine system, and circulatory system. Alterations in gut microbiota have been linked to the development of autism spectrum disorders, anxiety, depressive-like behavior, impaired physical performance, and motivation, as well as neurodegenerative diseases. This figure was created with BioRender (https://biorender.com/)
There is a growing recognition of the role of gut microbiome in neurodegenerative diseases. Notably, early microbiome changes were detected in preclinical Alzheimer’s disease (AD) patients and prodromal Parkinson’s disease (PD) patients.51,52,53 Moreover, studies on animal models have provided compelling evidence that the altered gut microbiome drives neurodegenerative disease pathogenesis, primarily through the modulation of microglial functions and activation.54,55,56,57 Microglial activation and neuroinflammation are pathological hallmarks of neurodegenerative diseases.58 The microbiota–gut–brain axis represents an important regulator of glial functions,59,60,61,62 making it an actionable target to ameliorate the development and progression of neurodegenerative diseases.
The purpose of this review is to update the current state of knowledge of the mechanisms governing the microbiota–gut–brain axis in neurodegenerative diseases, with a particular emphasis on the interactions between gut microbiome and glial cells (microglia, astrocytes, and oligodendrocytes). We next discuss the roles of gut microbiota-derived metabolites, gut microbiota-related neurotransmitters, and gut hormones in neurodegenerative diseases. While these elements are highly interconnected and interdependent, we present each element separately to enhance clarity and provide focused discussions on their distinct mechanisms and contributions. Subsequently, we examine the potential of targeting the intestinal barrier, blood–brain barrier (BBB), meninges, and peripheral immune system to modulate the microbiota–gut–brain axis and counteract glial dysfunction and neurodegeneration. Finally, we conclude by assessing the pre-clinical and clinical evidence of probiotics, prebiotics, and fecal microbiota transplantation (FMT) in neurodegenerative diseases. In addition, we provide a brief update on the current understanding of the roles of microglia in neurodegenerative diseases.
Roles of microglia in neurodegenerative diseases
Microglia are the primary innate immune cells of the CNS, accounting for nearly 10% of CNS cells. Although microglia were erroneously considered inert bystanders of CNS disorders, they possessed diverse context-dependent functions central to CNS development, homeostasis, and diseases.63,64,65 Under homeostatic conditions, microglia contribute to the regulation of numerous physiological functions, including neurogenesis,66,67 angiogenesis,68 maintaining BBB integrity,69 synaptic pruning and remodeling,70,71 synaptic transmission,72,73,74 myelin health,75,76 as well as phagocytosis and removal of apoptotic neurons and cellular debris.67,77,78 Microglia actively surveys and responds promptly to various environmental perturbations in the CNS by evoking a broad repertoire of cellular alterations to restore homeostasis.79,80
The importance of microglia in AD has been clearly illustrated in a recent spatiotemporal analysis. Among the three major glial cell types (microglia, astrocytes, and oligodendrocytes), microglia are the primary responder to beta-amyloid (Aβ) plaques and accumulate in close vicinity of the plaques (<10 µm).81 Several genome-wide association studies (GWAS) have also implicated microglia as the primary cell type expressing AD genes.82,83,84,85 In addition, growing evidence has implicated microglia in the pathogenesis of PD. Postmortem analysis of ventral midbrains from PD patients revealed a significantly increased number of microglia with an ameboid shape, suggestive of an activated state.86 Importantly, studies have identified a significant association between PD risk variants and microglia.86,87 However, conflicting results were reported in a single-nuclei transcriptomic atlas of the human substantia nigra (SN), which found no association between PD risk and microglia or astrocytes,88 underscoring the imperative for additional comprehensive studies. On the other hand, postmortem transcriptomic analysis of the amyotrophic lateral sclerosis (ALS) spinal cord has reported an increase in inflammatory reactions driven by microglia and astrocytes.89 Similarly, the involvement of microglia in frontotemporal dementia (FTD) and Huntington’s disease (HD) is also well documented.90,91,92
A core function of microglia is the efficient recognition and phagocytic clearance of protein aggregates and cellular debris without damaging surrounding tissue to maintain CNS homeostasis.93,94 The phagocytic activity of microglia is crucial for the removal of Aβ,94 tau,95 and α-synuclein.96 However, microglial phagocytic activity becomes dysfunctional during aging and neurodegenerative diseases, resulting in the gradual accumulation of toxic compounds and cognitive decline.93,94 Moreover, overactive microglial phagocytosis of stressed but viable neurons leads to neuronal loss and neurodegeneration.97 Several regulators of microglial phagocytosis have been identified, including but not limited to tyrosine kinase-binding protein (TYROBP),98 triggering receptor expressed on myeloid cells 2-apolipoprotein E (TREM2-APOE) pathway,99,100 spleen tyrosine kinase (SYK),101,102 classical complement system,103 purinergic system,104 sialic acid binding immunoglobin-like lectins (Siglecs) (CD22 and CD33),105,106,107 TAM system,77 and mechanosensor Piezo1.108,109
Mechanisms of microglial activation
Several genetically distinct subtypes of microglia have been discovered as they respond to signals or challenges in the brain microenvironment, namely homeostatic microglia and “disease-associated microglia” (DAM) or “microglial neurodegenerative phenotype” (MGnD).99,100 The DAM was first identified in a 5xFAD mouse model, an amyloid model harboring five mutations associated with familial AD, and was found to cluster in close proximity to the Aβ plaques.99,110 The transition from homeostatic state to DAM is associated with the downregulation of homeostatic markers and upregulation of genes related to AD and other neurodegenerative diseases, including APOE, TREM2, and TYROBP. Stage-1 DAM represents a transitory and functional subtype with a higher capacity of phagocytosis initiated by a TREM2-independent mechanism, whereas stage-2 DAM represents a dysfunctional state that contributes to AD pathology initiated by a TREM2-dependent mechanism.99,111 This transition also leads to considerable morphological changes, transforming microglia from thin cell bodies with highly ramified extensions into ameba-like cells with fewer branches (Fig. 2).65

Microglial activation and neurodegeneration. Aging induces microglial activation by activating the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) signaling pathway. Misfolded proteins and protein aggregates induce microglial activation by impairing microglial autophagy. Stage-1 DAM represents a transitory and functional subtype with a higher capacity of phagocytosis initiated by a TREM2-independent mechanism, whereas stage-2 DAM represents a dysfunctional state initiated by a TREM2-dependent mechanism. The microglial spleen tyrosine kinase (SYK) signaling provides metabolic support to facilitate microglial transition into stage-2 DAM. Maladaptive microglial-T-cell signaling drives neurodegeneration by releasing neurotoxic factors. Microglial activation creates a feed-forward vicious cycle that aggravates neurodegeneration as activated microglia contribute to the propagation of protein aggregates into unaffected brain regions. This figure was created with BioRender (https://biorender.com/)
A recent study on naturally aged mice found that in the absence of an additional trigger, the activation of cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) signaling pathway is sufficient to promote aging-related inflammation and neurodegeneration by triggering reactive microglial transcriptional states.112 The cGAS-STING signaling pathway is an innate immune sensing system that is capable of driving both acute and chronic low-grade inflammation, which is central to the development of neurodegenerative pathologies.113 In addition, intact autophagy is required for effective microglial transition into DAM phenotype and microglial proliferation in response to Aβ plaques in 5xFAD mice. Deletion of autophagy gene Atg7 led to impaired ability of microglia to engage Aβ plaques and promoted microglial senescence, which was reversed by administration of senolytic drugs.114 Nevertheless, the transcriptional states of microglia remain incompletely understood, as microglia may respond to multiple pathological stimuli in the brain simultaneously. For instance, microglia adopt two distinct DAM phenotypes when responding to amyloid pathology and myelin damage in 5xFAD mice with dysfunctional myelin (Cnp−/−5xFAD mice), which may reflect the comorbid state of the aged brain.115
More recently, two studies have identified a critical intracellular regulator of microglial activation that acts downstream of TREM2 and CD33 to counteract Aβ pathology, namely SYK.101,102 Microglial SYK signaling enables effective microglial response to Aβ by providing metabolic support via the phosphoinositide 3-kinase (PI3K)-AKT-glycogen synthase kinase-3β (GSK-3β)-mammalian target of rapamycin (mTOR) pathway, allowing microglia to acquire complete DAM phenotype. The loss of SYK signaling interfered with microglial clustering around Aβ plaques, microglial transition into complete DAM phenotype, and phagocytosis of Aβ following exposure to Aβ in 5xFAD mice.101,102 Conversely, replacing the mutant microglia of Trem2−/− 5xFAD mice with Trem2+/+ circulation-derived myeloid cells through hematopoietic cell transplantation effectively restored microglial activation in response to Aβ plaques. This effect is attributed to the restoration of microglial SYK signaling and the DAM transcriptional program.116 The observed favorable outcomes of SYK-mediated complete/stage-2 DAM phenotype in Aβ pathology are in contrast with the prevailing view that stage-2 DAM represents a dysfunctional and pro-inflammatory state. Thus, we recommend caution in oversimplifying DAM states similar to the previous nomenclatures (resting versus activated; M1 versus M2) to account for the inherent plasticity of microglia. More research is needed to elucidate the upstream and downstream mechanisms governing microglial transition across their many states, including a potential exploration of their reversibility.
Microglia-mediated neuroinflammation
Among the diverse microglial functions, microglia-mediated neuroinflammation has received attention due to its complex and dynamic role in health and disease. Early neuroinflammation is protective as it promotes tissue repair, cellular debris clearance, and pathogen removal.117 Furthermore, early neuroinflammation has been shown to be an adaptive mechanism by microglia that protects against AD pathology by reducing the levels of Aβ and tau.118,119,120 Similarly, early microglial activation assists in the clearance of neuronal human TAR DNA-binding protein 43 (hTDP-43) and motor neuron recovery in the ALS mouse model.121 However, microglia lose their homeostatic molecular signatures and become progressively activated with increasing age or during pathological conditions, transitioning into distinct disease-associated phenotypes with sustained release of pro-inflammatory cytokines and chemokines.122,123,124 Chronic microglial activation leads to persistent low-grade neuroinflammation that is detrimental to neurons and synapses, leading to neurodegeneration.125 Indeed, neuroinflammation and microglial activation are consistent features across neurodegenerative diseases, including AD,126,127,128 PD,86,129 HD,130,131 FTD,91,132 and ALS.89,133,134,135
Numerous studies have reported that misfolded proteins and protein aggregates, including tau,136,137,138,139 Aβ,138,140 α-synuclein,141,142,143,144 mutant huntingtin,145 TDP-43,132,146 superoxide dismutase 1 (SOD1),147,148,149 and fused in sarcoma (FUS)150 induce microglial activation and neuroinflammation. Autophagy deficiency induced by protein aggregates has been shown to be a major driver of microglial activation (Fig. 2). Prolonged exposure to Aβ impairs microglial autophagy by inducing lysosomal dysfunction, resulting in microglial activation.151 Autophagy deficiency disrupts microglial response to Aβ by inhibiting DAM development and inducing microglial senescence.114 Moreover, the loss of functional microglial autophagy is deleterious as it exacerbates tau pathology and spreading in PS19 tau transgenic mice,152 as well as contributes to elevated release of pro-inflammatory cytokines and NLR family pyrin domain-containing 3 (NLRP3) inflammasome activation in Becn1+/− APP/PS1 mice.153 Activated microglia also release chemokines that disrupt neuronal autophagy by altering the neuronal C-C chemokine receptor type 5 (CCR5)-mTORC1-autophagy pathway in HD and tauopathy mice.154
In PD models, α-synuclein inhibits microglial autophagy by triggering toll-like receptor 4 (TLR4)-dependent p38 mitogen-activated protein kinase (MAPK) phosphorylation and activating the AKT-mTOR signaling cascade.144,155 This leads to a self-perpetuating cycle that further exacerbates neuroinflammation in PD as microglia with impaired autophagy have elevated pro-inflammatory responses and lose the ability to clear α-synuclein, resulting in neurodegeneration.144,156,157 C9orf72 mutation, the leading genetic cause of ALS and FTD, disrupts microglial autophagy and triggers sustained activation of NLRP3 inflammasome and nuclear factor-κB (NF-κB) signaling in human-induced pluripotent stem cell-derived microglia-like cells (hiPSC-MG). The dysfunctional microglial autophagy aggravates motor neuron death in microglia-motor neurons co-culture following excitotoxic insult, a key pathomechanism in ALS.158 Ultimately, the activation of microglia by these protein aggregates creates a feed-forward vicious cycle that aggravates neurodegeneration as activated microglia contribute to the propagation of tau, Aβ, α-synuclein, and TDP-43 into unaffected brain regions.159,160,161,162,163,164
Recently, the intricate interplay between microglia, tauopathy, APOE, and T cells in driving neurodegeneration has been elucidated.165 APOE is a lipid and cholesterol transporter with numerous CNS-related functions, including regulation of microglial and astrocytic functions,100,166,167 cerebrovascular integrity,168 BBB integrity,169,170 myelin dynamics,166,171 and neuronal network activity.172 APOE exists in three isoforms, namely APOE2, APOE3, and APOE4, among which APOE4 isoform has been identified as the strongest genetic risk factor for late-onset AD.173 Studies have demonstrated that APOE4 drives Aβ- and tau-mediated neurodegeneration by inducing microglial and astrocytic activation.174,175,176,177,178,179 In addition, APOE4 genetic background drives an accelerated spread of α-synuclein pathology and neurodegeneration.180,181
In light of the strong correlation between tau pathology and brain atrophy in AD, rather than Aβ,182 a comparison was made between the immune responses of amyloid-depositing APP/PS1-21 (A/PE4) and 5xFAD (5xE4) mice, versus P301S tau transgenic (TE4) mice expressing human APOE4.165 Interestingly, the number of T cells was significantly increased only in TE4 mice in regions where brain atrophy occurred, and this increase was positively correlated with the number of microglia. Further sequencing analysis on the T cells revealed that TE4 mice carried increased activated CD8+ T cells and reduced exhausted T cells, suggesting that the T-cell activation drives tau-mediated neurodegeneration. The study also demonstrated that interfering with the immunological hub between activated microglia and T cells using cell-depleting treatments attenuated tau-mediated neurodegeneration. Microglial depletion reduced CD3+ and CD8+ T cells and attenuated tau pathology in TE4 mice. Conversely, T-cell depletion induced microglial transition from an activated state to a homeostatic state, along with reduced tau pathology in TE4 mice.165 These findings are in concordance with a recent study that reported a detrimental synergism between microglia and CD8+ T cells in exacerbating neuronal and glial damage.183
Similar maladaptive microglial-T-cell signaling also drives neurodegeneration in the α-synuclein-driven PD mouse model, which was ameliorated following genetic knockout or pharmacological depletion of T cells.184 Furthermore, infiltration of T cells in the CNS drives microglial and astrocytic activation in two different ALS mouse models (hSOD1G93A and TDP-43A315T mice). These pathological changes were largely prevented by reducing immune cell infiltration using natalizumab, accompanied by reduced motor neuron degeneration, delayed onset of paralysis, and prolonged survival.185 Together, these data indicate that microglial-T-cell signaling offers a prospective avenue for tackling neuroinflammation and neurodegeneration.
Microbiota–gut–brain axis in neurodegenerative diseases
Interaction between gut microbiota and microglia
The interaction between microglia and gut microbiota begins early in life. A recent study demonstrated that early-life administration of a broad-spectrum antibiotic cocktail led to altered microglial morphology and myelin-related gene expression in adolescent mice, accompanied by anxiety-like and compulsive-like behaviors.186 Throughout the host lifespan, the gut microbiome provides essential signals to microglia during health and disease.59,60,187,188 Notably, among the neuronal and glial cells, microglia are the most vulnerable to alterations in the gut microbiome.189
Under homeostatic conditions, the gut microbiome is responsible for regulating microglial maturation and activation via short-chain fatty acids (SCFAs) release.190,191 Erny and colleagues found that germ-free (GF) mice and antibiotic-treated mice suffered from impaired microglial immune responses when challenged with lipopolysaccharide (LPS) and lymphocytic choriomeningitis virus (LCMV) infection. However, the microglial defects and immaturity were partially restored by recolonization with complex microbiota and SCFAs supplementation.191 In a subsequent study, Erny et al. discovered that the host microbiota regulates microglia mitochondrial functions and identified acetate as the major SCFA-rescuing microglial homeostasis in GF mice.190 In addition, the gut microbiota plays a role in facilitating the transition of microglia to DAM phenotype during aging, as specific-pathogen-free (SPF) aged mice display higher expression of DAM-related genes than GF-aged mice.192 Antibiotic-induced gut microbiota depletion stimulates global reduction of Ly6Chi monocytes pool and promotes Ly6Chi monocytes transition towards a pro-inflammatory state. The elevated immune activation is coupled with microglial activation, impaired hippocampal synaptic transmission, and cholinergic gamma oscillations.193 Additional evidence suggests that reshaping the gut microbiome of high-fat diet (HFD)-fed obese mice with dietary fibers successfully mitigated the cognitive and social impairments of their offspring by alleviating the microglial maturation defects. SCFAs supplementation in the offspring with acetate and propionate promoted microglial maturation and reduced maternal obesity-induced cognitive and social deficits.194
Aside from the regulation of microglial homeostasis, gut microbiota-derived metabolites also play a crucial role in triggering microglial cell death.195 During aging, the increased level of gut microbial metabolite isoamylamine (IAA) crosses the BBB and induces microglial apoptosis by activating the S100 calcium-binding protein A8 (S100A8) signaling. Specifically, an increased abundance of Ruminococcaceae and reduced Ruminococcaceae-targeting bacteriophage family Myoviridae were observed in the gut of aged mice and elderly people, contributing to increased IAA. IAA binds to the promoter region of S100A8 and interrupts its hairpin structure, facilitating p53 access to the S100A8 promoter region. The study further demonstrated that IAA administration induced cognitive decline in young mice, whereas IAA reduction attenuated the neuronal loss and cognitive deficits of aged mice.195
In this section, we present evidence of the interaction between gut microbiota and microglia in different neurodegenerative diseases.
Alzheimer’s disease
Accumulating evidence has demonstrated the interaction between gut microbiota and microglia in AD. In the triple transgenic AD (3xTg-AD) mouse model, the development of AD pathologies, including Aβ plaque, hyperphosphorylated tau, synaptic dysfunction, and microglial activation appears to be influenced by the gut microbiome. This is evident as SPF 3xTg-AD mice exhibit greater AD pathologies compared to GF 3xTg-AD mice. Importantly, FMT from AD patients to GF 3xTg-AD mice restored the main AD pathologies and microglial activation.196
Similar findings have been reported in GF and antibiotic-treated amyloidogenic APP/PS1 mice.54,197,198,199 It was reported that GF condition confers protection against Aβ pathology and microglial activation in APPSWE/PS1L166P (APP/PS1-21) mice. However, this protection was diminished following FMT from 12-month-old conventionally raised APP/PS1-21 mice to 4-month-old GF APP/PS1-21 mice.198 Similar trends were also observed in APP/PS1-21 following gut microbiota depletion using long-term (5-week) and short-term (7-day) antibiotic treatment.54,197 Long-term perturbation of gut microbiome using antibiotic cocktail resulted in reduced Aβ deposition, reduced plaque-localized microglia and altered transcriptional profile of microglia (increased homeostatic microglial genes and decreased MGnD genes) in 7-week-old male APP/PS1-21 mice. Interestingly, these effects were absent in female mice, suggesting potential sexual dimorphism in their responses to gut microbiome manipulation. Importantly, the AD pathologies in antibiotic-treated male mice were partially restored after 3-week FMT from age-matched male APP/PS1-21 mice.197
Recently, growing studies have illustrated the presence of critical windows of microbial development, during which early-life modulation of the gut microbiome has a long-lasting impact on different aspects of physiology.186,200,201,202 To further validate the time-specific role of the gut microbiome in AD, Dodiya et al. repeated the experiment using short-term 7-day antibiotic treatment administered from postnatal day 14 to day 21, and sacrificed the mice at 9 weeks of age. Consistent with their previous findings,197 short-term antibiotic treatments effectively reduced Aβ pathology, reduced DAM population, and expression of microglial sensome genes in male APP/PS1-21 mice, which were reversed by FMT from age-matched mice. Interestingly, microglia depletion using colony-stimulating factor 1 receptor inhibitor, PLX5622, mitigated the protective effect of antibiotic treatment against amyloidosis. These results suggest that microglia are essential mediators of the microbiota–gut–brain axis in Aβ pathology.54
Similarly, the gut microbiota is required for microglial activation in 5xFAD mice.203,204,205 5xFAD mouse model expresses five familial AD mutations and develops Aβ accumulation and gliosis as early as 2 months of age, synaptic degeneration by 4 months of age, and cognitive impairment as early as 4–5 months of age.206,207 It was found that gut microbiota ablation using 5-month antibiotics treatment prevented microglial activation in 7-month-old 5xFAD mice by reducing immune cell infiltration.203 Moreover, 5-month antibiotics treatment alleviated AD pathologies and microglial activation in 6.5-month-old 5xFAD mice by inhibiting CCAAT/enhancer binding protein β/asparagine endopeptidase (C/EBPβ/AEP) signaling.204
The involvement of the gut microbiome in facilitating microglial activation is also evident in tau-mediated neurodegeneration. A recent study demonstrated the interplay between gut microbiota, tau and APOE in AD.56 Seo and colleagues genetically engineered P301S tau transgenic mice to express different isoforms of human APOE (APOE3 and APOE4) and raised them in conventional or GF environments. Compared to conventionally raised P301S mice expressing human APOE4 (TE4 mice), their GF counterparts showed reduced signs of neurodegeneration (brain atrophy) and neuroinflammation (microglial and astrocytic activation). However, FMT from sex-matched conventionally raised TE4 mice mitigated the neuroprotective effects of GF conditions, indicating that the gut microbiota is responsible for the emergence of tau-mediated neurodegeneration. To further investigate the role of gut microbiota, the researchers induced early-life gut microbiota perturbation through a short-term antibiotic treatment administered from postnatal day 16 to day 22, and sacrificed the mice at 40 weeks of age. Interestingly, they observed sex-dependent and APOE isoform-dependent neuroprotection as the neuroprotective effects of antibiotic treatment were more pronounced in male mice expressing human APOE3 (TE3 mice).56 The sex-dependent neuroprotective effects of microbiome perturbations in tau pathology are reminiscent of that observed in amyloid pathology,197 underscoring the need for future research to consider the gender effects.
Aging is the predominant risk factor for neurodegenerative diseases as a result of the lifetime accumulation of neuropathologies.208 Notably, the gut microbiome of centenarians is associated with “youth-like” signatures (depletion of inflammatory pathobionts and enrichment of beneficial commensals), showing high similarity to those of young individuals.209 To investigate whether the acquisition of “youth-like” microbiota could restore aging-induced neurocognitive and immune impairments, Boehme et al. compared the effects of FMT from young (3–4 months; yFMT) or old (19–20 months; oFMT) mice into old recipient mice. Hippocampal transcriptomic analysis revealed that yFMT reversed the aging-induced alterations in the expression of six microglial sensome genes in old mice. These genes included Trem2, Dap12, C1qb, Fcgr2b, Gpr84, and Tlr13.210 Of note, DAP12, also known as TYROBP, is an immunoreceptor tyrosine-based activation motifs (ITAM)-containing transmembrane adapter that associates with TREM2.98 Dap12, along with Apoe, were among the most robustly upregulated genes during the microglial transition from homeostatic state to DAM phenotype.99 On the other hand, complement component 1q (C1q) is the initiating protein of the classical complement pathway predominantly produced by microglia in the brain.211 Studies on P301S tau transgenic mice and plaque-bearing mouse models have shown that C1q binds to synapses and facilitates microglial phagocytosis of synapses.103,212 Recent evidence also revealed that the complement-dependent synapse elimination in P301S mice involves coordinated action between microglia and astrocytes.213
Altogether, the evidence from different mouse models consistently underscores the significance of the microbiota–gut–brain axis in AD pathologies.
Parkinson’s disease
Although GI symptoms and gut microbiota alterations are common in PD patients during the disease course,214,215 the underlying mechanisms linking the gut microbiome and PD have only been unveiled recently. The first corroboration arises from the study by Sampson et al., which demonstrated that the development of α-synuclein pathology, microglial activation, and motor deficits in α-synuclein-overexpressing (ASO) mice appear to be influenced by the gut microbiome. This is evident as SPF ASO mice exhibit greater PD pathologies compared to their GF and antibiotic-treated counterparts. Importantly, FMT from PD patients to GF ASO mice restored the main disease features, including α-synuclein-mediated motor dysfunction.55 In another study, transgenic rats overexpressing α-synuclein displayed progressive gut dysbiosis with aging, whereas a short-term antibiotic treatment mitigated α-synuclein expression in the forebrain.216 Furthermore, FMT from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice induced motor impairments and neurotransmitter loss in healthy mice. Conversely, FMT from healthy mice ameliorated gut dysbiosis and PD pathologies in MPTP-induced mice, including gut inflammation, glial activation, neurotransmitter abnormalities, and motor dysfunction.217 In addition, the development of GI dysfunction and motor symptoms following chronic rotenone administration occurs only in conventionally raised mice, but not in GF mice.218 These studies substantiated the significance of the microbiota–gut–brain axis in the pathogenesis of PD.
Amyotrophic lateral sclerosis
The motor neuron injury in ALS is thought to arise from multiple interacting pathophysiological mechanisms, including glial dysfunction and neuroinflammation.89,219,220 Compared to AD and PD, the roles of gut microbiota in ALS remain relatively understudied. Nevertheless, growing evidence has indicated alterations in the gut microbiome and impaired intestinal barrier integrity in both ALS patients and animal models.221,222,223,224,225
Notably, a gradual reduction in the abundance of Akkermansia muciniphila and reduced bacterial production of nicotinamide (NAM) were observed during the course of disease in SOD1G93A mice. Conversely, supplementation of A. muciniphila was associated with increased motor neurons in the spinal cord, improved motor function, reduced brain atrophy, and prolonged lifespan.223 The gut microbiome is also closely linked to C9orf72 function.57 The hexanucleotide (GGGGCC) repeat expansion in the C9orf72 gene has been identified as the leading genetic cause of ALS and FTD.226,227,228 It was found that the gut microbiota is a potent modifier of disease severity and microglial activation in C9orf72-mutant mice raised in different environments. Specifically, C9orf72−/− mice reared at pro-inflammatory environment (Harvard Institute) exhibit significantly different gut microbiota composition, greater autoimmune and inflammatory phenotypes, microglial activation, and shorter lifespan as compared to the C9orf72−/− mice reared at pro-survival environment (Broad Institute). Importantly, FMT from C9orf72(Broad)−/− mice significantly ameliorated the autoimmune and inflammatory phenotypes in C9orf72(Harvard)−/− mice.57
Alterations in the gut microbiome have been observed in SOD1G93A mice prior to the onset of motor dysfunction, muscle atrophy, and immune cell activation in the spinal cord.224 In addition, gut dysbiosis precedes the aggregation of human-SOD1G93A protein in the colon and intestine, along with the development of ENS dysfunction in SOD1G93A mice.222 These studies indicate an early microbial contribution to ALS disease pathology.
Interestingly, conflicting results have been reported regarding the consequences of gut microbiota depletion in ALS mouse models. Studies have demonstrated the beneficial effects of antibiotic treatment in ameliorating ALS pathologies. Antibiotic treatment has been shown to inhibit SOD1G93A aggregation in the intestines of SOD1G93A mice, coupled with improved enteric neuromuscular function.222 Similar beneficial effects were observed in C9orf72-mutant mice. The administration of antibiotics, either prior to the onset or after the establishment of inflammation, effectively suppressed the emergence of inflammatory and autoimmune phenotypes in C9orf72−/− mice. Moreover, lifelong antibiotics administration prevented the accumulation of infiltrating myeloid cells within the spinal cord and microglial activation.57
However, detrimental effects of antibiotic treatment have also been reported in SOD1G93A mice. Long-term antibiotic treatment induces motor neuron death in the spinal cord and brain atrophy, thereby exacerbating motor abnormalities in SOD1G93A mice.223 In addition, antibiotic-induced dysbiosis markedly worsened disease progression in SOD1G93A mice by downregulating homeostatic microglial genes and upregulating MGnD signatures in the spinal cord.229 Presently, there is a scarcity of research utilizing GF SOD1 mice due to the considerable challenge of rederivation, which is associated with high mortality rates.223 Further investigations are warranted to understand the roles of the gut microbiome and the implications of gut microbiota depletion in ALS. With the growing characterization of ALS-associated genes and the development of ALS animal models (extensively reviewed in ref. 230), we recommend future studies to move beyond SOD1 mice. This is relevant considering that SOD1-ALS accounts for only about 12% of familial and less than 2% of sporadic ALS cases.231
Huntington’s disease
HD is an autosomal dominant neurodegenerative disease caused by a CAG repeat expansion in the huntingtin (HTT) gene. This results in misfolding and accumulation of mutant huntingtin protein in brain cells, including neurons, microglia, and astrocytes.232 In addition to motor, cognitive, and psychiatric abnormalities, HD patients experience a range of GI disturbances, including nutrient deficiency, diarrhea, and unintended weight loss.233,234 However, it was only recently that gut dysbiosis has been revealed in preclinical HD models and HD patients, and studies examining the interaction between gut microbiota and microglia remain absent.
The initial evidence of gut dysbiosis emerged from the R6/1 transgenic mouse model of HD. A notable difference in gut microbiota composition was observed between R6/1 mice and wild-type (WT) mice at 12 weeks of age (early disease stage), which coincided with the manifestation of motor deficits and weight loss.235 Gut dysbiosis and intestinal barrier impairment were also detected in R6/2 mice. It was found that R6/2 mice exhibited increased intestinal permeability and reduced colon length at 16 weeks of age (early-mid disease stage), as compared to age-matched WT mice.236 Moreover, a higher relative abundance of Bacteroidetes and a lower relative abundance of Firmicutes were reported in both R6/1 and R6/2 mice, as compared to WT mice.235,236 However, a notable limitation of the current studies is the lack of metabolomic analyses, which greatly hinders our understanding of the metabolites that regulate HD pathogenesis.
A recent study on HD gene expansion carriers (HDGECs) has identified an altered gut microbiome compared to age-matched and gender-matched healthy controls. Moreover, a reduced abundance of Eubacterium hallii was associated with increased severity of motor deficits.237 E. hallii is a major butyrate-producing species with important health implications, and its depletion has been linked to several diseases.238,239,240 In addition, E. hallii has also been shown to influence BA metabolism.241,242 However, the study did not observe an increase in Bacteroidetes and a reduction in Firmicutes, as reported in both R6/1 and R6/2 mice.235,236
Interestingly, the degree of gut dysbiosis appears to be influenced by the gender of the mice. This difference is evident in male R6/1 mice, which develops greater gut microbiota alterations than female R6/1 mice at 8 weeks of age. Moreover, the plasma levels of acetate were elevated only in male R6/1 mice at 14 weeks of age.243 Similar sexual dimorphism was observed in the application of FMT. A recent study performed FMT from WT mice into R6/1 mice and found that male R6/1 mice exhibited greater resistance to FMT engraftment when compared to female R6/1 mice. Consequently, the cognitive function of male R6/1 mice showed no discernible improvement as compared to their female counterparts. However, FMT is ineffective in ameliorating gut dysfunction and motor functions of R6/1 mice.244
Interaction between gut microbiota and astrocytes
Astrocytes are the most abundant glial cells in the CNS with an expanding repertoire of functions, making them a subject of growing research interest. Astrocytes are integral to the maintenance of CNS homeostasis, and any disruptions in their functions contribute to the development of neuropathologies. Importantly, growing studies have revealed the bidirectional signaling between astrocytes and microglia in driving neuroinflammation and neurodegeneration.245,246
Emerging evidence has elucidated the communication between gut microbiota and astrocytes across health and disease. For instance, the gut microbiota metabolizes tryptophan into various indole derivatives that act as ligands for the aryl hydrocarbon receptor (AHR) expressed within astrocytes and microglia. The AHR activation suppresses NF-κB signaling and inhibits CNS inflammation in experimental autoimmune encephalomyelitis (EAE) mouse models of multiple sclerosis.247,248 In addition, the administration of indole-3-propionic acid attenuated the activity of neurotoxic reactive A1 astrocytes in a mouse model of ischemic stroke.249 Furthermore, the gut microbiota is involved in restricting neuroinflammation by promoting anti-inflammatory tumor necrosis factor-related apoptosis-inducing ligand-positive (TRAIL+) astrocytes and inducing T-cell apoptosis via TRAIL-death receptor 5 (DR5) signaling.250
A recent in vitro study has found that butyrate stimulated adenosine triphosphate release from astrocytes in a cytosolic Ca2+-dependent manner, suggesting a potential neuroprotective mechanism worth further exploration.251 The astrocytic calcium signaling underlies vital physiological functions, and its dysregulation has been associated with neuroinflammation and neurodegeneration.246,252 Gut microbiota manipulation has also been linked to alterations in astrocytic proliferation and functions.253,254,255,256 However, these studies are limited by the lack of tools to characterize the astrocytes and largely rely on glial fibrillary acidic protein (GFAP) densitometry, which provides limited insights and characterization of astrocytes. It was recommended to be cautious in interpreting the increased number of GFAP+ cells as an increase in reactive astrocytes, as GFAP content alone is not a definitive indicator of their reactivity or altered functions.257 For a comprehensive overview of the improved tools, approaches, and potential markers to unravel astrocyte biology, we recommend referring to the excellent reviews by Escartin et al.257 and Yu et al.258
Alzheimer’s disease
The effects of gut microbiota manipulation on astrocytes in AD have only emerged recently. Gut microbiota perturbation has also been shown to reduce reactive astrogliosis, promote a shift in astrocytes towards a more homeostatic-like state, and protect against amyloidosis and tau-mediated neurodegeneration. Interestingly, these effects appear to be more prominent in male mice.56,61 The sexual dimorphism in astrocytic responses to gut microbiome perturbation is reminiscent of that in microglia, further underscoring the importance of future research to account for gender effects. However, the neuroprotective effects of gut microbiota depletion were diminished following microbiota restoration and SCFA supplementation. In particular, FMT from age-matched control mice restored astrogliosis in antibiotic-treated APP/PS1-21 mice, while SCFAs supplementation restored the gliosis and tau pathology in GF TE4 mice.56,61 Although preliminary, these findings in animals demonstrated the involvement of gut microbiome in facilitating the development and progression of AD pathologies, including the modulation of astrocytic responses.
Parkinson’s disease
Our understanding of the roles of astrocytic dysfunction and pro-inflammatory glial responses in PD pathogenesis is expanding.246 The amelioration of gut dysbiosis in MPTP-induced mice via FMT, dietary intervention, and probiotic administration has been shown to alleviate neuroinflammation and dopaminergic neuron loss. These effects are attributed to the reduction of glial activation, as well as the restoration of metabolite and neurotransmitter abnormalities.217,259,260,261 In addition, it was found that FMT from healthy human controls mitigated MPTP-induced dysbiosis and neurotoxicity in MPTP-induced mice, whereas FMT from PD patients exacerbated these pathologies.261 Taken together, these findings corroborate the contributory roles of gut microbiota in driving glial activation and neuropathologies.
Interaction between gut microbiota and oligodendrocytes
Oligodendrocytes are myelin-forming glial cells in the CNS that myelinate axons to facilitate axonal conduction and provide metabolic support to axons. The axon-supporting functions of oligodendrocytes are critical for the maintenance of motor, sensory, and cognitive functions.262,263 Although oligodendrocyte pathology is extensively explored in demyelinating disorders, accumulating evidence suggests its involvement in the pathogenesis of neurodegenerative diseases, including AD,110 PD,86 ALS,89 and HD.264
Studies elucidating the interaction between gut microbiome and oligodendrocytes have only emerged recently. Notably, the gut microbiome has been shown to modulate oligodendrocyte maturation and myelin production. Perturbations in gut microbiome, including GF conditions and antibiotic treatment, have been shown to trigger excessive myelination in the prefrontal cortex by inducing oligodendrocyte maturation and upregulating myelin-related genes.62,265 Importantly, these alterations in GF mice were ameliorated by colonization with a conventional microbiota following weaning.265 Moreover, the administration of tributyrin, a prodrug of butyrate, rescued the myelin dysregulation and behavioral deficits in antibiotic-treated mice.62 The beneficial effects of butyrate on oligodendrocytes are similarly evident in cuprizone- and lysolecithin-induced demyelination.266 Furthermore, the gut microbiota facilitates the conversion of dietary tyrosine to 4-ethylphenol (4EP), which is further sulfated into 4-ethylphenyl sulfate that can enter the brain. Colonization of GF mice with 4EP-producing bacteria leads to reduced oligodendrocyte maturation and neuronal myelination, ultimately promoting anxiety-like behavior and altered social communication.39 Nevertheless, the specific impact of these modulations on oligodendrocyte functions within the context of other neurodegenerative diseases remains to be clarified. Given the growing recognition of the microglia-oligodendrocyte interplay and astrocyte-oligodendrocyte interplay in regulating myelin health,76,267 we anticipate that the gut microbiome may exerts its influence on oligodendrocytes by modulating microglia and astrocytes, thus suggesting the need for further investigation.
Emphasis should also be placed on oligodendrocyte precursor cells (OPCs). Aside from their canonical role in maturing into myelinating oligodendrocytes, recent evidence suggests that OPCs are also involved in the regulation of proper guidance of interneurons, axonal regeneration, angiogenesis, and inflammatory processes.268 The administration of antibiotics has been shown to impair OPC differentiation following lysolecithin-induced demyelination, resulting in fewer differentiated oligodendrocytes within the demyelinated lesions. However, the OPC differentiation and extent of remyelination were unaltered in cuprizone-treated GF mice when compared to their SPF counterparts.269 These findings underscore the complexity of microbiome perturbation on OPCs and the need for further research, including their relevance within the context of other neurodegenerative diseases.
Gut microbiota-derived metabolites in neurodegenerative diseases
The gut microbiota contributes to host physiology and brain health by generating a variety of metabolites through bacterial de novo metabolism and by modifying host-derived molecules.31,270 In this review, we discuss the mechanisms of the microbiota–gut–brain axis in neurodegenerative diseases using a metabolite-centric approach. The presence of a species possessing specific biosynthetic capabilities does not guarantee in vivo production of downstream metabolites in pharmacologically relevant quantities. Moreover, multiple gut microbes can produce the same metabolite. Thus, examining the gut microbiota through a functional metabolic lens (metabolite-centric), rather than focusing on taxonomic or phylogenetic aspects, is more valuable for understanding the intricate interactions between the microbiota and the host.271,272
Short-chain fatty acids mitigate neuroinflammation and neurodegeneration
The microbial fermentative activity of gut microbiota is vital for the production of SCFAs, including butyrate, acetate, and propionate, from non-digestible dietary fibers.271,273 SCFAs are saturated fatty acids composed of one to six carbon atoms. The predominant SCFAs found in the human body are acetate (C2), propionate (C3), and butyrate (C4), which comprise ~95% of the total SCFA pool.271 Numerous studies have illustrated the link between SCFAs and human physiological processes, including immunity,274,275 intestinal homeostasis,276,277,278 cholesterol metabolism,17 and control of glucose homeostasis and energy balance.279,280,281
SCFAs exert their physiological activities by acting as endogenous ligands for G-protein-coupled receptors (GPCRs), and modulating gene expression by inhibiting histone deacetylases (HDACs).282 GPCRs are the largest family of cell surface receptor proteins that regulate diverse physiological and pathological processes, and as such, are one of the most intensively studied targets for drug development.283 Moreover, GPCRs play a pivotal role in enabling the nervous system to accurately respond to external stimuli and internal states.284 SCFAs are endogenous ligands for a subset of GPCRs, including GPR43 and GPR41, which were subsequently renamed as free fatty acid receptor 2 (FFAR2) and FFAR3, respectively. Another important GPCR activated by SCFA is GPR109A, also known as hydroxycarboxylic acid receptor 2 (HCAR2), which is activated by butyrate and β-D-hydroxy butyrate.282 It was reported that the FFAR2-deficient SPF mice developed microglial defects resembling GF mice.191 In addition, an in vitro study has demonstrated that acetate exerts anti-inflammatory effects in Aβ-induced BV-2 microglial cells by upregulating the levels of GPR41 and inhibiting the ERK/JNK/NF-κB signaling pathway.285
On the other hand, HDACs are part of the epigenetic regulatory mechanisms that control gene expression. Histone deacetylation by HDACs is associated with transcriptional repression by inducing a closed chromatin structure. Dysregulated epigenetic regulations and the consequent impact on gene expression and cellular processes are important contributors to aging and age-related human pathologies, including neurodegenerative diseases.286,287 Among the SCFAs, butyrate is the most potent HDAC inhibitor that is generally thought to inhibit the activity of class I HDACs (HDAC1, −2, −3, and −8) and class IIa HDACs (HDAC4, −5, −7, and −9), but not class IIb HDACs (HDAC6 and HDAC10) and class III HDACs (sirtuins).288 Acetate and butyrate have been shown to inhibit the inflammatory response of LPS-stimulated primary microglia by inhibiting HDAC activity and NF-κB activation.289 Furthermore, the inhibition of microglial HDAC1 expression by propionate and butyrate has been shown to alleviate microglial activation and reduce the levels of pro-inflammatory factors in GF mice.290 Conversely, the anti-inflammatory effects of butyrate on LPS-induced BV-2 cells were blocked by HDAC3 agonist ITSA-1 and MCT1 inhibitor AZD3965.291
Alzheimer’s disease
Several studies have reported reduced SCFAs-producing species and SCFA levels in individuals with mild cognitive impairment (MCI) and AD patients.292,293,294,295 Notably, reduced fecal levels of SCFAs were negatively associated with Aβ deposition in patients with MCI.293 In addition, increased levels of HDAC2 and HDAC6 were detected in AD mouse models and AD patients.296,297 Thus, HDAC inhibition represents a promising approach for the treatment of AD. This is exemplified by the notable findings that the genetic deletion of microglial Hdac1 and Hdac2 substantially ameliorated the cognitive deficits of 5xFAD mice by enhancing microglial phagocytosis of Aβ.298 Despite numerous studies supporting the pivotal roles of SCFAs in mediating gut microbiota-microglia communication, mechanistic studies elucidating the underlying mechanisms of SCFAs in AD remain limited and yield conflicting results.
Studies have demonstrated the neuroprotective effects of sodium butyrate in 5xFAD mice by inhibiting microglial activation and promoting synaptic plasticity (Fig. 3a).299,300 Moreover, probiotic and prebiotic interventions aimed at elevating SCFA levels have demonstrated neuroprotective effects in AD mouse models by inhibiting glial activation and Aβ deposition.301,302,303,304,305,306 Elevating butyrate through probiotic intervention (Clostridium butyricum) has been shown to inhibit microglial activation and reduce the levels of levels pro-inflammatory cytokines in APP/PS1 mice. Furthermore, butyrate exerts anti-inflammatory effects by downregulating the levels of cyclooxygenase-2 (COX-2) and CD11b, and suppressing NF-κB signaling in Aβ-induced BV-2 cells.303 Notably, an oral combination therapy (AMX0035) comprising sodium phenylbutyrate and tauroursodeoxycholic acid (TUDCA) is currently undergoing a phase II clinical trial to evaluate its safety and biological activity in AD patients [NCT03533257].

Microbiota–gut–brain axis in Alzheimer’s disease. a Short-chain fatty acids (SCFAs) exert their neuroprotective effects by acting as endogenous ligands for G-protein-coupled receptors (GPCRs) and modulating gene expression by inhibiting histone deacetylases (HDACs). b Trimethylamine N-oxide (TMAO) promotes microglial activation, neuroinflammation, Aβ and tau pathology. c Neuroprotective bile acids (BAs), including UDCA and TUDCA, inhibit neuroinflammation via direct and indirect pathways. In the direct pathway, UDCA and TUDCA activate the nuclear receptor Farnesoid X receptor (FXR) and membrane receptor Takeda G-protein-coupled receptor 5 (TGR5) found in microglia and neurons. In the indirect pathway, UDCA and TUDCA provide signals to the central nervous system indirectly via intestinal TGR5-dependent glucagon-like peptide-1 (GLP-1) pathway and intestinal FXR-dependent fibroblast growth factor 15 or 19 (FGF15/19) pathway. d Tryptophan and indole derivatives activate microglial aryl hydrocarbon receptor (AHR) signaling to inhibit microglial activation and neuroinflammation. e Polyunsaturated fatty acids (PUFAs): omega-3 fatty acids exhibit neuroprotective effects in Alzheimer’s disease, whereas omega-6 fatty acid arachidonic acid and its pro-inflammatory metabolites induce microglial activation. This figure was created with BioRender (https://biorender.com/)
However, conflicting findings regarding the roles of SCFAs in AD have also been reported. A recent population-based study has revealed a positive association between serum propionic acid and cognitive decline in older adults, with potential mediation by hypercholesterolemia and diabetes.307 Interestingly, it has been demonstrated that SCFAs induce microglial activation and worsen Aβ pathology in both SPF and GF APP/PS1 mice.199 Furthermore, SCFA supplementation has been found to trigger C/EBPβ/AEP signaling activation and induce cognitive impairment in GF 3xTg-AD mice.196 The C/EBPβ is an inflammation-regulated transcription factor that regulates the expression of pro-inflammatory genes, thereby contributing to the pathogenesis of AD.204,308,309 The AEP is a lysosomal cysteine protease that cleaves tau at N255 and N368 residues, and amyloid precursor protein (APP) at N373 and N585 residues, resulting in amyloidogenic fragmentation and tau hyperphosphorylation.310,311 In GF 5xFAD mice, the administration of acetate aggravates hippocampal Aβ deposition by disrupting microglial phagocytosis of Aβ.190 The detrimental effects of SCFAs are similarly evident in a tauopathy mouse model. A recent study demonstrated that SCFA supplementation in GF TE4 mice mitigated the neuroprotective effects of GF rearing, resulting in increased gliosis and tau pathology. Conversely, the depletion of SCFAs-producing bacteria using antibiotic treatment conferred protection against tau-mediated neurodegeneration and neuroinflammation in TE3 mice.56 A recent study reported that intermittent fasting is effective in alleviating reactive microgliosis and astrogliosis, Aβ deposition, and cognitive impairment of 5xFAD mice by remodeling the microbiota–gut–brain axis. However, metabolomic analysis of cecal contents found that butyric acid was significantly downregulated in response to intermittent fasting, as compared to mice that were fed ad libitum.312
The considerable heterogeneity observed across distinct mouse models of AD, each characterized by distinct pathological pathways, poses a significant challenge in anticipating the implications of SCFAs. Moreover, the majority of studies that reported the detrimental effects of SCFAs were conducted using GF mice,56,190,196,199 which are functionally and structurally abnormal across various physiological functions.313 Thus, caution is warranted when extrapolating these findings to human diseases. In conclusion, it is evident that further investigation is warranted to elucidate the multifaceted nature and contextual significance of SCFAs in AD.
Parkinson’s disease
As with AD described previously, the roles of SCFAs in PD appear to be context-dependent and remain incompletely comprehended. Nevertheless, the majority of studies support the beneficial effects of SCFAs on microglial functions in the context of PD (Fig. 4a). Emerging evidence has revealed that epigenetic perturbation is an important contributor to PD, positioning it as a promising target for potential therapeutic interventions.286,314 For example, HDAC5 inhibition is effective in attenuating microglial activation and PD-related pathologies in 6-hydroxydopamine (6-OHDA)-lesioned rats.315 On the other hand, the activation of GPCRs has been demonstrated to exhibit neuroprotective properties in PD mouse models. Notably, the administration of probiotic Clostridium butyricum triggers the activation of colonic GPR41/43, resulting in the inhibition of microglial activation and mitigation of PD-related pathologies in MPTP-treated mice.316 Moreover, the activation of GPR41 in enteric neurons using AR420626 has been shown to mimic the neuroprotective effects of propionate in improving motor functions and preventing dopaminergic neuronal loss in 6-OHDA-induced PD mice.317

Microbiota–gut–brain axis in Parkinson’s disease. a Short-chain fatty acids (SCFAs) exert their neuroprotective effects by acting as endogenous ligands for G-protein-coupled receptors (GPCRs) and modulating gene expression by inhibiting histone deacetylases (HDACs). b Neuroprotective bile acids (BAs), including UDCA and TUDCA, inhibit neuroinflammation via direct and indirect pathways. In the direct pathway, UDCA and TUDCA activate the nuclear receptor Farnesoid X receptor (FXR) and membrane receptor Takeda G-protein-coupled receptor 5 (TGR5) found in microglia and neurons. In the indirect pathway, UDCA and TUDCA provide signals to the central nervous system indirectly via intestinal TGR5-dependent glucagon-like peptide-1 (GLP-1) pathway and intestinal FXR-dependent fibroblast growth factor 15 or 19 (FGF15/19) pathway. c Trimethylamine N-oxide (TMAO) promotes microglial activation and neuroinflammation. However, contradictory findings have been reported regarding the roles of TMAO in PD. d Tryptophan and indole derivatives activate microglial aryl hydrocarbon receptor (AHR) signaling to inhibit microglial activation and neuroinflammation. e Branched-chain amino acids (BCAAs) promote anti-inflammatory microglial phenotypes. This figure was created with BioRender (https://biorender.com/)
Boosting the levels of SCFAs with prebiotic intervention upregulates the neuroprotective phenotype of microglia in ASO mice, coupled with reduced motor deficits and α-synuclein aggregation in the SN.318 Similarly, the administration of probiotic Bifidobacterium breve CCFM1067 restored the levels of SCFAs in MPTP-induced PD mice, resulting in reduced glial activation, oxidative stress, and motor impairments.260 Additionally, studies have demonstrated the neuroprotective effects of sodium butyrate in MPTP-induced PD mice by attenuating microglial reactivity via the inhibition of TLR4/MyD88/NF-κB and MAPK signaling pathway.319,320,321 These findings are in concordance with a substantial body of literature that consistently reports diminished SCFAs-producing species and SCFA levels in prodromal stage of PD and PD patients compared to healthy controls, which may be correlated with the clinical severity of PD.52,322,323,324,325,326,327,328,329,330,331 The observed reduction in SCFAs in PD has inspired a proof-of-concept study to investigate the potential of prebiotic fibers in PD patients [NCT04512599]. Importantly, the trial reported an increased abundance of SCFAs-producing species and increased plasma SCFA levels, along with improved intestinal barrier integrity and reduced intestinal inflammation.332
Nonetheless, discrepant findings on the influence of SCFAs on microglial functions in PD have been documented. Of note, SCFA supplementation in GF ASO mice induced microglial activation, α-synuclein accumulation and motor dysfunction.55 Conversely, the reduction of SCFAs in MPTP-induced mice yielded beneficial effects, including alleviation of motor dysfunction, microglial and astrocytic activation in the SN.217,333 The intriguing duality of SCFAs in PD warrants further exploration. We speculate that the activation of C/EBPβ/AEP signaling might serve as a plausible mechanism underlying the detrimental effects of SCFAs, as demonstrated in GF 3xTg-AD mice.196 A recent study found that the C/EBPβ/AEP signaling is age-dependently activated in human α-synuclein transgenic mice and PD patients, which is responsible for mediating microglial activation and PD pathologies.334
In addition, growing evidence is shedding light on the distinct effects of different SCFAs on microglial functions. In particular, acetate has been found to restore microglial homeostasis in GF mice, while propionate and butyrate do not have the same restorative effect.190 This is further complicated by a recent study which reported that both reduced fecal propionic acid and butyric acid following low-dose maslinic acid treatment, as well as increased fecal acetic acid following high-dose maslinic acid treatment, demonstrated neuroprotective effects against PD pathologies in MPTP-treated mice. However, only high-dose maslinic acid treatment reduced microglial activation and neuroinflammation, and the effects are speculated to be mediated by acetic acid.335 On the other hand, a comparison between acetate, propionate, and butyrate in primary microglia has reported an abundant overlap between butyrate and propionate in microglial transcriptomic profile. However, individual SCFA failed to achieve comparable effects as the combined SCFA treatment.199 Thus, we recommend future studies to examine the effects of individual SCFA supplementation and combined SCFA supplementation on microglial function in PD mouse models. The beneficial effects of butyrate supplementation on PD pathologies might be attributed to its potent HDAC inhibitory activity and its ability to promote intestinal barrier and BBB integrity.336,337,338,339
Amyotrophic lateral sclerosis
Growing evidence has linked epigenetic dysregulations to ALS.340 Notably, genetic knockdown and pharmacological inhibition of HDACs have been shown to ameliorate ALS pathology in different ALS models.341,342,343,344 Furthermore, studies have identified reduced butyrate-producing species in ALS patients and SOD1G93A mice.221,222,345,346
Butyrate supplementation has demonstrated neuroprotective effects in SOD1G93A mice.222,347 The administration of butyrate has been shown to increase the abundance of butyrate-producing Butyrivibrio fibrisolvens, restore Paneth cell homeostasis and enhance the intestinal barrier integrity of SOD1G93A mice. This is accompanied by reduced SOD1G93A aggregation in the intestine, thereby slowing disease progression and prolonging the lifespan of SOD1G93A mice.347 Moreover, butyrate treatment reduces SOD1G93A aggregation and GFAP expression in the colon and lumbar spine of SOD1G93A mice, resulting in improved enteric neuromuscular function.222 Using motor neuron-like NSC34 cells with overexpression of hSOD1G93A, it was found that butyrate improved mitochondrial bioenergetics by improving mitochondrial network and upregulating the transcription of peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC1α).348 The PGC1α signaling is a master regulator of mitochondrial biogenesis and energy metabolism with significant therapeutic relevance in neurodegenerative diseases, including ALS.349
Bile acids regulate neuroinflammation and neurodegeneration
BAs are amphipathic cholesterol metabolites that serve diverse signaling functions. The primary BAs (cholic acid and chenodeoxycholic acid) are synthesized primarily in the liver, and released into the small intestine upon food intake, the majority of which are reabsorbed in the terminal ileum and recycled via enterohepatic circulation. However, non-reabsorbed primary BAs are converted by the gut microbiota into secondary BAs (deoxycholic acid, DCA, and lithocholic acid, LCA) via deconjugation and dehydroxylation, endowing them with new biological activities.20,350 This phenomenon is evidenced by the significantly higher concentration of secondary BAs in the intestines and fecal samples of SPF mice compared to GF mice, providing strong evidence for their microbial origin.351,352,353 Moreover, reduced concentrations of DCA and LCA were detected in the intestinal and fecal samples of individuals who had recently used antibiotics, albeit with the caveat of a small sample size.354
Alzheimer’s disease
Several studies have linked an altered BA profile to AD. Although studies in AD patients have yielded slightly heterogeneous results in BA composition, they have reported a consistent pattern of reduced levels of primary BAs and elevated levels of secondary BAs. Secondary BAs associated with AD and cognitive impairments include DCA, LCA, glycodeoxycholic acid, taurodeoxycholic acid, glycolithocholic acid, and taurolithocholic acid.355,356,357,358 Moreover, an elevated level of secondary BA taurohyodeoxycholic acid was reported in the serum of GF 3xTg-AD mice receiving FMT from AD patients.196 In another study, the administration of a traditional Chinese medicine decoction effectively ameliorated Aβ plaque pathology, neuroinflammation, and cognitive impairment in APP/PS1 mice. These beneficial effects were attributed to the suppression of gut dysbiosis and a reduction in the serum levels of secondary BAs, namely DCA and taurohyodeoxycholic acid.359
Nevertheless, it is noteworthy that certain secondary BAs serve significant biological functions, including the regulation of host metabolism, immunity, and resistance against intestinal pathogen expansion.21,360,361,362,363,364 More importantly, several secondary BAs have demonstrated pronounced neuroprotective and anti-inflammatory activities, particularly ursodeoxycholic acid (UDCA) and its amidated conjugate, TUDCA (Fig. 3c). They exert anti-inflammatory effects by activating the nuclear receptor Farnesoid X receptor (FXR) and membrane receptor Takeda G-protein-coupled receptor 5 (TGR5), both of which are found in microglia and neurons.365,366
The anti-inflammatory activity of TUDCA has been demonstrated in LPS-stimulated primary microglial cells and BV-2 microglial cells.367,368,369 In addition, TUDCA is effective in attenuating microglial reactivity in LPS-treated mice.367,370,371 Notably, TUDCA treatment has demonstrated the ability to inhibit Aβ deposition and glial activation in APP/PS1 mice and HFD-fed A7-Tg mice.372,373,374,375
Moreover, BAs have the potential to modulate the gut–brain axis by maintaining intestinal homeostasis, as TGR5 and FXR signaling are pivotal regulators of intestinal immune response and barrier function.365,376 Peripheral BAs provide signals to the CNS indirectly via TGR5-dependent glucagon-like peptide-1 (GLP-1) pathway and the FXR-dependent fibroblast growth factor 15 or 19 (FGF15/19) pathway.350 Currently, a phase II clinical trial is underway to examine the safety and biological activity of AMX0035, an oral combination of TUDCA and sodium phenylbutyrate, in the treatment of AD [NCT03533257].
Parkinson’s disease
The dysregulation of BA homeostasis has been implicated as a pivotal factor in the pathogenesis of PD. Studies have revealed elevated levels of secondary BAs in PD patients, while findings regarding primary BAs have yielded varying results.377,378,379,380 An elevated level of primary BA (cholic acid) was identified in the plasma of PD patients.379,380 Conversely, a reduction in glycine-conjugated primary BAs (glycocholic acid and glycochenodeoxycholic acid) was reported in post-mortem frontal cortex samples of PD patients, which was associated with the duration of PD diagnosis.377 However, another study reported increased levels of glycine- and taurine-conjugated primary BAs in plasma samples of PD patients.379 The discrepancy in BA profiles between plasma samples and brain samples warrants further investigation to understand the potential implications for PD pathogenesis. Notably, elevated levels of BAs were detected in the plasma samples of pre-PD patients, indicating that alterations in BA profile manifest many years before the onset of PD.381 Moreover, PD patients exhibited reduced plasma levels of neuroprotective BAs, namely UDCA and TUDCA.379 This observation aligns with the findings obtained in prodromal PD mice.382 Interestingly, a significant elevation in the risk of PD was observed among individuals who underwent cholecystectomy (removal of the gallbladder), a surgical procedure that has been associated with detrimental effects on both gut microbiota and BA composition.383,384,385 It was reported that mice that received FMT from patients with post-cholecystectomy diarrhea exhibited increased levels of secondary BAs, specifically DCA, LCA, and hyodeoxycholic acid.383
Similar to AD, the administration of UDCA and TUDCA represents a promising therapeutic approach to counteract microglial activation and neuroinflammation in PD (Fig. 4b). Multiple in vivo studies have demonstrated the neuroprotective effects of UDCA and TUDCA in counteracting mitochondrial dysfunction, oxidative stress, and neuroinflammation within PD mouse models.386,387,388,389,390,391,392 Moreover, the administration of a low-protein high-carbohydrate diet effectively mitigated PD pathologies in MPTP-treated mice by increasing the serum concentrations of TUDCA.393 These encouraging results have prompted two clinical trials, NCT03840005 (phase II) and NCT02967250 (phase I), which investigate the application of UDCA in PD patients. UDCA was found to be safe and well-tolerated in PD patients at a daily dose of 30 mg/kg.394
Aside from the direct effects of brain BAs, peripheral BAs provide signals to the CNS indirectly via the TGR5-dependent GLP-1 pathway and FXR-dependent FGF15/19 pathway.350 Notably, the modification of gut microbiota, BA metabolism and activation of intestinal TGR5 by dioscin treatment led to enhanced secretion of GLP-1 in both the intestine and brain of MPTP-treated mice. This is coupled with reduced glial activation and amelioration of motor deficits. Importantly, the co-administration of UDCA and dioscin further enhanced the neuroprotective effects of dioscin.390 GLP-1 is an incretin hormone that plays a pivotal role in stimulating insulin secretion and lowering blood glucose levels, making it highly relevant in the context of neurodegenerative diseases.395 The administration of GLP-1 receptor agonist inhibited microglial activation, prevented microglial-mediated conversion of astrocytes into the neurotoxic A1 phenotype, and effectively protected against α-synucleinopathy-induced neurodegeneration.396 Furthermore, the administration of probiotic Clostridium butyricum restored the levels of colonic GLP-1 and expression of cerebral GLP-1 receptor in MPTP-treated mice, leading to reduced microglial activation and alleviated motor deficits.316
Amyotrophic lateral sclerosis
Alterations in BA profiles have been documented in ALS patients and SOD1G93A mice, emphasizing the potential relevance of BAs to ALS pathology.397,398 A significant reduction in primary BAs and neuroprotective BA (TUDCA) was recently reported in the fecal samples of ALS patients with cognitive impairment compared to those with normal cognition.398 The existing evidence strongly supports the neuroprotective effects of TUDCA in ALS. TUDCA has been found to confer protection against cyclopiazonic acid-induced degeneration in both mouse and human stem cell-derived hSOD1G93A motor neurons. Furthermore, TUDCA enhances neuromuscular junction innervation in the tibialis anterior muscle of early-stage hSOD1G93A mice.399
Notably, an oral combination of TUDCA and sodium phenylbutyrate (AMX0035) is effective in slowing functional decline and prolonging the overall survival of ALS patients.400,401,402 The promising data from the phase II CENTAUR trial has recently led to its approval by the US FDA for the treatment of ALS.219 The phase III PHOENIX trial of this combination is ongoing [NCT05021536]. In addition, a phase III clinical trial is underway to evaluate the safety and efficacy of TUDCA as an add-on treatment to riluzole in ALS patients [NCT03800524].
Trimethylamine N-oxide promotes neuroinflammation and neurodegeneration
Trimethylamine N-oxide (TMAO) is a metabolite derived from dietary choline, betaine, and l-carnitine via a two-step pathway. Dietary choline is initially metabolized by the gut microbiota into trimethylamine (TMA), which is then absorbed and further oxidized in the liver into TMAO.403 A functional gut microbiota is required for the accumulation of TMAO in the brain tissue of aged mice, as aged GF mice displayed lower TMAO than aged SPF mice.404 This is consistent with another metabolomic analysis which reported significantly reduced levels of TMAO in the brain, serum, and feces of GF mice compared to conventionally raised mice, corroborating its microbial origin.405
Alzheimer’s disease
Two recent metabolomic studies have reported elevated TMAO levels in the brains of aged mice.406,407 In addition, the levels of TMAO in the plasma and brain of 18-month-old 3xTg-AD mice are markedly higher than those in 8-month-old mice.408 Importantly, elevated levels of TMAO have been observed in the plasma and cerebrospinal fluid (CSF) of individuals with MCI and AD.409,410 Several studies have shown that TMAO has the ability to traverse the BBB and contribute to neurodegeneration by inducing microglial and astrocytic activation.411,412,413,414
TMAO supplementation has been shown to induce cognitive impairment in APP/PS1 mice by promoting neuroinflammation, Aβ and tau pathology (Fig. 3b). Interestingly, the study also found that TMAO supplementation disrupted the integrity of the intestinal barrier and BBB.414 Moreover, TMAO induces inflammation and aggravates Aβ and tau pathology in D-galactose/AlCl3-induced AD mice by activating the PI3K/AKT/mTOR signaling pathway.415 Conversely, the reduction of TMAO following the administration of 3,3-dimethyl-1-butanol alleviated cognitive impairment of APP/PS1 mice by attenuating Aβ pathology and neuroinflammation.416 The administration of probiotic Lactobacillus plantarum is effective in remodeling the gut microbiota and reducing TMAO levels of APP/PS1 mice, resulting in amelioration of neuroinflammation and neurodegeneration.417 In addition, physical exercise has been shown to reduce the serum concentrations of TMAO, TMA, and betaine in APP/PS1 mice, conferring protection against neuroinflammation and AD pathologies.414
Parkinson’s disease
An evident increase in TMAO synthesis was detected in the post-mortem frontal cortex of PD patients with dementia, in comparison to patients with normal cognitive function and MCI.377 Moreover, elevated plasma TMAO levels in PD patients are correlated with disease severity and motor symptom progression. Patients with higher baseline TMAO levels are at higher risk of experiencing deterioration in motor symptoms and cognitive function.418 However, contradictory findings have been reported regarding the roles of TMAO in PD. A study on drug-naïve early-stage PD patients found that the plasma TMAO levels were lower in PD patients compared to healthy controls. Moreover, patients with lower plasma TMAO levels are associated with a higher rate of increase in levodopa-equivalent dose, as well as a higher risk of dementia conversion.419 Further research is needed to clarify the contribution of TMAO in the pathogenesis of PD.
Nevertheless, the existing evidence from PD models has indicated potential detrimental effects of TMAO (Fig. 4c). TMAO pre-treatment has been shown to worsen MPTP-induced microglial and astrocytic activation in the striatum, SN, and hippocampus of MPTP-treated mice.412,413 Using midbrain organoid models, it was found that TMAO induced pathological changes similar to PD, including loss of dopaminergic neurons, astrocytic activation, and phosphorylation of α-synuclein.420
Tryptophan and indole derivatives regulate neuroinflammation and neurodegeneration
Amino acids are vital precursors to numerous bioactive molecules, including neurotransmitters and neuromodulators, making them indispensable for optimal brain function and health.421 Growing evidence has elucidated the pivotal roles of gut microbiota in the metabolism and utilization of essential amino acids, particularly tryptophan. Alterations in tryptophan metabolism have been implicated in the pathogenesis of neurodegenerative diseases, including AD, PD, ALS, and HD. Tryptophan is an essential amino acid obtained through dietary sources and acts as a biosynthetic precursor to several microbial and host metabolites, including indole and its derivatives.422,423,424 Notably, significantly higher concentrations of indole derivatives were detected in the gut, serum, and brain of SPF mice compared to GF mice, thereby corroborating their microbial origin.351,405
The indole derivatives exert a wide range of immunological activities by binding to the AHR. The AHR is a ligand-dependent transcription factor expressed by epithelial cells, immune cells, microglia, and astrocytes.247,248,425 Previous studies on multiple sclerosis mouse models have demonstrated that the AHR acts as a negative regulator of NF-κB activation, and the specific deletions of Ahr in astrocytes and microglia resulted in CNS inflammation and neurodegeneration.247,248
Alzheimer’s disease
An altered composition of indole-producing bacteria was observed in APP/PS1 mice, concomitant with compromised intestinal barrier integrity and cognitive impairment. The administration of an indole mixture (indole, indole-3-acetic acid, and indole-3-propionic acid) attenuated the microglial reactivity and neuroinflammation by activating microglial AHR signaling and inhibiting NLRP3 inflammasomes. Consequently, the treatment led to reduced Aβ deposition, diminished tau hyperphosphorylation and improved cognitive function (Fig. 3d).426 Moreover, the administration of high-tryptophan diet proved effective in alleviating gut dysbiosis and inhibiting microglial reactivity in APP/PS1 mice by activating central AHR signaling and inhibiting NF-κB signaling.427
On the other hand, a significantly higher concentration of indoxyl-3-sulfate was reported in the brain and serum of GF 3xTg-AD mice receiving FMT from AD patients, when compared to their counterparts receiving healthy control microbiota.196 Indoxyl-3-sulfate is a uremic toxin adversely related to several mental and neurological conditions, including anxiety, anorexia nervosa, AD, and PD.377,428,429,430,431 An in vitro study revealed that it induces oxidative stress and inflammation in primary astrocytes and mixed glial cell cultures.432 Further studies are needed to comprehensively understand the roles of different indole derivatives in the pathogenesis of AD.
Parkinson’s disease
A recent metagenomic analysis of the gut microbiome of PD patients has revealed a reduction in the tryptophan biosynthesis pathway.331 Dietary supplementation of tryptophan has been shown to alleviate inflammation and motor deficits of rotenone‐induced PD rats by inhibiting NF‐κB activation, but the neuroprotective effects were blocked by an AHR inhibitor.433 In addition, studies have demonstrated the neuroprotective effects of indole-3 carbinol in rotenone-induced PD rats and LPS-treated rats (Fig. 4d).434,435 Notably, indole-3-carbinol effectively prevents rotenone-induced α-synuclein accumulation, astrocytic activation, neuroinflammation, and motor dysfunction by activating the SIRT1/AMP-activated protein kinase (AMPK) signaling pathway.434 Moreover, indole-3-carbinol is effective in counteracting LPS-induced oxidative stress and neuroinflammation.435
On the other hand, the uremic toxin indoxyl sulfate is increased in PD patients.377,429 An elevated CSF/plasma ratio of indoxyl sulfate was detected in PD patients, with notably higher concentrations observed in patients experiencing motor fluctuations compared to those without motor fluctuations.429 Furthermore, a marked upregulation of indoxyl sulfate was identified in the post-mortem frontal cortex samples of PD patients with dementia, compared to patients with normal cognitive function and MCI.377
Polyunsaturated fatty acids regulate neuroinflammation
Polyunsaturated fatty acids (PUFAs), encompassing omega-3 and omega-6 fatty acids, are essential fatty acids with diverse roles in human physiology, including CNS functioning. These include the regulation of neurogenesis, synaptic plasticity, microglial functions and neuroinflammation. The two predominant PUFAs in the brain are omega-3 fatty acid docosahexaenoic acid (DHA) and omega-6 fatty acid arachidonic acid (AA).436 The gut microbiota plays a vital role in the conversion of a subset of PUFAs into bioactive metabolites.196,437
Alzheimer’s disease
A recent lipidomic analysis of the brains of AD patients has revealed a significant reduction in the ratio of omega-3 to omega-6 fatty acids compared to age-matched cognitively normal individuals. In addition, the study found that omega-3 fatty acids are positively correlated with cognitive function and negatively correlated with Aβ, neurofibrillary tangles burden, and Braak stage. Conversely, the pro-inflammatory metabolites of omega-6 fatty acids are positively correlated with AD pathologies (Fig. 3e).438 Indeed, a higher dietary intake of omega-3 fatty acids is associated with a reduced risk of dementia and cognitive decline.439 The beneficial effects of omega-3 fatty acids may be attributed to their ability to stimulate microglial phagocytosis of Aβ42 and promote the transition of microglia to an anti-inflammatory phenotype, as demonstrated in human CHME-3 microglial cells.440
A recent study reported that GF 3xTg-AD mice exhibited reduced genes involved in AA metabolism, along with decreased levels of AA-associated inflammatory enzymes, in comparison to their SPF counterparts. However, FMT from AD patients to GF 3xTg-AD mice remarkably increased the relative abundance of Bacteroides strains involved in PUFA metabolism, resulting in increased concentrations of AA and its metabolites (leukotriene B4, prostaglandin E2, and 12-hydroxyheptadecatrienoic acid). Consequently, the elevated levels of AA-associated metabolites induced microglial activation and the development of AD pathologies in AD-humanized ex-GF 3xTg-AD mice by activating the C/EBPβ/AEP signaling pathway.196 Similarly in another study, FMT from AD patients to Thy1-C/EBPβ transgenic mice led to increased levels of prostaglandin E2 and 12-hydroxyheptadecatrienoic acid, resulting in C/EBPβ/AEP pathway activation, microglial activation, and AD pathologies.441 Indeed, elevated levels of leukotriene B4 and prostaglandin E2 were recently detected in the post-mortem brains of AD patients.438 Interestingly, the co-administration of glyceryl-conjugated prostaglandin E2 and SCFAs additively induced microglial activation in GF 3xTg-AD mice.196
Amyotrophic lateral sclerosis
PUFAs and their bioactive derivatives are vital regulators of neuronal function and neuroinflammation,436 making them highly relevant in the context of ALS. This is evident in two recent metabolomic analyses which revealed significant dysregulation of lipid metabolism, including PUFAs, among ALS patients compared to healthy controls.442,443 Notably, a higher plasma level of omega-3 fatty acid α-linolenic acid (ALA) was associated with slower functional decline and longer survival. In addition, higher plasma levels of omega-3 fatty acid eicosapentaenoic acid (EPA) and omega-6 fatty acid linoleic acid (LA) were associated with a lower risk of death, although they did not impact the rate of functional decline.444 The protective effects of ALA on ALS are similarly supported by another study, wherein males with higher pre-diagnostic plasma ALA levels are at a lower subsequent risk of developing ALS.445 These results are supported by a prospective study that observed a markedly reduced risk of ALS in individuals with higher dietary intakes of omega-3 fatty acids, including ALA and marine omega-3 fatty acids.446 On the other hand, an increased risk of ALS is observed in males with higher pre-diagnostic plasma levels of omega-3 fatty acid DHA, and in females with higher pre-diagnostic plasma levels of omega-6 fatty acid AA.445
Perturbation in lipid-related metabolism pathways, including AA metabolism, has been identified as a common metabolic signature in a multi-omics analysis of spinal motor neurons (sMNs) derived from four ALS hiPSC lines of various genetic background (SOD1A4V, C9ORF72, TDP-43Q343R, and sporadic).447 Importantly, the reduction of AA levels using 5-lipoxygenase inhibitor (caffeic acid) is effective in rescuing the reduced percentage of HB9::GFP+ cells (a marker of motor neurons) and increased percentage of 7AAD+ cells (a marker of cell death) in C9ORF72 and SOD1A4V ALS lines. Moreover, caffeic acid treatment is effective in delaying disease onset and prolonging the lifespan of SOD1G93A mice. These beneficial outcomes are attributed to the attenuation of microglial and astrocytic activation, and an increase in the number of motor neurons in the spinal cord.447
Branched-chain amino acids exert anti-inflammatory effects
Branched-chain amino acids (BCAAs) (leucine, isoleucine, and valine) are essential amino acids obtained through diet or gut microbial biosynthesis, as they are not synthesized by humans.448 Besides acting as building blocks for protein synthesis, BCAAs play a prominent role in diverse aspects of health and diseases by regulating numerous physiological processes.449,450 An early in vitro study has demonstrated the ability of BCAAs to alter microglial phenotypes, particularly towards the anti-inflammatory M2 phenotype (Fig. 4e).451
Parkinson’s disease
Reduced plasma levels of BCAAs and aromatic amino acids were detected in PD patients, which were significantly correlated with disease severity.452,453 This may be related to the reduction in Prevotella copri in PD patients,331,454 which is the species responsible for BCAA biosynthesis.448 A recent study observed reduced serum BCAA levels in rotenone-induced PD mice, accompanied by intestinal dysfunctions, motor deficits, α-synuclein accumulation, and loss of dopaminergic neurons in SN. Conversely, the administration of a high BCAA diet reversed the pathological changes induced by rotenone, along with reduced levels of pro-inflammatory cytokines in the colon and SN.455 Furthermore, administration of probiotic L. plantarum CCFM405 restored the biosynthesis of BCAAs, restored intestinal barrier integrity, and inhibited microglial and astrocytic activation in rotenone-induced PD mice.456
However, we recommend caution against excessive elevation of BCAAs as BCAAs imbalance is associated with detrimental health consequences, including hyperphagia, metabolic disorders, cardiovascular diseases, and reduced lifespan.450,457 Another factor warranting consideration is the potential competition between amino acids and levodopa, a commonly prescribed antiparkinsonian drug, in utilizing the intestinal and BBB transporters and consequent interference with levodopa pharmacokinetics.458,459
Nicotinamide mitigates neurodegeneration
Nicotinamide (NAM), also known as niacinamide, is a vitamin B3 derivative and constitutes one of the primary precursors of nicotinamide adenine dinucleotide (NAD+).460,461 A recent study has revealed a synergistic relationship in which the host-derived NAM is used by the gut microbiome for the synthesis of both NAD+ and nicotinic acid. The nicotinic acid is then taken up by the host tissue for NAD+ biosynthesis via the Preiss–Handler pathway. Importantly, NAM was not converted to NAD+ and nicotinic acid in GF mice and antibiotic-treated mice.462
NAD+ has been implicated in various aspects of health and disease due to its intricate involvement in numerous cellular processes and metabolic pathways. It is a central regulator of energy metabolism by acting as a coenzyme for redox reactions. Moreover, it serves as a cofactor for a wide variety of enzymes, including those involved in immune response and inflammation. These enzymes include sirtuins, poly(ADP-ribose) polymerase (PARP) protein family, and cyclic ADP-ribose (cADPR) synthases.460,461 However, the levels of NAD+ undergo progressive decline during aging, leading to the development and progression of age-related diseases.460
Amyotrophic lateral sclerosis
Neurodegenerative diseases, including ALS, are associated with disrupted NAD+ homeostasis and NAD+ depletion.461 A marked decrease in the levels of NAM has been observed in both the serum and CSF of ALS patients compared to healthy controls. The serum levels of NAM are positively associated with the ALS functional status score.223
A notable difference in the metagenomic NAM biosynthetic pathway has been reported between SOD1G93A mice and WT mice. The altered NAM biosynthesis was linked to A. muciniphila, which showed a gradual reduction in disease progression in SOD1G93A mice, but not in WT mice. Notably, the administration of A. muciniphila significantly improved the motor function and prolonged the lifespan of SOD1G93A mice. These beneficial effects of A. muciniphila were attributed to the enhanced NAM biosynthesis, which resulted in increased levels of NAM in the serum and CSF of SOD1G93A mice.223 In addition, NAM treatment has been shown to improve the survival of sMNs derived from both sporadic and familial ALS hiPSCs, as well as isogenic iPSC lines harboring SOD1L144F and TDP-43G298S mutations, by rescuing the mitochondrial respiration defects.463
Gut microbiota-related neurotransmitters in neurodegenerative diseases
The complex pathophysiology of neurodegenerative diseases involves multiple neurotransmitter systems dysfunction, including dopaminergic, cholinergic, serotonergic, glutamatergic, and GABAergic systems.464 Importantly, the gut microbiome has been shown to regulate brain functions by modulating these neurotransmitter systems.465 In this section, we provide an extensive review of the evidence regarding gut microbiota manipulation as a potential therapeutic approach to restore neurotransmitter systems in neurodegenerative diseases.
Serotonin
Serotonin, also known as 5-hydroxytryptamine (5-HT), is a neurotransmitter that serves a wide range of roles in the brain and gut, more notably its influence on the microbiota the microbiota–gut–brain axis.466 Approximately 90% of serotonin is produced and secreted by the gut enterochromaffin cells, which are strongly influenced by the gut microbiota.33,467,468 Moreover, the hippocampal serotonin level has been found to be affected by the gut microbiota, possibly by altering the peripheral availability of tryptophan.469 Although gut-derived serotonin is unable to cross the BBB, the serotonin precursor (5-hydroxytryptophan) and serotonin derivatives (N-acetylserotonin and melatonin) can cross the BBB to influence the CNS.34,403 Interestingly, the artificial elevation of intestinal serotonin levels enriches spore-forming bacteria, particularly Turicibacter sanguinis, which is involved in serotonin biosynthesis. These findings indicate a bidirectional host–microbial signaling that regulates gut microbiota colonization via the serotonergic system.470
Alzheimer’s disease
The dysregulation of the serotonergic system represents a complex pathological process in AD due to its involvement in multiple AD pathologies, including APP processing and Aβ deposition.471,472,473 A recent study reported lower brain serotonin transporter availability and higher cortical Aβ deposition in individuals with MCI compared to healthy controls.474 Furthermore, significantly reduced urine and serum serotonin concentrations have been reported in AD patients compared to controls.475 Interestingly, the administration of selective serotonin reuptake inhibitors (SSRIs) is effective in suppressing Aβ levels in human and AD mouse models.471,476,477
Differences in genes related to the serotonergic system have been reported between GF and SPF 3xTg-AD mice, which suggests higher hippocampal serotonin signaling in the GF mice.196 This study corroborates the involvement of gut microbiome in regulating serotonergic system in AD. However, there are currently sparse studies investigating the potential of gut microbiota manipulation to restore the serotonergic system in AD, and the available studies did not consistently show improvements in the serotonergic system. The co-administration of prebiotics fructo-oligosaccharides (FOS) and galacto-oligosaccharides (GOS) increased the concentration of serotonin in the brains of APP/PS1 mice.478 However, another study did not observe any improvement in the serotonergic system following prebiotic Sparassis crispa-1 polysaccharide administration in d-galactose/AlCl3-induced AD mice.479 Nevertheless, the available evidence suggests that probiotics, prebiotics and FMT hold immense potential in AD, as they have shown promising results in modulating the serotonergic system in different neurological disorders.34,480,481,482,483
Parkinson’s disease
Beyond the established dopaminergic dysfunction characterizing PD, growing evidence implicates serotonergic dysfunction in the progression of PD, which is associated with disease burden and may precede motor manifestations or dopaminergic dysfunction.484,485 Furthermore, the development of neuropsychiatric symptoms in PD patients, including apathy, anxiety, and depression, is associated with serotonergic dysfunction.486,487 In addition, studies have demonstrated the modulation of the serotonergic system by α-synucleinopathy. For instance, the overexpression of human α-synuclein in serotonin receptors in raphe nuclei in mice has been found to impair forebrain serotonin neurotransmission and trigger a depressive-like phenotype.488 Conversely, the serotonergic neurotransmission deficits were alleviated following antisense oligonucleotide-induced α-synuclein knockdown.488,489
A recent metagenomic analysis of the gut microbiome of PD patients has revealed a dysregulation in the synthesis and metabolism of multiple neurotransmitters, including serotonin. This dysregulation may be attributed to the observed reduction in the tryptophan biosynthesis pathway and sporulation genes.331 Additionally, a recent study found that healthy mice, upon receiving FMT from PD mice, exhibited impaired motor function and reduced striatal dopamine and serotonin levels.217 These findings underscore the potential significance of gut microbiota manipulation to restore the serotonergic system in PD. Notably, prebiotic polymannuronic acid administration led to the elevation of serotonin and its metabolite, 5-hydroxyindoleacetic acid (5-HIAA), in the striatum of MPTP-induced mice.490 Moreover, FMT from control mice mitigated MPTP-induced gut dysbiosis and the decline of striatal serotonin and 5-HIAA in recipient mice.217 Conversely, recent studies investigating the impact of probiotics on the serotonergic system in PD have shown less favorable outcomes. In a recent randomized controlled trial (RCT), no significant changes in serum serotonin levels were observed following a 3-month adjunctive probiotic Bifidobacterium animalis subsp. lactis treatment in PD patients receiving conventional regimen (Benserazide and dopamine agonists).491 Furthermore, the administration of L. plantarum PS128 was ineffective in restoring MPTP-induced serotonin reduction.492 Future studies may explore the potential benefits of multi-strain probiotic formulations for achieving the desired serotonergic effects in PD. These studies should emphasize on the importance of informed strain selection, including the potential synergy or additive effects of individual probiotic strains.493
Gamma-aminobutyric acid (GABA)
GABA is the main inhibitory neurotransmitter primarily generated and regulated by astrocytes and neurons. Tonic GABA current is a vital regulator of brain states and cognitive functions, including learning and memory, sensory processing, circadian rhythm, and vigilance state. Moreover, tonic GABA current is integral for normal motor functions, and its dysregulation has been associated with motor symptoms in PD and HD.494 The gut microbiome is involved in the production of GABA, as gut microbiota manipulation has been found to impact GABA levels.495,496,497 Several gut microbes have been identified as GABA producers, including the members of the Bacteroides, Bifidobacterium and Lactobacillus genera.37,495,498,499,500
Alzheimer’s disease
The AD pathologies, including Aβ and tau pathology, have been shown to induce GABAergic dysfunction and contribute to excitatory and inhibitory (E/I) imbalance.501,502,503 An abnormal increase in tonic inhibition has been reported in APP/PS1 mice and 5xFAD mice due to aberrant release of GABA by reactive astrocytes, leading to impaired synaptic plasticity and memory.504,505 However, recent evidence suggests that GABAergic hypoactivation potentiates neurodegeneration and cognitive impairment in AD patients.502,506 Further research is warranted to elucidate the observed discrepancies in the role of GABAergic system within AD pathogenesis, including the involvement of astrocytes.
A recent study has reported variations in the expression levels of genes related to GABAergic system between GF and SPF 3xTg-AD mice, suggesting an increase in hippocampal GABA production and GABA receptors in the GF counterparts.196 These findings suggest a potential avenue for manipulating the gut microbiota to address GABAergic dysfunction in AD. In an initial investigation with limited sample size, it was reported that FMT from an AD patient resulted in a reduction of GABA levels in the fecal metabolites of recipient mice.507 Conversely, yFMT effectively restored hippocampal GABA levels in aged recipient mice towards the levels observed in young mice, coinciding with improvements in cognitive behavior.210 Moreover, the administration of prebiotic Sparassis crispa-1 polysaccharide increased the hippocampal levels of GABA in D-galactose/AlCl3-induced AD mice, resulting in improved cognitive function.479 Future studies employing larger sample sizes and AD mouse models could provide valuable insights into the potential therapeutic implications of modulating the gut microbiome to target the GABAergic system in AD.
Parkinson’s disease
Growing evidence has reported alterations in the GABAergic system in PD, which are associated with motor symptoms, psychomotor symptoms, axial symptoms, cognitive impairment, olfactory dysfunction, and visual hallucinations.508,509,510 An alteration in the synthesis and metabolism of GABA was recently reported in a metagenomic analysis of the PD gut microbiome.331 Recent studies have explored the potential therapeutic role of probiotics and prebiotics in addressing these underlying pathologies. The administration of probiotic Pediococcus pentosaceus and prebiotic polymannuronic acid has shown promise in alleviating gut dysbiosis and reducing GABA levels in the brains of MPTP-induced mice, resulting in the amelioration of motor dysfunction and neuronal degeneration.490,511 A recent RCT found that the co-administration of probiotic and conventional regimen in PD patients increased the abundance of species-level genome bins (SGBs) involved in GABA synthesis and reduced the abundance of SGBs encoding GABA degradation, as compared to PD patients receiving conventional regimen alone.491
Dopamine
Dopamine is a multifaceted neurotransmitter involved in the regulation of learning, motivation, motor, and cognitive control, in concert with other neurotransmitters.512,513,514,515,516 Disruption of dopaminergic transmission leads to many debilitating CNS disorders, including PD.517 Accumulating evidence underscores the regulatory role of the gut microbiome in dopamine signaling.518 A recent study found that the exercise-triggered striatal dopamine response was blunted in antibiotic-treated mice due to an increase in the levels of monoamine oxidase, the enzyme responsible for dopamine degradation. Conversely, GF mice receiving FMT from vigorous mice exhibited elevations in dopamine levels.48
Parkinson’s disease
Aside from the aggregation of misfolded α-synuclein in the intracellular Lewy bodies, a major neuropathological hallmark of PD is the loss of dopaminergic neurons within the SN.519 Dopaminergic deficiency underlies the cardinal motor features of PD, including bradykinesia, rigidity, resting tremor, and postural instability.520 Thus, the mainstay pharmacological treatments for the management of PD motor symptoms are predominantly dopamine-related interventions, including levodopa preparations, dopamine agonists, and monoamine oxidase-B inhibitors.519
The gut microbiome influences multiple aspects of dopaminergic system within PD pathophysiology, as well as the efficacy of dopamine-related drugs.518,521,522,523 This influence is exemplified by a study in which FMT from PD mice reduced striatal levels of dopamine and its metabolites in recipient mice, accompanied by impaired motor function.217 Moreover, metagenomic analysis of the gut microbiome in PD patients has detected a reduced capacity to generate dopamine precursors, particularly tyrosine.331 Several gut microbes have shown detrimental effects on the dopaminergic system in PD mouse models.215 For instance, the administration of Proteus mirabilis during the premotor phase of MPTP-treated mice aggravated striatal dopaminergic neuronal damage and motor symptoms.524
A substantial body of literature has demonstrated the potential of microbiome-based therapeutics in alleviating dopaminergic damage and motor deficits in PD mouse models.217,261,321,393,492,525,526 Adjunctive probiotic administration was found to increase serum dopamine levels and improve motor function in PD patients receiving conventional regimen, compared to patients receiving conventional regimen alone.491 Moreover, the administration of berberine has been shown to stimulate the production of dopa and dopamine in the gut microbiota by activating tyrosine hydroxylase and dopa decarboxylase. Dopa, in turn, crosses the BBB and is converted into dopamine. Consequently, the increased levels of dopamine in the brain protected against MPTP-induced motor deficits.36
Acetylcholine
Acetylcholine is a major neurotransmitter released by cholinergic neurons. The cholinergic signaling is responsible for coordinating various cognitive processes in the brain, including memory, learning, attention, sleep, and other higher brain functions.527 Increasing evidence is elucidating the complex interactions between the gut microbiome and cholinergic signaling. A recent study found that antibiotic-induced gut dysbiosis and microglial activation resulted in marked reductions in hippocampal synaptic transmission and cholinergic gamma oscillations.193 A possible mechanism underlying the antibiotic-induced cholinergic dysfunction is the elevated activity of corticohippocampal acetylcholinesterase (AChE), which leads to increased acetylcholine breakdown.528,529 Moreover, the disruption of the gut microbiome in mice fed a high-fat high-sugar diet resulted in increased AChE expression in the brain.530
Alzheimer’s disease
The significance of cholinergic signaling in AD was long recognized.531 A hallmark of cognitive impairment is the loss of basal forebrain cholinergic neurons, which is driven by APP processing, Aβ deposition, tau hyperphosphorylation and dysregulated immune response.527 At present, there is a limited number of studies examining the potential of microbiome-based therapeutics for modulating the cholinergic system in AD, but the evidence available supports their beneficial effects on the cholinergic system. Notably, the administration of probiotic L plantarum MTCC1325 restored the levels of acetylcholine in the brains of D-galactose-induced AD rats by reducing AChE activity.532 Similarly, another study found that prebiotic FOS from Morinda officinalis was effective in restoring acetylcholine in the brains of d-galactose-induced AD rats by reducing AChE levels.533 Further investigation into the neuroprotective potential of microbiome-based therapeutics in the cholinergic system in other AD animal models is warranted.
Glutamate
The excitatory neurotransmitter glutamate is a critical regulator of neuronal excitability and synaptic plasticity, which are integral for synaptic transmission, learning, memory, and cognitive function.534 Increasing evidence also points towards the role of glutamate, released by astrocytes, as a gliotransmitter to regulate synaptic transmission and plasticity.535,536,537 However, excessive glutamate levels trigger neuronal death through excitotoxicity and have been implicated in the pathogenesis of neurodegenerative diseases.534 Similar to the neurotransmitter systems discussed previously, glutamatergic signaling is influenced by the gut microbiome.465 The influence of gut microbiome on glutamatergic signaling has been demonstrated across rodent models of different neurological disorders, including schizophrenia,538 depression,44 AD,196 and PD.331
Parkinson’s disease
Aberrant glutamatergic neurotransmission represents a key contributing factor to neurodegeneration and PD.377,510,539 However, our understanding of the influence of the gut microbiome on glutamate in PD remains limited, and the studies available primarily examine glutamate within the gut microbiome. Although PD is generally associated with glutamatergic hyperactivity, a recent metagenomic analysis of PD gut microbiome revealed a reduction in glutamate/glutamine synthesis genes and pathways, along with an increase in glutamate degradation pathway.331 These findings are in agreement with the results of an earlier meta-analysis of the PD gut microbiome.540 However, it is noteworthy that glutamate does not readily cross the BBB, which may limit its direct influence on brain glutamate levels.541,542 On the contrary, several transporters for glutamine, the direct precursor of glutamate, have been identified.543 Notably, the combined administration of probiotics with conventional regimen led to a reduction in serum glutamine concentrations in PD patients compared to the placebo group receiving the conventional regimen only.491 Further studies are warranted to examine the potential and underlying mechanisms of microbiome-based therapeutics in modulating glutamatergic signaling in PD.
Gut hormones in neurodegenerative diseases
The hormone-producing enteroendocrine cells (EECs) are specialized epithelial cells found throughout the GI tract.544,545 More than 20 gut hormones with overlapping targets and actions have been identified. Once released, the gut hormones exert local effects on neighboring cells within the mucosa and neuronal networks, as well as systemic effects on distant organs, including the CNS.544,546 Notably, the gut microbiota-derived metabolites, including SCFAs, secondary BAs, and indoles, have been demonstrated to modulate gut hormone secretion from EECs.546,547 In this section, we review gut hormones that have demonstrated significance in neurodegenerative diseases, specifically ghrelin, leptin, and GLP-1.
Ghrelin
Ghrelin is an octanoylated peptide hormone that exerts its biological activity by acting as an endogenous ligand for the growth hormone secretagogue receptor (GHSR), more commonly known as the ghrelin receptor. The highest expression of ghrelin receptors is found within the brain.544,548 The central ghrelin signaling plays a pivotal role in diverse physiological functions, including the regulation of food intake, hippocampal synaptic plasticity and neurogenesis, anxiety, depression, and cognitive function.544,549,550,551 Notably, age-related reductions in ghrelin signaling have been associated with a decline in cognitive function.551 The gut microbiome, along with its metabolites, has been reported to modulate ghrelin secretion and CNS functions.552,553
Alzheimer’s disease
Accumulating evidence has indicated dysfunctional ghrelin signaling in AD. It was found that Aβ binds to and inhibits hippocampal GHSR1α activity in 5xFAD mice, resulting in synaptic deficits and cognitive decline.554 In addition, plasma ghrelin levels reduce gradually in 3xTg-AD mice with aging, which is reversed by the administration of multi-strain probiotics formulation (SLAB51), leading to the attenuation of cognitive impairment.555 A recent study in the elderly reported an age-dependent reduction in plasma ghrelin and an elevated ratio of plasma liver-expressed antimicrobial peptide 2 (LEAP2)/ghrelin, which is associated with cognitive impairment. Subsequently, the study found that reducing the plasma LEAP2/ghrelin ratio reversed cognitive deficits in aged mice by restoring hippocampal neurogenesis, alleviating synaptic loss, and inhibiting neuroinflammation.551 Moreover, the activation of ghrelin signaling has been shown to attenuate microglial activation in mouse models of normal aging, accelerated aging and AD.556,557 The neuroprotective effects of ghrelin might be attributed to its ability to impact age-related pathways, including sirtuin-1 activation, with studies demonstrating its ability to prolong lifespan in different mouse models of aging.556,558
Parkinson’s disease
The ghrelin receptors are expressed in various brain regions, including the SN. Ghrelin participates in the regulation of motor functions by modulating the dopaminergic neuronal excitability within the SN.559,560 Additionally, ghrelin promotes the differentiation of midbrain neural stem cells into dopaminergic neurons by activating the Wnt/β-catenin pathway.561 Conversely, the inhibition of central GHSR1α activation via intra-SN administration of a selective GHSR1α inhibitor resulted in impaired motor coordination.562 Evidence from PD patients has identified reduced plasma ghrelin levels in the fasting state,563,564 as well as an impaired ghrelin response and secretion in the postprandial state.564,565 These findings have provided a pivotal basis for further investigations into the neuroprotective effects of ghrelin in PD. Indeed, accumulating evidence has demonstrated the neuroprotective effects of ghrelin in multiple rodent models of PD, including MPTP-induced PD mice,560,566,567,568 6-OHDA-induced PD rats,569,570 and A53T α-synuclein transgenic mice.571 These beneficial effects are attributed to its ability to promote autophagy, enhance mitochondrial biogenesis and bioenergetics, mitigate neuroinflammation, and inhibit apoptosis.560,566,567,568,569
Leptin
Leptin is an adipocyte-derived peptide hormone produced by the white adipose tissue. It conveys metabolic information to the leptin receptors primarily expressed in the CNS to regulate energy homeostasis and immune functions.572,573 Notably, impaired brain energetics is a core feature across neurodegenerative diseases, including AD, PD, ALS, FTD, and HD.574 Leptin signaling is involved in multiple signaling pathways, including the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) pathway, PI3K/AKT pathway, mTOR pathway, and AMPK pathway.575 These pathways are highly relevant in the context of aging, neuroinflammation, and neurodegeneration.245,576
Recent evidence has demonstrated an association between adiposity and reduced cognitive function,577,578,579 suggesting that leptin signaling represents a target to ameliorate cognitive impairment. Notably, several studies have demonstrated the significant role of gut microbiome in modulating leptin signaling and adiposity.580,581,582,583 In addition, a recent meta-analysis of 16 RCTs has revealed that the administration of probiotics or synbiotics (prebiotics + probiotics) leads to a significant reduction in circulating leptin levels.584
Alzheimer’s disease
Leptin is generally considered beneficial for cognitive function, as studies have consistently shown that individuals with higher leptin levels are associated with a reduced risk of dementia and AD.585,586,587 This is supported by a recent meta-analysis that has reported reduced blood leptin levels in AD patients compared to cognitively normal individuals.587 Moreover, leptin is involved in counteracting Aβ pathology and tau phosphorylation to prevent synaptic dysfunction.588,589 This is evident in a recent study that has revealed a robust association between low plasma leptin and increased CSF Aβ concentration in patients with cognitive impairment, which indicates increased brain amyloid deposition.590 Notably, the elevation of plasma leptin levels via chronic administration of multi-strain probiotics has been shown to ameliorate Aβ pathology in 3xTg-AD mice.555
Parkinson’s disease
Although leptin primarily acts on neurons in the hypothalamus, leptin signaling also has an important role in preserving the dopaminergic system.591,592,593 Leptin receptors are widely expressed in several extra-hypothalamic regions, including dopaminergic neurons in SN.594,595 However, our understanding of the role of leptin in PD remains incomplete, with limited evidence from clinical studies. Notably, a recent meta-analysis, including only 198 PD patients and 182 controls, found that the serum leptin levels in PD patients were slightly lower compared to those in the control group, although the difference did not reach statistical significance.596 These limited findings emphasize the need for more comprehensive research to understand the mechanisms and functions of leptin in the onset, progression, and management of PD.
Glucagon-like peptide-1
GLP-1 is an incretin hormone produced by the EECs in response to ingested nutrients to stimulate glucose-dependent insulin secretion and suppress appetite.544 GLP-1 has pleiotropic actions due to the extensive expression of GLP-1 receptors (GLP-1R) in multiple organs, including microglia and astrocytes across various brain regions, thus is being explored for its potential in neurodegenerative diseases.574,597,598 GLP-1R agonists, approved for the treatment of type 2 diabetes and obesity, have demonstrated promising neuroprotective effects in neurodegenerative diseases owing to their ability to suppress neuroinflammation.598 The secretion of GLP-1 is modulated by the gut microbiota,583,599 as well as multiple gut microbiota-derived metabolites, including SCFAs, secondary BAs, indoles, and PUFAs.437,547 Moreover, probiotics and prebiotics are capable of stimulating GLP-1 secretion and upregulating the expression of GLP-1R in the brain.316,600,601
Alzheimer’s disease
Diabetes is a well-established risk factor for cognitive impairment and dementia, with dementia now among the leading causes of death in the diabetic population.602,603,604 Given these connections, there is a growing interest in repurposing the anti-diabetic class of drug GLP-1R agonists for AD.605 Substantial preclinical evidence supports the neuroprotective effects of GLP-1R agonists on cognition in AD, which is attributed to the ability to reduce Aβ pathology, tau phosphorylation, glial activation, and neuroinflammation.598,606 Notably, GLP-1R agonist, NLY01, inhibits Aβ-induced microglial activation via microglial GLP-1R, resulting in reduced reactive astrocyte conversion in both 5xFAD and 3xTg-AD mice.607 In addition, the neuroprotective effects of GLP-1R agonists were substantiated by clinical studies that reported a reduction in cognitive impairment and lower dementia incidence among type 2 diabetes patients.608,609 These encouraging results have prompted two phase III RCTs, NCT04777396 and NCT04777409, to explore the efficacy of oral GLP-1R agonist semaglutide in early AD. Interestingly, semaglutide treatment has been shown to activate the gut intraepithelial lymphocytes GLP-1R and modulate the gut microbiota, contributing to a reduction in T cell-mediated inflammation.610
Parkinson’s disease
Diabetes is a well-established risk factor for PD, and has been linked to a faster disease progression, due to the overlapping underlying pathological mechanisms.611,612,613,614 It has been reported that diabetic patients receiving GLP-1R agonists are associated with a lower risk of developing PD.615 The neuroprotective effects of GLP-1R agonists in PD are supported in numerous preclinical studies and have been attributed to their ability to restore insulin signaling, reduce glial activation, neuroinflammation, and oxidative stress.395 Similar to the findings in AD mouse models,607 the administration of NLY01 prevented microglial activation and reactive astrocyte conversion, effectively protecting against α-synucleinopathy-induced neurodegeneration.396 Notably, exenatide, administered at the dose approved for type 2 diabetes treatment, crosses the BBB in appreciable quantities and improves the motor and cognitive function of PD patients.616
The amelioration of gut dysbiosis with microbiome-based therapeutics has been shown to restore GLP-1 signaling and attenuate PD pathologies. An engineered GLP-1-producing probiotic strain, Lactococcus lactis MG1363-pMG36e-GLP-1, counteracts MPTP-induced neurodegeneration by inhibiting glial activation, oxidative stress, and ferroptosis.617,618,619 In addition, probiotic C. butyricum restores colonic GLP-1 levels and upregulates GLP-1R expression in the brains of MPTP-induced mice, accompanied by reduced microglial activation and PD pathologies.316 The alleviation of MPTP-induced gut dysbiosis and BAs alterations by dioscin treatment activates the intestinal TGR5 to stimulate GLP-1 secretion and upregulate GLP-1R expression in the brain, thereby attenuating glial activation and neurodegeneration.390 Altogether, these studies underscore the potential of gut microbiome interventions to restore GLP-1 signaling and ameliorate PD pathogenesis.
Points of intervention to improve microbiota–gut–brain axis
In this section, we outline three potential points of interventions to regulate the microbiota–gut–brain axis, namely the intestinal barrier, BBB, and meninges. We provide evidence that dysfunctional barrier integrity induces glial activation and neurodegeneration. Thus, restoring the integrity of these biological barriers holds promise in counteracting dysfunctional glial states in neurodegenerative diseases. Among the biological barriers, targeting the intestinal barrier represents the most promising and straightforward approach, given the direct interactions between gut microbiota and the intestinal barrier. In addition, growing evidence suggests that peripheral immune system may influence neurodegenerative processes directly at the periphery,620 Thus, we discuss the evidence of immune regulation by the gut microbiota, specifically focusing on regulatory T (Treg) cells and T helper 17 (TH17) cells, which are increasingly recognized for their neuroimmune role within the context of gut–brain axis.621,622
Intestinal barrier restoration by microbial metabolites
A pivotal regulator of the microbiota–gut–brain axis is the multi-layered intestinal barrier. The intestinal barrier, comprising the mucus layer, epithelial barrier, and gut vascular barrier, collectively provides the host with excellent protection against external hazards. For a detailed overview of the structure and CNS-related functions of the intestinal barrier, please refer to the comprehensive review by Pellegrini et al.623 Gut microbiome alterations and intestinal barrier impairment have been observed in patients with MCI, AD, PD, and ALS patients.326,540,624,625,626 Thus, the restoration of intestinal barrier integrity represents a promising therapeutic strategy to disrupt the cascade of events leading to neurodegeneration.
The mucus layer is a partially penetrable and highly dynamic physical barrier that separates the luminal content from the underlying intestinal layers. Mucins, the primary structural element of the mucus layer, are a family of heavily glycosylated proteins produced and secreted by goblet cells in the epithelium. Apart from its classic GI-related functions, it is increasingly evident that the mucus layer also plays a role in modulating immunity and inflammation by regulating the microbiota composition and the interaction between microbiota and host. The influence of gut microbiota on mucus layer homeostasis has also been revealed.627,628 Early evidence has shown that GF mice exhibit characteristics of a defective mucus layer.629 As the task of producing large amounts of mucin is challenging to the goblet cells, a controlled unfolded protein response and endoplasmic reticulum (ER) expansion are required to prevent the accumulation of ER stress. An intact gut microbiota contributes to goblet cell maturation and mucus barrier assembly by activating the epithelial cell-specific ER stress sensor ERN2 and its downstream transcription factor X-box binding protein 1 (XBP1).336 ERN2 is one of the three major signaling branches of unfolded protein response specifically expressed in mucin-producing goblet cells of gastrointestinal and respiratory tracts. It is responsible for restoring goblet cell proteostasis.630,631
During aging, the intestinal barrier undergoes progressive breakdown, which facilitates the translocation of pro-inflammatory gut microbiota-derived metabolites and pathogenic bacterial products into the bloodstream (Fig. 5). This process triggers elevated systemic inflammatory responses that gradually impair BBB integrity, resulting in CNS inflammation and neurodegeneration.623 Microbiome-mediated disruption of the intestinal barrier during aging facilitates excessive translocation of N6-carboxymethyllysine across the intestinal mucosal barrier into the bloodstream. This increased translocation results in the accumulation of N6-carboxymethyllysine in the brain, which in turn contributes to microglial dysfunction by inducing oxidative stress and mitochondrial dysfunction.404 Notably, FMT from young mice to aged mice restored the intestinal epithelial barrier integrity and attenuated microglial activation in the aged mice.632

Improving microbiota–gut–brain axis via the intestinal barrier. a High-fiber diets contribute to a healthy gut microbiome and enhance intestinal barrier integrity by increasing SCFAs-producing species, and fiber-degrading species and promoting resistance to perturbations. Indole and its derivatives improve intestinal barrier integrity by activating epithelial aryl hydrocarbon receptors (AHR). b Low-fiber diets, aging and sleep deprivation contribute to dysbiosis and disrupt intestinal barrier integrity by reducing SCFAs-producing species and fiber-degrading species while increasing mucin-degrading species. Low-fiber diets induce mucosal and systemic immune depression by impairing the metabolic fitness of CD4+ T cells. This figure was created with BioRender (https://biorender.com/)
Interestingly, recent evidence has revealed the adverse effects of neuropathological changes in AD brains on the gut microbiome and intestinal barrier integrity.633,634,635 A recently developed transgenic AD mice model with amyloid and neurofibrillary tangles pathology (ADLPAPT mice) manifested altered gut microbiota, chronic intestinal inflammation, increased intestinal barrier permeability, and systemic inflammation. However, daily FMT from WT mice for 4 months successfully transformed the gut microbiota composition of ADLPAPT mice, leading to restored circulating Ly6Chi monocytes population, reduced Aβ deposition, tau aggregates, and microglial activation.635 Similar findings were reported in another study following intracerebroventricular injection of Aβ1–42 oligomers. Aβ1–42-treated mice began developing alterations in the composition and diversity of gut microbiota 4 weeks post-surgery. These changes were coupled with elevated intestinal permeability and intestinal inflammation via the inhibition of the cholinergic anti-inflammatory pathway.634 These findings underscore the necessity of future studies to delineate the temporal sequence of pathologies.
Short-chain fatty acids restore intestinal barrier integrity
The SCFAs produced by the gut microbiota during the fermentation of dietary fiber are critical in the reinforcement of the mucus layer (Fig. 5a). High-fiber intake increases dietary fiber metabolizers and SCFAs producers, which in turn stimulates mucus secretion to maintain a well-structured and intact mucus layer.636,637 Importantly, dietary fiber also promotes resistance of human gut microbiota to perturbations by limiting microbial growth and balancing the positive and negative interspecies interactions.638 A proof-of-concept trial conducted in PD patients has demonstrated that SCFAs-promoting prebiotic fiber intervention reduced plasma zonulin, indicating improved intestinal barrier integrity.332 Moreover, a recent randomized clinical trial reported that a 3-month high-fiber intervention with a Mediterranean diet greatly reduced plasma lipopolysaccharide-binding protein (LBP) and fecal zonulin concentrations in women with an impaired intestinal barrier. The study further performed model-based causal mediation analysis and revealed that the barrier-stabilizing effect of the Mediterranean diet was mediated by the propionate and butyrate.639
In contrast, a chronic or intermittent low-fiber diet in gnotobiotic mice stimulates the proliferation and activity of mucin-degrading bacteria, resulting in mucus layer erosion and increased pathogen susceptibility (Fig. 5b).640 In addition, feeding mice a western-style diet characterized by low dietary fiber, high fats, and high simple carbohydrates induces depletion of fiber-degrading bacteria, including Bacteroidetes (family S24-7) and Actinobacteria (Bifidobacterium). This was accompanied by reduced SCFAs production, reduced inner mucus growth rate, and increased mucus penetrability.641 Consumption of a western-style diet also drives the emergence of mutations in Bacteroides thetaiotaomicron, an anaerobe specialized in fermenting complex polysaccharides. These mutations promote the degradation of mucin O-glycans in the mouse gut.642 Consequently, the increased intestinal permeability facilitates the translocation of bacteria and their byproducts into the mucosa and bloodstream. This leads to aberrant activation of the intestinal and circulating immune and inflammatory cells, resulting in systemic inflammation, neuroinflammation, and neurodegeneration.623,643
Restoring the Bacteroidetes of obese mice fed a high-fat and fiber-deficient diet through oat β-glucan supplementation successfully restored the intestinal barrier integrity, including mucus thickness and levels of tight junction (TJ) proteins. In addition, β-glucan supplementation attenuated microglial activation and cognitive impairment in the obese mice. Importantly, the beneficial effects of β-glucan on cognitive function were lost upon antibiotics administration, indicating that the gut microbiota is a vital mediator of cognitive function.644 Similarly, reshaping the gut microbiome of HFD-fed mice using dimethyl itaconate restores intestinal barrier integrity by increasing SCFAs-producing bacteria and restoring intestinal immune homeostasis. This is accompanied by alleviation of microglial activation, neuroinflammation, synaptic impairment, and cognitive impairment induced by HFD.645
Recently, growing evidence has indicated that sleep deprivation (SD) induces gut dysbiosis and intestinal barrier disruption, leading to microglial activation and cognitive decline (Fig. 5b).646,647,648 Persistent SD (<6 h per night) during midlife is associated with a 30% elevated risk of dementia in a longitudinal study of 7959 participants.649 Chronic SD triggers gut microbiota alteration, leading to reduced mucus thickness and reduced levels of TJ proteins in mice colons, via NLRP3 inflammasome activation. This is accompanied by impaired BBB integrity, NLRP3 activation in the brain, microglial activation, and cognitive impairment. Moreover, FMT from chronically sleep-deprived mice to control mice largely recapitulated the pathological changes observed in chronic SD.646 Similar detrimental consequences of SD on cognitive function were observed in humans. SD in humans induced depletion of SCFAs-producing species, systemic inflammation via the activation of TLR4/NF-κB signaling pathway. This is coupled with increased serum levels of zonulin and S100β, indicating compromised intestinal barrier and BBB.647,650 Interestingly, GF sleep-deprived mice exhibited weaker inflammatory responses, reduced intestinal barrier damage, and milder cognitive impairment as compared to their SPF counterparts, highlighting the crucial role of gut microbiota in mediating SD-induced pathological changes. Indeed, FMT from SD human to GF mice significantly reduced SCFAs levels, promoted both peripheral and central inflammatory responses, and impaired cognitive function.647
Indole and its derivatives restore intestinal barrier integrity
Other major metabolites involved in the maintenance of intestinal barrier integrity are the indole and its derivatives, which are produced by bacterial fermentation of dietary tryptophan. Tryptophan is an essential amino acid obtained through dietary sources and acts as a biosynthetic precursor to several microbial and host metabolites, including indole.425,651 Notably, metagenomic analysis of PD gut microbiome has revealed a reduction in the tryptophan biosynthesis pathway.331 Several indole derivatives, including indole-3-ethanol, indole-3-pyruvate, and indole-3-aldehyde, maintain the integrity of the apical junctional complex and improve intestinal barrier integrity by activating the epithelial AHR (Fig. 5a).652 The AHR is a ligand-dependent transcription factor widely expressed in the intestinal microenvironment that has a profound influence on the preservation of intestinal barrier integrity.425 The AHR signaling participates in the regulation of epithelial cell differentiation and crypt stem cell proliferation. Dietary supplementation with indole-3-carbinol activates the AHR in intestinal epithelial cells to restore intestinal barrier integrity in mice infected with Citrobacter rodentium.653 Aside from its effects on epithelial AHR, dietary supplementation with indole-3-carbinol also triggers the activation of endothelial AHR in LPS-treated mice. This leads to reduced inflammatory activation in endothelial cells, resulting in improved gut vascular barrier integrity.654
A recent study has highlighted significant alterations in indole-producing bacteria in APP/PS1 mice, which were coupled with intestinal barrier dysfunction and cognitive dysfunction. Conversely, oral gavage with a mixture of indole, indole-3-acetic acid, and indole-3-propionic acid reversed the intestinal barrier impairment, reduced Aβ accumulation and tau hyperphosphorylation, leading to improved synaptic plasticity and cognitive function of APP/PS1 mice. In addition, the indoles inhibited microglial activation and NLRP3 inflammasome activation by activating microglial AHR.426
Blood–brain barrier (BBB) restoration by microbial metabolites
The BBB is a complex, multicellular, and dynamic interface crucial for maintaining CNS homeostasis and normal brain function. It stringently regulates the passage of countless molecules, including essential nutrients and deleterious xenobiotic molecules. It is also responsible for the removal of toxic metabolic waste products and endogenous endotoxins from the brain.655,656 Importantly, it acts as an immunological barrier by restricting the migration of immune cells into the brain.657 The BBB is composed of tightly packed, non-fenestrated endothelial cells linked by TJs and adherens junctions (AJs), which share a common basement membrane with pericytes and astrocytes. These cells, together with neurons and microglia, form the neurovascular unit (NVU) and collectively contribute to the maintenance of BBB integrity.655,656
However, the BBB undergoes progressive loss of integrity with age. Compromised BBB initiates the infiltration of peripheral immune cells and triggers microglial and astrocytic activation, ultimately resulting in synaptic and neuronal loss.658 The infiltration of blood-derived protein, fibrin, also induces microglial polarization to oxidative stress and neurodegenerative phenotypes in AD and multiple sclerosis mice.659 A recent single-nucleus RNA-sequencing analysis of the hippocampus and frontal cortex of AD patients found that 30 of the top 45 AD GWAS hits are expressed in brain vascular cells.85 Furthermore, BBB disruption is well characterized in other neurodegenerative diseases, including PD, ALS, FTD, and HD.658,660 The BBB disruption in neurodegenerative diseases makes it a clear target of intervention. Despite their anatomical separation, growing evidence has demonstrated the impact of intestinal perturbations on brain health. Ample evidence has shown that gut microbiota-derived metabolites are important regulators of BBB integrity,661,662,663,664,665 suggesting that the BBB may be an important communication interface between gut microbiota and glial cells.
Short-chain fatty acids improve BBB integrity
SCFAs are the most extensively studied microbial-derived metabolites with considerable influence on BBB integrity (Fig. 6a). An initial investigation revealed that GF mice exhibit increased BBB permeability and compromised TJs as compared to SPF mice. However, the BBB defects in GF mice were restored following mono-colonization with SCFA-producing bacteria or oral gavage with sodium butyrate.662 Similar trends were observed following gut microbiota depletion using antibiotic treatment. Antibiotic-treated mice displayed greater BBB permeability and reduced TJ proteins, which were partially ameliorated following FMT from SPF mice.665 Studies on in vitro BBB models have elucidated the protective mechanisms of SCFAs.663,666 Propionate was found to rescue LPS-induced impairment in the permeability of hCMEC/D3 monolayers by reducing oxidative stress.663 Another recent study on bEnd.3 endothelial cells demonstrated that butyrate and propionate improved BBB integrity by regulating the organization of the actin cytoskeleton and increasing the interaction between actin and TJ protein ZO-1. Moreover, butyrate and propionate reversed the LPS-induced reduction in ZO-1 and claudin-5 at the cell–cell junctions and restored the LPS-induced mitochondrial dysfunction.666 Further research is warranted to validate the impact of SCFAs on BBB integrity in neurodegenerative disease models.

Improving microbiota–gut–brain axis via the blood–brain barrier. a SCFAs and p-cresol glucuronide improve BBB integrity and prevent glial activation. b Elevated levels of trimethylamine-N-oxide (TMAO) has been reported in the plasma and cerebrospinal fluid of individuals with mild cognitive impairment, Alzheimer’s disease (AD) and Parkinson’s disease (PD). TMAO is detrimental to BBB integrity and induces glial activation. This figure was created with BioRender (https://biorender.com/)
Trimethylamine N-oxide disrupts BBB integrity
Another important metabolite that participates in the regulation of BBB integrity is TMAO (Fig. 6b). Studies have reported elevated levels of TMAO in the plasma and CSF of individuals with MCI, AD, and PD.409,410,667 Furthermore, the gene families involved in TMA production were found to be elevated in the PD gut microbiome, leading to increased choline metabolism and subsequent TMA production.331 In the brain, TMAO is detrimental to neuronal physiology as it induces neuronal senescence, oxidative stress, and alters synaptic plasticity.408,668,669,670 Moreover, TMAO is also associated with neuroinflammation by inducing microglial and astrocytic activation.411,412,413,414
However, conflicting results have emerged regarding the role of TMAO on BBB physiology. A recent study found that APP/PS1 mice fed with a TMAO-supplemented diet manifested reduced occludin and ZO-1 expression in the parietal cortex, along with increased microglial and astrocytic activation.414 The detrimental effects of TMAO on BBB integrity were also reported in chronic kidney disease patients, using resistance arteries in adipose tissue as a surrogate model of BBB.671 However, chronic low-dose TMAO supplementation has been found to prevent LPS-induced BBB alterations and memory impairment in C57Bl/6J mice.661 In an in vitro BBB model using hCMEC/D3 cells, it was demonstrated that TMA exposure increased paracellular permeability, whereas TMAO exposure enhanced barrier integrity and trans-endothelial electrical resistance (TEER).
The dichotomous roles of structurally similar TMA and TMAO are intriguing and demand further investigation on the choline-TMA-TMAO pathway. In the context of AD, choline is generally considered beneficial, as a low dietary choline intake has been associated with an elevated risk of dementia and AD.672 Lifelong choline supplementation has shown protective effects against Aβ pathology and microglial activation in female APP/PS1 mice.673 Conversely, choline deficiency has been found to exacerbate Aβ and tau pathologies in female 3xTg-AD mice.674 Furthermore, choline deficiency also impairs glucose metabolism in 3xTg-AD mice and non-transgenic control mice,674 which may be relevant to AD as hyperglycemia and diabetes are associated with BBB breakdown and dementia.675,676,677 More studies are needed to understand the mechanisms underlying the choline-TMA-TMAO pathway and determine the optimal gut microbiota manipulation that improves BBB integrity. Notably, a longitudinal study conducted on healthy men has identified ten gut microbial species that are associated with plasma TMAO concentration. Among them, the Bacteroidetes Alistipes shahii significantly strengthened the association between red meat/choline intake and plasma TMAO levels.678
p-Cresol glucuronide improves BBB integrity
Another bacterial metabolite involved in the regulation of BBB integrity is p-cresol glucuronide (pCG), which is a phenol derived from the microbial metabolism of phenylalanine and tyrosine (Fig. 6a).664 Recent evidence has demonstrated that pCG treatment improved the BBB integrity of male C57Bl/6 mice by upregulating several transporter-related pathways and downregulating inflammatory pathways. Subsequent examinations on hCMEC/D3 cells revealed that pCG counteracted the permeabilizing effects of LPS by inhibiting the TLR4 signaling.664 Future studies utilizing neurodegenerative disease models are necessary to examine the effects of pCG on BBB integrity during neurodegenerative disease pathology.
Meninges as an emerging target
The gut–meningeal axis has emerged as another significant aspect of the gut–brain interface that has garnered considerable attention. The meninges are a three-layered membranous barrier, comprising an outer dura mater, middle arachnoid mater, and inner pia mater, that cover the spinal cord and brain parenchyma. Rather than being an inert structural barrier, growing evidence indicates that the meninges actively participate in extensive neuro-immunological crosstalk with the brain. The meninges host diverse innate and adaptive immune cells, which are distributed in varying amounts across the distinct meningeal layers to enable tissue-specific functions.679 During aging, the meninges undergo a shift towards pro-inflammatory immune responses, characterized by an increased number of T cells and B cells.680,681,682,683 Dysfunctional meninges also represent an important contributor to neuroinflammation in neurodegenerative diseases by acting as a checkpoint for T-cell infiltration.684,685,686 Moreover, meningeal lymphatic dysfunction induces microglial activation and acquisition of DAM signature, accompanied by Aβ deposition in the brains of 5xFAD mice.687
While the causal relationship between the gut microbiome and meningeal impairment in neurodegenerative diseases has yet to be determined, recent evidence suggests that the microbiota-immune cell dialog has important implications on the meninges. A key mediator of the gut–meningeal axis is the mucosal-associated invariant T (MAIT) cells.688 MAIT cells are unconventional, innate-like T cells present abundantly in the human blood, liver, and mucosa. They recognize microbial-derived vitamin B2 (riboflavin) metabolites presented by non-polymorphic major histocompatibility complex (MHC) class I-related (MR1) molecule through their semi-invariant T-cell receptor (TCR).689,690 The development and maturation of MAIT cells are dependent on microbiota, as demonstrated by reduced numbers of MAIT cells in GF mice, which can be restored through microbial colonization.691,692,693 In addition, a stressed and unbalanced microbiota in HFD-fed obese mice decreased MAIT cell frequency by reducing MAIT cell agonist ligands. This is accompanied by increased gut inflammation, gut dysbiosis, and compromised gut integrity.694
The regulatory role of MAIT cells on meningeal barrier integrity and neuroinflammation was recently demonstrated following the detection of MAIT cells in the meninges and choroid plexus (CP) of mice.688 MAIT cells are responsible for preserving meningeal homeostasis by expressing antioxidant molecules, as evidenced by increased oxidative damage and impaired meningeal barrier integrity in MAIT cell-deficient Mr1–/– mice compared to their age-matched Mr1+/+ counterparts. Moreover, Mr1–/– mice exhibited extensive microgliosis in the cortex and hippocampus, along with impaired cognitive function. However, these defects were prevented by the adoptive transfer of MAIT cells from WT mice, supporting the protective functions of MAIT cells on the meningeal barrier.688 Future studies investigating the effects of gut microbiota manipulations on meningeal MAIT cells and meningeal barrier integrity using neurodegenerative disease mouse models are needed to ascertain the potential of targeting the MAIT cells.
Restoration of peripheral immune homeostasis
Dysregulation of the immune system, including innate and adaptive immune response, and the resultant chronic inflammation are important drivers of neurodegeneration.695 Although peripheral immune cells are generally thought to induce neurodegeneration after CNS infiltration, growing evidence suggests that they may influence neurodegenerative processes directly at the periphery.620
The gut microbiome participates in extensive bidirectional communication with the immune system.622,696 Notably, certain gut microbiota and their metabolites may stimulate or inhibit the differentiation of naive CD4+ T cells into TH17 cells. TH17 cells are highly abundant at mucosal barriers and play a critical role in regulating tissue homeostasis. A balanced ratio of Treg and TH17 cells is vital for maintaining an optimal intestinal immune system.697 Unhealthy dietary interventions, such as high-fat, high-sugar, and high-salt interventions, have been shown to negatively impact the gut microbiome and induce intestinal inflammation by impairing TH17 cell functions.698,699,700 In addition, the gut microbiome regulates the activity of retinoic acid receptor-related orphan receptor gamma-positive (RORγ+) Treg cells, dysregulation of which can lead to diseases such as colitis, food allergy, and colon cancer.622,701,702,703,704
The SCFAs have been shown to modulate the functions of various immune cells, including T cells, B cells, macrophages, dendritic cells, and innate lymphoid cells.274,705 For instance, butyrate has been shown to restrict inflammation by promoting the differentiation of Treg cells and supporting the functions of regulatory B cells.706,707,708 A recent study found that a short-term dietary switch from a fiber-rich diet to a low-fiber feast diet induced a transient depression of mucosal and systemic immunity by impairing the metabolic fitness of both mucosal and peripheral CD4+ T cells. These effects are associated with a rapid reduction of SCFAs, impaired mTOR activity, and mitochondrial function of CD4+ T cells.709 On the other hand, certain secondary BAs exert anti-inflammatory effects by stimulating Treg cell differentiation and inhibiting TH17 cell differentiation.361,363,364,702,710 While these findings shed light on the relationship between the gut microbiome and immune responses, further studies are needed to elucidate their intricate interplay in neurodegenerative diseases. Emerging evidence has underscored the neuroimmune role of Treg cells and TH17 cells in the gut–brain axis, extending to neuropathological conditions such as AD, suggesting potential areas for future research.621,622
Microbiome-based therapeutics
The alterations of the gut microbiome in neurodegenerative disease patients have prompted researchers to explore the clinical applications of microbiome-based therapeutics, including prebiotics, probiotics, and FMT. The rationale is further supported by several studies that demonstrated that the human gut microbiome is a major determinant of plasma metabolome, potentially playing a more dominant role than genetics.26,27,28
Probiotics and prebiotics
According to The International Scientific Association for Probiotics and Prebiotics (ISAPP), probiotics are defined as “live microorganisms that, when administered in adequate amounts, confer a health benefit on the host”.711 The administration of probiotics generally aims to introduce defined microbial strains to stimulate the health-promoting pathways in the microbiome and increase the production of beneficial metabolites.711,712 The development of new cultivation and sequencing technologies, along with the expansion of our knowledge of the human gut microbiota landscape has enabled probiotics to move beyond the traditional Lactobacillus and Bifidobacterium to the next-generation probiotics (NGPs). Examples of NGPs include A. muciniphila, Faecalibacterium prausnitzii, Roseburia intestinalis, and Bacteroides fragilis.712,713,714
On the other hand, a prebiotic is defined by ISAPP as “a substrate that is selectively utilized by host microorganisms conferring a health benefit”.715 Prebiotics are non-digestible substances that selectively stimulate the growth of beneficial bacteria to increase the production of associated metabolites. The most extensively documented prebiotics include inulin, FOS, and GOS, which promote the growth of Lactobacillus or Bifidobacterium spp. Nevertheless, technological advances have expanded the targets of prebiotics beyond Lactobacillus and Bifidobacterium, to other newly identified health-promoting gut microbes, such as Roseburia, Eubacterium, and Faecalibacterium spp.715,716
In this section, we have summarized the findings of in vivo studies investigating the applications of probiotics (Table 1) and prebiotics (Table 2) in neurodegenerative disease models, with a specific focus on their effects on the intestinal barrier, BBB, and glial cells. Overall, these studies have consistently demonstrated the beneficial effects of probiotics and prebiotics on cognitive functions in animal models. This is attributed to their ability to enhance the integrity of the intestinal barrier and BBB, while also promoting the resolution of intestinal inflammation. Probiotics and prebiotics prove effective in re-establishing beneficial gut microbiota and restoring metabolic functions, particularly the SCFA-producing species, resulting in elevated levels of SCFAs. The alleviation of biological barrier impairments, coupled with the reduction in systemic LPS levels, attenuates systemic inflammation and glial activation, resulting in reduced neurodegenerative disease pathology. Furthermore, the administration of probiotics and prebiotics appears to contribute to the restoration of neurotransmitter systems in neurodegenerative disease models.
In addition, these studies have reported an increased relative abundance of beneficial gut microbes and a reduced relative abundance of harmful gut microbes. However, concerns have been raised regarding the limitations and risk of bias associated with the use of relative abundance due to technical variation, which may lead to erroneous interpretations founded on proportional profiling.717,718 The changes in the relative abundance of a microbial taxon are inherently compensated by equivalent increases or decreases in the remaining taxa, resulting in correlation biases that adversely affect downstream analysis.719 Bias can be introduced throughout all stages of sample processing, including the selection of sample collection protocol, preservative choice, storage temperature, DNA extraction protocol, library preparation, sequencing platform, and bioinformatics analysis pipeline.720,721,722,723 Thus, it is recommended to harmonize study protocols and incorporate quantitative microbiome profiling (absolute quantification) to complement relative abundance data.718,719,722 This approach will provide a more comprehensive understanding of the impact of microbiome-based therapeutics on the gut microbiome and neurodegenerative disease pathologies.
Fecal microbiota transplantation (FMT)
FMT refers to the procedure of transferring a healthy donor’s microbial ecosystem to the gastrointestinal tract of a recipient, with the aim of modifying the recipient’s gut microbiome and treating diseases associated with gut dysbiosis.724,725 It is considered a broad and largely untargeted gut microbiota modulation strategy, pitting donor microbial ecosystems against those of the recipient.726 However, FMT has undergone significant advancements over the years, evolving from a relatively crude procedure of transferring fresh donor stool to a mainstream treatment option. This evolution is enabled by the development of standardized FMT products, with increased emphasis on their pharmaceutical formulation, pharmacokinetics, pharmacodynamics, and toxicity.727 A key strength of FMT is the ability to transfer both the favorable microbes, as well as their intricate supporting ecosystem.726 FMT is most commonly applied in the management of Clostridioides difficile infection and inflammatory bowel disease.728 Notably, FMT has achieved a significant milestone following the recent FDA approval of RBX2660 (Rebyota) and SER-109 (Vowst) for the prevention of recurrent Clostridioides difficile infection, highlighting the therapeutic potential of FMT in human disease.729,730,731 Interestingly, SER-109 is an oral microbiome therapeutic administered in the capsule dosage form, eliminating the need for endoscopy procedures.729
As discussed previously, the development of neurodegenerative diseases and glial activation are linked to gut dysbiosis and altered microbial metabolites. In this section, we summarize the findings of in vivo studies investigating the applications of FMT in neurodegenerative diseases (Table 3). In general, FMT from either healthy control mice or WT mice has demonstrated promising results in correcting gut dysbiosis and alleviating neurodegeneration in recipient mouse models of neurodegenerative diseases. These positive outcomes can be ascribed to the promotion of beneficial microbes and the elevation of beneficial metabolites, resulting in the restoration of intestinal barrier and BBB integrity. Consequently, the recipient mice exhibited a reduction in systemic inflammation, glial activation, and neuroinflammation.
Interestingly, the rearing environment of FMT donors may influence the outcomes of FMT. A recent study showed that FMT from C9orf72-mutant mice housed at pro-survival environment (Broad Institute) effectively ameliorated the autoimmune inflammatory phenotypes in recipient C9orf72-mutant mice, whereas FMT from those housed at pro-inflammatory environment (Harvard Institute) did not improve these phenotypes.57 In addition, the extent of FMT engraftment in recipient mice may be influenced by the gender of the recipients. This phenomenon is exemplified in the R6/1 HD mouse model, wherein male R6/1 mice exhibited greater resistance to FMT engraftment when compared to female R6/1 mice. Consequently, the cognitive function of male R6/1 mice showed no discernible improvement as compared to their female counterparts.244 Similar sexual dimorphism of FMT has been reported in a clinical trial of irritable bowel syndrome, wherein female patients exhibit significantly better responses compared to male patients.732 Thus, future studies should account for these factors, given their substantial influence on the efficacy and response to FMT.
Clinical applications of microbiome-based therapeutics
Given the accumulating evidence demonstrating the potential of gut microbiota manipulations on cognitive functions, intense interest exists in applying microbiome-based therapeutics to alleviate the progression of neurodegenerative diseases. In this section, we summarize the completed and ongoing clinical trials in Tables 4 and 5, respectively. A recent study has identified constipation/bowel movement frequency as a potential mediator in the relationship between gut microbiota and the likelihood of developing prodromal PD.52 Thus, we have included clinical trials that investigated the effects on constipation as an endpoint.
Despite advancements in understanding gut–brain communications and encouraging results in experimental models, the clinical trials of microbiome-based therapeutics have yielded mixed results. Microbiome-based therapeutics generally exhibit favorable safety and tolerability profiles, alongside some degree of clinical efficacy in improving cognitive function and motor function. Moreover, these interventions are effective in improving constipation-related parameters in PD patients, including bowel movements, gut transit time, stool consistency, and quality of life related to constipation. However, a notable limitation of the current studies is the lack of gut microbiome profiling, which hinders the comparison of taxonomic composition and metabolomic profiles before and after the intervention. In addition, there is a paucity of clinical trials exploring microbiome-based therapeutics in ALS, FTD, and HD.
The application of probiotics and prebiotics is limited by several factors. A notable limitation of probiotics is their suboptimal therapeutic efficacy, primarily attributed to their vulnerability to low gastric pH and exposure to various digestive enzymes, resulting in probiotic inactivation and impaired bioactivity. In addition, the low mucoadhesive capability and insufficient intestinal retention of probiotics greatly hamper their intestinal colonization.733 Several innovative strategies are currently being investigated to enhance the oral bioavailability and intestinal targeting capability of probiotics. These include microencapsulation,734 hydrogel encapsulation,735 nanoparticle encapsulation,733 integration with nanozyme,736 nanocoating,737 mineral coating,738 and optogenetic probiotic system.739,740
On the other hand, it is difficult to predict and control the outcomes of microbiome manipulation by prebiotics, as they affect the growth and activities of multiple microbial species.728,741 Nevertheless, ongoing efforts are focused on understanding how dietary fibers with different structures modulate the human gut microbiome.742 Furthermore, growing studies are exploring the strategy of precision manipulation of the gut microbiota to achieve specific health endpoints.742,743,744,745 Moreover, the effectiveness of prebiotics varies depending on the native microbiota. Prebiotics are more likely to be effective when the necessary bacterial strains are already present in the gut. However, these beneficial strains may be lacking in certain medical conditions. Thus, it is essential to administer the missing symbiotic bacterial strains to restore and support a healthy gut microbiota.728 A refined understanding of the mechanisms governing the metabolism and absorption of prebiotics will inform the rational design of clinical trials.
Another notable trend is the limited number of trials investigating the applications of FMT in the treatment of neurodegenerative diseases. An evident gap in our understanding pertains to the “ideal” gut microbiome that resists neurodegeneration, thereby significantly complicating the identification of the most suitable donors. Similar limitations are common in the application of FMT in anticancer treatment. Interestingly, increasing studies are investigating the use of fecal samples from cancer patients who exhibited complete and durable responses to immune checkpoint inhibitors.726 This approach holds promise for investigation in the context of neurodegenerative diseases.
In addition, FMT is limited by its variable degree of engraftment, limited scalability, and a lack of standardized protocols.725,726 The extent of microbial engraftment is an important proxy of the clinical success of FMT.746,747 To improve FMT engraftment, antibiotic preconditioning for the recipient has been recommended, based on two recent meta-analyses that reported a higher degree of donor microbiome engraftment in recipients with a precarious microbial community.746,747,748 Moreover, combined routes of FMT administration are an effective strategy to enhance strain engraftment.747 Another point of consideration is the necessity of repeated FMT administration to counteract the persistent disease-driving forces that affect the gut ecological niche and achieve long-term efficacy. This clinical requirement underscores the critical need for a standardized FMT product with excellent stability and reproducibility.746 FMT may also carry the risk of unintended introduction of pathogens and antibiotic-resistant microbes into the recipients.749,750 Thus, it is imperative to screen the donor samples prior to the FMT to mitigate these risks.728
Conclusions
The unabated rise in the global prevalence of neurodegenerative diseases,751 coupled with the suboptimal therapeutic outcomes of current FDA-approved drugs, emphasize the need for an alternative strategy to discover effective therapeutic targets. The microbiota–gut–brain axis represents an important regulator of glial functions, making it an actionable target for ameliorating the development and progression of neurodegenerative diseases. With the aid of cutting-edge technologies, we are beginning to delineate the intricate communication between gut microbiota and glial cells in neurodegenerative diseases. A dysregulated gut microbiome adversely affects glial cells by compromising the integrity of the intestinal barrier and BBB, with recent evidence also revealing the involvement of the meningeal barrier. The available preclinical evidence supports the use of probiotics, prebiotics, and FMT to attenuate glial activation and cognitive impairment by restoring the integrity of the intestinal barrier and BBB. Nevertheless, the clinical translation of microbiome-based therapeutics remains challenging, underscoring the need for continued research efforts to unravel the complexities of the microbiota–gut–brain axis and fully harness their potential.
Establishing a definitive causal relationship between the altered gut microbiome and disease remains challenging as it is difficult to determine whether the observed microbiome alterations are causative, consequential, or merely a bystander response to the disease. Moreover, existing animal models do not fully recapitulate the intricacies of the human microbiome and pathobiology. Thus, the excessively high rate (95%) of positive results and causal claims in human microbiota-associated rodents demands caution against overinterpreting and overstating the causal implications of these findings.752 Nevertheless, animal models, when combined with single-cell technologies and computational techniques, remain essential complementary tools as they offer valuable mechanistic insights that are difficult to obtain through human studies.
The increasing maturity of technical and methodological innovations has enabled us to unravel numerous aspects of the microbiota–gut–brain axis and discover opportunities for therapeutic development. Notably, the recent development of non-invasive, ingestible sampling devices has made it possible to collect luminal contents throughout the intestinal tract, potentially overcoming the limitations of stool samples in reflecting the regional variation of gut microbiota.354,753 In addition, we are beginning to understand the intricate cell-to-cell signaling mechanisms of gut microbiota that regulate community behaviors.754 Ultimately, the malleability of the human gut microbiome presents exciting opportunities for the development of personalized microbiome-based therapeutics for neurodegenerative diseases.
References
Caballero-Flores, G., Pickard, J. M. & Núñez, G. Microbiota-mediated colonization resistance: mechanisms and regulation. Nat. Rev. Microbiol. 21, 347–360 (2023).
Hoegenauer, C., Hammer, H. F., Mahnert, A. & Moissl-Eichinger, C. Methanogenic archaea in the human gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 19, 805–813 (2022).
Miyauchi, E. et al. The impact of the gut microbiome on extra-intestinal autoimmune diseases. Nat. Rev. Immunol. 23, 9–23 (2023).
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010).
Almeida, A. et al. A new genomic blueprint of the human gut microbiota. Nature 568, 499–504 (2019).
Lynch, S. V. & Pedersen, O. The human intestinal microbiome in health and disease. New Engl. J. Med. 375, 2369–2379 (2016).
Donald, K. & Finlay, B. B. Early-life interactions between the microbiota and immune system: impact on immune system development and atopic disease. Nat. Rev. Immunol. 23, 735–748 (2023).
Kennedy, K. M. et al. Fetal meconium does not have a detectable microbiota before birth. Nat. Microbiol. 6, 865–873 (2021).
Bäckhed, F. et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690–703 (2015).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012).
Stewart, C. J. et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588 (2018).
Roswall, J. et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe 29, 765–776.e763 (2021).
Jumpertz, R. et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 94, 58–65 (2011).
Schluter, J. et al. The gut microbiota is associated with immune cell dynamics in humans. Nature 588, 303–307 (2020).
Ghosh, S., Whitley, C. S., Haribabu, B. & Jala, V. R. Regulation of intestinal barrier function by microbial metabolites. Cell Mol. Gastroenterol. Hepatol. 11, 1463–1482 (2021).
Zhang, W. et al. Gut-innervating nociceptors regulate the intestinal microbiota to promote tissue protection. Cell 185, 4170–4189.e4120 (2022).
Haghikia, A. et al. Propionate attenuates atherosclerosis by immune-dependent regulation of intestinal cholesterol metabolism. Eur. Heart J. 43, 518–533 (2022).
Le, H. H. et al. Characterization of interactions of dietary cholesterol with the murine and human gut microbiome. Nat. Microbiol. 7, 1390–1403 (2022).
Yao, L. et al. A biosynthetic pathway for the selective sulfonation of steroidal metabolites by human gut bacteria. Nat. Microbiol. 7, 1404–1418 (2022).
Collins, S. L. et al. Bile acids and the gut microbiota: metabolic interactions and impacts on disease. Nat. Rev. Microbiol. 21, 236–247 (2023).
Sato, Y. et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 599, 458–464 (2021).
Sberro, H. et al. Large-scale analyses of human microbiomes reveal thousands of small, novel genes. Cell 178, 1245–1259.e1214 (2019).
Ma, Y. et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat. Biotechnol. 40, 921–931 (2022).
Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R. & Goodman, A. L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467 (2019).
Javdan, B. et al. Personalized mapping of drug metabolism by the human gut microbiome. Cell 181, 1661–1679.e1622 (2020).
Chen, L. et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat. Med. 28, 2333–2343 (2022).
Visconti, A. et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 10, 4505 (2019).
Diener, C. et al. Genome–microbiome interplay provides insight into the determinants of the human blood metabolome. Nat. Metab. 4, 1560–1572 (2022).
López-Otín, C. et al. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Ahmed, H. et al. Microbiota-derived metabolites as drivers of gut-brain communication. Gut Microbes 14, 2102878 (2022).
Morais, L. H., Schreiber, H. L. & Mazmanian, S. K. The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 19, 241–255 (2021).
Liu, L., Huh, J. R. & Shah, K. Microbiota and the gut-brain-axis: implications for new therapeutic design in the CNS. EBioMedicine 77, 103908 (2022).
Yano, JessicaM. et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161, 264–276 (2015).
Zhang, Z.-W. et al. Morinda officinalis oligosaccharides increase serotonin in the brain and ameliorate depression via promoting 5-hydroxytryptophan production in the gut microbiota. Acta Pharm. Sin. B 12, 3298–3312 (2022).
Engevik, M. A. et al. Human-derived Bifidobacterium dentium modulates the mammalian serotonergic system and gut–brain axis. Cell Mol. Gastroenterol. Hepatol. 11, 221–248 (2021).
Wang, Y. et al. Oral berberine improves brain dopa/dopamine levels to ameliorate Parkinson’s disease by regulating gut microbiota. Signal Transduct. Target Ther. 6, 77 (2021).
Strandwitz, P. et al. GABA-modulating bacteria of the human gut microbiota. Nat. Microbiol. 4, 396–403 (2019).
Buffington, S. A. et al. Dissecting the contribution of host genetics and the microbiome in complex behaviors. Cell 184, 1740–1756.e1716 (2021).
Needham, B. D. et al. A gut-derived metabolite alters brain activity and anxiety behaviour in mice. Nature 602, 647–653 (2022).
Wu, W.-L. et al. Microbiota regulate social behaviour via stress response neurons in the brain. Nature 595, 409–414 (2021).
Sharon, G. et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 177, 1600–1618.e1617 (2019).
Morton, J. T. et al. Multi-level analysis of the gut-brain axis shows autism spectrum disorder-associated molecular and microbial profiles. Nat. Neurosci. 26, 1208–1217 (2023).
Zhang, Y. et al. Bacteroides species differentially modulate depression-like behavior via gut-brain metabolic signaling. Brain Behav. Immun. 102, 11–22 (2022).
Mayneris-Perxachs, J. et al. Microbiota alterations in proline metabolism impact depression. Cell Metab. 34, 681–701.e610 (2022).
Bosch, J. A. et al. The gut microbiota and depressive symptoms across ethnic groups. Nat. Commun. 13, 7129 (2022).
Radjabzadeh, D. et al. Gut microbiome-wide association study of depressive symptoms. Nat. Commun. 13, 7128 (2022).
Valles-Colomer, M. et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 4, 623–632 (2019).
Dohnalová, L. et al. A microbiome-dependent gut-brain pathway regulates motivation for exercise. Nature 612, 739–747 (2022).
Fontana, F. et al. The human gut microbiome of athletes: metagenomic and metabolic insights. Microbiome 11, 27 (2023).
Scheiman, J. et al. Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat. Med. 25, 1104–1109 (2019).
Ferreiro, A. L. et al. Gut microbiome composition may be an indicator of preclinical Alzheimer’s disease. Sci. Transl. Med. 15, eabo2984 (2023).
Huang, B. et al. Gut microbiome dysbiosis across early Parkinson’s disease, REM sleep behavior disorder and their first-degree relatives. Nat. Commun. 14, 2501 (2023).
Palacios, N. et al. Metagenomics of the gut microbiome in Parkinson’s disease: prodromal changes. Ann. Neurol. 94, 486–501 (2023).
Dodiya, H. B. et al. Gut microbiota-driven brain Aβ amyloidosis in mice requires microglia. J. Exp. Med. 219, e20200895 (2022).
Sampson, T. R. et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480.e1412 (2016).
Seo, D.-o et al. ApoE isoform- and microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy. Science 379, eadd1236 (2023).
Burberry, A. et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 582, 89–94 (2020).
Wilson, D. M. et al. Hallmarks of neurodegenerative diseases. Cell 186, 693–714 (2023).
Cook, J. & Prinz, M. Regulation of microglial physiology by the microbiota. Gut Microbes 14, 2125739 (2022).
Zhou, R. et al. Microbiota-microglia connections in age-related cognition decline. Aging Cell 21, e13599 (2022).
Chandra, S. et al. The gut microbiome regulates astrocyte reaction to Aβ amyloidosis through microglial dependent and independent mechanisms. Mol. Neurodegener. 18, 45 (2023).
Keogh, C. E. et al. Myelin as a regulator of development of the microbiota-gut-brain axis. Brain Behav. Immun. 91, 437–450 (2021).
Borst, K., Dumas, A. A. & Prinz, M. Microglia: immune and non-immune functions. Immunity 54, 2194–2208 (2021).
Prinz, M., Masuda, T., Wheeler, M. A. & Quintana, F. J. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu. Rev. Immunol. 39, 251–277 (2021).
Paolicelli, R. C. et al. Microglia states and nomenclature: a field at its crossroads. Neuron 110, 3458–3483 (2022).
Diaz-Aparicio, I. et al. Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J. Neurosci. 40, 1453–1482 (2020).
Sierra, A. et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495 (2010).
Dudvarski Stankovic, N. et al. Microglia-blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol. 131, 347–363 (2016).
Ronaldson, P. T. & Davis, T. P. Regulation of blood-brain barrier integrity by microglia in health and disease: a therapeutic opportunity. J. Cereb. Blood Flow. Metab. 40, S6–s24 (2020).
Favuzzi, E. et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 184, 4048–4063.e4032 (2021).
Faust, T. E., Gunner, G. & Schafer, D. P. Mechanisms governing activity-dependent synaptic pruning in the developing mammalian CNS. Nat. Rev. Neurosci. 22, 657–673 (2021).
Pascual, O. et al. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 109, E197–E205 (2012).
Basilico, B. et al. Microglia control glutamatergic synapses in the adult mouse hippocampus. Glia 70, 173–195 (2022).
Corsi, G. et al. Microglia modulate hippocampal synaptic transmission and sleep duration along the light/dark cycle. Glia 70, 89–105 (2022).
McNamara, N. B. et al. Microglia regulate central nervous system myelin growth and integrity. Nature 613, 120–129 (2023).
Kent, S. A. & Miron, V. E. Microglia regulation of central nervous system myelin health and regeneration. Nat. Rev. Immunol. 24, 49–63 (2024).
Huang, Y. et al. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 22, 586–594 (2021).
Rudolph, M. et al. Microglia-mediated phagocytosis of apoptotic nuclei is impaired in the adult murine hippocampus after stroke. Glia 69, 2006–2022 (2021).
Augusto-Oliveira, M. et al. Plasticity of microglia. Biol. Rev. Camb. Philos. Soc. 97, 217–250 (2022).
Prinz, M., Jung, S. & Priller, J. Microglia biology: one century of evolving concepts. Cell 179, 292–311 (2019).
Zeng, H. et al. Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer’s disease. Nat. Neurosci. 26, 430–446 (2023).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Yang, A. C. et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892 (2022).
Smajić, S. et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 145, 964–978 (2021).
Langston, R. G. et al. Association of a common genetic variant with Parkinson’s disease is mediated by microglia. Sci. Transl. Med. 14, eabp8869 (2022).
Agarwal, D. et al. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 11, 4183 (2020).
Humphrey, J. et al. Integrative transcriptomic analysis of the amyotrophic lateral sclerosis spinal cord implicates glial activation and suggests new risk genes. Nat. Neurosci. 26, 150–162 (2023).
Crotti, A. & Glass, C. K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 36, 364–373 (2015).
Bright, F. et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 15, 540–555 (2019).
Gao, C., Jiang, J., Tan, Y. & Chen, S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct. Target Ther. 8, 359 (2023).
Deczkowska, A., Amit, I. & Schwartz, M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 21, 779–786 (2018).
Podleśny-Drabiniok, A., Marcora, E. & Goate, A. M. Microglial phagocytosis: a disease-associated process emerging from Alzheimer’s disease genetics. Trends Neurosci. 43, 965–979 (2020).
Odfalk, K. F., Bieniek, K. F. & Hopp, S. C. Microglia: friend and foe in tauopathy. Prog. Neurobiol. 216, 102306 (2022).
Scheiblich, H. et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 184, 5089–5106.e5021 (2021).
Brown, G. C. Cell death by phagocytosis. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-023-00921-6 (2023).
Haure-Mirande, J.-V., Audrain, M., Ehrlich, M. E. & Gandy, S. Microglial TYROBP/DAP12 in Alzheimer’s disease: transduction of physiological and pathological signals across TREM2. Mol. Neurodegener. 17, 55 (2022).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e1217 (2017).
Krasemann, S. et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e569 (2017).
Ennerfelt, H. et al. SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell 185, 4135–4152.e4122 (2022).
Wang, S. et al. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 185, 4153–4169.e4119 (2022).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Illes, P. et al. Regulation of microglial functions by purinergic mechanisms in the healthy and diseased CNS. Cells 9, 1108 (2020).
Pluvinage, J. V. et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192 (2019).
Aires, V. et al. CD22 blockage restores age-related impairments of microglia surveillance capacity. Front. Immunol. 12, 684430 (2021).
Eskandari-Sedighi, G., Jung, J. & Macauley, M. S. CD33 isoforms in microglia and Alzheimer’s disease: friend and foe. Mol. Asp. Med. 90, 101111 (2023).
Hu, J. et al. Microglial Piezo1 senses Aβ fibril stiffness to restrict Alzheimer’s disease. Neuron 111, 15–29.e18 (2023).
Jäntti, H. et al. Microglial amyloid beta clearance is driven by PIEZO1 channels. J. Neuroinflamm. 19, 147 (2022).
Murdock, M. H. & Tsai, L.-H. Insights into Alzheimer’s disease from single-cell genomic approaches. Nat. Neurosci. 26, 181–195 (2023).
Yin, Z. et al. Identification of a protective microglial state mediated by miR-155 and interferon-γ signaling in a mouse model of Alzheimer’s disease. Nat. Neurosci. 26, 1196–1207 (2023).
Gulen, M. F. et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 620, 374–380 (2023).
Decout, A., Katz, J. D., Venkatraman, S. & Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21, 548–569 (2021).
Choi, I. et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat. Cell Biol. 25, 963–974 (2023).
Depp, C. et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 618, 349–357 (2023).
Yoo, Y. et al. A cell therapy approach to restore microglial Trem2 function in a mouse model of Alzheimer’s disease. Cell Stem Cell 30, 1043–1053.e1046 (2023).
Kwon, H. S. & Koh, S.-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 9, 42 (2020).
Albrecht, D. S. et al. Early neuroinflammation is associated with lower amyloid and tau levels in cognitively normal older adults. Brain Behav. Immun. 94, 299–307 (2021).
Fan, Z., Brooks, D. J., Okello, A. & Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 140, 792–803 (2017).
Hamelin, L. et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264 (2016).
Spiller, K. J. et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 21, 329–340 (2018).
Sobue, A. et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol. Commun. 9, 1 (2021).
Olah, M. et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 9, 539 (2018).
Gerrits, E. et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 141, 681–696 (2021).
Hickman, S. et al. Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369 (2018).
Leng, F. & Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172 (2021).
Malpetti, M. et al. Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain 143, 1588–1602 (2020).
Johnson, E. C. B. et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 26, 769–780 (2020).
Tansey, M. G. et al. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 22, 657–673 (2022).
Rocha, N. P. et al. Microglia activation in basal ganglia is a late event in Huntington disease pathophysiology. Neurol. Neuroimmunol. Neuroinflamm. 8, e984 (2021).
Wilton, D. K. & Stevens, B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol. Dis. 143, 104963 (2020).
Bevan-Jones, W. R. et al. Neuroinflammation and protein aggregation co-localize across the frontotemporal dementia spectrum. Brain 143, 1010–1026 (2020).
Dols-Icardo, O. et al. Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 7, e829 (2020).
Beers, D. R. & Appel, S. H. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 18, 211–220 (2019).
Tam, O. H. et al. Postmortem cortex samples identify distinct molecular subtypes of ALS: retrotransposon activation, oxidative stress, and activated glia. Cell Rep. 29, 1164–1177.e1165 (2019).
Jin, M. et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 12, 6565 (2021).
Didonna, A. Tau at the interface between neurodegeneration and neuroinflammation. Genes Immun. 21, 288–300 (2020).
Dani, M. et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141, 2740–2754 (2018).
Finze, A. et al. Individual regional associations between Aβ-, tau- and neurodegeneration (ATN) with microglial activation in patients with primary and secondary tauopathies. Mol. Psychiatry https://doi.org/10.1038/s41380-023-02188-8 (2023).
De, S. et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 10, 1541 (2019).
Bido, S. et al. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 12, 6237 (2021).
Garcia, P. et al. Neurodegeneration and neuroinflammation are linked, but independent of alpha-synuclein inclusions, in a seeding/spreading mouse model of Parkinson’s disease. Glia 70, 935–960 (2022).
Sterling, J. K. et al. Interleukin-6 triggers toxic neuronal iron sequestration in response to pathological α-synuclein. Cell Rep. 38, 110358 (2022).
Tu, H. Y. et al. α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 20, e13522 (2021).
Crotti, A. et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 17, 513–521 (2014).
Zamudio, F. et al. TDP-43 mediated blood-brain barrier permeability and leukocyte infiltration promote neurodegeneration in a low-grade systemic inflammation mouse model. J. Neuroinflamm. 17, 283 (2020).
Roberts, K. et al. Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia 61, 409–419 (2013).
Zhao, W. et al. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 58, 231–243 (2010).
Deora, V. et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 68, 407–421 (2020).
Ajmone-Cat, M. A. et al. Increased FUS levels in astrocytes leads to astrocyte and microglia activation and neuronal death. Sci. Rep. 9, 4572 (2019).
Pomilio, C. et al. Microglial autophagy is impaired by prolonged exposure to β-amyloid peptides: evidence from experimental models and Alzheimer’s disease patients. GeroScience 42, 613–632 (2020).
Xu, Y. et al. Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proc. Natl. Acad. Sci. USA 118, e2023418118 (2021).
Houtman, J. et al. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J. 38, e99430 (2019).
Festa, B. P. et al. Microglial-to-neuronal CCR5 signaling regulates autophagy in neurodegeneration. Neuron 111, 2021–2037.e2012 (2023).
Choi, I. et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 11, 1386 (2020).
Cheng, J. et al. Microglial autophagy defect causes Parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 16, 2193–2205 (2020).
Qin, Y. et al. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease. Brain Behav. Immun. 91, 324–338 (2021).
Banerjee, P. et al. Cell-autonomous immune dysfunction driven by disrupted autophagy in C9orf72-ALS iPSC-derived microglia contributes to neurodegeneration. Sci. Adv. 9, eabq0651 (2023).
Brelstaff, J. H. et al. Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci. Adv. 7, eabg4980 (2021).
d’Errico, P. et al. Microglia contribute to the propagation of Aβ into unaffected brain tissue. Nat. Neurosci. 25, 20–25 (2022).
George, S. et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 14, 34 (2019).
Pascoal, T. A. et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 27, 1592–1599 (2021).
Guo, M. et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain 143, 1476–1497 (2020).
Svahn, A. J. et al. Nucleo-cytoplasmic transport of TDP-43 studied in real time: impaired microglia function leads to axonal spreading of TDP-43 in degenerating motor neurons. Acta Neuropathol. 136, 445–459 (2018).
Chen, X. et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 615, 668–677 (2023).
Wang, N. et al. Opposing effects of apoE2 and apoE4 on microglial activation and lipid metabolism in response to demyelination. Mol. Neurodegener. 17, 75 (2022).
Tcw, J. et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 185, 2213–2233.e2225 (2022).
Liu, C.-C. et al. Peripheral apoE4 enhances Alzheimer’s pathology and impairs cognition by compromising cerebrovascular function. Nat. Neurosci. 25, 1020–1033 (2022).
Montagne, A. et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 581, 71–76 (2020).
Sun, N. et al. Single-nucleus multiregion transcriptomic analysis of brain vasculature in Alzheimer’s disease. Nat. Neurosci. 26, 970–982 (2023).
Blanchard, J. W. et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611, 769–779 (2022).
Victor, M. B. et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell 29, 1197–1212.e1198 (2022).
Raulin, A.-C. et al. ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies. Mol. Neurodegener. 17, 72 (2022).
Shi, Y. et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 216, 2546–2561 (2019).
Shi, Y. et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017).
Mahan, T. E. et al. Selective reduction of astrocyte apoE3 and apoE4 strongly reduces Aβ accumulation and plaque-related pathology in a mouse model of amyloidosis. Mol. Neurodegener. 17, 13 (2022).
Koutsodendris, N. et al. Neuronal APOE4 removal protects against tau-mediated gliosis, neurodegeneration and myelin deficits. Nat. Aging 3, 275–296 (2023).
Ferrari-Souza, J. P. et al. APOEε4 associates with microglial activation independently of Aβ plaques and tau tangles. Sci. Adv. 9, eade1474 (2023).
Lee, S. et al. APOE modulates microglial immunometabolism in response to age, amyloid pathology, and inflammatory challenge. Cell Rep. 42, 112196 (2023).
Davis, A. A. et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 12, eaay3069 (2020).
Zhao, N. et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 12, eaay1809 (2020).
Giannakopoulos, P. et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500 (2003).
Jorfi, M. et al. Infiltrating CD8 + T cells exacerbate Alzheimer’s disease pathology in a 3D human neuroimmune axis model. Nat. Neurosci. 26, 1489–1504 (2023).
Williams, G. P. et al. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain 144, 2047–2059 (2021).
Garofalo, S. et al. Blocking immune cell infiltration of the central nervous system to tame neuroinflammation in amyotrophic lateral sclerosis. Brain Behav. Immun. 105, 1–14 (2022).
Lynch, C. M. K. et al. Critical windows of early-life microbiota disruption on behaviour, neuroimmune function, and neurodevelopment. Brain Behav. Immun. 108, 309–327 (2023).
Abdel-Haq, R., Schlachetzki, J. C. M., Glass, C. K. & Mazmanian, S. K. Microbiome-microglia connections via the gut-brain axis. J. Exp. Med. 216, 41–59 (2019).
Mossad, O. & Erny, D. The microbiota-microglia axis in central nervous system disorders. Brain Pathol. 30, 1159–1177 (2020).
Huang, Y. et al. The gut microbiome modulates the transformation of microglial subtypes. Mol. Psychiatry 28, 1611–1621 (2023).
Erny, D. et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 33, 2260–2276.e2267 (2021).
Erny, D. et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18, 965–977 (2015).
Matsudaira, T. et al. Cellular senescence in white matter microglia is induced during ageing in mice and exacerbates the neuroinflammatory phenotype. Commun. Biol. 6, 665 (2023).
Çalışkan, G. et al. Antibiotic-induced gut dysbiosis leads to activation of microglia and impairment of cholinergic gamma oscillations in the hippocampus. Brain Behav. Immun. 99, 203–217 (2022).
Liu, X. et al. High-fiber diet mitigates maternal obesity-induced cognitive and social dysfunction in the offspring via gut-brain axis. Cell Metab. 33, 923–938.e926 (2021).
Teng, Y. et al. Gut bacterial isoamylamine promotes age-related cognitive dysfunction by promoting microglial cell death. Cell Host Microbe 30, 944–960.e948 (2022).
Chen, C. et al. Gut microbiota regulate Alzheimer’s disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut 71, 2233–2252 (2022).
Dodiya, H. B. et al. Sex-specific effects of microbiome perturbations on cerebral Aβ amyloidosis and microglia phenotypes. J. Exp. Med. 216, 1542–1560 (2019).
Harach, T. et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 7, 41802 (2017).
Colombo, A. V. et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 10, e59826 (2021).
Olszak, T. et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493 (2012).
Tamburini, S., Shen, N., Wu, H. C. & Clemente, J. C. The microbiome in early life: implications for health outcomes. Nat. Med. 22, 713–722 (2016).
Chu, C. et al. The microbiota regulate neuronal function and fear extinction learning. Nature 574, 543–548 (2019).
Wang, X. et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 29, 787–803 (2019).
Chen, C. et al. Gut dysbiosis contributes to amyloid pathology, associated with C/EBPβ/AEP signaling activation in Alzheimer’s disease mouse model. Sci. Adv. 6, eaba0466 (2020).
Mezö, C. et al. Different effects of constitutive and induced microbiota modulation on microglia in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 8, 119 (2020).
Oakley, H. et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006).
Forner, S. et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci. Data 8, 270 (2021).
Hou, Y. et al. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581 (2019).
Pang, S. et al. Longevity of centenarians is reflected by the gut microbiome with youth-associated signatures. Nat. Aging 3, 436–449 (2023).
Boehme, M. et al. Microbiota from young mice counteracts selective age-associated behavioral deficits. Nat. Aging 1, 666–676 (2021).
Peng, L., Bestard-Lorigados, I. & Song, W. The synapse as a treatment avenue for Alzheimer’s disease. Mol. Psychiatry 27, 2940–2949 (2022).
Dejanovic, B. et al. Changes in the synaptic proteome in tauopathy and rescue of Tau-induced synapse loss by C1q antibodies. Neuron 100, 1322–1336.e1327 (2018).
Dejanovic, B. et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2, 837–850 (2022).
Warnecke, T. et al. Gastrointestinal involvement in Parkinson’s disease: pathophysiology, diagnosis, and management. NPJ Parkinsons Dis. 8, 31 (2022).
Cannon, T. & Gruenheid, S. Microbes and Parkinson’s disease: from associations to mechanisms. Trends Microbiol. 30, 749–760 (2022).
Singh, Y. et al. Overexpression of human alpha-Synuclein leads to dysregulated microbiome/metabolites with ageing in a rat model of Parkinson disease. Mol. Neurodegener. 18, 44 (2023).
Sun, M.-F. et al. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav. Immun. 70, 48–60 (2018).
Bhattarai, Y. et al. Role of gut microbiota in regulating gastrointestinal dysfunction and motor symptoms in a mouse model of Parkinson’s disease. Gut Microbes 13, 1866974 (2021).
Mead, R. J. et al. Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 22, 185–212 (2023).
Eshima, J. et al. Molecular subtypes of ALS are associated with differences in patient prognosis. Nat. Commun. 14, 95 (2023).
Wu, S. et al. Leaky intestine and impaired microbiome in an amyotrophic lateral sclerosis mouse model. Physiol. Rep. 3, e12356 (2015).
Zhang, Y. et al. Aberrant enteric neuromuscular system and dysbiosis in amyotrophic lateral sclerosis. Gut Microbes 13, 1996848 (2021).
Blacher, E. et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 572, 474–480 (2019).
Figueroa-Romero, C. et al. Temporal evolution of the microbiome, immune system and epigenome with disease progression in ALS mice. Dis. Model Mech. 13, dmm041947 (2019).
Zeng, Q. et al. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci. Rep. 10, 12998 (2020).
Renton, A. E. et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Majounie, E. et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330 (2012).
Cox, L. M. et al. The microbiota restrains neurodegenerative microglia in a model of amyotrophic lateral sclerosis. Microbiome 10, 47 (2022).
Todd, T. W. & Petrucelli, L. Modelling amyotrophic lateral sclerosis in rodents. Nat. Rev. Neurosci. 23, 231–251 (2022).
Akçimen, F. et al. Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat. Rev. Genet. 24, 642–658 (2023).
Tabrizi, S. J. et al. Potential disease-modifying therapies for Huntington’s disease: lessons learned and future opportunities. Lancet Neurol. 21, 645–658 (2022).
Sharma, G. et al. Gut microbiota dysbiosis and Huntington’s disease: exploring the gut-brain axis and novel microbiota-based interventions. Life Sci. 328, 121882 (2023).
McAllister, B. et al. Timing and impact of psychiatric, cognitive, and motor abnormalities in Huntington disease. Neurology 96, e2395–e2406 (2021).
Kong, G. et al. Microbiome profiling reveals gut dysbiosis in a transgenic mouse model of Huntington’s disease. Neurobiol. Dis. 135, 104268 (2020).
Stan, T. L. et al. Increased intestinal permeability and gut dysbiosis in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 10, 18270 (2020).
Wasser, C. I. et al. Gut dysbiosis in Huntington’s disease: associations among gut microbiota, cognitive performance and clinical outcomes. Brain Commun. 2, fcaa110 (2020).
Mukherjee, A., Lordan, C., Ross, R. P. & Cotter, P. D. Gut microbes from the phylogenetically diverse genus Eubacterium and their various contributions to gut health. Gut Microbes 12, 1802866 (2020).
Montalban-Arques, A. et al. Commensal Clostridiales strains mediate effective anti-cancer immune response against solid tumors. Cell Host Microbe 29, 1573–1588.e1577 (2021).
Worby, C. J. et al. Longitudinal multi-omics analyses link gut microbiome dysbiosis with recurrent urinary tract infections in women. Nat. Microbiol. 7, 630–639 (2022).
Udayappan, S. et al. Oral treatment with Eubacterium hallii improves insulin sensitivity in db/db mice. NPJ Biofilms Microbiomes 2, 16009 (2016).
Wang, D. et al. Characterization of gut microbial structural variations as determinants of human bile acid metabolism. Cell Host Microbe 29, 1802–1814.e1805 (2021).
Gubert, C. et al. Gene-environment-gut interactions in Huntington’s disease mice are associated with environmental modulation of the gut microbiome. iScience 25, 103687 (2022).
Gubert, C. et al. Faecal microbiota transplant ameliorates gut dysbiosis and cognitive deficits in Huntington’s disease mice. Brain Commun. 4, fcac205 (2022).
Patani, R., Hardingham, G. E. & Liddelow, S. A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 19, 395–409 (2023).
Lee, H. G., Wheeler, M. A. & Quintana, F. J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 21, 339–358 (2022).
Rothhammer, V. et al. Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018).
Rothhammer, V. et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597 (2016).
Xie, Y. et al. Indole-3-propionic acid alleviates ischemic brain injury in a mouse middle cerebral artery occlusion model. Exp. Neurol. 353, 114081 (2022).
Sanmarco, L. M. et al. Gut-licensed IFNγ + NK cells drive LAMP1 + TRAIL+ anti-inflammatory astrocytes. Nature 590, 473–479 (2021).
Karbownik, M. S., Sokołowska, P. & Kowalczyk, E. Gut microbiota metabolites differentially release gliotransmitters from the cultured human astrocytes: a preliminary report. Int. J. Mol. Sci. 24, 6617 (2023).
Brandebura, A. N., Paumier, A., Onur, T. S. & Allen, N. J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 24, 23–39 (2023).
Daly, C. M. et al. Sex differences in response to a high fat, high sucrose diet in both the gut microbiome and hypothalamic astrocytes and microglia. Nutr. Neurosci. 25, 321–335 (2022).
Lakosa, A. et al. Impact of the gut microbiome on nicotine’s motivational effects and glial cells in the ventral tegmental area in male mice. Neuropsychopharmacology 48, 963–974 (2023).
Xu, L. et al. Inhibitory effect of Dendrobium officinale polysaccharide on oxidative damage of glial cells in aging mice by regulating gut microbiota. Int. J. Biol. Macromol. 247, 125787 (2023).
Zhao, Q. et al. Microbiota from healthy mice alleviates cognitive decline via reshaping the gut-brain metabolic axis in diabetic mice. Chem. Biol. Interact. 382, 110638 (2023).
Escartin, C. et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325 (2021).
Yu, X., Nagai, J. & Khakh, B. S. Improved tools to study astrocytes. Nat. Rev. Neurosci. 21, 121–138 (2020).
Jiang, Z. et al. Ketogenic diet protects MPTP-induced mouse model of Parkinson’s disease via altering gut microbiota and metabolites. MedComm 4, e268 (2023).
Li, T. et al. Neuroprotective effects of Bifidobacterium breve CCFM1067 in MPTP-induced mouse models of Parkinson’s disease. Nutrients 14, 4678 (2022).
Xie, Z. et al. Healthy human fecal microbiota transplantation into mice attenuates MPTP-induced neurotoxicity via AMPK/SOD2 pathway. Aging Dis. 14, 2193–2214 (2023).
Bierhansl, L. et al. Thinking outside the box: non-canonical targets in multiple sclerosis. Nat. Rev. Drug Discov. 21, 578–600 (2022).
Knowles, J. K., Batra, A., Xu, H. & Monje, M. Adaptive and maladaptive myelination in health and disease. Nat. Rev. Neurol. 18, 735–746 (2022).
Ferrari Bardile, C., Radulescu, C. I. & Pouladi, M. A. Oligodendrocyte pathology in Huntington’s disease: from mechanisms to therapeutics. Trends Mol. Med. 29, 802–816 (2023).
Hoban, A. E. et al. Regulation of prefrontal cortex myelination by the microbiota. Transl. Psychiatry 6, e774–e774 (2016).
Chen, T. et al. Butyrate suppresses demyelination and enhances remyelination. J. Neuroinflamm. 16, 165 (2019).
Molina-Gonzalez, I. et al. Astrocyte-oligodendrocyte interaction regulates central nervous system regeneration. Nat. Commun. 14, 3372 (2023).
Xiao, Y. & Czopka, T. Myelination-independent functions of oligodendrocyte precursor cells in health and disease. Nat. Neurosci. 26, 1663–1669 (2023).
McMurran, C. E. et al. The microbiota regulates murine inflammatory responses to toxin-induced CNS demyelination but has minimal impact on remyelination. Proc. Natl. Acad. Sci. USA 116, 25311–25321 (2019).
Zhou, Y., Hu, G. & Wang, M. C. Host and microbiota metabolic signals in aging and longevity. Nat. Chem. Biol. 17, 1027–1036 (2021).
Krautkramer, K. A., Fan, J. & Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 19, 77–94 (2021).
Skelly, A. N., Sato, Y., Kearney, S. & Honda, K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat. Rev. Immunol. 19, 305–323 (2019).
de Vos, W. M., Tilg, H., Van Hul, M. & Cani, P. D. Gut microbiome and health: mechanistic insights. Gut 71, 1020–1032 (2022).
Kim, C. H. Control of lymphocyte functions by gut microbiota-derived short-chain fatty acids. Cell Mol. Immunol. 18, 1161–1171 (2021).
Yang, W. et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 11, 4457 (2020).
Zhao, Y. et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol. 11, 752–762 (2018).
van der Hee, B. & Wells, J. M. Microbial regulation of host physiology by short-chain fatty acids. Trends Microbiol. 29, 700–712 (2021).
Wang, R. et al. Treatment of peanut allergy and colitis in mice via the intestinal release of butyrate from polymeric micelles. Nat. Biomed. Eng. 7, 38–55 (2023).
Carmody, R. N. & Bisanz, J. E. Roles of the gut microbiome in weight management. Nat. Rev. Microbiol. 21, 535–550 (2023).
Palmnäs-Bédard, M. S. A. et al. The human gut microbiota and glucose metabolism: a scoping review of key bacteria and the potential role of SCFAs. Am. J. Clin. Nutr. 116, 862–874 (2022).
Zhao, L. et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 359, 1151–1156 (2018).
Dalile, B., Van Oudenhove, L., Vervliet, B. & Verbeke, K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 16, 461–478 (2019).
Hauser, A. S. et al. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017).
Wong, T.-S. et al. G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders. Signal Transduct. Target Ther. 8, 177 (2023).
Liu, J. et al. Anti-neuroinflammatory effect of short-chain fatty acid acetate against Alzheimer’s disease via upregulating GPR41 and inhibiting ERK/JNK/NF-κB. J. Agric Food Chem. 68, 7152–7161 (2020).
Park, J., Lee, K., Kim, K. & Yi, S.-J. The role of histone modifications: from neurodevelopment to neurodiseases. Signal Transduct. Target Ther. 7, 217 (2022).
Wang, K. et al. Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct. Target Ther. 7, 374 (2022).
Davie, J. R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 133, 2485s–2493s (2003).
Caetano-Silva, M. E. et al. Inhibition of inflammatory microglia by dietary fiber and short-chain fatty acids. Sci. Rep. 13, 2819 (2023).
Song, L. et al. Roseburia hominis alleviates neuroinflammation via short-chain fatty acids through histone deacetylase inhibition. Mol. Nutr. Food Res. 66, e2200164 (2022).
Wang, X. et al. Gut microbiota-derived metabolites mediate the neuroprotective effect of melatonin in cognitive impairment induced by sleep deprivation. Microbiome 11, 17 (2023).
Fan, K. C. et al. Altered gut microbiota in older adults with mild cognitive impairment: a case-control study. Front. Aging Neurosci. 15, 1162057 (2023).
Gao, C. et al. Early changes of fecal short-chain fatty acid levels in patients with mild cognitive impairments. CNS Neurosci. Ther. 29, 3657–3666 (2023).
Grabrucker, S. et al. Microbiota from Alzheimer’s patients induce deficits in cognition and hippocampal neurogenesis. Brain 146, 4916–4934 (2023).
Wu, L. et al. Altered gut microbial metabolites in amnestic mild cognitive impairment and Alzheimer’s disease: signals in host-microbe interplay. Nutrients 13, 228 (2021).
Gräff, J. et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226 (2012).
Ding, H., Dolan, P. J. & Johnson, G. V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 106, 2119–2130 (2008).
Datta, M. et al. Histone deacetylases 1 and 2 regulate microglia function during development, homeostasis, and neurodegeneration in a context-dependent manner. Immunity 48, 514–529.e516 (2018).
Fernando, W. et al. Sodium butyrate reduces brain amyloid-β levels and improves cognitive memory performance in an Alzheimer’s disease transgenic mouse model at an early disease stage. J. Alzheimers Dis. 74, 91–99 (2020).
Jiang, Y. et al. Sodium butyrate ameliorates the impairment of synaptic plasticity by inhibiting the neuroinflammation in 5XFAD mice. Chem. Biol. Interact. 341, 109452 (2021).
Liu, N. et al. Mesoporous silica nanoparticle-encapsulated Bifidobacterium attenuates brain Aβ burden and improves olfactory dysfunction of APP/PS1 mice by nasal delivery. J. Nanobiotechnol. 20, 439 (2022).
Zhu, G., Zhao, J., Wang, G. & Chen, W. Bifidobacterium breve HNXY26M4 attenuates cognitive deficits and neuroinflammation by regulating the gut–brain axis in APP/PS1 mice. J. Agric Food Chem. 71, 4646–4655 (2023).
Sun, J. et al. Effect of Clostridium butyricum against microglia-mediated neuroinflammation in Alzheimer’s disease via regulating gut microbiota and metabolites butyrate. Mol. Nutr. Food Res. 64, e1900636 (2020).
Liu, Q. et al. Mannan oligosaccharide attenuates cognitive and behavioral disorders in the 5xFAD Alzheimer’s disease mouse model via regulating the gut microbiota-brain axis. Brain Behav. Immun. 95, 330–343 (2021).
Zhang, Q. et al. β-Glucan attenuates cognitive impairment of APP/PS1 mice via regulating intestinal flora and its metabolites. CNS Neurosci. Ther. 29, 1690–1704 (2023).
Sun, Y. et al. Promotion of astrocyte-neuron glutamate-glutamine shuttle by SCFA contributes to the alleviation of Alzheimer’s disease. Redox Biol. 62, 102690 (2023).
Neuffer, J. et al. Exploration of the gut-brain axis through metabolomics identifies serum propionic acid associated with higher cognitive decline in older persons. Nutrients 14, 4688 (2022).
Liu, P. et al. High-fat diet-induced diabetes couples to Alzheimer’s disease through inflammation-activated C/EBPβ/AEP pathway. Mol. Psychiatry 27, 3396–3409 (2022).
Wang, Z. H. et al. C/EBPβ regulates delta-secretase expression and mediates pathogenesis in mouse models of Alzheimer’s disease. Nat. Commun. 9, 1784 (2018).
Zhang, Z. et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 20, 1254–1262 (2014).
Zhang, Z. et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 6, 8762 (2015).
Pan, R. Y. et al. Intermittent fasting protects against Alzheimer’s disease in mice by altering metabolism through remodeling of the gut microbiota. Nat. Aging 2, 1024–1039 (2022).
Luczynski, P. et al. Growing up in a bubble: using germ-free animals to assess the influence of the gut microbiota on brain and behavior. Int. J. Neuropsychopharmacol. 19, pyw020 (2016).
Zhang, D. et al. Targeting epigenetic modifications in Parkinson’s disease therapy. Med. Res. Rev. 43, 1748–1777 (2023).
Mazzocchi, M. et al. Peripheral administration of the Class-IIa HDAC inhibitor MC1568 partially protects against nigrostriatal neurodegeneration in the striatal 6-OHDA rat model of Parkinson’s disease. Brain Behav. Immun. 102, 151–160 (2022).
Sun, J. et al. Probiotic Clostridium butyricum ameliorated motor deficits in a mouse model of Parkinson’s disease via gut microbiota-GLP-1 pathway. Brain Behav. Immun. 91, 703–715 (2021).
Hou, Y. F. et al. Gut microbiota-derived propionate mediates the neuroprotective effect of osteocalcin in a mouse model of Parkinson’s disease. Microbiome 9, 34 (2021).
Abdel-Haq, R. et al. A prebiotic diet modulates microglial states and motor deficits in α-synuclein overexpressing mice. eLife 11, e81453 (2022).
Hou, Y. et al. Neuroprotective effects of short-chain fatty acids in MPTP induced mice model of Parkinson’s disease. Exp. Gerontol. 150, 111376 (2021).
Guo, T. T. et al. Neuroprotective effects of sodium butyrate by restoring gut microbiota and inhibiting TLR4 signaling in mice with MPTP-induced Parkinson’s disease. Nutrients 15, 930 (2023).
Srivastav, S. et al. Probiotics mixture increases butyrate, and subsequently rescues the nigral dopaminergic neurons from MPTP and rotenone-induced neurotoxicity. J. Nutr. Biochem. 69, 73–86 (2019).
Nishiwaki, H. et al. Short chain fatty acids-producing and mucin-degrading intestinal bacteria predict the progression of early Parkinson’s disease. NPJ Parkinsons Dis. 8, 65 (2022).
Unger, M. M. et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 32, 66–72 (2016).
Nuzum, N. D. et al. Gut microbiota differences between healthy older adults and individuals with Parkinson’s disease: a systematic review. Neurosci. Biobehav Rev. 112, 227–241 (2020).
Cirstea, M. S. et al. Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Mov. Disord. 35, 1208–1217 (2020).
Perez-Pardo, P. et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut 68, 829–843 (2019).
Aho, V. T. E. et al. Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol. Neurodegener. 16, 6 (2021).
Keshavarzian, A. et al. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 30, 1351–1360 (2015).
Chen, S. J. et al. Association of fecal and plasma levels of short-chain fatty acids with gut microbiota and clinical severity in patients with Parkinson disease. Neurology 98, e848–e858 (2022).
Yang, X. et al. Parkinson’s disease is associated with impaired gut-blood barrier for short-chain fatty acids. Mov. Disord. 37, 1634–1643 (2022).
Wallen, Z. D. et al. Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 13, 1–20 (2022).
Hall, D. A. et al. An open label, non-randomized study assessing a prebiotic fiber intervention in a small cohort of Parkinson’s disease participants. Nat. Commun. 14, 926 (2023).
Zhong, Z. et al. Fecal microbiota transplantation exerts a protective role in MPTP-induced Parkinson’s disease via the TLR4/PI3K/AKT/NF-κB pathway stimulated by α-synuclein. Neurochem. Res. 46, 3050–3058 (2021).
Wang, H. et al. C/EBPβ/AEP is age-dependently activated in Parkinson’s disease and mediates α-synuclein in the gut and brain. NPJ Parkinsons Dis. 9, 1 (2023).
Cao, X. et al. Low and high doses of oral maslinic acid protect against Parkinson’s disease via distinct gut microbiota-related mechanisms. Biomed. Pharmacother. 165, 115100 (2023).
Grey, M. J. et al. The epithelial-specific ER stress sensor ERN2/IRE1β enables host-microbiota crosstalk to affect colon goblet cell development. J. Clin. Investig. 132, e153519 (2022).
Liang, L. et al. Gut microbiota-derived butyrate regulates gut mucus barrier repair by activating the macrophage/WNT/ERK signaling pathway. Clin. Sci. 136, 291–307 (2022).
Wang, R. X., Lee, J. S., Campbell, E. L. & Colgan, S. P. Microbiota-derived butyrate dynamically regulates intestinal homeostasis through regulation of actin-associated protein synaptopodin. Proc. Natl. Acad. Sci. USA 117, 11648–11657 (2020).
Alpino, G. et al. Beneficial effects of butyrate on brain functions: a view of epigenetic. Crit. Rev. Food Sci. Nutr. https://doi.org/10.1080/10408398.2022.2137776 (2022).
Bennett, S. A., Tanaz, R., Cobos, S. N. & Torrente, M. P. Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 204, 19–30 (2019).
Rossaert, E. et al. Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol. Commun. 7, 107 (2019).
Guo, W. et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 8, 861 (2017).
Fazal, R. et al. HDAC6 inhibition restores TDP-43 pathology and axonal transport defects in human motor neurons with TARDBP mutations. EMBO J. 40, e106177 (2021).
Burg, T. et al. Histone deacetylase inhibition regulates lipid homeostasis in a mouse model of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 22, 11224 (2021).
Nicholson, K. et al. The human gut microbiota in people with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 22, 186–194 (2021).
Fang, X. et al. Evaluation of the microbial diversity in amyotrophic lateral sclerosis using high-throughput sequencing. Front. Microbiol. 7, 1479 (2016).
Zhang, Y. G. et al. Target intestinal microbiota to alleviate disease progression in amyotrophic lateral sclerosis. Clin. Ther. 39, 322–336 (2017).
Li, X. et al. Butyrate ameliorates mitochondrial respiratory capacity of the motor-neuron-like cell line NSC34-G93A, a cellular model for ALS. Biomolecules 12, 333 (2022).
Jamwal, S., Blackburn, J. K. & Elsworth, J. D. PPARγ/PGC1α signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharm. Ther. 219, 107705 (2021).
Perino, A. & Schoonjans, K. Metabolic messengers: bile acids. Nat. Metab. 4, 416–423 (2022).
Meier, K. H. U. et al. Metabolic landscape of the male mouse gut identifies different niches determined by microbial activities. Nat. Metab. 5, 968–980 (2023).
Brown, K. et al. Microbiota alters the metabolome in an age- and sex-dependent manner in mice. Nat. Commun. 14, 1348 (2023).
Zarrinpar, A. et al. Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat. Commun. 9, 2872 (2018).
Shalon, D. et al. Profiling the human intestinal environment under physiological conditions. Nature 617, 581–591 (2023).
Baloni, P. et al. Metabolic network analysis reveals altered bile acid synthesis and metabolism in Alzheimer’s disease. Cell Rep. Med. 1, 100138 (2020).
MahmoudianDehkordi, S. et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease—an emerging role for gut microbiome. Alzheimers Dement. 15, 76–92 (2019).
Nho, K. et al. Altered bile acid profile in mild cognitive impairment and Alzheimer’s disease: relationship to neuroimaging and CSF biomarkers. Alzheimers Dement. 15, 232–244 (2019).
Varma, V. R. et al. Bile acid synthesis, modulation, and dementia: a metabolomic, transcriptomic, and pharmacoepidemiologic study. PLoS Med. 18, e1003615 (2021).
Gu, X. et al. Huanglian Jiedu decoction remodels the periphery microenvironment to inhibit Alzheimer’s disease progression based on the “brain-gut” axis through multiple integrated omics. Alzheimers Res. Ther. 13, 44 (2021).
Wei, M. et al. A dysregulated bile acid-gut microbiota axis contributes to obesity susceptibility. EBioMedicine 55, 102766 (2020).
Campbell, C. et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479 (2020).
Buffie, C. G. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208 (2015).
Hang, S. et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148 (2019).
Paik, D. et al. Human gut bacteria produce ΤΗ17-modulating bile acid metabolites. Nature 603, 907–912 (2022).
Fogelson, K. A., Dorrestein, P. C., Zarrinpar, A. & Knight, R. The gut microbial bile acid modulation and its relevance to digestive health and diseases. Gastroenterology 164, 1069–1085 (2023).
Huang, F., Pariante, C. M. & Borsini, A. From dried bear bile to molecular investigation: a systematic review of the effect of bile acids on cell apoptosis, oxidative stress and inflammation in the brain, across pre-clinical models of neurological, neurodegenerative and neuropsychiatric disorders. Brain Behav. Immun. 99, 132–146 (2022).
Yanguas-Casás, N., Barreda-Manso, M. A., Nieto-Sampedro, M. & Romero-Ramírez, L. TUDCA: an agonist of the bile acid receptor GPBAR1/TGR5 with anti-inflammatory effects in microglial cells. J. Cell Physiol. 232, 2231–2245 (2017).
Romero-Ramírez, L., García-Rama, C., Wu, S. & Mey, J. Bile acids attenuate PKM2 pathway activation in proinflammatory microglia. Sci. Rep. 12, 1459 (2022).
Kim, S. J. et al. Anti-inflammatory effect of tauroursodeoxycholic acid in RAW 264.7 macrophages, bone marrow-derived macrophages, BV2 microglial cells, and spinal cord injury. Sci. Rep. 8, 3176 (2018).
Yanguas-Casás, N., Barreda-Manso, M. A., Nieto-Sampedro, M. & Romero-Ramírez, L. Tauroursodeoxycholic acid reduces glial cell activation in an animal model of acute neuroinflammation. J. Neuroinflamm. 11, 50 (2014).
Wu, X. et al. Protective effects of tauroursodeoxycholic acid on lipopolysaccharide-induced cognitive impairment and neurotoxicity in mice. Int. Immunopharmacol. 72, 166–175 (2019).
Dionísio, P. A. et al. Amyloid-β pathology is attenuated by tauroursodeoxycholic acid treatment in APP/PS1 mice after disease onset. Neurobiol. Aging 36, 228–240 (2015).
Lo, A. C. et al. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 50, 21–29 (2013).
Nunes, A. F. et al. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 45, 440–454 (2012).
Ochiai, T. et al. Tauroursodeoxycholic acid attenuates diet-induced and age-related peripheral endoplasmic reticulum stress and cerebral amyloid pathology in a mouse model of Alzheimer’s disease. J. Prev. Alzheimers Dis. 8, 483–494 (2021).
Fuchs, C. D. & Trauner, M. Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat. Rev. Gastroenterol. Hepatol. 19, 432–450 (2022).
Kalecký, K. & Bottiglieri, T. Targeted metabolomic analysis in Parkinson’s disease brain frontal cortex and putamen with relation to cognitive impairment. NPJ Parkinsons Dis. 9, 84 (2023).
Li, P. et al. Gut microbiota dysbiosis is associated with elevated bile acids in Parkinson’s disease. Metabolites 11, 29 (2021).
Shao, Y. et al. Comprehensive metabolic profiling of Parkinson’s disease by liquid chromatography-mass spectrometry. Mol. Neurodegener. 16, 4 (2021).
Yakhine-Diop, S. M. S. et al. Metabolic alterations in plasma from patients with familial and idiopathic Parkinson’s disease. Aging 12, 16690–16708 (2020).
Gonzalez-Riano, C. et al. Prognostic biomarkers of Parkinson’s disease in the Spanish EPIC cohort: a multiplatform metabolomics approach. NPJ Parkinsons Dis. 7, 73 (2021).
Graham, S. F. et al. Metabolomic profiling of bile acids in an experimental model of prodromal Parkinson’s disease. Metabolites 8, 71 (2018).
Xu, Y. et al. Gut microbiota alteration after cholecystectomy contributes to post-cholecystectomy diarrhea via bile acids stimulating colonic serotonin. Gut Microbes 15, 2168101 (2023).
Kim, R. et al. Cholecystectomy and subsequent risk of Parkinson’s disease: a nationwide retrospective cohort study. NPJ Parkinsons Dis. 7, 100 (2021).
Frost, F. et al. Carrying asymptomatic gallstones is not associated with changes in intestinal microbiota composition and diversity but cholecystectomy with significant dysbiosis. Sci. Rep. 11, 6677 (2021).
Moreira, S. et al. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 295, 77–87 (2017).
Rosa, A. I. et al. Tauroursodeoxycholic acid improves motor symptoms in a mouse model of Parkinson’s disease. Mol. Neurobiol. 55, 9139–9155 (2018).
Castro-Caldas, M. et al. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s disease. Mol. Neurobiol. 46, 475–486 (2012).
Cuevas, E. et al. Tauroursodeoxycholic acid (TUDCA) is neuroprotective in a chronic mouse model of Parkinson’s disease. Nutr. Neurosci. 25, 1374–1391 (2022).
Mao, Z. et al. Protective effects of dioscin against Parkinson’s disease via regulating bile acid metabolism through remodeling gut microbiome/GLP-1 signaling. J. Pharm. Anal. 13, 1153–1167 (2023).
Abdelkader, N. F., Safar, M. M. & Salem, H. A. Ursodeoxycholic acid ameliorates apoptotic cascade in the rotenone model of Parkinson’s disease: modulation of mitochondrial perturbations. Mol. Neurobiol. 53, 810–817 (2016).
Qi, H. et al. Ursodeoxycholic acid protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis in MPTP/MPP(+)-induced Parkinson’s disease. Neurosci. Lett. 741, 135493 (2021).
Chu, C. et al. A low-protein, high-carbohydrate diet exerts a neuroprotective effect on mice with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease by regulating the microbiota-metabolite-brain axis and fibroblast growth factor 21. J. Agric. Food Chem. 71, 8877–8893 (2023).
Payne, T. et al. A double-blind, randomized, placebo-controlled trial of ursodeoxycholic acid (UDCA) in Parkinson’s disease. Mov. Disord. 38, 1493–1502 (2023).
Nowell, J., Blunt, E. & Edison, P. Incretin and insulin signaling as novel therapeutic targets for Alzheimer’s and Parkinson’s disease. Mol. Psychiatry 28, 217–229 (2023).
Yun, S. P. et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 24, 931–938 (2018).
Dodge, J. C., Yu, J., Sardi, S. P. & Shihabuddin, L. S. Sterol auto-oxidation adversely affects human motor neuron viability and is a neuropathological feature of amyotrophic lateral sclerosis. Sci. Rep. 11, 803 (2021).
Gong, Z. et al. Gut microbiota links with cognitive impairment in amyotrophic lateral sclerosis: a multi-omics study. J. Biomed. Res. 37, 125–137 (2022).
Thams, S. et al. A stem cell-based screening platform identifies compounds that desensitize motor neurons to endoplasmic reticulum stress. Mol. Ther. 27, 87–101 (2019).
Paganoni, S. et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 63, 31–39 (2021).
Paganoni, S. et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. New Engl. J. Med. 383, 919–930 (2020).
Paganoni, S. et al. Survival analyses from the CENTAUR trial in amyotrophic lateral sclerosis: evaluating the impact of treatment crossover on outcomes. Muscle Nerve 66, 136–141 (2022).
Connell, E. et al. Microbial-derived metabolites as a risk factor of age-related cognitive decline and dementia. Mol. Neurodegener. 17, 43 (2022).
Mossad, O. et al. Gut microbiota drives age-related oxidative stress and mitochondrial damage in microglia via the metabolite N6-carboxymethyllysine. Nat. Neurosci. 25, 295–305 (2022).
Lai, Y. et al. High-coverage metabolomics uncovers microbiota-driven biochemical landscape of interorgan transport and gut-brain communication in mice. Nat. Commun. 12, 6000 (2021).
Ding, J. et al. A metabolome atlas of the aging mouse brain. Nat. Commun. 12, 6021 (2021).
Lu, Y. et al. Multi-omics analysis reveals neuroinflammation, activated glial signaling, and dysregulated synaptic signaling and metabolism in the hippocampus of aged mice. Front. Aging Neurosci. 14, 964429 (2022).
Govindarajulu, M. et al. Gut metabolite TMAO induces synaptic plasticity deficits by promoting endoplasmic reticulum stress. Front. Mol. Neurosci. 13, 138 (2020).
Buawangpong, N. et al. Increased plasma trimethylamine-N-oxide levels are associated with mild cognitive impairment in high cardiovascular risk elderly population. Food Funct. 13, 10013–10022 (2022).
Vogt, N. M. et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res. Ther. 10, 124 (2018).
Brunt, V. E. et al. The gut microbiome-derived metabolite trimethylamine N-oxide modulates neuroinflammation and cognitive function with aging. GeroScience 43, 377–394 (2021).
Qiao, C. M. et al. Orally induced high serum level of trimethylamine n-oxide worsened glial reaction and neuroinflammation on MPTP-induced acute Parkinson’s disease model mice. Mol. Neurobiol. 60, 5137–5154 (2023).
Quan, W. et al. Trimethylamine N-oxide exacerbates neuroinflammation and motor dysfunction in an acute MPTP mice model of Parkinson’s disease. Brain Sci. 13, 790 (2023).
Zhang, Y. et al. Trimethylamine N-oxide aggravated cognitive impairment from APP/PS1 mice and protective roles of voluntary exercise. Neurochem. Int. 162, 105459 (2023).
Hu, X. et al. TMAO promotes dementia progression by mediating the PI3K/Akt/mTOR pathway. Tissue Cell 81, 102034 (2023).
Gao, Q. et al. Decreased levels of circulating trimethylamine N-oxide alleviate cognitive and pathological deterioration in transgenic mice: a potential therapeutic approach for Alzheimer’s disease. Aging 11, 8642–8663 (2019).
Wang, Q. J. et al. Concomitant memantine and Lactobacillus plantarum treatment attenuates cognitive impairments in APP/PS1 mice. Aging 12, 628–649 (2020).
Chen, S. J. et al. The gut metabolite trimethylamine N-oxide is associated with Parkinson’s disease severity and progression. Mov. Disord. 35, 2115–2116 (2020).
Chung, S. J. et al. Gut microbiota-derived metabolite trimethylamine N-oxide as a biomarker in early Parkinson’s disease. Nutrition 83, 111090 (2021).
Lee, Y. et al. Gut metabolite trimethylamine N-oxide induces aging-associated phenotype of midbrain organoids for the induced pluripotent stem cell-based modeling of late-onset disease. Front. Aging Neurosci. 14, 925227 (2022).
Needham, B. D., Kaddurah-Daouk, R. & Mazmanian, S. K. Gut microbial molecules in behavioural and neurodegenerative conditions. Nat. Rev. Neurosci. 21, 717–731 (2020).
Dodd, D. et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652 (2017).
Agus, A., Planchais, J. & Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 23, 716–724 (2018).
Platten, M. et al. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 18, 379–401 (2019).
Stockinger, B., Shah, K. & Wincent, E. AHR in the intestinal microenvironment: safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 18, 559–570 (2021).
Sun, J. et al. Microbiota-derived metabolite Indoles induced aryl hydrocarbon receptor activation and inhibited neuroinflammation in APP/PS1 mice. Brain Behav. Immun. 106, 76–88 (2022).
Pan, S. et al. A high-tryptophan diet alleviated cognitive impairment and neuroinflammation in APP/PS1 mice through activating aryl hydrocarbon receptor via the regulation of gut microbiota. Mol. Nutr. Food Res. https://doi.org/10.1002/mnfr.202300601 (2023).
Kalecký, K., German, D. C., Montillo, A. A. & Bottiglieri, T. Targeted metabolomic analysis in Alzheimer’s disease plasma and brain tissue in non-hispanic whites. J. Alzheimers Dis. 86, 1875–1895 (2022).
Sankowski, B. et al. Higher cerebrospinal fluid to plasma ratio of p-cresol sulfate and indoxyl sulfate in patients with Parkinson’s disease. Clin. Chim. Acta 501, 165–173 (2020).
Brydges, C. R. et al. Indoxyl sulfate, a gut microbiome-derived uremic toxin, is associated with psychic anxiety and its functional magnetic resonance imaging-based neurologic signature. Sci. Rep. 11, 21011 (2021).
Fan, Y. et al. The gut microbiota contributes to the pathogenesis of anorexia nervosa in humans and mice. Nat. Microbiol. 8, 787–802 (2023).
Adesso, S. et al. Indoxyl sulfate affects glial function increasing oxidative stress and neuroinflammation in chronic kidney disease: interaction between astrocytes and microglia. Front. Pharm. 8, 370 (2017).
Wang, Y. et al. Tryptophan in the diet ameliorates motor deficits in a rotenone-induced rat Parkinson’s disease model via activating the aromatic hydrocarbon receptor pathway. Brain Behav. 11, e2226 (2021).
Mohamad, K. A., El-Naga, R. N. & Wahdan, S. A. Neuroprotective effects of indole-3-carbinol on the rotenone rat model of Parkinson’s disease: Impact of the SIRT1-AMPK signaling pathway. Toxicol. Appl. Pharm. 435, 115853 (2022).
Saini, N. et al. Protective effect of Indole-3-carbinol, an NF-κB inhibitor in experimental paradigm of Parkinson’s disease: in silico and in vivo studies. Brain Behav. Immun. 90, 108–137 (2020).
Bazinet, R. P. & Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 15, 771–785 (2014).
Miyamoto, J. et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat. Commun. 10, 4007 (2019).
Ebright, B. et al. Eicosanoid lipidome activation in post-mortem brain tissues of individuals with APOE4 and Alzheimer’s dementia. Alzheimers Res. Ther. 14, 152 (2022).
Wei, B. Z. et al. The relationship of omega-3 fatty acids with dementia and cognitive decline: evidence from prospective cohort studies of supplementation, dietary intake, and blood markers. Am. J. Clin. Nutr. 117, 1096–1109 (2023).
Hjorth, E. et al. Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-β42 by human microglia and decrease inflammatory markers. J. Alzheimers Dis. 35, 697–713 (2013).
Xia, Y. et al. Bacteroides fragilis in the gut microbiomes of Alzheimer’s disease activates microglia and triggers pathogenesis in neuronal C/EBPβ transgenic mice. Nat. Commun. 14, 5471 (2023).
Goutman, S. A. et al. Metabolomics identifies shared lipid pathways in independent amyotrophic lateral sclerosis cohorts. Brain 145, 4425–4439 (2022).
Goutman, S. A. et al. Untargeted metabolomics yields insight into ALS disease mechanisms. J. Neurol. Neurosurg. Psychiatry 91, 1329–1338 (2020).
Bjornevik, K. et al. Association of polyunsaturated fatty acids and clinical progression in patients with ALS: post hoc analysis of the EMPOWER trial. Neurology 101, e690–e698 (2023).
O’Reilly, É. J. et al. Prediagnostic plasma polyunsaturated fatty acids and the risk of amyotrophic lateral sclerosis. Neurology 94, e811–e819 (2020).
Fitzgerald, K. C. et al. Dietary ω-3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis. JAMA Neurol. 71, 1102–1110 (2014).
Lee, H. et al. Multi-omic analysis of selectively vulnerable motor neuron subtypes implicates altered lipid metabolism in ALS. Nat. Neurosci. 24, 1673–1685 (2021).
Pedersen, H. K. et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535, 376–381 (2016).
Ling, Z. N. et al. Amino acid metabolism in health and disease. Signal Transduct. Target Ther. 8, 345 (2023).
McGarrah, R. W. & White, P. J. Branched-chain amino acids in cardiovascular disease. Nat. Rev. Cardiol. 20, 77–89 (2023).
De Simone, R. et al. Branched-chain amino acids influence the immune properties of microglial cells and their responsiveness to pro-inflammatory signals. Biochim Biophys. Acta 1832, 650–659 (2013).
Zhang, Y. et al. Plasma branched-chain and aromatic amino acids correlate with the gut microbiota and severity of Parkinson’s disease. NPJ Parkinsons Dis. 8, 48 (2022).
Yan, Z. et al. Alterations of gut microbiota and metabolome with Parkinson’s disease. Micro. Pathog. 160, 105187 (2021).
Bedarf, J. R. et al. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 9, 39 (2017).
Yan, Z. et al. Role of gut microbiota-derived branched-chain amino acids in the pathogenesis of Parkinson’s disease: an animal study. Brain Behav. Immun. 106, 307–321 (2022).
Chu, C. et al. Lactobacillus plantarum CCFM405 against rotenone-induced Parkinson’s disease mice via regulating gut microbiota and branched-chain amino acids biosynthesis. Nutrients 15, 1737 (2023).
Solon-Biet, S. M. et al. Branched chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat. Metab. 1, 532–545 (2019).
Rusch, C., Flanagan, R., Suh, H. & Subramanian, I. To restrict or not to restrict? Practical considerations for optimizing dietary protein interactions on levodopa absorption in Parkinson’s disease. NPJ Parkinsons Dis. 9, 98 (2023).
Tărlungeanu, D. C. et al. Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell 167, 1481–1494.e1418 (2016).
Covarrubias, A. J., Perrone, R., Grozio, A. & Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 22, 119–141 (2021).
Katsyuba, E., Romani, M., Hofer, D. & Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2, 9–31 (2020).
Chellappa, K. et al. NAD precursors cycle between host tissues and the gut microbiome. Cell Metab. 34, 1947–1959.e1945 (2022).
Hor, J. H. et al. ALS motor neurons exhibit hallmark metabolic defects that are rescued by SIRT3 activation. Cell Death Differ. 28, 1379–1397 (2021).
Teleanu, R. I. et al. Neurotransmitters—key factors in neurological and neurodegenerative disorders of the central nervous system. Int. J. Mol. Sci. 23, 5954 (2022).
Chen, Y., Xu, J. & Chen, Y. Regulation of neurotransmitters by the gut microbiota and effects on cognition in neurological disorders. Nutrients 13, 2099 (2021).
Margolis, K. G., Cryan, J. F. & Mayer, E. A. The microbiota-gut-brain axis: from motility to mood. Gastroenterology 160, 1486–1501 (2021).
Reigstad, C. S. et al. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 29, 1395–1403 (2015).
Zhai, L. et al. Ruminococcus gnavus plays a pathogenic role in diarrhea-predominant irritable bowel syndrome by increasing serotonin biosynthesis. Cell Host Microbe 31, 33–44.e35 (2023).
Clarke, G. et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 18, 666–673 (2013).
Fung, T. C. et al. Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nat. Microbiol. 4, 2064–2073 (2019).
Cirrito, J. R. et al. Effect of escitalopram on Aβ levels and plaque load in an Alzheimer mouse model. Neurology 95, e2666–e2674 (2020).
Cirrito, J. R. et al. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. USA 108, 14968–14973 (2011).
Rodríguez, J. J., Noristani, H. N. & Verkhratsky, A. The serotonergic system in ageing and Alzheimer’s disease. Prog. Neurobiol. 99, 15–41 (2012).
Smith, G. S. et al. Molecular imaging of the association between serotonin degeneration and beta-amyloid deposition in mild cognitive impairment. Neuroimage Clin. 37, 103322 (2023).
Whiley, L. et al. Metabolic phenotyping reveals a reduction in the bioavailability of serotonin and kynurenine pathway metabolites in both the urine and serum of individuals living with Alzheimer’s disease. Alzheimers Res. Ther. 13, 20 (2021).
Sheline, Y. I. et al. Effect of escitalopram dose and treatment duration on CSF Aβ levels in healthy older adults: a controlled clinical trial. Neurology 95, e2658–e2665 (2020).
Reddy, A. P. et al. Selective serotonin reuptake inhibitor citalopram ameliorates cognitive decline and protects against amyloid beta-induced mitochondrial dynamics, biogenesis, autophagy, mitophagy and synaptic toxicities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 30, 789–810 (2021).
Zhang, S. et al. Prebiotics modulate the microbiota-gut-brain axis and ameliorate cognitive impairment in APP/PS1 mice. Eur. J. Nutr. 62, 2991–3007 (2023).
Zhang, W. et al. Neuroprotective effects of polysaccharide from Sparassis crispa on Alzheimer’s disease-like mice: involvement of microbiota-gut-brain axis. Int. J. Biol. Macromol. 225, 974–986 (2023).
Wang, J. et al. Fecal microbiota transplantation improves VPA-induced ASD mice by modulating the serotonergic and glutamatergic synapse signaling pathways. Transl. Psychiatry 13, 17 (2023).
Tian, P. et al. Multi-Probiotics ameliorate major depressive disorder and accompanying gastrointestinal syndromes via serotonergic system regulation. J. Adv. Res. 45, 117–125 (2023).
Kong, Q. et al. Daily intake of Lactobacillus alleviates autistic-like behaviors by ameliorating the 5-hydroxytryptamine metabolic disorder in VPA-treated rats during weaning and sexual maturation. Food Funct. 12, 2591–2604 (2021).
Wang, Y. et al. Probiotics and fructo-oligosaccharide intervention modulate the microbiota-gut brain axis to improve autism spectrum reducing also the hyper-serotonergic state and the dopamine metabolism disorder. Pharm. Res. 157, 104784 (2020).
Wile, D. J. et al. Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: cross-sectional studies. Lancet Neurol. 16, 351–359 (2017).
Wilson, H. et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T α-synuclein parkinsonism: a cross-sectional study. Lancet Neurol. 18, 748–759 (2019).
Maillet, A. et al. The prominent role of serotonergic degeneration in apathy, anxiety and depression in de novo Parkinson’s disease. Brain 139, 2486–2502 (2016).
Maillet, A. et al. Serotonergic and dopaminergic lesions underlying parkinsonian neuropsychiatric signs. Mov. Disord. 36, 2888–2900 (2021).
Miquel-Rio, L. et al. Human α-synuclein overexpression in mouse serotonin neurons triggers a depressive-like phenotype. Rescue by oligonucleotide therapy. Transl. Psychiatry 12, 79 (2022).
Alarcón-Arís, D. et al. Selective α-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: potential therapy for Parkinson’s disease. Mol. Ther. 26, 550–567 (2018).
Dong, X. L. et al. Polymannuronic acid prevents dopaminergic neuronal loss via brain-gut-microbiota axis in Parkinson’s disease model. Int. J. Biol. Macromol. 164, 994–1005 (2020).
Sun, H. et al. Probiotics synergized with conventional regimen in managing Parkinson’s disease. NPJ Parkinsons Dis. 8, 62 (2022).
Liao, J. F. et al. Lactobacillus plantarum PS128 alleviates neurodegenerative progression in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse models of Parkinson’s disease. Brain Behav. Immun. 90, 26–46 (2020).
Kwoji, I. D., Aiyegoro, O. A., Okpeku, M. & Adeleke, M. A. Multi-Strain Probiotics: Synergy Among Isolates Enhances Biological Activities. Biology 10, 322 (2021).
Koh, W., Kwak, H., Cheong, E. & Lee, C. J. GABA tone regulation and its cognitive functions in the brain. Nat. Rev. Neurosci. 24, 523–539 (2023).
Bravo, J. A. et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 108, 16050–16055 (2011).
Kootte, R. S. et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. 26, 611–619.e616 (2017).
Janik, R. et al. Magnetic resonance spectroscopy reveals oral Lactobacillus promotion of increases in brain GABA, N-acetyl aspartate and glutamate. Neuroimage 125, 988–995 (2016).
Barrett, E. et al. γ-Aminobutyric acid production by culturable bacteria from the human intestine. J. Appl. Microbiol. 113, 411–417 (2012).
Duranti, S. et al. Bifidobacterium adolescentis as a key member of the human gut microbiota in the production of GABA. Sci. Rep. 10, 14112 (2020).
Horvath, T. D. et al. Bacteroides ovatus colonization influences the abundance of intestinal short chain fatty acids and neurotransmitters. iScience 25, 104158 (2022).
Zheng, J. et al. Interneuron accumulation of phosphorylated tau impairs adult hippocampal neurogenesis by suppressing GABAergic transmission. Cell Stem Cell 26, 331–345.e336 (2020).
Bi, D., Wen, L., Wu, Z. & Shen, Y. GABAergic dysfunction in excitatory and inhibitory (E/I) imbalance drives the pathogenesis of Alzheimer’s disease. Alzheimers Dement. 16, 1312–1329 (2020).
Tang, Y. et al. The hippocampus associated GABAergic neural network impairment in early-stage of Alzheimer’s disease. Ageing Res. Rev. 86, 101865 (2023).
Wu, Z., Guo, Z., Gearing, M. & Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 5, 4159 (2014).
Jo, S. et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896 (2014).
Carello-Collar, G. et al. The GABAergic system in Alzheimer’s disease: a systematic review with meta-analysis. Mol. Psychiatry https://doi.org/10.1038/s41380-023-02140-w (2023).
Fujii, Y. et al. Fecal metabolite of a gnotobiotic mouse transplanted with gut microbiota from a patient with Alzheimer’s disease. Biosci. Biotechnol. Biochem. 83, 2144–2152 (2019).
Liu, X. Y. et al. Amelioration of olfactory dysfunction in a mouse model of Parkinson’s disease via enhancing GABAergic signaling. Cell Biosci. 13, 101 (2023).
Terkelsen, M. H., Hvingelby, V. S. & Pavese, N. Molecular imaging of the GABAergic system in Parkinson’s disease and atypical parkinsonisms. Curr. Neurol. Neurosci. Rep. 22, 867–879 (2022).
Khan, A. F. et al. Patient-specific models link neurotransmitter receptor mechanisms with motor and visuospatial axes of Parkinson’s disease. Nat. Commun. 14, 6009 (2023).
Pan, S. et al. Probiotic Pediococcus pentosaceus ameliorates MPTP-induced oxidative stress via regulating the gut microbiota-gut-brain axis. Front. Cell Infect. Microbiol. 12, 1022879 (2022).
Mohebi, A. et al. Dissociable dopamine dynamics for learning and motivation. Nature 570, 65–70 (2019).
Krok, A. C. et al. Intrinsic dopamine and acetylcholine dynamics in the striatum of mice. Nature 621, 543–549 (2023).
da Silva, J. A., Tecuapetla, F., Paixão, V. & Costa, R. M. Dopamine neuron activity before action initiation gates and invigorates future movements. Nature 554, 244–248 (2018).
Chantranupong, L. et al. Dopamine and glutamate regulate striatal acetylcholine in decision-making. Nature 621, 577–585 (2023).
Ott, T. & Nieder, A. Dopamine and cognitive control in prefrontal cortex. Trends Cogn. Sci. 23, 213–234 (2019).
Cramb, K. M. L., Beccano-Kelly, D., Cragg, S. J. & Wade-Martins, R. Impaired dopamine release in Parkinson’s disease. Brain 146, 3117–3132 (2023).
Hamamah, S. et al. Role of microbiota-gut-brain axis in regulating dopaminergic signaling. Biomedicines 10, 436 (2022).
Armstrong, M. J. & Okun, M. S. Diagnosis and treatment of Parkinson disease: a review. J. Am. Med. Assoc. 323, 548–560 (2020).
Blesa, J. et al. Motor and non-motor circuit disturbances in early Parkinson disease: which happens first? Nat. Rev. Neurosci. 23, 115–128 (2022).
van Kessel, S. P. et al. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat. Commun. 10, 310 (2019).
Maini Rekdal, V. et al. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323 (2019).
Hu, X. et al. Piperine improves levodopa availability in the 6-OHDA-lesioned rat model of Parkinson’s disease by suppressing gut bacterial tyrosine decarboxylase. CNS Neurosci. Ther. https://doi.org/10.1111/cns.14383 (2023).
Choi, J. G. et al. Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Sci. Rep. 8, 1275 (2018).
Zhao, Z. et al. Fecal microbiota transplantation protects rotenone-induced Parkinson’s disease mice via suppressing inflammation mediated by the lipopolysaccharide-TLR4 signaling pathway through the microbiota-gut-brain axis. Microbiome 9, 226 (2021).
Liu, X. et al. Polymannuronic acid prebiotic plus Lacticaseibacillus rhamnosus GG probiotic as a novel synbiotic promoted their separate neuroprotection against Parkinson’s disease. Food Res. Int. 155, 111067 (2022).
Ananth, M. R. et al. Basal forebrain cholinergic signalling: development, connectivity and roles in cognition. Nat. Rev. Neurosci. 24, 233–251 (2023).
Sarkar, S. R., Mazumder, P. M. & Banerjee, S. Oligosaccharide and flavanoid mediated prebiotic interventions to treat gut dysbiosis associated cognitive decline. J. Neuroimmune Pharm. 17, 94–110 (2022).
Roy Sarkar, S., Mitra Mazumder, P. & Banerjee, S. Probiotics protect against gut dysbiosis associated decline in learning and memory. J. Neuroimmunol. 348, 577390 (2020).
Guo, Y. et al. A diet high in sugar and fat influences neurotransmitter metabolism and then affects brain function by altering the gut microbiota. Transl. Psychiatry 11, 328 (2021).
Liu, P. P., Xie, Y., Meng, X. Y. & Kang, J. S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target Ther. 4, 29 (2019).
Nimgampalle, M. & Kuna, Y. Anti-Alzheimer properties of probiotic, Lactobacillus plantarum MTCC 1325 in Alzheimer’s disease induced Albino rats. J. Clin. Diagn. Res. 11, Kc01–kc05 (2017).
Chen, D. et al. Prebiotic effect of fructooligosaccharides from Morinda officinalis on Alzheimer’s disease in rodent models by targeting the microbiota-gut-brain axis. Front. Aging Neurosci. 9, 403 (2017).
Nimgampalle, M. et al. Neurotransmitter systems in the etiology of major neurological disorders: emerging insights and therapeutic implications. Ageing Res Rev. 89, 101994 (2023).
de Ceglia, R. et al. Specialized astrocytes mediate glutamatergic gliotransmission in the CNS. Nature 622, 120–129 (2023).
Noh, K. et al. Cortical astrocytes modulate dominance behavior in male mice by regulating synaptic excitatory and inhibitory balance. Nat. Neurosci. 26, 1541–1554 (2023).
Durkee, C., Kofuji, P., Navarrete, M. & Araque, A. Astrocyte and neuron cooperation in long-term depression. Trends Neurosci. 44, 837–848 (2021).
Zheng, P. et al. The gut microbiome from patients with schizophrenia modulates the glutamate-glutamine-GABA cycle and schizophrenia-relevant behaviors in mice. Sci. Adv. 5, eaau8317 (2019).
Campanelli, F. et al. Striatal glutamatergic hyperactivity in Parkinson’s disease. Neurobiol. Dis. 168, 105697 (2022).
Romano, S. et al. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis. 7, 27 (2021).
Chang, C. H., Lin, C. H. & Lane, H. Y. d-glutamate and gut microbiota in Alzheimer’s disease. Int. J. Mol. Sci. 21, 2676 (2020).
Boyko, M. et al. The integrity of the blood-brain barrier as a critical factor for regulating glutamate levels in traumatic brain injury. Int. J. Mol. Sci. 24, 5897 (2023).
Zaragozá, R. Transport of Amino Acids Across The Blood-brain Barrier. Front. Physiol. 11, 973 (2020).
Bany Bakar, R., Reimann, F. & Gribble, F. M. The intestine as an endocrine organ and the role of gut hormones in metabolic regulation. Nat. Rev. Gastroenterol. Hepatol. 20, 784–796 (2023).
Hickey, J. W. et al. Organization of the human intestine at single-cell resolution. Nature 619, 572–584 (2023).
Gribble, F. M. & Reimann, F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat. Rev. Endocrinol. 15, 226–237 (2019).
Masse, K. E. & Lu, V. B. Short-chain fatty acids, secondary bile acids and indoles: gut microbial metabolites with effects on enteroendocrine cell function and their potential as therapies for metabolic disease. Front. Endocrinol. 14, 1169624 (2023).
Priest, C. & Tontonoz, P. Inter-organ cross-talk in metabolic syndrome. Nat. Metab. 1, 1177–1188 (2019).
Brown, R. M. et al. Potential gut-brain mechanisms behind adverse mental health outcomes of bariatric surgery. Nat. Rev. Endocrinol. 17, 549–559 (2021).
Buntwal, L. et al. Ghrelin-mediated hippocampal neurogenesis: implications for health and disease. Trends Endocrinol. Metab. 30, 844–859 (2019).
Tian, J. et al. Liver-expressed antimicrobial peptide 2 elevation contributes to age-associated cognitive decline. JCI Insight 8, e166175 (2023).
Perry, R. J. et al. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 534, 213–217 (2016).
Han, H. et al. From gut microbiota to host appetite: gut microbiota-derived metabolites as key regulators. Microbiome 9, 162 (2021).
Tian, J. et al. Disrupted hippocampal growth hormone secretagogue receptor 1α interaction with dopamine receptor D1 plays a role in Alzheimer’s disease. Sci. Transl. Med. 11, eaav6278 (2019).
Bonfili, L. et al. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 7, 2426 (2017).
Fujitsuka, N. et al. Increased ghrelin signaling prolongs survival in mouse models of human aging through activation of sirtuin1. Mol. Psychiatry 21, 1613–1623 (2016).
Dhurandhar, E. J., Allison, D. B., van Groen, T. & Kadish, I. Hunger in the absence of caloric restriction improves cognition and attenuates Alzheimer’s disease pathology in a mouse model. PLoS ONE 8, e60437 (2013).
Kaiser, K. A. et al. The effect of a pharmaceutical ghrelin agonist on lifespan in C57BL/6J male mice: a controlled experiment. Aging Cell. 22, e13787 (2023).
Shi, L. et al. Peptide hormone ghrelin enhances neuronal excitability by inhibition of Kv7/KCNQ channels. Nat. Commun. 4, 1435 (2013).
Andrews, Z. B. et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J. Neurosci. 29, 14057–14065 (2009).
Gong, B. et al. Ghrelin promotes midbrain neural stem cells differentiation to dopaminergic neurons through Wnt/β-catenin pathway. J. Cell Physiol. 235, 8558–8570 (2020).
Suda, Y. et al. Down-regulation of ghrelin receptors on dopaminergic neurons in the substantia nigra contributes to Parkinson’s disease-like motor dysfunction. Mol. Brain 11, 6 (2018).
Song, N. et al. Assessments of plasma ghrelin levels in the early stages of Parkinson’s disease. Mov. Disord. 32, 1487–1491 (2017).
Hornsby, A. K. E. et al. Unacylated-ghrelin impairs hippocampal neurogenesis and memory in mice and is altered in Parkinson’s dementia in humans. Cell Rep. Med. 1, 100120 (2020).
Unger, M. M. et al. Postprandial ghrelin response is reduced in patients with Parkinson’s disease and idiopathic REM sleep behaviour disorder: a peripheral biomarker for early Parkinson’s disease? J. Neurol. 258, 982–990 (2011).
Jiang, H., Li, L. J., Wang, J. & Xie, J. X. Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp. Neurol. 212, 532–537 (2008).
Moon, M. et al. Neuroprotective effect of ghrelin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease by blocking microglial activation. Neurotox. Res. 15, 332–347 (2009).
Wang, H. et al. Ghrelin protects dopaminergic neurons against MPTP neurotoxicity through promoting autophagy and inhibiting endoplasmic reticulum mediated apoptosis. Brain Res. 1746, 147023 (2020).
He, X. et al. Acylated ghrelin is protective against 6-OHDA-induced neurotoxicity by regulating autophagic flux. Front. Pharm. 11, 586302 (2020).
Rees, D. et al. Acyl-ghrelin attenuates neurochemical and motor deficits in the 6-OHDA model of Parkinson’s disease. Cell Mol. Neurobiol. 43, 2377–2384 (2023).
Jiao, L. et al. Early low-dose ghrelin intervention via miniosmotic pumps could protect against the progressive dopaminergic neuron loss in Parkinson’s disease mice. Neurobiol. Aging 101, 70–78 (2021).
Myers, M. G., Affinati, A. H., Richardson, N. & Schwartz, M. W. Central nervous system regulation of organismal energy and glucose homeostasis. Nat. Metab. 3, 737–750 (2021).
Pérez-Pérez, A. et al. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 35, 71–84 (2017).
Cunnane, S. C. et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 19, 609–633 (2020).
Casado, M. E., Collado-Pérez, R., Frago, L. M. & Barrios, V. Recent advances in the knowledge of the mechanisms of leptin physiology and actions in neurological and metabolic pathologies. Int. J. Mol. Sci. 24, 1422 (2023).
Liu, G. Y. & Sabatini, D. M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203 (2020).
Song, Z. H. et al. Impact of ectopic fat on brain structure and cognitive function: a systematic review and meta-analysis from observational studies. Front. Neuroendocrinol. 70, 101082 (2023).
Anand, S. S. et al. Evaluation of adiposity and cognitive function in adults. JAMA Netw. Open 5, e2146324 (2022).
Deng, Y. T. et al. Association of life course adiposity with risk of incident dementia: a prospective cohort study of 322,336 participants. Mol. Psychiatry 27, 3385–3395 (2022).
Bäckhed, F., Manchester, J. K., Semenkovich, C. F. & Gordon, J. I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 104, 979–984 (2007).
Schéle, E. et al. The gut microbiota reduces leptin sensitivity and the expression of the obesity-suppressing neuropeptides proglucagon (Gcg) and brain-derived neurotrophic factor (Bdnf) in the central nervous system. Endocrinology 154, 3643–3651 (2013).
Hamamah, S. & Covasa, M. Gut microbiota restores central neuropeptide deficits in germ-free mice. Int. J. Mol. Sci. 23, 11756 (2022).
Heiss, C. N. et al. The gut microbiota regulates hypothalamic inflammation and leptin sensitivity in western diet-fed mice via a GLP-1R-dependent mechanism. Cell Rep. 35, 109163 (2021).
Noormohammadi, M. et al. The effect of probiotic and synbiotic supplementation on appetite-regulating hormones and desire to eat: a systematic review and meta-analysis of clinical trials. Pharm. Res. 187, 106614 (2023).
Lieb, W. et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. J. Am. Med. Assoc. 302, 2565–2572 (2009).
Mooldijk, S. S., Ikram, M. K. & Ikram, M. A. Adiponectin, leptin, and resistin and the risk of dementia. J. Gerontol. A Biol. Sci. Med. Sci. 77, 1245–1249 (2022).
García-García, I., Fernández-Andújar, M., Narváez, M. & García-Casares, N. Assessing adipokines as potential biomarkers of dementia, Alzheimer’s disease, and mild cognitive impairment: a systematic review and meta-analysis. Obes. Rev. 24, e13573 (2023).
Hamilton, K. & Harvey, J. The neuronal actions of leptin and the implications for treating Alzheimer’s disease. Pharmaceuticals 14, 52 (2021).
Hamilton, K., Morrow, K., Markantoni, E. & Harvey, J. Leptin prevents aberrant targeting of tau to hippocampal synapses via PI 3 kinase driven inhibition of GSK3β. J. Neurochem. 167, 520–537 (2023).
Lilamand, M. et al. Plasma leptin is associated with amyloid CSF biomarkers and Alzheimer’s disease diagnosis in cognitively impaired patients. J. Gerontol. A Biol. Sci. Med. Sci. 78, 645–652 (2023).
Yang, S. Q. et al. Leptin mediates protection of hydrogen sulfide against 6-hydroxydopamine-induced Parkinson’s disease: involving enhancement in Warburg effect. Neurochem. Int. 135, 104692 (2020).
Weng, Z. et al. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 282, 34479–34491 (2007).
Regensburger, M. et al. Emerging roles of leptin in Parkinson’s disease: chronic inflammation, neuroprotection and more? Brain Behav. Immun. 107, 53–61 (2023).
de Vrind, V. A. J. et al. Leptin receptor expressing neurons in the substantia nigra regulate locomotion, and in the ventral tegmental area motivation and feeding. Front. Endocrinol. 12, 680494 (2021).
Figlewicz, D. P. et al. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 964, 107–115 (2003).
Rahnemayan, S. et al. Leptin levels in patients with Parkinson’s disease: a systematic review and meta-analysis. Clin. Nutr. ESPEN 41, 104–109 (2021).
Hammoud, R. & Drucker, D. J. Beyond the pancreas: contrasting cardiometabolic actions of GIP and GLP1. Nat. Rev. Endocrinol. 19, 201–216 (2023).
Kopp, K. O., Glotfelty, E. J., Li, Y. & Greig, N. H. Glucagon-like peptide-1 (GLP-1) receptor agonists and neuroinflammation: implications for neurodegenerative disease treatment. Pharm. Res. 186, 106550 (2022).
Yoon, H. S. et al. Akkermansia muciniphila secretes a glucagon-like peptide-1-inducing protein that improves glucose homeostasis and ameliorates metabolic disease in mice. Nat. Microbiol. 6, 563–573 (2021).
Singh, A. et al. Inulin fiber dose-dependently modulates energy balance, glucose tolerance, gut microbiota, hormones and diet preference in high-fat-fed male rats. J. Nutr. Biochem. 59, 142–152 (2018).
Weninger, S. N. et al. Oligofructose improves small intestinal lipid-sensing mechanisms via alterations to the small intestinal microbiota. Microbiome 11, 169 (2023).
Pearson-Stuttard, J. et al. Trends in predominant causes of death in individuals with and without diabetes in England from 2001 to 2018: an epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol. 9, 165–173 (2021).
Dove, A. et al. The impact of diabetes on cognitive impairment and its progression to dementia. Alzheimers Dement. 17, 1769–1778 (2021).
Wang, Y. et al. Onset age of diabetes and incident dementia: a prospective cohort study. J. Affect Disord. 329, 493–499 (2023).
Ballard, C. et al. Drug repositioning and repurposing for Alzheimer disease. Nat. Rev. Neurol. 16, 661–673 (2020).
Kong, F. et al. Glucagon-like peptide 1 (GLP-1) receptor agonists in experimental Alzheimer’s disease models: a systematic review and meta-analysis of preclinical studies. Front. Pharm. 14, 1205207 (2023).
Park, J. S. et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol. Commun. 9, 78 (2021).
Cukierman-Yaffe, T. et al. Effect of dulaglutide on cognitive impairment in type 2 diabetes: an exploratory analysis of the REWIND trial. Lancet Neurol. 19, 582–590 (2020).
Nørgaard, C. H. et al. Treatment with glucagon-like peptide-1 receptor agonists and incidence of dementia: Data from pooled double-blind randomized controlled trials and nationwide disease and prescription registers. Alzheimers Dement. 8, e12268 (2022).
Wong, C. K. et al. Divergent roles for the gut intraepithelial lymphocyte GLP-1R in control of metabolism, microbiota, and T cell-induced inflammation. Cell Metab. 34, 1514–1531.e1517 (2022).
Rhee, S. Y. et al. Association between glycemic status and the risk of Parkinson disease: a nationwide population-based study. Diabetes Care 43, 2169–2175 (2020).
Han, K., Kim, B., Lee, S. H. & Kim, M. K. A nationwide cohort study on diabetes severity and risk of Parkinson disease. NPJ Parkinsons Dis. 9, 11 (2023).
De Pablo-Fernandez, E. et al. Association between diabetes and subsequent Parkinson disease: a record-linkage cohort study. Neurology 91, e139–e142 (2018).
Athauda, D. et al. The impact of type 2 diabetes in Parkinson’s disease. Mov. Disord. 37, 1612–1623 (2022).
Brauer, R. et al. Diabetes medications and risk of Parkinson’s disease: a cohort study of patients with diabetes. Brain 143, 3067–3076 (2020).
Athauda, D. et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 390, 1664–1675 (2017).
Yue, M. et al. Neurotrophic role of the next-generation probiotic strain L. lactis MG1363-pMG36e-GLP-1 on Parkinson’s disease via inhibiting ferroptosis. Nutrients 14, 4886 (2022).
Fang, X. et al. Neuroprotective effects of an engineered commensal bacterium in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine Parkinson disease mouse model via producing glucagon-like peptide-1. J. Neurochem. 150, 441–452 (2019).
Fang, X. et al. Therapeutic effect of GLP-1 engineered strain on mice model of Alzheimer’s disease and Parkinson’s disease. AMB Express 10, 80 (2020).
Berriat, F., Lobsiger, C. S. & Boillée, S. The contribution of the peripheral immune system to neurodegeneration. Nat. Neurosci. 26, 942–954 (2023).
Douglas, A., Stevens, B. & Lynch, L. Interleukin-17 as a key player in neuroimmunometabolism. Nat. Metab. 5, 1088–1100 (2023).
Ramanan, D. et al. Regulatory T cells in the face of the intestinal microbiota. Nat. Rev. Immunol. 23, 749–762 (2023).
Pellegrini, C. et al. The intestinal barrier in disorders of the central nervous system. Lancet Gastroenterol. Hepatol. 8, 66–80 (2023).
Chandra, S., Sisodia, S. S. & Vassar, R. J. The gut microbiome in Alzheimer’s disease: what we know and what remains to be explored. Mol. Neurodegener. 18, 9 (2023).
Martin, S., Battistini, C. & Sun, J. A gut feeling in amyotrophic lateral sclerosis: microbiome of mice and men. Front. Cell Infect. Microbiol. 12, 839526 (2022).
Wang, X. et al. C-type lectin-like receptor 2 and zonulin are associated with mild cognitive impairment and Alzheimer’s disease. Acta Neurol. Scand. 141, 250–255 (2020).
Gustafsson, J. K. & Johansson, M. E. V. The role of goblet cells and mucus in intestinal homeostasis. Nat. Rev. Gastroenterol. Hepatol. 19, 785–803 (2022).
Paone, P. & Cani, P. D. Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut 69, 2232–2243 (2020).
Johansson, MalinE. V. et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe 18, 582–592 (2015).
Hetz, C., Zhang, K. & Kaufman, R. J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21, 421–438 (2020).
Cloots, E. et al. Evolution and function of the epithelial cell-specific ER stress sensor IRE1β. Mucosal Immunol. 14, 1235–1246 (2021).
Parker, A. et al. Fecal microbiota transfer between young and aged mice reverses hallmarks of the aging gut, eye, and brain. Microbiome 10, 68 (2022).
Xu, Q. et al. Marked response of rat ileal and colonic microbiota after the establishment of Alzheimer’s disease model with bilateral intraventricular injection of Aβ (1-42). Front. Microbiol. 13, 819523 (2022).
Qian, X.-h et al. Injection of amyloid-β to lateral ventricle induces gut microbiota dysbiosis in association with inhibition of cholinergic anti-inflammatory pathways in Alzheimer’s disease. J. Neuroinflamm. 19, 236 (2022).
Kim, M. S. et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 69, 283–294 (2020).
Wang, D. D. et al. The gut microbiome modulates the protective association between a Mediterranean diet and cardiometabolic disease risk. Nat. Med. 27, 333–343 (2021).
Makki, K., Deehan, E. C., Walter, J. & Bäckhed, F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 23, 705–715 (2018).
Ostrem Loss, E. et al. Carbohydrate complexity limits microbial growth and reduces the sensitivity of human gut communities to perturbations. Nat. Ecol. Evol. 7, 127–142 (2023).
Seethaler, B. et al. Short-chain fatty acids are key mediators of the favorable effects of the Mediterranean diet on intestinal barrier integrity: data from the randomized controlled LIBRE trial. Am. J. Clin. Nutr. 116, 928–942 (2022).
Desai, M. S. et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e1321 (2016).
Schroeder, B. O. et al. Bifidobacteria or Fiber protects against diet-induced microbiota-mediated colonic mucus deterioration. Cell Host Microbe 23, 27–40.e27 (2018).
Dapa, T. et al. Diet leaves a genetic signature in a keystone member of the gut microbiota. Cell Host Microbe 30, 183–199.e110 (2022).
Więckowska-Gacek, A., Mietelska-Porowska, A., Wydrych, M. & Wojda, U. Western diet as a trigger of Alzheimer’s disease: from metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res. Rev. 70, 101397 (2021).
Shi, H. et al. β-glucan attenuates cognitive impairment via the gut-brain axis in diet-induced obese mice. Microbiome 8, 143 (2020).
Pan, W. et al. Dimethyl itaconate ameliorates cognitive impairment induced by a high-fat diet via the gut-brain axis in mice. Microbiome 11, 30 (2023).
Zhao, N. et al. Intestinal dysbiosis mediates cognitive impairment via the intestine and brain NLRP3 inflammasome activation in chronic sleep deprivation. Brain Behav. Immun. 108, 98–117 (2023).
Wang, Z. et al. Gut microbiota modulates the inflammatory response and cognitive impairment induced by sleep deprivation. Mol. Psychiatry 26, 6277–6292 (2021).
Li, N. et al. Akkermansia muciniphila supplementation prevents cognitive impairment in sleep-deprived mice by modulating microglial engulfment of synapses. Gut Microbes 15, 2252764 (2023).
Sabia, S. et al. Association of sleep duration in middle and old age with incidence of dementia. Nat. Commun. 12, 2289 (2021).
Shimizu, Y. et al. Shorter sleep time relates to lower human defensin 5 secretion and compositional disturbance of the intestinal microbiota accompanied by decreased short-chain fatty acid production. Gut Microbes 15, 2190306 (2023).
Yu, H. X. et al. Ongoing clinical trials in aging-related tissue fibrosis and new findings related to AhR pathways. Aging Dis. 13, 732–752 (2022).
Scott, S. A., Fu, J. & Chang, P. V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 117, 19376–19387 (2020).
Metidji, A. et al. The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 49, 353–362.e355 (2018).
Wiggins, B. G. et al. Endothelial sensing of AHR ligands regulates intestinal homeostasis. Nature 621, 821–829 (2023).
Terstappen, G. C., Meyer, A. H., Bell, R. D. & Zhang, W. Strategies for delivering therapeutics across the blood–brain barrier. Nat. Rev. Drug Discov. 20, 362–383 (2021).
Barisano, G. et al. Blood-brain barrier link to human cognitive impairment and Alzheimer’s Disease. Nat. Cardiovasc. Res. 1, 108–115 (2022).
Amann, L., Masuda, T. & Prinz, M. Mechanisms of myeloid cell entry to the healthy and diseased central nervous system. Nat. Immunol. 24, 393–407 (2023).
Knox, E. G. et al. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 27, 2659–2673 (2022).
Mendiola, A. S. et al. Defining blood-induced microglia functions in neurodegeneration through multiomic profiling. Nat. Immunol. 24, 1173–1187 (2023).
Sweeney, M. D., Sagare, A. P. & Zlokovic, B. V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150 (2018).
Hoyles, L. et al. Regulation of blood–brain barrier integrity by microbiome-associated methylamines and cognition by trimethylamine N-oxide. Microbiome 9, 235 (2021).
Braniste, V. et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 6, 263ra158–263ra158 (2014).
Hoyles, L. et al. Microbiome–host systems interactions: protective effects of propionate upon the blood–brain barrier. Microbiome 6, 55 (2018).
Stachulski, A. V. et al. A host-gut microbial amino acid co-metabolite, p-cresol glucuronide, promotes blood-brain barrier integrity in vivo. Tissue Barriers 11, 2073175 (2023).
Sun, N. et al. Antibiotic-induced microbiome depletion in adult mice disrupts blood-brain barrier and facilitates brain infiltration of monocytes after bone-marrow transplantation. Brain Behav. Immun. 92, 102–114 (2021).
Knox, E. G. et al. Microbial-derived metabolites induce actin cytoskeletal rearrangement and protect blood-brain barrier function. iScience 25, 105648 (2022).
Voigt, R. M. et al. Gut microbial metabolites in Parkinson’s disease: association with lifestyle, disease characteristics, and treatment status. Neurobiol. Dis. 170, 105780 (2022).
Li, D. et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell 17, e12768 (2018).
Deng, Y. et al. Higher circulating trimethylamine N-oxide aggravates cognitive impairment probably via downregulating hippocampal SIRT1 in vascular dementia rats. Cells 11, 3650 (2022).
Zhou, S. et al. Dietary choline metabolite TMAO impairs cognitive function and induces hippocampal synaptic plasticity declining through the mTOR/P70S6K/4EBP1 pathway. Food Funct. 14, 2881–2895 (2023).
Hernandez, L. et al. Blood–brain barrier and gut barrier dysfunction in chronic kidney disease with a focus on circulating biomarkers and tight junction proteins. Sci. Rep. 12, 4414 (2022).
Yuan, J. et al. Is dietary choline intake related to dementia and Alzheimer’s disease risks? Results from the Framingham Heart Study. Am. J. Clin. Nutr. 116, 1201–1207 (2022).
Velazquez, R. et al. Lifelong choline supplementation ameliorates Alzheimer’s disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell 18, e13037 (2019).
Dave, N. et al. Dietary choline intake is necessary to prevent systems-wide organ pathology and reduce Alzheimer’s disease hallmarks. Aging Cell 22, e13775 (2023).
Xue, M. et al. Diabetes mellitus and risks of cognitive impairment and dementia: a systematic review and meta-analysis of 144 prospective studies. Ageing Res. Rev. 55, 100944 (2019).
Jang, M., Choi, N. & Kim, H. N. Hyperglycemic neurovasculature-on-a-chip to study the effect of SIRT1-targeted therapy for the type 3 diabetes “Alzheimer’s disease”. Adv. Sci. 9, e2201882 (2022).
Jeong, J.-h, Lee, D. H. & Song, J. HMGB1 signaling pathway in diabetes-related dementia: Blood-brain barrier breakdown, brain insulin resistance, and Aβ accumulation. Biomed. Pharmacother. 150, 112933 (2022).
Li, J. et al. Interplay between diet and gut microbiome, and circulating concentrations of trimethylamine N-oxide: findings from a longitudinal cohort of US men. Gut 71, 724–733 (2022).
Rustenhoven, J. & Kipnis, J. Brain borders at the central stage of neuroimmunology. Nature 612, 417–429 (2022).
Rustenhoven, J. et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 184, 1000–1016.e1027 (2021).
Neutzner, M. et al. Impact of aging on meningeal gene expression. Fluids Barriers CNS 20, 12 (2023).
Brioschi, S. et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science 373, eabf9277 (2021).
Da Mesquita, S. et al. Aging-associated deficit in CCR7 is linked to worsened glymphatic function, cognition, neuroinflammation, and β-amyloid pathology. Sci. Adv. 7, eabe4601 (2021).
Unger, M. S. et al. CD8 + T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav. Immun. 89, 67–86 (2020).
Schläger, C. et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 530, 349–353 (2016).
Gate, D. et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020).
Da Mesquita, S. et al. Meningeal lymphatics affect microglia responses and anti-Aβ immunotherapy. Nature 593, 255–260 (2021).
Zhang, Y. et al. Mucosal-associated invariant T cells restrict reactive oxidative damage and preserve meningeal barrier integrity and cognitive function. Nat. Immunol. 23, 1714–1725 (2022).
Godfrey, D. I., Koay, H.-F., McCluskey, J. & Gherardin, N. A. The biology and functional importance of MAIT cells. Nat. Immunol. 20, 1110–1128 (2019).
Kjer-Nielsen, L. et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491, 717–723 (2012).
Koay, H.-F. et al. A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat. Immunol. 17, 1300–1311 (2016).
Legoux, F. et al. Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science 366, 494–499 (2019).
Constantinides, M. G. et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science 366, eaax6624 (2019).
Toubal, A. et al. Mucosal-associated invariant T cells promote inflammation and intestinal dysbiosis leading to metabolic dysfunction during obesity. Nat. Commun. 11, 3755 (2020).
Zhang, W., Xiao, D., Mao, Q. & Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target Ther. 8, 267 (2023).
Zheng, D., Liwinski, T. & Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 30, 492–506 (2020).
Schnell, A., Littman, D. R. & Kuchroo, V. K. TH17 cell heterogeneity and its role in tissue inflammation. Nat. Immunol. 24, 19–29 (2023).
Li, D. et al. Gut microbial metabolite deoxycholic acid facilitates Th17 differentiation through modulating cholesterol biosynthesis and participates in high-fat diet-associated colonic inflammation. Cell Biosci. 13, 186 (2023).
Wilck, N. et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 551, 585–589 (2017).
Kawano, Y. et al. Microbiota imbalance induced by dietary sugar disrupts immune-mediated protection from metabolic syndrome. Cell 185, 3501–3519.e3520 (2022).
Sefik, E. et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997 (2015).
Song, X. et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 577, 410–415 (2020).
Ohnmacht, C. et al. The microbiota regulates type 2 immunity through RORγt + T cells. Science 349, 989–993 (2015).
Hanna, B. S. et al. The gut microbiota promotes distal tissue regeneration via RORγ(+) regulatory T cell emissaries. Immunity 56, 829–846.e828 (2023).
Kim, C. H. Complex regulatory effects of gut microbial short-chain fatty acids on immune tolerance and autoimmunity. Cell Mol. Immunol. 20, 341–350 (2023).
Rosser, E. C. et al. Microbiota-derived metabolites suppress arthritis by amplifying aryl-hydrocarbon receptor activation in regulatory B cells. Cell Metab. 31, 837–851.e810 (2020).
Arpaia, N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Siracusa, F. et al. Short-term dietary changes can result in mucosal and systemic immune depression. Nat. Immunol. 24, 1473–1486 (2023).
Yan, Y. et al. Bacteroides uniformis-induced perturbations in colonic microbiota and bile acid levels inhibit TH17 differentiation and ameliorate colitis developments. NPJ Biofilms Microbiomes 9, 56 (2023).
Hill, C. et al. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 11, 506–514 (2014).
Cunningham, M. et al. Shaping the future of probiotics and prebiotics. Trends Microbiol. 29, 667–685 (2021).
Hitch, T. C. A. et al. Microbiome-based interventions to modulate gut ecology and the immune system. Mucosal Immunol. 15, 1095–1113 (2022).
Cani, P. D. et al. Akkermansia muciniphila: paradigm for next-generation beneficial microorganisms. Nat. Rev. Gastroenterol. Hepatol. 19, 625–637 (2022).
Gibson, G. R. et al. Expert consensus document: the International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 14, 491–502 (2017).
Lordan, C., Thapa, D., Ross, R. P. & Cotter, P. D. Potential for enriching next-generation health-promoting gut bacteria through prebiotics and other dietary components. Gut Microbes 11, 1–20 (2020).
Nearing, J. T., Comeau, A. M. & Langille, M. G. I. Identifying biases and their potential solutions in human microbiome studies. Microbiome 9, 113 (2021).
Vandeputte, D. et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511 (2017).
Lloréns-Rico, V. et al. Benchmarking microbiome transformations favors experimental quantitative approaches to address compositionality and sampling depth biases. Nat. Commun. 12, 3562 (2021).
Allali, I. et al. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol. 17, 194 (2017).
Costea, P. I. et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 35, 1069–1076 (2017).
Maghini, D. G. et al. Quantifying bias introduced by sample collection in relative and absolute microbiome measurements. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01754-3 (2023).
Poulsen, C. S., Ekstrøm, C. T., Aarestrup, F. M. & Pamp, S. J. Library preparation and sequencing platform introduce bias in metagenomic-based characterizations of microbiomes. Microbiol. Spectr. 10, e00090–00022 (2022).
Cammarota, G. et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 66, 569–580 (2017).
Lopetuso, L. R. et al. The first international Rome consensus conference on gut microbiota and faecal microbiota transplantation in inflammatory bowel disease. Gut 72, 1642–1650 (2023).
Simpson, R. C., Shanahan, E. R., Scolyer, R. A. & Long, G. V. Towards modulating the gut microbiota to enhance the efficacy of immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 20, 697–715 (2023).
Khoruts, A., Staley, C. & Sadowsky, M. J. Faecal microbiota transplantation for Clostridioides difficile: mechanisms and pharmacology. Nat. Rev. Gastroenterol. Hepatol. 18, 67–80 (2021).
Sorbara, M. T. & Pamer, E. G. Microbiome-based therapeutics. Nat. Rev. Microbiol. 20, 365–380 (2022).
Feuerstadt, P. et al. SER-109, an oral microbiome therapy for recurrent Clostridioides difficile infection. New Engl. J. Med. 386, 220–229 (2022).
Khanna, S. et al. Efficacy and safety of RBX2660 in PUNCH CD3, a phase III, randomized, double-blind, placebo-controlled trial with a Bayesian primary analysis for the prevention of recurrent Clostridioides difficile infection. Drugs 82, 1527–1538 (2022).
Sims, M. D. et al. Safety and tolerability of SER-109 as an investigational microbiome therapeutic in adults with recurrent Clostridioides difficile Infection: a phase 3, open-label, single-arm trial. JAMA Netw. Open 6, e2255758 (2023).
Holvoet, T. et al. Fecal microbiota transplantation reduces symptoms in some patients with irritable bowel syndrome with predominant abdominal bloating: short- and long-term results from a placebo-controlled randomized trial. Gastroenterology 160, 145–157.e148 (2021).
Xu, C. et al. Novel nano-encapsulated probiotic agents: encapsulate materials, delivery, and encapsulation systems. J. Control Release 349, 184–205 (2022).
Razavi, S. et al. Microencapsulating polymers for probiotics delivery systems: preparation, characterization, and applications. Food Hydrocoll. 120, 106882 (2021).
Wang, R. et al. Poly‐γ‐glutamic acid microgel‐encapsulated probiotics with gastric acid resistance and smart inflammatory factor targeted delivery performance to ameliorate colitis. Adv. Funct. Mater. 32, 2113034 (2022).
Cao, F. et al. Artificial-enzymes-armed Bifidobacterium longum probiotics for alleviating intestinal inflammation and microbiota dysbiosis. Nat. Nanotechnol. 18, 617–627 (2023).
Pan, J. et al. A single-cell nanocoating of probiotics for enhanced amelioration of antibiotic-associated diarrhea. Nat. Commun. 13, 2117 (2022).
Geng, Z. et al. Biointerface mineralization generates ultraresistant gut microbes as oral biotherapeutics. Sci. Adv. 9, eade0997 (2023).
Pan, H. et al. Light-sensitive Lactococcus lactis for microbe–gut–brain axis regulating via upconversion optogenetic micro-nano system. ACS Nano 16, 6049–6063 (2022).
Zhang, X. et al. A red light-controlled probiotic bio-system for in-situ gut-brain axis regulation. Biomaterials 294, 122005 (2023).
Woo, A. Y. M. et al. Targeting the human gut microbiome with small-molecule inhibitors. Nat. Rev. Chem. 7, 319–339 (2023).
Deehan, E. C. et al. Precision microbiome modulation with discrete dietary fiber structures directs short-chain fatty acid production. Cell Host Microbe 27, 389–404.e386 (2020).
Lancaster, S. M. et al. Global, distinctive, and personal changes in molecular and microbial profiles by specific fibers in humans. Cell Host Microbe 30, 848–862.e847 (2022).
Zeng, X. et al. Gut bacterial nutrient preferences quantified in vivo. Cell 185, 3441–3456.e3419 (2022).
Oliveira, R. A. & Pamer, E. G. Assembling symbiotic bacterial species into live therapeutic consortia that reconstitute microbiome functions. Cell Host Microbe 31, 472–484 (2023).
Porcari, S. et al. Key determinants of success in fecal microbiota transplantation: from microbiome to clinic. Cell Host Microbe 31, 712–733 (2023).
Ianiro, G. et al. Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat. Med. 28, 1913–1923 (2022).
Schmidt, T. S. B. et al. Drivers and determinants of strain dynamics following fecal microbiota transplantation. Nat. Med. 28, 1902–1912 (2022).
DeFilipp, Z. et al. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. New Engl. J. Med. 381, 2043–2050 (2019).
Zellmer, C. et al. Shiga toxin-producing Escherichia coli transmission via fecal microbiota transplant. Clin. Infect. Dis. 72, e876–e880 (2021).
Nichols, E. et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease 2019. Lancet Public Health 7, E105–E125 (2022).
Walter, J., Armet, A. M., Finlay, B. B. & Shanahan, F. Establishing or exaggerating causality for the gut microbiome: lessons from human microbiota-associated rodents. Cell 180, 221–232 (2020).
Folz, J. et al. Human metabolome variation along the upper intestinal tract. Nat. Metab. 5, 777–788 (2023).
Oliveira, R. A., Cabral, V., Torcato, I. & Xavier, K. B. Deciphering the quorum-sensing lexicon of the gut microbiota. Cell Host Microbe 31, 500–512 (2023).
Lee, H.-J., Lee, K.-E., Kim, J.-K. & Kim, D.-H. Suppression of gut dysbiosis by Bifidobacterium longum alleviates cognitive decline in 5XFAD transgenic and aged mice. Sci. Rep. 9, 11814 (2019).
Ou, Z. et al. Protective effects of Akkermansia muciniphila on cognitive deficits and amyloid pathology in a mouse model of Alzheimer’s disease. Nutr. Diabetes 10, 12 (2020).
Chen, L.-H. et al. Lacticaseibacillus paracasei PS23 effectively modulates gut microbiota composition and improves gastrointestinal function in aged SAMP8 mice. Nutrients 13, 1116 (2021).
Yang, X. et al. Probiotics modulate the microbiota–gut–brain axis and improve memory deficits in aged SAMP8 mice. Acta Pharm. Sin. B 10, 475–487 (2020).
Wang, Y. et al. Modulation of the gut microbiota and glycometabolism by a probiotic to alleviate amyloid accumulation and cognitive impairments in AD rats. Mol. Nutr. Food Res. 66, e2200265 (2022).
Sancandi, M. et al. Effects of a probiotic suspension Symprove™ on a rat early-stage Parkinson’s disease model. Front. Aging Neurosci. 14, 986127 (2022).
Labarre, A. et al. Fatty acids derived from the probiotic Lacticaseibacillus rhamnosus HA-114 suppress age-dependent neurodegeneration. Commun. Biol. 5, 1340 (2022).
Han, D. et al. Prebiotics regulation of intestinal microbiota attenuates cognitive dysfunction induced by surgery stimulation in APP/PS1 mice. Aging Dis. 11, 1029–1045 (2020).
Luo, S. et al. A monomeric polysaccharide from Polygonatum sibiricum improves cognitive functions in a model of Alzheimer’s disease by reshaping the gut microbiota. Int. J. Biol. Macromol. 213, 404–415 (2022).
Fu, J. et al. In-depth investigation of the mechanisms of Schisandra chinensis polysaccharide mitigating Alzheimer’s disease rat via gut microbiota and feces metabolomics. Int. J. Biol. Macromol. 232, 123488 (2023).
Sun, Y. et al. Dendrobium officinale polysaccharide attenuates cognitive impairment in circadian rhythm disruption mice model by modulating gut microbiota. Int. J. Biol. Macromol. 217, 677–688 (2022).
Song, L., Gao, Y., Zhang, X. & Le, W. Galactooligosaccharide improves the animal survival and alleviates motor neuron death in SOD1G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 246, 281–290 (2013).
Sun, J. et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 9, 189 (2019).
Xiao, J. et al. Probiotic Bifidobacterium breve in improving cognitive functions of older adults with suspected mild cognitive impairment: a randomized, double-blind, placebo-controlled trial. J. Alzheimers Dis. 77, 139–147 (2020).
Kobayashi, Y., Kuhara, T., Oki, M. & Xiao, J. Z. Effects of Bifidobacterium breve A1 on the cognitive function of older adults with memory complaints: a randomised, double-blind, placebo-controlled trial. Benef. Microbes 10, 511–520 (2019).
Hwang, Y.-H. et al. Efficacy and safety of Lactobacillus Plantarum C29-fermented soybean (DW2009) in individuals with mild cognitive impairment: a 12-week, multi-center, randomized, double-blind, placebo-controlled clinical trial. Nutrients 11, 305 (2019).
Akbari, E. et al. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer’s disease: a randomized, double-blind and controlled trial. Front. Aging Neurosci. 8, 256 (2016).
Akhgarjand, C. et al. Effects of probiotic supplements on cognition, anxiety, and physical activity in subjects with mild and moderate Alzheimer’s disease: a randomized, double-blind, and placebo-controlled study. Front. Aging Neurosci. 14, 1032494 (2022).
Tamtaji, O. R. et al. Probiotic and selenium co-supplementation, and the effects on clinical, metabolic and genetic status in Alzheimer’s disease: a randomized, double-blind, controlled trial. Clin. Nutr. 38, 2569–2575 (2019).
Ibrahim, A. et al. Multi-strain probiotics (Hexbio) containing MCP BCMC strains improved constipation and gut motility in Parkinson’s disease: a randomised controlled trial. PLoS ONE 15, e0244680 (2020).
Tamtaji, O. R. et al. Clinical and metabolic response to probiotic administration in people with Parkinson’s disease: a randomized, double-blind, placebo-controlled trial. Clin. Nutr. 38, 1031–1035 (2019).
Yang, X. et al. Effect of Lacticaseibacillus paracasei strain Shirota supplementation on clinical responses and gut microbiome in Parkinson’s disease. Food Funct. 14, 6828–6839 (2023).
Agahi, A. et al. Does severity of Alzheimer’s disease contribute to its responsiveness to modifying gut microbiota? A double blind clinical trial. Front. Neurol. 9, 662 (2018).
Asaoka, D. et al. Effect of probiotic Bifidobacterium breve in improving cognitive function and preventing brain atrophy in older patients with suspected mild cognitive impairment: results of a 24-week randomized, double-blind, placebo-controlled trial. J. Alzheimers Dis. 88, 75–95 (2022).
Borzabadi, S., Oryan, S., Eidi, A. & Asemi, Z. The effects of probiotic supplementation on gene expression related to inflammation, insulin and lipid in patients with Parkinson’s disease: a randomized, double-blind, placebocontrolled trial. Arch. Iran. Med. 21, 289–295 (2018).
Barichella, M. et al. Probiotics and prebiotic fiber for constipation associated with Parkinson disease: An RCT. Neurology 87, 1274–1280 (2016).
Tan, A. H. et al. Probiotics for constipation in Parkinson disease: a randomized placebo-controlled study. Neurology 96, e772–e782 (2021).
Lu, C. S. et al. The add-on effect of Lactobacillus plantarum PS128 in patients with Parkinson’s disease: a pilot study. Front Nutr. 8, 650053 (2021).
Di Gioia, D. et al. A prospective longitudinal study on the microbiota composition in amyotrophic lateral sclerosis. BMC Med. 18, 153 (2020).
Aljumaah, M. R. et al. The gut microbiome, mild cognitive impairment, and probiotics: a randomized clinical trial in middle-aged and older adults. Clin. Nutr. 41, 2565–2576 (2022).
Mehrabani, S. et al. The effects of synbiotic supplementation on oxidative stress markers, mental status, and quality of life in patients with Parkinson’s disease: a double-blind, placebo-controlled, randomized controlled trial. J. Funct. Foods 100, 105397 (2023).
Chen, X. et al. Preliminary evidence for developing safe and efficient fecal microbiota transplantation as potential treatment for aged related cognitive impairments. Front. Cell Infect. Microbiol. 13, 1103189 (2023).
DuPont, H. L. et al. Fecal microbiota transplantation in Parkinson’s disease—a randomized repeat-dose, placebo-controlled clinical pilot study. Front. Neurol. 14, 1104759 (2023).
Segal, A. et al. Fecal microbiota transplant as a potential treatment for Parkinson’s disease—a case series. Clin. Neurol. Neurosurg. 207, 106791 (2021).
Kuai, X. Y. et al. Evaluation of fecal microbiota transplantation in Parkinson’s disease patients with constipation. Micro. Cell Fact. 20, 98 (2021).
Acknowledgements
This work was supported by the Fundamental Research Grant Scheme [FRGS/1/2021/SKK0/MUSM/03/4]. All figures were created with Biorender.com.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.Y.K., J.S.L., and W.Q.M. Writing—original draft: J.S.L. and W.Q.M. Figure preparation: W.Q.M. and J.S.L. Writing—review and editing: L.K.S.T., C.X.N., H.H.C., S.H.Y., J.B.F., Y.S.O., C.W.H., and K.Y.K. Funding acquisition: C.W.H. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Loh, J.S., Mak, W.Q., Tan, L.K.S. et al. Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases. Sig Transduct Target Ther 9, 37 (2024). https://doi.org/10.1038/s41392-024-01743-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01743-1
