
Abstract
Specific cell states in metazoans are established by the symphony of gene expression programs that necessitate intricate synergic interactions between transcription factors and the co-activators. Deregulation of these regulatory molecules is associated with cell state transitions, which in turn is accountable for diverse maladies, including developmental disorders, metabolic disorders, and most significantly, cancer. A decade back most transcription factors, the key enablers of disease development, were historically viewed as ‘undruggable’; however, in the intervening years, a wealth of literature validated that they can be targeted indirectly through transcriptional co-activators, their confederates in various physiological and molecular processes. These co-activators, along with transcription factors, have the ability to initiate and modulate transcription of diverse genes necessary for normal physiological functions, whereby, deregulation of such interactions may foster tissue-specific disease phenotype. Hence, it is essential to analyze how these co-activators modulate specific multilateral processes in coordination with other factors. The proposed review attempts to elaborate an in-depth account of the transcription co-activators, their involvement in transcription regulation, and context-specific contributions to pathophysiological conditions. This review also addresses an issue that has not been dealt with in a comprehensive manner and hopes to direct attention towards future research that will encompass patient-friendly therapeutic strategies, where drugs targeting co-activators will have enhanced benefits and reduced side effects. Additional insights into currently available therapeutic interventions and the associated constraints will eventually reveal multitudes of advanced therapeutic targets aiming for disease amelioration and good patient prognosis.
Similar content being viewed by others
Introduction
Transcription factors are the principal drivers of multiple diseases.1 Numerous studies have highlighted that targeting transcription factors can be exceedingly beneficial in disease diagnosis as well as prognosis.2,3 However, most transcription factors are notoriously ‘undruggable’ due to an intrinsic disorder in their structure owing to convex DNA binding interface and flatter protein binding interface, rendering difficulties in targeting their functional associations with DNA or proteins.4,5
In principle, transcription of a particular gene can be regulated by modulation of the activity of any component that affects this process.1 Transcription factors, in association with transcriptional co-regulators, form multiprotein complexes to translate cellular signals, thereby facilitating transcription of different genes.6 The structurally and functionally diverse co-regulators can activate or repress transcription in a cell state-specific manner.7 Current advances in research have suggested that co-regulators not only work as transcriptional effectors, but also as delicate metabolic sensors that perceive discrete changes in nutrient and metabolite availability and reproduce transcriptional responses.8,9 The co-regulators have been perennially classified into two types, transcriptional co-activators and co-repressors.10,11 Amongst them, the co-activators possess the potential to bind transcription factors anchored to DNA in association with catalytic multiprotein complexes and regulate certain epigenetic modifications such as acetylation,12 demethylation,13,14 allowing effective transcription to take place.15 Co-repressors, on the other hand, dock to the transcriptional complexome, and generally mediate deacetylation,16 methylation,17 thereby, suppressing the transcription of its target genes.
The process of transcription encompasses intricately regulated combinatorial effects of transcription co-activators and co-repressors, as well as time-dependent flexibility, to translate cellular signals maintaining homeostasis.18 Even a modest change in either of these factors can disrupt the equilibrium, subsequently inducing series of malevolent traits in the cells and ultimately leading to various disease conditions, including cancer.19,20 Researchers have shown that the upregulation of disease-promoting transcription factors is one of the major impulsions of disease progression, propelled by deregulated transcriptional programs involving diverse interweaved actions of transcriptional co-activators.6,21 Hence, this transcriptional addiction offers us an alternate art-of-war that can be adopted to reduce the function of the disease-associated transcription factors.
In this review, we begin with an abridgement of co-activator involvement in transcriptional circuitry, followed by the regulation of their activity, expression, and multilateral contributions, in several pathophysiological conditions including developmental disorders, metabolic disorders, with special emphasis on cancer. This accumulated knowledge will enlighten us with recent advances in comprehending the control of gene expression, thereby, rendering novel and attractive opportunities to develop new therapeutic strategies, consequentially targeting the core transcription machinery to curtail disease progression.
A historical perspective of transcriptional co-activators
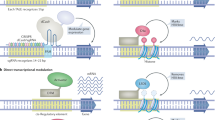
Co-activators, the essential components of cellular functioning, are known to modulate development, cell differentiation, maintenance of stem cells, aging, and their active involvement was recorded in developmental defects, metabolic disorders, and cancer.22,23 In the year 1942, Conrad H. Waddington coined the term “epigenetics” to describe the new branch of biology, which describes the regulation of gene transcription and genomic stability without involving alterations in the DNA sequence.24 Later, in the 1990s, studies were designed to elucidate the functional roles of the coactivators, initially in yeast.25 However, a few years later, the existence of co-factors, the principal epigenetic regulators, was first connoted by transcriptional squelching between estrogen and progesterone receptors.26 Since the discovery of SRC-1 (steroid receptor coactivator-1), vast increase in the understanding of the transcriptional control mechanisms of the co-activators have taken place. Numerous co-activators have been isolated, their biochemical properties and molecular mechanisms have been critically evaluated.27 Several, non-enzymatic cofactors like TAFs, mediators, and numerous enzymatic cofactors like the histone-modifying cofactors (histone deacetylase, histone acetyltransferase, histone methyltransferase, histone demethylase) and ATP-dependent chromatin-remodeling cofactors (SWI/SNF, ISWI, Mi-2/NuRD, and INO80/SWR1 families) have been discovered since.28 Deciphering the functional role of these co-activators has significantly enhanced our understanding of transcriptional co-activator biology.29 Based on the significant influence of the co-activators in transcriptional regulation,6 more co-activators and their mode of action are yet to be discovered, which will not only foster a better understanding of transcriptional regulation but will also potentiate the development of therapeutic targets across diverse pathological conditions. Timeline of notable findings are illustrated in Fig. 1a.

Transcriptional co-activators: history and classification based on mechanism of action. a Historical timeline of key events in significant developments of co-activators. b Transcriptional co-activators employ diverse mechanistic approaches to augment transcription of target genes. (I) The first class of transcriptional co-activators comprise the proteins that induce posttranslational changes like histone acetylation, methylation and ubiquitination to facilitate euchromatinization and accelerated transcription. (II) The second class facilitates transcription through its ATP-dependent motor activities that induce DNA unwinding activities. (III) This class of co-activators promotes transcription augmentation by enabling the recruitment of RNA polymerase II on the transcriptional machinery. (IV) The final class consists of the secondary co-activators that enhance transcription by serving as scaffolds for the recruitment of other co-regulators. Co-A Co-activator, TBP TATA-box binding protein, Pol polymerase, TF transcription factor, SRC-1 steroid receptor co-activator 1, HDAC histone deacetylase, HMT histone methyltransferase, HAT histone acetyltransferase, UTF1 undifferentiated embryonic cell transcription factor 1. This figure was created using BioRender (https://biorender.com/)
Mechanism of transcriptional co-activation
Cell-specific transcription activation is largely regulated by functional interplay between transcriptional co-activators and transcription factors.6,30 Therefore, understanding the mechanisms of appropriate co-activator recruitment to facilitate effective transcription is of paramount importance. There are quite a few reports on co-activator recruitment at different stages of transcription.31,32,33,34 However, the complexity of the mechanism involving co-activators is beginning to be understood. Based on accumulated evidences, different stages of transcription activation and the cross-talk with co-activators during the process, have been summarized.
Orchestration of co-activators and the transcriptional machinery: symphony of transcription
Co-activators are recruited sequentially during eukaryotic transcription. Removal of repressor complexes marks the initiation of transcription activation.31 This is followed by recruitment of DNA-binding transcriptional activators to specific DNA sequences termed as transcription factor binding sites (TFBSs), located in the promoter or enhancer regions of the target gene.35 Immediately after recruitment of the activator, the large conformationally flexible mediator complex, which functions as transcriptional co-activator, is recruited to the promoter.36 Mediator recruitment eventually promotes docking of chromatin remodeling transcription co-activators. One such family of co-activators is SWI/SNF (SWItch/Sucrose Non-Fermentable), that physically interacts with the mediator to establish nucleosome-depleted regions through nucleosome clearing.37,38 SWI/SNF binds to nucleosomal DNA with its translocase domain which is composed of torsion domain and tracking domain. The torsion domain, upon ATP hydrolysis, leads to directional DNA translocation, destroying histone-DNA contacts and creating a transient DNA loop. The DNA loop then propagates around the nucleosome and resolves on the exit site of the nucleosome, inducing nucleosomal repositioning.39 After the removal of the nucleosomes from the promoter, the open chromatin state facilitates general transcription factor recruitment, preinitiation complex formation, and RNA polymerase II (RNA Pol II) binding.40 The co-activator-mediated nucleosome remodeling is discussed in succeeding sections of this review.
At the next step of transcriptional activation, the general transcription factors (GTFs) are recruited at the promoter.41,42 In concordance with the conventional wisdom, the first GTF that is recruited to the promoter DNA is TFIID. Most human promoter DNA contains at least one of the TFIID binding sites: a TATA box sequence upstream of the transcription start site, the initiator element at the transcription start site and the downstream promoter element. The interaction is mediated by TFIID subunits: the TATA-binding protein (TBP) and the TATA-binding protein associated factors (TAFs).43 Numerous studies have documented that TAFs function as co-activators and facilitate the interaction between general transcription machinery and the activators.44 The GTFs TFIIA and TFIIB are subsequently recruited leading to stable interaction between TBP and the promoter. RNA Pol II is then recruited to the pre-initiation complex probably in association with TFIIF. TFIIE and TFIIH are finally recruited to facilitate DNA melting and formation of transcriptionally competent pre-initiation complex (PIC).45 Contrary to the accepted perception, the amino terminus of the mediator subunit MED26 directly interacts with the TFIID subunit TAF7, transforming TFIID to an active structural and functional state.46 The mediator complex interacts extensively with Pol II stalk, dock domain and CTD (C-terminal domain) thereby, facilitating the incorporation of Pol II, creating an entire PIC (pre-initiation complex) structure.47 TFIIA and TBIIB bind to opposite sides of TBP (TFIID subunit). TBIIB, TFIIE, and TFIIF then directly bind to RNA Pol II.47 Owing to its large size, TFIIH interacts simultaneously with TFIIE at the base of the Pol II stalk and position X-box binding protein (XPB) on the DNA. TFIIB linker helix aligns with TFIIF arm at the promoter melting start site, probably facilitating the separation of the DNA strands. The clamp domain starts to swing down during strand separation, prompting the TFIIF arm domain to come closer to the TFIIB B-linker and Pol II rudder, thereby forming a physical barrier for DNA re-annealing. XBP acts as a DNA translocase and inserts the melted single-stranded DNA into the Pol II active site, consequently establishing the open-promoter state of Pol II, which is ready for RNA synthesis.48 Eventually, Pol II dissociates from the promoter once the newly synthesized transcript is about 30 nucleotides. Serine 5 on the Pol II CTD is phosphorylated by TFIIH, leading to the recruitment of capping enzyme (CE).49 The transcript further undergoes 5’ capping, which protects it from exonuclease-mediated degradation.42 This stage is known as early transcript elongation stage. Eukaryotic inactive genes halt at this stage and this process is denoted as Pol II pausing.50
Transition from initiation to elongation requires the dissociation of mediator complexes from the promoter. Conceivably, acetylation of lysine 16 residue on histone 4 facilitates mediator complex dissociation from the promoters after completion of its major task in transcription initiation.51 For productive elongation to take place, additional modifications at the serine 2 in the CTD (C-tail domain) of the RNA Pol II is required.52 The eukaryotic transcription co-activator complex SAGA (Spt-Ada-Gcn5 acetyltransferase) is involved in this process. SAGA has four functionally independent modules: histone acetyl transferase (HAT) module, deubiquitinating module (DUBm), transcription factor (TF) binding module and TBP module. DUBm mediates deubiquitination of H2B which in turn facilitates the recruitment of Ctk1. Ctk1-mediated phosphorylation of Ser2 of RNAPII CTD allows the release of paused Pol II, thereby facilitating elongation of mRNA transcripts.53 Subsequently, cleavage of the new transcript and template independent polyadenylation at 3’ end marks transcription termination.54
The mRNA export pathway and co-transcriptional mRNP surveillance is regulated by the Sgf73 (a component of SAGA HAT module).55 Sgf73 interacts with Sem1p, which is a proteasomal subunit of Sac3p-Thp1p mRNA export complex TREX-2.56,57 This interaction induces the separation of the deubiquitylation module from the SAGA complex. The separation facilitates localization of mRNA export factor Mex67-Mtr2 and TREX-2 to the transcriptional machinery, consequently leading to mRNA export.58 Another component of the DUB module, Sus1p, which is also associated with TREX-2, is responsible for targeting of genes to the NPCs (nuclear pore complexes).59
Modulation of chromatin looping by the co-activators
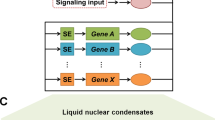
Prior to the emergence of topological associating domains (TADs), the precise control of transcriptional activation relies on the interaction between remote cis-regulatory modules (e.g., enhancers, and the promoter. The formation of chromatin loops before the recruitment of the activators facilitates the communication between the promoters and enhancers.60 The transcriptional co-activators have also been reported to play significant role in this process. The transcriptional co-activators YAP and TAZ promote recruitment of the mediator complex at the enhancer, thereby establishing long range chromatin looping and facilitating enhancer-promoter contacts to recruit lineage-specific transcription factors.61 The mediator co-activator complex further acts as a bridge to relay information from the enhancers to the promoters. The tail module of mediator complex associates with the enhancer bound transcription factors while the other modules bind to Pol II and PIC at the promoter to dynamically link the promoter and the enhancer.62
Co-activator interaction with transcription factors
Transcription factors can bind to DNA in a sequence-specific manner63; however, these principal regulators need assistance from several other factors to regulate chromatin remodeling, DNA unwinding, and RNA polymerase II recruitment, which are necessary for effective transcription to take place. These biochemical activities are the speciality of the transcriptional co-activators, which are multiprotein complexes that dock on the DNA-binding activators.64 The sequence-specific transcription factors contain variable and intrinsically disordered transcription activation domains (TADs).65 Interaction with the TAD domain of site-specific transcription factors, mediates the positioning of the transcriptional co-activators at promoter regions,66 where they induce chromatin remodeling and act as bridges between general transcription machinery and the activator, hence promoting transcription activation.67 For example, CBP/p300 histone acetyl transferase interacts directly with C-terminal transactivation domain of E2F transcription factor.68
Moreover, the TAD domain of transcription factor exhibit “structural plasticity” which propels an adaptive association with multiple co-regulatory molecules.69,70 A study by Marceau et al.71 has reported that the transcription factor FOXM1 contains a disordered TAD. When in association with negative regulatory domain (NRD), the TAD domain attains order in its structure. However, dissociation from NRD restores the disordered conformation. The disordered TAD is then capable of binding the transcriptional co-activator CBP.
The conventional model of co-activator mediated gene transcription indicates passive role of the transcription factor, where they are only responsible for localizing the co-activator complexes to the genes.71 However, some studies have also reported that the docking of the co-activator on the transcription factor switches on the co-activator activity. A study has reported that CBP and P/CAF when bound to mutant HNF-1α transcription factor do not exhibit HAT activity, indicating that the transcription factors not only recruit the co-activators at the promoter region but also modulate their enzymatic activity.72 Another study has demonstrated that the transcriptional co-activator PGC-1α exhibits a quiescent stage when not bound to transcription factor. However, interaction with transcription factors induces a conformational change and promotes the interaction of PGC-1α with SRC-1 and CBP/p300.73
Modulation of chromatin structure by co-activators
Chromatin has been reported to be an instructive DNA scaffold that can respond to intracellular and extracellular cues, and act to regulate the many uses of DNA.74 One of the most significant ways to regulate transcription has been to influence chromatin packaging, which determines the availability of DNA elements.75 This is achieved by two major type of modifications, covalent histone-modifications and ATP-dependent chromatin remodeling.76 Methylation,17 acetylation,12 ubiquitination,77 demethylation13 and deubiquitination,78 the crucial histone modifications introduced by the co-activators, are principle regulators of chromatin structure and are involved in the manipulation and expression of gene.79 On the other hand, ATP-dependent chromatin-remodeling complexes guide gene expression by restructuring the nucleosome.80 Complicated integration and synchronization of these modifications not only regulate chromatin structure but recruit the transcriptional machinery and govern target gene expression.81
Methylation
Methylation of the histone proteins is one of the important phenomena regulating chromatin structure and gene transcription.17 Nucleosomes, the fundamental unit of chromatin, is composed of a stretch of DNA wrapped around a protein octamer consisting of two copies each of the four histone proteins: H2A, H2B, H3, and H4.82 All of these proteins possess a tail extension, which is targeted for methylation.82 Histones can be methylated on two amino acid residues, lysine (K) and arginine (R); however, lysine residues of histone tails are mostly of preference.83 Several transcriptional co-activators possess the methyl transferase activity and are known to modulate the histone architecture to promote transcription.84 Histone methylation has been found to be associated with both compact and relaxed chromatin structure, depending on the methylation sites.85 For instance, higher H3K4me1/2/3, H3K36me3 and H3K79me1/2/3 helps in euchromatinization; on the other hand, heterochromatinization is characterized by higher levels of H3K9me2/3, H3K27me2/3 and H4K20me3.86,87 The lysine methyl transferase KMT DOT1L demethylates histone H3 at lysine 79 (H3K79me2) which promotes lineage-specific gene expression to regulate TH cell function.88 Another lysine methyl transferase (SET8/KMT5A) is the only mammalian enzyme known to monomethylate histone H4 at lysine 20 (H4K20me1). This particular histone modification plays important roles in DNA damage repair by recruiting signaling proteins like 53BP1 to the site of double-stranded DNA breaks.89 In addition, protein arginine N-methyltransferases (PRMTs) asymmetrically dimethylate 3 (R3) residue of H4, potentiating subsequent histone acetylation, and contributes to the maintenance of an active chromatin structure. This suggests that such histone modifications can function as a transcriptional activation mark.84 Methylation of core nucleosomal histones can either activate or repress transcription depending on which amino acid residues in the histones are methylated and how many methyl groups are attached. Deregulation of methylation has been found to cause neurodegenerative diseases, metabolic disorders, and cancer.90,91,92
Acetylation
Acetylation of histones is highly dynamic and regulated by the opposing action of two family of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs).12,16 Transcriptional co-activators utilize acetyl CoA as cofactor and catalyse the transfer of an acetyl group to the ε-amino group of lysine side chains,93 thereby neutralizing the positive charge of lysine that weakens the non-covalent electrostatic interactions between histones and negatively charged phosphate groups of DNA.94 As a consequence, the condensed chromatin state is transformed into a more relaxed euchromatin to enable greater accessibility of DNA and promotes transcription of related genes.95 Each of these molecules can modify multiple sites within the histone N-terminal tails, which in turn dictates the subsequent histone modifications.79
A study demonstrated that the transcriptional co-activator p300 facilitates acetylation of H3K122 in the globular domain via interacting with BRG1 (Brahma-related gene 1). This in turn destabilizes histone-DNA binding and assists transcription.96 Hassan et al.97 reported that histone acetylation by SAGA complex stabilizes binding of SWI/SNF binding to the nucleosome to mediate ATP-dependent chromatin remodeling. SWI/SNF has also been shown to interact with p300 and regulate H3K27 acetylation to enhance transcription.98 In addition to the histone tails there are also other sites of acetylation present within the globular histone core, such as acetylation at H3K56 in humans by hGCN5.99 Apart from hGCN5, the transcriptional co-activator p300 is also reported to be associated with H3K56ac. Strikingly, knockdown of p300 induces loss of H3K56ac and increase in DNA damage, establishing a prominent role of the transcriptional coactivator-mediated histone acetylation in nucleosome remodeling.100
Ubiquitination
In addition to the above, ubiquitination is another type of reversible histone modification. Ubiquitination is a process that ligates ubiquitin, a 76 amino acid protein, on lysine residues of histones, by covalent interaction through an isopeptide bond between its C-terminal glycine and the ϵ-amino group of a lysine residue.101 The mechanisms by which histone ubiquitination affect transcription are multiple. Histone ubiquitination can alter higher-order chromatin folding and provide greater access of the underlying DNA, which may function as a signal for the recruitment of transcription regulatory molecules.102 It is also possible for ubiquitination to act as an integrator of different post transcriptional modifications on histones.103
Mono-ubiquitination of histone H2B at lysine 123 in Saccharomyces cerevisiae or at lysine 120 in mammals is necessary for maintaining stable altered nucleosome state for transcription.104,105 Functional human homologs of the yeast BRE1 E3 ubiquitin ligase are the transcriptional co-activators RNF20 and RNF40. The co-activator molecule RNF20 enhances the global level of ubiquitylation at lysine 120 of histone H2B, thereby promoting activator-dependent transcription.106 A study by Krajewski et al.107 has shown that ubiquitination of H2BK34, which is surrounded by two coils of DNA superhelix, directly influences nucleosome conformation via steric hindrances. Petty et al.108 have demonstrated that H2B is ubiquitylated by co-activators RAD6 and BRE1 which is associated with gene activation in yeast and mammals. Histone H3B monoubiquitylation has also emerged as a new regulator for heterochromatinization in metazoans.109 The study indicates that co-activator-mediated ubiquitination is definitively associated with gene activation.
Demethylation
Histone demethylation is dynamically regulated by the activity of histone demethylases that are categorized into two families: KDM1 family/Lysine specific demethylase 1 (LSD1) and Jumonji C (JmJC) domain-containing histone demethylases.110 KDM1 family of nuclear amine oxidase homolog removes mono- and di-methylated lysine of H3 at lysine 4 or 9 in a cell-state specific context. In contrast, the JmJC domain-containing demethylases, belonging to the 2-oxoglutarate-dependent dioxygenases, are capable of removal of trimethylations. Wissmann et al.111 reported that KDM1A demethylates H3K9me1 and H3K9me2 when complexed with androgen receptor, leading to transcription activation, and Cloos et al.112 reiterated that KDM4C, in particular, increases euchromatin available for transcription. Several in vitro differentiation studies have established the necessity for the KDM6 H3K27me2/3 demethylases, KDM6A/UTX and KDM6B/JMJD3, in overcoming the repressive chromatin state and initiate normal transcription.113,114,115,116 Another fascinating study by Tsai et al.117 showed the interaction between lncRNA HOTAIR and KDM1A/CoREST complex, which recruits the demethylase complexes to the target site, creates a repressed chromatin state. However, histone demethylases have been shown to have enigmatic biological interactions and current studies indicate contradictory function in transcription activation. Further studies are imperative to establish their role in promoting transcription.
Deubiquitination
The process of deubiquitination involves the removal of ubiquitin molecule from the target proteins and dissolution of ubiquitin complexes.17 BAP1, a ubiquitin C-terminal hydrolase (UCH) domain-containing protein, promote gene expression by catalyzing removal of monoubiquitination on lysine 119 of histone H2A (H2AK119ub1) through a multiprotein complex.118 Deubiquitination module of the SAGA complex, that comprises Usp22, Eny2, and Atxn7, deubiquitinates H2BK120ub following DNA damage, which is critical for class switch recombination.119 USP22, a well-known role co-activator of VEGF-A, specifically plays a crucial role by reversing the ubiquitination by ubiquitylating enzymes. It serves as ubiquitin hydrolase and catalyzes the deubiquitination of H2A and H2B, thereby counteracting heterochromatin silencing and promoting gene transcription.120 Another study by Ducker et al.121 has reported that USP17 induces deubiquitination of the transcription factor ELK-1 at lysine 35, consequently upregulating its transcription. However, the mechanism underlying deubiquitination-mediated transcription activation is yet to be defined with clarity.
ATP-dependent chromatin remodeling
Chromatin remodeling involves changing the histone-DNA interactions by disrupting, assembling or nucleosome sliding.122 This process is carried out by a family of enzymes with ATPase and helicase ancestry, the chromatin remodeling enzyme complexes. These remodeling enzymes induce partial dismantling of nucleosomes, liberating segments of DNA and rendering them accessible to the interacting proteins.123
Till date, 4 classes of these remodeling enzymes have been identified: SWI/SNF, ISWI, CHD, and INO80.124 Coincidentally all these enzymes function as transcriptional co-activators. SWI/SNF is one of the first described chromatin remodeler enzymes that is recruited to the promoter at the same time as the transcription activators. Upon ATP hydrolysis, SWI/SNF carries out a directional DNA translocation, which destroys DNA-histone binding, causing the nucleosome to reposition.125 The ISWI family of remodelers regulate DNA accessibility by mobilizing nucleosomes and controlling the length of linker DNA separating nucleosomes by a mechanism that is not very lucid till date.126 The evolutionarily conserved INO80 family of ATP-dependent chromatin-remodeling enzymes modify chromatin in a number of ways including nucleosome sliding and exchange of variant histones. INO80, along with SWI/SNF remodelers, promotes nucleosomal clearing of PHO5 gene promoter.127 However, information about the role of INO80 in transcriptional co-activation is limited; therefore, conclusive statement about the mechanism of their execution cannot be stated. Biochemical analyses revealed that chromodomain helicase DNA (CHD)-binding proteins affect DNA-histone interactions within the nucleosome in a manner that is distinct from the yeast SWI/SNF complex.128 Interestingly, they are often linked with maintenance of pluripotency in embryonic stem cells.129
Types of transcriptional co-activators
Depending on their mechanism of action, transcriptional co-activators can be broadly categorized into four different classes.30 The first class of co-activator proteins performs histone modifications, resulting in dispersed structure of chromatin, thereby rendering it accessible to transcription factors.130 The second class comprises proteins that possess ATP-dependent DNA chromatin remodeling activity, thus augmenting transcription.131 The third class interacts with general transcription apparatus and recruits RNA pol II to promote transcription initiation and elongation.132 The fourth class of co-activators, known as the secondary co-activators, interacts with transcription factors and function as scaffolding non-enzymatic proteins to recruit other co-activators containing enzymatic activities on the target gene promoter.30 Furthermore, several transcriptional co-activators exhibit the properties of both primary and secondary co-activators in variable contexts133,134 (Fig. 1b).
Overview of various co-activator families
Several families of proteins have been characterized and classified as transcriptional co-activators. Here, we have attempted to elaborate the structural conformation and the cell-specific functions of different co-activator families.
BET family
The four conserved members of the BET (bromodomain and extraterminal domain) family of proteins in the mammals are BRD2 (also known as FSRG1, RING3, RNF3, FSH, or D6S113E), BRD3 (also known as ORFX or RING3L), BRD4 (also known as MCAP or HUNK1) and BRDT (also known as BRD6, CT9, or SPGF21).135 The bromodomain-containing proteins (BRDs) have been recognized to function as epigenetic readers.136 Epigenetic readers are a group of specialized docking domain containing proteins that identify and bind to various covalent modifications on histones, non-histone proteins and DNA. BRDs specifically recognize acetylated lysine residues in histone H3 and H4.137 For instance, a study has reported that IL1β or TNF-induced acetylation of H4K5Ac, H4K8Ac, and H4K12Ac mediates the recruitment of the BET proteins, BRD3 and BRD4, to the matrix degrading enzyme gene promoter, consequently upregulating their expression in human chondrosarcoma cells.138 Histone H3 acetylation, especially at H3K18Ac, facilitates the recruitment of BRD3 and BRD4 to the promoter of CXCL8 gene which encodes interleukin-8 protein. This promotes the expression of IL-8 in airway smooth muscle cells and drives steroid-resistant neutrophilic airway inflammation in asthmatic individuals.139
BRD4 can also function as an atypical histone acetyl transferase. However, the mode of acetylation is distinct from other HATs as BRD4 has the property to induce acetylation of histone H3 on Lys residue 122 (H3K122Ac), leading to destabilization of nucleosome structure and chromatin destruction.140 The HAT activity of BRD4 has been documented in inflammation-driven airway remodeling.141 BETs can also interact with transcription elongation complexes and transcription factors through lysine acetylation-dependent or independent mechanisms.142 The positive transcription elongation factor, P-TEFb is a cyclin-dependent kinase comprising CDK9 and other cyclin subunits like cyclin T1.143 The BETs are responsible for recruiting CDK9 and cyclin T1 to RNA Pol II.138 This interaction mediates the phosphorylation of Ser2 and Ser5 of Pol II C-terminal domains, thus allowing productive elongation.144
SRC family
The steroid receptor co-activators of p160 family consisting of three homologous members SRC-1 (also known as NCOA1), SRC-2 (also known as TIF2, GRIP1 and NCOA2) and SRC-3 (also known as p/CIP, RAC3, AIB1, ACTR, TRAM1, and NCOA3) has been recognized to regulate a plethora of physiological processes. The SRC family of proteins possess three structural domains.145 The conserved basic helix-loop-helix-Per/ARNT/Sim (bHLH-PAS) domain, located in the N-terminal, is required for interaction with transcription factors and contains a canonical NLS (nuclear localization signal).146 The central region consists of three LXXLL motifs (X is any amino acid). This region mediates interaction with transcription factors and the nuclear receptors. Central region also contains a serine/threonine-rich domain which upon phosphorylation influences the SRC activity.145 Two transcriptional activation domains (ADA1 and ADA2) are located in the C-terminal.147 The ADA1 activation domain is involved in binding with the transcriptional co-activators CBP/p300. The SRCs exerts their role in chromatin modification through this interaction with CBP/p300.148 The ADA2 activation domain binds to the histone methyltransferases CARM1 (co-activator-associated arginine methyltransferase 1) and PRMT1 (protein arginine N-methyltransferase 1) to facilitate transcription activation.149
SRC-1 co-activators are involved in regulating carbohydrate metabolism. SRC-1 has been reported to initiate gluconeogenic program through transactivating pyruvate carboxylase by modulating the activity of C/EBPα.150 SRC-1 has also been reported to control insulin signaling by modulating the expression of insulin receptor substrate 1 (IRS1).151 SRC-2 has been determined to be a positive regulator of mammalian circadian rhythm as they function as transcriptional co-activators of the brain and muscle ARNT-Like 1 (BAML1) and circadian locomotor output cycles kaput (CLOCK).152 SRC-3 has been widely reported to be amplified in tumors.153 SRC-3 modulates the AKT signaling pathway to stimulate prostate and ovarian cancer cell growth and promote glycolysis in urinary bladder cancer, by upregulating the expression of GLUT1 and PGK1 genes via its interaction with HIF1α (hypoxia inducible factor 1α).154 Given their role in coordinating energy accretion and utilization in the context of normal physiology and malignancy, the SRC-family of transcriptional co-activators is an emerging area of concern.
KMT family
The lysine methyltransferase (KMT) family of transcriptional co-activators methylates histones and consists of 23 different SET proteins and one 7βS protein (a total of 24 different enzymes).155 The methyl transferases contain a SET domain, and flanking the SET domains are a pre-SET domain and a post-SET domain. Pre-SET domain stabilizes the structure by forming triangular zinc clusters using cysteine residues. The SET domain contains a catalytic core composed of β-strands.156 The lysine residues in the histone tail of the substrate and the S-adenosyl methionine (SAM) are bound and oriented into the SET domain to initiate methylation.157 This promotes SN2 nucleophilic attack of the ε-amine that leads to transfer of methyl group from SAM to lysine, thereby introducing monomethyl-lysine.158 Following an initial round of methylation, the monomethyl or dimethyl lysine residues are oriented for subsequent methylation events.159
Several studies have reported the role of KMT family of proteins in transcriptional regulation. KMT2C/D COMPASS complex of methyl transferases and their interacting partners promote active euchromatic conformations by modification of histone-3 tail residues.160 Cyclin D1-mediated recruitment of lysine methyltransferase (KMT) G9a/EHMT2 induces H3K9me2 that promotes positioning of chromosomes by facilitating the interaction between nuclear lamina (NL) and the lamina-associated domains (LAD).161 However, accumulated evidences indicate that methylation-mediated transcription suppression is also predominant. Tanaka et al.162 suggested that SETD8/PR-SET7-mediated mono methylation of histone H4 at lysine 20 leads to repression of p16INK4A and ribosome-associated genes that are associated with senescence. SET7/9-mediated methylation of FoxO3 K270 prevented FoxO3 interaction with its target genes and prevented the transcriptional activation of FoxO3, indicating that the site of methylation regulates diverse biological processes.163
CBP/p300 family
Two paralogous acetyl transferases that have been widely recognized to function as transcriptional co-activators to enhance transcriptional activation are CREB binding protein (CBP) and p300. CBP, also known as KAT3A, is encoded by the CREBBP gene and p300, also known as KAT3B, is encoded by EP300 gene.164 Both the paralogous transcriptional co-activators contain highly conserved modular structure that encompasses an acetyltransferase domain, acetyl lysine-binding bromodomain (BD) and diverse structured modules like KIX domain, the cysteine/histidine regions (TAZ1 and TAZ2), the interferon response binding domain (iBID) and the nuclear receptor interaction domain (RID).165 According to Shikama et al.166 nucleosome assembly protein/template activating protein (NAP/TAF), which functions as histone chaperones, can functionally interact with p300 co-activator proteins. The histone 3 lysine 27 acetylation (H3K27ac) activity at regulatory elements such as enhancers and promoters, that is mediated by the acetyl transferases CBP and p300 is required for cell type-specific gene expression patterns.167 At specific regions of the genome in the mouse embryonic stem cells, p300 is responsible for maintaining H3K27ac, according to Martire et al.168 p300 interacts with Glut2 promoter and the transcription factor HNF1α to upregulate the expression of Glut2, a major glucose transporter in the hepatocytes and the pancreatic β-cells.169 Owing to their diverse gene regulatory role, deregulation of p300/CBP contributes to various pathological conditions.
CRTC family
The cAMP response element binding protein (CREB) has been documented to function in association with a family of co-activators known as cAMP-regulated transcriptional co-activators (CRTCs).170 They are also referred to as transducer of regulated CREB activity (TORC) or mucoepidermoid carcinoma translocated protein (MECT).171 CRTCs are highly phosphorylated at basal conditions and are retained in the cytoplasm through interactions with 14-3-3 proteins. Rise in cAMP and calcium level induces calcineurin-mediated dephosphorylation of CRTC that facilitates its release from 14-3-3 complexes.170,171 CRTC family of co-activators comprise three members: CRTC1, CRTC2, and CRTC3.172 Mutational analyses have also showed that the CRTCs contain distinct functional domains that are responsible for regulating pre-mRNA splicing.173 CRTC family of CREB regulated transcription co-activators are involved in cAMP-pathway-mediated melanocyte differentiation. CRTC3 binds to a conserved enhancer of CREB and leads to upregulation of oculocutaneous albinism 2 (OCA2) protein expression, which then promotes melanosome maturation. CREB/CRTC1 pathway further influences the neuronal activity-dependent gene transcription.174 CRTC1 upon dephosphorylation due to neuronal activity is translocated to nucleus, where it binds to the transcriptional complexome in CRE/TATA promoters to promote neuronal-activity dependent transcription.175 CRTCs have also been suggested to be involved in ACTH-induced transcription of StAR (Steroidogenic Acute Regulatory) protein, where ACTH mediates the recruitment of CRTC2 and CRTC3 to the StAR promoter leading to increased levels of Star heteronuclear RNA,176 indicating that these co-activators modulate context-specific activation of diverse genes.
CITED family
CITED (CBP/p300-interacting transactivators with E (glutamic acid)/D (aspartic acid)-rich carboxyl-terminal domain) family of transcriptional co-activators are 22–27 kDa proteins177 that interact directly with the CBP/p300 family of transcriptional co-activators through a conserved C-terminal domain known as CR2 (conserved region 2).178 All the known members of CITED family undergo nuclear translocation where they interact with sequence-specific DNA binding proteins and activate transcription in a CBP/p300 dependent manner.179 CITED2 has also been reported to be essential for embryonic development.180 The embryonic fibroblasts of CITED2-/- mouse had defective proliferation, senescence-associated cellular morphology and increase in expression of cell growth inhibitors p16INK4a, p19ARF, and p15INK4b.181 CBP/p300 interacts with HIF1α through its CH1 domain to activate transcription of hypoxia responsive genes and promote tumor angiogenesis. CITED2/CITED4 interacts with CBP/p300 at the CH1 domain, preventing association with HIF1α and functions as an inhibitor of hypoxia signaling.182 Accumulated evidence, therefore, indicates involvement of the CITED family in various biological activities to regulate CBP/p300-dependent transcription.
TRIM family
Tripartite motif-containing (TRIM) protein super family is associated with a wide range of biological processes.183 The TRIM motif (also known as RBCC motif), which identifies this superfamily, consists of a RING domain, one or two B-box domains, N-terminal-associated coiled-coil domain and C-terminal domain. In humans, ~70 TRIM genes have been identified which have been further subclassified on the basis of their C-terminal domain.183 The RING domain mediates conjugation with ubiquitin, with SUMO (small ubiquitin-like modifier), or with ISG15 (IFN-stimulated protein of 15 kDa).184 The zinc-binding motif containing RING domain, is the catalytic center which provides biological flexibility to the TRIM family of proteins.185 The RING domains of TRIM5α, TRIM8, TRIM11, TRIM21, TRIM22 and TRIM25 mediate ubiquitylation events owing to the E3 ubiquitin ligase activity.186 This E3 ubiquitin ligase activity has been established to be crucial for anti-HIV functions.184 Some members of the TRIM family contain a COS box which is located immediately downstream of the coiled-coil domain. The COS box mediates binding to microtubules.187 C-terminal domains, like the ADP ribosylation factor-like (ARF) domain, are associated with vesicular trafficking, whereas fibronectin type 3 (FN3) domains, might be involved in actin crosslinking. Owing to the presence of bromodomain, the TRIM family members (TRIM24, TRIM28, and TRIM33) can act as chromatin remodelers.188 An example of the kind is the regulation of self-renewal transcription network by TRIM28. TRIM28, together with other pluripotency markers like CNOT3, ZFX, and c‐MYC, co-occupies putative gene promoters to promote self-renewal.189 TRIM24-mediated regulation of glioma stem cell proliferation and self‐renewal has also been reported. In response to EGFR, TRIM24 recruits STAT3 and stabilizes STAT3-chromatin interaction to promote cancer stem cell proliferation and maintenance.190 Given their role as transcriptional co-activators, the TRIMs have the potential to emerge as therapeutic targets in different pathological conditions.
MRTF family
The mechano-sensitive myocardin family of transcriptional co-activators comprising myocardin, MRTF-A/MKL1/MAL, and MRTF-B/MKL2 are associated with the MADS box transcription factor SRF (serum response factor) to activate transcription of genes responsible for myogenesis, cell proliferation, migration and creation of transcriptional–cytoskeletal regulatory circuit by encoding components of actin cytoskeleton.191 The MRTFs contains several conserved domains that are essential for actin-binding, chromatin organization, homo- and hetero-dimerization and transcriptional activation.192 Esnault et al.193 identified 960 serum-responsive SRF-linked genes and majority of these genes were regulated by MRTF-mediated RNA polymerase recruitment and promoter escape. In the context of pathology of the intervertebral disc (IVD), Fearing et al.194 reported that transcriptional co-activator MRTF-A regulates nucleus pulposus cell phenotype. MRTF-A and transcription co-activators YAP/TAZ promotes pathologic and fibroblastic phenotype of the adult human NP cells in association with F-actin stress fibers, indicating that the MRTF-family of co-activators are principal regulators of cytoskeletal dynamics and mechano-sensing, both under normal and diseased physiological conditions.
DExD/H box family
The DExD/H (Asp-Glu-x-Asp/His) box family of proteins are known to play major roles in RNA synthesis and function.195 Owing to the homology with DNA helicases, the prototypic members of the family exhibits ATP-dependent RNA helicase activity. These proteins also act as RNA chaperones and promote local RNA unwinding to mediate the formation of optimal RNA structures.196 The DExD/H proteins contain N- and C-terminal extensions through which they interact with several components of the transcriptional machinery to regulate transcription. For instance, DDX5 (p68) has been demonstrated to act as transcriptional co-activator of Polo-like kinase-1 (PLK1) by stimulating the transcription from PLK1 responsive promoter.197 DHx9 interacted with CBP with its N-terminal domain, while the helicase domain and an overlapping region of the N-terminal domain was found to interact with Pol II, thereby enhancing the enforcement of the transcriptional complex at responsive promoters.198 DDX3 has been depicted to co-activate transcription from p21 promoter.199 Furthermore, DDX3 also facilitates the interaction of IRF3 with the transcriptional co-activators CBP/p300, hence guiding an antiviral signaling-induced transcription factor complex formation on target gene promoters.200 Owing to the accelerating importance of DExD/H-box family of proteins in transcriptional regulation, further descriptive studies to decipher their significance in normal physiology and disease pathology, may provide alternate therapeutic options.
PGC-1 family
The members of peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1 (PGC-1) family of transcriptional co-activators have been reported to exert several biological functions like energy metabolism, skeletal muscle fiber type switching, heart development, adaptation to thermogenesis, and endurance-type exercise.201,202 The founding member of this family is PGC-1α. This small family of co-activators also includes PGC-1β, the close homolog of PGC-1α and PGC-1-related coactivator (PRC).203 The N-terminus contains the major nuclear hormone receptor-interacting motif (LXXLL), which facilitates ligand-dependent interactions with different transcription factors like ER,204 PPARα,205 RXRα,206 glucocorticoid receptor and HNF4α.207 The C-terminal region contains the RNA processing motifs like the serine-arginine-rich (RS) domain and a RNA-binding motif (RMM). The presence of the transcription activation domain along with the RNA processing motifs is a unique feature of the PGC-1 family.73
PGC-1 family of proteins acts as secondary co-activators by serving as a docking platform for other co-regulatory molecules.30 In humans, PGC-1α is a master regulator of energy metabolism and mitochondrial homeostasis. PGC-1α co-activates the expression of nuclear respiratory factors 1 and 2 (NRF1 and NRF2) which further facilitates the transcription of genes associated with mitochondrial respiratory chain complexes.208 Human PGC-1α also interacts directly with RNA and with NXF1 (Nuclear RNA export factor 1) to promote nuclear export of co-activated transcripts, essential for age-related telomere maintenance.209 PGC-1β has been reported to upregulate expression of genes associated with oxidative phosphorylation and electron transport chain.210 Moreover, PGC1β KO mice demonstrated decreased activity during the dark cycle and less response to physiological stresses, like adrenergic stimulation in BAT (brown adipose tissue), cold exposure in BAT, and hepatic steatosis.211 Altogether it can be stated that the PGC-1 family of co-activators play a non-redundant role in the basal and stress-related mitochondrial activity regulation.
Regulation of transcriptional co-activators
Regulation of co-activator activity and expression
Activity and expression of co-activators can determine the fate of a cell by modulating an immensity of physiological processes.212,213 The state of normalcy in a cell is determined by the delicate maintenance of several essential factors, including the co-activators, failure of which will eventually lead to a diseased condition.214,215 The mechanisms for molecular regulation of these co-activators are described below.
Signal transduction
Transient signals induced by interactions of cell surface receptors and ligands are translated into prolonged alterations in the gene expression profile by various signaling pathways, entailing reversible assembly of numerous factors.216,217 These signal transductions control expression and activity of transcription factors, as well as co-regulators, thereby modulating cellular transcriptional program.218 Heretofore, countless studies have predicted the possibility of regulation of co-activators by signaling pathways. Willert et al.219 found CBP/p300 to be one of the target genes of WNT signaling pathway by microarray analysis. Moreover, CBP/p300 has been found to act as a co-activator of β-catenin, indicating towards a possible feedback loop mechanism.220 27 of the 72 TRIM family genes are reported to be sensitive to interferon signaling.221 In skeletal muscles, PGC-1α activity is governed partly by p38 MAPK and CaMKII222 and in liver by LIPIN1.223 Another study reported PGC-1α is regulated by TLR2 signaling in mice with Staphylococcal aureus sepsis.224 In head and neck cancer, the WNT pathway effector protein, β-catenin, was found to play important role in MLL1 transcription regulation.225 In diabetic nephropathy, transcription co-activator SET7 is regulated by the TGF-β pathway.226 Multiple studies have reported the hippo signaling pathway to be the prime regulator of YAP/TAZ expression and activity.227
Epigenetic regulation
The genome of all the cells in an organism essentially consists of the same DNA. However, their functions vary depending on the quantitative difference in their gene expression profile.228 This form of regulation renders an additional adaptive switch that helps the organism to exquisitely regulate expression and function of different factors and sustain under unfavorable conditions.229 Activity of transcriptional co-activators has also been documented to be regulated by such epigenetic modulations.230 For example, YAP is monomethylated at lysine 494 by another co-activator SET7, which helps in cytoplasmic retention of YAP.231 Methylation at arginine residue of KIX domain of CBP by coactivator-associated arginine methyltransferase 1 (CARM1) inhibits the interaction of CBP with CREB, thereby, blocking their downstream activity.232 Rieger et al.233 proved that phosphorylation of p300 at serine 89 by protein kinase C (PKC) regulates its interaction with β-catenin.234 It was observed that in early anaphase, cyclin dependent kinase-1 (CDK1)/Cyclin B complex stabilizes SET7 by phosphorylation at the serine 29 residue. In addition, acetylation of MLL1 at two conserved residues, K1130 and K1133, by sirtuin1 (SIRT1) affects its methyltransferase activity.235 Liu et al.236 found that BRD4 methylation at R179, R181, and R183 residue by protein arginine methyltransferase2/4 (PRMT2/4) is essential to selectively control the transcriptional program by facilitating BRD4 recruitment to histones or chromatin. Regulation of a co-activator activity by another co-activator, where BRD4 was found to be methylated at lysine 99 residue by SETD6, which in turn negatively regulates target gene expression, was also reported.237 Activity of transcription co-activator TRIM5α is restricted by autoubiquitination, wherein, E2 Ub-conjugating enzyme Ube2W is employed to anchor the Lys63-linked polyUb chains.238 A research article by Mersaoui et al.239 provided evidence that arginine methyltransferase 5 (PRMT5) methylates DDX5 at its RGG/RG motif by direct interaction. This motif is necessary for DDX5 to interact with XRN2 and repress formation of cellular R-loops, which is essential for transcriptional termination. In accordance, Wu et al.240 proposed a unique regulation of SRC3 by a coordinated phosphorylation dependent ubiquitination mechanism.
Protein–protein interactions
The context-dependent activation and inactivation of transcription co-activator function is often determined by the proteins they interact with. BRD4, for example, interacts with different proteins under specific circumstances and therefore, regulate multiple cellular pathways.241 Mechanistically, Yu et al.241 revealed that in hepatocellular carcinoma, DDX5 forms transcriptional regulatory complex in association with BRD4 to positively regulate transcription of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA). BRD4, via its extra-terminal domain, interacts with arginine demethylase Jumonji domain-containing 6 (JMJD6), lysine methyltransferase nuclear receptor-binding SET domain 3 (NSD3), and the nucleosome remodeling enzymes SWIF/SNF and CDH4, to perform context specific functions.242 The Human Papillomavirus Type 16 E6 oncoprotein physically binds to CBP/p300 and downregulates p53 transcriptional activity.243 PIMT (PRIP-interacting protein with methyltransferase domain), a RNA-binding protein, strongly binds to CBP/p300 through its cysteine-histidine rich C/H1 and C/H3 domains and regulate their activity.244 Sheppard et al.245 unveiled the importance of the interaction between CBP/p300 and SRC1 through its activation domain 1 (AD1) in assisting the recruitment of CBP/p300 to the estrogen receptor.
Non-coding RNAs
Non-coding RNAs (ncRNAs) are functional RNA molecules that do not have protein coding region, and are therefore not translated into protein.246 However, they actively take part in expression and activity regulation of diverse proteins, including transcription co-activators.247 Most extensively studied ncRNAs in co-activator modulation are microRNAs (miRNAs) and long non-coding RNA (lncRNAs).248 MicroRNAs generally regulate co-activator gene expression by direct interaction with the mRNA but their role in activity regulation is not explicitly understood.249 However, lncRNAs can regulate both co-activator activity and expression owing to their diverse mode of action.250 There are multitudinous studies unveiling the interplay between co-activators and ncRNAs. For instance, sequencing (ChIRP-seq) together with CRISPR/Cas9 mutagenesis of the target sites proved that p300 is recruited to the enhancer region by lncSMAD7 to trigger enhancer acetylation and transcriptional activation of its target gene.251 Lagos et al.252 showed that miR-132 suppresses p300 activity during antiviral innate immune response. NEAT1 lncRNA forms a complex with BRD4 and WDR5 and maintains them in a less-active state.253 In multiple myeloma, dual luciferase reporter assay showed that H19 inhibits miR-152-3p to enhance BRD4 expression.254 There is also evidence suggesting negative regulation of BRD4 by miR-141-3p.255 In ovarian cancer, SET7 has been shown to be modulated by miR-153, and lncRNA SNHG6 has been found to downregulate SETD7 by posttranscriptional destabilization.256
Modulation of co-activator function by signaling pathways
Signal transduction pathways can be defined as coordinated interdependencies amongst structurally and functionally diverse class of biomolecules that conjointly dictate the response of a given cell to a particular cue received by endocrine, paracrine and cytokine signaling.257 The preponderance of transcriptional co-activator-related research articles and knowledge bases has recognized them as one of the pivotal molecules that are being actively regulated by signaling pathways. Gusterson et al.258 demonstrated that in cardiac cells, activation of CBP/p300 upon phenylephrine (PE) treatment is dependent on p42/p44 MAPK pathway. CBP/p300 has been reported to be degraded by murine double minute 2 (MDM2) in NIH-3T3 cells, which is regulated by the MAPK pathway.259 In addition, under certain circumstances, MAP4K downstream kinase nuclear dbf2-related 1/2 kinases (NDR1/2) directly phosphorylate and inhibit YAP.260 YAP has also been found to be ubiquitinated and degraded by PARK2, an important downstream factor of PLCE1-SNAIL axis.261 PYGO2, a WNT signaling downstream protein, facilitates the recruitment of MLL1/MLL2 complex to WNT target gene promoters.262 Transcriptional co-activator TRIM37 activation during Hepatitis B virus (HBV) infection-associated hepatic fibrosis, is mediated by reactive oxygen species (ROS)-induced nuclear factor κB (NF-κB) signaling.263 Meerson et al.264 have shown that leptin and insulin signaling indirectly modulates nuclear receptor co-activator 1 (NCOA1) through miR-4443. During prostate cancer progression, SRC-1 is phosphorylated by MAPK on Ser1185 and Thr1179 and thereby, increases its binding affinity to androgen receptors (AR).265 p38 MAPK and GSK3 have also been reported to phosphorylate SRC-3 on ser869 and ser505, which not only enhances its binding ability with AR but also determines the mode of action through ubiquitination.240 GSK3β has been observed to negatively regulate PGC-1α through inhibition of transcription factor EB (TFEB), which has an established role in PGC-1α gene expression.266 Puigserver et al.267 discovered many cytokines that stimulate activating phosphorylation of PGC-1 through p38 MAPK pathway, ultimately resulting in heightened respiration and energy expenditure in muscle cells. One of the most significantly upregulated miRNAs in response to elevated WNT signaling cascade, miR-150, is found to markedly suppress CREB signaling pathway by targeting its core transcription factors CREB1 and EP300.268 Jun N-terminal kinase (JNK) inhibits CRTC3 activity by mediating their phosphorylation and cytoplasmic retention.269 The AMPK signaling is another well-known phosphorylation-dependent inducer of CRTC activity.270 These context-dependent diverse modes of regulation of transcription co-activators provide a rational platform for effective disease diagnosis and therapeutics.
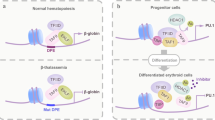
Interplay between co-activators and co-repressors
Cellular homeostasis is maintained by a perplexing complexity of transcriptional networks that regulate gene expression programs within a cell.271,272 The cycling behavior of the transcriptional network that alternates between on/off stages balances the transcriptional output. The key regulators of these alternate cycling events are the transcriptional co-activator and co-repressor molecules that function as “accelerator and brake”, respectively, to control target gene expression, in association with specific transcription factors.273 The transcriptional active and inactive states are significantly reinforced through different mechanisms like acetylation/deacetylation and methylation/demethylation, which are mediated by the collaborative interplay between transcriptional co-activators and co-repressors in relation to cell-specific chromatin contexts.274 In a normal physiological system, the dynamic equilibrium between the expression of transcriptional co-activators and co-repressors controls transcriptional plasticity, to regulate waves of transcription cycling which delicately equipoise homeostasis.275 One such example is of the thyroid hormone (TH)-mediated gene transcription. The thyroid hormone receptors (TRs) can bind to thyroid hormone response elements (TREs) in both liganded and unliganded conformation. When bound to TH, the receptor undergoes a conformational change that promotes the recruitment of transcriptional co-activators with histone acetyl transferase (HAT) activity that generates a permissive chromatin environment to promote target gene expression. However, in the absence of TH, due to a different structural conformation in the unliganded state, the TRs recruit a co-repressor complex (Co-R) with histone deacetylase activity (HDAC) that induces a repressive chromatin environment to prevent transcription of target genes. Thus, the co-ordinated action of transcriptional co-activators and co-repressors tightly control the TH-mediated gene transcription in cells.276 Another recent study conducted by Zaghet et al.277 has revealed that the interaction between the co-activators and co-repressors play an important role in preserving germ cell identity and immortality in C. elegans. H3K36 and H3K27 methylation propagated by methyltransferases is essential for germ cell maintenance. JMJD-5/KDM8, Jumonji C domain-containing demethylase/hydroxylase, which has been documented to function as context-dependent transcriptional co-activator or co-repressor,278 does not constrain H3K36me2 regions or remove H3K36me2 deposition. However, JMJD-5 blocks H3K36me2 accumulation in the regions that are normally associated with this modification. Therefore, a precise balance of methylation regulated by the methyltransferases and histone demethylates is essential for maintaining equilibrium.277
Contrary to the conventional regulatory mechanism, a myriad of evidences suggests that during malignant transformation, distorted transcriptional regulation is observed due to transcriptional rigidity.279 Cancer cell systems exhibit restricted plasticity due to which anti-mitotic inputs are disrupted, whereas the proliferative and anti-apoptotic signals are enhanced280 (Fig. 2a). For instance, the gain of function or loss of function mutations of transcriptional co-activators upregulate oncogenic transcriptional signaling, by facilitating permissive chromatin environment. One-third of cutaneous squamous cell carcinoma documents the loss of function mutations of CBP/p300 lysine acetyltransferases. Loss of function of these co-activators leads to enhanced HrasS35-mediated epidermal thickening, which initiates the formation of skin papillomas.281 However, gain of function of HAT/TAZ2 domain mutants have been observed in head and neck cancer patients. These CREBBP and EP300 mutations promoted a hyperacetylated state and enhanced DNA damage repair and radioresistance.282 Cancer progression also involves altered expression of transcriptional co-repressors. For instance, C-terminal binding proteins 1 and 2 (CtBP1 and CtBP2) are known to interact with polycomb group complexes, including components such as REST/CoREST, HDAC1 and HDAC2, to mediate transcriptional repression.283 However, CtBP1 is deregulated in malignancy. The elevated levels of CtBP expression across different cancer types have indicated that this co-repressor plays a key role in epigenetic regulation of cancer by repressing the transcription of a multitude of tumor suppressor genes.284.285 The loss-of-function of co-repressors has also been illustrated in oncogenic process. One such example is that of downregulation of the co-repressor breast cancer metastasis suppressor 1 (BRMS1). Loss of BRMS1 promotes carcinogenesis by facilitating the recruitment of RelA/p65 to NF-κB-dependent anti-apoptotic genes.286 Scaffold/Matrix-Associated Region-1 (SMAR1) deregulation in cancer is another example of co-repressor loss of function. Downregulation of SMAR1 promotes CCND1 transcriptional activation that promotes cancer cell proliferation.287 This deregulation of transcriptional co-regulators highlights the distortion of co-activator/co-repressor balance in disease pathology.

Transcriptional co-activators: Interplay with co-repressors and involvement in developmental and metabolic disorders. a In healthy individuals, cellular homeostasis is perpetuated by a dynamic equilibrium between the transcriptional co-activators (Co-A) and co-repressors (Co-R), that fine tunes the balance between cell proliferation and cell death signals. However, during disease conditions, like malignant transformation, the balance is skewed towards those co-regulators (both co-A and co-R) that mediate cell proliferation signals. Context-specific gain-of-function or loss-of-function mutations of transcriptional co-regulators mediate upregulation of oncogenic transcriptional signaling, thereby facilitating cancer promotion and progression. b Involvement of transcriptional co-activators in three common developmental disorders (ASD autism spectrum disorder, ADHD attention deficit/hyperactivity disorder, ID intellectual disability) and two of the most prevalent metabolic disorders, diabetes and obesity. This figure was created using BioRender (https://biorender.com/)
Diseases associated with mutation of transcription co-activator families
Considering the compendium of previously stated facts on transcriptional co-activators, it is now quite evident that co-activators are indispensable for establishing homeostasis during gene expression. Therefore, exquisite regulation of these factors is imperative to maintain normal physiological conditions, derangement of which will cause manifestation of diseases. There are several reports that have delineated the mutations in these co-activator genes as the major force driving disease progression. Examples of diseases that are associated with the mutations of the co-activators have been summarized in Table 1. Involvement of co-activators in developmental disorders, metabolism-related diseases and cancer has been elaborated below.
Co-activator involvement in developmental disorders
Developmental disorders are known to be heterogeneous conditions that have been reported to affect a significant population of children worldwide.288 The most frequently diagnosed developmental conditions throughout the world are, autism spectrum disorders (ASD), attention-deficit/hyperactivity disorder (ADHD) and intellectual disability (ID).289,290 Wealth of evidences have suggested that chromatin remodeling and transcriptional regulation plays a crucial part in the development of these diseases.291 Here we have briefed the co-activator mediated transcriptional deregulations that lead to developmental disorders (Fig. 2b).
Autism spectrum disorder (ASD)
A component in mammalian SWI/SNF complex, BAF53b is essential for neuronal development, function and cell identity.292 Loss of function of BAF53b has been associated with increased risk of developing ASD.293 BCL9 and CBP deletion have also been reported in ASD.294 De novo mutation leading to an amino acid substitution of the transcriptional co-activator MKL2 or MRTFB has been associated with ASD. However, the mechanism of this mutation mediated AD development is yet to be elucidated.295 SETDB1 has been shown to influence embryological development by promoting the maintenance of pluripotency and suppressing the differentiation of embryonic SCs,296 and therefore, is required for nervous system development and function while dysregulation of SETDB1 is implicated in the pathogenesis of CNS disorders including ASD.297 Altered expression or deletion of KDM4C has been linked to altered methylation patterns leading to autism.298 Another histone methyltransferase KMT2E (MLL5) haploinsufficiency has been linked to manifestation of autism like behavior in mice.299 3% of individuals with ASD were found to exhibit multiple de novo frameshift insertion and deletion mutations in this gene. Moreover, a cohort of 2500 patients has been reported to contain de novo missense and nonsense mutation of histodemethylase KDM5B.300 Missense variant dead box helicase 5 (DDX5) have been shown to affect protein-protein interactions and, increase the risk of ASD.301 A study by Crider et al.302 provided evidence that a significant decrease in the expression of ER co-activators, SRC1 (34%), CBP (77%), PCAF (52%) was observed in the middle frontal gyrus of ASD patients. Benito et al.303 found that pharmacological inhibition of BET/BRD leads to autism-like behavior in mice. A significant proportion of ASD cases have been observed to possess mitochondrial metabolic dysfunction.304 Hypermethylation of PGC-1α promoter-induced mitochondrial dysfunction has also been found to cause ASD.305 Hence, it can be stated that the co-activator molecules have a critical role in autism spectrum disorder (ASD) and therefore, can be a good therapeutic target for amelioration of ASD and related diseases.
Attention deficit/hyperactivity disorder (ADHD)
Transcription co-activator CITED2 has been found to contribute in maintaining proper somatosensory neocortical length, neuronal connectivity, and neocortical development.306 Conditional knockout of CITED2 in the forebrain of mice led to aberrant neocortical development, which can be associated with ADHD.307 Gao et al.308 proved that haploinsufficiency of KDM6B can be linked to ADHD related behaviors in mice. Olfson et al.309 conducted whole exome sequencing of 152 parent–child trios and identified KDM5B to be one of the high-risk genes in ADHD. Rare copy number variation in TRIM32 gene and single nucleotide polymorphism of TRIM31 gene are the drivers of ADHD development.310 Mutation in a SWI/SNF chromatin remodeling complex protein ARID2, has been found in the patients with ADHD.311 Analyzing whole-genome sequencing data from 272 patient samples, Zhou et al.312 showed that one of the top candidate genes that are linked with ADHD is KMT2D. An epigenome-wide association study revealed the association of co-activator ZNF544 with ADHD during early childhood.313 Geneviève et al.314 found that 44% of the individuals with DEAD-box RNA helicase 3 (DDX3)-related disorders suffer from attention deficit/hyperactivity disorder (ADHD) symptoms. Though, evidence indicating the connection between co-activator deregulation and developmental disorders is abundant, very few, if any, systemic study deciphers the detailed molecular mechanism.
Intellectual disability (ID)
Acetylation status of proteins is exquisitely regulated in neuronal plasticity and cognition behavior regulation. One of the main regulators of this status, CBP/p300, has been found to have a link in ID progression.315 Mutation at 3p25.3 on SETD5 gene, which is expressed throughout the brain, is suggested to facilitate ID.316 KMT2D and KDM6A gene mutations lead to defective methylation pattern and as a consequence, drive Kabuki Syndrome-related ID.317 Lebrun et al.318 studied KMT2A gene in a cohort of 200 patients and found deletion and missense mutation in Wiedemann-Steiner syndrome related IDs. Mutations in lysine demethylase 1A (KDM1A) affect their active site residues and catalytic activity, which in turn limits their binding affinity to TFs. These mutations are reported to promote intellectual ability impairment.319 YAP1 loss-of-function mutations were observed in patients with Colomoba, an eye abnormality that is often associated with intellectual disability.320 A rare neurodevelopmental disorder caused by variation in the genes, encoding members of SWI/SNF family of transcriptional co-activators, is SWI/SNF-related intellectual disability disorders (SSRIDDs). The most common cause of SSRIDD is mutation in ARID1B, which is a core component of SWI/SNF complexes.321 Barish et al.322 reported that SSRIDDS is also associated with mutations in BICRA (BRD4 interacting chromatin remodeling complex-associated protein) gene. Similar to ADHD, mutations in DDX3X have been associated with intellectual disability. Blok et al.323 reported that in females, mutations in DEAD box helicase protein DDX3X accounts for 1–3% of unexplained intellectual disabilities. De novo mutations and segregating missense mutations were also observed in males. Through their study Blok et al.323 established that DDX3X mutation possess an X-linked recessive inheritance pattern. Balak et al.324 further reported that de novo missense mutation of DDX6 is also associated with intellectual disbility. X-linked intellectual disability (XLID) contains TRIM1 missense mutations (p.R347Q and p.N343S) in affected as well as obligate carriers. Moreover TRIM1 mutation (p.Asn343Ser) was found in 480 patients with idiopathic intellectual disability,325 whereas mutations in TRIM18 led to X-linked form of Opitz Syndrome.326
Co-activator involvement in metabolic disorders
Metabolic disorders can be described as a constellation of intertwingled pathophysiological abnormalities arise from metabolic origin.327 The most commonly occurring metabolic disorders are diabetes and obesity.328 The need for identification and characterization followed by therapeutic implementation is also rising. Metabolic disorders are genetically diverse disease and a myriad of gene regulation complexes have been linked with it.329 Transcription co-activators are one of the multiple factors that closely govern the process of transcription and metabolic disorder progression.330 Here we have summarized the co-activators that are reported frequently in the context of diabetes and obesity (Fig. 2b).
Diabetes
In the last few decades, diabetes has been emerged as one of the most diagnosed metabolic disorders with almost 463 million cases worldwide. Progressive loss of β-cell identity and insulin resistance is generally associated with type 2 diabetes.331 It has been observed that downregulation of CBP/p300-mediated H3K27 deacetylation promoted β cell failure in type 2 diabetes in islets of prediabetic db/db mice.332 Moreover, in hyperglycemia, loss of p300 histone acetyl transferase activity promotes β cell apoptosis.333 However, unbalanced levels of histone acetylation have been found to be involved with diabetic retinopathy, one of the major causes of diabetes-associated morbidity. Significant increase in acetylation of retinal histone H3 at lysine 9 (H3K9) and lysine 23 (H3K23) was observed in experimental diabetic animals. It was also observed that in the retina, HAT p300-mediated acetylation is associated with proinflammatory molecule induction, suggesting that transcriptional co-activator-mediated acetylation is a major contributor of diabetic retinopathy334; hence, a tissue-specific role of CBP/p300 is predominant in diabetes manifestation. Sakai et al.335 further established that disruption of the GCN5 and CITED2 ameliorates diabetes and also dampens gluconeogenesis. The p160 co-activators (p/CIP and SRC-1) have also been found to negatively regulate insulin sensitivity and the levels of insulin receptor substrate (IRS) proteins. Moreover, downregulation of p/CIP and SRC-1 was found to enhance insulin sensitivity and increase glucose uptake in both regular and high fat diet-fed p/CIP and SRC-1 double knockout (DKO) mice,336 indicating that targeting these diverse co-activators, is a promising pharmacological target for treatment of both type 2 diabetes and obesity. Role of TRIM family of transcriptional co-activators has also been implicated in diabetes mellitus. Wan et al.337 reported that when compared to healthy control, elevated expression of TRIM32 was observed in the type 2 diabetes mellitus patients. In vitro experiments further revealed that under high glucose conditions, marked increase in the expression of TRIM32 along with a concomitant downregulation in the AKT and mTOR phosphorylation levels was observed, which further exacerbated pancreatic cell autophagy and hampered insulin secretion, thereby promoting development of type 2 diabetes. The Hippo pathway transcriptional co-activators YAP/TAZ has also been documented to mediate insulin resistance by promoting phosphorylation of IRS1. Combination treatment with YAP/TAZ inhibitor (verteporfin) and metformin led to complete inhibition of the insulin and IGF1 signaling.338 Collectively, more elaborate studies are essential to discern the role of transcriptional co-activators in diabetes, which will direct the development of new therapeutic strategies in future.
Obesity
Obesity is typically defined as a multifactorial chronic disease with several causes resulting in excessive body fat accumulation, which sometimes is associated with poor health conditions.339 Zhou et al.340 found that selective inhibition of the HAT domain of CBP/p300 histone acetyltransferases, by A-485, markedly decreased the fat mass in obese mice. Contrarily, another study reported that the loss of CBP in the hypothalamus resulted in obesity.341 Hu et al.342 predicted a mechanism of BRD4-induced obesity through peroxisome proliferator-activated receptor ɣ (PPARɣ)-dependent growth differentiation factor 3 (GDF3) regulation. Obese conditions in mice has been observed to activate CRTC2/3 by decreasing the expression of salt-inducible kinases (SIK), a Ser/Thr kinase that phosphorylates and inhibits CRTCs.343 Tumor necrosis factor α (TNF-α) mediated PGC-1α downregulation has been reported in obesity in rodents. Similar reduction is also reported in obese human patient samples.344 Deletion of TRIM 28 and deficiency of SRC1 has also been associated with obese condition.345
Co-activator involvement in different cancers
A rampant occurrence
With the rising burden of cancer, it has become imperative to develop new therapeutic approaches to curb disease progression. According to Globocan (2020), based on estimated worldwide age-standardized mortality rates, including all gender and all ages, lung cancer (18%), breast cancer (13.6%), colorectal cancer (9%), liver cancer (8.7%), stomach cancer (7.7%), prostate cancer (7.7%), cervical cancer (7.3%), esophageal cancer (5.6%), pancreatic cancer (4.5%) and ovarian cancer (4.2%) accounts for a substantial amount of cancer-related deaths.346 Moreover, based on age-standardized mortality rates, head and neck cancer (including lip, oral cavity, larynx, oropharynx, hypopharynx, salivary glands and nasopharynx) and leukemia, contributes to 8.59% and 4%, respectively, of the total cancer-related deaths.346 Hence, with the rising burden of cancer, it has become imperative to develop new therapeutic approaches to curb disease progression. Based on the Globocan statistics, this review attempts to abridge the involvement of several transcriptional co-activators that are responsible for the deregulated activity of several transcription factors across these twelve cancer types, which contribute substantially to cancer-related mortality worldwide. Such an approach will consequentially unearth novel therapeutic targets to curtail tumor progression, thereby reducing the burden of cancer.
Co-activator involvement in the deadliest forms of cancer
Breast Cancer
Breast cancer has been reported to be the most commonly diagnosed cancer with nearly 2.3 million new cases in the year 2020 (Globocan, 2020).346 In addition, breast cancer is the leading cause of cancer related deaths in women worldwide.347 Diverse histopathological subtypes make it more difficult to predict the progression of the disease. Hence, despite the progress in its detection and treatment, it seems necessary to unravel the roots of breast cancer so that new therapeutic approaches can be designed for its proper abrogation348 (Fig. 3a).

Transcriptional co-activators effectuate transcription of oncogenes in cancers of endocrine organs. (a) Several transcriptional co-activators, namely SRC, CITED2, SET7, DDX17, and ADA3 have been identified to work in association with the hormone-regulated transcription factor, estrogen receptor (ER), to promote breast cancer. Apart from ER co-regulators, several other transcriptional co-activators like MRTF, BCL9/BCL9L, EYA2, TRIM24 and YAP/TAZ promote breast cancer tumorigenesis. (b) In prostate cancer, the involvement of transcriptional co-activators CBP/p300,BCL9, PC4and ARA70 have been documented. (c) Enhanced expression of YAP/TAZ, CBP/p300, CRTC2, TRIM24, and BRD4 have been observed to be associated with ovarian cancer metastasis, therapy resistance and poor patient prognosis (d) The coordinated interaction between VGLL1 and TEAD1 promotes cervical cancer growth by mediating transcription of HPV early genes. Other transcriptional co-activators associated with cervical cancer are KMT2A, TRIM24 and TRIM28. CRPC, castrate-resistant prostate cancer; APIC, androgen-independent prostate cancer; HRPC, hormone-refractory prostate cancer; HGSC, ovarian high-grade serous carcinoma. This figure was created using BioRender (https://biorender.com/)
MRTF
Myocardin-related transcription factors (MRTFs) are a family of functionally related transcription co-activators that include myocardin, MRTF-A/MKL1/MAL and MRTF-B/MKL2, etc. This family of proteins associate with the MADS box transcription factors like serum response factor (SRF) and induce transcription of genes responsible for Rho-dependent cytoskeletal processes like cell motility, adhesion, and spread of breast cancer cells.349 Microtubule-associated serine/threonine-protein kinase-like (MASTL) acts as an activator of MRTF-A/SRF (myocardin-related transcription factor A/serum response factor) signaling. Taskinen et al.350 observed that mechanistically, MASTL associated with MRTF-A and increased its nuclear retention and transcriptional activity. This MASTL/MRTF-A signaling promotes breast cancer cell motility and invasion. Another study has reported that, P-cadherin upregulates MRTF/SRF signaling in early stages of breast carcinogenesis to promote self-renewal, proliferation and invasion.351
BCL9 and BCL9L
The aberrant activation of WNT/β-CATENIN signaling pathway leads to early events in carcinogenesis.352 Increasing evidences indicate that BCL9 and BCL9L transcriptional co-activators are over expressed in a significant population of breast cancer patients,353 and modulate the expression of β-CATENIN to promote tumor growth, cell migration, and metastasis in TNBC models.354 Targeting BCL9/BCL9L has been reported to have efficient anti-tumor effect through the inhibition of WNT and TGF-β signaling pathways, suggesting a viable therapeutic approach for TNBC treatment.355
SRC
The transcriptional activity of the estrogen receptor (ER) is regulated by its ligands as well as the co-regulators.356 Thus, changes in the expression of ER co-activators may be of utmost importance for the response to endocrine therapy.357 SRC (Steroid Receptor Coactivator) family of co-activator proteins including SRC-1, SRC-2, and SRC-3 are the most well-known ER co-regulators. SRC-1 and SRC-3 are of particular importance since high levels of these two transcriptional co-activators have been found in a number of breast cancer studies358 and their increased levels are associated with nodal positivity and endocrine resistance.359
CITED2
Another ER transcriptional co-activator in breast cancer is CBP/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 2 (CITED2) which is overexpressed in breast cancer tissues and is associated with worse clinical outcome.360 Increased expression of this protein may result in estrogen-independent ER activation, thereby reducing estrogen dependence and response to hormone therapies.361 Jayaraman et al.362 has also documented that CITED2 can modulate macrophage recruitment to influence breast cancer growth.
SET7
Yet another ERα transcriptional co-activator that has been recognized for several years is SET7, a protein lysine methyltransferase (PKMT) encoded by the SETD7 gene which is a key regulatory enzyme that mediates methylation of lysine residues of histone and non-histone proteins.363 SET7 has been documented to be over expressed in clinical breast cancer samples and the over expression of SET7 has been associated with tumor size, weight and expression of VEGF.364 SET7 stabilizes ER by methylating lysine 302 (K302) residue which is essential for recruitment of the transcription factor to its target genes and their transactivation.365 Gene Ontology (GO) analysis suggests that the ERα and SET7 co-activated target genes are primarily involved in the regulation of cell migration, but the precise molecular mechanism is undetermined till date.366
DDX17
The DEAD-box RNA helicase p72 (DDX17) has been shown to act as transcriptional co-activator for ERα.367 Studies have reported that knockdown of DDX17 results in a significant inhibition of estrogen-dependent transcription of endogenous ERα-responsive genes and estrogen-dependent growth of breast cancer cells.368 Moreover, in ER-positive breast cancer, DDX17 also acts as transcriptional co-activator of SOX2 and upregulates SOX2-mediated stem cell like features.369
ADA3
One of the kingpins that is associated with chromatin modification for transcriptional activation is the ADA3 (Alteration/Deficiency in Activation 3) protein which is an adaptor component of several lysine acetyltransferase complexes.370 Mirza et al.371 have reported that ADA3 is over expressed in breast cancer patients and as a transcriptional co-activator, human ADA3 (hADA3) interacts with ERα, thereby transactivating its downstream targets leading to breast cancer progression.372
EYA2
Amongst the different evolutionarily conserved transcription factors that impact carcinogenesis, sineoculis homeobox homolog (SIX) family proteins have been shown to play important roles in cell proliferation, migration, and apoptosis.373 SIX1, the most extensively studied of the SIX family members, is known to promote tumor invasion, metastasis, and paclitaxel resistance in breast cancer cells.374 In breast cancer patients, overexpression of co-activator EYA2 has been associated with poor prognosis.375 It has been reported that EYA2 (eyes absent 2) transcriptional co-activator, mandatorily forms a bipartite transcription initiation complex with SIX1 transcription factor and enhances proliferation, metastasis and DNA damage repair of breast cancer cells, ultimately promoting breast cancer progression.376
TRIM24
Tripartite motif 24 protein (TRIM24) also known as transcriptional intermediary factor 1α (TIF1α) is a transcriptional co-activator that has a N-terminal TRIM domain with three zinc-binding domains – a RING, a B-box type 1 and a B-box type 2 and also a coiled region with potential self-assembly properties,377 a C-terminal region containing a PHD finger, a bromodomain, and a nuclear receptor interaction box.377 TRIM24 transcriptional co-activator has also been shown to turnaround transcriptional networks to induce breast cancer progression and is associated with poor survival in breast cancer patients.378 It has been observed that TRIM24 directly activates MET gene expression, which in turn upregulates c-MET-PI3K-mTOR pathway in metaplastic breast cancer (MpBC).379 It has also been observed that TRIM24 interacts with SMAD3 and dissociates its interaction with tumor suppressor TRIM33. The TRIM24-SMAD3 complex is further recruited to chromatin, which enhances SMAD3 activation and immune response-related cytokine expression, thereby promoting enhanced breast cancer stemness, MDSC (myeloid-derived suppressor cell) recruitment, and metastasis.380
YAP/TAZ
The conventional wisdom suggests that YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif) are transcriptional co-activators that majorly interact with TEAD family of transcription factors to promote tumorigenesis.381 However, recent evidences suggest, YAP and TAZ can also function as co-activators for AP-1 and STAT3 transcription factors leading to poor survival of triple negative breast cancer patients, but have meager effect on survival of patients suffering from other forms of breast cancers.382 Furthermore, TAZ can also act as a co-activator of hypoxia-inducible factor-1 (HIF-1α), which results in enhanced transcriptional activity of HIF-1α.383 Moreover, YAP in association with another transcriptional co-activator AIB1 (amplification of the p160 nuclear hormone receptor co-activator amplified in breast cancer-1; also known as NCOA3, SRC3, or TRAM3) has been shown to physically interact with TEAD4. This AIB1-YAP-TEAD4 interaction is essential for cell invasiveness in mammospheres.384
Prostate Cancer
According to Globocan (2020), a total of 1,414,259 new cases of prostate cancer and 375,304 prostate cancer-related deaths were reported globally. The increasing incidences of prostate cancer associated with the alarming mortality rates emphasizes the need to develop alternate therapeutic strategies to curb prostate cancer.385 The major co-activators involved are described (Fig. 3b).
CBP/p300
In primary and metastatic castration-resistant prostate cancer tissues, CBP/p300 are over expressed at mRNA levels.386 CBP/p300 has firmly been established to act as transcriptional co-activators of androgen receptor (AR).387 p300 has been documented to interact directly with AR N-terminal domain and AR-Ligand binding domain.388 Upon interaction, CBP/p300 acetylates AR and promotes AR stability.389 Ji et al.390 reported that CUB-domain-containing protein 1 (CDCP1) is highly expressed in late-stage and castrate-resistant prostate cancer (CRPC). In CRPC tumorigenesis, the co-activators BRD4 and CBP/p300 co-regulates the transcriptional activity of CDCP1.
CITED2
The transcriptional co-activator CITED2 has enhanced expression in metastatic prostate cancer and its expression is also correlated with poor survival. Shin et al.391 have reported that in prostate cancer, CITED2 acts as a molecular chaperone and guides p300 and PRMT5 to nucleolin, consequently inducing nucleolin activation. This CITED2-nucleolin axis is associated with prostate cancer metastasis.391
BCL9
BCL9 is highly expressed in clinical prostate specimens in comparison to the benign prostate tissues.392 In prostate cancer group, the positive rate of BCL9 was 53.1% (52/98), whereas in benign group the positivity rate was 25.0% (5/20; P = 0.022). Moreover, it was observed that the higher expression of BCL9 was correlated with shorter biochemical recurrence-free survival (P = 0.037) as indicated by Kaplan-Meier survival analysis.393 Detailed mechanism of the BCL9-mediated prostate cancer progression is yet to be elucidated.
PC4
The transcriptional co-activator PC4 is highly upregulated in prostate cancer and is associated with metastasis, progression and prognosis. PC4 has also been found to be significantly upregulated in androgen-independent prostate cancer (AIPC) when compared with Androgen-dependent prostate cancer (ADPC). It has been observed that PC4 suppress c-MYC/p21 pathway to inhibit cell growth and cell cycle arrest at G1/S phase. Moreover, PC4 also promotes the expression of HIF-1α and activates β-catenin signaling to exert its oncogenic activities.394 Further it has been recorded by Chakravarthi et al.395 that PC4 binds to the promoter region of several oncogenes like Polo-like kinase 1(PLK1), C-MYC, serine-threonine kinase BUB1B to regulate their expression.
ARA70
The first AR co-regulator that was identified is androgen receptor (AR)-associated coregulator 70 (ARA70). It has been observed that ARA70 interacts with ARA70-N2 domain via the consensus FXXLF motif to promote AR activity. Moreover, ARA70 is highly expressed in prostate cancer specimens (91.74%) than in benign tissues (64.64%, p < 0.0001). In addition, ARA70 is also upregulated in high-grade prostate cancer tissues and in hormone-refractory LNCaP xenografts.396,397 However, elaborate studies involving ARA70-mediated signaling has not been documented till date.
Ovarian Cancer
Ovarian cancer is one of the most common cancer in women and it accounts for 4.2% of total cancer related death in females. Though various reproductive and hormonal factors including parity, oral contraceptive use, and lactation may lead to lower risk, however several other causes like menopause at older age and hormone replacement therapy confer escalated risks of ovarian cancer.398 Therefore, identification of alternative therapeutic targets is necessary (Fig. 3c).
YAP/TAZ
The transcriptional co-activators YAP and TAZ have been reported to promote ovarian cancer tumorigenesis.399,400 Moreover, mRNA and protein levels of TAZ has been reported to be upregulated in ovarian cancer and a meta-analysis of microarray datasets of ovarian cancer has identified that increased expression of TAZ mRNA is correlated with poor prognosis in patients with ovarian cancer.401 Furthermore, YAP is highly expressed in inflammatory and cancerous fallopian tube tissues and YAP interacts with FGF-FGFR pathway to regulate fallopian tube umbilical epithelial cell activity. Recent studies have indicated that ovarian high-grade serous carcinoma (HGSC) might originate from fallopian tube umbilical epithelial cells primarily the secretory epithelial cells of fallopian tubes. Association of YAP with cancerous fallopian tube tissues further indicates involvement of YAP in HGSC.402
CBP/p300
Using the ovarian cancer cell line SKOV3, it has been reported that Staphylococcal nuclease domain-containing protein 1 (SND1) regulates the gene transcriptional activation of SLUG (an epithelial-to-mesenchymal transition marker) by increasing chromatin accessibility through the recruitment of the acetyltransferases GCN5 and CBP/p300 to the SLUG promoter proximal region.403 Moreover, physical association of BRCA1 was observed with the transcriptional co-activators/acetyltransferases p300 and CBP. Endogenous as well as overexpressed BRCA1 and p300 were found to associate in a phosphorylation-independent manner. BRCA1 interacts with the cAMP response element-binding protein (CREB) domain of p300/CBP via both its amino and carboxyl termini to mediate BRCA1-dependent transactivation.404
CRTC2
CRTC2 is over-expressed in chemo-resistant tissues of ovarian cancer. It has been observed that ovarian cancer patients with high expression of CRTC2 has poor prognosis. In addition, CRTC2 regulates the autophagic flux partially through PI3K-AKT pathway. Thus, CRTC2 might be a potential predictor as well as target for ovarian cancer.405 Furthermore, CRTC2 in association with CREB is also involved in the transcriptional activation of BCRP (Breast Cancer Resistant Protein)/ ABCG2, which further promotes ovarian cancer.406
BRD4
It has been observed that the transcriptional co-activator BRD4 is the fourth most amplified gene in HGSC, the most aggressive type of ovarian cancer. Overexpression of BRD4 has also been associated with poor patient prognosis. Moreover, increased expression of BRD4 is associated with upregulated expression of several oncogenes, such as MYC, NOTCH3, and NRG1. These oncogenes enhance tumor cell proliferation, genomic instability, metastasis, and resistance to chemotherapy.407,408
TRIM24
Zhang et al.409 have reported that TRIM24 is over-expressed in ovarian carcinoma in comparison to normal ovarian tissues. Upregulated expression of TRIM24 was observed to be closely correlated with serum CA-125 (P = 0.0294), metastasis (P = 0.0022), FIGO (International Federation of Gynecology and Obstetrics) stage (P = 0.0068) and Ki-67 level (P = 0.0395). Moreover, high expression of TRIM24 predicted worse prognosis in ovarian cancer patients. Furthermore, TRIM24 has been documented to promote AKT-phosphorylation, which in turn regulates metastasis. Another study by Zhou et al.410 has revealed that elevated expression of TRIM24 was linked to higher rates of lymphatic and distant metastasis. Moreover, TRIM24 negatively regulates the activity of FOXM1 to promote ovarian cancer progression.
Cervical Cancer
Cervical cancer, a malignant tumor of the lowermost part of the cervix, belongs to one of subsets of cancer with very high incidence and mortality rates. In the year 2020, the estimated number of new cases of cervical cancer was 604,000 with 342,000 deaths worldwide. It contributes to 7.3% cancer-related deaths in women worldwide.346 Identification of frequently deregulated factors is therefore necessary to design new therapeutic approaches to manage cervical cancer (Fig. 3d).
VGLL1
The key etiological agents responsible for the development of cervical cancer are human papillomaviruses (HPVs).411 It has been established that TEAD1 transcription factor activates the early promoter of human papillomaviruses.412 In addition, a study reported that TEAD1 mediated HPV early gene expression is regulated at the transcriptional level by VGLL1 (Vestigial-Like Family Member 1), which is a TEAD-interacting transcriptional co-activator. VGLL1/TEAD1 complex has been shown to interact with HPV16 long control region (LCR) and downregulation of VGLL1 and/or TEAD1 significantly decreases viral early gene expression, suggesting that VGLL1/TEAD1 is essential for efficient transcription of HPV early genes.413 Moreover, contrary to TEAD1, VGLL1 exhibits tissue-specific expression and is associated with development and differentiation of epithelial lineage tissues in concordance with HPV gene expression, thereby indicating that VGLL1 might facilitate epithelial specificity of HPV gene expression.413
KMT2A
KMT2A (histone-lysine N-methyltransferase 2A, former MLL) is a transcriptional coactivator with histone H3 lysine 4 (H3K4) methyltransferase activity.414 KMT2A is popularly known to be associated with acute leukemias, especially in infants, where it mostly interacts with six partner genes (AFF1, MLLT3, MLLT10, MLLT1, ELL, AFDN).415 However, a recent study has highlighted that KMT2A is also prevalent in cervical cancer, where it promotes cancer cell growth by regulating VADC1 (Voltage-dependent anion-selective channel 1). Downregulation of KMT2A was further shown to suppress cervical cancer cell proliferation and migration, accompanied with an activation of PARP/Caspase pathway and inhibition of VADC1, whereas overexpression of VDAC1 leads to a reversal of the KMT2A knockdown mediated changes, indicating that KMT2A/VDAC1 signaling axis might be a new therapeutic target for cervical cancer prevention.416
TRIM24
Cervical cancer has also been reported to have higher expression of TRIM24 transcriptional co-activator. It has been demonstrated that TRIM24 regulates the NF-κB and AKT signaling pathways, thereby contributing to cancer progression and metastasis.417
TRIM28
In comparison to their normal counterparts, cervical cancer cell lines and tissues also show an upregulated expression of TRIM28 transcriptional co-activator.418 TRIM28 has been found to significantly increase the phosphorylation of mTOR and its downstream molecule S6K1, leading to mTOR mediated cervical cancer growth and progression.419
Lung Cancer
With an estimated 1.8 million deaths, lung cancer is the leading cause of cancer related deaths worldwide.346 In spite of numerous developments in treatment modality, the survival outcomes are discouraging. For the low- and middle-income countries, lung cancer is emerging as a serious health concern.420 So, identifying new therapeutic avenues to combat the disease is a prerogative. Accordingly, the following transcriptional regulators have been identified to act as masterminds modulating lung cancer at large (Fig. 4a).

Role of transcriptional co-activators in lung cancer, head and neck cancer and leukemia. (a) In lung cancer, non-small cell lung cancer (NSCLC) growth is facilitated by the activity of several transcriptional co-activators including CRTC2, CBP/p300, PC4 and MRTF. The transcriptional co-activator COLCA1 interacts with the transcription factor POU2F3 to promote small cell lung cancer (SCLC) growth. (b) In head and neck cancers (HNC), YAP and AIB1 interact with TEAD family of transcription factors to facilitate tumorigenesis. TAZ, on the other hand, forms a complex with TEAD4, binds to the promoter region of the pluripotency gene SOX2, consequentially initiating its transcription, thereby upregulating self-renewal and maintenance of CSC population. The co-activator molecules CRTC2, TRIM24 and BRD4 have also been reported to be actively involved in head and neck cancer growth, proliferation and CSC maintenance. (c) In leukemia, deregulation of the co-activators MKL/MRTF-A, CBP/p300, CITED2, BRD4 and DDX5 have been observed. CSC, cancer stem cells; AMKL, acute megakaryoblastic leukemia; AML, acute myeloid leukemia; CML, chronic myelogenous leukemia; ALL, acute lymphocytic leukemia. This figure was created using BioRender (https://biorender.com/)
POU2AF2 (C11orf53)/POU2AF3 (COLCA2)
POU2F3 (POU class 2 homeobox 3; also known as SKN-1a/OCT-11) is the master regulator of cell identity in the neuroendocrinelow variant of small cell lung cancer (SCLC).421 Co-immunoprecipitation assay performed by Zhou et al.422 revealed that the transcriptional co-activator COLCA2 (POU2AF3) and C11orf53 (POU2AF2) physically interacts with transcription factor POU2F3 to regulate tuft cell-like SCLC cell growth. Furthermore, mutation in N-terminal binding domain of COLCA2 reduced its interaction with POU2F3. In addition, ectopic expression of POU2F3 with COLCA2 in HEK293T cells activated the expression of AVIL, a POU2F3 direct target. However, when each factor was expressed individually, the expression of AVIL was not activated, indicating that physical interaction between transcriptional co-activator COLCA2 with transcription factor POU2F3 is essential to facilitate POU2F3-mediated gene transcription. Moreover, Colca2–/– mice and C11orf53–/– were reported to be viable. Therefore, disruption of this co-activator/transcription factor physical interaction is predicted to be a potential therapeutic strategy to selectively inhibit tuft cell-like SCLCs with minimal toxicities.422,423
CRTC2
Non-small cell lung cancer (NSCLC) accounts for about 80–85% of all lung cancers424 and about 20% of NSCLC report alterations in LKB1 (Liver Kinase B1).425 It has been reported that LKB1-deficient NSCLC (LKBC) is associated with aberrant dephosphorylation and activation of the transcriptional co-activator CRTC2. It has been reported that CRTC2 is highly expressed in lung cancer tissues compared to adjacent normal.426 Active CRTC2 shuttles to the nucleus, binds to CREB and stimulates their downstream signaling cascade.427 ID1 (Inhibitor of DNA Binding 1) is a canonical CREB target and the constitutive upregulation of CREB/CRTC2 pathway in LKBC promotes oncogenesis by enhancing the expression of ID1 which upon activation regulates the expression of genes responsible for extracellular matrix and cytoskeleton modulation, cell-cell interactions, anchorage-independent growth and lung colonization, thereby promoting a more aggressive tumor phenotype.428
CBP/p300
CREB-binding protein (CBP) and its paralog, E1A-binding protein (p300) are highly conserved transcriptional coactivator with four transactivation domains that mediate interaction with transcription factors.429 These co-activators contain histone acetyltransferase (HAT) activity, which enables them to acetylate various non-histone transcription-related proteins such as p53.430 Overexpression of CBP/p300 is considered as a poor prognosis indicator for lung cancer patients.431 Activation and upregulation of human telomerase reverse transcriptase (hTERT) is a hallmark of lung cancer. It has been reported that, upregulation of hTERT expression and tumor growth in lung adenocarcinoma cells is mediated by CBP, which binds to hTERT promoter and upregulates its transcription.432 Another study conducted by Zhang et al.433 reported that lncRNA LINC01977 interacts with SMAD3 and induces its nuclear transport. The nuclear SMAD3 interacts with CBP/p300 to regulate the transcription of ZEB1, thereby promoting malignancy of early-stage lung adenocarcinoma.433 Moreover, it has also been documented that p300 in association with CREB suppress lipid peroxidation by binding to the GPX4 (glutathione peroxidase 4) promoter region, which further inhibits ferroptosis in lung adenocarcinoma.434 Hence, overexpression of CBP corresponds to poor prognosis in lung cancers.
PC4
Large scale microarray data integration across 21 major cancer types identified the transcription co-activator PC4 (positive co-factor 4) amongst the 46 common cancer signatures.435,436 It has been reported that PC4 is highly expressed in NSCLC cells and tissues.432 Moreover, PC4 has also been associated with lymphatic metastasis and poor prognosis in lung adenocarcinoma.437 PC4 has been shown to mediate transcriptional activation of several oncogenes. Studies have indicated that in lung adenocarcinoma, PC4 functions as an upstream inducer of VEGF-C, VEGF-D, and VEGF-R3, which are necessary for promoting angiogenesis.437 Moreover, the downregulation of PC4 led to anti-tumorigenic effects on NSCLC cells involving induction of cancer cell death and differentiation. Thus, PC4 could be a potential therapeutic target for non-small cell lung carcinoma.438
MRTF
Du et al.439 reported that in NSCLC, the transcriptional co-activator MRTF-A (myocardin-related transcription factor-A), interacts with NF-κB/p65 rather than its common binding partner SRF (serum response factor). This interaction facilitates NF-κB/p65 binding to the PD-L1 promoter, thereby promoting the transcription activation and expression of PD-L1. This further potentiates immune escape of NSCLC cells. Moreover, overexpression of MRTF-A has also been reported to regulate the activity of HOTAIR promoter, thereby promoting proliferation and migration of NSCLC cells through lncRNA HOTAIR.440
Head and Neck Cancer
Head and neck cancer (HNC) is one of the most common cancers worldwide by its incidence rate and accounts for over 800,000 new cases annually.346 It contributes to almost 8.59% of total cancer-related deaths amongst male worldwide. HNC comprises of the cancers at various sites, including the lip, oral cavity, larynx, nasopharynx, oropharynx, hypopharynx, salivary glands, nasal and paranasal cavity. The most common sites of occurrence in HNC vary by geographic distribution, because the etiology of HNC is associated with the components of modern lifestyle like tobacco, alcohol, areca nut etc.441 For this reason, understanding basic molecular crosstalk in the pathogenesis of HNC is one of the most important aspects of management of this cancer (Fig. 4b).
YAP/TAZ
YAP and TAZ are transcriptional co-activators with various upstream signals, which are mainly regulated by the Hippo signaling pathway.442 Studies have reported that both YAP and TAZ are over-expressed in head and neck squamous cell carcinoma (HNSCC).443 In HNSCC, the WNT signaling pathway and the ERBB4 ICD (erbb2 receptor tyrosine kinase 4 intracellular domain) are known to elicit YAP activation. The active YAP and its functional paralog TAZ then migrate to the nucleus, form a transcriptional complex with their DNA-binding partner TEAD (transcriptional enhanced associate domain) and promotes transcription of YAP/TAZ target genes (CTGF, CYR61, ANKRD1 etc.), ultimately aiding tumorigenesis.444 Moreover, the transcriptional complex formed by TAZ and TEAD4 has two binding sites in SOX2 promoter, which in turn facilitates transcription of SOX2, leading to the self-renewal property and maintenance of the cancer stem cell population in HNSCC cells. This further increases the risk of tumor recurrence and poor patient prognosis.445 Thus, targeting YAP/TAZ co-activators have high potential as targeted therapy for HNSCC treatment.
CRTCs
The family of cAMP-regulated transcriptional coactivators (CRTCs) is known to associate with the transcription factor cAMP response element–binding protein (CREB), which has proto-oncogenic properties.446 In HNSCCs, mitogen-activated kinase kinase1 (MEKK1) constitutively activates and overexpresses CRTC2 (cAMP-responsive element-binding protein (CREB)-regulated transcription coactivator 2) via a non-canonical MEKK1-p38 signaling axis. As a matter of fact, overexpression of CRTC2 leads to higher physical interactions between CREB and CRCT2, accentuating the CREB downstream signaling, which is essential for a several cancer-associated adaptive response like glucose metabolism, cell growth, survival, immune evasion, and the maintenance of cancer stem cells.447 It has also been reported that in salivary mucoepidermoid carcinoma (MEC), frequent CRTC1/3-MAML2 fusions were observed. This CRTC1-MAML2 interaction promotes salivary MEC development and maintenance.448,449
TRIM24
Transcriptional co-activator TRIM24 has been shown to drive cell cycle progression and upregulate CYCLIN D1 and p-Rb expression in HNSCC, suggesting that TRIM24 is involved in HNSCC progression through regulation of cell cycle related proteins.450 Cui et al.451 reported that TRIM24 variants were highly expressed in 56 HNSCC samples (P < 0.001). Furthermore, 54.95% (50/91) of HNSCC samples showed upregulated expression of TRIM24 by immunohistochemistry. In addition, univariate analysis indicated that high TRIM24 expression correlated with worse overall survival (P = 0 .020). Moreover, in multivariate analysis, TRIM24 was also recognized as an independent predictor of overall survival (P = 0 .030). In addition, TRIM24 was able to induce upregulation of GLUT3, a glucose transporter that further confirms the fact that TRIM24 regulates glucose metabolism, thereby promoting cancer metabolism.450,452
BRD4
Bromodomain containing 4 (BRD4) is a protein that associates with acetylated histones through its double bromodomains and facilitates transcription of the downstream genes.453 A significant overexpression of BRD4 in primary HNSCC samples as well as 4-nitroquinoline 1-oxide (4NQO)-induced HNSCC animal model was found to assist cell proliferation, migration, and invasion.454 Another study revealed that BRD4 and MMP2 expression levels were correlated in oral squamous cell carcinoma (OSCC), and both were highly expressed in lymph node metastasis cases, including delayed metastasis, and that suggests its potential use as novel predictor of metastasis.455 BRD4 has also been found to facilitate spheroid formation and invasion through a BRD4/EZH2 pathway which non-canonically activates STAT3 transcription factor, thereby promoting tumor progression through ΔNp63α-mediated transcription, which strongly suggests its possible involvement in cancer stem cell maintenance.456
Leukemia
Leukemia is a malignancy that is characterized by transformed hematopoietic progenitors and by bone marrow infiltration.457 According to Globocan 2020, leukemia accounted for 4% of total cancer-related death in males and 2.7% of total cancer-related death in females.346 Therefore, with the rapid increase in leukemic burden, addressing challenges in curbing the disease is imperative (Fig. 4c).
MKL1/MRTF-A
MKL1/MRTF-A has first been identified as a part of recurrent t (1;22) chromosomal translocation in acute megakaryoblastic leukemia. This translocation is specific to infantile AMKL and has majorly been diagnosed in patients younger than 6 months of age.457 Studies concerning MKL1/MRTF-A requires further investigation.
CBP/p300
Mixed lineage leukemia (MLL) gene has been reported to recruit p300/CBP through its transcriptional activation domain, which further promotes acetylation of histone H3 at lysines 9, 18, and 27. The AF4 family/ENL family/P-TEFb complex (AEP) binds to acetylated H3K9/18/27 to activate transcription, consequently activating the cellular machinery required for aberrant self-renewal of leukemia stem cells.458 CBP/p300 has also been reported to mediate the leukemic functions of MYB.459
CITED2
Elevated expression levels of the co-activator CITED2 (CBP/p300-interacting-transactivator-with-an-ED-rich-tail 2) has been associated with maintenance of both normal and leukemic hematopoietic stem and progenitor cells (HSPCs).460 Moreover, a subset of AML patients displayed higher expression levels of CITED2 in CD34(+) cells as compared with normal CD34(+) HSPCs.461 CITED2 also regulates p53 activity to promote AML, and therefore it can be a potential target for AML therapy.
BRD4
BRD4 has been found to be expressed in primary CML cells, CD34+/CD38− leukemic stem cells (LSC), and in the CML cell lines KU812, K562, KCL22, and KCL22T315I.462 Collaboration between BRD4 and DOT1L has been reported to be important in highly transcribed genes in proximity to super enhancers. By means of dimethylated histone H3 K79, DOTL1 facilitates histone H4 acetylation, consequently regulating the binding of BRD4 to chromatin Moreover, inhibition of BRD4 activity was found to suppress proliferation in the majority of patients with chronic phase CML.463
DDX5
An important oncogenic mechanism for T cell acute lymphoblastic leukemia (T-ALL) is aberrant Notch signaling. It has been observed that the transcriptional co-activator DDX5 acts as a component of MAML1 protein complex to facilitate NOTCH1 transcription activation complex in human T-ALL leukemic cells.464 Human T-cell leukemia virus type 1 (HTLV-1) is a causative agent of adult T-cell leukemia/lymphoma (ATL). Moreover, DDX5 and its paralog DDX17, has been reported to promote alternative splicing of cellular genes after NF-κB activation by HTLV-1, to facilitate the initiation of leukemic state.465
Colorectal cancer
In the year 2020, more than 1.9 million new cases of colorectal cancer (including anus) and 935,000 deaths were estimated to occur, which represents about 10% of total cancer cases and cancer-related deaths. Overall, colorectal cancer ranks third in terms of incidence, but second in terms of mortality,346 and is therefore of immense concern as far as novel and specific therapeutic strategies are concerned (Fig. 5a).

Pleiotropic influence of transcriptional co-activators in driving gastrointestinal cancers. The malignancies associated with GI tract contribute to one-third of all cancer-related deaths, with colorectal cancer, liver cancer, stomach cancer, esophageal cancer, and pancreatic cancer being the main contributors. a Transcriptional co-activators DDX27, KMT2A, CBP, BRD4, and YAP interacts with diverse transcription factors to facilitate colorectal cancer progression. b YAP, TAZ, CBP/p300, PPM1G, and HBx are significantly associated with liver cancer progression. c Gastric/Stomach cancer progression is modulated chiefly by YAP/TAZ and TRIM24-mediated regulation of WNT/β-CATENIN pathway, DDX5-mediated upregulation of mTOR/S6K1 pathway, and BRD4/E2F1-mediated upregulation of miR-106b-5p. d Transcriptional co-activators CRTC1 and ZNF282 have been reported to physically interact with transcription factors CREB and E2F1, respectively, fostering esophageal cancer growth and metastasis. e The co-activators PIWIL1, CBP/p300, SETD8, YAP, and BRD4 have been established to be key regulators of pancreatic cancer promotion and proliferation. PDAC pancreatic ductal carcinoma, APC/C anaphase promoting complex/cyclosome. This figure was created using BioRender (https://biorender.com/)
DDX27
Chromosomal instability (CIN) is a hallmark of colorectal cancer, which results in copy number alterations (CNAs).466 DDX27, transcriptional co-activator is significantly upregulated and has extremely high frequency of copy number gain in colorectal cancer (CRC), and has also been found to be upregulated in CRC tissues.467 A study by Tang et al.468 has identified NF-κB pathway as the principal target of DDX27 in CRC. DDX27 binds with NPM1 (nucleophosmin1) to interact with NF-κB in the nucleus leading to increased binding of NF-κB to the target gene promoters, triggering enhanced expression of VIMENTIN and SLUG, thereby promoting metastasis in CRC. Thus, in the context of colorectal cancer, the functional interaction between DDX27-NPM1-NF-κB is essential for tumor progression and metastasis.468
CBP/p300
The transcriptional co-activators CBP (CREB-binding protein) and p300 are histone acetyltransferases (HATs) that regulate tumor initiation and progression. It has been observed that prolonged poor prognosis was associated with high expression of CBP/p300. This finding indicated that CBP/p300 could be a potential therapeutic target for CRC treatment.469 Xu et al.470 further reported that mesenchymal stem cells in the tumor microenvironment secretes CCL7. The CCL7/CCR1 in turn activates CBP/p300, which upon activation acetylates KLF5 and promotes CRC proliferation and metastasis.
KMT2A
The KMT family of histone modification enzymes contain the SET domain that regulates gene transcription by promoting methylation of H3K4. KMT2A/KMT2D, KMT2C/KMT2B, SETd1A/SETd1B are the three pairs of KMT members that play significant role in tumorigenesis.471 KMT2 family mutations has been positively correlated with CRC progression.472 It has been documented that KMT2A interacts with p65 transcription factor (p65 is also known as nuclear factor NF-kappa-B), which is essential for its recruitment on the promoter region of CATHEPSIN Z (CTSZ), which is one of the important downstream targets of KMT2A. Upon recruitment on the promoter, KMT2A trimethylates H3K4, that in turn promotes CTSZ transcriptional activation, leading to enhanced epithelial-to-mesenchymal transition in CRC cells.473
BRD4
Bromodomain-containing protein 4 (BRD4) mediates its role as transcriptional co-activator by acting both as a passive scaffold to promote recruitment of transcription factors and as an active kinase to phosphorylate RNA polymerase II, thereby regulating transcription.474 Upregulated expression of transcriptional regulator BRD4 is frequently observed in CRC. Targeting BRD4 resulted in significant downregulation in the expression of MYC proto-oncogene, restraining colon cancer progression.475 Wang et al.476 observed that in CRC, BRD4 phosphorylation has been reported to promote interaction with STAT3 to subsequently induce chromatin remodeling through enhanced binding interactions with enhancers and super-enhancers, thereby supporting a tumor-promoting transcriptional program. Moreover, it has also been reported that upon loss of mediator kinase, MED12 and BRD4 cooperate to sustain colorectal cancer growth.477
YAP1
In sporadic CRC, a SNP located in the YAP1 gene has been identified as a common genetic risk variant with a hazard ratio of 1.05 and over expression of YAP1 is associated with shorter survival.478 It has been documented in β-CATENIN-driven colorectal cancer, that YES1 (a tyrosine kinase) phosphorylates YAP1 on Y357, subsequently promoting YAP1 nuclear localization and activation. The active YAP1 transcriptional co-activator then interacts and forms a ternary complex with the transcription factor TBX5 and β-CATENIN to promote CRC survival and progression.479 Furthermore, YAP/TAZ-TEAD4 complex has been documented to transcriptionally upregulate the expression of CCBE1 (collagen and calcium-binding EGF domain 1) by binding to CCBE1 enhancer region of both CRC cells and cancer-associated fibroblasts. This in turn upregulates VEGFC proteolysis and induces lymphangiogenesis in a CRC cell-derived xenograft model in vivo.480
Liver cancer
In 2020, an estimated 830,200 people died from liver cancer globally. Global age-standardized mortality for liver cancer was 8.7 per 100,000 people and was highest in the eastern part of Asia.481 Hepatocarcinogenesis involves the synergistic action of several cellular mechanisms including the transcription of several factors associated with inflammation, oxidative stress, hypoxia, along with other molecular mechanisms.482 Therefore, a proper understanding of the mechano-molecular aspects of hepatocarcinogenesis and identification of appropriate target molecules and signaling pathways responsible for tumor progression is crucial in order to develop effective therapies against hepatic cancers (Fig. 5b).
PPM1G
The most commonly identified coactivators that are associated with modification of epigenetic landscape are histone acetyltransferases (HATs), deacetylases (HDACs), kinases, and phosphatases.483 PPM1G/PP2Cγ phosphatase (one member of a family of metal-dependent Ser/Thr phosphatases) has been identified as a NF-κB transcriptional coactivator. This particular co-activator mediates its function by binding to the NF-κB target gene promoters in association with the RELA subunit of the NF-κB family, thereby facilitating the transition between initiation and elongation.484 Intriguingly, high levels of PPM1G were noted in advanced hepatocellular carcinoma stages. Further experimentation revealed that in hepatocellular carcinoma, MYC/MAX and EP300 activate PPM1G which in turn dephosphorylates SRSF3, triggering the alternative splicing of genes related to cell cycle and transcriptional regulation.485
YAP/TAZ
YAP and TAZ have been identified as key players associated with Sorafenib resistance in hepatocellular carcinoma (HCC). In a TEAD- and ATF4-dependent manner, YAP/TAZ enables HCC cells to overcome Sorafenib-induced ferroptosis. Mechanistically, in a TEAD-dependent manner, YAP/TAZ induces the expression of SLC7A11, a key transporter maintaining intracellular glutathione homeostasis. At the same time, YAP/TAZ sustains protein stability, nuclear localization, and transcriptional activity of ATF4, which in turn cooperatively induce SLC7A11 expression.486 It has also been reported, upon MYC/β-CATENIN activation, YAP/TAZ accumulated in HCC cells to promote mitogenic activation, tumor growth and survival.487 Another recent study has further highlighted that TAZ is a direct transcriptional target of c-MYC, which further promotes c-MYC-driven murine hepatocarcinogenesis by regulating the expression of the anti-apoptotic BCL2L12 gene.488 Moreover, pertaining to ubiquitous activation of YAP and TAZ in human liver cancers,489 YAP/TAZ-based rewiring strategies can be potential approaches to overcome HCC therapy resistance.
CBP/p300
The transcriptional co-activators CBP/p300 mediates increased acetylation of H3K18 and H3K27 in HCC tissues.490 It has been reported that the transcription factors HIF-1α and STAT3 can maximally induce transcription of VEGF when in association with CBP/p300 co-activator. Moreover, interruption of this transcriptional complex by melatonin prevented HIF-1α occupancy of the VEGF promoter and prevented HCC progression. Thus, administering pharmacological doses of melatonin, a well-known dietary supplement, may be highly beneficial in inhibiting liver cancer by disrupting the HIF-1α/STAT3/CBP/p300 complex.491
HBx
Hepatitis B virus X protein (HBx) has been found to be overexpressed in liver cancer tissues.492 It acts as a transcriptional co-activator through direct interaction with various proteins, such as the TATA-binding protein, RPB5 subunit of RNA polymerase II, TF IIH, TF IIB, and the proteins of basic domain-leucine zipper (bZIP) family, including the cyclic AMP-response element (CRE)-binding protein (CREB).493 HBx interacts with the co-activators CBP/p300 and cooperates with CBP/p300 in the CREB-mediated activation. Thus, HBx may be considered as a potentiator of the signal mediated by CREB, and this mechanism may be involved in HBV-mediated oncogenesis.494 HBx has also been found to act as co-activator of heat shock factor 1 (HSF1) to upregulate the expression of HSPA8 in liver cancer cells. HSPA8 upon expression, enhanced HBV replication and dampened ferroptosis-mediated cell death by upregulating the expression of SLC7A11/GPX4 and by decreasing the erastin-mediated ROS (reactive oxygen species) and Fe2+ accumulation in cells, thereby supporting liver cancer progression.495 Moreover, HBx mediates H3K4me3 modification in WDR5-dependent manner496 and it also has been documented to interact with MYH9, to induce its expression by modulating GSK3β/β-catenin/c-Jun signaling.497 Futhermore, HBx interacts with ARRB1 and the autophagic core protein MAP1LC3/LC3 to induce ARRB1-mediated autophagy. This autophagic induction drives G1/S cycle and promotes HCC.498
Gastric cancer
Gastric cancer remains an important cancer worldwide as it was responsible for over one million new cases and an estimated 769,000 deaths in 2020. Incidence rates are twofold higher in males than in females. In males, it is one of the most commonly diagnosed cancers and the leading cause of cancer-related deaths in several Asian countries346 (Fig. 5c).
YAP/TAZ
One of the main risk factor for gastric cancer is Helicobacter pylori infection. It has been reported that deregulation of the Hippo pathway in the gastrointestinal tissues is one of the prime causes of H. pylori-mediated gastric carcinogenesis. Upon H. pylori infection, an increase in both TAZ nuclear expression and transcriptional activity of transcriptional enhancer TEA domain (TEAD) transcription factors was observed, which in turn induced EMT, invasion, and cancer stem cell-like properties.499 It has also been observed that a ubiquitously expressed protein tyrosine phosphatase, SHP2 (Src homology-2 domain-containing protein tyrosine phosphatase-2) interacts with the transcriptional co-activators YAP/TAZ, which in turn promotes its nuclear localization. In the nucleus, SHP2 mediates parafibromin/β-catenin complex formation, stimulating WNT-target gene activation.500 Based on the gene ontology (GO) analysis, it was determined that blood microparticle, platelet alpha granule lumen, and chylomicron are common cellular locations of YAP and TAZ. However, a functional divergence between YAP and TAZ was perceived owing to the GO terms focal adhesion (FA) and cell-substrate junction, which were particularly enriched in YAP-targets, suggesting that in gastric cancer cells, YAP plays a crucial role in regulating cell-substrate junctions.501
TRIM 24
Gastric cancer cell lines and tissues frequently manifest abnormally upregulated expression of TRIM-24 transcriptional co-activator. A study has reported that downregulated expression of miR-511 is essential for sustained expression of TRIM24.502 TRIM24, when active, promotes cell proliferation, migration and invasion by activating the WNT/β-CATENIN signaling pathway.503
BRD4
Epigenetic regulation requires the involvement of three different types of proteins. First are the enzymes that modify histones or DNA, and are known as writers. Second are enzymes that remove modifications on histone or DNA, the erasers, and third are the proteins that recognize these modifications, known as readers.504 BRD4, the bromodomain containing transcriptional co-activators belong to the class of epigenetic readers. It has been reported that the expression of BRD4 in human GC tissues correlates with shortened metastasis-free gastric cancer patient survival.505 It has been observed that BRD4 associates with the transcription factor E2F1 via its two bromodomains. This association promotes the recruitment of BRD4 to the promoter of miR-106b-5p, thereby facilitating its transcription. An active miR-106b-5p targets 3′-UTR of p21, eventually to regulate cellular senescence.506 Qin et al.507 further reported that the epigenetic reader BRD4 recognizes acetylated lysine 146 (K146) and K187 on Snail. This prevents Snail recognition by its E3 ubiquitin ligases FBXL14 and β-Trcp1, consequently promoting metastasis by inhibiting Snail polyubiquitination and proteasomal degradation.
DDX5
DDX5, DEAD (Asp-Glu-Ala-Asp) box helicase 5 is a transcriptional co-activator that is overexpressed in different malignancies and associated with progression of cancer.508 It has been reported that in gastric cancer tissues, DDX5 is dramatically upregulated, and its overexpression correlates with gastric cancer cell growth and invasion.509 It has also been observed that DDX5 promotes cell proliferation by upregulating mTOR/S6K1 signaling activity, stipulating that targeting DDX5/mTOR/S6K1 might be a novel therapeutic approach for the treatment of gastric cancer.510
MRTF
The transcriptional co-activator MRTF-A upregulates the expression of miR-155 promoter by inducing histone acetylation and RNA polymerase II recruitment. Subsequently, miR-155 suppresses the expression of SOX1 to promote gastric cancer cell migration.511 Furthermore, Wang et al.512 showed that MICALC2-mediated upregulation of nuclear MRTF-A promotes CDC42 activation, MMP9 expression, and gastric cancer cell migration.512
Esophageal cancer
Approximately 604,000 new cases of esophageal cancer have been reported with almost 544,000 deaths only in the year 2020. Esophageal cancer is responsible for one in every 18 cancer-related deaths in 2020. Approximately 70% of cases occur in men, and there is a twofold to threefold difference in incidence and mortality rates between the sexes. It is responsible for 8.3% of cancer-related deaths in males throughout the world346 (Fig. 5d).
CRTCs
Liver kinase B1 (LKB1) is an essential serine/threonine kinase that is downregulated in a subset of esophageal tumor. Owing to this downregulation, LKB1 is unable to downregulate the expression of CREB-regulated transcription co-activator 1 (CRTC1), leading to their aberrant activation. Mechanistically, upon activation, CRTC1 interacts with the CREB transcription factor and enhances the expression of CREB target genes like LYPD3, a high-glycosylated cell surface protein,513 ultimately augmenting cell migration and invasion, which contributes to esophageal cancer progression.514 On the other hand, CRTC2 in cooperation with CBP/p300 deposits acetylation marks on histones at inflammatory gene loci, consequently promoting active transcription and cytokine expression. This CRTC2-CBP/p300-mediated histone modification, links metabolic and epigenetic states to inflammatory potential in esophageal cancer.515
ZNF282
E2F1 transcription factor is a key player that modulates cell cycle, DNA damage response, and apoptosis.516 It has been observed that ZNF282 (Zinc finger protein 282) functions as an E2F1 co-activator in esophageal squamous-cell carcinoma (ESCC), inducing accelerated transcription of E2F1 target genes like CCND2, CCNA1, CDC2, and CDC6, facilitating G1/S transition and cell cycle progression. Moreover, in comparison to normal esophageal epithelium, ZNF282 has been frequently reported to be overexpressed in ESCC tissues and ZNF282 depletion increased apoptosis and promoted cell cycle arrest at G1/S, suggesting that ZNF282 transcriptional co-activator, plays pivotal role in controlling E2F1-mediated ESCC progression.517
Pancreatic cancer
According to Globocan (2020), pancreatic cancer accounts for 5.3% and 3.8% of total cancer-related deaths in males and females, respectively.346 The most common concern of pancreatic cancer is the detection of the disease at advanced stages as the patients seldom exhibit symptoms at the earlier stages. Alarmingly, as sufficient causes of pancreatic cancer have not been deciphered yet, therefore identifying potential therapeutic targets might assist in the abrogation of this disease518 (Fig. 5e).
PIWIL1
Piwi-like protein 1 (PIWIL1) is encoded by the PIWIL1 gene in humans and the expression of this gene is generally restricted to germ cells. Li et al.519 have shown that human PIWIL1 in apo state (without piRNA binding), acts as a co-activator of anaphase promoting complex/cyclosome (APC/C) E3 complex, which in turn selectively targets a cell adhesion-related protein, Pinin, for degradation and enhances pancreatic ductal carcinoma (PDAC) metastasis. Moreover, at mRNA and protein levels, the expression of PIWIL1 was found to be associated with progenitor molecular subtype of pancreatic cancer, indicating that in resectable pancreatic cancer, PIWIL1 can be considered as a potential prognostic marker.520
CBP/p300
CBP/p300 is highly expressed in pancreatic tissues in comparison to normal tissues. Manegold et al.521 demonstrated that when CBP is active, it acts as a co-activator of β-CATENIN and induces PDAC progression. On the contrary, upon pharmacological inhibition of CBP, its homologous co-activator p300 interacts with β-CATENIN to promote differentiation and renders the cancer cells susceptible to therapy. Inhibition of p300 by XP-524 has also been reported to increase oncogenic KRAS, which is found to be expressed in 90% of the PDAC cases. This indicates that p300 might play an important role in PDAC progression.522
SETD8
The methyl transferase SETD8 has been documented to be upregulated in pancreatic cancer. Liu et al.523 have reported that SETD8 interacts with STAT3 and induces monomethylation of H4K20 on DUSP10 promoter, thereby promoting epigenetic silencing of DUSP10 (Dual Specificity Phosphatase 10). Inhibition of DUSP10, consequently promotes the upregulation of ERK1/2, consequently promoting pancreatic adenocarcinoma. It was observed that SETD8 interacts with promoter region of RRAD to reduce the levels of lipid peroxidation, which further inhibits ferroptosis-mediated death of pancreatic cancer cells.524
YAP
A recent study by Zhou et al.525 has documented that in comparison to normal controls, YAP1 is the most highly expressed protein in pancreatic cancer tissues (log2 fold change 6.4; p = 5E−06). Moreover, YAP has also been demonstrated as an independent prognostic marker in pancreatic cancer (hazard ratio 1.870, 95% confidence interval (CI) 1.224–2.855, p = 0.004). Liu et al.526 reported that YAP as a transcriptional co-activator interacts with ZEB1 to promote transcription of ITGA3, consequently enhancing EMT plasticity and spheroid formation. Furthermore, Unc-51 like kinase 1 (ULK1) interacts with YAP in the nucleus and promotes its phosphorylation-mediated stabilization. Upon stabilization, YAP facilitates PKM2 (pyruvate kinase M2) transcription, glycolysis, and PDAC cell proliferation and growth.527 Owing to its diverse regulatory role in pancreatic cancer, YAP can be a potential therapeutic target.
BRD4
Jiao et al.528 reported that in comparison to the adjacent non-cancerous tissues, elevated expression of BRD4 is observed in pancreatic cancer. It was also observed that BRD4 interacted with the promoter region of CAVEOLIN-2, subsequently promoting transcriptional activation of CAVEOLIN-2. Clinical studies further indicated that in pancreatic cancer patients, BRD4 (high)/caveolin-2 (high) was associated with shorter disease-free survival. Another study by Yamazaki et al.529 further reported that YAP/BRD4 binding at the enhancer region is associated with transcriptional activation of receptor tyrosine kinase-like orphan receptor 1 (ROR1), thereby promoting tumor growth and metastasis.
This study so far has highlighted the transcriptional co-activators that have been depicted to be deregulated across most prevalent cancer types. However, considerate inspection also reveals an involvement of common co-activators across several cancer types. Therefore, profound understanding of the molecular mechanisms by which these master transcriptional regulators exert their function will potentiate the development of a pan-disease therapeutic regime. Table 2 attempts to collate the predominance of frequently deregulated co-activators across the deadliest forms of cancer.
Co-activator involvement in cancer stemness
Though multitudinal therapeutic interventions for treating cancer are used worldwide, high rate of metastasis, recurrence, and relevant mortality still persists.530 One of the major role-players in this are the small population of pluripotent cells residing within the tumor, known as cancer stem cells (CSCs).531 Within the tumor, the cancer cells and CSCs remain in a dynamic equilibrium state which is maintained by two opposing phenomena, differentiation and de-differentiation.532 Upon receiving certain cues, cancer stem cells can differentiate to give rise to the cancer cells or cancer cells can de-differentiate back to their CSC state.533 CSCs accomplish this dynamic state by re-wiring their transcriptional machinery which further determines the aggressiveness and recurrence rate of the cancer.534 Researchers have shown the involvement of co-activators in cancer stem cell self-renewal and maintenance. Some of these findings are discussed below.
Using a mouse model of glioma, Pietras et al.535 experimentally proved that Osteopontin-CD44 signaling facilitates the maintenance of the CSCs phenotypes via CBP/p300-dependent enhancement of HIF-2α activity. The CBP/p300-interacting transactivator with ED-rich tail 2 (CITED2) further suppresses the CSC markers and reduces the cancer stem cell population in NSCLC.536 Integrated transcriptome and protein-protein interaction studies revealed that the arginine methyltransferase PRMT6 can regulate stemness properties via MEK/ERK pathway in hepatocellular carcinoma.537 Zhu et al.538 performed Spearman correlation test using TCGA pan cancer data and found the KMT2 family genes to be associated with cancer stemness and drug sensitivity. Recently, in intestinal tumorigenesis, co-activator MLL1 was observed to govern WNT/β-Catenin induced cancer stemness.539 KDM2A in breast cancer enhances stemness and angiogenesis by Jagged1 (JAG1) dependent mechanism.540 Another lysine demethylase KDM6B has been reported to enhance stemness related genes like SOX2, SOX9, and OCT4.541 Li et al.542 identified JMJD3 as one of the main drivers of esophageal squamous cell carcinoma pathogenesis through JMJD3/MYC/miR-17–92 pathway and regulate stemness and sensitivity to therapy. Steroid receptor co-activator 1 and 3 (SRC1/3) plays a crucial role in CSC state maintenance and metastasis in breast cancer cell lines. In addition, it was also observed that siRNA mediated knockdown of SRC1/3 significantly reduced the CSC population.543 Jaworska et al.544 in their study have summarized the role of different proteins of the TRIM co-activator family in modulating different signaling pathways associated with self-renewal of CSCs. One more study found TRIM29 stabilizes interferon-stimulated gene 15 (ISG15) and promote cancer stem cell-like phenotype in pancreatic ductal adenocarcinomas (PDACs).545 Recent findings have suggested the involvement of YAP/TAZ in stemness maintenance and their deregulation may induce transformation of the cancer cells into CSCs.546 It is now quite evident that the co-activators not only take part in tumor growth but are also responsible for cancer stem cell fueled metastasis, drug resistance and unresponsiveness to therapies. Oncologists are still in search of therapies to abrogate both the cancer stem cell population along with the differentiated cancer cell population. Due to their active collaboration in almost all aspects of cancer progression, the possibility of co-activator-based CSC targeting strategies in successfully eliminating both the CSC and non-CSC components of cancer, will retrench tumor relapse or secondary tumorigenesis.
Applications in biomedical research and targeted therapy
Importance of co-activators in disease research and drug discovery
TFs, in order to swiftly integrate cellular stimuli, must have the ability to rapidly recruit multiple proteins associated with transcription machinery using single short domains. By the course of evolution, TFs have adapted to perform such function by promptly producing an ensemble of malleable structures that are modified depending on its binding partners.1,6 As a consequence, they do not possess specific structural integrity, making it difficult to target them. Researchers have attempted to device alternative targeting strategies, specifically involving the transcriptional co-activators. It is well-established that co-activators are modulated by signal transduction, and depending on the received information, they drive TF activity in the context of gene expression.16,17 Hence, development of strategies to modulate the co-activators is worth exploring since they can be easily and safely targeted, and can effectively alter the function of the TFs. Such ideas have now been practically implemented in the field of drug discovery and multiple new strategies such as small molecule inhibitors (SMIs), proteolysis-targeting chimeras (PROTACs) and molecular glue degraders targeting the co-activators are being considered through extensive research (Fig. 6).

Current therapeutic strategies to target transcriptional co-activators. a Targeting the co-activators (CoA) with small molecule inhibitors (SMIs) is a widely used strategy to inhibit the function of the CoAs. The SMI interacts with binding residues on the target protein surface and mediates either orthosteric or allosteric inhibition. During orthosteric inhibition, SMIs directly block protein-protein interactions with their binding partners. Allosteric inhibition is achieved when the SMIs bind to the target proteins and induce conformational changes on the binding surface, thereby dampening its activity. SMIs have also been reported to bind to functional domain (FD), like the bromodomains, of the transcriptional CoAs, consequently preventing the interaction between the acetyl groups and the bromodomains to inhibit transcriptional activation. b Molecular glue degraders are another potential therapeutic strategy. The interaction between an E3 ubiquitin ligase and the transcriptional co-activators are induced by the molecular glue, which promotes ubiquitination-mediated degradation of the co-activators. c The most rapidly growing heterobifunctional protein degrading system is proteolysis-targeting chimeras (PROTACs). The degradation system of PROTACs comprises an anchor and a warhead, which is connected by a linker molecule. The warhead binds to the protein of interest (POI), while the anchor recruits E3 ubiquitin ligase, thereby hijacking the ubiquitin proteasome system of the cell to degrade the POI. This figure was created using BioRender (https://biorender.com/)
Previously, in this review, involvement of co-activators in different mechanomolecular aspects of transcriptional regulation and their association with multiple disease phenotypes have been elaborated. Numerous studies have also identified co-activator gene mutations as drivers of multiple diseases (Table 1). Altogether, these findings suggest that targeting the co-activators will not only reduce the transcription factor-mediated malicious gene expression, but will also disrupt the molecular interconnections between multiple disease-causing pathways, leading to better patient prognosis.
Small molecule inhibitors of transcriptional co-activators
Small molecule inhibitors (SMIs) are the chemical compounds having the molecular weight of <500 Da and they have been reported to interact with the binding pockets present on the surface of the target proteins to disrupt their functionality547 (Fig. 6a). Use of SMIs has gained wide popularity owing to several selective advantages. First, the SMIs can be easily prepared and structurally modified based on conceptually straightforward techniques. New compounds with greater potency can be readily generated by modulating the structural conformation of the old compounds. Moreover, due to the well-known chemical groups in their molecular composition, SMIs with high penetration property and rapid metabolism can be created effortlessly.547,548 Therefore, treatment with SMIs is a lucrative therapeutic strategy to target diseases associated with deregulated co-activator function. The selective SMIs of transcriptional co-activators across different disease phenotypes, along with their mechanism of action, have been summarized in Table 3.
Analogous to chemotherapeutic drugs, SMIs often confer resistance by inducing shift in cellular state. For instance, a study by Sun et al.549 have reported that TEAD-YAP protein-protein interaction inhibition using TEAD auto-palmitoylation inhibitor MGH-CP1 induces a transient static cell state instead of abrogating the cell population. Contrarily, some inhibitors can perform multiple targeting that can minimize drug resistance; however, it can increase significant risk of toxicity.550 Drug efflux is another concern that disables the functionality of SMIs.551 Moreover, inability to selectively target the diseased cells and off-target toxicity are the areas of particular concern, leading to the surge for more consistent alternatives.
Alternate strategies in targeting co-activators
The overreliance on stereotypical idea of antagonistic or agonistic pharmacological perception has limited the reach of small molecule-based strategies, causing substantial stagnation in therapeutic innovation, because of the lack of pursuing some of the best characterized potential target molecules in life-threatening diseases. A promising alternative to this can be the modulation of the disease-causing protein by chemically redirecting them towards the cellular ubiquitin proteasome system for degradation. This approach has been practically implemented via development of proteolysis-targeting chimeras (PROTACs) and molecular glue degraders, the latest discoveries in the field of biomedical research. Although these techniques are still at the bench, rigorous research is being implemented in order to bring them to the bedside.
Molecular glue degraders
Molecular glue degraders offer an intriguing targeting strategy by sub-stoichiometrically catalysing the rapid degradation of inaccessible targets. Molecular glues are monovalent molecules of <500 Da that induces interaction between an E3 ubiquitin ligase and the target protein by reshaping the surface of the E3 ligase substrate receptor. As a consequence of this interaction, the target protein degradation takes place552 (Fig. 6b). Molecular glue degraders show great potential for treating diseases such as developmental diseases, molecular disorders and also cancer. Brownsey et al.553 have explored the application of linkage vector on A-485 conjugated with molecular glue pomalidomide, in targeted degradation of CBP/p300 in myeloma cell line. Using CRISPR/Cas9 knockout screens, a study established a JQ1 based monovalent degrader compound for BRD4 degradation.554 Ling et al.555 confirmed that the small molecule inhibitor FL118 can act as a molecular glue degrader by interacting with the co-activator DDX5 to promote tyrosine dephosphorylation and subsequent proteasomal degradation without affecting the mRNA levels of the target protein. Moreover, studies have stressed on the importance of molecular glue degraders as a remedy for neurodegenerative diseases,556 though their broad-scale implementation requires pervasive research efforts in the fields of molecular biology and medicinal chemistry.
PROTAC
Proteolysis-targeting chimeras (PROTACs) are one of the most rapidly growing heterobifunctional targeted protein degradation systems that principally contains two functional ligands, an anchor and a warhead, connected by one chemical linker molecule.557 The warhead binds to the protein of interest (POI), while the anchor recruits E3 ubiquitin ligase, thereby hijacking the ubiquitin proteasome system of the cell to degrade the POI557 (Fig. 6c). Since its discovery in 2001 by Craig M. Crews, PROTAC has progressed to combat several diseases. Two of the proteolytic targeting chimeras, ARV-110 (NCT03888612) and ARV-471 (NCT04072952) have successfully made it to phase-II clinical trials, ARV-110 for metastatic castration-resistant prostate cancer, and ARV-471 for metastatic ER+/HER2− breast cancer.558
Concomitantly, scientists have successfully proved the efficacy of PROTACs against the following TFs in the context of cancer: nuclear factor-κB (NF-κB),559 androgen receptor (AR),560 estrogen receptor (ER),561 c-MYC,562 p53,563 STAT3,564 STAT5,565 and SMAD3.566 In addition to targeting the TFs, PROTACS have also been developed against transcription co-activators with an aim to indirectly regulate TF-associated disease phenotypes. Thomas et al.567 provided evidence of JET-209 based PROTAC degrader for CBP/p300 in leukemia. Another CBP/p300 targeting PROTAC named “JQAD1” has also been established in neuroblastoma.386 Huang et al.568 constructed a biologically inspired PROTAC against TRIM24 by coating the poly lactic-co-glycolic acid (PLGA) nanoparticles containing PROTAC degrader with M2 macrophage membrane (MELT) for atherosclerosis. Lee et al.569 developed PROTAC for targeting SRC1 through N-degron pathway in vivo, suggesting the usefulness of N-degron pathway-based degraders of disease-relevant proteins. Otto et al.570 designed dBET, a PROTAC against BRD4 for reducing BRD4 mediated c-MYC gene expression in colorectal cancer. Till date, most of the studies relevant to PROTAC development against transcription factors and co-activators have concentrated around cancer. However, in the near future, the advent of this strategy will be intended to target developmental and metabolic disorders.
Like SMIs, there are certain limitations associated with the broad spectrum use of PROTACs in disease therapy. For example, it exhibits poor blood-brain barrier permeability, degradation of off-target proteins, differential expression of E3 ubiquitin ligase in different parts of the body, and poor pharmacokinetics.569,570 These facts will necessitate further improvements in PROTACs to increase their efficacy and target specificity.
Drug repurposing
Repurposing approved drugs is currently a novel approach for disease treatment and is gaining immense popularity, since it may be implemented without facing the impediment imposed by extensive trials and delayed approvals. Strategizing the use of clinically approved drugs not only minimizes the timely and costly endeavours associated with drug development but also provides effective, safer, and cheaper drugs.571 Amongst the inhibitors of TF co-activators, there are several FDA-approved drugs currently used for different diseases. Verteporfin, an inhibitor of YAP/TAZ, is an FDA-approved drug for the treatment of age-related macular degeneration, pathologic myopia or presumed ocular histoplasmosis.572 Carnosic acid, a component of rosemary extract that is FDA-approved for use as food additives, is an inhibitor of transcriptional co-activators BCL9/9L.572 Under inhibitors of CBP/p300 histone acetyl transferases, melatonin is a recognized dietary supplement that falls under FDA’s Dietary Health and Education Act, and cyproheptadine is a clinically approved drug for perennial and seasonal allergic rhinitis, vasomotor rhinitis and allergic conjunctivitis.572 Therapeutic repurposing of these drugs for targeting the transcriptional co-activators can thereby be an alternate and effective pharmacological strategy across several pathological conditions.
Conclusion
Efficient transcriptional signaling mediated by transcription factors is often dependent on the transcriptional co-factors that either physically associate with and/or biochemically modify the genome to reinforce target gene activation or repression.1,7 This “soft-wiring” integration of different biological pathways via co-activator action is responsible for modulation of transcriptional activity of several transcription factors across different pathological conditions.16 Transcription factors are foundations of the regulatory circuits responsible for selective gene expression.2,3,6 Gene expression deregulation is perhaps an inevitable driver of a variety of physiological disorders, including developmental disorders, metabolic disorders, and cancer.1 However, modulation of transcription factors that regulate disease-driving genes is one of the most arduous ventures in the field of drug discovery because of its high degree of intrinsic complexity in terms of both structure and interactions.4,5 Therefore, we sought to identify their ‘partners-in-crime’, the transcriptional co-activators, and the molecular correspondence underpinning the regulatory circuit of gene regulation. This will aid the development of indirect yet effective targeting strategies against disease-driving transcription factors and will eventually help in better patient prognosis. An up-to-date account on co-activator involvement in diverse disease phenotype and several therapeutic strategies like small molecule inhibitors, molecular glue degrader and PROTACs for targeting the activity of these co-activators have been discussed extensively in this review.
Strikingly, the co-activators can be of immense use to treat diseases where a significant upregulation in their expression has been observed or the primary driver of the disease is not known. Against this backdrop, development of therapeutic strategies to modulate the activity of transcriptional co-activators like PGC-1α that mediates cellular and mitochondrial homeostasis can be a promising therapeutic approach.208,209 Angiogenesis is another important regulator of several disease conditions like neurodevelopmental disease and cancer. CRTC family of transcriptional co-activators that have been reported to maintain vascular physiology can be promising therapeutic target.573 Hence, a better understanding of the interplay between key cellular compartments, cellular niche and the transcriptional co-activators will help identify alternative therapeutic targets.
However, targeting co-activators may pose certain impediments since they not only play crucial role in disease progression, but also regulate other physiological mechanisms that are necessary for the survival of the organism. For example, CBP/p300 serves as a co-activator for multiple transcription factors like ER,387 AR,574 NF-κB575 amongst others. This addiction of the co-activators for multiple transcription factors poses a challenge for the researchers in the field of drug discovery.576 Presently, a multitude of new modifications in the inhibitors are being incorporated to facilitate specific and targeted delivery to the cells of interest. For example, in the context of cancer, delivery of the drugs to the tumor through exosomes,577 liposomes578 or nanoparticle-mediated delivery systems579 may have the potential to achieve desired anti-tumor effects without major risk of off-target toxicity.
Looking towards the future, more co-activators are yet to be discovered, especially in the arena of the cancer stem cells, enabling us to improve our ability to modulate this particular class of regulatory molecules. This review has been an attempt to address an issue that has not been dealt with in a comprehensive manner and hopes to direct attention towards future research that will encompass patient-friendly therapeutic strategies, where drugs will have enhanced benefits and reduced side effects. This will be of considerable potential since utilization of these modulators in combination with conventional chemotherapeutic drugs will overcome the frequently observed phenomenon of cancer recurrence, and additionally treat various developmental and metabolic disorders with elan and success.
References
Lee, T. I. & Young, R. A. Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013).
Lambert, M., Jambon, S., Depauw, S. & David-Cordonnier, M. H. Targeting transcription factors for cancer treatment. Molecules 23, 1479 (2018).
Santos‐Terra, J. et al. Transcription factors in neurodevelopmental and associated psychiatric disorders: a potential convergence for genetic and environmental risk factors. Int. J. Dev. Neurosci. 81, 545–578 (2021).
Arkin, M. R., Tang, Y. & Wells, J. A. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem. Biol. 21, 1102–1114 (2014).
Su, B. G. & Henley, M. J. Drugging fuzzy complexes in transcription. Front. Mol. Biosci. 8, 795743 (2021).
Stallcup, M. R. & Poulard, C. Gene-specific actions of transcriptional coregulators facilitate physiological plasticity: evidence for a physiological coregulator code. Trends Biochem. Sci. 45, 497–510 (2020).
Rogatsky, I., Luecke, H. F., Leitman, D. C. & Yamamoto, K. R. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc. Natl Acad. Sci. USA 99, 16701–16706 (2002).
Dentin, R., Hedrick, S., Xie, J., Yates, J. III & Montminy, M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science 319, 1402–1405 (2008).
Oosterveer, M. H. & Schoonjans, K. Hepatic glucose sensing and integrative pathways in the liver. Cell. Mol. Life Sci. 71, 1453–1467 (2014).
Siddappa, M. et al. Identification of transcription factor co-regulators that drive prostate cancer progression. Sci. Rep. 10, 20332 (2020).
Kelly, S. N., McKenna, T. J. & Young, L. S. Coregulatory protein–orphan nuclear receptor interactions in the human adrenal cortex. J. Endocrinol. 186, 33–42 (2005).
Nagy, Z. & Tora, L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 26, 5341–5357 (2007).
Eberharter, A. & Becker, P. B. Histone acetylation: a switch between repressive and permissive chromatin. EMBO Rep. 3, 224–229 (2002).
Garcia-Bassets, I. et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 128, 505–518 (2007).
Velthuijs, N. et al. Integration of transcription coregulator complexes with sequence-specific DNA-binding factor interactomes. Biochim. Biophys. Acta Gene Regul. Mech. 1864, 194749 (2021).
Delcuve, G. P., Khan, D. H. & Davie, J. R. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin. Epigenetics 4, 1–13 (2012).
Hervouet, E., Peixoto, P., Delage-Mourroux, R., Boyer-Guittaut, M. & Cartron, P. F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenetics 10, 1–18 (2018).
Mouchiroud, L., Eichner, L. J., Shaw, R. J. & Auwerx, J. Transcriptional coregulators: fine-tuning metabolism. Cell Metab. 20, 26–40 (2014).
Rosen, E., Eguchi, J. & Xu, Z. Transcriptional targets in adipocyte biology. Expert Opin. Ther. Targets 13, 975–986 (2009).
Burandt, E. et al. Prognostic relevance of AIB1 (NCoA3) amplification and overexpression in breast cancer. Breast Cancer Res. Treat. 137, 745–753 (2013).
Wang, L. et al. Transcriptional coregulator NUPR1 maintains tamoxifen resistance in breast cancer cells. Cell Death Dis. 12, 149 (2021).
Fong, Y. W., Cattoglio, C., Yamaguchi, T. & Tjian, R. Transcriptional regulation by coactivators in embryonic stem cells. Trends Cell Biol. 22, 292–298 (2012).
Anderson, R. & Prolla, T. PGC-1α in aging and anti-aging interventions. Biochim. Biophys. Acta Gen. Sub. 1790, 1059–1066 (2009).
Waddington, C. H. The epigenotype. Endeavour 1, 18–20 (1942).
Baniahmad, A. et al. Interaction of human thyroid hormone receptor beta with transcription factor TFIIB may mediate target gene derepression and activation by thyroid hormone. Proc. Natl Acad. Sci. USA 90, 8832–8836 (1993).
Shibata, H. et al. Role of co-activators and co-repressors in the mechanism of steroid/thyroid receptor action. Recent Prog. Horm. Res. 52, 141–64 (1997).
Onate, S. A., Tsai, S. Y., Tsai, M. J. & O’Malley, B. W. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270, 1354–1357 (1995).
Wang, Z. et al. Interplay between cofactors and transcription factors in hematopoiesis and hematological malignancies. Signal Transduct. Target. Ther. 6, 24 (2021).
Näär, A. M., Lemon, B. D. & Tjian, R. Transcriptional coactivator complexes. Annu. Rev. Biochem. 70, 475–501 (2001).
Spiegelman, B. M. & Heinrich, R. Biological control through regulated transcriptional coactivators. Cell 119, 157–167 (2004).
Krasnov, A. N. et al. On the way of revealing coactivator complexes cross-talk during transcriptional activation. Cell Biosci. 6, 15 (2016).
Scholes, N. S. & Weinzierl, R. O. Molecular dynamics of “Fuzzy” transcriptional activator-coactivator interactions. PLoS Comput. Biol. 12, 1004935 (2016).
Featherstone, M. Coactivators in transcription initiation: here are your orders. Curr. Opin. Genet. Dev. 12, 149–155 (2002).
Fischer, V., Schumacher, K., Tora, L. & Devys, D. Global role for coactivator complexes in RNA polymerase II transcription. Transcription 10, 29–36 (2019).
Boeva, V. Analysis of genomic sequence motifs for deciphering transcription factor binding and transcriptional regulation in eukaryotic cells. Front. Genet. 7, 24 (2016).
Harper, T. M. & Taatjes, D. J. The complex structure and function of Mediator. J. Biol. Chem. 293, 13778–13785 (2018).
Tolkunov, D. et al. Chromatin remodelers clear nucleosomes from intrinsically unfavorable sites to establish nucleosome-depleted regions at promoters. Mol. Biol. Cell 22, 2106–2118 (2011).
André, K. M. et al. Functional interplay between Mediator and RSC chromatin remodeling complex controls nucleosome-depleted region maintenance at promoters. Cell Rep. 42, 112465 (2023).
Rawal, Y. et al. SWI/SNF and RSC cooperate to reposition and evict promoter nucleosomes at highly expressed genes in yeast. Genes Dev. 32, 695–710 (2018).
Yu, J. et al. Analysis of local chromatin states reveals gene transcription potential during mouse neural progenitor cell differentiation. Cell Rep. 32, 107953 (2020).
Murakami, K. et al. Architecture of an RNA polymerase II transcription pre-initiation. Complex. Sci. 342, 1238724 (2013).
Petrenko, N., Jin, Y., Dong, L., Wong, K. H. & Struhl, K. Requirements for RNA polymerase II preinitiation complex formation in vivo. eLife 8, 43654 (2019).
Chen, X. et al. Structural insights into preinitiation complex assembly on core promoters. Science 372, 8490 (2021).
Felinski, E. A. & Quinn, P. G. The coactivator dTAFII110/hTAFII135 is sufficient to recruit a polymerase complex and activate basal transcription mediated by CREB. Proc. Natl Acad. Sci. USA 98, 13078–13083 (2001).
Goodrich, J. A., Cutler, G. & Tjian, R. Contacts in context: promoter specificity and macromolecular interactions in transcription. Cell 84, 825–830 (1996).
Nguyen, V. Q. et al. Spatiotemporal coordination of transcription preinitiation complex assembly in live cells. Mol. Cell 81, 3560–3575 (2021).
Rengachari, S., Schilbach, S., Aibara, S., Dienemann, C. & Cramer, P. Structure of human mediator-RNA polymerase II pre-initiation complex. Nature 594, 129–133 (2021).
Fishburn, J., Tomko, E., Galburt, E. & Hahn, S. Double-stranded DNA translocase activity of transcription factor TFIIH and the mechanism of RNA polymerase II open complex formation. Proc. Natl Acad. Sci. USA 112, 3961–3966 (2015).
Mandal, S. S. et al. Functional interactions of RNA-capping enzyme with factors that positively and negatively regulate promoter escape by RNA polymerase II. Proc. Natl Acad. Sci. USA 101, 7572–7777 (2004).
Topisirovic, I., Svitkin, Y. V., Sonenberg, N. & Shatkin, A. J. Cap and cap‐binding proteins in the control of gene expression. Wiley Interdiscip. Rev. RNA 2, 277–298 (2011).
Adelman, K. & Lis, J. T. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet. 13, 720–731 (2012).
Zhu, X. et al. Histone modifications influence mediator interactions with chromatin. Nucleic Acids Res. 39, 8342–8354 (2011).
Ahn, S. H., Kim, M. & Buratowski, S. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol. Cell 13, 67–76 (2004).
Cheon, Y., Kim, H., Park, K., Kim, M. & Lee, D. Dynamic modules of the coactivator SAGA in eukaryotic transcription. Exp. Mol. Med. 52, 991–1003 (2020).
Larochelle, M. et al. Common mechanism of transcription termination at coding and noncoding RNA genes in fission yeast. Nat. Commun. 9, 4364 (2018).
Kim, M., Choi, Y., Kim, H. & Lee, D. SAGA DUBm-mediated surveillance regulates prompt export of stress-inducible transcripts for proteostasis. Nat. Commun. 10, 2458 (2019).
Faza, M. B. et al. Sem1 is a functional component of the nuclear pore complex-associated messenger RNA export machinery. J. Cell Biol. 184, 833–846 (2009).
Park, H. S., Lee, J., Lee, H. S., Ahn, S. H. & Ryu, H. Y. Nuclear mRNA export and aging. Int. J. Mol. Sci. 23, 5451 (2022).
Garcia-Oliver, E. et al. A novel role for Sem1 and TREX-2 in transcription involves their impact on recruitment and H2B deubiquitylation activity of SAGA. Nucleic Acids Res. 41, 5655–5668 (2013).
Schneider, M. et al. The nuclear pore-associated TREX-2 complex employs mediator to regulate gene expression. Cell 162, 1016–1028 (2015).
Espinola, S. M. et al. Cis-regulatory chromatin loops arise before TADs and gene activation, and are independent of cell fate during early Drosophila development. Nat. Genet. 53, 477–486 (2021).
Galli, G. G. et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol. Cell 60, 328–337 (2015).
Panigrahi, A. & O’Malley, B. W. Mechanisms of enhancer action: the known and the unknown. Genome Biol. 22, 1–30 (2021).
Suter, D. M. Transcription factors and DNA play hide and seek. Trends Cell Biol. 30, 491–500 (2020).
Aoyagi, S. & Archer, T. K. Dynamics of coactivator recruitment and chromatin modifications during nuclear receptor mediated transcription. Mol. Cell. Endocrinol. 280, 1–5 (2008).
Ravarani, C. N. et al. High‐throughput discovery of functional disordered regions: investigation of transactivation domains. Mol. Syst. Biol. 14, 8190 (2018).
Baughman, H. E. et al. An intrinsically disordered transcription activation domain increases the DNA binding affinity and reduces the specificity of NFκB p50/RelA. J. Biol. Chem. 298, 102349 (2022).
Xu, L., Glass, C. K. & Rosenfeld, M. G. Coactivator and corepressor complexes in nuclear receptor function. Curr. Opin. Genet. Dev. 9, 140–147 (1999).
Fry, C. J. et al. Activation of the murine dihydrofolate reductase promoter by E2F1: a requirement for CBP recruitment. J. Biol. Chem. 274, 15883–15891 (1999).
Darling, A. L. & Uversky, V. N. Intrinsic disorder and posttranslational modifications: the darker side of the biological dark matter. Front. Genet. 9, 158 (2018).
Marceau, A. H. et al. An order-to-disorder structural switch activates the FoxM1 transcription factor. eLife 8, 46131 (2019).
Puigserver, P. et al. Activation of PPARγ coactivator-1 through transcription factor docking. Science 286, 1368–1371 (1999).
Puigserver, P. & Spiegelman, B. M. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr. Rev. 24, 78–90 (2003).
Soutoglou, E. et al. Transcription factor-dependent regulation of CBP and P/CAF histone acetyltransferase activity. EMBO J. 20, 1984–1992 (2001).
Gsell, C., Richly, H., Coin, F. & Naegeli, H. A chromatin scaffold for DNA damage recognition: how histone methyltransferases prime nucleosomes for repair of ultraviolet light-induced lesions. Nucleic Acids Res. 48, 1652–1668 (2020).
Gamarra, N. & Narlikar, G. J. Collaboration through chromatin: motors of transcription and chromatin structure. J. Mol. Biol. 433, 166876 (2021).
Dinant, C., Houtsmuller, A. B. & Vermeulen, W. Chromatin structure and DNA damage repair. Epigenetics Chromatin 1, 1–13 (2008).
Kim, J., An, Y. K., Park, S. & Lee, J. S. Bre1 mediates the ubiquitination of histone H2B by regulating Lge1 stability. FEBS Lett. 592, 1565–1574 (2018).
Klose, R. J. & Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8, 307–318 (2007).
Zhang, T., Cooper, S. & Brockdorff, N. The interplay of histone modifications–writers that read. EMBO Rep. 16, 1467–1481 (2015).
Wanior, M., Krämer, A., Knapp, S. & Joerger, A. C. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene 40, 3637–3654 (2021).
Swygert, S. G. & Peterson, C. L. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta Gene Regul. Mech. 1839, 728–736 (2014).
Bendandi, A., Patelli, A. S., Diaspro, A. & Rocchia, W. The role of histone tails in nucleosome stability: an electrostatic perspective. Comput. Struct. Biotechnol. J. 18, 2799–2809 (2020).
Bannister, A. J. & Kouzarides, T. Reversing histone methylation. Nature 436, 1103–1106 (2005).
Liaw, A., Liu, C., Ivanovski, S. & Han, P. The relevance of DNA methylation and histone modification in periodontitis: a scoping review. Cells 11, 3211 (2022).
Kim, S. & Kaang, B. K. Epigenetic regulation and chromatin remodeling in learning and memory. Exp. Mol. Med. 49, 281–281 (2017).
Miller, J. L. & Grant, P. A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell. Biochem. 61, 289–317 (2012).
Scheer, S. et al. The methyltransferase DOT1L controls activation and lineage integrity in CD4+ T cells during infection and inflammation. Cell Rep. 33, 108505 (2020).
Milite, C. et al. The emerging role of lysine methyltransferase SETD8 in human diseases. Clin. Epigenetics 8, 1–15 (2016).
Wen, K. X. et al. The role of DNA methylation and histone modifications in neurodegenerative diseases: a systematic review. PLoS ONE 11, 0167201 (2016).
Audia, J. E. & Campbell, R. M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 8, 019521 (2016).
Chen, Y. et al. The role of histone methylation in the development of digestive cancers: a potential direction for cancer management. Signal Transduct. Target. Ther. 5, 143 (2020).
Sun, X. J., Man, N., Tan, Y., Nimer, S. D. & Wang, L. The role of histone acetyltransferases in normal and malignant hematopoiesis. Front. Oncol. 5, 108 (2015).
Ghoneim, M., Fuchs, H. A. & Musselman, C. A. Histone tail conformations: a fuzzy affair with DNA. Trends Biochem. Sci. 46, 564–578 (2021).
Portillo-Ledesma, S. et al. Nucleosome clutches are regulated by chromatin internal parameters. J. Mol. Biol. 433, 166701 (2021).
Uehara, S. & Morimoto, T. The interaction of the transcriptional coactivator P300 with the chromatin remodeling factor Brg1 increases the acetylation of the histone H3 globular domain in heart failure. Circulation 144, 10994–10994 (2021).
Hassan, A. H., Neely, K. E. & Workman, J. L. Histone acetyltransferase complexes stabilize swi/snf binding to promoter nucleosomes. Cell 104, 817–827 (2001).
Alver, B. H. et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8, 14648 (2017).
Tjeertes, J. V., Miller, K. M. & Jackson, S. P. Screen for DNA‐damage‐responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 28, 1878–1889 (2009).
Das, C., Lucia, M. S., Hansen, K. C. & Tyler, J. K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117 (2009).
Zhang, Y. Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev. 17, 2733–2740 (2003).
Oss-Ronen, L., Sarusi, T. & Cohen, I. Histone mono-ubiquitination in transcriptional regulation and its mark on life: emerging roles in tissue development and disease. Cells 11, 2404 (2022).
Ai, H. et al. Chemical synthesis of post-translationally modified H2AX reveals redundancy in interplay between histone phosphorylation, ubiquitination, and methylation on the binding of 53BP1 with nucleosomes. J. Am. Chem. Soc. 144, 18329–18337 (2022).
Leung, A. et al. Histone H2B ubiquitylation and H3 lysine 4 methylation prevent ectopic silencing of euchromatic loci important for the cellular response to heat. Mol. Biol. Cell 22, 2741–2753 (2011).
Espinosa, J. M. Histone H2B ubiquitination: the cancer connection. Genes Dev. 22, 2743–2749 (2008).
Zhou, S. et al. Role of H2B mono-ubiquitination in the initiation and progression of cancer. Bull. Cancer 108, 385–398 (2021).
Krajewski, W. A., Li, J. & Dou, Y. Effects of histone H2B ubiquitylation on the nucleosome structure and dynamics. Nucleic Acids Res. 46, 7631–7642 (2018).
Petty, E. & Pillus, L. Balancing chromatin remodeling and histone modifications in transcription. Trends Genet. 29, 621–629 (2013).
Chen, J. J., Stermer, D. & Tanny, J. C. Decoding histone ubiquitylation. Front. Cell Dev. Biol. 10, 968398 (2022).
Zhou, X. & Ma, H. Evolutionary history of histone demethylase families: distinct evolutionary patterns suggest functional divergence. BMC Evol. Biol. 8, 1–16 (2008).
Wissmann, M. et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 9, 347–353 (2007).
Cloos, P. A. et al. The putative oncogene GASC1 demethylates tri-and dimethylated lysine 9 on histone H3. Nature 442, 307–311 (2006).
Dimitrova, E., Turberfield, A. H. & Klose, R. J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 16, 1620–1639 (2015).
De Santa, F. et al. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094 (2007).
Burgold, T. et al. The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS ONE 3, 3034 (2008).
Park, D. H. et al. Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep. 8, 1290–1299 (2014).
Tsai, M. C. et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693 (2010).
Campagne, A. et al. BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat. Commun. 10, 348 (2019).
Ramachandran, S. et al. The SAGA deubiquitination module promotes DNA repair and class switch recombination through ATM and DNAPK-mediated γH2AX formation. Cell Rep. 15, 1554–1565 (2016).
Zhang, X. Y., Pfeiffer, H., Thorne, A. & McMahon, S. B. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle 7, 1522–1524 (2008).
Ducker, C. et al. De-ubiquitination of ELK-1 by USP17 potentiates mitogenic gene expression and cell proliferation. Nucleic Acids Res. 47, 4495–4508 (2019).
Bowman, G. D. Mechanisms of ATP-dependent nucleosome sliding. Curr. Opin. Struct. Biol. 20, 73–81 (2010).
Becker, P. B. & Workman, J. L. Nucleosome remodeling and epigenetics. Cold Spring Harb. Perspect. Biol. 5, 017905 (2013).
Reyes, A. A., Marcum, R. D. & He, Y. Structure and Function of Chromatin Remodelers. J. Mol. Biol. 433, 166929 (2021).
Havas, K. et al. Generation of superhelical torsion by ATP-dependent chromatin remodeling activities. Cell 103, 1133–1142 (2000).
Bartholomew, B. ISWI chromatin remodeling: one primary actor or a coordinated effort? Curr. Opin. Struct. Biol. 24, 150–155 (2014).
Barbaric, S. et al. Redundancy of chromatin remodeling pathways for the induction of the yeast PHO5 promoter in vivo. J. Biol. Chem. 282, 27610–27621 (2007).
Tran, H. G., Steger, D. J., Iyer, V. R. & Johnson, A. D. The chromo domain protein chd1p from budding yeast is an ATP-dependent chromatin-modifying factor. EMBO J. 19, 2323–2331 (2000).
Sharma, A. Modeling congenital heart disease using pluripotent stem cells. Curr. Cardiol. Rep. 22, 1–6 (2020).
Kim, T. H. et al. Correlating histone acetylation with nucleosome core particle dynamics and function. Proc. Natl Acad. Sci. USA 120, 2301063120 (2023).
Osley, M. A., Tsukuda, T. & Nickoloff, J. A. ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat. Res. 618, 65–80 (2007).
Farnung, L. & Vos, S. M. Assembly of RNA polymerase II transcription initiation complexes. Curr. Opin. Struct. Biol. 73, 102335 (2022).
Bittencourt, D. et al. G9a functions as a molecular scaffold for assembly of transcriptional coactivators on a subset of glucocorticoid receptor target genes. Proc. Natl Acad. Sci. USA 109, 19673–19678 (2012).
Abe, Y. et al. JMJD1A is a signal-sensing scaffold that regulates acute chromatin dynamics via SWI/SNF association for thermogenesis. Nat. Commun. 6, 7052 (2015).
Zhang, Q. & Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 19, 417–432 (2019).
Wang, N., Wu, R., Tang, D. & Kang, R. The BET family in immunity and disease. Signal Transduct. Target. Ther. 6, 23 (2021).
Marmorstein, R. & Zhou, M. M. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 6, 018762 (2014).
Dai, J. et al. Recruitment of Brd3 and Brd4 to acetylated chromatin is essential for proinflammatory cytokine-induced matrix-degrading enzyme expression. J. Orthop. Surg. Res. 14, 1–10 (2019).
Clifford, R. L. et al. CXCL8 histone H3 acetylation is dysfunctional in airway smooth muscle in asthma: regulation by BET. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, 962–972 (2015).
Devaiah, B. N. et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 23, 540–548 (2016).
Tian, B. et al. Efficacy of novel highly specific bromodomain-containing protein 4 inhibitors in innate inflammation–driven airway remodeling. Am. J. Respir. Cell Mol. Biol. 60, 68–83 (2019).
Borck, P. C., Guo, L. W. & Plutzky, J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ. Res. 126, 1190–1208 (2020).
Baumli, S. et al. The structure of P‐TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 27, 1907–1918 (2008).
Bowman, E. A. & Kelly, W. G. RNA polymerase II transcription elongation and Pol II CTD Ser2 phosphorylation: a tail of two kinases. Nucleus 5, 224–236 (2014).
Leo, C. & Chen, J. D. The SRC family of nuclear receptor coactivators. Gene 245, 1–11 (2000).
Greb-Markiewicz, B. & Kolonko, M. Subcellular localization signals of bHLH-PAS proteins: Their significance, current state of knowledge and future perspectives. Int. J. Mol. Sci. 20, 4746 (2019).
Mahajan, M. A., Murray, A., Levy, D. & Samuels, H. H. Nuclear receptor coregulator (NRC): mapping of the dimerization domain, activation of p53 and STAT-2, and identification of the activation domain AD2 necessary for nuclear receptor signaling. Mol. Endocrinol. 21, 1822–1834 (2007).
Yi, M., Tong, G. X., Murry, B. & Mendelson, C. R. Role of CBP/p300 and SRC-1 in transcriptional regulation of the pulmonary surfactant protein-A (SP-A) gene by thyroid transcription factor-1 (TTF-1). J. Biol. Chem. 277, 2997–3005 (2002).
Feng, Q. et al. Biochemical control of CARM1 enzymatic activity by phosphorylation. J. Biol. Chem. 284, 36167–36174 (2009).
Kapadia, B. et al. ERK2-mediated phosphorylation of transcriptional coactivator binding protein PIMT/NCoA6IP at Ser298 augments hepatic gluconeogenesis. PLoS ONE 8, 83787 (2013).
White, M. F. in Textbook of Diabetes 5th edn, (eds Holt, R. I. G. et al.) Ch. 8 (John Wiley & Sons, Ltd., 2017).
Stashi, E. et al. SRC-2 is an essential coactivator for orchestrating metabolism and circadian rhythm. Cell Rep. 6, 633–645 (2014).
Li, L., Deng, C. X. & Chen, Q. SRC-3, a steroid receptor coactivator: implication in cancer. Int. J. Mol. Sci. 22, 4760 (2021).
Zhao, W. et al. Steroid receptor coactivator-3 regulates glucose metabolism in bladder cancer cells through coactivation of hypoxia inducible factor 1α. J. Biol. Chem. 289, 11219–11229 (2014).
Bhat, K. P., ÜmitKaniskan, H., Jin, J. & Gozani, O. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 20, 265–286 (2021).
Poulard, C., Noureddine, L. M., Pruvost, L. & Le Romancer, M. Structure, activity, and function of the protein lysine methyltransferase G9a. Life 11, 1082 (2021).
Trievel, R. C., Beach, B. M., Dirk, L. M., Houtz, R. L. & Hurley, J. H. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell 111, 91–103 (2002).
Smith, B. C. & Denu, J. M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta Gene Regul. Mech. 1789, 45–57 (2009).
Couture, J. F., Hauk, G., Thompson, M. J., Blackburn, G. M. & Trievel, R. C. Catalytic roles for carbon-oxygen hydrogen bonding in SET domain lysine methyltransferases. J. Biol. Chem. 281, 19280–19287 (2006).
Lavery, W. J., Barski, A., Wiley, S., Schorry, E. K. & Lindsley, A. W. KMT2C/D COMPASS complex-associated diseases [K CD COM-ADs]: an emerging class of congenital regulopathies. Clin. Epigenetics 12, 1–20 (2020).
Li, Z. et al. Cyclin D1 integrates G9a-mediated histone methylation. Oncogene 38, 4232–4249 (2019).
Tanaka, H. et al. The SETD8/PR-Set7 methyltransferase functions as a barrier to prevent senescence-associated metabolic remodeling. Cell Rep. 18, 2148–2161 (2017).
Xie, Q. et al. Lysine methylation of FOXO3 regulates oxidative stress‐induced neuronal cell death. EMBO Rep. 13, 371–377 (2012).
Polansky, H. & Schwab, H. Latent viruses can cause disease by disrupting the competition for the limiting factor p300/CBP. Cell. Mol. Biol. Lett. 23, 1–6 (2018).
Liu, X. et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850 (2008).
Shikama, N. et al. Functional interaction between nucleosome assembly proteins and p300/CREB-binding protein family coactivators. Mol. Cell. Biol. 20, 8933–8943 (2000).
Wang, M., Chen, Z. & Zhang, Y. CBP/p300 and HDAC activities regulate H3K27 acetylation dynamics and zygotic genome activation in mouse preimplantation embryos. EMBO J. 41, 112012 (2022).
Martire, S., Nguyen, J., Sundaresan, A. & Banaszynski, L. A. Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol. Cell Biol. 21, 1–12 (2020).
Ban, N. et al. Hepatocyte nuclear factor-1α recruits the transcriptional co-activator p300 on the GLUT2 gene promoter. Diabetes 51, 1409–1418 (2002).
Song, Y. et al. Structural insights into the CRTC2–CREB complex assembly on CRE. J. Mol. Biol. 430, 1926–1939 (2018).
Luo, Q. et al. Mechanism of CREB recognition and coactivation by the CREB-regulated transcriptional coactivator CRTC2. Proc. Natl Acad. Sci. USA 109, 20865–20870 (2012).
Ma, L. et al. The CREB regulated transcription coactivator 2 suppresses HIV-1 transcription by preventing RNA Pol II from binding to HIV-1 LTR. Virol. Sin. 36, 796–809 (2021).
Amelio, A. L., Caputi, M. & Conkright, M. D. Bipartite functions of the CREB co‐activators selectively direct alternative splicing or transcriptional activation. EMBO J. 28, 2733–2747 (2009).
Ostojić, J. et al. Transcriptional co-activator regulates melanocyte differentiation and oncogenesis by integrating cAMP and MAPK/ERK pathways. Cell Rep. 35, 109136 (2021).
Parra-Damas, A., Rubió-Ferrarons, L., Shen, J. & Saura, C. A. CRTC1 mediates preferential transcription at neuronal activity-regulated CRE/TATA promoters. Sci. Rep. 7, 18004 (2017).
Smith, L. I. Involvement of CREB-regulated transcription coactivators (CRTC) in transcriptional activation of steroidogenic acute regulatory protein (Star) by ACTH. Mol. Cell. Endocrinol. 499, 110612 (2020).
Ferraz-de-Souza, B. et al. CBP/p300-interacting transactivator, with Glu/Asp-rich C-terminal domain, 2, and pre-B-cell leukemia transcription factor 1 in human adrenal development and disease. J. Clin. Endocrinol. Metab. 94, 678–683 (2009).
Ng, P. K. et al. Functional characterization of two CITED3 homologs (gcCITED3a and gcCITED3b) in the hypoxia-tolerant grass carp, Ctenopharyngodon idellus. BMC Mol. Biol. 10, 1–14 (2009).
Fernandes, M. T., Calado, S. M., Mendes-Silva, L. & Bragança, J. CITED2 and the modulation of the hypoxic response in cancer. World J. Clin. Oncol. 11, 260 (2020).
Yin, Z. et al. The essential role of Cited2, a negative regulator for HIF-1α, in heart development and neurulation. Proc. Natl Acad. Sci. USA 99, 10488–10493 (2002).
Kranc, K. R. et al. Transcriptional coactivator Cited2 induces Bmi1 and Mel18 and controls fibroblast proliferation via Ink4a/ARF. Mol. Cell. Biol. 23, 7658–7666 (2003).
Fox, S. B. et al. CITED4 inhibits hypoxia-activated transcription in cancer cells, and its cytoplasmic location in breast cancer is associated with elevated expression of tumor cell hypoxia-inducible factor 1α. Cancer Res. 64, 6075–6081 (2004).
Ozato, K., Shin, D. M., Chang, T. H. & Morse, H. C. III TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860 (2008).
Watanabe, M. & Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 161, 135–144 (2017).
Stevens, R. V., Esposito, D. & Rittinger, K. Characterisation of class VI TRIM RING domains: linking RING activity to C-terminal domain identity. Life Sci. Alliance 2, e201900295 (2019).
Rajsbaum, R., García-Sastre, A. & Versteeg, G. A. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 426, 1265–1284 (2014).
Wright, K. M., Du, H., Dagnachew, M. & Massiah, M. A. Solution structure of the microtubule‐targeting COS domain of MID1. FEBS J. 283, 3089–3102 (2016).
Zaware, N. & Zhou, M. M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 26, 870–879 (2019).
Hu, G. et al. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 23, 837–848 (2009).
Lv, D. et al. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat. Commun. 8, 1454 (2017).
Pipes, G. T., Creemers, E. E. & Olson, E. N. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 20, 1545–1556 (2006).
Velasquez, L. S. et al. Activation of MRTF-A–dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc. Natl Acad. Sci. USA 110, 16850–16855 (2013).
Esnault, C. et al. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 28, 943–958 (2014).
Fearing, B. V. et al. Mechanosensitive transcriptional coactivators MRTF-A and YAP/TAZ regulate nucleus pulposus cell phenotype through cell shape. FASEB J. 33, 14022 (2019).
Fuller-Pace, F. V. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 34, 4206–421 (2006).
Silverman, E., Edwalds-Gilbert, G. & Lin, R. J. DExD/H-box proteins and their partners: helping RNA helicases unwind. Gene 312, 1–16 (2003).
Iyer, R. S. et al. The RNA helicase/transcriptional co-regulator, p68 (DDX5), stimulates expression of oncogenic protein kinase, Polo-like kinase-1 (PLK1), and is associated with elevated PLK1 levels in human breast cancers. Cell Cycle 13, 1413–1423 (2014).
Nakajima, T. et al. RNA helicase A mediates association of CBP with RNA polymerase II. Cell 90, 1107–1112 (1997).
Chao, C. H. et al. DDX3, a DEAD box RNA helicase with tumor growth–suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res. 66, 6579–6588 (2006).
Saikruang, W. et al. The RNA helicase DDX3 promotes IFNB transcription via enhancing IRF-3/p300 holocomplex binding to the IFNB promoter. Sci. Rep. 12, 3967 (2022).
Schuler, M. et al. PGC1α expression is controlled in skeletal muscles by PPARβ, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 4, 407–414 (2006).
Lin, J., Handschin, C. & Spiegelman, B. M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370 (2005).
De Vitto, H., Bode, A. M. & Dong, Z. The PGC-1/ERR network and its role in precision oncology. NPJ Precis. Oncol. 3, 9 (2019).
Tcherepanova, I., Puigserver, P., Norris, J. D., Spiegelman, B. M. & McDonnell, D. P. Modulation of estrogen receptor-α transcriptional activity by the coactivator PGC-1. J. Biol. Chem. 275, 16302–16308 (2000).
Vega, R. B., Huss, J. M. & Kelly, D. P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 20, 1868–1876 (2000).
Delerive, P., Wu, Y., Burris, T. P., Chin, W. W. & Suen, C. S. PGC-1 functions as a transcriptional coactivator for the retinoid X receptors. J. Biol. Chem. 277, 3913–3917 (2002).
Knutti, D., Kaul, A. & Kralli, A. A tissue-specific coactivator of steroid receptors, identified in a functional genetic screen. Mol. Cell. Biol. 20, 2411–2422 (2000).
Wu, Z. et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 (1999).
Mihaylov, S. R. et al. The master energy homeostasis regulator PGC-1α exhibits an mRNA nuclear export function. Nat. Commun. 14, 5496 (2023).
Srivastava, S., Barrett, J. N. & Moraes, C. T. PGC-1α/β upregulation is associated with improved oxidative phosphorylation in cells harboring nonsense mtDNA mutations. Hum. Mol. Genet. 16, 993–1005 (2007).
Sonoda, J., Mehl, I. R., Chong, L. W., Nofsinger, R. R. & Evans, R. M. PGC-1β controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl Acad. Sci. USA 104, 5223–5228 (2007).
Islam, H., Edgett, B. A. & Gurd, B. J. Coordination of mitochondrial biogenesis by PGC-1α in human skeletal muscle: a re-evaluation. Metabolism 79, 42–51 (2018).
Hussein, S. M., Duff, E. K. & Sirard, C. Smad4 and β-catenin co-activators functionally interact with lymphoid-enhancing factor to regulate graded expression of Msx2. J. Biol. Chem. 278, 48805–48814 (2003).
Edginton‐White, B. & Bonifer, C. The transcriptional regulation of normal and malignant blood cell development. FEBS J. 289, 1240–1255 (2022).
Lin, J. D. Minireview: the PGC-1 coactivator networks: chromatin-remodeling and mitochondrial energy metabolism. Mol. Endocrinol. 23, 2–10 (2009).
Nussinov, R., Tsai, C. J. & Jang, H. Allostery, and how to define and measure signal transduction. Biophys. Chem. 283, 106766 (2022).
Westermarck, J., Ivaska, J. & Corthals, G. L. Identification of protein interactions involved in cellular signaling. Mol. Cell. Proteom. 12, 1752–1763 (2013).
Tang, H. & Xue, G. Major physiological signaling pathways in the regulation of cell proliferation and survival. Handb. Exp. Pharmacol. 249, 13–30 (2018).
Willert, J., Epping, M., Pollack, J. R., Brown, P. O. & Nusse, R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev. Biol. 2, 1–7 (2002).
Teo, J. L. & Kahn, M. The Wnt signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv. Drug Deliv. Rev. 62, 1149–1155 (2010).
Carthagena, L. Human TRIM gene expression in response to interferons. PLoS ONE 4, 4894 (2009).
Zhang, Y. et al. Multiple signaling pathways regulate contractile activity‐mediated PGC‐1α gene expression and activity in skeletal muscle cells. Physiol. Rep. 2, 12008 (2014).
Finck, B. N. et al. Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab. 4, 199–210 (2006).
Sweeney, T. E., Suliman, H. B., Hollingsworth, J. W., Welty-Wolf, K. E. & Piantadosi, C. A. A toll-like receptor 2 pathway regulates the Ppargc1a/b metabolic co-activators in mice with Staphylococcal aureus sepsis. PLoS ONE 6, 25249 (2011).
Zhu, Q. et al. The Wnt-driven Mll1 epigenome regulates salivary gland and head and neck cancer. Cell Rep. 26, 415–428 (2019).
Yuan, H. et al. Epigenetic histone modifications involved in profibrotic gene regulation by 12/15-lipoxygenase and its oxidized lipid products in diabetic nephropathy. Antioxid. Redox Sign. 24, 361–375 (2016).
Werneburg, N., Gores, G. J. & Smoot, R. L. The Hippo pathway and YAP signaling: emerging concepts in regulation, signaling, and experimental targeting strategies with implications for hepatobiliary malignancies. Gene Expr. 20, 67 (2020).
Pisco, A. O., Tojo, B. & McGeever, A. Single-cell analysis for whole-organism datasets. Annu. Rev. Biomed. Data Sci. 4, 207–226 (2021).
López-Maury, L., Marguerat, S. & Bähler, J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat. Rev. Genet. 9, 583–593 (2008).
Kramer, D. A. Commentary: Gene-environment interplay in the context of genetics, epigenetics, and gene expression. J. Am. Acad. Child Adolesc. Psychiatry 44, 19–27 (2005).
Oudhoff, M. J. et al. Control of the hippo pathway by Set7-dependent methylation of Yap. Dev. Cell 26, 188–194 (2013).
Xu, W. et al. A transcriptional switch mediated by cofactor methylation. Science 294, 2507–2511 (2001).
Rieger, M. E. et al. p300/β-catenin interactions regulate adult progenitor cell differentiation downstream of WNT5a/protein kinase C (PKC). J. Biol. Chem. 291, 6569–6582 (2016).
Wu, S. et al. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 24, 2531–2542 (2010).
Aguilar-Arnal, L., Katada, S., Orozco-Solis, R. & Sassone-Corsi, P. NAD+-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat. Struct. Mol. Biol. 22, 312–318 (2015).
Liu, L. et al. Arginine methylation of BRD4 by PRMT2/4 governs transcription and DNA repair. Sci. Adv. 8, 8928 (2022).
Vershinin, Z. et al. BRD4 methylation by the methyltransferase SETD6 regulates selective transcription to control mRNA translation. Sci. Adv. 7, 5374 (2021).
Fletcher, A. J. et al. TRIM 5α requires Ube2W to anchor Lys63‐linked ubiquitin chains and restrict reverse transcription. EMBO J. 34, 2078–2095 (2015).
Mersaoui, S. Y. et al. Arginine methylation of the DDX 5 helicase RGG/RG motif by PRMT 5 regulates resolution of RNA: DNA hybrids. EMBO J. 38, 100986 (2019).
Wu, R. C., Feng, Q., Lonard, D. M. & O’Malley, B. W. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell 129, 1125–1140 (2007).
Yu, B. et al. DDX55 promotes hepatocellular carcinoma progression by interacting with BRD4 and participating in exosome‐mediated cell‐cell communication. Cancer Sci. 113, 3002–3017 (2022).
Liu, W. et al. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 155, 1581–1595 (2013).
Zimmermann, H., Degenkolbe, R., Bernard, H. U. & O’Connor, M. J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 73, 6209–6219 (1999).
Misra, P. et al. Interaction of PIMT with transcriptional coactivators CBP, p300, and PBP differential role in transcriptional regulation. J. Biol. Chem. 277, 20011–20019 (2002).
Sheppard, H. M., Harries, J. C., Hussain, S., Bevan, C. & Heery, D. M. Analysis of the steroid receptor coactivator 1 (SRC1)-CREB binding protein interaction interface and its importance for the function of SRC1. Mol. Cell. Biol. 21, 39–50 (2001).
Li, J. & Liu, C. Coding or noncoding, the converging concepts of RNAs. Front. Genet. 10, 496 (2019).
Statello, L., Guo, C. J., Chen, L. L. & Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118 (2021).
Grixti, J. M. & Ayers, D. Long noncoding RNAs and their link to cancer. Noncoding RNA Res. 5, 77–82 (2020).
Jin, J., Martin, M., Hartley, A. V. & Lu, T. PRMTs and miRNAs: functional cooperation in cancer and beyond. Cell Cycle 18, 1676–1686 (2019).
Gao, Y. et al. Long non-coding RNA FGD5-AS1 regulates cancer cell proliferation and chemoresistance in gastric cancer through miR-153-3p/CITED2 axis. Front. Genet. 11, 715 (2020).
Maldotti, M. et al. The acetyltransferase p300 is recruited in trans to multiple enhancer sites by lncSmad7. Nucleic Acids Res. 50, 2587–2602 (2022).
Lagos, D. et al. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 12, 513–519 (2010).
Pistoni, M. et al. Long noncoding RNA NEAT1 acts as a molecular switch for BRD4 transcriptional activity and mediates repression of BRD4/WDR5 target genes. Mol. Cancer Res. 19, 799–811 (2021).
Zheng, J. F., Guo, N. H., Zi, F. M. & Cheng, J. Long noncoding RNA H19 promotes tumorigenesis of multiple myeloma by activating BRD4 signaling by targeting microRNA 152-3p. Mol. Cell. Biol. 40, 00382–19 (2020).
Zhang, H., Tan, M., Zhang, J., Han, X. & Ma, Y. Propofol inhibits thyroid cancer cell proliferation, migration, and invasion by suppressing SHH and PI3K/AKT signaling pathways via the miR-141-3p/BRD4 axis. J. Health Eng. 2021, 2704753 (2021).
Zhou, J. et al. MicroRNA-153 functions as a tumor suppressor by targeting SET7 and ZEB2 in ovarian cancer cells. Oncol. Rep. 34, 111–120 (2015).
Perrimon, N., Pitsouli, C. & Shilo, B. Z. Signaling mechanisms controlling cell fate and embryonic patterning. Cold Spring Harb. Perspect. Biol. 4, 5975 (2012).
Gusterson, R. et al. The transcriptional co-activators CBP and p300 are activated via phenylephrine through the p42/p44 MAPK cascade. J. Biol. Chem. 277, 2517–2524 (2002).
Sánchez-Molina, S. et al. The histone acetyltransferases CBP/p300 are degraded in NIH 3T3 cells by activation of Ras signalling pathway. Biochem. J. 398, 215–224 (2006).
Rausch, V. & Hansen, C. G. The Hippo pathway, YAP/TAZ, and the plasma membrane. Trends Cell Biol. 30, 32–48 (2020).
Zhou, X. et al. Regulation of Hippo/YAP signaling and Esophageal Squamous Carcinoma progression by an E3 ubiquitin ligase PARK2. Theranostics 10, 9443 (2020).
Zhou, C. et al. Pygo2 functions as a prognostic factor for glioma due to its up-regulation of H3K4me3 and promotion of MLL1/MLL2 complex recruitment. Sci. Rep. 6, 22066 (2016).
Xie, H. et al. ROS/NF-κB signaling pathway-mediated transcriptional activation of TRIM37 promotes HBV-associated hepatic fibrosis. Mol. Ther. Nucleic Acids 22, 114–123 (2020).
Meerson, A. & Yehuda, H. Leptin and insulin up-regulate miR-4443 to suppress NCOA1 and TRAF4, and decrease the invasiveness of human colon cancer cells. BMC Cancer 16, 1–9 (2016).
Xu, J., Wu, R. C. & O’malley, B. W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 9, 615–630 (2009).
Theeuwes, W. F., Gosker, H. R., Schols, A. M. W. J., Langen, R. C. J. & Remels, A. H. V. Regulation of PGC-1α expression by a GSK-3β-TFEB signaling axis in skeletal muscle. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118610 (2020).
Puigserver, P. et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 8, 971–982 (2001).
Guo, Y. H. et al. Wnt/β-catenin pathway transactivates microRNA-150 that promotes EMT of colorectal cancer cells by suppressing CREB signaling. Oncotarget 7, 42513 (2016).
Kim, J. H. et al. JNK suppresses melanogenesis by interfering with CREB-regulated transcription coactivator 3-dependent MITF expression. Theranostics 10, 4017 (2020).
Lee, J. M. et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 285, 32182–32191 (2010).
Hogan, N. T. et al. Transcriptional networks specifying homeostatic and inflammatory programs of gene expression in human aortic endothelial cells. eLife 6, 22536 (2017).
Gomes, A. P. & Blenis, J. A nexus for cellular homeostasis: the interplay between metabolic and signal transduction pathways. Curr. Opin. Biotech. 34, 110–117 (2015).
Rosenfeld, M. G., Lunyak, V. V. & Glass, C. K. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 20, 1405–1428 (2006).
Torchia, J., Glass, C. & Rosenfeld, M. G. Co-activators and co-repressors in the integration of transcriptional responses. Curr. Opin. Cell Biol. 10, 373–383 (1998).
Wolffe, A. P. Chromatin remodeling regulated by steroid and nuclear receptors. Cell Res. 7, 127–142 (1997).
Singh, B. K. & Yen, P. M. A clinician’s guide to understanding resistance to thyroid hormone due to receptor mutations in the TRα and TRβ isoforms. Clin. Diabetes Endocrinol. 3, 1–11 (2017).
Zaghet, N. et al. Coordinated maintenance of H3K36/K27 methylation by histone demethylases preserves germ cell identity and immortality. Cell Rep. 37, 110050 (2021).
Youn, M. Y. et al. JMJD5, a Jumonji C (JmjC) domain-containing protein, negatively regulates osteoclastogenesis by facilitating NFATc1 protein degradation. J. Biol. Chem. 287, 12994–13004 (2012).
Bradner, J. E., Hnisz, D. & Young, R. A. Transcriptional addiction in cancer. Cell 168, 629–643 (2017).
Feinberg, A. P. & Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 4, 143–153 (2004).
Anastasi, S., Alemà, S. & Segatto, O. Making sense of Cbp/p300 loss of function mutations in skin tumorigenesis. J. Pathol. 250, 3–6 (2020).
Kumar, M. et al. Inhibition of histone acetyltransferase function radiosensitizes CREBBP/EP300 mutants via repression of homologous recombination, potentially targeting a gain of function. Nat. Commun. 12, 6340 (2021).
Stossi, F., Madak-Erdogan, Z. & Katzenellenbogen, B. S. Estrogen receptor alpha represses transcription of early target genes via p300 and CtBP1. Mol. Cell. Biol. 29, 1749–1759 (2009).
Deng, Y. et al. Protocatechuic aldehyde represses proliferation and migration of breast cancer cells through targeting C-terminal binding protein 1. J. Breast Cancer 23, 20–35 (2020).
Wu, Y. & Zhao, H. CTBP1 strengthens the cisplatin resistance of gastric cancer cells by upregulating RAD51 expression. Oncol. Lett. 22, 1–9 (2021).
Liu, Y., Smith, P. W. & Jones, D. R. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol. Cell. Biol. 26, 8683–8696 (2006).
Rampalli, S., Pavithra, L., Bhatt, A., Kundu, T. K. & Chattopadhyay, S. Tumor suppressor SMAR1 mediates cyclin D1 repression by recruitment of the SIN3/histone deacetylase 1 complex. Mol. Cell. Biol. 25, 8415–8429 (2005).
Olusanya, B. O. et al. Developmental disabilities among children younger than 5 years in 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Glob Health 6, 1100–1121 (2018).
Lord, C., Cook, E. H., Leventhal, B. L. & Amaral, D. G. Autism spectrum disorders. Neuron 28, 355–363 (2000).
Francés, L. et al. Current state of knowledge on the prevalence of neurodevelopmental disorders in childhood according to the DSM-5: a systematic review in accordance with the PRISMA criteria. Child Adolesc. Psychiatry Ment. Health 16, 27 (2022).
Mossink, B., Negwer, M., Schubert, D. & NadifKasri, N. The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective. Cell. Mol. Life Sci. 78, 2517–2563 (2021).
López, A. J. & Wood, M. A. Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front. Behav. Neurosci. 9, 100 (2015).
Rowland, M. E., Jajarmi, J. M., Osborne, T. S. & Ciernia, A. V. Insights into the emerging role of Baf53b in Autism Spectrum Disorder. Front. Mol. Neurosci. 15, 805158 (2022).
Kalkman, H. O. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol. Autism 3, 1–12 (2012).
Tabuchi, A. & Ihara, D. Regulation of dendritic synaptic morphology and transcription by the SRF cofactor MKL/MRTF. Front. Mol. Neurosci. 14, 767842 (2021).
Wu, K. et al. SETDB1-mediated cell fate transition between 2C-like and pluripotent states. Cell Rep. 30, 25–36 (2020).
Markouli, M., Strepkos, D., Chlamydas, S. & Piperi, C. Histone lysine methyltransferase SETDB1 as a novel target for central nervous system diseases. Prog. Neurobiol. 200, 101968 (2021).
Kato, H. et al. Rare genetic variants in the gene encoding histone lysine demethylase 4 C (KDM4C) and their contributions to susceptibility to schizophrenia and autism spectrum disorder. Transl. Psychiatry 10, 421 (2020).
Li, Y. J. KMT2E haploinsufficiency manifests autism-like behaviors and amygdala neuronal development dysfunction in mice. Mol. Neurobiol. 60, 1609–1625 (2023).
Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014).
Chen, S. et al. De novo missense variants disrupting protein–protein interactions affect risk for autism through gene co-expression and protein networks in neuronal cell types. Mol. Autism 11, 1–16 (2020).
Crider, A., Thakkar, R., Ahmed, A. O. & Pillai, A. Dysregulation of estrogen receptor beta (ERβ), aromatase (CYP19A1), and ER co-activators in the middle frontal gyrus of autism spectrum disorder subjects. Mol. Autism 5, 1–10 (2014).
Benito, E. et al. The BET/BRD inhibitor JQ1 improves brain plasticity in WT and APP mice. Transl. Psychiatry 7, 1239–1239 (2017).
Citrigno, L. et al. The mitochondrial dysfunction hypothesis in autism spectrum disorders: current status and future perspectives. Int. J. Mol. Sci. 21, 5785 (2020).
Bam, S., Buchanan, E., Mahony, C. & O’Ryan, C. DNA methylation of PGC-1α is associated with elevated mtDNA copy number and altered urinary metabolites in autism spectrum disorder. Front. Cell Dev. Biol. 9, 696428 (2021).
Fame, R. M., MacDonald, J. L., Dunwoodie, S. L., Takahashi, E. & Macklis, J. D. Cited2 regulates neocortical layer II/III generation and somatosensory callosal projection neuron development and connectivity. J. Neurosci. 36, 6403–6419 (2016).
Wagner, N. R. & MacDonald, J. L. Atypical neocortical development in the Cited2 conditional knockout leads to behavioral deficits associated with neurodevelopmental disorders. Neuroscience 455, 65–78 (2021).
Gao, Y., Aljazi, M. B. & He, J. Kdm6b haploinsufficiency causes ASD/ADHD-like behavioral deficits in mice. Front. Behav. Neurosci. 16, 905783 (2022).
Olfson, E. et al. Ultra-rare de novo damaging coding variants are enriched in attention-deficit/hyperactivity disorder and identify risk genes. Preprint at https://www.medrxiv.org/content/10.1101/2023.05.19.23290241v1 (2023).
Rizzi, T. S. et al. The ATXN1 and TRIM31 genes are related to intelligence in an ADHD background: evidence from a large collaborative study totaling 4,963 subjects. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156, 145–157 (2011).
Wang, X., Wu, H., Sun, H., Wang, L. & Chen, L. ARID2, a rare cause of Coffin–Siris Syndrome: a clinical description of two cases. Front. Pediatr. 10, 911954 (2022).
Zhou, A. et al. Common genetic risk factors in ASD and ADHD co-occurring families. Hum. Genet. 142, 217–230 (2023).
Walton, E. et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol. Psychiatry 22, 250–256 (2017).
Geneviève, D. Lessons from two series by physicians and caregivers’ self-reported data, and DNA methylation profile in DDX3X-related disorders. Preprint at https://www.researchsquare.com/article/rs-2760508/v1 (2023).
Valor, L. M., Viosca, J., Lopez-Atalaya, J. P. & Barco, A. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 19, 5051–5064 (2013).
Grozeva, D. et al. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 94, 618–624 (2014).
Bjornsson, H. T. The Mendelian disorders of the epigenetic machinery. Genome Res. 25, 1473–1481 (2015).
Lebrun, N. et al. Molecular and cellular issues of KMT2A variants involved in Wiedemann-Steiner syndrome. Eur. J. Hum. Genet. 26, 107–116 (2018).
Pilotto, S. et al. LSD1/KDM1A mutations associated to a newly described form of intellectual disability impair demethylase activity and binding to transcription factors. Hum. Mol. Genet. 25, 2578–2587 (2016).
Williamson, K. A. et al. Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am. J. Hum. Genet. 94, 295–302 (2014).
Bögershausen, N. & Wollnik, B. Mutational landscapes and phenotypic spectrum of SWI/SNF-related intellectual disability disorders. Front. Mol. Neurosci. 11, 252 (2018).
Barish, S. et al. BICRA, a SWI/SNF complex member, is associated with BAF-disorder related phenotypes in humans and model organisms. Am. J. Hum. Genet. 107, 1096–1112 (2020).
Blok, L. S. et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am. J. Hum. Genet. 97, 343–352 (2015).
Balak, C. et al. Rare de novo missense variants in RNA helicase DDX6 cause intellectual disability and dysmorphic features and lead to P-body defects and RNA dysregulation. Am. J. Hum. Genet. 105, 509–525 (2019).
Geetha, T. S. et al. Targeted deep resequencing identifies MID 2 mutation for X‐linked intellectual disability with varied disease severity in a large kindred from India. Hum. Mutat. 35, 41–44 (2014).
Di Rienzo, M., Romagnoli, A., Antonioli, M., Piacentini, M. & Fimia, G. M. TRIM proteins in autophagy: selective sensors in cell damage and innate immune responses. Cell Death Differ. 27, 887–902 (2020).
Hoffman, D. J., Powell, T. L., Barrett, E. S. & Hardy, D. B. Developmental origins of metabolic diseases. Physiol. Rev. 101, 739–795 (2021).
Al-Goblan, A. S., Al-Alfi, M. A. & Khan, M. Z. Mechanism linking diabetes mellitus and obesity. Diabetes Metab. Syndr. Obes. 7, 587–591 (2014).
Barroso, I. & McCarthy, M. I. The genetic basis of metabolic disease. Cell 177, 146–161 (2019).
Ibar, C. & Irvine, K. D. Integration of Hippo-YAP signaling with metabolism. Dev. Cell 54, 256–267 (2020).
Talchai, C., Xuan, S., Lin, H. V., Sussel, L. & Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234 (2012).
Zhang, L. et al. CBP/p300 HAT maintains the gene network critical for β cell identity and functional maturity. Cell Death Dis. 12, 476 (2021).
Kadiyala, C. S. R. et al. Acetylation of retinal histones in diabetes increases inflammatory proteins: effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J. Biol. Chem. 287, 25869–25880 (2012).
Ruiz, L. et al. Proteasomal degradation of the histone acetyl transferase p300 contributes to beta-cell injury in a diabetes environment. Cell Death Dis. 9, 600 (2018).
Sakai, M. et al. The GCN5-CITED2-PKA signalling module controls hepatic glucose metabolism through a cAMP-induced substrate switch. Nat. Commun. 7, 13147 (2016).
Wang, Z., Shah, O. J. & Hunter, T. The transcriptional coactivators p/CIP and SRC-1 control insulin resistance through IRS1 in obesity models. PLoS ONE 7, 36961 (2012).
Wan, T. et al. Overexpression of TRIM32 promotes pancreatic β‐cell autophagic cell death through Akt/mTOR pathway under high glucose conditions. Cell Biol. Int. 46, 2095–2106 (2022).
Wang, C. et al. YAP/TAZ regulates the insulin signaling via IRS1/2 in endometrial cancer. Am. J. Cancer Res. 6, 996 (2016).
Lin, X. & Li, H. Obesity: epidemiology, pathophysiology, and therapeutics. Front. Endocrinol. 12, 706978 (2021).
Zhou, F. et al. Selective inhibition of CBP/p300 HAT by A-485 results in suppression of lipogenesis and hepatic gluconeogenesis. Cell Death Dis. 11, 745 (2020).
Moreno, C. L. et al. Role of hypothalamic creb-binding protein in obesity and molecular reprogramming of metabolic substrates. PLoS ONE 11, 0166381 (2016).
Hu, X. et al. Brd4 modulates diet-induced obesity via PPARγ-dependent Gdf3 expression in adipose tissue macrophages. JCI Insight 6, 143379 (2021).
Yoon, Y. S. et al. Activation of the adipocyte CREB/CRTC pathway in obesity. Commun. Biol. 4, 1214 (2021).
Kobayashi, M., Deguchi, Y., Nozaki, Y. & Higami, Y. Contribution of PGC-1α to obesity-and caloric restriction-related physiological changes in white adipose tissue. Int. J. Mol. Sci. 22, 6025 (2021).
Bond, S. T. et al. Deletion of Trim28 in committed adipocytes promotes obesity but preserves glucose tolerance. Nat. Commun. 12, 74 (2021).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Smolarz, B., Nowak, A. Z. & Romanowicz, H. Breast cancer—epidemiology, classification, pathogenesis and treatment (review of literature). Cancers 14, 2569 (2022).
Kashyap, D. et al. Global increase in breast cancer incidence: risk factors and preventive measures. Biomed. Res. Int. 2022, 9605439 (2022).
Medjkane, S., Perez-Sanchez, C., Gaggioli, C., Sahai, E. & Treisman, R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat. Cell Biol. 11, 257–268 (2009).
Taskinen, M. E. et al. MASTL promotes cell contractility and motility through kinase-independent signaling. J. Cell Biol. 219, 201906204 (2020).
Faria, L. et al. Activation of an actin signaling pathway in pre-malignant mammary epithelial cells by P-cadherin is essential for transformation. Dis. Model Mech. 16, 049652 (2023).
Moghe, A. & Monga, S. P. BCL9/BCL9L in hepatocellular carcinoma: will it or Wnt it be the next therapeutic target? Hepatol. Int. 14, 460–462 (2020).
Elsarraj, H. S. et al. BCL9/STAT3 regulation of transcriptional enhancer networks promote DCIS progression. NPJ Breast Cancer 6, 12 (2020).
Sustmann, C., Flach, H., Ebert, H., Eastman, Q. & Grosschedl, R. Cell-type-specific function of BCL9 involves a transcriptional activation domain that synergizes with β-catenin. Mol. Cell. Biol. 28, 3526–3537 (2008).
Wang, X. et al. BCL9/BCL9L promotes tumorigenicity through immune-dependent and independent mechanisms in triple negative breast cancer. Oncogene 40, 2982–2997 (2021).
Swahn, M. L. et al. The effect of RU 486 administered during the early luteal phase on bleeding pattern, hormonal parameters and endometrium. Hum. Reprod. 5, 402–408 (1990).
Haque, M. M. & Desai, K. V. Pathways to endocrine therapy resistance in breast cancer. Front. Endocrinol. 10, 573 (2019).
Bleach, R. & McIlroy, M. The divergent function of androgen receptor in breast cancer; analysis of steroid mediators and tumor intracrinology. Front. Endocrinol. 9, 594 (2018).
Flågeng, M. H. et al. Nuclear receptor co-activators and HER-2/neu are upregulated in breast cancer patients during neo-adjuvant treatment with aromatase inhibitors. Br. J. Cancer 101, 1253–1260 (2009).
Minemura, H. et al. CITED2 in breast carcinoma as a potent prognostic predictor associated with proliferation, migration and chemoresistance. Cancer Sci. 107, 1898–1908 (2016).
Lau, W. M., Doucet, M., Huang, D., Weber, K. L. & Kominsky, S. L. CITED2 modulates estrogen receptor transcriptional activity in breast cancer cells. Biochem. Biophys. Res. Commun. 437, 261–266 (2013).
Jayaraman, S., Doucet, M. & Kominsky, S. L. CITED2 attenuates macrophage recruitment concordant with the downregulation of CCL20 in breast cancer cells. Oncol. Lett. 15, 871–878 (2018).
Keating, S. & El-Osta, A. Transcriptional regulation by the Set7 lysine methyltransferase. Epigenetics 8, 361–372 (2013).
Gu, Y., Zhang, X., Yu, W. & Dong, W. Oncogene or tumor suppressor: the coordinative role of lysine methyltransferase SET7/9 in cancer development and the related mechanisms. J. Cancer 13, 623 (2022).
Subramanian, K. et al. Regulation of estrogen receptor α by the SET7 lysine methyltransferase. Mol. Cell 30, 336–347 (2008).
Cerutti, C. et al. Computational identification of new potential transcriptional partners of ERRα in breast cancer cells: specific partners for specific targets. Sci. Rep. 12, 3826 (2022).
Wilson, B. J. et al. The p68 and p72 DEAD box RNA helicases interact with HDAC1 and repress transcription in a promoter-specific manner. BMC Mol. Biol. 5, 1–15 (2004).
Wortham, N. C. et al. The DEAD-box protein p72 regulates ERα-/oestrogen-dependent transcription and cell growth, and is associated with improved survival in ERα-positive breast cancer. Oncogene 28, 4053–4064 (2009).
Alqahtani, H. et al. DDX17 (P72), a Sox2 binding partner, promotes stem-like features conferred by Sox2 in a small cell population in estrogen receptor-positive breast cancer. Cell. Signal. 28, 42–50 (2016).
Mohibi, S. et al. Mammalian alteration/deficiency in activation 3 (Ada3) is essential for embryonic development and cell cycle progression. J. Biol. Chem. 287, 29442–29456 (2012).
Mirza, S. et al. Cytoplasmic localization of alteration/deficiency in activation 3 (ADA3) predicts poor clinical outcome in breast cancer patients. Breast Cancer Res. Treat. 137, 721–731 (2013).
Chand, V., Nandi, D., Mangla, A. G., Sharma, P. & Nag, A. Tale of a multifaceted co-activator, hADA3: from embryogenesis to cancer and beyond. Open Biol. 6, 160153 (2016).
Kumar, J. P. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cell. Mol. Life Sci. 66, 565–583 (2009).
Reichenberger, K. J., Coletta, R. D., Schulte, A. P., Varella-Garcia, M. & Ford, H. L. Gene amplification is a mechanism of Six1 overexpression in breast cancer. Cancer Res. 65, 2668–2675 (2005).
Xu, H., Jiao, Y., Yi, M., Zhao, W. & Wu, K. EYA2 correlates with clinico-pathological features of breast cancer, promotes tumor proliferation, and predicts poor survival. Front. Oncol. 9, 26 (2019).
Blevins, M. A., Towers, C. G., Patrick, A. N., Zhao, R. & Ford, H. L. The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Expert Opin. Ther. Targets 19, 213–225 (2015).
Tisserand, J. et al. Tripartite motif 24 (Trim24/Tif1α) tumor suppressor protein is a novel negative regulator of interferon (IFN)/signal transducers and activators of transcription (STAT) signaling pathway acting through retinoic acid receptor α (Rarα) inhibition. J. Biol. Chem. 286, 33369–33379 (2011).
Tsai, W. W. et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 468, 927–932 (2010).
Shah, V. V. et al. Mammary-specific expression of Trim24 establishes a mouse model of human metaplastic breast cancer. Nat. Commun. 12, 5389 (2021).
Yu, B. et al. KAT6A acetylation of SMAD3 regulates myeloid‐derived suppressor cell recruitment, metastasis, and immunotherapy in triple‐negative breast cancer. Adv. Sci. 8, 2100014 (2021).
Cunningham, R. & Hansen, C. G. The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin. Sci. 136, 197–222 (2022).
He, L. et al. YAP and TAZ are transcriptional co-activators of AP-1 proteins and STAT3 during breast cellular transformation. eLife 10, 67312 (2021).
Xiang, L. et al. HIF-1α and TAZ serve as reciprocal co-activators in human breast cancer cells. Oncotarget 6, 11768 (2015).
Zhang, C. & Moberg, K. One repressor to rule them all: ANCO 1 links YAP and AIB 1. EMBO Rep. 21, 49647 (2020).
Wang, L. et al. Prostate cancer incidence and mortality: global status and temporal trends in 89 countries from 2000 to 2019. Front. Public Health 10, 176 (2022).
Chen, Q. et al. Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents. Theranostics 12, 4935 (2022).
Waddell, A. R., Huang, H. & Liao, D. CBP/p300: critical co-activators for nuclear steroid hormone receptors and emerging therapeutic targets in prostate and breast cancers. Cancers 13, 2872 (2021).
Yu, X. et al. Structural insights of transcriptionally active, full-length androgen receptor coactivator complexes. Mol. Cell 79, 812–823 (2020).
Zhong, J. et al. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer Res. 74, 1870–1880 (2014).
Ji, D. et al. Targeting CDCP1 gene transcription coactivated by BRD4 and CBP/p300 in castration-resistant prostate cancer. Oncogene 41, 3251–3262 (2022).
Shin, S. H. et al. Aberrant expression of CITED2 promotes prostate cancer metastasis by activating the nucleolin-AKT pathway. Nat. Commun. 9, 4113 (2018).
Ling, X. H. et al. BCL9, a coactivator for Wnt/β-catenin transcription, is targeted by miR-30c and is associated with prostate cancer progression. Oncol. Lett. 11, 2001–2008 (2016).
He, G. et al. Association of BCL9 expression with prostate cancer clinicopathological features and survival. Zhonghua Yi Xue Za Zhi 95, 2603–2606 (2015).
Luo, P. et al. The human positive cofactor 4 promotes androgen-independent prostate cancer development and progression through HIF-1α/β-catenin pathway. Am. J. Cancer Res. 9, 682 (2019).
Chakravarthi, B. V. et al. MicroRNA-101 regulated transcriptional modulator SUB1 plays a role in prostate cancer. Oncogene 35, 6330–6340 (2016).
Hu, Y. C. et al. Functional domain and motif analyses of androgen receptor coregulator ARA70 and its differential expression in prostate cancer. J. Biol. Chem. 279, 33438–33446 (2004).
Shang, Z. et al. Human kallikrein 2 (KLK2) promotes prostate cancer cell growth via function as a modulator to promote the ARA70-enhanced androgen receptor transactivation. Tumor Biol. 35, 1881–1890 (2014).
Reid, B. M., Permuth, J. B. & Sellers, T. A. Epidemiology of ovarian cancer: a review. Cancer Biol. Med. 14, 9 (2017).
Hall, C. A. et al. 2010. Hippo pathway effector Yap is an ovarian cancer oncogene. Cancer Res. 70, 8517–8525 (2010).
Zhang, X. et al. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene 30, 2810–2822 (2011).
Chen, G., Xie, J., Huang, P. & Yang, Z. Overexpression of TAZ promotes cell proliferation, migration and epithelial-mesenchymal transition in ovarian cancer. Oncol. Lett. 12, 1821–1825 (2016).
Hua, G. et al. YAP induces high-grade serous carcinoma in fallopian tube secretory epithelial cells. Oncogene 35, 2247–2265 (2016).
Xin, L. et al. SND1 acts upstream of SLUG to regulate the epithelial–mesenchymal transition (EMT) in SKOV3 cells. FASEB J. 33, 3795–3806 (2019).
Pao, G. M., Janknecht, R., Ruffner, H., Hunter, T. & Verma, I. M. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl Acad. Sci. USA 97, 1020–1025 (2000).
Ou, C. et al. CRTC2 promotes paclitaxel resistance by inducing autophagy in ovarian cancer in part via the PI3K-AKT signaling axis. J. Cancer 14, 1011 (2023).
Xie, Y. et al. Functional cyclic AMP response element in the breast cancer resistance protein (BCRP/ABCG2) promoter modulates epidermal growth factor receptor pathway-or androgen withdrawal-mediated BCRP/ABCG2 transcription in human cancer cells. Biochim. Biophys. Acta Gene Regul. Mech. 1849, 317–327 (2015).
Drumond-Bock, A. L. & Bieniasz, M. The role of distinct BRD4 isoforms and their contribution to high-grade serous ovarian carcinoma pathogenesis. Mol. Cancer 20, 145 (2021).
Petersen, S. et al. CCNE1 and BRD4 co-amplification in high-grade serous ovarian cancer is associated with poor clinical outcomes. Gynecol. Oncol. 157, 405–410 (2020).
Zhang, L., Chen, H., Ding, B. & Jiang, W. High expression of TRIM24 predicts worse prognosis and promotes proliferation and metastasis of epithelial ovarian cancer. J. Ovarian Res. 15, 19 (2022).
Zhou, H. E., Pan, S. S. & Han, H. TRIM24 aggravates the progression of ovarian cancer through negatively regulating FOXM1 level. Eur. Rev. Med. Pharmacol. Sci. 23, 10647–10656 (2019).
Kombe Kombe, A. J. et al. Epidemiology and burden of human papillomavirus and related diseases, molecular pathogenesis, and vaccine evaluation. Front. Public Health 8, 552028 (2021).
Ishiji, T. et al. Transcriptional enhancer factor (TEF)‐1 and its cell‐specific co‐activator activate human papillomavirus‐16 E6 and E7 oncogene transcription in keratinocytes and cervical carcinoma cells. EMBO J. 11, 2271–2281 (1992).
Mori, S., Takeuchi, T., Ishii, Y. & Kukimoto, I. The transcriptional cofactor VGLL1 drives transcription of human papillomavirus early genes via TEAD1. J. Virol. 94, 01945–19 (2020).
Bochynska, A., Lüscher-Firzlaff, J. & Lüscher, B. Modes of interaction of KMT2 histone H3 lysine 4 methyltransferase/COMPASS complexes with chromatin. Cells 7, 17 (2018).
Zerkalenkova, E. et al. BTK, NuTM2A, and PRPF19 are novel KMT2A partner genes in childhood acute leukemia. Biomedicines 9, 924 (2021).
Zhang, C. et al. KMT2A regulates cervical cancer cell growth through targeting VDAC1. Aging 12, 9604 (2020).
Lin, L., Zhao, W., Sun, B., Wang, X. & Liu, Q. Overexpression of TRIM24 is correlated with the progression of human cervical cancer. Am. J. Transl. Res. 9, 620 (2017).
Liu, B. et al. Expression and significance of TRIM 28 in squamous carcinoma of esophagus. Pathol. Oncol. Res. 25, 1645–1652 (2019).
Li, F., Wang, Z. & Lu, G. TRIM28 promotes cervical cancer growth through the mTOR signaling pathway. Oncol. Rep. 39, 1860–1866 (2018).
Nath, A., Sathishkumar, K., Das, P., Sudarshan, K. L. & Mathur, P. A clinicoepidemiological profile of lung cancers in India–Results from the National Cancer Registry Programme. Indian J. Med. Res. 155, 264–272 (2022).
Huang, Y. H. et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 32, 915–928 (2018).
Zhou, C., Huang, H., Wang, Y., Sendinc, E. & Shi, Y. Selective regulation of tuft cell-like small cell lung cancer by novel transcriptional co-activators C11orf53 and COLCA2. Cell Discov. 8, 112 (2022).
Wu, X. S. et al. OCA-T1 and OCA-T2 are coactivators of POU2F3 in the tuft cell lineage. Nature 607, 169–175 (2022).
Molina, J. R., Yang, P., Cassivi, S. D., Schild, S. E. & Adjei, A. A. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 83, 584–594 (2008).
Ndembe, G. et al. LKB1: can we target a hidden target? Focus on NSCLC. Front. Oncol. 12, 889826 (2022).
He, Y. et al. Identification and validation of PROM1 and CRTC2 mutations in lung cancer patients. Mol. Cancer 13, 1–9 (2014).
Zhou, X. et al. Dependency of human and murine LKB1-inactivated lung cancer on aberrant CRTC-CREB activation. eLife 10, 66095 (2021).
Rodón, L. et al. The CREB coactivator CRTC2 promotes oncogenesis in LKB1-mutant non–small cell lung cancer. Sci. Adv. 5, 6455 (2019).
Henry, R. A., Kuo, Y. M. & Andrews, A. J. Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry 52, 5746–5759 (2013).
Kaspe, L. et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol. Cell. Biol. 26, 789 (2006).
Ansari, M. S. Z. Pharmacological targeting of CBP/p300 drives a redox/autophagy axis leading to senescence-induced growth arrest in non-small cell lung cancer cells. Cancer Gene Ther. 30, 124–136 (2023).
Guo, W. et al. Transcriptional coactivator CBP upregulates hTERT expression and tumor growth and predicts poor prognosis in human lung cancers. Oncotarget 5, 9349 (2014).
Zhang, T. et al. Super-enhancer hijacking LINC01977 promotes malignancy of early-stage lung adenocarcinoma addicted to the canonical TGF-β/SMAD3 pathway. J. Hematol. Oncol. 15, 114 (2022).
Wang, Z. et al. CREB stimulates GPX4 transcription to inhibit ferroptosis in lung adenocarcinoma. Oncol. Rep. 45, 1–12 (2021).
Xu, L., Geman, D. & Winslow, R. L. Large-scale integration of cancer microarray data identifies a robust common cancer signature. BMC Bioinform. 8, 1–13 (2007).
Zhang, T. Inhibition of PC 4 radiosensitizes non‐small cell lung cancer by transcriptionally suppressing XLF. Cancer Med. 7, 1326–1337 (2018).
Tao, S., Wang, X., Wu, L., Shen, C. & Tan, Q. High expression of PC4 is associate with lymphatic metastasis and predicts poor prognosis in lung adenocarcinoma probably via CCR7/VEGF- C/VEGFR-3 cascade. Preprint at https://assets.researchsquare.com/files/rs-761143/v1/0c09a74d-48e6-4477-9f00-70ff384004e9.pdf?c=1631887729 (2021).
Zhang, Y. et al. Efficacy of a small molecule inhibitor of the transcriptional cofactor PC4 in prevention and treatment of non-small cell lung cancer. PLoS ONE 15, 0230670 (2020).
Du, F. et al. MRTF-A-NF-κB/p65 axis-mediated PDL1 transcription and expression contributes to immune evasion of non-small-cell lung cancer via TGF-β. Exp. Mol. Med. 53, 1366–1378 (2021).
Zhang, K., Zhou, Y., Feng, G. & Zeng, F. MRTF-A regulates the proliferation and migration of non-small cell lung cancer cells of A549 through HOTAIR. Chin. Zhongguo Fei Ai Za Zhi 22, 82–89 (2021).
Kawakita, D. et al. Trends in the incidence of head and neck cancer by subsite between 1993 and 2015 in Japan. Cancer Med. 11, 1553–1560 (2022).
Mo, J. S., Park, H. W. & Guan, K. L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 15, 642–656 (2014).
Faraji, F., Ramirez, S. I., Anguiano Quiroz, P. Y., Mendez-Molina, A. N. & Gutkind, J. S. Genomic hippo pathway alterations and persistent YAP/TAZ Activation: new hallmarks in head and neck cancer. Cells 11, 1370 (2022).
Segrelles, C., Paramio, J. M. & Lorz, C. The transcriptional co-activator YAP: a new player in head and neck cancer. Oral. Oncol. 86, 25–32 (2018).
Li, J. et al. The Hippo effector TAZ promotes cancer stemness by transcriptional activation of SOX2 in head neck squamous cell carcinoma. Cell Death Dis. 10, 603 (2019).
Steven, A. et al. What turns CREB on? And off? And why does it matter? Cell. Mol. Life Sci. 77, 4049–4067 (2020).
Carper, M. B. et al. Activation of the CREB coactivator CRTC2 by aberrant mitogen Signaling promotes oncogenic functions in HPV16 positive head and neck cancer. Neoplasia 29, 100799 (2022).
Nakano, S. et al. Salivary mucoepidermoid carcinoma: histological variants, grading systems, CRTC1/3‐MAML2 fusions, and clinicopathological features. Histopathology 80, 729–735 (2022).
Birkeland, A. C. et al. Correlation of Crtc1/3-Maml2 fusion status, grade and survival in mucoepidermoid carcinoma. Oral. Oncol. 68, 5–8 (2017).
Wang, H., Xue, W. & Jiang, X. Overexpression of TRIM24 stimulates proliferation and glucose metabolism of head and neck squamous cell carcinoma. BioMed. Res. Int. 2018, 6142843 (2018).
Cui, Z. et al. TRIM24 overexpression is common in locally advanced head and neck squamous cell carcinoma and correlates with aggressive malignant phenotypes. PLoS ONE 8, 63887 (2013).
Chang, Y. C. et al. Glucose transporter 4 promotes head and neck squamous cell carcinoma metastasis through the TRIM24-DDX58 axis. J. Hematol. Oncol. 10, 1–12 (2017).
Dey, A., Chitsaz, F., Abbasi, A., Misteli, T. & Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl Acad. Sci. USA 100, 8758–8763 (2003).
Wu, Y. et al. Therapeutic targeting of BRD4 in head neck squamous cell carcinoma. Theranostics 9, 1777 (2019).
Yamamoto, T. et al. BRD4 promotes metastatic potential in oral squamous cell carcinoma through the epigenetic regulation of the MMP2 gene. Br. J. Cancer 123, 580–590 (2020).
Fisher, M. L. et al. BRD4 regulates transcription factor ΔNp63α to drive a cancer stem cell phenotype in squamous cell carcinomas BRD4 control of ΔNp63α. Cancer Res. 81, 6246–6258 (2021).
Gruber, T. A. & Downing, J. R. The biology of pediatric acute megakaryoblastic leukemia. Blood 126, 943–949 (2015).
Yokoyama, A. Leukemogenesis via aberrant self‐renewal by the MLL/AEP‐mediated transcriptional activation system. Cancer Sci. 112, 3935–3944 (2021).
Takao, S. et al. Convergent organization of aberrant MYB complex controls oncogenic gene expression in acute myeloid leukemia. eLife 10, 65905 (2021).
Mattes, K. et al. Transcriptional regulators CITED2 and PU.1 cooperate in maintaining hematopoietic stem cells. Exp. Hematol. 73, 38–49 (2019).
Korthuis, P. M. CITED2-mediated human hematopoietic stem cell maintenance is critical for acute myeloid leukemia. Leukemia 29, 625–635 (2015).
Peter, B. et al. BRD4 degradation blocks expression of MYC and multiple forms of stem cell resistance in Ph+ chronic myeloid leukemia. Am. J. Hematol. 97, 1215–1225 (2022).
Gilan, O. et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat. Struct. Mol. Biol. 23, 673–681 (2016).
Lin, S. et al. DDX5 is a positive regulator of oncogenic NOTCH1 signaling in T cell acute lymphoblastic leukemia. Oncogene 32, 4845–4853 (2013).
Shallak, M. et al. The endogenous HBZ interactome in ATL leukemic cells reveals an unprecedented complexity of host interacting partners involved in RNA splicing. Front. Immunol. 13, 939863 (2022).
Camps, J. et al. Integrative genomics reveals mechanisms of copy number alterations responsible for transcriptional deregulation in colorectal cancer. Genes Chromosomes Cancer 48, 1002–1017 (2009).
Sugai, T. et al. A genome‐wide study of the relationship between chromosomal abnormalities and gene expression in colorectal tumors. Genes Chromosomes Cancer 60, 250–262 (2021).
Tang, J. et al. DEAD-box helicase 27 promotes colorectal cancer growth and metastasis and predicts poor survival in CRC patients. Oncogene 37, 3006–3021 (2018).
Ishihama, K. et al. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J. Clin. Pathol. 60, 1205–1210 (2007).
Xu, Z. et al. CCL7 and TGF-β secreted by MSCs play opposite roles in regulating CRC metastasis in a KLF5/CXCL5-dependent manner. Mol. Ther. 30, 2327–2341 (2022).
Rao, R. C. & Dou, Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 15, 334–346 (2015).
Liao, C. et al. Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer. Int. J. Biol. Markers 37, 149–157 (2022).
Fang, Y. et al. KMT2A histone methyltransferase contributes to colorectal cancer development by promoting cathepsin Z transcriptional activation. Cancer Med. 8, 3544–3552 (2019).
Devaiah, B. N., Gegonne, A. & Singer, D. S. Bromodomain 4: a cellular Swiss army knife. J. Leukoc. Biol. 100, 679–686 (2016).
Zhang, P. et al. BRD4 inhibitor AZD5153 suppresses the proliferation of colorectal cancer cells and sensitizes the anticancer effect of PARP inhibitor. Int. J. Biol. Sci. 15, 942 (2019).
Wang, W. et al. Stromal induction of BRD4 phosphorylation results in chromatin remodeling and BET inhibitor resistance in colorectal cancer. Nat. Commun. 12, 4441 (2021).
Sooraj, D. et al. MED12 and BRD4 cooperate to sustain cancer growth upon loss of mediator kinase. Mol. Cell 82, 123–139 (2022).
Mouillet-Richard, S. & Laurent-Puig, P. YAP/TAZ signalling in colorectal cancer: lessons from consensus molecular subtypes. Cancers 12, 3160 (2020).
Rosenbluh, J. et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473 (2012).
Song, J. et al. The YAP–TEAD4 complex promotes tumor lymphangiogenesis by transcriptionally upregulating CCBE1 in colorectal cancer. J. Biol. Chem. 299, 103012 (2023).
Rumgay, H. et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 77, 1598–1606 (2022).
Alqahtani, A. et al. Hepatocellular carcinoma: molecular mechanisms and targeted therapies. Medicina 55, 526 (2019).
Näär, A. M. et al. Composite co-activator ARC mediates chromatin-directed transcriptional activation. Nature 398, 828–832 (1999).
Hyder, U. et al. The ARF tumor suppressor targets PPM1G/PP2Cγ to counteract NF-κB transcription tuning cell survival and the inflammatory response. Proc. Natl Acad. Sci. USA 117, 32594–32605 (2020).
Chen, D. et al. PPM1G promotes the progression of hepatocellular carcinoma via phosphorylation regulation of alternative splicing protein SRSF3. Cell Death Dis. 12, 722 (2021).
Gao, R. et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol. Med. 13, 14351 (2021).
Bisso, A. et al. Cooperation between MYC and β‐Catenin in liver tumorigenesis requires Yap/Taz. Hepatology 72, 1430–1443 (2020).
Wang, H. et al. TAZ is indispensable for c-MYC-induced hepatocarcinogenesis. J. Hepatol. 76, 123–134 (2022).
Wang, H. et al. Distinct and overlapping roles of hippo effectors YAP and TAZ during human and mouse hepatocarcinogenesis. Cell. Mol. Gastroenterol. Hepatol. 11, 1095–1117 (2021).
Cai, L. Y. et al. Targeting p300/CBP attenuates hepatocellular carcinoma progression through epigenetic regulation of metabolism. Cancer Res. 81, 860–872 (2021).
Carbajo-Pescador, S. et al. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br. J. Cancer 109, 83–91 (2013).
Huang, C., Liu, W., Zhao, X., Zhao, L. & Wang, F. Downregulation of HBx restrains proliferation, migration, and invasion of HepG2 cells. Anal. Cell. Pathol. 2021, 1–11 (2021).
Bouchard, M. J. & Schneider, R. J. The enigmatic X gene of hepatitis B virus. J. Virol. 78, 12725–12734 (2004).
Cougot, D. et al. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J. Biol. Chem. 282, 4277–4287 (2007).
Wang, Y. et al. HBx-induced HSPA8 stimulates HBV replication and suppresses ferroptosis to support liver cancer progression. Cancer Res. 83, 1048–1061 (2023).
Gao, W. et al. HBx protein contributes to liver carcinogenesis by H3K4me3 modification through stabilizing WD repeat domain 5 protein. Hepatology 71, 1678–1695 (2020).
Lin, X. et al. Silencing MYH9 blocks HBx-induced GSK3β ubiquitination and degradation to inhibit tumor stemness in hepatocellular carcinoma. Signal Transduct. Target. Ther. 5, 13 (2020).
Lei, Y. et al. HBx induces hepatocellular carcinogenesis through ARRB1-mediated autophagy to drive the G1/S cycle. Autophagy 17, 4423–4441 (2021).
Tiffon, C. et al. TAZ controls Helicobacter pylori-induced epithelial–mesenchymal transition and cancer stem cell-like invasive and tumorigenic properties. Cells 9, 1462 (2020).
Tsutsumi, R. et al. YAP and TAZ, Hippo signaling targets, act as a rheostat for nuclear SHP2 function. Dev. Cell 26, 658–665 (2013).
Lee, Y. et al. Common and unique transcription signatures of YAP and TAZ in gastric cancer cells. Cancers 12, 3667 (2020).
He, S. et al. MicroRNA-511 inhibits cellular proliferation and invasion in colorectal cancer by directly targeting hepatoma-derived growth factor. Oncol. Res. 26, 1355 (2018).
Fang, Z. et al. TRIM24 promotes the aggression of gastric cancer via the Wnt/β-catenin signaling pathway. Oncol. Lett. 13, 1797–1806 (2017).
Dawson, M. A., Kouzarides, T. & Huntly, B. J. Targeting epigenetic readers in cancer. N. Engl. J. Med. 367, 647–657 (2012).
Zhang, M. et al. Overexpression of BRD4 in gastric cancer and its clinical significance as a novel therapeutic target. Curr. Cancer Drug Targets https://doi.org/10.2174/1568009623666230606164030 (2023).
Dong, X., Hu, X., Chen, J., Hu, D. & Chen, L. F. BRD4 regulates cellular senescence in gastric cancer cells via E2F/miR-106b/p21 axis. Cell Death Dis. 9, 203 (2018).
Qin, Z. Y. et al. BRD4 promotes gastric cancer progression and metastasis through acetylation-dependent stabilization of snail. Cancer Res. 79, 4869–4881 (2019).
Fuller-Pace, F. V. The DEAD box proteins DDX5 (p68) and DDX17 (p72): multi-tasking transcriptional regulators. Biochim. Biophys. Acta 1829, 756–763 (2013).
Wang, J., Han, C., Wang, J. & Peng, Q. RNA helicase DDX5-induced circPHF14 promotes gastric cancer cell progression. Aging 15, 2525 (2023).
Du, C. et al. DDX5 promotes gastric cancer cell proliferation in vitro and in vivo through mTOR signaling pathway. Sci. Rep. 7, 42876 (2017).
Yin, L. et al. The MRTF-A/miR-155/SOX1 pathway mediates gastric cancer migration and invasion. Cancer Cell Int. 20, 1–13 (2020).
Wang, Y. et al. MICAL2 facilitates gastric cancer cell migration via MRTF-A-mediated CDC42 activation. Front. Mol. Biosci. 8, 568868 (2021).
Gu, Y. et al. Altered LKB1/CREB-regulated transcription co-activator (CRTC) signaling axis promotes esophageal cancer cell migration and invasion. Oncogene 31, 469–479 (2012).
Hu, T., Zhang, Y., Yang, T., He, Q. & Zhao, M. LYPD3, a new biomarker and therapeutic target for acute myelogenous leukemia. Front. Genet. 13, 795–820 (2022).
Compton, S. E. et al. LKB1 controls inflammatory potential through CRTC2-dependent histone acetylation. Mol. Cell 83, 1872–1886 (2023).
Denechaud, P. D., Fajas, L. & Giralt, A. E2F1, a novel regulator of metabolism. Front. Endocrinol. 8, 311 (2017).
Yeo, S. Y. et al. ZNF282 (Zinc finger protein 282), a novel E2F1 co-activator, promotes esophageal squamous cell carcinoma. Oncotarget 5, 12260 (2014).
Rawla, P., Sunkara, T. & Gaduputi, V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J. Oncol. 10, 10 (2019).
Li, F. et al. piRNA-independent function of PIWIL1 as a co-activator for anaphase promoting complex/cyclosome to drive pancreatic cancer metastasis. Nat. Cell Biol. 22, 425–43 (2020).
Li, W. et al. The prognosis value of PIWIL1 and PIWIL2 expression in pancreatic cancer. J. Clin. Med. 8, 1275 (2019).
Manegold, P. et al. Differentiation therapy targeting the β-catenin/CBP interaction in pancreatic cancer. Cancers 10, 95 (2018).
Principe, D. R. et al. XP-524 is a dual-BET/EP300 inhibitor that represses oncogenic KRAS and potentiates immune checkpoint inhibition in pancreatic cancer. Proc. Natl Acad. Sci. USA 119, 2116764119 (2022).
Liu, M. et al. SETD8 potentiates constitutive ERK1/2 activation via epigenetically silencing DUSP10 expression in pancreatic cancer. Cancer Lett. 499, 265–278 (2021).
Lu, Z. et al. SETD8 inhibits ferroptosis in pancreatic cancer by inhibiting the expression of RRAD. Cancer Cell Int. 23, 1–16 (2023).
Zhou, Q. et al. YAP1 is an independent prognostic marker in pancreatic cancer and associated with extracellular matrix remodeling. J. Transl. Med. 18, 1–10 (2020).
Liu, M. et al. Zinc-dependent regulation of ZEB1 and YAP1 coactivation promotes epithelial-mesenchymal transition plasticity and metastasis in pancreatic cancer. Gastroenterology 160, 1771–1783 (2021).
Jia, Y. et al. Crosstalk between hypoxia-sensing ULK1/2 and YAP-driven glycolysis fuels pancreatic ductal adenocarcinoma development. Int. J. Biol. Sci. 17, 2772 (2021).
Jiao, F. et al. Caveolin-2 is regulated by BRD4 and contributes to cell growth in pancreatic cancer. Cancer Cell Int. 20, 1–14 (2020).
Yamazaki, M. et al. YAP/BRD4‐controlled ROR1 promotes tumor‐initiating cells and hyperproliferation in pancreatic cancer. EMBO J. 42, 112614 (2023).
Dillekås, H., Rogers, M. S. & Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 8, 5574–5576 (2019).
Islam, F., Gopalan, V., Smith, R. A. & Lam, A. K. Y. Translational potential of cancer stem cells: a review of the detection of cancer stem cells and their roles in cancer recurrence and cancer treatment. Exp. Cell Res. 335, 135–147 (2015).
Jewer, M. et al. Translational control of breast cancer plasticity. Nat. Commun. 11, 2498 (2020).
Walcher, L. et al. Cancer stem cells—origins and biomarkers: perspectives for targeted personalized therapies. Front. Immunol. 11, 1280 (2020).
Yang, L. et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 5, 8 (2020).
Pietras, A. et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 14, 357–369 (2014).
He, Z. et al. A novel regulatory role of CITED2 in non-small cell lung cancer (NSCLC) stem cells and in modulating the effect of HDACi treatment on NSCLC. Int. J. Radiat. Oncol. Biol. Phys. 87, 662 (2013).
Chan, L. H. et al. PRMT6 regulates RAS/RAF binding and MEK/ERK-mediated cancer stemness activities in hepatocellular carcinoma through CRAF methylation. Cell Rep. 25, 690–701 (2018).
Zhu, J. et al. A pan-cancer study of KMT2 family as therapeutic targets in cancer. J. Oncol. 2022, 3982226 (2022).
Grinat, J. et al. The epigenetic regulator Mll1 is required for Wnt-driven intestinal tumorigenesis and cancer stemness. Nat. Commun. 11, 6422 (2020).
Chen, J. Y. et al. Lysine demethylase 2A promotes stemness and angiogenesis of breast cancer by upregulating Jagged1. Oncotarget 7, 27689 (2016).
Shait Mohammed, M. R. et al. The histone H3K27me3 demethylases KDM6A/B resist anoikis and transcriptionally regulate stemness-related genes. Front. Cell Dev. Biol. 10, 780176 (2022).
Li, S. M. et al. JMJD3 promotes esophageal squamous cell carcinoma pathogenesis through epigenetic regulation of MYC. Signal Transduct. Target. Ther. 5, 165 (2020).
Rohira, A. D. et al. Targeting SRC coactivators blocks the tumor-initiating capacity of cancer stem-like cells. Cancer Res. 77, 4293–4304 (2017).
Jaworska, A. M., Wlodarczyk, N. A., Mackiewicz, A. & Czerwinska, P. The role of TRIM family proteins in the regulation of cancer stem cell self-renewal. Stem Cells 38, 165–173 (2020).
Sun, J. et al. Loss of TRIM29 suppresses cancer stem cell-like characteristics of PDACs via accelerating ISG15 degradation. Oncogene 39, 546–559 (2020).
Li, Y. W. et al. Apigenin suppresses the stem cell-like properties of triple-negative breast cancer cells by inhibiting YAP/TAZ activity. Cell Death Dis. 4, 105 (2018).
Li, Q. & Kang, C. Mechanisms of action for small molecules revealed by structural biology in drug discovery. Int. J. Mol. Sci. 21, 5262 (2020).
Lucero, B., Francisco, K. R., Liu, L. J., Caffrey, C. R. & Ballatore, C. Protein–protein interactions: developing small-molecule inhibitors/stabilizers through covalent strategies. Trends Pharmacol. Sci. 44, 474–488 (2023).
Sun, Y. et al. Pharmacological blockade of TEAD–YAP reveals its therapeutic limitation in cancer cells. Nat. Commun. 13, 6744 (2022).
Lin, A. et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 11, 8412 (2019).
Housman, G. et al. Drug resistance in cancer: an overview. Cancers 6, 1769–1792 (2014).
Domostegui, A., Nieto-Barrado, L., Perez-Lopez, C. & Mayor-Ruiz, C. Chasing molecular glue degraders: screening approaches. Chem. Soc. Rev. 51, 5498–5517 (2022).
Brownsey, D. K. et al. Identification of ligand linkage vectors for the development of p300/CBP degraders. RSC Med. Chem. 13, 726–730 (2022).
Shergalis, A. G. CRISPR screen reveals BRD2/4 molecular glue-like degrader via recruitment of DCAF16. ACS Chem. Biol. 18, 331–339 (2023).
Ling, X. et al. FL118, acting as a ‘molecular glue degrader’, binds to, dephosphorylates and degrades the oncoprotein DDX5 (p68) to control c‐Myc, survivin and mutant Kras against colorectal and pancreatic cancer with high efficacy. Clin. Transl. Med. 12, 881 (2022).
Li, Z. et al. Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature 575, 203–209 (2019).
Han, X., Wei, W. & Sun, Y. PROTAC degraders with ligands recruiting MDM2 E3 ubiquitin ligase: an updated perspective. Acta Mater. Med. 1, 244 (2022).
Henley, M. J. & Koehler, A. N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 20, 669–688 (2021).
Liu, J. et al. TF-PROTACs enable targeted degradation of transcription factors. J. Am. Chem. Soc. 143, 8902–8910 (2021).
Jia, X. & Han, X. Targeting androgen receptor degradation with PROTACs from bench to bedside. Biomed. Pharmacother. 158, 114112 (2023).
Zhang, X. et al. PROTAC degrader of estrogen receptor α targeting DNA-binding domain in breast cancer. ACS Pharmacol. Transl. Sci. 5, 1109–1118 (2022).
Li, X. et al. c-Myc-targeting PROTAC based on a TNA-DNA bivalent binder for combination therapy of triple-negative breast cancer. J. Am. Chem. Soc. 145, 9334–9342 (2023).
Kong, L. et al. Selective degradation of the p53‐R175H oncogenic hotspot mutant by an RNA aptamer‐based PROTAC. Clin. Transl. Med. 13, 1191 (2023).
Jin, J. et al. Small-molecule PROTAC mediates targeted protein degradation to treat STAT3-dependent epithelial cancer. JCI Insight 7, 160606 (2022).
Kaneshige, A. et al. Discovery of a potent and selective STAT5 PROTAC degrader with strong antitumor activity in vivo in acute myeloid leukemia. J. Med. Chem. 66, 2717–2743 (2023).
Yang, J. et al. VHL-recruiting PROTAC attenuates renal fibrosis and preserves renal function via simultaneous degradation of Smad3 and stabilization of HIF-2α. Cell Biosci. 12, 203 (2022).
Thomas, J. E. et al. Discovery of exceptionally potent, selective, and efficacious PROTAC degraders of CBP and p300 proteins. J. Med. Chem. 66, 8178–8199 (2023).
Huang, J. H. et al. Bioinspired PROTAC-induced macrophage fate determination alleviates atherosclerosis. Acta Pharmacol. Sin. https://doi.org/10.1038/s41401-023-01088-5 (2023)
Lee, Y. et al. Targeted degradation of transcription coactivator SRC‐1 through the N‐degron pathway. Angew. Chem. Int. Ed. Engl. 59, 17548–17555 (2022).
Otto, C. et al. Targeting bromodomain-containing protein 4 (BRD4) inhibits MYC expression in colorectal cancer cells. Neoplasia 21, 1110–1120 (2019).
Pushpakom, S. et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58 (2019).
Drugs@FDA Data Files. https://www.fda.gov/drugs/drug-approvals-and-databases/drugsfda-data-files/ (2023).
Kanki, H. et al. CREB coactivator CRTC2 plays a crucial role in endothelial function. J. Neurosci. 40, 9533–9546 (2020).
Ianculescu, I., Wu, D. Y., Siegmund, K. D. & Stallcup, M. R. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J. Biol. Chem. 287, 4000–4013 (2012).
Gerritsen, M. E. et al. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl Acad. Sci. USA 94, 2927–2932 (1997).
Nestler, E. J. Transcriptional mechanisms of drug addiction. Clin. Psychopharmacol. Neurosci. 10, 136 (2012).
Chen, H. et al. Exosomes, a new star for targeted delivery. Front. Cell Dev. Biol. 9, 751079 (2021).
Liu, P., Chen, G. & Zhang, J. A review of liposomes as a drug delivery system: current status of approved products, regulatory environments, and future perspectives. Molecules 27, 1372 (2022).
Pala, R., Anju, V. T., Dyavaiah, M., Busi, S. & Nauli, S. M. Nanoparticle-mediated drug delivery for the treatment of cardiovascular diseases. Int. J. Nanomed. 15, 3741–3769 (2020).
Roelfsema, J. H. et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 76, 572–580 (2005).
Jiang, H. et al. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 23, 543–551 (2006).
Gang, E. J. et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 33, 2169–2178 (2014).
Jia, D. et al. Crebbp loss drives small cell lung cancer and increases sensitivity to HDAC inhibition. Cancer Discov. 8, 1422–1437 (2018).
Rühlmann, F. et al. The prognostic capacities of CBP and p300 in locally advanced rectal cancer. World J. Surg. Oncol. 17, 1–8 (2019).
Peck, B. et al. 3D functional genomics screens identify CREBBP as a targetable driver in aggressive triple-negative breast cancer CREBBP and CDK4/6 inhibition. Cancer Res. 81, 847–859 (2021).
Ogiwara, H. et al. Targeting p300 addiction in CBP-deficient cancers causes synthetic lethality by apoptotic cell death due to abrogation of MYC expression targeting p300 addiction in CBP-deficient cancers. Cancer Discov. 6, 430–445 (2016).
Garcia‐Pelaez, J., Barbosa‐Matos, R., Gullo, I., Carneiro, F. & Oliveira, C. Histological and mutational profile of diffuse gastric cancer: current knowledge and future challenges. Mol. Oncol. 15, 2841–2867 (2021).
Cheng, G. et al. Loss of p300 accelerates MDS-associated leukemogenesis. Leukemia 31, 1382–1390 (2017).
Chen, Z., Chen, C., Li, L., Zhang, T. & Wang, X. Pan-cancer analysis reveals that E1A binding protein p300 mutations increase genome instability and antitumor immunity. Front. Cell Dev. Biol. 9, 729927 (2021).
Gayther, S. A. et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 24, 300–303 (2000).
Bi, Y. et al. EP300 as an oncogene correlates with poor prognosis in esophageal squamous carcinoma. J. Cancer 10, 5413 (2019).
Tlemsani, C. et al. SETD2 and DNMT3A screen in the Sotos-like syndrome French cohort. J. Med. Genet. 53, 743–751 (2016).
Steiner, C. E. & Marques, A. P. Growth deficiency, mental retardation and unusual facies. Clin. Dysmorphol. 9, 155–156 (2000).
Khodaeian, M. et al. Kabuki syndrome: identification of two novel variants in KMT2D and KDM6A. Mol. Syndromol. 12, 118–126 (2021).
Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576 (2015).
Miao, C., Zhang, X., Ma, T. & Zhu, H. Investigating the potential relationship between KMT2C/KMT2D mutations and immune checkpoint inhibitor (ICI) in colorectal cancer (CRC). Ann. Oncol. 31, 451 (2020).
Ardeshir-Larijani, F. et al. KMT2D mutation is associated with poor prognosis in non–small-cell lung cancer. Clin. Lung Cancer 19, 489–501 (2018).
Goldsworthy, M. Mutations in Mll2, an H3K4 methyltransferase, result in insulin resistance and impaired glucose tolerance in mice. PLoS ONE 8, 61870 (2013).
Damásio, J. et al. Novel KMT2B mutation causes cerebellar ataxia: expanding the clinical phenotype. Clin. Genet. 100, 743–747 (2021).
Mun, J. K. et al. Successful pallidal stimulation in a patient with KMT2B-related dystonia. J. Mov. Disord. 13, 154 (2020).
Wong, W. H., Junck, L., Druley, T. E. & Gutmann, D. H. NF1 glioblastoma clonal profiling reveals KMT2B mutations as potential somatic oncogenic events. Neurology 93, 1067–1069 (2019).
Xue, C. et al. Phase I study of LZM005 in patients with HER2-positive metastatic breast cancer. NPJ Breast Cancer 8, 132 (2022).
Siano, M. A. et al. De novo mutation in KMT2C manifesting as Kleefstra Syndrome 2: Case report and literature review. Pediatr. Rep. 14, 131–139 (2022).
Fagan, R. J. & Dingwall, A. K. COMPASS ascending: emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. 458, 56–65 (2019).
Simigdala, N. et al. Loss of Kmt2c in vivo leads to EMT, mitochondrial dysfunction and improved response to lapatinib in breast cancer. Cell. Mol. Life Sci. 80, 100 (2023).
Wang, X. X. et al. Somatic mutations of the mixed-lineage leukemia 3 (MLL3) gene in primary breast cancers. Pathol. Oncol. Res. 17, 429–433 (2011).
Jorge, S. D. et al. Integrative modeling, molecular mechanics, and molecular dynamics evaluation of genomics variants in KMT2C (MLL3), a gene involved in Kleefstra Syndrome Type 2. FASEB J. 36, abstr (2022).
Senapati, D., Kumari, S. & Heemers, H. V. Androgen receptor co-regulation in prostate cancer. Asian J. Urol. 7, 219–232 (2020).
Taylor, B. S. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010).
Dasgupta, S., Lonard, D. M. & O’Malley, B. W. Nuclear receptor coactivators: master regulators of human health and disease. Annu. Rev. Med. 65, 279–292 (2014).
Shinriki, S. & Matsui, H. Unique role of DDX41, a DEAD-box type RNA helicase, in hematopoiesis and leukemogenesis. Front. Oncol. 12, 992340 (2022).
Churpek, J. E. & Smith-Simmer, K. in Gene Reviews (eds Adam, M. P. et al.) (University of Washington, Seattle, 2021).
Choi, E. J. et al. DDX41 mutation in patients with idiopathic cytopenia of undetermined significance, myelodysplastic syndrome, and acute myeloid leukemia. Blood 134, 3002 (2019).
Stransky, N. et al. The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160 (2011).
Pugh, T. J. et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106–110 (2012).
Wang, L. et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 365, 2497–2506 (2011).
Unal, A., Dursun, A., Emre, U., Tascilar, N. F. & Ankarali, H. Evaluation of common mutations in the Mediterranean fever gene in Multiple Sclerosis patients: is it a susceptibility gene? J. Neurol. Sci. 294, 38–42 (2010).
Arra, M. et al. The M694V variant of the familial Mediterranean fever gene is associated with sporadic early-onset Alzheimer’s disease in an Italian population sample. Dement. Geriatr. Cogn. Disord. 23, 55–59 (2006).
Hämäläinen, R. H. et al. Novel mutations in the TRIM37 gene in Mulibrey Nanism. Hum. Mutat. 23, 522–522 (2004).
Xu, B. et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 43, 864–868 (2011).
Cossée, M. et al. Use of SNP array analysis to identify a novel TRIM32 mutation in limb-girdle muscular dystrophy type 2H. Neuromuscul. Disord. 19, 255–260 (2009).
Vianna, C. R. et al. Hypomorphic mutation of PGC-1β causes mitochondrial dysfunction and liver insulin resistance. Cell Metab. 4, 453–464 (2006).
Selicorni, A., Mariani, M., Lettieri, A. & Massa, V. Cornelia de Lange syndrome: from a disease to a broader spectrum. Genes 12, 1075 (2021).
Gorvin, C. M. et al. Mice with a Brd4 mutation represent a new model of Nephrocalcinosis. J. Bone Miner. Res. 34, 1324–1335 (2019).
Dianatpour, S., Khatami, M., Heidari, M. M. & Hadadzadeh, M. Novel point mutations of CITED2 gene are associated with non-familial congenital heart disease (CHD) in sporadic pediatric patients. Appl. Biochem. Biotechnol. 190, 896–906 (2020).
Hughes, D., Gordon-Weeks, A. & Silva, M. Hereditary pancreatitis–a review of current concepts and management. OBM Hepatol. Gastroenterol. 3, 1–25 (2019).
Yun, M. R. et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK‐rearranged lung cancer. EMBO Mol. Med. 11, 10581 (2019).
Calvo, F. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15, 637–646 (2013).
Sorrentino, G. et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 16, 357–366 (2014).
Chan, P. et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 12, 282–289 (2016).
Pobbati, A. V. et al. Targeting the central pocket in human transcription factor TEAD as a potential cancer therapeutic strategy. Structure 23, 2076–2086 (2015).
Bum-Erdene, K. et al. Small-molecule covalent modification of conserved cysteine leads to allosteric inhibition of the TEAD⋅ Yap protein-protein interaction. Cell Chem. Biol. 26, 378–389 (2019).
Wu, X. The General Hospital Corporation. Tead transcription factor autopalmitoylation inhibitors (2017).
Kaneda, A. et al. The novel potent TEAD inhibitor, K-975, inhibits YAP1/TAZ-TEAD protein-protein interactions and exerts an anti-tumor effect on malignant pleural mesothelioma. Am. J. Cancer Res. 10, 4399 (2020).
Pobbati, A. V. & Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 10, 3622 (2020).
Heinrich, T. et al. Optimization of TEAD P-site binding fragment hit into in vivo active lead MSC-4106. J. Med. Chem. 65, 9206–9229 (2022).
Hsu, P. C. et al. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 7, 51922 (2016).
Quan, Y., Li, Z., Zhu, K. & Liang, J. Transcatheter arterial chemoembolization combined with Hippo/YAP inhibition significantly improve the survival of rats with transplanted hepatocellular carcinoma. Lipids Health Dis. 20, 1–10 (2021).
Giraud, J. et al. Verteporfin targeting YAP1/TAZ‐TEAD transcriptional activity inhibits the tumorigenic properties of gastric cancer stem cells. Int. J. Cancer 146, 2255–2267 (2020).
Liu, K., Du, S., Gao, P. & Zheng, J. Verteporfin suppresses the proliferation, epithelial-mesenchymal transition and stemness of head and neck squamous carcinoma cells via inhibiting YAP1. J. Cancer 10, 4196 (2019).
Shin, Y. Y. et al. Melatonin and verteporfin synergistically suppress the growth and stemness of head and neck squamous cell carcinoma through the regulation of mitochondrial dynamics. J. Pineal Res. 72, 12779 (2022).
Wei, C. & Li, X. Verteporfin inhibits cell proliferation and induces apoptosis in different subtypes of breast cancer cell lines without light activation. BMC Cancer 20, 1–8 (2020).
El-Sahli, S. et al. A triple-drug nanotherapy to target breast cancer cells, cancer stem cells, and tumor vasculature. Cell Death Dis. 12, 8 (2021).
Oku, Y. et al. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 5, 542–549 (2015).
Hagenbeek, T. J. et al. An allosteric pan-TEAD inhibitor blocks oncogenic YAP/TAZ signaling and overcomes KRAS G12C inhibitor resistance. Nat. Cancer 4, 812–828 (2023).
Yuan, J. Q., Ding, N. H. & Xiao, Z. The Hippo transducer YAP/TAZ as a biomarker of therapeutic response and prognosis in trastuzumab-based neoadjuvant therapy treated HER2-positive breast cancer patients. Front. Pharmacol. 11, 537265 (2020).
Neoadjuvant Zoledronate and Atorvastatin in Triple Negative Breast Cancer (YAPPETIZER) https://classic.clinicaltrials.gov/ct2/show/NCT03358017.
Testing a New Anti-cancer Drug Combination, Entinostat and GSK525762C, for Advanced and Refractory Solid Tumors and Lymphomas. https://classic.clinicaltrials.gov/ct2/show/NCT03925428.
Tang, T. T. et al. Small molecule inhibitors of TEAD auto-palmitoylation selectively inhibit proliferation and tumor growth of NF2-deficient mesothelioma. Mol. Cancer Ther. 20, 986–998 (2021).
Kim, M. H. et al. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 35, 462–478 (2016).
da Motta, L. L. et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 36, 122–132 (2017).
Wang, H. et al. BET inhibitor JQ1 enhances anti-tumor immunity and synergizes with PD-1 blockade in CRC. J. Cancer 13, 2126 (2022).
Chen, H. et al. BET inhibitors target the SCLC-N subtype of small-cell lung cancer by blocking NEUROD1 transactivation. Mol. Cancer Res. 21, 91–101 (2023).
Choi, H. I. et al. BET inhibitor suppresses migration of human hepatocellular carcinoma by inhibiting SMARCA4. Sci. Rep. 11, 11799 (2021).
Song, S. et al. Targeting Hippo coactivator YAP1 through BET bromodomain inhibition in esophageal adenocarcinoma. Mol. Oncol. 14, 1410–1426 (2020).
Zhou, S. et al. BET protein inhibitor JQ1 downregulates chromatin accessibility and suppresses metastasis of gastric cancer via inactivating RUNX2/NID1 signaling. Oncogenesis 9, 33 (2020).
Ni, M. et al. BRD4 inhibition sensitizes cervical cancer to radiotherapy by attenuating DNA repair. Oncogene 40, 2711–2724 (2021).
Chen, N. et al. YAP1 maintains active chromatin state in head and neck squamous cell carcinomas that promotes tumorigenesis through cooperation with BRD4. Cell Rep. 39, 110970 (2022).
Baek, M. et al. X Comprehensive transcriptome profiling of BET inhibitor-treated HepG2 cells. PLoS ONE 17, 0266966 (2020).
Chen, X. et al. Discovery of 2-((2-methylbenzyl) thio)-6-oxo-4-(3, 4, 5-trimethoxyphenyl)-1, 6-dihydropyrimidine-5-carbonitrile as a novel and effective bromodomain and extra-terminal (BET) inhibitor for the treatment of sepsis. Eur. J. Med. Chem. 238, 114423 (2022).
Hilton-Proctor, J. P. et al. Substituted 1-methyl-4-phenylpyrrolidin-2-ones–Fragment-based design of N-methylpyrrolidone-derived bromodomain inhibitors. Eur. J. Med. Chem. 191, 112120 (2020).
Hill, M. D. et al. Development of BET inhibitors as potential treatments for cancer: optimization of pharmacokinetic properties. ACS Med. Chem. Lett. 13, 1165–1171 (2022).
Wyce, A. et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS ONE 8, 72967 (2013).
Dong, R. et al. Design, synthesis and anticancer evaluation of 3-methyl-1H-indazole derivatives as novel selective bromodomain-containing protein 4 inhibitors. Bioorg. Med. Chem. 55, 116592 (2022).
Fang, L. et al. Discovery of 3, 5-dimethylisoxazole derivatives as novel, potent inhibitors for bromodomain and extraterminal domain (BET) family. Bioorg. Med. Chem. 39, 116133 (2021).
Zhao, Y. et al. Structure-based discovery of CF53 as a potent and orally bioavailable bromodomain and extra-terminal (BET) bromodomain inhibitor. J. Med. Chem. 61, 6110–6120 (2018).
Morrison-Smith, C. D. et al. Combined targeting of the BRD4–NUT–p300 axis in NUT midline carcinoma by dual selective bromodomain inhibitor, NEO2734. Mol. Cancer Ther. 19, 1406–1414 (2020).
Picaud, S. et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. 75, 5106–5119 (2015).
Kopytko, P., Piotrowska, K., Janisiak, J. & Tarnowski, M. Garcinol—a natural histone acetyltransferase inhibitor and new anti-cancer epigenetic drug. Int. J. Mol. Sci. 22, 2828 (2021).
Liu, Y. et al. Histone acetyltransferase (HAT) P300/CBP inhibitors induce synthetic lethality in PTEN-deficient colorectal cancer cells through destabilizing AKT. Int. J. Biol. Sci. 16, 1774 (2020).
Bowers, E. M. et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 17, 471–482 (2010).
Yang, Y. et al. Discovery of highly potent, selective, and orally efficacious p300/CBP histone acetyltransferases inhibitors. J. Med. Chem. 63, 1337–1360 (2020).
Zou, L. J. et al. Y08197 is a novel and selective CBP/EP300 bromodomain inhibitor for the treatment of prostate cancer. Acta Pharmacol. Sin. 40, 1436–1447 (2019).
Tezil, T. et al. Lifespan-increasing drug nordihydroguaiaretic acid inhibits p300 and activates autophagy. NPJ Aging Mech. Dis. 5, 7 (2019).
Oike, T. et al. C646, a selective small molecule inhibitor of histone acetyltransferase p300, radiosensitizes lung cancer cells by enhancing mitotic catastrophe. Radiother. Oncol. 111, 222–227 (2014).
Caligiuri, M. et al. FT-6876, a potent and selective inhibitor of CBP/p300, is active in preclinical models of androgen receptor-positive breast cancer. Target. Oncol. 18, 269–285 (2023).
Welti, J. et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov. 11, 1118–1137 (2021).
Nicosia, L. et al. Potent pre-clinical and early phase clinical activity of EP300/CBP bromodomain inhibitor CCS1477 in multiple myeloma. Blood 140, 852–853 (2022).
Jenwitheesuk, A., Nopparat, C., Mukda, S., Wongchitrat, P. & Govitrapong, P. Melatonin regulates aging and neurodegeneration through energy metabolism, epigenetics, autophagy and circadian rhythm pathways. Int. J. Mol. Sci. 15, 16848–16884 (2014).
Tsuboyama, N. et al. Therapeutic targeting of BAP1/ASXL3 sub-complex in ASCL1-dependent small cell lung cancer. Oncogene 41, 2152–2162 (2022).
Wang, L. et al. Artepillin C time−dependently alleviates metabolic syndrome in obese mice by regulating CREB/CRTC2 − BMAL1 signaling. Nutrients 15, 1644 (2023).
Chen, Y. et al. A propolis-derived small molecule ameliorates metabolic syndrome in obese mice by targeting the CREB/CRTC2 transcriptional complex. Nat. Commun. 13, 246 (2022).
Kost, G. C. et al. A novel anti‐cancer agent, 1‐(3, 5‐dimethoxyphenyl)‐4‐ [(6‐fluoro‐2‐methoxyquinoxalin‐3‐yl) aminocarbonyl] piperazine (rx‐5902), interferes with β‐catenin function through Y593 phospho‐p68 RNA helicase. J. Cell. Biochem. 116, 1595–1601 (2015).
Cai, W. et al. Wanted DEAD/H or alive: helicases winding up in cancers. J. Natl Cancer Inst. 109, 278 (2017).
Diamond, J. et al. Phase 1 study of RX-5902, a novel orally bioavailable inhibitor of phosphorylated P68, which prevents β-catenin translocation in advanced solid tumors. Ann. Oncol. 28, 83 (2017).
Capasso, A. et al. First-in-class phosphorylated-p68 inhibitor RX-5902 inhibits β-Catenin signaling and demonstrates antitumor activity in triple-negative breast cancer. Mol. Cancer Ther. 18, 1916–1925 (2019).
Qiu, Y. et al. Simvastatin suppresses renal cell carcinoma cells by regulating DDX5/DUSP5. Scand. J. Urol. 55, 337–343 (2021).
Bennett, J. et al. Discovery of a chemical tool inhibitor targeting the bromodomains of TRIM24 and BRPF. J. Med. Chem. 59, 1642–1647 (2016).
Leal, A. S., Misek, S. A., Lisabeth, E. M., Neubig, R. R. & Liby, K. T. The Rho/MRTF pathway inhibitor CCG-222740 reduces stellate cell activation and modulates immune cell populations in KrasG12D; Pdx1-Cre (KC) mice. Sci. Rep. 9, 7072 (2019).
De la Roche, M. et al. An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 3, 680 (2012).
Keating, S. T. et al. The Set7 lysine methyltransferase regulates plasticity in oxidative phosphorylation necessary for trained immunity induced by β-glucan. Cell Rep. 31, 107548 (2020).
Takemoto, Y. et al. Identification of cyproheptadine as an inhibitor of SET domain containing lysine methyltransferase 7/9 (Set7/9) that regulates estrogen-dependent transcription. J. Med. Chem. 59, 3650–3660 (2016).
Acknowledgements
The authors are thankful to the Department of Science and Technology and Biotechnology, GoWB, India for support to U.C. The authors are thankful to CSIR, GoI, for funding the fellowship of P.D.T. (File No.-09/028(1066)/2018-EMR-I). We thank Kunal Pramanik, Tiyasa Saha, Shrestha Ghosh, and Faruque Siddique for reviewing the manuscript.
Author information
Authors and Affiliations
Contributions
P.D.T. compiled the original manuscript and sketched the figures. U.C. conceptualized the review and added critical revisions. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Talukdar, P.D., Chatterji, U. Transcriptional co-activators: emerging roles in signaling pathways and potential therapeutic targets for diseases. Sig Transduct Target Ther 8, 427 (2023). https://doi.org/10.1038/s41392-023-01651-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01651-w
