
Abstract
Astroglia are a broad class of neural parenchymal cells primarily dedicated to homoeostasis and defence of the central nervous system (CNS). Astroglia contribute to the pathophysiology of all neurological and neuropsychiatric disorders in ways that can be either beneficial or detrimental to disorder outcome. Pathophysiological changes in astroglia can be primary or secondary and can result in gain or loss of functions. Astroglia respond to external, non-cell autonomous signals associated with any form of CNS pathology by undergoing complex and variable changes in their structure, molecular expression, and function. In addition, internally driven, cell autonomous changes of astroglial innate properties can lead to CNS pathologies. Astroglial pathophysiology is complex, with different pathophysiological cell states and cell phenotypes that are context-specific and vary with disorder, disorder-stage, comorbidities, age, and sex. Here, we classify astroglial pathophysiology into (i) reactive astrogliosis, (ii) astroglial atrophy with loss of function, (iii) astroglial degeneration and death, and (iv) astrocytopathies characterised by aberrant forms that drive disease. We review astroglial pathophysiology across the spectrum of human CNS diseases and disorders, including neurotrauma, stroke, neuroinfection, autoimmune attack and epilepsy, as well as neurodevelopmental, neurodegenerative, metabolic and neuropsychiatric disorders. Characterising cellular and molecular mechanisms of astroglial pathophysiology represents a new frontier to identify novel therapeutic strategies.
Similar content being viewed by others

Neuropathology: from neuronal doctrine to a glial inclusive view
Disorders of the central nervous system (CNS), in particular those leading to cognitive deficits, are the main challenge facing medicine in the 21st century. Pathophysiologically based cures of CNS disorders do not yet exist; at best contemporary medicine is limited to symptomatic treatments. This status quo reflects the complexity of the human brain and spinal cord and lack of fundamental knowledge of multiple pathophysiological mechanisms underlying neurological disorders. Another cardinal problem faced by experimental medicine is a conspicuous translational failure of animal models of human diseases.
The human brain and spinal cord are composed of multiple cell types, including neural parenchymal cells (neurones and neuroglia) that form the active networks responsible for the functional output and a variety of supporting stromal cells (endothelial, pericytes, fibroblasts etc.) (Fig. 1).1 Within this active milieu, all cells are linked by numerous feed-back and feed-forward connections that stipulate coordinated interactions of all elements of the nervous tissue. At the same time different neural cells perform distinct functions: more that five hundred million years of nervous system evolution segregated neural cells into electrically excitable neurones responsible for input/output information transfer and information processing and electrically non-excitable neuroglia,2,3,4 which provide homoeostatic support and defence of the nervous tissue.3,5,6,7,8,9 Neuroglia responses and changes upon pathology are fundamental for defining the progression and outcome of neurological diseases. Neurones are highly specialised cells with limited self-protective capabilities and contribute little to adaptive nervous tissue responses to damage. When stressed, neurones limit their activity to preserve energy; if the stress continues neurones die. In contrast, when facing pathological attack, neuroglial cells upregulate neuroprotection and mount an evolutionary conserved active defensive response known as reactive gliosis. These complex adaptive glial changes counteract pathological insults. For example, reactive microglia phagocyte pathogens and cellular debris, reactive astrocytes limit damage by erecting barriers to spread of inflammation and, together with the oligodendroglial lineage cells responsible for remyelinating axons, support postlesional regeneration.

The nervous system with sensory input and motor output and multicellular active milieu of the nervous tissue. Reproduced from ref. 6
The central contribution of neuroglia to neuropathology was already recognised by Rudolf Virchow, who considered the ‘interstitial tissue (i.e. neuroglia) of the brain and spinal marrow is one of the most frequent seats of morbid change’10 cited from an English edition.11 This view on a primary pathological role of neuroglia was shared by many neuropathologists of late 19th and early 20th century.12,13,14,15,16,17 Unfortunately, for much of the subsequent 20th century, these concepts became superseded by the neurone doctrine, such that glial responses to CNS disorders were considered to be non-specific, stereotypic, always subsequent to primary neuronal damage and of little functional consequence. The pathological potential of neuroglia resurrected only relatively recently, while the recognition of the central role of neuroglia in neuropathology began to be universally acknowledged.8,18,19,20,21,22,23,24,25,26,27 Pathology of neuroglia is complex, disorder- and context-specific, and includes various forms of reactivity, atrophy, loss and gain of function. Different pathological glial phenotypes may co-exist in the same pathological process or can be associated with different stages of the disease or disorder. Glial cells to a very great extent define neuropathology, its progression and outcome: as long as glial defence prevails, the pathological process is resolved, whereas the failure of glial defence results in neuronal death and neurological deficits. Glial contributions to pathology can be primary (for example, astrocytic expression of mutant glial fibrillary acidic protein, GFAP, causing Alexander disease) or secondary, when glial cells react to pathology by mounting context-specific defensive responses. Pathological insults may also cause the death of glial cells resulting in loss of function with subsequent neuronal damage. Glial responses to pathology also reflect the degree of damage. Acute insults often cause multi-level damage including structural damage, metabolic stress and impairment of molecular homoeostasis, which trigger widespread homoeostatic failure. In chronic disorders, homoeostatic failures progress and multiply, triggering sequential and heterogeneous glial responses.
It is important to note that widespread views on neuroglia as a latent toxic cells, as dormant killers, which, when triggered, eat up healthy nervous tissue, are incorrect; as a rule it is the loss of glial supportive or protective functions which damages neurones. Similarly erroneous are widely popularised views that reactive glia polarise into simple opposing functional states that are either good or bad, neuroprotective or neurotoxic, pro-inflammatory or anti-inflammatory, A1 or A2, and M1 or M2. Such oversimplifications are incorrect and misleading (see refs. 20,24 for detailed discussion). At a fundamental level, most glial responses to pathology are adaptive and allostatic favouring recovery and regeneration rather than destruction. This is in keeping with the premise that ‘nothing makes sense in biology except in the light of evolution’.28,29 In this regard, it is notable that astrocyte reactivity is an ancient response among vertebrates and has been essentially conserved across over 100 million years of divergent mammalian evolution that separate rodents, carnivores, herbivores and primates including humans. This argues that in the context of CNS disorders, that shaped glial responses during evolution, such as responses to microbial infections and traumatic injury, astrocyte reactivity exerts essential beneficial functions. Nevertheless, astrocytes and other glia can also mediate detrimental effects in neurological disorders either through downregulation of essential functions or through gain of inappropriate functions such as promoting excess inflammation. Moreover, various pathological changes can emerge together, in sequence or in isolation being disease-, stage-, and context-specific, and are influenced by age and systemic pathologies.
Principles of astroglial pathophysiology
Astroglia are primary homoeostatic cells of the CNS
Astroglia (Fig. 2) are a heterogeneous class of neuroglial cells unified by their common neuroepithelial origin and their common function, which is the preservation of CNS homoeostasis. Astroglia include (i) protoplasmic astrocytes of the grey matter, (ii) fibrous astrocytes of the white matter, (iii) perimeningeal astrocytes; (iv) velate astrocytes of the cerebellum, (v) radial astrocytes (radial stem astrocytes of neurogenic niches, Müller retinal glia, cerebellar Bergmann glia and tanycytes localised mainly in the hypothalamus and in some parts of the spinal cord), (vi) pituicytes in the neurohypophysis, (vii) perivascular astrocytes, (viii) marginal astrocytes, (ix) Gomori astrocytes (rich in iron and localised in the arcuate nucleus of the hypothalamus and in the hippocampus), (x) ependymocytes, (xi) choroid plexus cells, and (xii) retinal pigment epithelial cells.6,30,31 Hominid primates contain several types of astroglia (interlaminar, polarised and varicose projection astrocytes) absent in the brains of all other animals.32,33,34,35

Diversity of astroglia
The main role of astroglial cells in the healthy CNS (Fig. 3) is the maintenance of tissue homoeostasis at all the levels of CNS organisation, from molecular (ions, metabolites, and neurotransmitters, etc.), to network (regulation of synaptic connectivity), to organ (formation and maintenance of blood–brain barrier and glymphatic clearance system) and systemic (chemosensing blood oxygen, Na+, CO2 or glucose). Parenchymal astroglia (protoplasmic astrocytes, retinal Müller glia, cerebellar Bergmann glia) contact synapses with thin peripheral processes known as leaflets or appendages,1,9,36,37 which form the synaptic cradle.38,39 These membrane structures contain high densities of transporters that support homoeostasis in the synaptic cleft.40 Astroglial cells also secrete numerous factors controlling synaptogenesis, synaptic maturation and synaptic extinction.41,42,43,44 Many protoplasmic astrocytes in grey matter of rodents occupy individual domains and interact with neighbouring astrocytes only at the edges of these domains, with little intermingling of process among different astrocytes.45 Such cyto-architecture however, may not be common for all species; in particular substantial overlap of astrocytic territories was found in cortex of ferret46 and human.47 The functional logic behind this organisation is not yet understood. There is now tremendous interest in the heterogeneity of astrocytes across the CNS and there is an ongoing explosion of studies that are expanding and correlating information about structural, genetic and functional diversity of astrocytes across the healthy CNS.9,48,49,50,51,52,53,54,55,56

Functions of astroglia. The image of astrocyte is drawn based on 3D EM reconstruction kindly provided by Prof. Min Zhou, Ohio State University
Classifying astroglial pathophysiology
Astroglia play diverse roles in CNS disorders. Through their homoeostatic cascades, they are indispensable elements of neuroprotection that define the resilience of the nervous tissue to injury and disease. Homoeostatic systems associated with astrocytes in healthy tissue also support neuroprotection after insults, for example by supplying neurones with energy substrates in ischaemic conditions, scavenging reactive oxygen species (astroglia are the main source of glutathione), removing excess glutamate and buffering K+ ions, thus containing excitotoxicity, and actively taking up or detoxifying various toxic agents.6,23,57 Contributions of astroglia to neuropathology are however not limited to homoeostatic neuroprotection but can in some circumstances contribute to disorder progression. In pathological conditions astroglial cells undergo multiple progressive and/or regressive changes, which can to a significant extent determine the progression and outcome of neurological diseases as discussed below. Astroglial pathophysiology can be broadly classified into: (i) Astroglial reactivity or reactive astrogliosis; (ii) astroglial atrophy with loss of function; (iii) astroglial degeneration and death; and (iv) astrocytopathies with aberrant pathological astrocytes (Fig. 4).6,23,57

Classification of astrogliopathology
Astroglial reactivity or reactive astrogliosis
The concept of astrocytic reactivity or reactive astrogliosis (we shall use these two terms interchangeably) as an almost universal part of neuropathology is deeply rooted. Astrocyte responses to CNS trauma and disease have been recognised since the time of Andriezen (1895), Cajal (1913) and Alzheimer (1911). Nearly a century ago, the formation by astrocytes of a protective barrier around the fibrotic scar tissue that replaces damaged neural tissue at the lesion core after traumatic injuries was characterised by Pio del-Rio-Hortega and Wilder Penfield58,59 and named gliosis of astrocytes.60 Since that time, the terms reactive astrogliosis and astroglial reactivity (etymology: glia and osis in Greek means ‘glial process’; in Latin the suffix -osis acquired the additional meaning of ‘disease’ and so astrogliosis may also carry a connotation of ‘glial disorder’) have become widely and interchangeably used to describe astroglial responses to pathology.20
Astroglial reactivity can now be defined as an evolutionarily conserved, graded, and multi-stage primarily defensive reaction of astrocytes to neuropathology.20 Thus, by definition, astroglial reactivity is always secondary, being a response of astroglial cells to a pathological process. Astrocytic reactivity reflects activation of complex molecularly defined programmes which define remodelling of biochemical, morphological, metabolic, and physiological properties of astroglia leading to an upregulation or loss of homoeostatic cascades, or in gain of new protective or regenerative functions.20 Astrocytic reactivity is highly context dependent and is manifested by many different reactive phenotypes or transient states. Astrocytic transcriptomes and molecular signatures in various neurological diseases are highly diverse, again highlighting the heterogeneity of this process.61,62,63,64,65,66
Astrocyte reactivity phenotypes can at present be broadly classified into two major categories of (i) non-proliferative astrogliosis, which is isomorphic with mainly preserved domain organisation; and (ii) proliferative astrogliosis, which is anisomorphic with loss of domain, substantial structural reorganisation and can either be diffuse or can result in the formation of new compact ‘limitans’ borders around overt fibrotic tissue lesions (Fig. 5). Notably, within these broad categories, reactive astrocytes can exhibit substantial differences in molecular signatures and functional states that can vary with tissue region and disorder context as discussed below. Many details of reactive astrocytic remodelling remain to be revealed and characterised.

Classification of reactive astrogliosis
(i) Non-proliferative astrogliosis. In the healthy adult CNS, astrocytes rarely divide and are essentially post-mitotic.67 Non-proliferative astrogliosis typically occurs in neural tissue that is responding to a pathology but is not overtly damaged and retains its basic tissue architecture, for example (i) during diffuse neuroinflammation caused by peripheral exposure to microbial antigens such as lipopolysaccharide (LPS),68,69 (ii) in tissue regions that are at a distance from focal lesions (diaschisis) caused by stroke, trauma or autoimmune attack,8,70 or (iii) in tissue undergoing neurodegenerative changes.71 Non-proliferative reactive astrogliosis can vary in intensity but is isomorphic such that the astrocytes mainly retain the discrete, non-overlapping cellular domains found in the healthy grey matter but with variable degrees of cellular hypertrophy and reorganisation of their processes.72 These astrocytes maintain their interactions with local neurones, synapses, oligodendrocytes and vascular cells but may alter these interactions in accordance with context-specific reactive changes in molecular expression and functions.65,66 Notably, molecular changes exhibited by non-proliferative reactive astrocytes can be fully reversible over time after single exposures for example to neuroinflammation caused by LPS.68
(ii) Proliferative astrogliosis. Astrocytes can re-enter the cell cycle and proliferate in response to overt tissue damage such as is caused by stroke, severe trauma, infection, foreign bodies (including medical implants), autoimmune inflammation, neoplasm or severe neurodegeneration.19,67,73,74 Newly proliferated reactive astrocytes form borders that separate damaged, inflamed, and fibrotic tissue from adjacent viable neural tissue, and during this border formation newly proliferated astrocytes become transcriptionally reprogrammed to adopt new cellular interactions with non-neural cells.19,65 Although most border-forming reactive astrocytes in narrow zones immediately abutting tissue lesions are newly proliferated, proliferation drops off rapidly with increased distance from lesions, which are surrounded by large areas of intermingled proliferative and non-proliferative reactive astrocytes as well.67,75 Most border forming astrocytes derive from local astrocytes,67 with a small contribution from proliferation of adult OPC.76,77 Proliferation of astrocytes is an indispensable part of reactive astrogliosis, and suppression of proliferation exacerbates damage and delays wound closure. There is a widespread and popular belief regarding reactive astrogliosis and astrocytic perilesional border as a harmful reaction that limits regenerative capacity of the nervous tissue. This is an incorrect view; reactive astrogliosis is fundamentally protective and is indispensable not only for wound closure and formation of a barrier separating fibrotic scar from the healthy tissue but also for postlesional regeneration. Suppression of normally occurring reactive astrogliosis worsens neurological outcome and inhibits postlesional plasticity, regeneration and repair, as well as a restoration of the blood-brain barrier, which all determine functional tissue remodelling and post-traumatic rehabilitation.65,78,79,80,81,82,83,84,85,86
Thus, at present, non-proliferative and proliferative astrocyte reactivity represent two broad categories that can be readily differentiated and are associated with diverse molecular and functional differences. Notably, neither of these categories should be regarded as homogenous or stereotypic, and differences among reactive astrocytes within them are being identified. There is accumulating evidence for diverse changes in molecular expression of reactive astrocytes in different types and different severities of tissue pathology,62,65,68,69 but there is not yet a good synthesis of how molecular, metabolic,87 structural, and functional changes relate to one another to generate precisely definable phenotypes.
(iii) Scar tissue in the CNS is fibrotic and not glial. In the CNS as in any other tissue, focal tissue damage is closed through rapid proliferation of stromal cells and formation of a fibrotic scar, which is the replacement of the lost parenchymal cells (i.e. neurones and neuroglia) with stromal cells (fibroblasts and pericytes). In this regard, CNS tissue is not different from any other tissue. It is primarily the stromal cells that produce extracellular matrix and form fibrotic scar tissue. The CNS differs in that the formation of this fibrotic scar develops in parallel and in coordination with reactive astrogliosis, which generates a perilesional astrocyte border that has similarities in appearance and function (and in molecular mechanisms involved) to the glia limitans formed by perimeningeal astrocytes that interface with stromal cells of the meninges around the entire CNS.19,88 Like perimeningeal astrocyte borders, the newly formed perilesional astrocyte borders also serve to separate neural tissue from non-neural tissue.85 The main scar-forming cells in the CNS are perivascular fibroblasts89 and type A pericytes,90 which produce the fibrotic extracellular matrix that cements the scar.91 There is a long history of referring to the glia that surround CNS as ‘glial scars’ or as ‘astrocyte scars’, but multiple lines of evidence challenge this usage. In no other organ are parenchymal cells that proliferate after injury referred to as scar tissue. Astrocytes are neural parenchymal cells that proliferate after injury to replace lost neural tissue, form borders, and protect and preserve neural parenchyma in different disorder contexts as discussed in multiple places in this article. We suggest that it is time to stop referring to these structures as ‘glial scars’ or ‘astrocyte scars’ and instead refer to them as ‘astrocyte borders’ or ‘glial borders’.19,88
(iv) Markers of astrocyte reactivity. Morphological changes of reactive astrocytes have long been recognised not only after traumatic injuries but also in many other pathological contexts.23 In addition, for many decades increased immunostaining with antibodies against glial fibrillary acidic protein (GFAP) has been regarded as a universal molecular marker of astrocyte reactivity. It must be noted, however, that increased GFAP expression and GFAP-positive astrocytic profiles are not always associated with pathology. Physiological stimulation (such as, for example, physical activity, environmental enrichment, exposure to various diets or even circadian rhythmicity) may significantly change GFAP levels and morphometric parameters of GFAP-positive cellular profiles.92,93,94 There is now strong interest in identifying molecular markers associated with astrocyte reactivity. As noted above, astrocyte reactivity is associated with diverse changes in molecular expression that can vary from mild to pronounced and that are highly context dependent. Increases in the expression of various molecules that have been noted in reactive astrocytes across multiple contexts include GFAP, vimentin, nestin (probably labelling proliferating astrocytes), synemin plectin, α-crystallin B chain, monoaminoxodase-B (MAO-B), heat shock factor binding protein 1, complement C3, lipocalin 2, C-X-C motif chemokine ligand 10, SerpinA3N, LCN2 and others,20,95,96,97,98,99,100,101,102,103 but it important to note that no single molecular marker (including GFAP) is an absolute, required and sufficient, indicator of astrocyte reactivity, and no molecular markers have yet been identified that reliably distinguish amongst different reactive astrocyte phenotypes. In the future, rather than look for additional global markers of astrogliosis, it will likely be more useful to look for molecules upregulated by astrocytes in specific contexts and that are associated with specific functions or effects of astrogliosis.
To summarise, normally occurring reactive astrogliosis is in the first instance an intrinsic and evolutionary conserved set of diverse astrocyte responses that are aimed at neuroprotection, maintenance of tissue homoeostasis and preservation of nervous tissue integrity.
Astroglial atrophy and loss of function
A widespread class of astrocytic changes observed in many neurological diseases and in the majority of neuropsychiatric disorders are represented by structural atrophy and functional asthenia and manifested in the loss of key homoeostatic functions, such as for example glutamate clearance. This atrophy and loss of function are often a primary cause of neuropathology, as for example is caused by substantial decreases in expression and function of glutamate transporters in Wernicke-Korsakoff encephalopathy or toxic brain damage.104,105 Functional asthenia of astrocytes is frequently associated with morphological atrophy, decrease in territorial domain and associated reduction in astrocytic synaptic coverage. These morphological changes decrease astrocytic presence in the neuropil and hence diminish their homoeostatic support of the nervous tissue. In particular, morphological atrophy of astrocytes is prominently presented in neuropsychiatric diseases including mood disorders, post-traumatic stress disorders, addiction, and in some forms of autistic spectrum disorders.106,107,108,109,110,111 Similarly, morphological atrophy and functional asthenia of astrocytes contribute to the pathophysiology of various neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD).112,113,114,115,116,117 Arguably, atrophic astrocytes cannot properly support synaptic transmission, which results in cognitive and psychiatric syndromes.
Astroglial degeneration and death (Clasmatodendrosis)
Many neuropathologies, including for example traumatic lesions, stroke or age-dependent diseases are associated with direct damage of astrocytes, causing their degeneration and death. Morphologically, degeneration and death of astrocytes are known as clasmatodendrosis (from Greek ‘κλάσμα’, fragment, ‘δένδρον’, tree, ‘ωσις’, process), described, for the first time, by Alois Alzheimer, who found disintegrating, fragmented processes of astrocytes and oligodendrocytes in epilepsy, neurosyphilis and dementia.14 Ramon y Cajal considered clasmatodendrosis as an early post-mortem artefact.118 Nonetheless, many studies demonstrated clasmatodendrosis as an outcome of pathologies, which occur in the pre-mortem tissue.119 Clasmatodendrosis (as the name suggests) is manifested by fragmentation of astrocytic processes, disappearance of distal processes, together with swelling and vacuolation of the cell body (Fig. 6). Clasmatodendrosis was described in traumatic brain injury, cerebral ischaemia, post-stroke dementia, status epilepticus, demyelinating diseases, cerebral oedema, toxic encephalopathies, small-vessel disease and neuroinfection.119,120 Prominent astrocyte degeneration and fragmentation appears as a common feature of fronto-temporal dementia; notably astrocytic degeneration correlates with the severity of the disease.121 Clasmatodendrosis of astrocytes was also found in the white matter of some post-mortem brains of AD and cerebrovascular pathology.122 All in all, the degree to which degeneration and loss of astrocytes may contribute to different neurological disorders is understudied and deserves more attention.

Clasmatodendrosis of astrocytes. a Clasmatodendrosis as seen and drawn by Ramon y Cajal.118 A. Cell with preserved processes. B. Astrocyte with fragmentations. C, D, E. Astrocyte with disrupted cytoplasmic expansions, but with preservation of perikaryon. a. capillary. b. disaggregated end feet. b Clasmatodendrosis of astrocytes in the aged brain of mouse (stratum radiatum of dorsal hippocampus). Upper panel Astrocytes with distinctive enlarged soma and vacuolisation of processes distinctive to clasmatodendrosis for a representative cluster of astrocytes. Lower panel: Imaris surface render of a confocal z-stack of GFAP (blue), S100β (green), and Vimentin (Red) demonstrates an astrocyte with clasmatodendrosis (left) showing the co-localisation of S100β+Vimentin+ beads along GFAP+ processes, and a reactive astrocyte with non-degenerative morphology adjacent to it (right). Reproduced from ref. 120
Astrocytopathies with aberrant pathological astrocytes
Aberrant astrocytes that may act as instigators and propagators of neuropathology have been described in several diseases; these aberrant forms are covered under the umbrella term of astrocytopathies.18,23,123 The prototypical example of a genetic primary astrocytopathy is Alexander disease, a leukomalacia in which astrocytes express mutant GFAP, although how the expression of this mutant gene translates into severe damage of the white matter remains unknown.124,125 Another example of genetic primary astrocytopathy is the Duchenne muscular dystrophy (DMD) caused by mutations in the gene encoding dystrophin, which in the CNS is present almost exclusively in astrocytes. Expression of this mutated gene greatly reduces expression and operation of astrocytic glutamate transporters, thus leading to an excitotoxicity linked to psychosocial abnormalities and impaired cognition.126,127 Familial genetic mutations and polymorphisms can also alter astrocyte functions in ways that contribute to disorder progression as noted in Huntington’s disease,117,128 familial amyotrophic lateral sclerosis (ALS) mutations,129,130 or familial AD mutations.131 In addition, gene polymorphisms can alter astrocyte responses to pathologies and contribute to disorder progression, as for example with APOE polymorphisms in AD or traumatic injuries,132,133,134,135 or with CD38 polymorphisms in PD.136 In addition, acquired astrocytopathies are prominent in hepatic encephalopathy (in which astrocytes lose their homoeostatic capabilities137) and in neuromyelitis optica, in which the astrocyte protein, AQP4 is the subject of autoimmune attack, leading to dysfunction and death of astrocytes resulting in pronounced inflammation and degeneration of neural tissue.85,138 Aberrant astrocytes expressing markers of both astrocyte and microglia have been detected in ALS, in stroke and in dementia with Lewy bodies.139,140
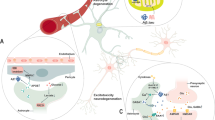
Astroglial detrimental effects through loss or gain of functions
There is mounting interest in how astrocytes that are reactive or diseased might have detrimental effects on the outcome of disorders. Although astrocyte reactivity is likely in the first instance to be targeted at maintaining CNS homoeostasis and circuit functions, various mechanisms could alter astrocyte functions in potentially detrimental ways, including (i) persistent reactivity may contribute to chronic neurodegenerative disorders or chronic inflammation, (ii) ageing and cellular senescence which may alter and reduce astrocyte functional capacities, or (iii) genetic mutations in diseased astrocytes or polymorphisms may alter normal astrocyte functions or responses. Such mechanisms could detrimentally impair astrocyte functions either through the loss or down-regulation of essential homoeostatic functions or through the gain of detrimental effects, or combinations of both. The potential loss of essential homoeostatic astrocyte functions includes (i) down-regulation of glutamate uptake that disturbs circuit function and increases excitotoxic potential141,142,143; deficient glutamate clearance is the primary element of several severe neurodegenerative diseases such as for example in ALS,144,145 and Wernicke’s encephalopathy146; (ii) down-regulation of K+ buffering resulting a neuronal hyper-excitability that disturbs circuit function and increases excitotoxic potential128; (iii) disruption of metabolic support of neurones and oligodendrocytes147,148; (iv) down-regulation of production of glutathione scavengers of cytotoxic reactive oxygen species (ROS)149,150; (v) reduced synapse support in ageing151 (Fig. 7). Gain of potential detrimental functions or effects could include: (i) increased pro-inflammatory signalling which at first is adaptive but can also be chronic and contribute to excess inflammation and degeneration152,153; (ii) increased GABA production resulting in disruption of circuit functions154,155; (iii) increased production of cytotoxic ROS155,156; (iv) increased accumulation or production of potentially toxic saturated very long chain fatty acids153,157 (Fig. 7). It has to be noted however that gain of function may also develop in parallel with loss of function, with reactive astrocytes for example losing their ability to clear glutamate in the context of motor neurone diseases.158 Similarly, modifications of astrocyte sphingolipid metabolism in reactive astrocytes affect their metabolic support of neurones and have indirect effects in this manner.

Potential detrimental effects of diseased, atrophied, reactive or aged astrocytes that can occur in specific contexts. See text for further explanation
Notably, many if not most, gains of potentially detrimental effects come about through the loss or malfunction of physiological astrocytic processes, rather than through the activation of pre-programmed and intentionally ‘toxic’ mechanisms. For example, transcriptional regulatory evaluations found no evidence for a programme of gain of function changes common across different degenerative disorders.65 Thus, gain of detrimental effects is likely to be context-dependent and unique to specific situations and effective treatment will require understanding the distinctive situations under which they arise. It deserves emphasis that to date there is no rigorous evidence for a programmed ‘neurotoxic’ astrocyte phenotype that is somehow activated in a common manner across multiple disorders and represents a ‘universal’ target, in spite of certain claims to this effect. Understanding the molecular and cellular mechanisms underlying the loss or gain of disorder-related detrimental effects mediated by astrocytes in different contexts has the potential to identify new treatment strategies for a wide variety of CNS disorders.
Conclusions
Astrogliopathology is complex, heterogeneous and context dependent with regard to disorder, disorder-stage, comorbidities, age, and sex. Reactive astrogliosis is in the first instance an evolutionary conserved protective response of the nervous tissue. In traumatic injuries (neurotrauma, neuroinfection, stroke or immune attack), irreversible proliferative astrogliosis is essential for wound closure and formation of a glial border that separates fibrotic scar tissue from the healthy neural tissue and promotes postlesional regeneration. Non-proliferative astrogliosis occurs in neural tissue responding to surrounding tissue pathology; astrocytes are not overtly damaged and maintain their interactions with local neurones, synapses, oligodendrocytes and vascular cells.
Although astrocyte reactivity is likely to be aimed at maintaining CNS homoeostasis and circuit functions, various mechanisms could alter astrocyte functions in potentially detrimental ways through the loss or down-regulation of essential homoeostatic astrocyte functions or through the gain of detrimental effects. Of note, the decrease in homoeostatic capacity of astrocytes seems to be the prevailing mechanism across various neuropathologies. In addition, cell autonomous primary astrocytopathies can give rise to aberrant astrocytes which drive neuropathological progression, and there is now mounting interest in how astrocytes that are reactive or diseased might have detrimental effects on the outcome of disorders.
Astrocytes control CNS damage: neurotrauma, stroke, neuroinfection and autoimmune attack
Neurotrauma
Acute focal traumatic brain injury
Acute focal traumatic brain injury (TBI) can be caused by penetrating lesions to the brain parenchyma that trigger cell death and haemorrhage, or by external force impact against the head resulting in brain contusion with intra-parenchymal haemorrhage, cell death and axonal damage. Fractures of the skull, vasogenic oedema, epi-, or subdural or intracerebral haematoma are frequently accompanied by focal TBI.159 The size and localisation of these focal injuries can vary widely, and this variability defines immediate and long-term neurological consequences.
The response to acute focal TBI with tissue damage develops in the following phases: (i) cell death and inflammation, (ii) cell proliferation, tissue replacement with fibrotic scar and wound closure and (iii) tissue remodelling and neuroplasticity aimed at restoration and functional compensation26,160 (Fig. 8). The first phase of CNS response to acute focal TBI is the disruption of the blood-brain barrier and traumatic injury of parenchymal cells. Mechanical forces trigger primary cell death, cellular lysis and cytotoxic oedema.161 Overactivation of ionotropic receptors as well as mechanoporation of cellular membranes162 lead to a massive Ca2+ entry that mediates cellular death.163 At the lesion core therefore, all neural cells die by necrosis, which leads to a massive release of damage-associated molecular patterns (DAMPs), including release of glutamate and ATP, which propagate excitotoxicity.164,165,166 Breach of blood-brain barrier results in the infiltration of blood-borne cells (erythrocytes, leucocytes, macrophages, and platelets) and molecules (such as fibrin, fibrinogen, collagen, or albumin). Invading white blood cells secrete pro-inflammatory factors, which trigger reactive response of neural parenchymal cells, most notably reactive astrogliosis and microgliosis. Similarly, fibrin, fibrinogen, collagen and other blood-derived molecules, together with DAMPs released from dying cells signal to neuroglia and instigate reactive gliosis.19,167,168

Stages of neuroinflammation and scar formation following traumatic brain injury. See text for explanation. Modified from ref. 6
Reactive gliosis, which starts within hours after the lesion, is a key and CNS-specific response to the TBI. The first responding cells are microglia and adult oligodendrocyte precursor cells (OPC), which migrate towards the lesion, with astrocyte responses to follow. There is a gradient of reactive morphotypes, with the most reactive cells (border-forming astrocytes and amoeboid microglia) concentrating at the lesion perimeter, or even entering (reactive microglia) the lesion core.75,169,170 Reactive microglia first position themselves between the infiltrating lymphocytes and newly proliferating reactive astrocytes that will form the perilesional border of astrocytes.171,172,173 Less prominent reactive morphotypes represented by polarised cells, which extend their processes toward the lesion area, are positioned more distantly, whereas healthy looking neuroglia demarcate the undamaged tissue. Adult OPC also undergo several forms of peculiar reactivity: some of them transform form into bipolar cells resembling foetal OPC, which migrate towards the lesion; adult OPC located distantly become hypertrophic with larger somata and more complex processes.77,174,175 Of note, GFAP expressed by reactive astrocytes following TBI, or released by damaged or dead astrocytes, can be also detected in the blood and spinal fluid, and the levels of GFAP in these fluids can reflect the severity of trauma and can be used as a diagnostic clinical tool.176
The second phase of the nervous tissue response is cell proliferation, and this is manifested by proliferation of fibroblasts and pericytes, which form extracellular matrix to produce the fibrotic scar177,178 that rapidly replaces lost neural parenchyma. In addition, reactive astrocytes at the perimeter of the fibrotic scar proliferate to form perilesional barrier that protects the adjacent viable nervous tissue. In mice, the peak in glial proliferation is observed during days 2 to 7 days after trauma, and subsequently proliferation gradually subsides.67,179 This second phase of acute focal TBI is complete about a month after the insult, when the mature lesion, composed of the central fibrotic scar (fully devoid of neural elements) and the surrounding astroglial limitans border have formed.26,160 Subsequently, the final phase of tissue remodelling plasticity starts; this phase may last for months and years during which the reshaping of neuronal ensembles provides for functional recovery. Perilesional astrocytic border (again contrary to a widespread beliefs) is permissive for axonal outgrowth and tissue recovery; ablation of glial limitans prevents functional rehabilitation.78,86,180 Neurotrauma boosts neurogenesis and migration of neuroblasts into the perilesional penumbra, where they arguably contribute to the neuronal circuitry repair.181
Diffuse traumatic brain injury
Diffuse (also known as mild) TBI results from the action of acceleration or deceleration forces on the head, which leads to a strain and concussion of the brain tissue. These mechanical forces induce sub-lethal damage to the cellular elements and may cause local disruption of the blood–brain barrier, with subsequent extravasation of blood cell and blood-born factors triggering focal inflammation.182 Histopathology of diffuse brain injury also includes diffuse axonal damage, axonal swelling, and disconnection.183 In about 50% of cases diffuse traumatic brain injury is complicated by long-term neurological consequences including cognitive decline, sleep disturbances and depression.184 Astrocytic reactivity in diffuse brain injury is quite distinct from full blown reactive astrogliosis in focal TBI. In cortex for example, astrocytes in response to diffuse injury up-regulate GFAP and become mildly hypertrophic, however they retain their territorial domain and generally do not proliferate. At the same time these astrocytes reduce expression of several key homoeostatic proteins such as glutamate transporters, glutamine synthetase, Kir4.1 inward rectifying channels involved in K+ buffering, and connexion 43 responsible for syncytial coupling.185 These asthenic astrocytes emerge shortly after the trauma and remain in the damaged area for months after injury. These malfunctional astrocytes are characterised by a prominent functional asthenia and lose key homoeostatic proteins, such as glutamate transporters, Kir4.1 channels, glutamine synthetase and gap junctional protein connexin 43, which results in an uncoupling of astrocytic syncytia.185 The aberrant asthenic astrocytes remain in the brain for months after the initial injury arguably delaying the recovery.185,186
Chronic traumatic encephalopathy
Chronic traumatic encephalopathy (CTE), which results from multiple and repeated mild traumatic injury (experienced for example by professional footballers, hockey players or boxers—hence its name ‘dementia pugilistica’187; pugilator - boxer in Latin), is a progressive neurodegenerative condition characterised by memory deficits, disorientation, confusion, aggression, and improper behaviours. In this pathology astrocytes undergo mild reactive changes and often develop astro-tauopathy (see below). Prominent clasmatodendrosis of astrocytes was observed in ~70% of post-mortem brains of patients diagnosed with CTE.188 CTE can also lead to atypical astrocyte responses that contribute to recurrent seizures.185 CTE is of steadily growing interest as a risk factor for various neurodegenerative disorders.
Spinal cord injury
For many decades, since the 1940s, based purely on correlative observations, reactive astrocytes that surround tissue lesions after traumatic injuries or stroke were regarded as ‘glial scars’ that were the primary cause for the failure of axon regeneration and functional recovery after spinal cord injury (SCI) and other causes of axotomy in the CNS.189 Recent studies overturned this long-standing dogma by showing that multiple experimental approaches to removing or genetically attenuating the astrocyte borders that form around lesions all failed to result in spontaneous axon regeneration.86 Moreover, other experimental approaches showed that substantial axon regeneration through lesions can be achieved by providing growth stimulating and chemoattractive factors, and this stimulated growth is attenuated (and not augmented) by disrupting astrocyte borders.86,190 There are now multiple lines of evidence that the failure of axon regeneration after SCI and other CNS injuries is due to multiple factors, including the failure of mature CNS neurones to reactivate and sustain developmental growth programmes, combined with a lack of appropriate chemoattraction.86,189,190,191,192,193 Indeed, there is increasing evidence that astrocyte borders around lesions can in fact support regrowing axons,86,190,194 suggesting that it is time to retire the term ‘glial scar’ when referring to astroglial borders around CNS tissue lesions.19,189
Ischaemia and stroke
Brain ischaemia can be caused by a systemic fall in blood supply (for example due to heart failure) or by occlusion of blood vessels through gradual thrombus formation or by acute embolism. Ischaemia can also be caused by the rupture of intracranial vessels, resulting in intracerebral haemorrhage. Ischaemia can be global or focal and acute or chronic. Global ischaemia causes widespread damage and cell death; about 10 minutes of global ischaemia usually is lethal. Focal ischaemia can trigger local damage to the nervous tissue ranging from cell death at the ischaemic core and various degrees of functional cellular deficits in the ischaemic penumbra. Ischaemia that causes acute functional deficits is commonly referred to as a stroke. The pathophysiology of ischaemic stroke is defined by the limitation of oxygen supply (hypoxia or anoxia), and restricted supply of metabolic substrates. The degree of damage is directly proportional to the degree of blood flow restriction. Decrease of blood flow below 1 ml/g/min causes total cellular death in the affected area. Cell death is a consequence of the decrease in ATP production which rapidly compromises ion (mainly due to halting Na+-K+ pump) and acid-base homoeostasis.195,196,197 At the cellular level, this translates into a massive increase in cytoplasmic Na+ concentration, membrane depolarisation and opening of voltage-gated Ca2+ channels, which in turn promotes massive release of glutamate that results in even large depolarisation enlarging Na+ and Ca2+ influx thus completing the vicious circle of excitotoxic damage, as Ca2+ overload triggers necrotic cell death.163,164,198,199 Breakdown of ion homoeostasis ion gradients, uncontrolled neurotransmitter release, oedema and mitochondrial failure are thus the key mechanisms behind pathophysiology of the ischaemic stroke.
The ischaemic core, where all neural cells are dead, is surrounded by the penumbra, in which cells are functionally compromised, but still surviving and can potentially be rescued. ATP production in the cells in the penumbra is reduced by ~50–70%, which supports some semblance of ion homoeostasis. At the same time, cells in the penumbra are subjected to periodical transient ischaemic depolarisations,200 mechanisms of which are essentially similar to spreading depression. Survival or death of neural cells in the penumbra correlates with the frequency of these transient ischaemic depolarisations. In essence, the balance between survival and death depends on neuroprotection and energy state of the tissue.201 This neuroprotection and support are mainly provided by astrocytes. First, astrocytes protect neurones in the penumbra, and second, after the end of infarct expansion, astrocytes form a peri-infarct barrier very similar to the perilesional barrier in the TBI.
In general, astrocytes resist ischaemic attacks better than neurones. Oxygen-glucose deprivation in cell cultures kills all neurones within an hour, whereas astrocytes survive for several hours more.202,203 In vivo, astrocytes are more sensitive to periods of ischaemia, although they still tolerate them better than neurones.200,204 Astrocytes survive even better in the penumbra, which is exposed to a lesser ischaemia. Astrocytes can at least temporarily switch to the glycolytic pathway to support their own energetics, and can use their glycogen pool (of which they are the sole possessors in the CNS205,206) to produce lactate and support energy substrate-deprived neurones.207 It must be remembered however, that an increase in lactate synthesis results in acidosis, which may severely damage astrocytes, and by proxy neurones. Hence, delivering glucose to the stroke affected brains exacerbates the infarction.208,209 In addition to providing energy support, astrocytes buffer glutamate overload through their glutamate transporters: ablation of the latter exacerbates ischaemic damage.210 Furthermore astrocytes are key elements for anti-oxidative defence being the main scavengers of reactive oxygen species.211
Astrocytes also protect the brain tissue through reactive astrogliosis. The stroke is invariably accompanied by the breach of the blood-brain barrier and infiltration of blood-borne elements into the brain parenchyma. This, together with cell death leads to a massive release of release of DAMPs including ATP, heat shock proteins, peroxiredoxins, and many others.212,213,214 These DAMPs trigger gliotic responses, initially represented by migration of microglia and OPCs as described previously, and second, after the end of infarct expansion, reactive astrogliosis. The infarct core is infiltrated with macrophages, dendritic cells and reactive microglia, which by combined effort, clear cellular debris. Reactive astrocytes proliferate and form, around the inflammatory cells and fibrosis of the infarct core, a barrier that protects the adjacent neural tissue (Fig. 9).160 A gradient of astrocytes at different reactive states is observed in a penumbra zone towards the healthy tissue, very much similar to that observed in the TBI and SCI.78,215,216 With time the dead tissue is replaced with fibrotic scar surrounded by glia limitans barrier that fences the healthy tissue and assists post-stroke regeneration.78 Grafts of neural progenitors that generate astrocytes can reduce stroke lesion volumes and promote repair.78,217 Notably, some reactive astrocytes, in the context of ischaemic brain damage, become actively phagocytic, thus contributing to the removal of damaged cells and assisting post-stroke regeneration.218 Conceptually, astrocytes (similarly to microglia) express several major phagocytic receptors allowing them to identify dead or dying cells and debris.219 Ischaemia arguably upregulates some of these receptors, although astrocytic phagocytosis is tightly coordinated with microglial one; with astrocytes removing small dendrites and microglia engulfing and scavenging soma and main processes.220

Reactive astrogliosis and protective astrocyte border formation in experimental stroke. Images show an ischaemic infarct in mouse striatum at 14 days after injection of the vasoconstrictive agent, L-NIO (N5-(1-Iminoethyl)-L-ornithine) in a manner similar to that described previously.78 Immunohistochemistry shows GFAP-expressing astrocytes stained red and NeuN-expressing neurones stained light blue. The left image shows the border of newly-proliferated reactive astrocytes that surround the fibrotic tissue (unstained) of the infarct core and isolate it from adjacent neural tissue. The right image shows a normal density of healthy neurones immediately adjacent to the protective astrocyte border. Images are courtesy of Dr. Shinong Wang and Dr. Yan Ao
There is also now a steadily growing interest in understanding and beneficially augmenting the neural plasticity and circuit reorganisation that occurs in areas of spared neural tissue after stroke and that can contribute to substantial recovery of functions.221,222 Astrocytes are increasingly recognised as playing important roles in synapse formation, maintenance and turnover in the healthy CNS and also in synapse remodelling after stroke.44,223,224,225 After stroke, astrocytes can undergo variable changes that may result in glucose hypometabolism that reduces the function neighbouring neurones70 but may also protect synapses and promote plasticity.226,227
Neuroinfection
Bacterial infection
Infectious diseases of the central nervous system are represented by meningitis, encephalitis, myelitis (infection of the spinal cord) and local abscesses. Many different types of pathogens, including bacteria, fungi, protozoa, viruses, and parasites may cause infectious damage to the CNS; however not all of them (or actually only a few of them) can cross CNS defences with ease. Various functional barriers, erected in particular by astrocytes, are highly effective in preventing CNS infection.228 When these barriers are compromised, pathogens enter. Some may cross the blood-brain barrier by the paracellular route, by transcytotic mechanisms, inside entering monocytes (the Trojan horse hypothesis), or by other mechanisms, such as hijacking of β-adrenergic receptors, as done for example by N. meningitides.229,230
The leading response of astrocytes to infectious agents is reactive astrogliosis, which erects parenchymal barriers and encapsulates brain abscesses, thus preventing infection spread. Reactive astrogliosis in neuroinfection is thus defensive and neuroprotective: inhibition of astrocytic reactivity, for example by knocking out GFAP,231 or by genetic deletion of NK-1R receptor for substance P, which suppresses reactive astrogliosis that defines resistance to N. meningitidis exacerbates the spread of infection and worsens neurological output.232 Pathogen-associated molecular patterns (PAMPs) trigger reactive astrogliosis by stimulating pathogen-recognition receptors including several types of Toll-like receptors, TLRs.233 Human astrocytes express TLR1 - 5 and TLR9, mouse astrocytes seem to posess all 9 TLRs;234,235 expression of TLRs was reported to increase in reactive astrocytes.236 Bacteria-derived lipopolysaccharide (which is a canonical PAMP often used to instigate reactive astrogliosis and microgliosis) act as agonists of TLR2 and 4.237 Breach of the blood-brain barrier, which accompanies neuroinfection, leads to an extravasation of blood-borne factors which also instigate reactive astrogliosis.238 Reactive astrocytes regulate entry and retention of leucocytes, thus controlling inflammatory response. In addition, reactive astrocytes secrete molecules attracting immune cells to the injured region as well as anti- and pro-inflammatory factors regulating neuroinflammation evoked by bacterial invasion.238 In focal brain infection, brain abscesses instigate classical inflammatory responses, with reactive astrogliosis, infiltration of macrophages and stromal cells, formation of fibrotic scar and erection of glial perilesional barrier.239,240
Parasites
Astrocytes are targets for the infection by neurotropic protozoa such as Toxoplasma gondii and Plasmodium falciparum. Astrocytes infected by T. gondii show complex response, including reactive remodelling, secretion of interleukins that reduce parasite burden241,242 and start to produce and release kynurenic acid, which is as an endogenous antagonist of NMDA and acetylcholine receptors. Increased production of kynurenic acid may be responsible for an increased risk of schizophrenia in infected patients.243 Deletion of astrocyte IL-6 receptor and down-stream JAK-STAT signalling exacerbates the spread of toxoplasma infection and worsens neurological output.244 In neuroinfection caused by the malaria parasite P. falciparum, astrocytes become damaged which causes a loss of glia limitans, facilitating the spread of infection.245
Systemic infection and inflammation
Systemic infection associated with septicaemia, is manifested by systemic inflammatory response syndrome, a non-specific complex reaction of the organism to any severe infection, mechanical or thermal injury, or pancreatitis.246 Sepsis often triggers a sepsis-associated encephalopathy (SAE) which presents itself with wide symptomatology including reduced attention, disrupted sleep-wakefulness balance, impaired speech and orientation, deficient leaning and memory, numerous perception disorders, focal neurological deficits, seizures, and, in terminal stages, coma.247 SAE may develop in two basic scenario: with and without disruption of the blood-brain barrier; in both cases astrocytes, their reactivity and enforcement of the glia limitans are critical.240,248 Early stages SAE are often manifested with a ‘sickness behaviour’, an adaptive body response aimed at preservation of energy; signs and symptoms include anorexia, anxiety, irritability, depression, anhedonia, decreased social communication and environmental interest, cognitive changes, including decreased concentration, learning ability and memory.249,250
Acute systemic injections of the bacterial antigen lipopolysaccharide (LPS) in mice induces diffuse inflammatory changes in the CNS and mimics this sickness behaviour and is often used as a model to study effects of inflammation on CNS functions. Acute systemic LPS injections induce pronounced but reversible changes in astrocyte gene expression in the prefrontal cortex, along with an approximate 25% decrease in astrocyte branch and process volumes, but with few currently detectable changes in astrocyte physiology and basic preservation of astrocyte core homoeostatic functions such a potassium buffering.68 The effects of prolonged LPS exposure on astroglia are less well studied. It deserves mention that LPS is a model for sepsis and should not be extrapolated as a generalised model that is somehow representative of different forms of CNS inflammation. Reactive astrocyte changes induced by LPS differ markedly from those induced by other forms of CNS inflammation such as is associated with autoimmune attack or traumatic injury.65
Viral infections of the brain
Viral infection targets all cells in the brain, with reactive gliosis dominating the tissue response.245,251,252,253,254 In addition, astrocytes can be infected and can serve as a viral reservoir. The human immunodeficiency virus (HIV) has significant neurotropism with frequent occurrence of neurological and cognitive symptoms and even HIV-associated dementia.255 Although HIV mainly infects and affects microglia, infected astrocytes show decreased homoeostatic capacity.256 In contrast, the herpes simplex virus (HSV) preferentially infects astrocytes and oligodendrocytes, whereas microglia become reactive and neuroprotective through release of inflammatory mediators, such as interferon-γ, TNF-α, IL-1, IL-6 and IL-8, RANTES and chemokine CXCL10.257,258,259,260 Microglia-derived TNF-α, for example inhibits viral replication in astrocytes,259 while microglial IL-6 reduces neuronal death. Human cytomegalovirus also affects astrocytes and reduces astrocytic production of thrombospondins,261 such affecting synaptogenesis, and suppresses astrocytic expression of glutamate transporters,262 thus decreasing neuroprotection again excitotoxic damage. The flaviviruses, such as ZIKA virus and the tick-borne encephalitis virus, selectively infect astrocytes, which become viral reservoirs.245 In human astrocytes neurotropic flaviviruses increase autophagy, although viral replication is autophagy-independent.263
Clinical manifestations of Coronavirus Disease 2019 (COVID-19), which results from infection with acute respiratory syndrome coronavirus 2 (SARS-CoV-2), include neurological, cognitive and psychiatric manifestations.264,265,266 Pathophysiology of the brain damage caused by SARS-2 includes: (i) direct viral infection of neural cells; (ii) severe systemic inflammation (cytokine storm), with damage to the blood-brain barrier and immune infiltration; (iii) hypoxia associated with respiratory failure; (iv) widespread thrombosis and stroke; and (v) psychological stress linked to disease experience (a kind of post-traumatic stress disorder) and epidemiological interventions.267 Both astrocytes and microglia contribute to the pathophysiology of COVID-19.268 Astrocytic reactivity was deduced from increased levels of GFAP in the blood plasma of COVID-19 patients269 and increased GFAP levels in the white mater in post-mortem tissues of COVID-19 victims with disseminated encephalomyelitis.270,271 Post-mortem analysis also revealed clasmatodendrotic astrocytes suggesting that COVID-19 may directly damage astrocytes.272 Interrogation of stem-cell derived organoids and organotypic slice cultures revealed preferential infection of astrocytes with SARS-2.273 Astrocytes infected with the virus demonstrated signs of reactivity, increased cytokine production and cellular stress. Incidentally these astrocytes did not possess ACE-2, known as a canonical SARS-CoV-2 gate into cells; arguably coronavirus entry factors DPP4 and BSG/CD147 could be involved.273
Prions
Prion diseases are neurodegenerative conditions represented by Creutzfeldt Jakob’s disease, Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, and Kuru disease.274 Prion diseases are caused by accumulation of a pathological prion, known as Prion PrPSc, which are misfolded polypepetides converted form physiologically significant cell-surface glycoprotein PrPC encoded by the PRNP gene.275 PrPSc and other prion-like polypepetides have the interesting properties of being infectious and able to induce the generation of more prion particles when seeded from one CNS region to another or from even from one individual to another either by ingestion via the diet, or via implantation into the CNS for example from surgical instruments or tissue grafts.276 Pathological prion instigates neuronal death, which in turn causes secondary astrocytic reactivity predominantly in the white matter, but the roles of reactive astroglia in prion diseases are poorly understood and hampered by the emerging complexity of astrocyte responses to prion induced pathologies.277
Autoimmune attack
Neuromyelitis optica
Neuromyelitis optica spectrum disorders (NMOSD) are autoimmune diseases that primarily affect and damage myelin and axons in the optic nerve and spinal cord. These disorders include neuromyelitis optica (NMO, also known as unilateral optic neuritis), isolated or recurrent transverse myelitis, longitudinally extensive transverse myelitis or isolated brain lesions with or without detectable anti AQP4-IgG autoantibody. All NMOSD are classified (according to the leading pathophysiological mechanisms) into diseases with identifiable antibodies against aquaporin 4 AQP4-IgG (NMOSD-AQP4), NMOSD without AQP4-IgG or with unknown AQP4-IgG status and NMOSD with identified antibodies against Myelin Oligodendrocyte Glycoprotein, (NMOSD-MOG).278,279,280 About 70% of NMOSD cases are caused by AQP4 auto-antibodies, which after entering the nervous tissue damage astrocytic endfeet rich with AQP4 channels. The remaining 30% of NMOSD are caused by MOG auto-antibodies that target and injure oligodendrocytes. These are neurodegenerative diseases, which besides optic nerve and spinal cord also affect the brain parenchyma and cause neurological and cognitive presentations.
NMO, also known as Devic’s syndrome (described by Eugene Devic281), is the most frequent NMOSD. NMO is mainly caused by auto-anti-AQP4 antibodies282 that initially attack astrocytic endfeet,283 leading to complement-mediated lysis and destruction of the glial limitans and astrocyte degeneration, which in turn cause the other pathophysiological sequalae (Fig. 10). Thus, NMO can be classified as a primary astrocytopathy. Astrocytic demise is manifested in classical clasmatodendrosis with cell swelling and fragmentation of processes. Massive astrocytic loss precedes demyelination and tissue damage.284 Disintegration of glia limitans translates into the widespread damage to the blood–brain barrier and degranulation of natural killer cells that release perforins and granzyme, which in turn, further injure astrocytes and endothelium.285 Disruption of the barrier leads to an extravasation of leucocytes and macrophages, reactive microgliosis, neuronal death and full-flown inflammatory response.85 In the MOG-associated variant of the disease auto-antibodies attack oligodendrocytes thus causing their death and secondary astro- and microgliotic response.286 Incidentally, the anti-AQP antibodies also attack kidneys, and patients with AQP4 auto-antibodies form of NMOSD demonstrate lover glomerular filtration rate.287

Pathophysiology of the AQP4 form of the neuromyelitis optica. See text for explanation. Modified from ref. 6
Multiple sclerosis
Multiple sclerosis (MS) is a chronic autoimmune inflammatory disease with antibodies primarily attacking myelin sheaths and causing multiple demyelinated areas in white and grey matter throughout the CNS (with most frequent localisation in the optic nerve, spinal cord, brain stem, periventricular white matter, and the grey matter near the subarachnoid space); these lesions are considered to be histopathological hallmarks of the disease.288,289,290 Immune attack on the nervous tissue begins from the entry of auto-reactive T lymphocytes and accumulation of auto-antibodies. With breach of the blood-brain barrier, immune cells infiltration as well as extravasation of blood-borne molecules such as fibronectin triggers, together with the death of oligodendrocytes, degeneration and transection of axons and starts a multicellular inflammatory response associated with reactive astro- and microgliosis.291,292,293,294 Focal inflammation evolves into fibrotic scars surrounded by glial borders and barriers; which are the substrate of mature sclerotic lesions. Interrogation of specific cell-cell interactions using new tools such as bar-coded viral tracing technology, is beginning to identify specific molecular interactions, for example that Sema4d and EphrinB3 expressed by microglia control astrocyte responses respectively through PlexinB2 and EphB3 receptors.295
Astrocytes play central and heterogeneous roles in the regulation of CNS inflammation in autoimmune diseases such as MS and its experimental models.152,296 Astrocytes in MS undergo both reactive and atrophic changes. Reactive astrogliosis is a prominent feature of MS lesions with reactive astrocytes surrounding active demyelinating foci.291,297 In the animal model of MS, experimental autoimmune encephalomyelitis (EAE), astrocytes are found around demyelinating areas but may even occur before emergence of the latter.298,299 Reactive astrocytes secrete various cytokines and are involved in recruitment of leucocytes traffic into the brain parenchyma.300,301 Recent findings suggest that exposure to certain environmental factors such as pesticides can augment astrocyte proinflammatory signalling and thereby promote CNS autoimmune inflammation.302 Notably, microbial metabolites produced by commensal gut flora can limit pathogenic activities of microglia and astrocytes and suppress CNS inflammation in MS experimental models303 and can contribute to the induction of specific LAMP1+TRAIL+ astrocytes that limit CNS inflammation by inducing T cell apoptosis.304
Fibrotic scar is a common component of MS mature lesions; active lesions are enriched in collagen-producing cells, aggregates of fibronectin and collagen, mesenchymal perivascular aggregates of platelet-derived growth factor receptor (PDGFR)β-bearing cells, and pro-fibrotic factors such as biglycan and decorin.305,306 In contrast to TBI, the MS scar is mainly produced by resident fibroblasts and not by vascular infiltrating stromal cells.89 Development of MS is also associated with direct damage to astrocytes. Astrocytes with swollen somata and processes surround blood cells infiltration, with some of them showing signs of clasmatodendrosis.297,307 In addition, astrocytes lose AQP4 channels in their endfeet and down-regulate expression of glutamate transporters, thus adding to excitotoxic damage of oligodendrocytes.308 Experimentally-induced depletion or attenuation of reactive astroglia markedly exacerbates the clinical progression, spread of inflammation and tissue loss in EAE during the induction phases of the disorder,81,309,310 whereas similarly induced depletion of proliferating reactive astrocytes at later times during the progressive phase ameliorates disease, and this amelioration is at least in part due to removal of astrocyte Ccl2 production.310 Similar effects are observed after transgenic deletion of Ccl2 specifically from astrocytes, which showed little effects early in EAE but demonstrated that astrocyte Ccl2 sustains disease symptoms and inflammation during chronic EAE, making it a therapeutic target.311 Such findings point towards different roles and different effects of reactive astrocytes at different times during the progression of autoimmune inflammation and suggest that different therapeutic approaches involving astrocytes may be required at different times. These observations also highlight that therapeutic approaches should be directed at specific aspects of astrocyte reactivity, for example astrocyte Ccl2 production, rather than at attenuating reactivity per se, which may lead to unexpected and undesirable consequences. New technologies are allowing selective identification of subsets or sub-states of reactive astrocytes, and have identified a role for a potentially therapeutically targetable mechanism that limits XBP1-driven pathogenic astrocyte responses.312
Conclusions
Injury or damage to CNS tissue of a mechanical, vascular, infectious or auto-immune nature triggers diverse inflammatory responses. Reactive astrogliosis plays essential roles in both attracting and containing neuroinflammation, and is critical for wound closure, fibrotic scar formation, erecting glia limitans barrier delineating the damaged tissue and supporting postlesional regeneration and plasticity.
Genetic astrocytopathies
Alexander disease
Alexander disease, or AxD (named after its discoverer W. Steward Alexander313) is an incurable genetic astrocytopathy caused by sporadic mutations in the GFAP gene.124 Astrocytic expression of mutant GFAP proteins results in a profound loss of white matter, i.e. AxD is a genetic leukomalacia. Histological hallmarks of AxD are the emergence of perivascular Rosenthal fibres consisting of ubiquinated aggregates of GFAP, vimentin, small heat shock proteins αβ-crystallin and Hsp27, and plectin.124,314 How expression of mutated GFAP in astrocytes translates into severe white matter deficiency remains unknown.315 Astrocytes in AxD show profound alterations in morphology and function, with evidence of abnormal glutamate clearance, substantial cell stress in the form of upregulated expression of heat shock protein, MAPK pathways, JNK and p38 kinases, as well as increased autophagy and proteasomal activity.124,316
Duchenne muscular dystrophy
Duchenne muscular dystrophy (named after Guillaume-Benjamin-Amand Duchenne de Boulogne who provided early and comprehensive description of clinical presentation and histopathology of this disorder317) is an X-linked recessive neuromuscular disorder caused by mutations in the DMD gene that encodes the protein dystrophin. Dystrophin is a main part of the dystrophin-associated protein complex (also known as a costamere) essential for contraction of the striated muscle.318,319 Expression of mutant DMD gene results in progressive muscle degeneration leading to various problems with locomotion, ambulation, as well as deficits of cardio-vascular and respiratory systems.320 In addition to muscular manifestations, Duchenne muscular dystrophy is often associated with psychosocial abnormalities and cognitive impairment, while histopathologically neuronal death and dendritic abnormalities are frequently observed post-mortem.126 In the CNS, dystrophin is mainly expressed in protoplasmic astrocytes in neocortex and in velate astrocytes and Bergmann glia in the cerebellum; with particularly high expression in perivascular astrocytic endfeet.321,322 At the cellular level dystrophin scaffolding network supports morphologically complex astrocytic processes and defines correct localisation, clustering and density of numerous channels, receptors and transporters. In particular, dystrophin-associated protein complex links AQP4 aquaporin channels and Kir4.1 channels in the endfeet: one of the key components of astrocytic homoeostatic hardware.323,324 Astrocytes differentiated from pluripotent stem cells isolated from Duchenne muscular dystrophy patients (and carrying mutant DMD gene) were characterised by abnormal cytoskeleton, severely deficient glutamate uptake, and compromised K+ buffering.127 Thus, the Duchenne muscular dystrophy is a primary genetic astrocytopathy that impairs synaptic transmission, causes excitotoxicity and secondary neurodegeneration.
Conclusions
The whole range of primary genetic astrocytopathies is yet to be fully characterised. Mutations in genes encoding astrocyte-specific proteins can lead either to loss of functions with subsequent secondary neural injury or to the emergence of aberrant cellular phenotypes damaging nervous tissue in yet unidentified manner.
Epilepsy and migraine
Epilepsy
Epilepsy manifested by seizures originates from an uncontrolled over-excitation of motor brain areas.325 At the cellular level this overexcitation stems from a slow synchronous depolarisation of neurones, known as paroxysmal depolarisation shift (PDS) within the epileptic foci. The PDS develops from large and relatively slow excitatory postsynaptic potentials mediated by AMPA and NMDA glutamate receptors activated by aberrant and large glutamate release within the foci.326 Astrocytes regulate glutamate presence in the interstitium,327 tune neuronal excitability through K+ buffering,328 and influence inhibitory/excitatory balance through tonic release of GABA329 and by supplying Cl- to inhibitory synapses.330 Astrocytes are severely affected in epileptic foci and abnormal astrocytic homoeostasis contributes to ictogenesis.331,332 In mesial temporal lobe epilepsy or tuberous sclerosis, astrocytes demonstrate a specific form of reactivity characterised by an increased expression of GFAP333 and morphological atrophy manifested by reduced complexity of arbour and loss of distal leaflets.334 These morphological aberrations are paralleled with loss of function. Loss-of-function missense mutations as well as single nucleotide polymorphisms in the genes encoding Kir4.1 and AQP4 (which is selectively astroglial and operates in concert with Kir4.1) are linked to epilepsy in humans.335 Astrocytes in post-mortem samples of patients with mesial temporal lobe epilepsy show significant down-regulation of Kir4.1 channels critical for K+ buffering.335 In experimental settings, conditional knockout of Kir4.1 results in epileptic phenotype.336,337 Failure of glutamate homoeostasis in epilepsy reflects significant down-regulation of astrocytic glutamate transporters and glutamine synthetase, the two principal components of the glutamate (GABA)-glutamine shuttle. In temporal lobe epilepsy, levels of astrocytic glutamate transporters in hippocampus are reduced by up to 40%.338 Similar decreases in astrocytic expression of glutamate transports are characteristic for animal models of epilepsy,339,340 and genetic ablation of EAAT1/2 triggers seizures.338,341 Astrocytic glutamine synthetase is reduced in post-mortem tissues from epilepsy patients.342 Arguably, decreased glutamine supply limits GABA release, thus increasing neuronal excitability.343 Reduced astrocytic homoeostatic support is also evidenced by the loss of endfeet polarisation of AQP4344 and decreased expression of monocarboxylate transporter 1345 responsive for lactate supply of neurones. Increased APOE expression and excessive lipid accumulation in astrocytes promote neuronal hyperexcitability and disease progression in temporal lobe epilepsy.346 Another astrocyte-specific mechanism involved in ictogenesis is linked to gap junction forming connexion Cx43 channels, which are down-regulated in epilepsy.347,348 Uncoupling of astrocytic syncytia or knocking out Cx43 in experimental models instigates seizures.348,349 It is also noteworthy that CTE can lead to atypical astrocyte responses that contribute to recurrent seizures.185
Familial hemiplegic migraine
Familial hemiplegic migraine FHM type 2, clinically manifested as migraine with aura, is linked to loss-of function mutations of theATP1A2 gene encoding the astrocyte-specific α2 subunit of Na+/K+ ATPase (NKA). Astrocytic NKA is central for K+ buffering350,351 and for astrocytic Na+ signalling.352 In the context of FHM type 2 deficient NKA not only results in the impaired K+ buffering but is also linked to down-regulation of astrocytic EAAT2 glutamate transporters, which dives pathophysiology of this form of migraine.353
Conclusions
Loss of astrocytic homoeostatic support is the primary mechanism underlying neuronal hyperexcitability in epilepsy and in migraine. In the case of epilepsy, impaired K+ buffering, glutamate clearance, lipid accumulation and malfunctioning of the glutamate (GABA)-glutamine shuttle emerge as leading pathophysiological processes. Familial hemiplegic migraine type 2 is linked to the loss of function mutations of astrocytic Na+/K+ ATPase which leads to an abnormal regulation of interstitial K+ and glutamate.
Astrocytes as main target of toxic encephalopathies
Hyperammonaemia and hepatic encephalopathy
Hepatic encephalopathy is a primary astrocytopathy caused by an increased level of blood ammonium. This increase is observed in several diseases including congenital deficits in urea cycle enzymes, Reyes syndrome in children, uraemic encephalopathy, diabetic encephalopathy, or hypoglycaemic encephalopathy, although the most frequent cause for hyperammonaemia is associated with acute (due to poisons or drugs) and chronic (cirrhosis) liver failure.354 Ammonium is mainly produced in the gut by amino acid deamination with subsequent conversion to ammonium by urease containing bacteria and is detoxified in the liver by the urea cycle enzymes and by glutamine synthetase, highly expressed in hepatocytes.355 Physiological levels of ammonium in the blood are about 10–20 μM, but following liver failure ammonium rise to millimolar levels.356,357 Ammonium crosses the blood-brain barrier with ease (in gaseous NH3 form), and in this manner liver failure causes its massive increase in the brain. Hyperammonaemia triggers numerous neurological and neuropsychiatric symptoms, including poor concentration, impaired memory and cognition, psychotic presentations, sleep disturbance and lethargy, and stupor and coma in severe cases.358 In its terminal phase, hyperammonaemia leads to brain oedema which may even cause brain herination, and this progressive oedema is the main case of death.
Astrocytes are the exclusive possessors of glutamine synthetase in the brain,137 which makes them the main target for ammonium. Most of ammonium entering the brain is converted to glutamine, which causes glutaminosis359 and effectively lowers brain parenchymal ammonium concentrations, but which also maintains the concentration gradient and favours additional entry of ammonium from the blood to the brain.360 At the cellular level, excess of ammonium mainly affects astrocytes with neuronal injury being secondary, resulting mainly from failure of astrocytic homoeostatic cascades and oedema. The histopathological hallmark of ammonium toxicity is manifested by the emergence of aberrant astrocytes, known as Alzheimer type II astrocytes. These pathological astrocytes were initially described by Carl Von Hösslin and Alois Alzheimer361 during post-mortem analysis of the brain of a patient with toxic copper encephalopathy, known today as a Wilson’s disease. The term Alzheimer astrocytes type II was introduced in 1942.362 Alzheimer’s type II astrocytes are characterised by (i) increased size of somata, primary processes and swollen endfeet; (ii) increased nuclei; (iii) decreased electron density of the cytoplasmic matrix in perikaryon, processes, and endfeet; (iv) increased density of mitochondria and enlarged endoplasmic reticulum in soma and primary processes; (v) less compact bundles of intermediate filaments.357,363 In addition, there are mild signs for astrocytic reactivity mainly in cerebral neocortices.364 The affected astrocytes demonstrate profound loss of function, in particular impaired K+ buffering,365,366 compromised H+ transport,367 and substantial decrease in glutamate uptake.368 Astrocytes exposed to excessive ammonium start to generate aberrant Ca2+ signals, which may cause excitotoxic release of glutamate.369,370
Increased activity of glutamine synthetase and increased production of glutamine are key pathophysiological features of ammonium neurotoxicity. Excessive glutamine is accumulated in astrocytes and converted back to glutamate and ammonium by mitochondrial phosphate-activated glutaminase. Overload of mitochondria with ammonium instigates over-production of reactive oxygen species and opening of mitochondrial permeability transition pore, which further damages astrocytes and induce astrocyte swelling; this sequence of events is known as the ‘Trojan horse’ hypothesis of hepatic encephalopathy.371 High ammonium also down-regulates astrocytic expression of glutamine exporting SNAT3/SLC38a3 transporter thus exacerbating glutamine retention.372 All these astrocytic changes result in considerable and often irreversible neuronal damage, with about 50% of patients demonstrating persistent neurological manifestations even after liver replacement and normalisation of blood ammonium.355
Trace metals toxic encephalopathies
Contact with many metals, such as arsenic (As), manganese (Mn), mercury (Hg), lead (Pb), aluminium (Al), nickel (Ni), bismuth (Bi), cadmium (Cd), zinc (Zn), copper (Cu) and iron (Fe), may evoke acute or chronic toxic encephalopathies; in addition, chronic accumulation of such metals is known to increase the risk for neurodegenerative disorders, particularly of AD and PD.105 Neurological symptoms of metal encephalopathies are quite variable and may include sleep abnormalities, disorientation, visual abnormalities, sensory lesions, cerebellar ataxia, hearing loss, weakness, tremor, memory problems and cognitive decline. In the brain, the excess of heavy metal is almost completely removed by astrocytes373; accumulation of metals in astrocytes impairs their homoeostatic capabilities, which in turn, damages neurones and results in neurological symptoms.
Iron is probably one of the most important metals sustaining critical physiological processes, but also with the potential to cause neurotoxicity when in excess. In the CNS, iron is accumulated mainly by astrocytes and to a somewhat lesser extent by microglia. This accumulation is mediated by plasmalemmal transporters predominantly expressed in glial cell, such as divalent metal transporter 1 DMT1/SLC11A2 or zinc transporter ZIP14/SLC39A14,374,375 which both transport Fe2+ (ferrous), whereas Fe3+ (ferric) is accumulated through transferrin receptors.375 In physiological contexts, astrocytes provide iron storage in the form of ferritin, and when needed, iron is released by ferroportin and ceruloplasmin ferroxidase.376 Ceruloplasmin deficiency results in brain iron overload and neurotoxicity.377
Iron overload, following increased intake, which may in particular result from the use of iron-containing implants widely employed in orthopaedic surgery,378 or can occur after haemorrhagic stroke, results in compensatory upregulation of glial iron transporters, and down-regulation of neuronal iron transporters, both aimed at neuroprotection.378 Astrocytic overload with iron however significantly reduces glutamate clearance by down-regulation expression of EAAT1 glutamate transporters379 and impairs operation of the glymphatic system.380
Another primary astrocytopathy linked to dysregulated iron homoeostasis is aceruloplasminemia, an autosomal recessive neurodegenerative disease caused by a loss of function mutations of the gene encoding ceruloplasmin, an enzyme that converts ferrous iron to ferric. Neurological presentations of aceruloplasminemia include ataxia, involuntary movement, cognitive dysfunctions and parkinsonism.381 In the CNS, ceruloplasmin is expressed almost exclusively in astrocytes, and its deficiency results in iron overload and formation of iron deposits. Oversized and deformed astrocytes are the histopathological hallmark of the disease.382 Impaired astrocytic iron homoeostasis triggers secondary neuronal iron toxicity, which together with loss of astrocytic homoeostatic support, triggers neuronal death.
Mercury causes severe brain abnormalities, for example in Minamata disease.383 Molecular pathophysiology of mercury toxicity is defined by an induction of reactive oxygen species production and down-regulation of anti-oxidant defence. Astrocytes are the main depository for mercury,384 which they accumulate by large neutral amino acid transporter LAT1/SLC7A5.385 Astrocytic overload with mercury causes cell swelling, it severally impairs glutamate uptake, K+ buffering and glutathione production,386 this causing secondary excitotoxicity and oxidative damage to neurones. The neurotoxicity of other metals, such as lead, aluminium or manganese are similarly mediated through astrocytic loss of function, as all these metals are primarily accumulated into astrocytes. In particular, exposure to lead results in a significant down-regulation of astrocytic glutamate transporters387 and in cell swelling possibly because of increased water transport through AQP4 channels.388 Similarly, aluminium and manganese reduce astrocytic glutamate uptake, thus leading to excitotoxicity and secondary neuronal death.389,390,391
Environmental toxins and neurodegenerative disorders
Exposure to various types of environmental toxins including trace metals as just discussed, but also pesticides such as organophosphates, substances of abuse such as methamphetamine, organic solvents, air pollutants, and others are increasingly recognised as risk factors in neurodegenerative disorders such as PD and others.392,393 Many of these substances induce widespread changes in astrocyte functions, including loss of functions and dysregulated astrogliosis, which in turn may serve as comorbidities in degenerative conditions such as AD, PD, and others.105,393,394,395
Conclusions
In summary, toxic encephalopathies are primary astrocytopathies, in which toxic substances are accumulated by and damage astrocytes, with neuronal loss being secondary to astroglial homoeostatic failure. Given the essential role of astrocytes in clearing and detoxifying many substances and the changes in astrocyte functions that such substances can evoke, it is interesting to consider (and worthy of more extensive investigation) that chronic exposure to trace amounts of such substances might act as risk factors and comorbidities in a variety of CNS neurodegenerative conditions.
Neuropsychiatric disorders: predominance of astrocytic atrophy and loss of function
Mood disorders
Major depressive disorder (MDD)
Decreases in the total number of glial cells and astrocytes are the main and most consistent histopathological hallmark of MDD post-mortem brains. Such decreases have been documented by both stereological and immunocytochemical studies. Decreases in astrocyte densities were observed in many brain regions including hippocampus, amygdala, prefrontal and anterior cingulate cortex.106,108,396,397,398,399 Astrocytes in post-mortem brains and in animal models of depression show atrophic morphology. Vimentin-positive astrocytes in the prefrontal white matter show fewer primary processes400; astrocytes in the grey matter lose ~ 50% of their perivascular endfeet.401 In addition, MDD is associated with significant decreases in astrocytic glutamate transporters and glutamine synthetase, impairing neurotransmission.402,403
In experimental models of depression, which are mainly based on exposure to unpredictable or social stress, numbers of GFAP-positive astrocytes, as well as GFAP levels in cortex and hippocampus, are significantly reduced.404,405 These morphological changes are accompanied by decreased astrocytic metabolism and glutamate uptake.406,407 Detailed analysis of astrocytic morphology using genetic reports (either ALDH1L1-eGFP reporter mice or viral transfection with astrocyte-targeted mCherry) revealed significant decrease of astrocytic complexity and shrinkage of astrocytic territorial domains in prefrontal cortex (Fig. 11,408,409). This structural atrophy was associated with a decrease in astrocytic expression of linker ezrin409; ezrin is essential for astrocytic morphological plasticity and extension of astrocytic leaflets.410,411 Anti-depressant treatment with fluoxetine or with specific acupuncture alleviated depressive behaviours, rescued astrocytic atrophy and restored ezrin expression.409 In Flinders Sensitive Line rats, which spontaneously develop depression-like phenotype, hippocampal GFAP-positive astrocytes are smaller and have atrophic arborisation.412 Likewise, astrocytes with small cell bodies and less complex processes were identified in Rhesus macaques with a self-injurious behaviour.413

Exposure to Chronic Unpredictable Stress (CUMS) induces morphological atrophy in prefrontal cortex astrocytes in mice. a Representative 3D reconstruction of astrocyte in control and CUMS groups. b Sholl analysis of astrocytic morphology for control and CUMS groups shows the number of intersections of astrocytic branches with concentric spheres centred in the middle of cell soma. c Maximal number of intersections for astrocytes in control and CUMS groups. d Average length of astrocytic processes in control and CUMS groups. b–d n = 15 for each group. e Representative examples of astrocytic territorial domains obtained as a projection of astrocytes along the z-axis projection for control and CUMS animals. f Average astrocytic domain area (E) and average length of astrocytic processes for control and CUMS group. All data are presented as mean ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001. The number of experiments is indicated on each column. Reproduced from ref. 409
Partial ablation of astrocytes in the prefrontal cortex of healthy rats (by injection of gliotoxin L-α-aminoadipic acid) resulted in the development of depressive-like behaviours, whereas injection of neurotoxin ibotenate did not have such an effect.404 Ablation of astrocytes also caused secondary damage to neurones translated into impaired working memory and learning.414 Very similar effects were caused by inhibiting glial glutamate transporters in the prefrontal cortex.415 At the same time, boosting astrocytic glutamate uptake with riluzole406 or ceftriaxone restored depressive behaviours triggered by chronic stress.416 Experimental models of depression also demonstrate uncoupling of astrocytic syncytia, whereas pharmacological inhibition of gap junctions by intracerebral infusion of carbenoxolone induced anhedonia and anxiety-like behaviour.417 Chronic stress additionally down-regulates astrocytic expression of AQP4.418
Bipolar disorder (BP)
Both the total number of glial cells and astrocytic densities are substantially (up to 40%) decreased in the post-mortem BP tissues, whereas numbers of neurones are not changed.419,420,421,422 Loss of astrocytes leads to an overall decrease in neuroprotection and affects neurotransmission.423 Astrocytic atrophy and reduced synaptic coverage might be responsible for hyperactive glutamatergic transmission often observed in BP, especially during mania phases.424,425,426,427
Post-traumatic stress disorder (PTSD)
A substantial (and increasing) number of people experience PTSD after various forms of traumatic experience. Pathological changes in astrocytes in various models of PTSD are similar to those in models of depression.110 For example, GFAP levels as well as densities of GFAP-positive astrocytic profiles are decreased in a single prolonged stress model of PTSD.428 Astrocytes in experimental PTSD model show significant atrophy with decreased processes complexity and the appearance of fusiform morphotypes.429
Anxiety disorders
In anxiety, astrocytes display generalised atrophy similar to that seen in other mood disorders. For example, about 60% of astrocytic perivascular endfeet are lost in the prefrontal cortex in high anxiety-like behaviour rats developing endogenous anxiety.430 Astrocytes in these rats have fewer processes and less complex arborisation.430,431 Anxiety may also be linked to astrocytic glutamate uptake deficiency, as intra-amygdala injections of glutamate transporter inhibitor dihydro-kainic acid causes anxiety behaviour.415
Antidepressants target astrocytes
Antidepressant drugs include (i) selective serotonin reuptake inhibitors (SSRI), of which fluoxetine (Prozac) is the most popular, (ii) serotonin/noradrenaline reuptake inhibitors (SNRI), (iii) tricyclic antidepressants, (iv) inhibitors of monoaminoxidase, (v) lithium, and (vi) ketamine. Lithium, valproic acid (VPA), and carbamazepine (CBZ) are used as anti-bipolar drugs. The SSRI, which inhibit serotonin transporter SERT/SLC6A4 (expressed in both neurones and astroglia) and thus increase bioavailability of serotonin, are probably the most widely used in the treatment of depression. Besides acting on neurones, fluoxetine and other SSRIs affect astrocytes; in the context of major depression, SSRI treatment reverses both astrocytic atrophy and the decrease in astrocyte numbers in animal models.405,432 Incidentally, acupuncture at the Zusanli (ST36) acupoint that is used to treat depressive symptoms, prevented the development of both astrocytic atrophy and depressive-like behaviours in mice subjected to chronic stress regimen.409 Surprisingly, the main molecular target of SSRIs in astrocytes is not the SERT but rather 5-HT2B serotonin receptors. Treatment with fluoxetine upregulates expression of 5-HT2B receptors (which is decreased by chronic stress) selectively in astrocytes, and not in neurones.433 More importantly however, fluoxetine acts as a powerful and specific agonist of astrocytic 5-HT2B receptors. Activation of these receptors by fluoxetine triggers several signalling cascades.434,435,436 In addition, fluoxetin-induced activation of astrocytic 5-HT2B receptors induces transactivation of epidermal growth factor receptors, which, in turn, recruit MAPK/ERK or PI3K/AKT signalling cascades to regulate expression of several genes (such as Ca2+-dependent phospholipase A2, cPLA2, subtype 2 of adenosine deaminases acting on RNA’s, ADAR2, or subtype 2 of kainate receptors, GluK2) related to mood disorders.437,438 Fluoxetine also normalises interstitial pH, which is affected in mood disorders, by phosphorylating and stimulating astrocytic Na+-H+ transporter NHE/SLC9a1.439
Lithium, which is an effective treatment against acute mania and depression in the context of BP, has several cellular targets. In particular, lithium inhibits glycogen synthase kinase 3β,440 which translates in an increase of the density and complexity of astrocytes in the animal models of depression.441 Lithium also regulates astrocytic morphology through the extracellular matrix regulatory enzyme lysyl oxidase (LOX) and peroxisome proliferator-activated receptor γ, with LOX being the most highly regulated lithium-responsive astroglial gene.441
The central anaesthetic ketamine is an inhibitor of NMDA receptors, with surprisingly potent and rapid anti-depressant action.442 Research into ketamine actions in the CNS revealed multiple effects, including potentiation of synaptogenesis, increased density of dendritic spines and upregulation of postsynaptic levels of AMPA receptors.443 In a rodent model of depression, treatment with ketamine alleviated aberrant behaviours and restored astrocytic atrophy.412 Ketamine action on the morphology of astrocytes is mediated through cAMP intracellular signalling: ketamine directly increases cytoplasmic cAMP without activation of metabotropic receptors.444 Ketamine also potentiates cholesterol transport from astrocytes to neurones, thus stimulating synaptogenesis and morphological plasticity.444
Obsessive compulsive disorders (OCD)
Obsessive compulsive disorders (OCD), such as for example Tourette syndrome or grooming disorders, are clinically presented by repetitive behaviours or compulsions and intrusive thoughts, or obsessions.445 The pathophysiology of OCD revolves around aberrant excitatory-inhibitory balance.446 Astrocytes are fundamental for both excitatory and inhibitory transmission through glutamate glutamate (GABA)-glutamine shuttle447 and chloride ionostasis 330 and are thus well positioned to contribute to OCD pathophysiology.448 Indeed, deletion of EAAT1 astrocytic glutamate transporter translates into OCD-like phenotype manifested by excessive self-grooming and repetitive tic-like head shake449 arguably linked to hyperactive resting state activity and aberrant functional connectivity of the cortico-striatal-thalamic circuitry as revealed by diffusion functional magnetic resonance imaging.450 Experimentally reducing astrocyte calcium signalling in striatal circuits induces OCD-like repetitive behaviours in rodents.451 Aberrant astrocytic glutamate homoeostasis may also be linked to Tourette syndrome.452 Recently the role of astrocytic protein SAPAP3, encoded by Dlgap3 gene, in pathophysiology of OCD was suggested based on in depth analysis of astrocytic proteome. The SAPAP3 regulates astrocytic morphology, their territorial domains and notably the volume and extension of perisynaptic leaflets.453
Schizophrenia
Astrocytic changes in schizophrenia are rather mild. There are no signs of reactivity, instead there are observations of a generalised decrease in the density of astrocytes in cortical and subcortical areas, and even more prominent astrocytic depletion in the white matter.454,455,456 Dystrophic and swollen astrocytes were detected in electron microscopy of post-mortem brains of schizophrenic patients.457 Similarly post-mortem analysis revealed decreased levels of astrocytic EAAT1/2 glutamate transporters.458,459 Animals with genetic deletion of astrocytic glutamate transporters show some schizophrenia-like behaviours.460 Astrocytes may contribute to the pathophysiology of schizophrenia through an increased production of NMDA receptor blocker kynurenic acid, which may also link risk of schizophrenia with infection as described in the previous chapter. Finally, there is evidence for an abnormal and delayed maturation of astrocytes derived from stem cells obtained from schizophrenia patients. These astrocytes show less complex morphology and functional asthenia461 and may be a part of generalised glial deficiency in embryonic development that is linked to schizophrenia.462
Addictive disorders
Glutamatergic transmission regulates reward circuitry and dopaminergic connectivity. Glutamatergic transmission is controlled and regulated by astrocytic glutamate-glutamine shuttle, thereby providing a means by which astrocytes may contribute to the pathophysiology of addictive disorders through glutamatergic pathways.463 Experimental models of addictive disorders demonstrate significant morphological atrophy of astrocytes in nucleus accumbens.109,464 Both GFAP-positive astrocytic profiles and 3D reconstructions of astrocytes expressing green florescent protein in the nucleus accumbens of rats addicted to cocaine show atrophic morphology, with the volume of reconstructed cells decreasing by 19% and surface area by 21% as well as reduced synaptic coverage.107 Reduction of the association of astrocytic leaflets with synapses is linked to a decreased expression of ezrin (which is responsible for leaflets extension), while deletion of ezrin increased addictive behaviour.464 In addition to morphological dystrophy, development of addiction is associated with significant decrease in expression of astrocytic glutamate transporters, reduced glutamate clearance, increased levels of glutamate and increased glutamate spillover in nucleus accumbens.465,466,467 Moreover, addiction build-up seems to be linked to astrocytic NMDA receptors, such that deletion of the NR2C subunit specifically from astrocytes potentiated extinction of cocaine preference memory.468 Generalised astrocytic atrophy is also characteristic for alcoholism, in which astrocytes display loss of function, such as for example reduction in synthesis and secretion of thrombospondins.469,470,471,472
Conclusions
Neuropsychiatric diseases are characterised by widespread astroglial asthenia, with decreased cell numbers, morphological atrophy, and loss of function. Ablation of astrocytes, or inhibition of astrocyte specific homoeostatic molecules in experimental animals trigger behavioural abnormalities that resemble neuropsychiatric symptomatology in humans. Despite widely popularised views of the inflammatory nature of psychiatric diseases, there is little evidence corroborating neuroinflammatory changes of CNS tissue.
Neurodevelopmental disorders
Down syndrome
Down syndrome (named so after John Langdon Down who described this syndrome under the name of ‘Mongolism’ in 1866473) is the genetic disorder caused by an extra copy of chromosome 21. Clinically Down syndrome is manifested by hypothyroidism, malformation of the cardiac system and gastrointestinal tract, abnormal hearing and vision, and aberrant development of the brain resulting in severe intellectual disability.474 The neocortex of Down syndrome individuals is characterised by profound (30–50%) decrease in the number of neurones and neuroglia.475 Aberrant and premature developmental gliogenesis (which ultimately affects both neurones and glia) lies at the core of the cellular pathophysiology of Down syndrome.476,477 This abnormal gliogenesis is linked to a deficit in Sonic hedgehog (Shh) signalling. Even a single injection of an Shh agonist was shown to rescue neuronal numbers and behavioural deficits in an animal model of the syndrome.478 The gene encoding astrocyte-specific protein S100B is located in chromosome 21 and increased levels of S100B are characteristic for Down syndrome.479 Another cascade linking astrocytes to the pathophysiology of Down syndrome is an increased production of hydrogen sulphide, which is catalysed by cystathionine-β-synthase expressed, more or less exclusively, in astrocytes, and upregulated in a rat model of the disease. Inhibition of this enzyme was claimed to rescue pathological behaviours.480
Spina bifida
Spina bifida (the split spine in Latin) arises from pathological embryogenesis, in which the neural tube fails to close. Clinical manifestations are multifaceted and include disabilities with motor, urinary, intestinal, sexual and mental presentations.481 Neurodegeneration and abnormal nervous tissue development is, arguably, triggered by the invasion of amniotic fluid into the developing spinal cord and adding to the mechanical trauma. In particular, spina bifida is characterised by premature astrogliogenesis, which affects neuronal development and instigates reactive astrogliosis.482
Astrocytic RASopathy
Mutations of rat sarcoma virus or RAS proteins are behind a class of disorders known as RASopathies.483 Frequently, RASopathies present cognitive deficits and delayed cognitive development. The pathophysiology of RASopathies is centred on excessive foetal astrogliogenesis and prevalence of functionally deranged astrocytes.484,485 In Costello syndrome, which is a form of RASopathy caused by mutations of the gene encoding HRAS protein, astrocytes are hypertrophic and demonstrate increased proliferative activity; it was argued that these astrocytes increase synaptogenesis through elevated secretion of synaptogenic factors and premature formation of perineuronal net.486
Intellectual disability
Intellectual disability (ID) is a heterogeneous genetic disorder in children with an incidence of 1–3%, representing a substantial public health burden.487,488 Familial studies have revealed a relatively large number of X-linked ID (XLID) forms which may explain the excess of affected males.489 In families affected by XLID, a number of loss-of-function mutations were found in the GDI1 gene,490 which encodes a cytosolic protein αGDI, the RAB GDP dissociation inhibitor, involved in the control of the guanosine diphosphate (GDP)-bound form of the brain RAB GTPases491,492 and regulates vesicle dynamics.493,494 Two isoforms of GDI are present: GDI1 encodes for αGDI, principally enriched in the brain, whilst GDI2 encodes for βGDI that is ubiquitously expressed.495
Initial studies of the role of GDI1 were focused on neurones. Neuronal migration and differentiation were impaired in GDI1-null mice, while synaptic vesicle dynamics was also affected.490 Moreover, the deletion of GDI likely affected neuronal vesicle trafficking, thereby altering short-term synaptic plasticity and causing short-term memory deficits.496 Conditional and neurone specific deletion of GDI1 (CamkII-Cre+-Gdi1flox/Y model), showed that the down regulation of αGDI in neurones in adult forebrain regions was sufficient to recapitulate the learning deficits previously shown in Gdi1-null mice.497
Astrocytes, however, also express αGDI and vesicle traffic in astrocytes is similarly impaired in the absence of GDI1.498 Proteomic analysis of astrocytes from astrocyte-specific knock-out GDI1 animals499 revealed a significant change in the expression of genes responsible for glucose homoeostasis. Specifically, glycogen phosphorylase, the enzyme mediating glycogenolysis, was significantly decreased in GDI1 deficient astrocytes Glycogen is in the brain predominantly, if not exclusively, expressed in astrocytes.206 In addition, expression of the mitochondrial isoform of phosphoenolpyruvate carboxykinase, involved in gluconeogenesis from non-carbohydrate carbon substrates such as pyruvate, lactate, glycerol and glucogenic amino acids, was significantly increased,499 indicating an elevated demand for free glucose through enhancing astrocytic glucose metabolism in GDI1-null mice. Indeed, measurements of noradrenaline (NA)-induced aerobic glycolysis by FRET nanosensors in individual astrocytes demonstrated an increased glucose utilisation, manifested as reduced NA-elevated free cytosolic glucose concentration, whereas the production of lactate by NA was unaltered.499 Measurements of NA-induced changes in cyclic adenosine monophosphate (cAMP) revealed an increased sensitivity of this second messenger to changes in extracellular lactate, indicating altered signalling landscape at the plasma membrane in relation to lactate homoeostasis. Behaviour in mice with astrocyte-specific GDI1 deletion showed a selective and significant impairment in working memory, which was rescued by inhibiting glycolysis by 2-deoxy-D-glucose injection. These results indicate that astrocytes contribute to pathophysiology of ID and cognitive impairment.499
Autistm spectrum disorders, ASD
Autism spectrum disorders (ASD; from Greek αυτός that means ‘being alone with yourself’) is a hypernym embracing the extended group of polyaetiological pathologies manifested by deviant social interactions, impaired language skills, and restrictive behaviours. The ASD-related nosological entities are many, yet they all seem to reflect an abnormal development of the CNS in pre- and early postnatal periods, linked to either genetic heritage or environmental insults.500,501,502 Glial contribution to the pathophysiology of ASD remains to be fully characterised.503 The transcriptome of brain samples from ASD patients demonstrated changes in expression of glial genes related to regulation of synaptogenesis and glial reactivity.504 Increased number of GFAP-positive astrocytes, oligodendrocytes and microglia were detected in the striatum, with no changes in glial numbers in the amygdala.111 Functionally, astrocytes derived from stem cells obtained from ASD patients and grafted into the rodent brain generated abnormal Ca2+ signals, which are arguably linked to impaired long-term potentiation and repetitive behaviours.505 Some evidence suggests that mutations in neuroligins expressed mainly in astrocytes and involved in regulation of synaptogenesis, can be related to ASD pathophysiology.506
A specific form of ASD, known as Rett syndrome (clinically manifested with microcephaly, loss of motor coordination, stereotypic hand wringing, ataxia, seizures, and sleep disturbances) is caused by a loss of function mutation of the methyl-CpG-binding protein 2 or MECP2 gene, expressed in both neurones and neuroglia.507 Deletion of this gene from astrocytes decreases glutamate clearance and produces ASD-like behavioural phenotypes including aberrant locomotion and anxiety, whereas re-expression of wild type MECP2 gene in astrocytes alleviated these symptoms.508,509 Clinical presentation of Rett syndrome often includes aberrant breathing patterns, which can be mimicked in mice by astrocyte-specific deletion of MECP2,510 whereas re-introduction of the gene rescued abnormal breathing.511 A most common single-gene form of ASD is the fragile X syndrome, also known as Martin–Bell, or Escalante’s syndrome. Fragile X syndrome is caused by mutations in FMR1 gene (which encodes fragile X mental retardation protein, FMRP) expressed in both neurones and astrocytes. Deletion of this gene in mice leads to the decrease in synaptic coverage by astrocytic leaflets, which translates into reduced densities of glutamatergic synapses.512 Deficits of astrocytic support of synaptogenesis can also be linked to a decrease in secretion of astrocyte-specific synaptogenic molecules such as hevin, SPARC proteins and thrombospondin-1.513,514 Astrocytes from FMR1 knockout mouse model of fragile X syndrome were characterised by an enhanced secretion pf interleukin-6 and tenascin C, as well as by increased purinergic signalling.515
Conclusions
Neurodevelopmental disorders are primarily associated with aberrant differentiation of neural cells. Notably, a premature shift of neural stem cells away from neuronogenesis and to gliogenesis seems to play a leading role in several syndromes. Furthermore, functional deficiencies of astrocytes promote neuronal damage, thus contributing to the cognitive deficits.
Neurodegenerative diseases
Astroglial decline in the ageing brain
Ageing is the main risk factor for many diseases, including an extended spectrum of neurodegenerative disorders leading to senile dementia. Conceptually, ageing reduces the functional capacity of all organs and systems, ultimately weakening the whole organism, reducing its adaptability, wearing out its defensive systems and bringing it to death through age-dependent diseases. In the process of physiological ageing, both systemic and tissue-specific systems show functional downfalls, which, when projected to the brain, translate into weakening defence and metabolic strain resulting from age-dependent declines of the cardio-vascular system and overall decreases in metabolism. The nervous system withstands ageing better than other organs and systems (a phenomenon recognised long ago516), which arguably reflects the high plastic potential allowing constant remodelling of the nervous tissue (the process which underlines life-long learning and adaptation) and high defensive and regenerative potential of neuroglia defining nervous system resilience and regeneration mending various insults accumulating during life-span.115,517 Indeed, even though human intelligence remains high long after 5th decade of life, other systems show substantial decline in the physical capacities of youth.
Neuroglial changes in physiological brain ageing are relatively subtle, with white matter being the most affected: in healthy old age, white matter volume is decreased by ~ 10%, whereas the grey matter is reduced by a mere 3%.518 This reflects age-dependent decrease in myelin, linked to a decrease in the number of oligodendrocytes and their precursors.519,520 Microglia in aged human brain show substantial morphofunctional deterioration with ~ 40% of all microglial cells becoming morphologically dystrophic and functionally asthenic.521,522 Likewise ageing astrocytes became atrophic, thus reducing their homoeostasis and defensive support.115 These changes in neuroglia define the susceptibility of the brain to pathology and hence glial performance defines physiological (with cognitive preservation) versus pathological (cognitive decline) brain ageing.
Studying the transcriptome of the ageing brain revealed substantially more prominent changes in neuroglia, and in astroglia in particular, when compared to neurones. Transcriptome profiling of human post-mortem tissues (obtained from people aged between 16 and 102 years) found prominent and complex changes in gene expression in oligodendrocytes and astrocytes, whereas the neuronal transcriptome remained more or less undisturbed.523 Analysis of gene expression in cortical astrocytes from old mice identified an increase in genes linked to an immune response with a decrease in expression of GFAP and genes related to neuroprotection and neuronal support,524 and the prominent up-regulation of genes contributing to synapse elimination in astrocytes from aged hippocampus and cerebellum.151 Single-cell RNA sequencing of ~50,000 transcriptomes from young (3 - 4 months) and old (21–23 months) mice demonstrated substantial regional heterogeneity of age-dependent changes indicating that ageing of neurones and glia may develop through distinct molecular pathways.525
Age-dependent changes of astrocytic morphology have been understudied, with some controversial observations reported when only the morphology of GFAP-stained profiles was analysed, which can easily be misinterpreted. Using combinatorial approaches, total numbers of astrocytes seem not to change in physiological ageing neither in humans nor in rodents.71,115,526 Expression of GFAP is generally increased with brain ageing, which was regarded as a sign of widespread astroglial reactivity and age-dependent neuroinflammation.527,528 At the same time, morphometric analysis of GFAP-positive profiles delivered contradictory results, as both increases and decreases in size and complexity of GFAP-positive astrocytic profiles in ageing were reported (see ref. 115 for details). Golgi staining of aged astrocytes did not show major morphological changes529, whereas age-dependent changes in astrocytes stained with antibodies against S100B and glutamine synthetase are characterised by complex region-dependent changes ranging from atrophy to hypertrophy.530 In marmoset, GFAP-positive astrocytic profiles showed remarkable atrophy in advanced ages.531
More accurate studies of morphology of astrocytes in aged mice in situ using intracellular injections of fluorescent dye Alexa Fluor 594 with subsequent two-photon imaging and 3D reconstruction identified a substantial reduction of astrocytic size and complexity with particular degradation of perisynaptic leaflets (Fig. 12).114 Very similar atrophic changes were found in human tissues obtained during surgery: again, substantial decreases in size, complexity and territorial domains of protoplasmic cortical astrocytes were documented (Fig. 12, ref. 532). Reduced astrocytic presence impaired glutamate clearance and K+ buffering thus affecting synaptic plasticity.114 Of note, supplying the old brains with young of stem cell-derived astrocytes in experimental rodents was beneficial, once more confirming the fundamental role of astrocytic homoeostatic support in brain ageing.533

Astrocytic atrophy in ageing. Upper panel shows 3D reconstructions of Alexa 594 filled cortical astrocytes of mice of different ages. Reproduced from ref. 114 Lower panel shows 3D of Alexa 594 filled astrocytes from the cortex of human patients (tissue obtained during neurosurgery) of different ages. Reproduced from ref. 532
Functionally aged astrocytes retain their receptors and capacity of generating Ca2+ signalling in response to neurochemical stimulation.534,535 Resting membrane of aged astrocytes remains highly hyperpolarised (~−80 mV), the input resistance increases somewhat with age, thus reflecting morphological shrinkage.114 The density of AMPA, NMDA and P2X receptors as well as the density of plasmalemmal glutamate transporter currents demonstrates bell-shaped age dependency, with maximal densities observed in adult animals, whereas ageing is accompanied by a 2-3 fold decrease of density of respective ion currents.534 Ageing seems to affect astrocytic spontaneous Ca2+ oscillations, which occur ~20 times more frequently in 20 months old (aged) mice when compared to young adult mice of 2.5 months of age.536 Astrocytic syncytial coupling is also decreased with age, probably reflecting age-dependent down-regulation of connexins expression.114,537 Overall, fundamental physiological properties of astrocytes seem to reflect generalised atrophy of these cells in ageing. All major astroglia functions decline with age (Fig. 13). This decline arguably represents the principal mechanism for age-dependent neurological disorders. Glial paralysis opens the gate for neurodegeneration and other diseases of old age.

Decline in astrocytic functions in ageing
Homoeostasis of major neurotransmitters, which is the most fundamental function of astroglial cells, is impaired in old nervous tissue. Ageing of the brain is paralleled by an increase of the glutamate to glutamine ratio indicating abnormal operation of the glutamate (GABA)-glutamine shuttle.538 Aged astrocytes indeed express fewer glutamate transporters, resulting in age-dependent decreases in glutamate clearance.539 Glutamate transporter currents, which are a direct measure of the glutamate transport, decrease to ~15% of their level in young adult astrocytes.534 Astrocytic catabolism of catecholamines is also affected by ageing. Astrocytic expression of MAO-B rises 2–3 times in old nervous tissue,540 which limits catecholamine bioavailability and may contribute to neurodegeneration.541,542 Finally, aged astrocytes were reported to increase synthesis and release of GABA, which may impair excitation-inhibition balance on the neuronal networks.543,544
Ageing also affects astrocytic endfeet and glia limitans perivascularis. Ageing is associated with increased thickness of capillary walls, basement membranes, and astroglial endfeet vascular coverage.545 In the ageing brain, AQP4 migrates away from endfeet, which reduces operational capacity of the glymphatic system by ~40%.546 Morphological atrophy of astrocytes also reduces their presence in the neuropil, which leads to an increase of extracellular diffusion channels and hence to an increase in mean diffusivity of the grey matter, detected in elderly humans with diffusion tensor imaging.547 Metabolic decline of aged astrocytes is also well documented.548,549 In addition, ageing may affect astrocyte-dependent neurotransmitter clearance, thus further limiting neuronal support.550 Old astrocytes substantially down-regulate synthesis of cholesterol due to a decreased expression of the main cholesterol synthesising enzyme HMG-CoA reductase,151 which limits synaptogenesis and membrane repair. Ageing is also associated with a prominent decrease in the neurogenic potential reflecting functional asthenia of stem radial astrocytes.551,552,553 Finally ageing impairs the ability of astrocytes to mount protective reactive astrogliosis120 thus limiting brain protection against pathological insults.
To summarise, ageing is accompanied by significant decline in astrocytic haemostatic and neuroprotective capacities, which may directly affect nervous tissue and increase it vulnerability to pathological attacks and neurodegeneration.
Amyotrophic lateral sclerosis, ALS
Amyotrophic lateral sclerosis is a malignant and rapid degeneration of upper and lower motor neurones located in the cortex, brain stem and spinal cord. Clinically, ALS presents itself as a progressive ascending paralysis and muscle atrophy leading ultimately to respiratory failure and death.554 Astrocyte pathology is a fundamental, if not leading, factor in neuronal death: astrocytes in ALS lose their capability to sustain neurones, to clear glutamate and they some aberrant astrocytic forms are generated, which may cause direct damage.555,556,557 In addition, astrocytic vesicular traffic in ALS context is enhanced, which is linked to aberrant Ca2+ signalling and homoeostasis.558 Although most ALS is sporadic, several familial genetic forms also exist and have provided animal models. Studies of a mouse model of familial ALS that express human mutant superoxide dismutase 1 (SOD1G93A) support a primary role of astrocytes. Expression of mutant SOD1 in neurones did not result in ALS symptoms.559 In contrast, silencing of this gene in astrocytes in global SOD1 expressing mice with ALS-like symptoms, arrested pathological progression.560,561 Similarly, grafting astrocytes hosting SOD1G93A mutant into the spinal cord of the healthy mice promoted motor neurones demise resulting in ALS symptoms.562 At the same time transplanting healthy astrocytes into the spinal cord of globally SOD1G93A mutant expressing rats delayed pathological progression.563
At least three major pathological astrocytic phenotypes have been identified in ALS: these are degenerating/atrophic, reactive and aberrant morphotypes. Functionally all of these phenotypes are characterised by loss of several homoeostatic molecules, which decreases neuronal support and neuroprotection ultimately resulting in neuronal death.
Pathological astrocytes, when mixed with healthy neurones, can, on their own precipitate neuronal death, again indicating a leading role of astrocytes in pathophysiology of ALS. Astrocytes derived from stem cells generated from ALS patients cannot sustain neurones in co-culture; instead, they decrease neuronal survival.564 Likewise, primary astrocytes prepared from ALS post-mortem samples induced necroptosis of neurones in co-culture,565 and transplantation of diseased astrocytes into the spinal cord triggered motor deficits and promoted motor neurones degeneration.566 Neurotoxicity is likely to be associated with aberrant proliferating astrocytes, which express astrocytic and microglial markers, and are characterised by the presence of lipid droplets and many secretary vesicles. Aberrant astrocytes cannot provide homoeostatic support, but instead generate reactive oxygen species that may mediate neurotoxicity.139,158,557,567
Atrophic and reactive astrocytes in ALS demonstrate profound loss of homoeostatic and supportive function. One of the leading mechanisms of astrocyte-associated neurotoxicity is associated with severe decrease in the expression of glutamate transporters, the presence of which is reduced, in ALS patients, to only ~10% of the levels in healthy subjects.144 Comparable decline in glutamate transporters is observed in the SOD1G93A mouse model of ALS.568,569 Deceased expression of astrocytic glutamate transporters was also detected in SOD1G93A mice,568,569 whereas transgenic overexpression of EAAT2570 or pharmacological stimulation of its expression with riluzole571,572 delayed progression of experimental APS. Astrocytes in ALS show many signs of functional decline, for example they demonstrate abnormal Ca2+ signalling,573 impaired secretion of neuroprotective glial-derived neurotrophic factor,574 as well as reduced lactate production.575
Fronto-temporal dementia, FTD
The role and contribution of astroglia to FTD remains largely unexplored. It seems however, (at least based on a limited number of studies) that degeneration, loss of function, dysregulated metabolism, impaired second messenger signalling and death of astrocytes are predominant pathophysiological manifestations.576,577 Astrocytes in FTD up-regulate apoptotic markers and show signs of apoptotic death577,578; moreover the prevalence of apoptotic astrocytes seems to correlate with the severity of the dementia.579 Clasmatodendrosis of astrocytes was observed in the majority of post-mortem samples from FTD patients.121 Mutations in the progranulin gene, GRN, which cause a genetic familial FTD, downregulate expression of EAAT2 astrocyte glutamate transporter, which mediates synaptic degeneration.143
Alzheimer’s disease
Alzheimer’s disease is a complex pathology that appears in early onset familial forms (~1–2% of all cases; hereditary disease linked to mutated genes encoding amyloid precursor protein APP, presenilins1 and 2, and tau protein) and late onset sporadic forms for which age is the main risk factor.580,581 Clinical progression of AD is defined first by massive synapse loss and subsequently by neuronal death, which ultimately results in profound brain atrophy and is clinically manifested by severe dementia. Senile plaques (extracellular depositions) and neuronal tangles (intracellular accumulation of misphosphorylated tau protein), are the main histopathological hallmarks of the AD, critical for post-mortem diagnosis. The once widely accepted primary role for β-amyloid in sporadic AD (known as the amyloid cascade hypothesis) is currently criticised as not being the sole cause of AD, but nevertheless, amyloid accumulation is clearly a part of AD pathology and evokes multifaceted glial responses.582,583,584
Astrocytic changes in AD are complex, with disease stage and brain region specificity and include reactivity, degeneration and clasmatodendrotic death, and atrophy with loss of function.34,585 Astrocytic reactivity in AD is arguably triggered by β-amyloid deposits; although at the advanced stages of the disease the dying neurones and compromised blood-brain barrier may act as instigators. In post-mortem specimens from AD victims, reactive, hypertrophic astrocytes as well as reactive microglia surround senile plaques, thus creating a protective barrier.586,587 Similarly, reactive astrocytes are associated with β-amyloid deposits in the brains of various transgenic AD models (most of which are models of hyperamyloidosis), although sometimes reactive remodelling of astrocytes precedes formation of β-amyloid plaques.588,589 Astrocytic reactivity in the vicinity of senile plaques is, as a rule, of anisotropic, non-proliferative, mild variety; territorial domains of astrocytes surrounding amyloid plaques do not overlap, and astrocytes never form a barrier resembling that around traumatic or ischaemic lesions. There are also no indications for fibrotic changes in the nervous tissue of AD patients and AD animal models. Reactive astrocytes in AD are characterised by up-regulated GFAP expression, enlarged soma and thickened main processes.71,590,591 At the same time neither GFAP levels nor astrocytic reactivity correlates with the severity of pathology592; and no significant difference in GFAP was found in the brains of demented and cognitively preserved people of advanced age.593 Reactive astrocytes play predominantly protective role, and inhibition of reactivity exacerbates pathology in AD animal models.594 Astrocytic reactivity is most likely triggered by β-amyloid595 and subsequent Ca2+ release from the ER.596 Notably, memory loss in the PS2APP mouse AD model has been linked to astrocyte Ca2+ hypoactivity due to reduced expression by astrocytes of the calcium sensor, STIM1; and overexpression of STIM1 selectively in astrocytes fully recovers astrocyte Ca2+ activity and synaptic plasticity in this model.597
Clasmatodendrotic astrocytes were found both in human post-mortem samples122,598 and in APP-SweDI (Swedish-Dutch-Iowa mutation of APP) expressing mice.599 The interlaminar astrocytes (which populate brains of high primates and are not present in other species32,33) are highly vulnerable to AD pathology: at the advanced stages of the disease interlaminar astrocytes disappear,600 although how this impacts on the disease pathophysiology remains unknown. Protoplasmic astrocytes with atrophic morphology were found in triple-transgenic (3xTG-AD; harbour mutant APP, PS1 and tau) and PDAPP-J20 (expressing Swedish-Indiana APP mutations). These astrocytes, characterised by reduced somata volume, thinner and less complex processes, are present in cortical regions and in the hippocampus.71,533,601,602,603,604 Morphological atrophy of astrocytes develops in a brain-region-linked fashion, starting first in the entorhinal and prefrontal cortices (which are, incidentally, the most vulnerable to AD) and then spreading to hippocampus.116,605,606 Reminiscent of the animal models, astrocytes differentiated from induced pluripotent stem cells obtained from patients with both familial and sporadic forms of AD are smaller and less complex compared to astrocytes form healthy controls; in addition, these ‘diseased’ astrocytes demonstrate abnormal localisation of key astrocytic markers.607 In functional studies, astrocytes differentiated from stem cells obtained from familial AD showed aberrant Ca2+ signalling,608,609 increased production of reactive oxygen species, reduced lactate supply, compromised neuroprotection, and inability to support neurones in co-cultures.610,611 Furthermore, APOE4-expressing astrocytes differentiated from stem cells could not internalise β-amyloid and had abnormal cholesterol accumulation.612 Astrocytic dystrophy and glucose hypometabolism were also observed in brain imaging of AD patients.595 Transcriptome analyses suggest both gain and loss of functions in astrocytes in AD mouse models, with down-regulation of genes associated with neuronal support and upregulation of inflammation related genes.613,614
Astrocytes in AD animal models, as well as astrocytes differentiated from stem cells isolated from AD patients, are characterised by decreased energy metabolism, decreased glucose uptake and glucose consumption.615,616 Deficient glycolysis limits production of lactate for neuronal support and L-serine, an obligatory precursor of D-serine, which neurones secrete to sustain synaptic plasticity.617 Exposure of astrocytes to β-amyloid down-regulates expression of glutamine synthetase,618 and in animal models, glutamine synthetase expression is reduced in astrocytes surrounding β-amyloid plaques.619 Likewise, the activity of glutamine synthetase is profoundly decreased in the brain tissue of AD patients due to protein oxidation.620 Astrocytic production of glutamine and associated glutamate-glutamine shuttle operation are reduced in AD, affecting synaptic transmission as well as detoxification of ammonium.447 Furthermore, astrocytic atrophy and reduced presence of astrocytes around synapses affects synaptic maintenance, glutamate homoeostasis, and K+ buffering thus affecting neuronal excitability and limiting synaptic plasticity.116 Astrocytic asthenia is also manifested by the limited reactivity in the most vulnerable brain regions such as entorhinal and prefrontal cortices in which astrocytes do not mount a defence response in the presence of β-amyloid depositions.116,605,606 Similarly, astrocytic reactivity seems to fade at the advanced stages of AD in human patients.621
Astrocytic remodelling in AD also includes an increased synthesis and secretion of GABA: high GABA concentrations were found in astrocytes in animal models and in AD patients.154,543,544 Increased GABA synthesis reflects up-regulation of glutamic acid decarboxylase GAD67 and monoaminoxidase-B (MAO-B), the latter producing GABA from putrescine.154 Increase in expression of MAO-B is arguably linked to an aberrant activity of the urea cycle linked β-amyloid.622 Secretion of GABA from astrocytes, likely mediated by Best1 anion channels,623 may increase tonic inhibition thus counteracting neuronal hyperexcitability characteristic for AD.624 In particular, astrocyte-derived released GABA activates neuronal GABAA and GABAB receptors, which, in turn, inhibit neuronal activity. Thus, GABA from reactive astrocytes diminishes the spike probability of the perforant-path-to-dentate-granule-cell synapse, leading to impairment in synaptic plasticity and memory function.155 The by-product of the putrescine catabolism is hydrogen peroxide, which is released from reactive astrocytes in the AD thus adding to the damage of the nervous tissue. Treatment of animals with excessive β-amyloidosis with newly developed reversible MAO-B inhibitor KDS2010 or the potent H2O2 scavenger AAD-2004 ameliorated neurodegeneration.155 At the molecular levels astrocytic switch to putrescine catabolism and GABA synthesis is linked to the urea cycle expressed in astrocytes and generally responsible for detoxification of brain ammonium. Astrocytes in AD model animals demonstrated up-regulated expression of genes (CPS1, OCT, ASL, ARG1, and ODC1) and metabolites (aspartate, ammonia, urea, putrescine, and GABA) of the urea cycle. In healthy CNS tissue, urea metabolism is non-cyclic, whereas it becomes fully cyclic upon exposure to β-amyloid. Astrocytic uptake of β-amyloid leads to it autophagic degradation, entrance of excess aspartate and ammonia into the urea cycle, increased putrescine and GABA production, with increased H2O2 production and neuronal damage leading to cognitive deficits. Inhibition or down-regulation of ODC1 breaks this vicious cycle and promotes astrocytic detoxification of β-amyloid.625 This demonstrates how physiologically relevant metabolic pathways employed by astrocytes for maintaining the homoeostasis of the nervous tissue may acquire detrimental proportions under conditions of excessive or prolonged stress.
All in all, astrocytic responses in AD are complex and disease stage-specific: in the early, compensated phase of AD (manifested by mild cognitive impairment) astrocytes, together with other neuroglia become reactive and arguably protect brain tissue. At advanced AD stages, glial paralysis contributes to brain tissue atrophy and clinical dementia (Fig. 14).

Glial reactivity, decline and paralysis define the pathophysiology of AD. At the prodromal and early stages of the disease astrocytes display atrophic morphology possibly indicating limited homoeostatic support, which may lead to the early synaptic malfunction. After emergence of the plaques reactive astrocytes and microglia surround β-amyloid depositions to protect the nervous tissue. Progressive decline in glial neuroprotection and homoeostatic support culminates in glial paralysis which permits neuronal death and brain atrophy manifested in dementia
Astrocytes as targets for AD management and therapy
At present is seems unlikely that a single magic bullet will be found to cure AD or other neurodegenerative diseases leading to dementia. Nevertheless, various approaches that may help sustain cognitive resilience during aging are emerging. For example, lifestyle modifications in the widest possible context can prolong cognitive longevity; in particular dieting, intellectual and physical activity, as well as social engagement were shown to delay or even improve age-dependent cognitive impairments.626,627,628 Experimental evidence is accumulating that astrocytes readily respond to lifestyle modifications, and may be instrumental in conferring clinical benefits. For example, exposure of mouse AD model to either enriched environment or voluntary physical activity, or combination of both reversed astrocytic atrophy and decreased β-amyloid load.92,603 Environmental stimulation and voluntary exercise also restores neurogenesis that is severely inhibited in AD mice.552,629 Dieting is another easily modifiable lifestyle factor. Caloric restriction in particular may increase cognitive resilience and sustain cognitive longevity.630 Caloric restriction regimen translates into increased astrocytic complexity, size of astrocytic territorial domains and volume of perisynaptic leaflets, paralleled with an improved glutamate clearance and K+ buffering, all improving synaptic plasticity.631 Special diets with high intake polyunsaturated fatty acid 2-hydroxy-docosahexaenoic were also shown to rescue astroglial atrophy, restore adult neurogenesis and improve cognitive performance in 5xTG AD mouse model.632
Astrocytes are primary targets for noradrenergic innervation633 that are provided by widespread projections of neurones located in locus coeruleus and are critical for cognitive functions. Neurones of this nucleus are particularly vulnerable to ageing and neurodegeneration and locus coeruleus is arguably the first location being affected in AD. Enhancing bioavailability of noradrenaline or adrenergic responsiveness of astrocytes could be a valid therapeutic strategy.541,542 This can be achieved by inhibiting astrocytic MAO-B, and indeed deperenil (aka selegiline) showed some efficacy in improving memory and clinical progression of AD.634 Inhibitors of MAO-B can also reduce hydrogen peroxide production linked to the putrescine catabolism. The transcranial direct current stimulation, which improves cognitive symptoms in AD patients, is mediated through α1-adreniceptors-mediated massive Ca2+ signalling in cortical astrocytes.635 Further exploration of such potential mechanisms that involve astrocyte is warranted.
Parkinson’s disease
Parkinson’s disease named after James Parkinson who provided the first description of this disorder,636 is characterised by progressive degeneration of dopaminergic midbrain neurones of the brainstem. Clinically the disease is manifested after ~70% of dopaminergic neurones die.637
Both astrocytic reactivity and astrocytic atrophy with loss of function are observed in affected brain regions in PD. Astrocytic reactivity,638,639 might be of secondary nature as a response to neuronal damage, especially in toxic animal models of PD. Reactive astrocytes in the context of PD increase production of ROS and decrease antioxidative protection, which may translate into neuronal damage.640,641 At the same time astrocytes in brain samples and in organoids made from iPSCs-differentiated astrocytes form Parkin-mutation familial PD had a much decreased GFAP expression compared to controls.642 Astrocytes obtained form stem cells isolated from patients with PD linked to LRRK2/dardarin mutation showed prominent morphological atrophy,113 as well as decreased expression of glutamate transporters,643 and prominent mitochondrial deficiencies.113,644 Astrocyte-selective expression of PD related A53T mutant α-synuclein causes severe downregulation of glutamate transporters expression, resulting in neuronal damage, aberrant microgliosis and paralysis.645 Protoplasmic (but not fibrous) astrocytes protect against α-synuclein toxicity by removing this latter through endocytosis,646,647 and hence astrocytic atrophy reduces neuroprotection.
Mitochondrial insufficiency in astrocytes is a hallmark of PD, leading to a profound deterioration of astrocytic homoeostatic, supportive, and protective capabilities.648 Maintenance of metabolic support and mitochondria pool of striatal neurones seems to be one of the astrocytic functions which possibly define the pathophysiology of PD. In particular astrocytes can be a central element in maintaining mitochondrial function of dopaminergic neurones thorough transmitophagy, the process when astrocytes receive and degrade damaged neuronal mitochondria,649 or even supply healthy mitochondria.650 Astrocyte-neuronal mitochondrial exchange, which alleviated neuronal damage was recently demonstrated in the co-cultures systems651,652; in particular mitochondrial donation rescued dopaminergic neurones.652
Huntington’s disease
Huntington’s disease (HD, or Huntington’s chorea; named after George Huntington who was the first to describe it653) is caused by a single dominant allele of the huntingtin gene containing an expanded number of CAG repeats; the disease develops when the number of repeats exceeds 40.654 Of note, astrocyte-specific deletion of mutant huntingtin in mice globally expressing this gene eased disease symptoms and delayed its progression, indicating a role for astrocytes in the pathophysiology of the disease.655 Astrocytes in HD undergo morphological atrophy and loss of many homoeostatic functions. In animal models of HD, atrophic astrocytes in striatum retract their leaflets from cortico-striatal synapses, which are known to be affected in the early HD.656 Similar atrophy was found in human astrocytes expressing mutant huntingtin and transplanted into the corpus callosum of mice.657 Astrocytic loss of function includes compromised K+ buffering,128 glutamate transport and Ca2+ signalling.117 Significant decrease in expression of Kir4.1 channels in striatal astrocytes was also detected in human tissue.658 Likewise, expression of glutamate transporters is reduced in astrocytes from human tissue and animal models of HD112,659; in particular astrocyte-selective expression of mutated huntingtin with 160 GAG repeats triggered profound down-regulation of EAAT2 and HD-like phenotype in mice.660
Astrotauopathies
Astrotauopathies are a distinct class of neurodegenerative diseases caused by an abnormal accumulation of tau exclusively in astrocytes.661,662 Astrotauopathies result in astrocytes with various morphotypes classified as (i) astrocytic plaques, (ii) tufted astrocytes, (iii) ramified astrocytes, (iv) globular astroglial inclusions, (v) thorn-shaped astrocytes and (vi) granular/fuzzy astrocytes (Fig. 15,661). Astrotauopathies drive the pathophysiology of progressive supranuclear palsy, corticobasal degeneration, Picks disease, argyrophilic grain disease, globular glial tauopathies, and ageing-related tau astrogliopathy (ARTAG).

Histopathology of astrocytes in astrotauopathies. See text for explanation. Modified, and reproduced from the images kindly provided by Professor Gabor Kovacs, Toronto University
Tufted astrocytes, characterised by accumulation of tau in proximal astrocytic processes, represent the histopathological hallmark of progressive supranuclear palsy, a rare neurodegenerative disorder with progressive impairment of balance, walking, eye movement, muscular rigidity, dysarthria, and dysphagia.663,664 Corticobasal degeneration is a primary astrocytopathy characterised by the accumulation of astrocytic plaques. These are visualised as fuzzy short argyrophilic processes arranged annularly with fine collaterals at vertical or sharp angles with tau is accumulated mainly at the distal parts of enlarged astrocytic processes.664,665 Clinically, corticobasal degeneration is manifested by movement deficits, impaired swallowing, speech, and memory. As the disease progresses, tau spreads to neurones causing cell death.
Another example of a primary astrocytopathy is the frontotemporal lobar degeneration or Picks dementia, leading to progressive loss of memory, primary progressive aphasia, and social misbehaviour. The onset of the disease is associated with massive dystrophy and death of astrocytes, which directly correlate to the severity of the disease.579,666 Ramified astrocytes with tau depositions in soma and in processes are the histopathological hallmark of the Pick’s disease.667 Global primary tauopathies, characterised by widespread 4-repeat tau inclusions in astrocytes and oligodendrocytes result in dementia dues to neurodegeneration in frontal and temporal lobes.661,668 Finally, ARTAG, which represents a spectrum of age-dependent dementias caused by tau accumulation in astrocytes. Pathological astrocytic morphotypes are (i) thorn-shaped astrocytes with tau inclusions in soma and proximal processes, and in the subpial and perivascular endfeet and (ii) granular-fuzzy astrocytes with fine granular tau inclusions mainly in the perinuclear region.662 This latter type of astrocyte was also detected in argyrophilic grain disease, in AD and HD.662
Conclusions
The contribution of neuroglial cells to pathophysiology of neurodegenerative diseases is complex and heterogeneous with substantial disease and disease-stage stage specificity. Aberrant and diseased astrocytes promote death of motor neurones in amyotrophic lateral sclerosis, whereas loss of astrocyte homoeostatic support contributes to neuronal malfunction and demise in AD, PD, and HD.
Future perspectives: astrocyte targets as a frontier for new therapies for neurological disorders
Astroglia (as well as all neuroglial cells) provide for comprehensive support of the CNS; together with other neuroglia, astroglia are indispensable elements of the nervous tissue maintaining its normal function in health and guarding it against disease. The pathophysiology of astroglia is complex, highly heterogeneous and mutable: multiple pathology-associated phenotypes may co-exist and emerge and disappear during the progression of a neuropathology. In the absence of detrimental genetic mutations or polymorphisms, any lesion of the brain triggers astroglia responses that are in the first instance aimed at the preservation and/or restoration of homoeostasis. Profound lesions resulting in the death of parenchymal cells and inflammation instigate reactive astrogliosis that forms a border between damaged non-neural lesion core and surrounding neural tissue. This border is of paramount importance for the survival of adjacent neural tissue and is ultimately required for post-lesional tissue regeneration. Most chronic diseases of the CNS are associated with loss of function of astrocytes, and such loss of function is emerging as a leading mechanism of disorder-related neurological dysfunctions ranging from mild changes in synapse regulation to potential overt neurotoxicity in extreme cases. Contrary to a popular belief, astrocytes do not, through a specific pre-programmed gain of function, generate a specific toxin(s) or adopt a specific toxic phenotype proposed to be common across many diseases that may represent a ‘universal’ therapeutic target. There is as yet no rigorous evidence for this. Instead, the disease and context specific loss or disruption of astrocyte functions can result in the accumulation of various potentially toxic metabolites or in the loss of support functions for neurones or synapses or in (the deadliest) combination of both. Notably, astrocyte loss of functions can lead to elaboration of potentially toxic molecules such as ROS or certain lipids and this may be mistaken for a ‘purposeful’ gain of detrimental function. Astrocytic functional paralysis translates into neuronal damage or death because neurones themselves are incapable of preserving tissue homoeostasis. Thus, understanding, and counteracting disease-associated astrocyte loss of functions should be a major therapeutic goal. As yet, we are not in a possession of astrocyte-specific therapies or drugs; nonetheless we know that many medicines or lifestyle changes do affect astroglia. Commonly positive outcomes are linked to increases in astrocyte presence and or up-regulation of key homoeostatic cascades (the glutamate transporters being most singular targets). In this article we have highlighted many different astrocyte-associated molecules that are emerging as potential therapeutic targets in different disorder contexts. The extensive breadth of these different molecules highlights the diversity of astrocyte reactivity not only across disorders, but within the same disorder at different timepoints or in different CNS locations. This observation in turn emphasises the importance that therapies aimed at modulating astrocyte reactivity will need to modulate specific molecules and specific aspects of reactivity in a disorder and context specific manner. The notion that astrocyte reactivity is somehow globally harmful per se and should therefore be blocked in its entirety is likely to do more harm than good and is no longer tenable. Notably, in addition to trying to block potential downstream detrimental effects, new therapies need to be designed that preserve and boost astrocytic defences and improve astrocytic homoeostasis; such therapies may turn the tide by ultimately developing pathophysiology-based treatments of CNS diseases.
References
Semyanov, A. & Verkhratsky, A. Astrocytic processes: from tripartite synapses to the active milieu. Trends Neurosci. 44, 781–792 (2021).
Verkhratsky, A., Arranz, A. M., Ciuba, K. & Pekowska, A. Evolution of neuroglia. Ann. N.Y. Acad. Sci. 1518, 120–130 (2022).
Verkhratsky, A. & Nedergaard, M. The homeostatic astroglia emerges from evolutionary specialization of neural cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, 20150428 (2016).
Verkhratsky, A., Parpura, V., Vardjan, N. & Zorec, R. Physiology of astroglia. Adv. Exp. Med. Biol. 1175, 45–91 (2019).
Kettenmann, H. & Ransom, B. R. (Oxford University Press, Oxford, 2013).
Verkhratsky, A. & Butt, A. Neuroglia: Function and Pathology. (Academic Press, Elsevier, 2023).
Yeh, C. Y., Wu, K. Y., Huang, G. J. & Verkhratsky, A. Radial stem astrocytes (aka neural stem cells): Identity, development, physio-pathology, and therapeutic potential. Acta Physiol. 238, e13967 (2023).
Sofroniew, M. V. & Vinters, H. V. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 (2010).
Khakh, B. S. & Sofroniew, M. V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 18, 942–952 (2015).
Virchow, R. Die Cellularpathologie in ihrer Begründung auf physiologische and pathologische Gewebelehre. Zwanzig Vorlesungen gehalten während der Monate Februar, März und April 1858 im pathologischen Institut zu Berlin. First edition edn, (August Hirschwald, 1858).
Virchow, R. L. K. Cellular Pathology (John Churchill, 1860).
Marinesco, M. G. Lesions des centres nerveux produites par la toxine du Bacillus Botulinus. C. R. Soc. Biol. 48, 989–991 (1896).
Achucarro, N. Some pathological findings in the neuroglia and in the ganglion cells of the cortex in senile conditions. Bull. Gov. Hosp. Insane 2, 81–90 (1910).
Alzheimer, A. in Histologische und histopathologische Arbeiten über die Grosshirnrinde mit besonderer Berücksichtigung der pathologischen Anatomie der Geisteskrankheiten. Vol. 3 (eds F. Nissl & A. Alzheimer) 401–562 (Gustav Fischer, 1910).
Frommann, C. Untersuchungen über die Gewebsveränderungen bei der Multiplen Sklerose des Gehirns und Rückenmarks. (Verlag von Gustav Fischer, 1878).
Nissl, F. Über einige Beziehungen zwischen Nervenzellerkrankungen und gliösen Erscheinungen bei verschiedenen Psychosen. Arch. Psychiatr. 32, 1–21 (1899).
Frommann, C. Untersuchungen über die normale und pathologische Anatomie des Rückenmarks. Teil 1., (Friedrich Frommann, 1864).
Verkhratsky, A., Zorec, R. & Parpura, V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol. 27, 629–644 (2017).
Sofroniew, M. V. Astrocyte reactivity: subtypes, states, and functions in CNS innate immunity. Trends Immunol. 41, 758–770 (2020).
Escartin, C. et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325 (2021).
Courtine, G. & Sofroniew, M. V. Spinal cord repair: advances in biology and technology. Nat. Med. 25, 898–908 (2019).
O’Shea, T. M., Burda, J. E. & Sofroniew, M. V. Cell biology of spinal cord injury and repair. J. Clin. Investig. 127, 3259–3270 (2017).
Pekny, M. et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345 (2016).
Paolicelli, R. C. et al. Microglia states and nomenclature: a field at its crossroads. Neuron 110, 3458–3483 (2022).
Patel, D. C., Tewari, B. P., Chaunsali, L. & Sontheimer, H. Neuron–glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 20, 282–297 (2019).
Burda, J. E., Bernstein, A. M. & Sofroniew, M. V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 275, 305–315 (2016).
Verkhratsky, A. et al. Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro. 4, e00082 (2012).
Dobzhansky, T. Biology, molecular and organismic. Am. Zool. 4, 443–452 (1964).
Dobzhansky, T. Nothing in biology makes sense except in the light of evolution. Am. Biol. Teach. 35, 125–129 (1973).
Reichenbach, A. & Bringmann, A. in Evolutionary Neuroscience (Second Edition) (ed J. H. Kaas) 397–439 (Academic Press, 2020).
Verkhratsky, A. & Nedergaard, M. Physiology of astroglia. Physiol. Rev. 98, 239–389 (2018).
Colombo, J. A. & Reisin, H. D. Interlaminar astroglia of the cerebral cortex: a marker of the primate brain. Brain Res. 1006, 126–131 (2004).
Oberheim, N. A. et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287 (2009).
Preman, P., Alfonso-Triguero, M., Alberdi, E., Verkhratsky, A. & Arranz, A. M. Astrocytes in Alzheimer’s disease: pathological significance and molecular pathways. Cells 10, 540 (2021).
Verkhratsky, A., Oberheim Bush, N. A., Nedergaard, M. & Butt, A. M. The special case of human astrocytes. Neuroglia 1, 21–29 (2018).
Grosche, J. et al. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat. Neurosci. 2, 139–143 (1999).
Salmon, C. K. et al. Organizing principles of astrocytic nanoarchitecture in the mouse cerebral cortex. Curr. Biol. 33, 957–972.e955 (2023).
Nedergaard, M. & Verkhratsky, A. Artifact versus reality–how astrocytes contribute to synaptic events. Glia 60, 1013–1023 (2012).
Verkhratsky, A. & Nedergaard, M. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130595 (2014).
Verkhratsky, A. & Rose, C. R. Na+-dependent transporters: the backbone of astroglial homeostatic function. Cell Calcium 85, 102136 (2020).
Risher, W. C. & Eroglu, C. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biol. 31, 170–177 (2012).
Sancho, L., Contreras, M. & Allen, N. J. Glia as sculptors of synaptic plasticity. Neurosci. Res. 167, 17–29 (2021).
Augusto-Oliveira, M. et al. Astroglia-specific contributions to the regulation of synapses, cognition and behaviour. Neurosci. Biobehav. Rev. 118, 331–357 (2020).
Allen, N. J. & Eroglu, C. Cell biology of astrocyte-synapse interactions. Neuron 96, 697–708 (2017).
Bushong, E. A., Martone, M. E., Jones, Y. Z. & Ellisman, M. H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192 (2002).
Lopez-Hidalgo, M., Hoover, W. B. & Schummers, J. Spatial organization of astrocytes in ferret visual cortex. J. Comp. Neurol. 524, 3561–3576 (2016).
Shapson-Coe, A. et al. A connectomic study of a petascale fragment ofhuman cerebral cortex. bioRxiv Preprint, (2021).
Batiuk, M. Y. et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 11, 1220 (2020).
Ben Haim, L. & Rowitch, D. H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 18, 31–41 (2017).
Chai, H. et al. Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95, 531–549.e539 (2017).
Falcone, C. et al. Cortical interlaminar astrocytes are generated prenatally, mature postnatally, and express unique markers in human and nonhuman primates. Cereb. Cortex 31, 379–395 (2021).
John Lin, C. C. et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405 (2017).
Khakh, B. S. & Deneen, B. The emerging nature of astrocyte diversity. Annu Rev. Neurosci. 42, 187–207 (2019).
Kronschlager, M. T. et al. Lamina-specific properties of spinal astrocytes. Glia 69, 1749–1766 (2021).
Torres-Ceja, B. & Olsen, M. L. A closer look at astrocyte morphology: development, heterogeneity, and plasticity at astrocyte leaflets. Curr. Opin. Neurobiol. 74, 102550 (2022).
Zhang, Y. et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016).
Verkhratsky, A., Ho, M. S., Vardjan, N., Zorec, R. & Parpura, V. General pathophysiology of astroglia. Adv. Exp. Med Biol. 1175, 149–179 (2019).
del Río-Hortega, P. & Penfield, W. G. Cerebral cicatrix: the reaction of neuroglia and microglia to brain wounds,. Bull. John Hopkins Hosp. 41, 278–303 (1927).
Penfiled, W. & Buckley, R. C. Punctires of the brain. Arch. Neurol. Psychiat 20, 1–13 (1928).
Penfield, W. in Special Cytology. The Form and Functions of teh Cell in Health and Disease (ed E. V. Cowdry) 1033–1068 (Paul B Hoeber, Inc, 1928).
Al-Dalahmah, O. et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 8, 19 (2020).
Itoh, N. et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: focus on astrocytes. Proc. Natl Acad. Sci. USA 115, E302–E309 (2018).
Kamphuis, W. et al. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia 63, 1036–1056 (2015).
Zamanian, J. L. et al. Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410 (2012).
Burda, J. E. et al. Divergent transcriptional regulation of astrocyte reactivity across disorders. Nature 606, 557–564 (2022).
Yu, X. et al. Context-specific striatal astrocyte molecular responses are phenotypically exploitable. Neuron 108, 1146–1162.e1110 (2020).
Wanner, I. B. et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 33, 12870–12886 (2013).
Diaz-Castro, B., Bernstein, A. M., Coppola, G., Sofroniew, M. V. & Khakh, B. S. Molecular and functional properties of cortical astrocytes during peripherally induced neuroinflammation. Cell Rep. 36, 109508 (2021).
Hasel, P., Rose, I. V. L., Sadick, J. S., Kim, R. D. & Liddelow, S. A. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat. Neurosci. 24, 1475–1487 (2021).
Nam, M. H. et al. Excessive astrocytic GABA causes cortical hypometabolism and impedes functional recovery after subcortical stroke. Cell Rep. 32, 107861 (2020).
Olabarria, M., Noristani, H. N., Verkhratsky, A. & Rodriguez, J. J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58, 831–838 (2010).
Wilhelmsson, U. et al. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc. Natl Acad. Sci. USA 103, 17513–17518 (2006).
Krawczyk, M. C. et al. Human astrocytes exhibit tumor microenvironment-, age-, and sex-related transcriptomic signatures. J. Neurosci. 42, 1587–1603 (2022).
O’Shea, T. M. et al. Foreign body responses in mouse central nervous system mimic natural wound responses and alter biomaterial functions. Nat. Commun. 11, 6203 (2020).
Conforti, P. et al. Fibrinogen regulates lesion border-forming reactive astrocyte properties after vascular damage. Glia 70, 1251–1266 (2022).
Hackett, A. R. et al. Injury type-dependent differentiation of NG2 glia into heterogeneous astrocytes. Exp. Neurol. 308, 72–79 (2018).
Yi, C., Verkhratsky, A. & Niu, J. Pathological potential of oligodendrocyte precursor cells: Terra Incognita. Trends Neurosci. 46, 81–596 (2023).
O’Shea, T. M. et al. Lesion environments direct transplanted neural progenitors towards a wound repair astroglial phenotype in mice. Nat. Commun. 13, 5702 (2022).
Mills, W. A. III et al. Astrocyte plasticity in mice ensures continued endfoot coverage of cerebral blood vessels following injury and declines with age. Nat. Commun. 13, 1794 (2022).
Toft-Hansen, H., Fuchtbauer, L. & Owens, T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia 59, 166–176 (2011).
Voskuhl, R. R. et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci. 29, 11511–11522 (2009).
Aswendt, M. et al. Reactive astrocytes prevent maladaptive plasticity after ischemic stroke. Prog. Neurobiol. 209, 102199 (2022).
Bush, T. G. et al. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23, 297–308 (1999).
Faulkner, J. R. et al. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 24, 2143–2155 (2004).
Sofroniew, M. V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 16, 249–263 (2015).
Anderson, M. A. et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 (2016).
Morita, M. et al. Metabolic plasticity of astrocytes and aging of the brain. Int. J. Mol. Sci. 20, 941 (2019).
Wahane, S. & Sofroniew, M. V. Loss-of-function manipulations to identify roles of diverse glia and stromal cells during CNS scar formation. Cell Tissue Res. 387, 337–350 (2022).
Dorrier, C. E. et al. CNS fibroblasts form a fibrotic scar in response to immune cell infiltration. Nat. Neurosci. 24, 234–244 (2021).
Dias, D. O. et al. Pericyte-derived fibrotic scarring is conserved across diverse central nervous system lesions. Nat. Commun. 12, 5501 (2021).
Di Carlo, S. E. & Peduto, L. The perivascular origin of pathological fibroblasts. J. Clin. Investig. 128, 54–63 (2018).
Rodriguez, J. J., Terzieva, S., Olabarria, M., Lanza, R. G. & Verkhratsky, A. Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis. 4, e678 (2013).
Carvalho-Paulo, D. et al. Hippocampal astrocytes in migrating and wintering semipalmated sandpiper Calidris pusilla. Front. Neuroanat. 11, 126 (2017).
Santos, J. W. Q. et al. Circadian variation in GFAP immunoreactivity in the mouse suprachiasmatic nucleus. Biol. Rhythm Res. 36, 141–150 (2005).
Jing, R. et al. Synemin is expressed in reactive astrocytes in neurotrauma and interacts differentially with vimentin and GFAP intermediate filament networks. J. Cell Sci. 120, 1267–1277 (2007).
Pekny, T. et al. Synemin is expressed in reactive astrocytes and Rosenthal fibers in Alexander disease. APMIS 122, 76–80 (2014).
Potokar, M., Morita, M., Wiche, G. & Jorgacevski, J. The diversity of intermediate filaments in astrocytes. Cells 9, 1604 (2020).
Chun, H., Lim, J., Park, K. D. & Lee, C. J. Inhibition of monoamine oxidase B prevents reactive astrogliosis and scar formation in stab wound injury model. Glia 70, 354–367 (2022).
Hartmann, K. et al. Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol. Commun. 7, 83 (2019).
Pekna, M. et al. Astrocyte responses to complement peptide C3a are highly context-dependent. Neurochem. Res. 48, 1233–1241 (2023).
Suk, K. Lipocalin-2 as a therapeutic target for brain injury: an astrocentric perspective. Prog. Neurobiol. 144, 158–172 (2016).
Phares, T. W., Stohlman, S. A., Hinton, D. R. & Bergmann, C. C. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J. Virol. 87, 3382–3392 (2013).
Potokar, M. & Jorgacevski, J. Plectin in the central nervous system and a putative role in brain astrocytes. Cells 10, 2353 (2021).
Hazell, A. S. Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem. Int. 55, 129–135 (2009).
Li, B., Xia, M., Zorec, R., Parpura, V. & Verkhratsky, A. Astrocytes in heavy metal neurotoxicity and neurodegeneration. Brain Res. 1752, 147234 (2021).
Rajkowska, G. & Stockmeier, C. A. Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr. Drug Targets 14, 1225–1236 (2013).
Scofield, M. D. et al. Cocaine self-administration and extinction leads to reduced glial fibrillary acidic protein expression and morphometric features of astrocytes in the nucleus accumbens core. Biol. Psychiatry 80, 207–215 (2016).
Scuderi, C., Verkhratsky, A., Parpura, V. & Li, B. Neuroglia in psychiatric disorders. Adv. Neurobiol. 26, 3–19 (2021).
Kruyer, A. & Scofield, M. D. Astrocytes in addictive disorders. Adv. Neurobiol. 26, 231–254 (2021).
Li, B., Zhang, D. & Verkhratsky, A. Astrocytes in post-traumatic stress disorder. Neurosci. Bull. 38, 953–965 (2022).
Scuderi, C. & Verkhratsky, A. The role of neuroglia in autism spectrum disorders. Prog. Mol. Biol. Transl. Sci. 173, 301–330 (2020).
Jiang, R., Diaz-Castro, B., Looger, L. L. & Khakh, B. S. Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci. 36, 3453–3470 (2016).
Ramos-Gonzalez, P. et al. Astrocytic atrophy as a pathological feature of Parkinson’s disease with LRRK2 mutation. NPJ Parkinsons Dis. 7, 31 (2021).
Popov, A. et al. Astrocyte dystrophy in ageing brain parallels impaired synaptic plasticity. Aging Cell 20, e13334 (2021).
Verkhratsky, A. et al. Astroglial asthenia and loss of function, rather than reactivity, contribute to the ageing of the brain. Pflug. Arch. 473, 753–774 (2021).
Verkhratsky, A., Rodrigues, J. J., Pivoriunas, A., Zorec, R. & Semyanov, A. Astroglial atrophy in Alzheimer’s disease. Pflug. Arch. 471, 1247–1261 (2019).
Diaz-Castro, B., Gangwani, M. R., Yu, X., Coppola, G. & Khakh, B. S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 11, eaaw8546 (2019).
Ramón Y Cajal, S. Contribucion al conocimiento de la neuroglia del cerebro humano. Trab. Lab. Investig. Biol. 11, 255–315 (1913).
Balaban, D., Miyawaki, E. K., Bhattacharyya, S. & Torre, M. The phenomenon of clasmatodendrosis. Heliyon 7, e07605 (2021).
Early, A. N., Gorman, A. A., Van Eldik, L. J., Bachstetter, A. D. & Morganti, J. M. Effects of advanced age upon astrocyte-specific responses to acute traumatic brain injury in mice. J. Neuroinflamm. 17, 115 (2020).
Martin, J. A., Craft, D. K., Su, J. H., Kim, R. C. & Cotman, C. W. Astrocytes degenerate in frontotemporal dementia: possible relation to hypoperfusion. Neurobiol. Aging 22, 195–207 (2001).
Tomimoto, H. et al. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 94, 146–152 (1997).
Ferrer, I. et al. Aging-related tau astrogliopathy (ARTAG): not only tau phosphorylation in astrocytes. Brain Pathol. 28, 965–985 (2018).
Messing, A., Brenner, M., Feany, M. B., Nedergaard, M. & Goldman, J. E. Alexander disease. J. Neurosci. 32, 5017–5023 (2012).
Messing, A. Refining the concept of GFAP toxicity in Alexander disease. J. Neurodev. Disord. 11, 27 (2019).
Hendriksen, R. G. F., Vles, J. S. H., Aalbers, M. W., Chin, R. F. M. & Hendriksen, J. G. M. Brain-related comorbidities in boys and men with Duchenne Muscular Dystrophy: a descriptive study. Eur. J. Paediatr. Neurol. 22, 488–497 (2018).
Patel, A. M. et al. Dystrophin deficiency leads to dysfunctional glutamate clearance in iPSC derived astrocytes. Transl. Psychiatry 9, 200 (2019).
Tong, X. et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 17, 694–703 (2014).
Nagai, M. et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622 (2007).
Di Giorgio, F. P., Boulting, G. L., Bobrowicz, S. & Eggan, K. C. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3, 637–648 (2008).
Kamphuis, W. et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One 7, e42823 (2012).
Mahan, T. E. et al. Selective reduction of astrocyte apoE3 and apoE4 strongly reduces Abeta accumulation and plaque-related pathology in a mouse model of amyloidosis. Mol. Neurodegener. 17, 13 (2022).
Jackson, R. J. et al. APOE4 derived from astrocytes leads to blood-brain barrier impairment. Brain 145, 3582–3593 (2022).
Giarratana, A. O. et al. APOE4 genetic polymorphism results in impaired recovery in a repeated mild traumatic brain injury model and treatment with Bryostatin-1 improves outcomes. Sci. Rep. 10, 19919 (2020).
Yu, T. S. et al. Astrocytic ApoE underlies maturation of hippocampal neurons and cognitive recovery after traumatic brain injury in mice. Commun. Biol. 4, 1303 (2021).
Zhou, S., Tian, Y., Song, X., Xiong, J. & Cheng, G. Brain proteome-wide and transcriptome-wide association studies, Bayesian colocalization and Mendelian randomization analyses revealed causal genes of Parkinson’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 78, 563–568 (2022).
Rose, C. F., Verkhratsky, A. & Parpura, V. Astrocyte glutamine synthetase: pivotal in health and disease. Biochem Soc. Trans. 41, 1518–1524 (2013).
Wingerchuk, D. M., Lennon, V. A., Lucchinetti, C. F., Pittock, S. J. & Weinshenker, B. G. The spectrum of neuromyelitis optica. Lancet Neurol. 6, 805–815 (2007).
Trias, E., Barbeito, L. & Yamanaka, K. Phenotypic heterogeneity of astrocytes in motor neuron disease. Clin. Exp. Neuroimmunol. 9, 225–234 (2018).
Wilhelmsson, U. et al. Injury leads to the appearance of cells with characteristics of both microglia and astrocytes in mouse and human brain. Cereb. Cortex 27, 3360–3377 (2017).
Rothstein, J. D. et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686 (1996).
Todd, A. C. & Hardingham, G. E. The regulation of astrocytic glutamate transporters in health and neurodegenerative diseases. Int. J. Mol. Sci. 21, 9607 (2020).
Marsan, E. et al. Astroglial toxicity promotes synaptic degeneration in the thalamocortical circuit in frontotemporal dementia with GRN mutations. J. Clin. Investig. 133, e164919 (2023).
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J. & Kuncl, R. W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84 (1995).
Foran, E. & Trotti, D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid. Redox Signal 11, 1587–1602 (2009).
Hazell, A. S. et al. Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia 58, 148–156 (2009).
Shigetomi, E., Saito, K., Sano, F. & Koizumi, S. Aberrant calcium signals in reactive astrocytes: a key process in neurological disorders. Int. J. Mol. Sci. 20, 996 (2019).
Mulica, P., Grunewald, A. & Pereira, S. L. Astrocyte-neuron metabolic crosstalk in neurodegeneration: a mitochondrial perspective. Front Endocrinol. 12, 668517 (2021).
Dringen, R., Brandmann, M., Hohnholt, M. C. & Blumrich, E. M. Glutathione-dependent detoxification processes in astrocytes. Neurochem. Res. 40, 2570–2582 (2015).
Schreiner, B. et al. Astrocyte depletion impairs redox homeostasis and triggers neuronal loss in the adult CNS. Cell Rep. 12, 1377–1384 (2015).
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N. & Allen, N. J. The aging astrocyte transcript .ome from multiple regions of the mouse brain. Cell Rep. 22, 269–285 (2018).
Linnerbauer, M., Wheeler, M. A. & Quintana, F. J. Astrocyte crosstalk in CNS inflammation. Neuron 108, 608–622 (2020).
Han, R. T., Kim, R. D., Molofsky, A. V. & Liddelow, S. A. Astrocyte-immune cell interactions in physiology and pathology. Immunity 54, 211–224 (2021).
Jo, S. et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896 (2014).
Chun, H. et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2- production. Nat. Neurosci. 23, 1555–1566 (2020).
Vandenberg, G. G., Dawson, N. J., Head, A., Scott, G. R. & Scott, A. L. Astrocyte-mediated disruption of ROS homeostasis in Fragile X mouse model. Neurochem. Int. 146, 105036 (2021).
Guttenplan, K. A. et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 599, 102–107 (2021).
Yamanaka, K. & Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 126, 31–38 (2018).
McKee, A. C. & Daneshvar, D. H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 127, 45–66 (2015).
Burda, J. E. & Sofroniew, M. V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248 (2014).
Baracaldo-Santamaria, D. et al. Revisiting excitotoxicity in traumatic brain injury: from bench to bedside. Pharmaceutics 14, 152 (2022).
Farkas, O., Lifshitz, J. & Povlishock, J. T. Mechanoporation induced by diffuse traumatic brain injury: an irreversible or reversible response to injury? J. Neurosci. 26, 3130–3140 (2006).
Szydlowska, K. & Tymianski, M. Calcium, ischemia and excitotoxicity. Cell Calcium 47, 122–129 (2010).
Dong, X. X., Wang, Y. & Qin, Z. H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharm. Sin. 30, 379–387 (2009).
Sowers, J. L. et al. Traumatic brain injury induces region-specific glutamate metabolism changes as measured by multiple mass spectrometry methods. iScience 24, 103108 (2021).
Cisneros-Mejorado, A., Perez-Samartin, A., Gottlieb, M. & Matute, C. ATP signaling in brain: release, excitotoxicity and potential therapeutic targets. Cell Mol. Neurobiol. 35, 1–6 (2015).
Schachtrup, C. et al. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J. Neurosci. 30, 5843–5854 (2010).
Bradbury, E. J. & Burnside, E. R. Moving beyond the glial scar for spinal cord repair. Nat. Commun. 10, 3879 (2019).
Jonas, R. A. et al. The spider effect: morphological and orienting classification of microglia in response to stimuli in vivo. PLoS One 7, e30763 (2012).
Popovich, P. G. & Hickey, W. F. Bone marrow chimeric rats reveal the unique distribution of resident and recruited macrophages in the contused rat spinal cord. J. Neuropathol. Exp. Neurol. 60, 676–685 (2001).
Bellver-Landete, V. et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 10, 518 (2019).
Zhou, X. et al. Microglia and macrophages promote corralling, wound compaction and recovery after spinal cord injury via Plexin-B2. Nat. Neurosci. 23, 337–350 (2020).
Brennan, F. H. et al. Microglia coordinate cellular interactions during spinal cord repair in mice. Nat. Commun. 13, 4096 (2022).
Hampton, D. W., Rhodes, K. E., Zhao, C., Franklin, R. J. & Fawcett, J. W. The responses of oligodendrocyte precursor cells, astrocytes and microglia to a cortical stab injury, in the brain. Neuroscience 127, 813–820 (2004).
Niu, J. et al. Aberrant oligodendroglial–vascular interactions disrupt the blood–brain barrier, triggering CNS inflammation. Nat. Neurosci. 22, 709–718 (2019).
Abdelhak, A. et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 18, 158–172 (2022).
Goritz, C. et al. A pericyte origin of spinal cord scar tissue. Science 333, 238–242 (2011).
Soderblom, C. et al. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J. Neurosci. 33, 13882–13887 (2013).
Mira, R. G., Lira, M. & Cerpa, W. Traumatic brain injury: mechanisms of glial response. Front Physiol. 12, 740939 (2021).
Myer, D. J., Gurkoff, G. G., Lee, S. M., Hovda, D. A. & Sofroniew, M. V. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129, 2761–2772 (2006).
Kokaia, Z., Martino, G., Schwartz, M. & Lindvall, O. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat. Neurosci. 15, 1078–1087 (2012).
Ng, S. Y. & Lee, A. Y. W. Traumatic brain injuries: pathophysiology and potential therapeutic targets. Front. Cell Neurosci. 13, 528 (2019).
Johnson, V. E., Stewart, W. & Smith, D. H. Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43 (2013).
Kenzie, E. S. et al. The dynamics of concussion: mapping pathophysiology, persistence, and recovery with causal-loop diagramming. Front. Neurol. 9, 203 (2018).
Shandra, O. et al. Repetitive diffuse mild traumatic brain injury causes an atypical astrocyte response and spontaneous recurrent seizures. J. Neurosci. 39, 1944–1963 (2019).
George, K. K., Heithoff, B. P., Shandra, O. & Robel, S. Mild traumatic brain injury/concussion initiates an atypical astrocyte response caused by blood-brain barrier dysfunction. J. Neurotrauma 39, 211–226 (2022).
Millspaugh, J. A. Dementia pugilistica. US Nav. Med. Bull. 35, 297–226 (1937).
Hsu, E. T. et al. Astrocytic degeneration in chronic traumatic encephalopathy. Acta Neuropathol. 136, 955–972 (2018).
Sofroniew, M. V. Dissecting spinal cord regeneration. Nature 557, 343–350 (2018).
Anderson, M. A. et al. Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature 561, 396–400 (2018).
Sun, F. et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 480, 372–375 (2011).
He, Z. & Jin, Y. Intrinsic control of axon regeneration. Neuron 90, 437–451 (2016).
Poplawski, G. H. D. et al. Injured adult neurons regress to an embryonic transcriptional growth state. Nature 581, 77–82 (2020).
Zukor, K. et al. Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J. Neurosci. 33, 15350–15361 (2013).
Kuriakose, D. & Xiao, Z. Pathophysiology and treatment of stroke: present status and future perspectives. Int. J. Mol. Sci. 21 (2020).
Woodruff, T. M. et al. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 6, 11 (2011).
Deb, P., Sharma, S. & Hassan, K. M. Pathophysiologic mechanisms of acute ischemic stroke: an overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 17, 197–218 (2010).
Lau, A. & Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. 460, 525–542 (2010).
Choi, D. W. Excitotoxicity: still hammering the ischemic brain in 2020. Front. Neurosci. 14, 579953 (2020).
Rossi, D. J., Brady, J. D. & Mohr, C. Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci. 10, 1377–1386 (2007).
Liu, S., Levine, S. R. & Winn, H. R. Targeting ischemic penumbra: part I - from pathophysiology to therapeutic strategy. J. Exp. Stroke Transl. Med. 3, 47–55 (2010).
Giffard, R. G. & Swanson, R. A. Ischemia-induced programmed cell death in astrocytes. Glia 50, 299–306 (2005).
Almeida, A., Delgado-Esteban, M., Bolanos, J. P. & Medina, J. M. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J. Neurochem. 81, 207–217 (2002).
Chen, Y. & Swanson, R. A. Astrocytes and brain injury. J. Cereb. Blood Flow. Metab. 23, 137–149 (2003).
Brown, A. M. & Ransom, B. R. Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271 (2007).
Oe, Y., Baba, O., Ashida, H., Nakamura, K. C. & Hirase, H. Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns. Glia 64, 1532–1545 (2016).
Tekkok, S. B., Brown, A. M., Westenbroek, R., Pellerin, L. & Ransom, B. R. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res. 81, 644–652 (2005).
Zhao, Y. & Rempe, D. A. Targeting astrocytes for stroke therapy. Neurotherapeutics 7, 439–451 (2010).
Giffard, R. G., Monyer, H. & Choi, D. W. Selective vulnerability of cultured cortical glia to injury by extracellular acidosis. Brain Res. 530, 138–141 (1990).
Mitani, A. & Tanaka, K. Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J. Neurosci. 23, 7176–7182 (2003).
Desagher, S., Glowinski, J. & Premont, J. Astrocytes protect neurons from hydrogen peroxide toxicity. J. Neurosci. 16, 2553–2562 (1996).
Petrovic-Djergovic, D., Goonewardena, S. N. & Pinsky, D. J. Inflammatory disequilibrium in stroke. Circ. Res 119, 142–158 (2016).
Garcia-Bonilla, L. & Iadecola, C. Peroxiredoxin sets the brain on fire after stroke. Nat. Med. 18, 858–859 (2012).
Gulke, E., Gelderblom, M. & Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 11, 1756286418774254 (2018).
Sims, N. R. & Yew, W. P. Reactive astrogliosis in stroke: contributions of astrocytes to recovery of neurological function. Neurochem. Int. 107, 88–103 (2017).
Choudhury, G. R. & Ding, S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol. Dis. 85, 234–244 (2016).
Llorente, I. L. et al. Patient-derived glial enriched progenitors repair functional deficits due to white matter stroke and vascular dementia in rodents. Sci. Transl. Med. 13, eaaz6747 (2021).
Morizawa, Y. M. et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 8, 28 (2017).
Konishi, H., Koizumi, S. & Kiyama, H. Phagocytic astrocytes: emerging from the shadows of microglia. Glia 70, 1009–1026 (2022).
Damisah, E. C. et al. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 6, eaba3239 (2020).
Joy, M. T. et al. CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell 176, 1143–1157.e1113 (2019).
Joy, M. T. & Carmichael, S. T. Encouraging an excitable brain state: mechanisms of brain repair in stroke. Nat. Rev. Neurosci. 22, 38–53 (2021).
Gleichman, A. J. & Carmichael, S. T. Astrocytic therapies for neuronal repair in stroke. Neurosci. Lett. 565, 47–52 (2014).
Carmichael, S. T., Kathirvelu, B., Schweppe, C. A. & Nie, E. H. Molecular, cellular and functional events in axonal sprouting after stroke. Exp. Neurol. 287, 384–394 (2017).
Pekny, M., Wilhelmsson, U., Tatlisumak, T. & Pekna, M. Astrocyte activation and reactive gliosis—a new target in stroke? Neurosci. Lett. 689, 45–55 (2019).
Yamagata, K. Astrocyte-induced synapse formation and ischemic stroke. J. Neurosci. Res 99, 1401–1413 (2021).
Fomitcheva, I. V., Sword, J., Shi, Y. & Kirov, S. A. Plasticity of perisynaptic astroglia during ischemia-induced spreading depolarization. Cereb. Cortex 33, 5469–5483 (2022).
Klein, R. S. & Hunter, C. A. Protective and pathological immunity during central nervous system infections. Immunity 46, 891–909 (2017).
Combes, V., Guillemin, G. J., Chan-Ling, T., Hunt, N. H. & Grau, G. E. The crossroads of neuroinflammation in infectious diseases: endothelial cells and astrocytes. Trends Parasitol. 28, 311–319 (2012).
Coureuil, M. et al. Meningococcus hijacks a b2-adrenoceptor/beta-Arrestin pathway to cross brain microvasculature endothelium. Cell 143, 1149–1160 (2010).
Stenzel, W., Soltek, S., Schluter, D. & Deckert, M. The intermediate filament GFAP is important for the control of experimental murine Staphylococcus aureus-induced brain abscess and Toxoplasma encephalitis. J. Neuropathol. Exp. Neurol. 63, 631–640 (2004).
Chauhan, V. S., Sterka, D. G. Jr., Gray, D. L., Bost, K. L. & Marriott, I. Neurogenic exacerbation of microglial and astrocyte responses to Neisseria meningitidis and Borrelia burgdorferi. J. Immunol. 180, 8241–8249 (2008).
Farina, C., Aloisi, F. & Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145 (2007).
Bsibsi, M. et al. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 53, 688–695 (2006).
Carty, M. & Bowie, A. G. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem. Pharm. 81, 825–837 (2011).
Trudler, D., Farfara, D. & Frenkel, D. Toll-like receptors expression and signaling in glia cells in neuro-amyloidogenic diseases: towards future therapeutic application. Mediators Inflamm. 2010, 497987 (2010).
Li, L., Acioglu, C., Heary, R. F. & Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav. Immun. 91, 740–755 (2021).
Zorec, R. & Verkhratsky, A. Astrocytes in the pathophysiology of neuroinfection. Essays Biochem. 67, 131–145 (2022).
Kielian, T. Immunopathogenesis of brain abscess. J. Neuroinflamm. 1, 16 (2004).
Shulyatnikova, T. & Verkhratsky, A. Astroglia in sepsis associated encephalopathy. Neurochem. Res. 45, 83–99 (2020).
Suzuki, Y. et al. Impaired resistance to the development of toxoplasmic encephalitis in interleukin-6-deficient mice. Infect. Immun. 65, 2339–2345 (1997).
Suzuki, Y., Sa, Q., Gehman, M. & Ochiai, E. Interferon-γ- and perforin-mediated immune responses for resistance against Toxoplasma gondii in the brain. Expert Rev. Mol. Med. 13, e31 (2011).
Schwarcz, R. & Hunter, C. A. Toxoplasma gondii and schizophrenia: linkage through astrocyte-derived kynurenic acid? Schizophr. Bull. 33, 652–653 (2007).
Drogemuller, K. et al. Astrocyte gp130 expression is critical for the control of Toxoplasma encephalitis. J. Immunol. 181, 2683–2693 (2008).
Zorec, R., Zupanc, T. A. & Verkhratsky, A. Astrogliopathology in the infectious insults of the brain. Neurosci. Lett. 689, 56–62 (2019).
Balk, R. A. Systemic inflammatory response syndrome (SIRS): where did it come from and is it still relevant today? Virulence 5, 20–26 (2014).
Sonneville, R. et al. Understanding brain dysfunction in sepsis. Ann. Intens. Care 3, 15 (2013).
Varatharaj, A. & Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 60, 1–12 (2017).
Dantzer, R. & Kelley, K. W. Twenty years of research on cytokine-induced sickness behavior. Brain Behav. Immun. 21, 153–160 (2007).
Hart, B. L. Biological basis of the behavior of sick animals. Neurosci. Biobehav Rev. 12, 123–137 (1988).
Jorgacevski, J. et al. ZIKV strains differentially affect survival of human fetal astrocytes versus neurons and traffic of ZIKV-laden endocytotic compartments. Sci. Rep. 9, 8069 (2019).
Potokar, M., Korva, M., Jorgacevski, J., Avsic-Zupanc, T. & Zorec, R. Tick-borne encephalitis virus infects rat astrocytes but does not affect their viability. PLoS One 9, e86219 (2014).
Lindqvist, R. et al. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflamm. 13, 277 (2016).
Stefanik, M. et al. Characterisation of Zika virus infection in primary human astrocytes. BMC Neurosci. 19, 5 (2018).
Hong, S. & Banks, W. A. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav. Immun. 45, 1–12 (2015).
Gray, L. R. et al. HIV-1 entry and trans-infection of astrocytes involves CD81 vesicles. PLoS One 9, e90620 (2014).
Conrady, C. D. et al. Microglia and a functional type I IFN pathway are required to counter HSV-1-driven brain lateral ventricle enlargement and encephalitis. J. Immunol. 190, 2807–2817 (2013).
Kumaraswamy, G. K., Fu, M. M. & Docherty, J. J. Innate and adaptive host response during the initial phase of herpes simplex virus encephalitis in the neonatal mouse. J. Neurovirol. 12, 365–374 (2006).
Lokensgard, J. R. et al. Robust expression of TNF-α, IL-1β, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J. Neurovirol. 7, 208–219 (2001).
Chucair-Elliott, A. J. et al. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia 62, 1418–1434 (2014).
Zhang, L. et al. Human cytomegalovirus infection modulates thrombospondins 1 and 2 in primary fetal astrocytes. Neuroreport 24, 526–535 (2013).
Zhang, L. et al. HCMV induces dysregulation of glutamate uptake and transporter expression in human fetal astrocytes. Neurochem. Res. 39, 2407–2418 (2014).
Tavcar Verdev, P. et al. In human astrocytes neurotropic flaviviruses increase autophagy, yet their replication is autophagy-independent. Cell Mol. Life Sci. 79, 566 (2022).
Steardo, L. Jr, Steardo, L. & Verkhratsky, A. Psychiatric face of COVID-19. Transl. Psychiatry 10, 261 (2020).
Steardo, L., Steardo, L. Jr, Zorec, R. & Verkhratsky, A. Neuroinfection may contribute to pathophysiology and clinical manifestations of COVID-19. Acta Physiol. 229, e13473 (2020).
Zhou, Z., Kang, H., Li, S. & Zhao, X. Understanding the neurotropic characteristics of SARS-CoV-2: from neurological manifestations of COVID-19 to potential neurotropic mechanisms. J. Neurol. 267, 2179–2184 (2020).
Verkhratsky, A., Li, Q., Melino, S., Melino, G. & Shi, Y. Can COVID-19 pandemic boost the epidemic of neurodegenerative diseases? Biol. Direct 15, 28 (2020).
Tremblay, M. E., Madore, C., Bordeleau, M., Tian, L. & Verkhratsky, A. Neuropathobiology of COVID-19: the role for glia. Front. Cell Neurosci. 14, 592214 (2020).
Kanberg, N. et al. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology 95, e1754–e1759 (2020).
Reichard, R. R. et al. Neuropathology of COVID-19: a spectrum of vascular and acute disseminated encephalomyelitis (ADEM)-like pathology. Acta Neuropathol. 140, 1–6 (2020).
Crunfli, F. et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl Acad. Sci. USA 119, e2200960119 (2022).
Tachibana, M. et al. Clasmatodendrosis is associated with dendritic spines and does not represent autophagic astrocyte death in influenza-associated encephalopathy. Brain Dev. 41, 85–95 (2019).
Andrews, M. G. et al. Tropism of SARS-CoV-2 for human cortical astrocytes. Proc. Natl Acad. Sci. USA 119, e2122236119 (2022).
Geschwind, M. D. Prion diseases. Contin. 21, 1612–1638 (2015).
Westergard, L., Christensen, H. M. & Harris, D. A. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim. Biophys. Acta 1772, 629–644 (2007).
Prusiner, S. B. Prions. Proc. Natl Acad. Sci. USA 95, 13363–13383 (1998).
Tahir, W., Thapa, S. & Schatzl, H. M. Astrocyte in prion disease: a double-edged sword. Neural Regen. Res. 17, 1659–1665 (2022).
Dos Passos, G. R. et al. MOG-IgG-associated optic neuritis, encephalitis, and myelitis: lessons learned from neuromyelitis optica spectrum disorder. Front. Neurol. 9, 217 (2018).
Bruscolini, A. et al. Diagnosis and management of neuromyelitis optica spectrum disorders—an update. Autoimmun. Rev. 17, 195–200 (2018).
Paul, S., Mondal, G. P., Bhattacharyya, R., Ghosh, K. C. & Bhat, I. A. Neuromyelitis optica spectrum disorders. J. Neurol. Sci. 420, 117225 (2021).
Devic, E. in PCongrès français de médecine (Premiere Session; Lyon, 1894; procès-verbaux, mémoires et discussions; publiés par M. le Dr L. Bard) 434–439 (Asselin et Houzeau, Louis Savy, 1895).
Lennon, V. A., Kryzer, T. J., Pittock, S. J., Verkman, A. S. & Hinson, S. R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 202, 473–477 (2005).
Roemer, S. F. et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 130, 1194–1205 (2007).
Misu, T. et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 125, 815–827 (2013).
Ratelade, J. et al. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol. 123, 861–872 (2012).
Lucchinetti, C. F. et al. Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 365, 2188–2197 (2011).
Jiang, Y. et al. Renal function in patients with AQP4 antibody-positive neuromyelitis optica spectrum disorder. Reserach Sqaure Preprint, (2020).
Popescu, B. F. & Lucchinetti, C. F. Pathology of demyelinating diseases. Annu Rev. Pathol. 7, 185–217 (2012).
Dendrou, C. A., Fugger, L. & Friese, M. A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558 (2015).
Reich, D. S., Lucchinetti, C. F. & Calabresi, P. A. Multiple sclerosis. N. Engl. J. Med. 378, 169–180 (2018).
Ponath, G., Park, C. & Pitt, D. The role of astrocytes in multiple sclerosis. Front. Immunol. 9, 217 (2018).
Guerrero, B. L. & Sicotte, N. L. Microglia in multiple sclerosis: friend or foe? Front. Immunol. 11, 374 (2020).
Antel, J. P., Becher, B., Ludwin, S. K., Prat, A. & Quintana, F. J. Glial cells as regulators of neuroimmune interactions in the central nervous system. J. Immunol. 204, 251–255 (2020).
Absinta, M. et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714 (2021).
Clark, I. C. et al. Barcoded viral tracing of single-cell interactions in central nervous system inflammation. Science 372, eabf1230 (2021).
Wheeler, M. A. & Quintana, F. J. Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb. Perspect. Med. 9 (2019).
Brosnan, C. F. & Raine, C. S. The astrocyte in multiple sclerosis revisited. Glia 61, 453–465 (2013).
Pham, H. et al. The astrocytic response in early experimental autoimmune encephalomyelitis occurs across both the grey and white matter compartments. J. Neuroimmunol. 208, 30–39 (2009).
Wang, D. et al. Astrocyte-associated axonal damage in pre-onset stages of experimental autoimmune encephalomyelitis. Glia 51, 235–240 (2005).
Sorensen, T. L. et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J. Clin. Investig. 103, 807–815 (1999).
Ponath, G. et al. Enhanced astrocyte responses are driven by a genetic risk allele associated with multiple sclerosis. Nat. Commun. 9, 5337 (2018).
Wheeler, M. A. et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176, 581–596.e518 (2019).
Rothhammer, V. et al. Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018).
Sanmarco, L. M. et al. Gut-licensed IFNγ+ NK cells drive LAMP1+ TRAIL+ anti-inflammatory astrocytes. Nature 590, 473–479 (2021).
D’Ambrosi, N. & Apolloni, S. Fibrotic scar in neurodegenerative diseases. Front. Immunol. 11, 1394 (2020).
Yahn, S. L. et al. Fibrotic scar after experimental autoimmune encephalomyelitis inhibits oligodendrocyte differentiation. Neurobiol. Dis. 134, 104674 (2020).
Yokote, H. & Mizusawa, H. Multiple sclerosis and neuromyelitis optica spectrum disorders: some similarities in two distinct diseases. Neural Regen. Res. 11, 410–411 (2016).
Pitt, D., Werner, P. & Raine, C. S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 6, 67–70 (2000).
Haroon, F. et al. Gp130-dependent astrocytic survival is critical for the control of autoimmune central nervous system inflammation. J. Immunol. 186, 6521–6531 (2011).
Mayo, L. et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 20, 1147–1156 (2014).
Kim, R. Y. et al. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J. Neuroimmunol. 274, 53–61 (2014).
Clark, I. C. et al. Identification of astrocyte regulators by nucleic acid cytometry. Nature 614, 326–333 (2023).
Alexander, W. S. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 72, 373–381 (1949).
Lanciotti, A. et al. Astrocytes: emerging stars in leukodystrophy pathogenesis. Transl. Neurosci. 4, 144–164 (2013).
Sosunov, A., Olabarria, M. & Goldman, J. E. Alexander disease: an astrocytopathy that produces a leukodystrophy. Brain Pathol. 28, 388–398 (2018).
Tian, R. et al. Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J. Neuropathol. Exp. Neurol. 69, 335–345 (2010).
Duchenne, G. B. A. Recherches sur la paralysie musculaire pseudohypertrophique myo-sclerosique. Arch. Gen. Med. 11, 5–25 (1868).
Estrella, N. L. & Naya, F. J. Transcriptional networks regulating the costamere, sarcomere, and other cytoskeletal structures in striated muscle. Cell Mol. Life Sci. 71, 1641–1656 (2014).
Belhasan, D. C. & Akaaboune, M. The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci. Lett. 722, 134833 (2020).
Nowak, K. J. & Davies, K. E. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep. 5, 872–876 (2004).
Hendriksen, R. G. et al. Dystrophin distribution and expression in human and experimental temporal lobe epilepsy. Front. Cell Neurosci. 10, 174 (2016).
Mancarci, B. O. et al. Cross-laboratory analysis of brain cell type transcriptomes with applications to interpretation of bulk tissue data. eneuro 4, ENEURO.0212-0217.2017 (2017).
Connors, N. C., Adams, M. E., Froehner, S. C. & Kofuji, P. The potassium channel Kir4.1 associates with the dystrophin-glycoprotein complex via alpha-syntrophin in glia. J. Biol. Chem. 279, 28387–28392 (2004).
Na, I. et al. Ordered disorder of the astrocytic dystrophin-associated protein complex in the norm and pathology. PLoS One 8, e73476 (2013).
Jenrow, K. & Elisevich, K. in Understanding Epilepsy: A Study Guide for the Boards (eds M. V. Spanaki & V. S. Wasade) 1–18 (Cambridge University Press, 2019).
Kubista, H., Boehm, S. & Hotka, M. The paroxysmal depolarization shift: reconsidering its role in epilepsy, epileptogenesis and beyond. Int. J. Mol. Sci. 20, 577 (2019).
Danbolt, N. C. Glutamate uptake. Prog. Neurobiol. 65, 1–105 (2001).
MacAulay, N. Molecular mechanisms of K+ clearance and extracellular space shrinkage. Glia cells as the stars. Glia 68, 2192–2211 (2020).
Kwak, H. et al. Astrocytes control sensory acuity via tonic inhibition in the thalamus. Neuron 108, 691–706.e610 (2020).
Untiet, V. et al. Astrocytic chloride is brain state dependent and modulates inhibitory neurotransmission in mice. Nat. Commun. 14, 1871 (2023).
Verhoog, Q. P., Holtman, L., Aronica, E. & van Vliet, E. A. Astrocytes as guardians of neuronal excitability: mechanisms underlying epileptogenesis. Front. Neurol. 11, 591690 (2020).
Binder, D. K. & Steinhauser, C. Astrocytes and epilepsy. Neurochem. Res. 46, 2687–2695 (2021).
Binder, D. K. Astrocytes: stars of the sacred disease. Epilepsy Curr. 18, 172–179 (2018).
Plata, A. et al. Astrocytic atrophy following status epilepticus parallels reduced Ca2+ activity and impaired synaptic plasticity in the rat hippocampus. Front. Mol. Neurosci. 11, 215 (2018).
Bedner, P. & Steinhauser, C. Altered Kir and gap junction channels in temporal lobe epilepsy. Neurochem. Int. 63, 682–687 (2013).
Chever, O., Djukic, B., McCarthy, K. D. & Amzica, F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 30, 15769–15777 (2010).
Haj-Yasein, N. N. et al. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia 59, 1635–1642 (2011).
Sarac, S. et al. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS 117, 291–301 (2009).
Gorter, J. A. et al. Glutamate transporters alterations in the reorganizing dentate gyrus are associated with progressive seizure activity in chronic epileptic rats. J. Comp. Neurol. 442, 365–377 (2002).
Ueda, Y. et al. Collapse of extracellular glutamate regulation during epileptogenesis: down-regulation and functional failure of glutamate transporter function in rats with chronic seizures induced by kainic acid. J. Neurochem. 76, 892–900 (2001).
Watanabe, T. et al. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. 845, 92–96 (1999).
Eid, T. et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37 (2004).
Ortinski, P. I. et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 13, 584–591 (2010).
Amiry-Moghaddam, M. et al. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc. Natl Acad. Sci. USA 100, 13615–13620 (2003).
Lauritzen, F. et al. Altered expression of brain monocarboxylate transporter 1 in models of temporal lobe epilepsy. Neurobiol. Dis. 45, 165–176 (2012).
Chen, Z. P. et al. Lipid-accumulated reactive astrocytes promote disease progression in epilepsy. Nat. Neurosci. 26, 542–554 (2023).
Steinhauser, C., Seifert, G. & Bedner, P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60, 1192–1202 (2012).
Bedner, P. et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222 (2015).
Pannasch, U. et al. Astroglial networks scale synaptic activity and plasticity. Proc. Natl Acad. Sci. USA 108, 8467–8472 (2011).
Barros, L. F. How expensive is the astrocyte? J. Cereb. Blood Flow. Metab. 42, 738–745 (2022).
Larsen, B. R. et al. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 62, 608–622 (2014).
Rose, C. R. & Verkhratsky, A. Principles of sodium homeostasis and sodium signalling in astroglia. Glia 64, 1611–1627 (2016).
Capuani, C. et al. Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol. Med. 8, 967–986 (2016).
Jayakumar, A. R. & Norenberg, M. D. Hyperammonemia in hepatic encephalopathy. J. Clin. Exp. Hepatol. 8, 272–280 (2018).
Ochoa-Sanchez, R., Tamnanloo, F. & Rose, C. F. Hepatic encephalopathy: from metabolic to neurodegenerative. Neurochem. Res. 46, 2612–2625 (2021).
Clemmesen, J. O., Larsen, F. S., Kondrup, J., Hansen, B. A. & Ott, P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 29, 648–653 (1999).
Brusilow, S. W., Koehler, R. C., Traystman, R. J. & Cooper, A. J. Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 7, 452–470 (2010).
Vilstrup, H. et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the study of the liver. Hepatology 60, 715–735 (2014).
Zielinska, M., Albrecht, J. & Popek, M. Dysregulation of astrocytic glutamine transport in acute hyperammonemic brain edema. Front. Neurosci. 16, 874750 (2022).
Ott, P. & Larsen, F. S. Blood-brain barrier permeability to ammonia in liver failure: a critical reappraisal. Neurochem. Int. 44, 185–198 (2004).
Von Hösslin, C. & Alzheimer, A. Ein Beitrag zur Klinik und pathologischen Anatomie der Westphal-Struempellschen Pseudosklerose. Z. Gesamt. Neurol. Psychiatr. 8, 183–209 (1912).
Waggoner, R. W. & Malamud, N. Wilson’s disease in the light of cerebral changes following ordinary acquired liver disorders. J. Nerv. Ment. Dis. 96, 410–423 (1942).
Norenberg, M. D. & Lapham, L. W. The astrocyte response in experimental portal-systemic encephalopathy: an electron microscopic study. J. Neuropathol. Exp. Neurol. 33, 422–435 (1974).
Tallis, S. et al. Changes in CNS cells in hyperammonemic portal hypertensive rats. J. Neurochem. 128, 431–444 (2014).
Obara-Michlewska, M., Ruszkiewicz, J., Zielinska, M., Verkhratsky, A. & Albrecht, J. Astroglial NMDA receptors inhibit expression of K4.1 channels in glutamate-overexposed astrocytes in vitro and in the brain of rats with acute liver failure. Neurochem. Int. 88, 20–25 (2014).
Rangroo Thrane, V. et al. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med. 19, 1643–1648 (2013).
Kelly, T., Kafitz, K. W., Roderigo, C. & Rose, C. R. Ammonium-evoked alterations in intracellular sodium and pH reduce glial glutamate transport activity. Glia 57, 921–934 (2009).
Galland, F. et al. Hyperammonemia compromises glutamate metabolism and reduces BDNF in the rat hippocampus. Neurotoxicology 62, 46–55 (2017).
Wang, F., Du, T., Liang, C., Verkhratsky, A. & Peng, L. Ammonium increases Ca2+ signalling and upregulates expression of Cav1.2 gene in astrocytes in primary cultures and in the in vivo brain. Acta Physiol. 214, 261–274 (2015).
Montana, V., Verkhratsky, A. & Parpura, V. Pathological role for exocytotic glutamate release from astrocytes in hepatic encephalopathy. Curr. Neuropharmacol. 12, 324–333 (2014).
Albrecht, J. & Norenberg, M. D. Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology 44, 788–794 (2006).
Zielinska, M., Popek, M. & Albrecht, J. Roles of changes in active glutamine transport in brain edema development during hepatic encephalopathy: an emerging concept. Neurochem. Res. 39, 599–604 (2014).
Tiffany-Castiglion, E. & Qian, Y. Astroglia as metal depots: molecular mechanisms for metal accumulation, storage and release. Neurotoxicology 22, 577–592 (2001).
Codazzi, F., Pelizzoni, I., Zacchetti, D. & Grohovaz, F. Iron entry in neurons and astrocytes: a link with synaptic activity. Front. Mol. Neurosci. 8, 18 (2015).
Guan, W. et al. Iron induces two distinct Ca2+ signalling cascades in astrocytes. Commun. Biol. 4, 525 (2021).
Wu, L. J. et al. Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res. 1001, 108–117 (2004).
Jeong, S. Y. & David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 278, 27144–27148 (2003).
Xia, M. et al. Iatrogenic iron promotes neurodegeneration and activates self-protection of neural cells against exogenous iron attacks. Function. 2, zqab003 (2021).
Mishra, M., Singh, R., Mukherjee, S. & Sharma, D. Dehydroepiandrosterone’s antiepileptic action in FeCl3-induced epileptogenesis involves upregulation of glutamate transporters. Epilepsy Res. 106, 83–91 (2013).
Liang, S. et al. Iron aggravates the depressive phenotype of stressed mice by compromising the glymphatic system. Neurosci. Bull. 36, 1542–1546 (2020).
Miyajima, H. Aceruloplasminemia, an iron metabolic disorder. Neuropathology 23, 345–350 (2003).
Kono, S. & Miyajima, H. Molecular and pathological basis of aceruloplasminemia. Biol. Res. 39, 15–23 (2006).
McAlpine, D. & Araki, S. Minamata disease: an unusual neurological disorder caused by contaminated fish. Lancet 2, 629–631 (1958).
Aschner, M., Rising, L. & Mullaney, K. J. Differential sensitivity of neonatal rat astrocyte cultures to mercuric chloride (MC) and methylmercury (MeHg): studies on K+ and amino acid transport and metallothionein (MT) induction. Neurotoxicology 17, 107–116 (1996).
Yin, Z. et al. The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J. Neurochem. 107, 1083–1090 (2008).
Aschner, M., Eberle, N. B., Miller, K. & Kimelberg, H. K. Interactions of methylmercury with rat primary astrocyte cultures: inhibition of rubidium and glutamate uptake and induction of swelling. Brain Res. 530, 245–250 (1990).
Struzynska, L., Chalimoniuk, M. & Sulkowski, G. The role of astroglia in Pb-exposed adult rat brain with respect to glutamate toxicity. Toxicology 212, 185–194 (2005).
Gunnarson, E. et al. Lead induces increased water permeability in astrocytes expressing aquaporin 4. Neuroscience 136, 105–114 (2005).
De Keyser, J., Mostert, J. P. & Koch, M. W. Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J. Neurol. Sci. 267, 3–16 (2008).
Struys-Ponsar, C. & Guillard, O. & van den Bosch de Aguilar, P. Effects of aluminum exposure on glutamate metabolism: a possible explanation for its toxicity. Exp. Neurol. 163, 157–164 (2000).
Suarez-Fernandez, M. B. et al. Aluminum-induced degeneration of astrocytes occurs via apoptosis and results in neuronal death. Brain Res. 835, 125–136 (1999).
Goldman, S. M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharm. Toxicol. 54, 141–164 (2014).
Nabi, M. & Tabassum, N. Role of environmental toxicants on neurodegenerative disorders. Front. Toxicol. 4, 837579 (2022).
O’Callaghan, J. P., Kelly, K. A., VanGilder, R. L., Sofroniew, M. V. & Miller, D. B. Early activation of STAT3 regulates reactive astrogliosis induced by diverse forms of neurotoxicity. PLoS One 9, e102003 (2014).
Michalovicz, L. T. et al. Astrocyte-specific transcriptome analysis using the ALDH1L1 bacTRAP mouse reveals novel biomarkers of astrogliosis in response to neurotoxicity. J. Neurochem. 150, 420–440 (2019).
Cotter, D. et al. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 12, 386–394 (2002).
Sild, M., Ruthazer, E. S. & Booij, L. Major depressive disorder and anxiety disorders from the glial perspective: etiological mechanisms, intervention and monitoring. Neurosci. Biobehav. Rev. 83, 474–488 (2017).
Cotter, D. R., Pariante, C. M. & Everall, I. P. Glial cell abnormalities in major psychiatric disorders: the evidence and implications. Brain Res. Bull. 55, 585–595 (2001).
Cobb, J. A. et al. Density of GFAP-immunoreactive astrocytes is decreased in left hippocampi in major depressive disorder. Neuroscience 316, 209–220 (2016).
O’Leary, L. A. et al. Widespread decrease of cerebral vimentin-immunoreactive astrocytes in depressed suicides. Front. Psychiatry 12, 640963 (2021).
Rajkowska, G., Hughes, J., Stockmeier, C. A., Javier Miguel-Hidalgo, J. & Maciag, D. Coverage of blood vessels by astrocytic endfeet is reduced in major depressive disorder. Biol. Psychiatry 73, 613–621 (2013).
Choudary, P. V. et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl Acad. Sci. USA 102, 15653–15658 (2005).
Bernstein, H. G. et al. Reduced density of glutamine synthetase immunoreactive astrocytes in different cortical areas in major depression but not in bipolar I disorder. Front. Cell Neurosci. 9, 273 (2015).
Banasr, M. & Duman, R. S. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 64, 863–870 (2008).
Czeh, B., Simon, M., Schmelting, B., Hiemke, C. & Fuchs, E. Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 31, 1616–1626 (2006).
Banasr, M. et al. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol. Psychiatry 15, 501–511 (2010).
Gomez-Galan, M., De Bundel, D., Van Eeckhaut, A., Smolders, I. & Lindskog, M. Dysfunctional astrocytic regulation of glutamate transmission in a rat model of depression. Mol. Psychiatry 18, 582–594 (2013).
Aten, S. et al. Chronic stress impairs the structure and function of astrocyte networks in an animal model of depression. Neurochem. Res. 48, 1191–1210 (2022).
Lin, S. S. et al. Electroacupuncture prevents astrocyte atrophy to alleviate depression. Cell Death Dis. 14, 343 (2023).
Derouiche, A. & Geiger, K. D. Perspectives for ezrin and radixin in astrocytes: kinases, functions and pathology. Int. J. Mol. Sci. 20, 3776 (2019).
Badia-Soteras, A. et al. Retraction of astrocyte leaflets from the synapse enhances fear memory. Biol. Psychiatry 94, 226–238 (2023).
Ardalan, M., Rafati, A. H., Nyengaard, J. R. & Wegener, G. Rapid antidepressant effect of ketamine correlates with astroglial plasticity in the hippocampus. Br. J. Pharm. 174, 483–492 (2017).
Ramsey, J., Martin, E. C., Purcell, O. M., Lee, K. M. & MacLean, A. G. Self-injurious behaviours in rhesus macaques: potential glial mechanisms. J. Intellect. Disabil. Res. 62, 1008–1017 (2018).
Lima, A. et al. Astrocyte pathology in the prefrontal cortex impairs the cognitive function of rats. Mol. Psychiatry 19, 834–841 (2014).
John, C. S. et al. Blockade of the GLT-1 transporter in the central nucleus of the amygdala induces both anxiety and depressive-like symptoms. Neuropsychopharmacology 40, 1700–1708 (2015).
Mineur, Y. S., Picciotto, M. R. & Sanacora, G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol. Psychiatry 61, 250–252 (2007).
Sun, J. D., Liu, Y., Yuan, Y. H., Li, J. & Chen, N. H. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology 37, 1305–1320 (2012).
Xia, M., Yang, L., Sun, G., Qi, S. & Li, B. Mechanism of depression as a risk factor in the development of Alzheimer’s disease: the function of AQP4 and the glymphatic system. Psychopharmacology 234, 365–379 (2017).
Ongur, D., Drevets, W. C. & Price, J. L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl Acad. Sci. USA 95, 13290–13295 (1998).
Rajkowska, G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol. Psychiatry 48, 766–777 (2000).
Harrison, P. J., Colbourne, L. & Harrison, C. H. The neuropathology of bipolar disorder: systematic review and meta-analysis. Mol. Psychiatry 25, 1787–1808 (2020).
Hercher, C., Chopra, V. & Beasley, C. L. Evidence for morphological alterations in prefrontal white matter glia in schizophrenia and bipolar disorder. J. Psychiatry Neurosci. 39, 376–385 (2014).
Butt, A. M. & Rivera, A. D. Astrocytes in bipolar disorder. Adv. Neurobiol. 26, 95–113 (2021).
Li, C. T., Yang, K. C. & Lin, W. C. Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: evidence from clinical neuroimaging studies. Front Psychiatry 9, 767 (2018).
de Sousa, R. T. et al. Genetic studies on the tripartite glutamate synapse in the pathophysiology and therapeutics of mood disorders. Neuropsychopharmacology 42, 787–800 (2017).
Eastwood, S. L. & Harrison, P. J. Markers of glutamate synaptic transmission and plasticity are increased in the anterior cingulate cortex in bipolar disorder. Biol. Psychiatry 67, 1010–1016 (2010).
Ongur, D. et al. Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol. Psychiatry 64, 718–726 (2008).
Han, F., Xiao, B. & Wen, L. Loss of glial cells of the hippocampus in a rat model of post-traumatic stress disorder. Neurochem Res 40, 942–951 (2015).
Saur, L. et al. Experimental post-traumatic stress disorder decreases astrocyte density and changes astrocytic polarity in the CA1 hippocampus of male rats. Neurochem Res 41, 892–904 (2016).
Di Benedetto, B. et al. Fluoxetine requires the endfeet protein aquaporin-4 to enhance plasticity of astrocyte processes. Front. Cell Neurosci. 10, 8 (2016).
Malik, V. A. et al. GDF15 promotes simultaneous astrocyte remodeling and tight junction strengthening at the blood-brain barrier. J. Neurosci. Res. 98, 1433–1456 (2020).
Verkhratsky, A., Parpura, V., Scuderi, C. & Li, B. Astroglial serotonin receptors as the central target of classic antidepressants. Adv. Neurobiol. 26, 317–347 (2021).
Dong, L., Li, B., Verkhratsky, A. & Peng, L. Cell type-specific in vivo expression of genes encoding signalling molecules in the brain in response to chronic mild stress and chronic treatment with fluoxetine. Psychopharmacology 232, 2827–2835 (2015).
Peng, L., Verkhratsky, A., Gu, L. & Li, B. Targeting astrocytes in major depression. Expert Rev. Neurother. 15, 1299–1306 (2015).
Hertz, L. et al. Signal transduction in astrocytes during chronic or acute treatment with drugs (SSRIs, antibipolar drugs, GABA-ergic drugs, and benzodiazepines) ameliorating mood disorders. J. Signal Transduct. 2014, 593934 (2014).
Hertz, L., Rothman, D. L., Li, B. & Peng, L. Chronic SSRI stimulation of astrocytic 5-HT2B receptors change multiple gene expressions/editings and metabolism of glutamate, glucose and glycogen: a potential paradigm shift. Front. Behav. Neurosci. 9, 25 (2015).
Li, B. et al. Down-regulation of GluK2 kainate receptor expression by chronic treatment with mood-stabilizing anti-convulsants or lithium in cultured astrocytes and brain, but not in neurons. Neuropharmacology 57, 375–385 (2009).
Li, B. et al. Cell type-specific gene expression and editing responses to chronic fluoxetine treatment in the in vivo mouse brain and their relevance for stress-induced anhedonia. Neurochem. Res. 37, 2480–2495 (2012).
Ren, J., Song, D., Bai, Q., Verkhratsky, A. & Peng, L. Fluoxetine induces alkalinization of astroglial cytosol through stimulation of sodium-hydrogen exchanger 1: dissection of intracellular signaling pathways. Front. Cell Neurosci. 9, 61 (2015).
Alda, M. Lithium in the treatment of bipolar disorder: pharmacology and pharmacogenetics. Mol. Psychiatry 20, 661–670 (2015).
Rivera, A. D. & Butt, A. M. Astrocytes are direct cellular targets of lithium treatment: novel roles for lysyl oxidase and peroxisome-proliferator activated receptor-gamma as astroglial targets of lithium. Transl. Psychiatry 9, 211 (2019).
Domino, E. F. Taming the ketamine tiger. 1965. Anesthesiology 113, 678–684 (2010).
Stenovec, M., Li, B., Verkhratsky, A. & Zorec, R. Ketamine action on astrocytes provides new insights into rapid antidepressant mechanisms. Adv. Neurobiol. 26, 349–365 (2021).
Lasic, E. et al. Astrocyte specific remodeling of plasmalemmal cholesterol composition by ketamine indicates a new mechanism of antidepressant action. Sci. Rep. 9, 10957 (2019).
Aouizerate, B. et al. Pathophysiology of obsessive-compulsive disorder: a necessary link between phenomenology, neuropsychology, imagery and physiology. Prog. Neurobiol. 72, 195–221 (2004).
Rubenstein, J. L. & Merzenich, M. M. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267 (2003).
Andersen, J. V., Schousboe, A. & Verkhratsky, A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: integration of the glutamate/GABA-glutamine cycle. Prog. Neurobiol. 217, 102331 (2022).
Tanaka, K. Astroglia and obsessive compulsive disorder. Adv. Neurobiol. 26, 139–149 (2021).
Aida, T. et al. Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology 40, 1569–1579 (2015).
Abe, Y. et al. Diffusion functional MRI reveals global brain network functional abnormalities driven by targeted local activity in a neuropsychiatric disease mouse model. Neuroimage 223, 117318 (2020).
Yu, X. et al. Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 99, 1170–1187.e1179 (2018).
de Leeuw, C. et al. Involvement of astrocyte metabolic coupling in Tourette syndrome pathogenesis. Eur. J. Hum. Genet 23, 1519–1522 (2015).
Soto, J. S. et al. Astrocyte-neuron subproteomes and obsessive-compulsive disorder mechanisms. Nature 616, 764–773 (2023).
Williams, M. R. et al. Astrocyte decrease in the subgenual cingulate and callosal genu in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 263, 41–52 (2013).
Bernstein, H. G., Steiner, J., Guest, P. C., Dobrowolny, H. & Bogerts, B. Glial cells as key players in schizophrenia pathology: recent insights and concepts of therapy. Schizophr. Res. 161, 4–18 (2015).
Rajkowska, G. et al. Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophr. Res. 57, 127–138 (2002).
Oifa, A. I. & Uranova, N. A. Electron-microscopic analysis of cytoarchitectonic disorders in the cerebral cortex in schizophrenia.Zh. Nevropatol. Psikhiatr. Im. S S Korsakova 91, 48–52 (1991).
Katsel, P. et al. Astrocyte and glutamate markers in the superficial, deep, and white matter layers of the anterior cingulate gyrus in schizophrenia. Neuropsychopharmacology 36, 1171–1177 (2011).
McCullumsmith, R. E. et al. Cell-specific abnormalities of glutamate transporters in schizophrenia: sick astrocytes and compensating relay neurons? Mol. Psychiatry 21, 823–830 (2016).
Karlsson, R. M. et al. Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 34, 1578–1589 (2009).
Windrem, M. S. et al. Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21, 195–208.e196 (2017).
Dietz, A. G., Goldman, S. A. & Nedergaard, M. Glial cells in schizophrenia: a unified hypothesis. Lancet Psychiatry 7, 272–281 (2020).
Kalivas, P. W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 10, 561–572 (2009).
Kruyer, A., Scofield, M. D., Wood, D., Reissner, K. J. & Kalivas, P. W. Heroin cue-evoked astrocytic structural plasticity at nucleus accumbens synapses inhibits heroin seeking. Biol. Psychiatry 86, 811–819 (2019).
Knackstedt, L. A., Melendez, R. I. & Kalivas, P. W. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol. Psychiatry 67, 81–84 (2010).
Shen, H. W., Scofield, M. D., Boger, H., Hensley, M. & Kalivas, P. W. Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J. Neurosci. 34, 5649–5657 (2014).
Gipson, C. D. et al. Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc. Natl Acad. Sci. USA 110, 9124–9129 (2013).
Shelkar, G. P., Gandhi, P. J., Liu, J. & Dravid, S. M. Cocaine preference and neuroadaptations are maintained by astrocytic NMDA receptors in the nucleus accumbens. Sci. Adv. 8, eabo6574 (2022).
Miguel-Hidalgo, J. J. et al. Glia pathology in the prefrontal cortex in alcohol dependence with and without depressive symptoms. Biol. Psychiatry 52, 1121–1133 (2002).
Miguel-Hidalgo, J. J., Overholser, J. C., Meltzer, H. Y., Stockmeier, C. A. & Rajkowska, G. Reduced glial and neuronal packing density in the orbitofrontal cortex in alcohol dependence and its relationship with suicide and duration of alcohol dependence. Alcohol Clin. Exp. Res. 30, 1845–1855 (2006).
Wilhelm, C. J. & Guizzetti, M. Fetal alcohol spectrum disorders: an overview from the glia perspective. Front. Integr. Neurosci. 9, 65 (2015).
Risher, M. L. et al. Adolescent intermittent alcohol exposure: dysregulation of thrombospondins and synapse formation are associated with decreased neuronal density in the adult hippocampus. Alcohol Clin. Exp. Res. 39, 2403–2413 (2015).
Down, J. L. H. Observations on an ethnic classification of idiots. Lond. Hosp. Rep. 3, 259–262 (1866).
Maatta, T., Tervo-Maatta, T., Taanila, A., Kaski, M. & Iivanainen, M. Mental health, behaviour and intellectual abilities of people with Down syndrome. Downs Syndr. Res Pr. 11, 37–43 (2006).
Karlsen, A. S. & Pakkenberg, B. Total numbers of neurons and glial cells in cortex and basal ganglia of aged brains with Down syndrome–a stereological study. Cereb. Cortex 21, 2519–2524 (2011).
Guidi, S. et al. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 18, 180–197 (2008).
Chen, C. et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 5, 4430 (2014).
Das, I. et al. Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndrome mouse model. Sci. Transl. Med. 5, 201ra120 (2013).
Lu, J. et al. S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors. PLoS One 6, e22126 (2011).
Panagaki, T. et al. Overproduction of hydrogen sulfide, generated by cystathionine beta-synthase, disrupts brain wave patterns and contributes to neurobehavioral dysfunction in a rat model of down syndrome. Redox Biol. 51, 102233 (2022).
Mitchell, L. E. et al. Spina bifida. Lancet 364, 1885–1895 (2004).
Oria, M. et al. Premature neural progenitor cell differentiation into astrocytes in retinoic acid-induced spina bifida rat model. Front. Mol. Neurosci. 15, 888351 (2022).
Xing, L., Li, X. & Snider, W. D. Neurodevelopment. "RASopathic" astrocytes constrain neural plasticity. Science 348, 636–637 (2015).
Wang, Y. et al. ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell 150, 816–830 (2012).
Hegedus, B. et al. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 1, 443–457 (2007).
Krencik, R. et al. Dysregulation of astrocyte extracellular signaling in Costello syndrome. Sci. Transl. Med 7, 286ra266 (2015).
Leonard, H. & Wen, X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 8, 117–134 (2002).
Roeleveld, N., Zielhuis, G. A. & Gabreels, F. The prevalence of mental retardation: a critical review of recent literature. Dev. Med. Child Neurol. 39, 125–132 (1997).
Lubs, H. A., Stevenson, R. E. & Schwartz, C. E. Fragile X and X-linked intellectual disability: four decades of discovery. Am. J. Hum. Genet. 90, 579–590 (2012).
D’Adamo, P. et al. Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat. Genet. 19, 134–139 (1998).
Pfeffer, S. & Aivazian, D. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 5, 886–896 (2004).
Regazzi, R., Kikuchi, A., Takai, Y. & Wollheim, C. B. The small GTP-binding proteins in the cytosol of insulin-secreting cells are complexed to GDP dissociation inhibitor proteins. J. Biol. Chem. 267, 17512–17519 (1992).
Pylypenko, O. et al. Structure of doubly prenylated Ypt1:GDI complex and the mechanism of GDI-mediated Rab recycling. EMBO J. 25, 13–23 (2006).
Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 (2009).
Alory, C. & Balch, W. E. Organization of the Rab-GDI/CHM superfamily: the functional basis for choroideremia disease. Traffic 2, 532–543 (2001).
Bianchi, V. et al. Cognitive impairment in Gdi1-deficient mice is associated with altered synaptic vesicle pools and short-term synaptic plasticity, and can be corrected by appropriate learning training. Hum. Mol. Genet. 18, 105–117 (2009).
Bianchi, V. et al. Forebrain deletion of αGDI in adult mice worsens the pre-synaptic deficit at cortico-lateral amygdala synaptic connections. PLoS One 7, e29763 (2012).
Potokar, M. et al. Impaired αGDI function in the X-linked intellectual disability: the impact on astroglia vesicle dynamics. Mol. Neurobiol. 54, 2458–2468 (2017).
D’Adamo, P. et al. Inhibiting glycolysis rescues memory impairment in an intellectual disability Gdi1-null mouse. Metabolism 116, 154463 (2021).
Abrahams, B. S. & Geschwind, D. H. Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355 (2008).
Krakowiak, P. et al. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 129, e1121–e1128 (2012).
Zeidan-Chulia, F. et al. Altered expression of Alzheimer’s disease-related genes in the cerebellum of autistic patients: a model for disrupted brain connectome and therapy. Cell Death Dis. 5, e1250 (2014).
Zeidan-Chulia, F. et al. The glial perspective of autism spectrum disorders. Neurosci. Biobehav. Rev. 38, 160–172 (2014).
Johnson, M. B. et al. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509 (2009).
Allen, M. et al. Astrocytes derived from ASD individuals alter behavior and destabilize neuronal activity through aberrant Ca2+ signaling. Mol. Psychiatry 27, 2470–2484 (2022).
Jamain, S. et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29 (2003).
Lyst, M. J. & Bird, A. Rett syndrome: a complex disorder with simple roots. Nat. Rev. Genet. 16, 261–275 (2015).
Okabe, Y. et al. Alterations of gene expression and glutamate clearance in astrocytes derived from an MeCP2-null mouse model of Rett syndrome. PLoS One 7, e35354 (2012).
Lioy, D. T. et al. A role for glia in the progression of Rett’s syndrome. Nature 475, 497–500 (2011).
Zhang, X. et al. The disruption of central CO2 chemosensitivity in a mouse model of Rett syndrome. Am. J. Physiol. Cell Physiol. 301, C729–C738 (2011).
Garg, S. K., Lioy, D. T., Knopp, S. J. & Bissonnette, J. M. Conditional depletion of methyl-CpG-binding protein 2 in astrocytes depresses the hypercapnic ventilatory response in mice. J. Appl Physiol. 119, 670–676 (2015).
Simhal, A. K. et al. Multifaceted changes in synaptic composition and astrocytic involvement in a mouse model of fragile X syndrome. Sci. Rep. 9, 13855 (2019).
Risher, W. C. et al. Astrocytes refine cortical connectivity at dendritic spines. Elife 3, e04047 (2014).
Wallingford, J., Scott, A. L., Rodrigues, K. & Doering, L. C. Altered developmental expression of the astrocyte-secreted factors hevin and SPARC in the fragile X mouse model. Front Mol. Neurosci. 10, 268 (2017).
Reynolds, K. E., Krasovska, V. & Scott, A. L. Converging purinergic and immune signaling pathways drive IL-6 secretion by Fragile X cortical astrocytes via STAT3. J. Neuroimmunol. 361, 577745 (2021).
Humphry, G. M. The Old Age. The Results of Information Received Respecting Nearly Nine Hundred Persons Who Had Attained the Age of Eighty Years, Including Seventy Four Centenarians. (MacMillan & Bowes, 1889).
Rodriguez-Arellano, J. J., Parpura, V., Zorec, R. & Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 323, 170–182 (2016).
Haug, H. & Eggers, R. Morphometry of the human cortex cerebri and corpus striatum during aging. Neurobiol. Aging 12, 336–338 (1991). discussion 352-335.
Peters, A. & Sethares, C. Oligodendrocytes, their progenitors and other neuroglial cells in the aging primate cerebral cortex. Cereb. Cortex 14, 995–1007 (2004).
Vanzulli, I. et al. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 94, 130–139 (2020).
Neumann, P., Lenz, D. E., Streit, W. J. & Bechmann, I. Is microglial dystrophy a form of cellular senescence? An analysis of senescence markers in the aged human brain. Glia 71, 377–390 (2023).
Streit, W. J., Sammons, N. W., Kuhns, A. J. & Sparks, D. L. Dystrophic microglia in the aging human brain. Glia 45, 208–212 (2004).
Soreq, L. et al. Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 18, 557–570 (2017).
Orre, M. et al. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 35, 1–14 (2014).
Ximerakis, M. et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 22, 1696–1708 (2019).
Pakkenberg, B. & Gundersen, H. J. Neocortical neuron number in humans: effect of sex and age. J. Comp. Neurol. 384, 312–320 (1997).
Nichols, N. R., Day, J. R., Laping, N. J., Johnson, S. A. & Finch, C. E. GFAP mRNA increases with age in rat and human brain. Neurobiol. Aging 14, 421–429 (1993).
David, J. P. et al. Glial reaction in the hippocampal formation is highly correlated with aging in human brain. Neurosci. Lett. 235, 53–56 (1997).
Castiglioni, A. J. Jr, Legare, M. E., DL, B. & Tiffany-Castiglioni, E. Morphological changes in astrocytes of aging mice fed normal or caloric restricted diets. Age 14, 102–106 (1991).
Rodriguez, J. J. et al. Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol. Aging 35, 15–23 (2014).
Rodríguez-Callejas, J. D., Fuchs, E. & Perez-Cruz, C. Atrophic astrocytes in aged marmosets present tau hyperphosphorylation, RNA oxidation, and DNA fragmentation. Neurobiol. Aging 129, 121–136 (2023).
Popov, A. et al. Aging impairs astrocytes in the human cerebral cortex. bioRxiv, 2022.2010.2031.514523, (2022).
Yang, Z. et al. Engrafted glial progenitor cells yield long-term integration and sensory improvement in aged mice. Stem Cell Res. Ther. 13, 285 (2022).
Lalo, U., Palygin, O., North, R. A., Verkhratsky, A. & Pankratov, Y. Age-dependent remodelling of ionotropic signalling in cortical astroglia. Aging Cell 10, 392–402 (2011).
Gomez-Gonzalo, M. et al. Neuron-astrocyte signaling is preserved in the aging brain. Glia 65, 569–580 (2017).
Mathiesen, C., Brazhe, A., Thomsen, K. & Lauritzen, M. Spontaneous calcium waves in Bergman glia increase with age and hypoxia and may reduce tissue oxygen. J. Cereb. Blood Flow. Metab. 33, 161–169 (2013).
Peters, O. et al. Astrocyte function is modified by Alzheimer’s disease-like pathology in aged mice. J. Alzheimers Dis. 18, 177–189 (2009).
Duarte, J. M., Do, K. Q. & Gruetter, R. Longitudinal neurochemical modifications in the aging mouse brain measured in vivo by 1H magnetic resonance spectroscopy. Neurobiol. Aging 35, 1660–1668 (2014).
Potier, B. et al. Reduction in glutamate uptake is associated with extrasynaptic NMDA and metabotropic glutamate receptor activation at the hippocampal CA1 synapse of aged rats. Aging Cell 9, 722–735 (2010).
Kumar, M. J. & Andersen, J. K. Perspectives on MAO-B in aging and neurological disease: where do we go from here? Mol. Neurobiol. 30, 77–89 (2004).
Zorec, R., Parpura, V. & Verkhratsky, A. Preventing neurodegeneration by adrenergic astroglial excitation. FEBS J. 285, 3645–3656 (2018).
Zorec, R. & Vardjan, N. Adrenergic regulation of astroglial aerobic glycolysis and lipid metabolism: towards a noradrenergic hypothesis of neurodegeneration. Neurobiol. Dis. 182, 106132 (2023).
Wu, Z., Guo, Z., Gearing, M. & Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat. Commun. 5, 4159 (2014).
Brawek, B. et al. A bell-shaped dependence between amyloidosis and GABA accumulation in astrocytes in a mouse model of Alzheimer’s disease. Neurobiol. Aging 61, 187–197 (2018).
Bors, L. et al. Age-dependent changes at the blood-brain barrier. A Comparative structural and functional study in young adult and middle aged rats. Brain Res. Bull. 139, 269–277 (2018).
Kress, B. T. et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861 (2014).
Salminen, L. E. et al. Regional age differences in gray matter diffusivity among healthy older adults. Brain Imaging Behav. 10, 203–211 (2016).
Acevedo, A. et al. Metabolic switch in the aging astrocyte supported via integrative approach comprising network and transcriptome analyses. Aging 15, On-line ahead of print (2023).
Camandola, S. & Mattson, M. P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 36, 1474–1492 (2017).
Gasiorowska, A. et al. The biology and pathobiology of glutamatergic, cholinergic, and dopaminergic signaling in the aging brain. Front. Aging Neurosci. 13, 654931 (2021).
Nicaise, A. M., Willis, C. M., Crocker, S. J. & Pluchino, S. Stem cells of the aging brain. Front. Aging Neurosci. 12, 247 (2020).
Rodriguez, J. J. et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS One 3, e2935 (2008).
Miranda, C. J. et al. Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell 11, 542–552 (2012).
Geevasinga, N., Menon, P., Ozdinler, P. H., Kiernan, M. C. & Vucic, S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 12, 651–661 (2016).
Barbeito, L. Astrocyte-based cell therapy: new hope for amyotrophic lateral sclerosis patients? Stem Cell Res. Ther. 9, 241 (2018).
Valori, C. F., Guidotti, G., Brambilla, L. & Rossi, D. Astrocytes in motor neuron diseases. Adv. Exp. Med. Biol. 1175, 227–272 (2019).
Diaz-Amarilla, P. et al. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA 108, 18126–18131 (2011).
Stenovec, M. et al. Amyotrophic lateral sclerosis immunoglobulins G enhance the mobility of Lysotracker-labelled vesicles in cultured rat astrocytes. Acta Physiol. 203, 457–471 (2011).
Lino, M. M., Schneider, C. & Caroni, P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22, 4825–4832 (2002).
Boillee, S. et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392 (2006).
Yamanaka, K. et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253 (2008).
Papadeas, S. T., Kraig, S. E., O’Banion, C., Lepore, A. C. & Maragakis, N. J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl Acad. Sci. USA 108, 17803–17808 (2011).
Lepore, A. C. et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 11, 1294–1301 (2008).
Haidet-Phillips, A. M. et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828 (2011).
Re, D. B. et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81, 1001–1008 (2014).
Qian, K. et al. Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Rep. 8, 843–855 (2017).
Jimenez-Riani, M. et al. Ultrastructural features of aberrant glial cells isolated from the spinal cord of paralytic rats expressing the amyotrophic lateral sclerosis-linked SOD1G93A mutation. Cell Tissue Res. 370, 391–401 (2017).
Bruijn, L. I. et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338 (1997).
Howland, D. S. et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl Acad. Sci. USA 99, 1604–1609 (2002).
Guo, H. et al. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet. 12, 2519–2532 (2003).
Miller, R. G., Mitchell, J. D., Lyon, M. & Moore, D. H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neurondisease (MND). Cochrane Database Syst. Rev. 2002, CD001447 (2002).
Carbone, M., Duty, S. & Rattray, M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem. Int. 60, 31–38 (2012).
Kawamata, H. et al. Abnormal intracellular calcium signaling and SNARE-dependent exocytosis contributes to SOD1G93A astrocyte-mediated toxicity in amyotrophic lateral sclerosis. J. Neurosci. 34, 2331–2348 (2014).
Brambilla, L. et al. Disruption of the astrocytic TNFR1-GDNF axis accelerates motor neuron degeneration and disease progression in amyotrophic lateral sclerosis. Hum. Mol. Genet. 25, 3080–3095 (2016).
Madji Hounoum, B. et al. Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia 65, 592–605 (2017).
Velebit, J. et al. Astrocytes with TDP-43 inclusions exhibit reduced noradrenergic cAMP and Ca(2+) signaling and dysregulated cell metabolism. Sci. Rep. 10, 6003 (2020).
Peteri, U. K., Niukkanen, M. & Castren, M. L. Astrocytes in neuropathologies affecting the frontal cortex. Front. Cell Neurosci. 13, 44 (2019).
Su, J. H. et al. DNA damage and activated caspase-3 expression in neurons and astrocytes: evidence for apoptosis in frontotemporal dementia. Exp. Neurol. 163, 9–19 (2000).
Broe, M., Kril, J. & Halliday, G. M. Astrocytic degeneration relates to the severity of disease in frontotemporal dementia. Brain 127, 2214–2220 (2004).
Scheltens, P. et al. Alzheimer’s disease. Lancet 388, 505–517 (2016).
Selkoe, D. J. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 (2001).
Kametani, F. & Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 12, 25 (2018).
Castellani, R. J. & Smith, M. A. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is ‘too big to fail’. J. Pathol. 224, 147–152 (2011).
Puzzo, D. & Conti, F. Conceptual and methodological pitfalls in experimental studies: an overview, and the case of Alzheimer’s disease. Front. Mol. Neurosci. 14, 684977 (2021).
Verkhratsky, A., Marutle, A., Rodriguez-Arellano, J. J. & Nordberg, A. Glial asthenia and functional paralysis: a new perspective on neurodegeneration and Alzheimer’s disease. Neuroscientist 21, 552–568 (2015).
Serrano-Pozo, A. et al. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J. Neuropathol. Exp. Neurol. 72, 462–471 (2013).
Beach, T. G. & McGeer, E. G. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res. 463, 357–361 (1988).
Verkhratsky, A., Zorec, R., Rodriguez, J. J. & Parpura, V. Astroglia dynamics in ageing and Alzheimer’s disease. Curr. Opin. Pharm. 26, 74–79 (2016).
Heneka, M. T. et al. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APPV717I transgenic mice. J. Neuroinflamm. 2, 22 (2005).
Osborn, L. M., Kamphuis, W., Wadman, W. J. & Hol, E. M. Astrogliosis: an integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 144, 121–141 (2016).
Smit, T. et al. Reactive astrocytes as treatment targets in Alzheimer’s disease—systematic review of studies using the APPswePS1dE9 mouse model. Glia 69, 1852–1881 (2021).
Simpson, J. E. et al. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 31, 578–590 (2010).
Wharton, S. B. et al. Population variation in glial fibrillary acidic protein levels in brain ageing: relationship to Alzheimer-type pathology and dementia. Dement Geriatr. Cogn. Disord. 27, 465–473 (2009).
Kraft, A. W. et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27, 187–198 (2013).
Livingston, N. R. et al. Relationship between astrocyte reactivity, using novel 11C-BU99008 PET, and glucose metabolism, grey matter volume and amyloid load in cognitively impaired individuals. Mol. Psychiatry 27, 2019–2029 (2022).
Alberdi, E. et al. Ca2+ -dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 12, 292–302 (2013).
Lia, A. et al. Rescue of astrocyte activity by the calcium sensor STIM1 restores long-term synaptic plasticity in female mice modelling Alzheimer’s disease. Nat. Commun. 14, 1590 (2023).
Sahlas, D. J., Bilbao, J. M., Swartz, R. H. & Black, S. E. Clasmatodendrosis correlating with periventricular hyperintensity in mixed dementia. Ann. Neurol. 52, 378–381 (2002).
Daschil, N. & Humpel, C. Green-fluorescent protein+ astrocytes attach to β-amyloid plaques in an Alzheimer mouse model and are sensitive for clasmatodendrosis. Front. Aging Neurosci. 8, 75 (2016).
Colombo, J. A., Quinn, B. & Puissant, V. Disruption of astroglial interlaminar processes in Alzheimer’s disease. Brain Res. Bull. 58, 235–242 (2002).
Diniz, D. G. et al. Age, environment, object recognition and morphological diversity of GFAP-immunolabeled astrocytes. Behav. Brain Funct. 12, 28 (2016).
Polis, B., Srikanth, K. D., Elliott, E., Gil-Henn, H. & Samson, A. O. L-Norvaline reverses cognitive decline and synaptic loss in a murine model of Alzheimer’s disease. Neurotherapeutics 15, 1036–1054 (2018).
Beauquis, J. et al. Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp. Neurol. 239, 28–37 (2013).
Beauquis, J. et al. Neuronal and glial alterations, increased anxiety, and cognitive impairment before hippocampal amyloid deposition in PDAPP mice, model of Alzheimer’s disease. Hippocampus 24, 257–269 (2014).
Yeh, C. Y., Vadhwana, B., Verkhratsky, A. & Rodriguez, J. J. Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro 3, 271–279 (2012).
Kulijewicz-Nawrot, M., Verkhratsky, A., Chvatal, A., Sykova, E. & Rodriguez, J. J. Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J. Anat. 221, 252–262 (2012).
Jones, V. C., Atkinson-Dell, R., Verkhratsky, A. & Mohamet, L. Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis. 8, e2696 (2017).
Lim, D., Ronco, V., Grolla, A. A., Verkhratsky, A. & Genazzani, A. A. Glial calcium signalling in Alzheimer’s disease. Rev. Physiol. Biochem Pharm. 167, 45–65 (2014).
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T. & Bacskai, B. J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215 (2009).
Mohamet, L., Jones, V. C., Dayanithi, G. & Verkhratsky, A. Pathological human astroglia in Alzheimer’s disease: opening new horizons with stem cell technology. Future Neurol. 13, 87–99 (2018).
Oksanen, M. et al. PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 9, 1885–1897 (2017).
Lin, Y. T. et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e1147 (2018).
Habib, N. et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23, 701–706 (2020).
Sadick, J. S. et al. Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 110, 1788–1805 e1710 (2022).
Dematteis, G. et al. Proteomic analysis links alterations of bioenergetics, mitochondria-ER interactions and proteostasis in hippocampal astrocytes from 3xTg-AD mice. Cell Death Dis. 11, 645 (2020).
Ryu, W. I. et al. Brain cells derived from Alzheimer’s disease patients have multiple specific innate abnormalities in energy metabolism. Mol. Psychiatry 26, 5702–5714 (2021).
Le Douce, J. et al. Impairment of glycolysis-derived l-serine production in astrocytes contributes to cognitive deficits in Alzheimer’s disease. Cell Metab. 31, 503–517.e508 (2020).
Zheng, J. et al. GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol. Metab. 47, 101180 (2021).
Olabarria, M., Noristani, H. N., Verkhratsky, A. & Rodriguez, J. J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 6, 55 (2011).
Butterfield, D. A. et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol. Dis. 22, 223–232 (2006).
Carter, S. F. et al. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 53, 37–46 (2012).
Ju, Y. H. et al. Astrocytic urea cycle detoxifies Aβ-derived ammonia while impairing memory in Alzheimer’s disease. Cell Metab. 34, 1104–1120 (2022).
Lee, S. et al. Channel-mediated tonic GABA release from glia. Science 330, 790–796 (2010).
Garaschuk, O. & Verkhratsky, A. GABAergic astrocytes in Alzheimer’s disease. Aging 11, 1602–1604 (2019).
Ju, Y. H. et al. Astrocytic urea cycle detoxifies Abeta-derived ammonia while impairing memory in Alzheimer’s disease. Cell Metab. 34, 1104–1120.e1108 (2022).
Augusto-Oliveira, M. & Verkhratsky, A. Mens sana in corpore sano: lifestyle changes modify astrocytes to contain Alzheimer’s disease. Neural Regen. Res. 16, 1548–1549 (2021).
Bredesen, D. E. Reversal of cognitive decline: a novel therapeutic program. Aging 6, 707–717 (2014).
Buchman, A. S. et al. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology 78, 1323–1329 (2012).
Rodriguez, J. J. et al. Voluntary running and environmental enrichment restores impaired hippocampal neurogenesis in a triple transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 8, 707–717 (2011).
Mattson, M. P. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab. 16, 706–722 (2012).
Popov, A. et al. Caloric restriction triggers morphofunctional remodeling of astrocytes and enhances synaptic plasticity in the mouse hippocampus. Cell Death Dis. 11, 208 (2020).
Fiol-deRoque, M. A. et al. Cognitive recovery and restoration of cell proliferation in the dentate gyrus in the 5XFAD transgenic mice model of Alzheimer’s disease following 2-hydroxy-DHA treatment. Biogerontology 14, 763–775 (2013).
Ding, F. et al. α1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 54, 387–394 (2013).
Tariot, P. N., Schneider, L. S., Patel, S. V. & Goldstein, B. Inhibitors of Monoamine Oxidase B: Pharmacology and Clinical Use in Neurodegenerative Disorders (ed I. Szelenyi) 301–317 (Birkhäuser Basel, 1993).
Monai, H. et al. Calcium imaging reveals glial involvement in transcranial direct current stimulation-induced plasticity in mouse brain. Nat. Commun. 7, 11100 (2016).
Parkinson, J. An Essay on the Shaking Palsy (Sherwood, Neely, and Jones, 1817).
Hornykiewicz, O. Biochemical aspects of Parkinson’s disease. Neurology 51, S2–S9 (1998).
Yun, S. P. et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 24, 931–938 (2018).
Booth, H. D. E., Hirst, W. D. & Wade-Martins, R. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 40, 358–370 (2017).
Alam, Z. I. et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 69, 1196–1203 (1997).
Gelders, G., Baekelandt, V. & Van der Perren, A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 4784268 (2018).
Kano, M. et al. Reduced astrocytic reactivity in human brains and midbrain organoids with PRKN mutations. NPJ Parkinsons Dis. 6, 33 (2020).
Iovino, L. et al. Trafficking of the glutamate transporter is impaired in LRRK2-related Parkinson’s disease. Acta Neuropathol. 144, 81–106 (2022).
Sonninen, T. M. et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 10, 14474 (2020).
Gu, X. L. et al. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 3, 12 (2010).
Kuter, K. Z., Cenci, M. A. & Carta, A. R. The role of glia in Parkinson’s disease: emerging concepts and therapeutic applications. Prog. Brain Res. 252, 131–168 (2020).
Song, Y. J. et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 68, 1073–1083 (2009).
Bantle, C. M., Hirst, W. D., Weihofen, A. & Shlevkov, E. Mitochondrial dysfunction in astrocytes: a role in Parkinson’s disease? Front. Cell Dev. Biol. 8, 608026 (2020).
Morales, I. et al. Neuroglial transmitophagy and Parkinson’s disease. Glia 68, 2277–2299 (2020).
Hayakawa, K. et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555 (2016).
English, K. et al. Astrocytes rescue neuronal health after cisplatin treatment through mitochondrial transfer. Acta Neuropathol. Commun. 8, 36 (2020).
Cheng, X.-Y. et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 9, 13 (2020).
Huntington, G. On chorea. Med Surg. Rep. 26, 317–321 (1872).
Gusella, J. F. et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 234–238 (1983).
Wood, T. E. et al. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet. 28, 487–500 (2019).
Octeau, J. C. et al. An optical neuron-astrocyte proximity assay at synaptic distance scales. Neuron 98, 49–66.e49 (2018).
Osipovitch, M. et al. Human ESC-derived chimeric mouse models of Huntington’s disease reveal cell-intrinsic defects in glial progenitor cell differentiation. Cell Stem Cell 24, 107–122.e107 (2019).
Hodges, A. et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 15, 965–977 (2006).
Lievens, J. C. et al. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol. Dis. 8, 807–821 (2001).
Bradford, J. et al. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl Acad. Sci. USA 106, 22480–22485 (2009).
Kovacs, G. G. Astroglia and Tau: new perspectives. Front. Aging Neurosci. 12, 96 (2020).
Kovacs, G. G. et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 131, 87–102 (2016).
Dickson, D. W., Ahmed, Z., Algom, A. A., Tsuboi, Y. & Josephs, K. A. Neuropathology of variants of progressive supranuclear palsy. Curr. Opin. Neurol. 23, 394–400 (2010).
Yoshida, M. Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 34, 555–570 (2014).
Hattori, M. et al. Distribution of astrocytic plaques in the corticobasal degeneration brain and comparison with tuft-shaped astrocytes in the progressive supranuclear palsy brain. Acta Neuropathol. 106, 143–149 (2003).
Kersaitis, C., Halliday, G. M. & Kril, J. J. Regional and cellular pathology in frontotemporal dementia: relationship to stage of disease in cases with and without Pick bodies. Acta Neuropathol. 108, 515–523 (2004).
Kovacs, G. G. et al. Neuropathology of the hippocampus in FTLD-Tau with Pick bodies: a study of the BrainNet Europe Consortium. Neuropathol. Appl. Neurobiol. 39, 166–178 (2013).
Ferrer, I. et al. Familial globular glial tauopathy linked to MAPT mutations: molecular neuropathology and seeding capacity of a prototypical mixed neuronal and glial tauopathy. Acta Neuropathol. 139, 735–771 (2020).
Acknowledgements
A.V., P.I., A.S. and Y.T. were supported by grants from NSFC-RSF (82261138557), the Innovation Team and Talents Cultivation Program of the National Administration of Traditional Chinese Medicine (ZYYCXTD-D-202003), and the Sichuan Science and Technology Program (2022YFH0006); Y.T. was supported by NSFC (82274668, 82230127) and Sichuan Provincial Administration of Traditional Chinese Medicine (2023zd024). Work in the M.V.S. laboratory is supported by National Institutes of Health (NS084030) and by the Dr. Miriam and Sheldon G. Adelson Medical Foundation. R.Z. is thanking the Slovenian Research Agency for funding the research through the Core programme ‘Cell Physiology’ P3 310. The work of A.S. was supported by the RSF grant 23-74-30008 for the NSFC-RSF project.
Author information
Authors and Affiliations
Contributions
M.V.S. and A.V. developed the concept and wrote the paper, Y.T., A.B., B.L., P.I., R.Z., A.S. discussed, edited, and contributed to the writing. All schematics were drawn by A.V. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
Authors declare no competing interest. Alexey Semyanov, Alexei Verkhratsky and Peter Illes are the editors of STTT.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verkhratsky, A., Butt, A., Li, B. et al. Astrocytes in human central nervous system diseases: a frontier for new therapies. Sig Transduct Target Ther 8, 396 (2023). https://doi.org/10.1038/s41392-023-01628-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01628-9
This article is cited by
-
Astrocyte cells in the brain have immune memory
Nature (2024)
-
A double inducible cell ablation system for eliminating senescent astrocytes via apoptosis
Molecular Biology Reports (2024)
