
Abstract
The endoplasmic reticulum (ER) functions as a quality-control organelle for protein homeostasis, or “proteostasis”. The protein quality control systems involve ER-associated degradation, protein chaperons, and autophagy. ER stress is activated when proteostasis is broken with an accumulation of misfolded and unfolded proteins in the ER. ER stress activates an adaptive unfolded protein response to restore proteostasis by initiating protein kinase R-like ER kinase, activating transcription factor 6, and inositol requiring enzyme 1. ER stress is multifaceted, and acts on aspects at the epigenetic level, including transcription and protein processing. Accumulated data indicates its key role in protein homeostasis and other diverse functions involved in various ocular diseases, such as glaucoma, diabetic retinopathy, age-related macular degeneration, retinitis pigmentosa, achromatopsia, cataracts, ocular tumors, ocular surface diseases, and myopia. This review summarizes the molecular mechanisms underlying the aforementioned ocular diseases from an ER stress perspective. Drugs (chemicals, neurotrophic factors, and nanoparticles), gene therapy, and stem cell therapy are used to treat ocular diseases by alleviating ER stress. We delineate the advancement of therapy targeting ER stress to provide new treatment strategies for ocular diseases.
Similar content being viewed by others
Introduction
Proteostasis is fundamental to cell survival, and an imbalance will result in diseases including metabolic, neurodegenerative, oncological, and cardiovascular disorders.1 The endoplasmic reticulum (ER) functions as a quality-control organelle for the proteins it produces, allowing only normal proteins to exit its vesicles.2 Protein quality control systems are responsible for maintaining proteostasis, including chaperones, ATPases, glucose-regulated protein 94 (Grp94), BiP (an Hsp70 family member), and two proteolytic systems: the ubiquitin–proteasome and the lysosome–autophagy systems.3 Misfolded proteins produced under stress conditions are then removed from the folding machinery, relocated from the ER into the cytosol, and degraded in the ubiquitin-proteasome via a series of pathways collectively referred to as ER-associated degradation (ERAD).4,5 However, persistent misfolded proteins will accumulate and lead to ER stress, which can elicit an adaptive response called the unfolded protein response (UPR). The consequences of UPR include protein synthesis inhibition, regulation of gene expression,6,7 and cell fate decisions like apoptosis8 to meet the cell’s demands.
Increasing evidence indicates that ER stress is crucial in the pathology of ocular diseases (Fig. 1). Proteostasis imbalance-induced ER stress and its related mutations have been broadly reported in the field of ophthalmology, making them a promising treatment target in ocular diseases. In this review, we will discuss the latest advances, focusing on the molecular mechanisms and therapeutic targets between ER stress and ocular diseases.

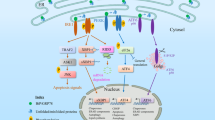
Overview of the regulatory mechanisms of ER stress in ocular diseases. ER stress plays an important role in several ocular diseases, including glaucoma, diabetic retinopathy, age-related macular dystrophy (AMD), retinitis pigmentosa (RP), achromatopsia (ACHM), cataract, corneal diseases, DES, myopia, uveitis, and uveal melanoma. The concrete mechanisms of ER stress in ocular diseases include regulation of gene mutations, epigenic modifications, impaired autophagy, oxidative stress, mitochondria dysfunction, and metabolism. The figure was created with BioRender.com (https://www.biorender.com/). AMD age-related macular dystrophy, RP retinitis pigmentosa, ACHM achromatopsia, DES dry eye syndrome
Unfolded protein response
The UPR is initiated and regulated by three ER sensors: inositol-requiring enzyme 1 (IRE1), double-stranded RNA-activated protein kinase R (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6). Owing to the binding of BiP, these sensors remain inactive (Fig. 2). The unfolded protein is considered to compete with the BiP binding receptor and results in the activation of three sensors during BiP dissociation, which triggers the UPR.9 Typical target genes of UPR can be correlated with protein folding, ERAD, oxidative stress, autophagy, mitochondrial dysfunction, and metabolic pathways that are induced differently due to tissue differences.10,11

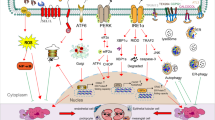
Unfolded protein response signaling pathways. Accumulation of misfolded and unfolded protein in endoplasmic reticulum (ER) will replace the BiP binding on PERK, ATF6 and IRE1, and activate them. PERK causes the phosphorylation of eIF2α, which leads to a reduction of ER protein accumulation and translation of the ATF4 mRNA. ATF4 then interacts with CHOP, which controls the expression of the target genes, such as GADD34 and ERO-1α. GADD34 encodes a regulatory subunit of an eIF2α-directed phosphatase complex, which in turn dephosphorylates eIF2α and recovers protein synthesis. The consequence of PERK pathway can be cell apoptosis. Under ER stress, IRE1 becomes dimerized and activated. The activated IRE1 excises an intron from XBP1 and transforms it into spliced XBP1 (XBP1s). XBP1s is transported to the nucleus, where it facilitates gene translation. Facing ER stress, ATF6 is transported to the Golgi apparatus and cleaved by Site-1 (S1P) and Site-2 (S2P) proteases. After the cleavage, ATF6 releases a cytosolic fragment (ATF6f), which directly controls the genes encoding ERAD components such as the basic transcription of leucine zipper (bZip) family and XBP1. The figure was created with BioRender.com (https://www.biorender.com/). ATF4 activating transcription factor 4, eukaryotic translation initiation factor 2α (eIF2α), C/EBP-homologous protein (CHOP), PERK PKR-like ER kinase, ATF6 activating transcription factor 6, IRE1 inositol requiring enzyme 1, XBP1 X-box binding protein 1, ERAD ER-associated degradation, bZip basic transcription of leucine zipper, GADD34 growth arrest and DNA damage-inducible 34, ERO-1α endoplasmic reticulum oxidoreductase 1 alpha
Unfolded protein response pathway
PERK-eIF2α-C/EBP-homologous protein
The unfolded protein binds to PERK and causes conformational changes, meaning PERK multimerizes and phosphorylates itself.12 Then, eukaryotic translation initiation factor 2α (eIF2α), a ubiquitous translation initiation factor, is inactivated by phosphorylation under the activation of PERK, alleviating translation, reducing protein synthesis, and contributing to protein load reduction.13 If ER stress persists, ATF4 mRNA translation activates the C/EBP-homologous protein (CHOP) promoter, which controls the target gene expression.6
IRE1-XBP1
IRE1 is a single-spanning transmembrane protein with dual protein kinase and ribonuclease activities.14,15 Once IRE1 is activated, it dimerizes and/or becomes oligomerized, leading to trans-phosphorylation of positive regulatory sites within the protein kinase domain (IRE1 becomes IRE1p), whose changes require adenosine nucleotides (ATP/ADP) as cofactors to exhibit nuclease activity.14,15,16 After activation of the nuclease character of IRE1, it excises an intron (a 26-nucleotide segment) from mRNA encoding a UPR-specific transcription factor called XBP1 (X-box binding protein 1) in metazoans, which transforms the unspliced XBP1 (XBP1u) to the spliced XBP1 (XBP1s).
ATF6-ATF6f-bZip
ATF6 is a 90-kDa protein constitutively expressed in cells and is a single-pass type 2 transmembrane protein with a large ER-luminal domain.17 It has a cytosolic NH2-terminal domain, which can act as a transcription factor of the basic-leucine-zipper (bZip) family.18 Site-1 and Site-2 proteases cleave ATF6 under ER stress. After cleavage, ATF6 releases a cytosolic fragment (ATF6f) that directly controls the transcription of XBP.19
Downstream effect of endoplasmic reticulum stress
ER stress activates all UPR signaling pathways, including protective and pro-apoptosis pathways. However, if the protein level increases before homeostasis restoration, ER stress will be prolonged and the stressed cells will undergo apoptosis.8,13 The downstream effects of ER stress can be involved in ERAD, protein synthesis, autophagy, oxidative stress, mitochondrial dysfunction, and metabolism.
Protein synthesis and ERAD
ERAD is a part of the ER-mediated protein quality control system, which manipulates the restoration of protein conformation and the clearance of abnormal proteins located on the ER membrane or cytoplasm. The ERAD degradation mechanism can be divided into four steps: substrate recognition by chaperones and lectin, dislocation across the ER membrane driven by VCP/p97, polyubiquitination by E3 ligases, and degradation by the 26S proteasome.20 ERAD-L, ERAD-M, and ERAD-C refer to different proteasome degradation substrates of proteins with folding problems or degradation signals, respectively, existing in the ER lumen, transmembrane, or cytoplasmic domain.21 ERAD can alleviate ER stress, which can be either induced or inhibited under UPR. Also, prolonged UPR affects protein synthesis, which further aggravates the ERAD deficiency. ER stress can modulate the phosphorylation of eIF2α, leading to the attenuation of protein synthesis, whereas the subsequent activation of ATF4/CHOP can increase protein synthesis, triggering apoptosis.13 CHOP encodes a regulatory subunit of an eIF2α-directed phosphatase complex that helps ER-stressed cells recover protein synthesis. Meanwhile, ATF6f released by the ATF6 pathway directly controls the genes encoding ERAD components like Derlin-3.19,22 The IRE1/XBP1 pathway is responsible for efficient protein folding, maturation, and degradation in the ER and encodes protein chaperones like ERdj4, p58IPK, EDEM, RAMP-4, PDI-P5, and HEDJ.23
Oxidative stress
Reactive oxygen species (ROS) can be produced in every aerobic cell. Antioxidant systems in cells can significantly prevent ROS production by direct action on radical chain reactions and through detoxifying enzymes like superoxide dismutase (SOD) and catalase, which produce peroxidases.24 The generation of ROS relies on several enzymes like nicotinamide adenine dinucleotide phosphate oxidase (NADPH, transforming electrons to molecular oxygen), xanthine oxidoreductase and peroxidases, and mitochondria containing electron transport systems.24 When the balance between ROS generation and antioxidant systems is disturbed, oxidative stress (OS) occurs.25 ROS links ER stress with oxidative stress. Oxidative stress and ER stress are responsible for cell death resulting from mitochondrial permeability, autophagy impairment, and inflammation. ROS directly activates NF-kB, which facilitates the transcription of inflammation-related cytokines. Growth arrest and DNA damage-inducible 34 (GADD34) is a direct CHOP target gene, that can generate ROS in cells by increasing protein synthesis.13,26 ER oxidoreductase (ERO-1α) is vital for disulfide bond formation, which helps proteins fold and transport electrons to molecular oxygen, and facilitates the oxidation of ER proteins. CHOP can upregulate ERO-1α and cause cell apoptosis.13 Increased ROS will increase Ca2+ to induce apoptosis by activating ITPR3/IP3R (inositol 1,4,5-triphosphate receptor type 3), which is an ER calcium channel.
Autophagy
Autophagy can be classified into three types: macroautophagy, microautophagy, and chaperone-mediated autophagy.27 Autophagy here mainly refers to macroautophagy. UPR regulates and interacts with autophagy through adenosine monophosphate-activated protein kinase (AMPK), Akt1-MTOR, and MAPK8 transduction.28 In particular, mitochondria (mitophagy) and ER (reticulophagy) are involved in ER stress. Under ER stress, an enlarged ER membrane contributes to autophagosome formation. To avoid protein accumulation, ATF4 will activate reticulophagy by facilitating the interaction between ER surface proteins like CCPG1 and ATF8.29,30 In reticulophagy, the DDRGK-dependent UFMylation process of ER surface proteins is suppressed by upstream ER stress.31 In aging diseases, removing impaired mitochondria by mitophagy in time is vital for cell survival. The PINK2/Parkin pathway involved in mitophagy can be inhibited by eIF2α/ATF4 knockout (KO).32 Under ER stress, the eIF2α/ATF4 pathway is essential for autophagy gene transcription, including p62, Nbr1, Atg7, Atg10, Gabarap, and Atg5.33 Mammalian oligomerized IRE1 not only cleaves XBP1 mRNA but also activates the stress-induced Jun N-terminal kinase (JNK) through inhibition of autophagy, which interacts with caspase 12.7 Inhibiting autophagy can facilitate IRE1 binding to tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2), which stabilizes its conformation and then interacts with apoptosis signal-regulating kinase 1 (ASK1).34 This indicates the IRE1-ASK1-JNK axis is activated in a pro-apoptosis process. Autophagy induced in ER stress can be toxic. Under prolonged ER stress, three branches of UPR are activated, which leads to cell death via a complex consisting of pro-caspase-8 and fas-associating protein with a novel death domain (FADD). This kind of apoptosis is independent of mitochondria and relies on ATG5, which means the involvement of autophagy.35
Mitochondria dysfunction
Mitochondria dysfunction can manifest as mitochondrial fusion, mitochondrial membrane permeability, transition, pore, and dynamic changes, which will result in NOD-like receptor protein 3 (NLRP3) inflammasome activation, intrinsic apoptosis, oxidative stress, and ER stress. Evidence indicates that the consequences of ER stress can be associated with mitochondrial fusion. The ER and mitochondria are adjacent, and they maintain lipid and Ca2+ homeostasis together. The sites where the ER membrane contacts the mitochondrial membrane are called mitochondria-associated ER membranes (MAMs).36 Any ER or mitochondrial disturbance can affect the other and initiate a cell response. Under ER stress, IP3R opening leads to the active Ca2+ transition between the ER and mitochondria, which facilitates NLRP3 inflammasome activation.37 As aforementioned, the Ca2+ released from mitochondria results in ER stress. This indicates that MAMs act as the bridge between NLRP3-induced inflammation and ER stress. Mitofusin 2 (Mfn2) is an upstream molecule that suppresses PERK activation and is the bond between UPR and mitochondrial metabolism.38 In melanoma, XBP1 facilitates the ubiquitination and degradation of Mfn2, which attributes to mitochondrial fission and mitophagy under ER stress.39 Activated CHOP immensely decreases Bcl2, in which BH4-Tat can alleviate the mitochondria membrane potential under ER stress, increases pro-apoptotic protein Bim, and activates caspases like caspase-9, -2, and -3.40,41 Subsequently, mitochondrial outer membrane permeabilization facilitates the release of cytochrome c.35 Bcl2 can also regulate BH3-only protein expressions (like BAX and BAK), which can bind to mitochondria and cause mitochondrial permeabilization. Therefore, CHOP regulates mitochondrial dysfunction and mitochondria-related intrinsic apoptosis via Bcl2, Bim, and caspases.
Metabolism
ER can act as not only the protein quality-control organelle but also the organelle for sterol and phospholipid synthesis and glucose metabolism.42 In particular, the cleavage process of ATF6 resembles that of the sterol response element binding protein (SREBP), which is involved in lipid metabolism.43 In liver cells, cleaved ATF6 binds to SREBP to form a complex and recruits HDAC1 to downregulate the transcription activity of SREBP.44 ATF6 is involved in fatty acid oxidation by interacting with PPARα.45 Choline cytidylyltransferase is the limited enzyme in the CDP-choline pathway and can be activated by XBP1s, which presents the lipid biosynthesis induced by IRE1/XBP1.46 Furthermore, IRE1/XBP1 regulates normal fatty acid synthesis and β-oxidation by indirectly activating PPARα.47,48 PERK/eIF2α regulates glucose and lipid metabolism through C/EBPβ and C/EBPα which directly regulate glucose production and PPARγ.49
ER stress and glaucoma
Glaucoma is a heterogeneous group of diseases characterized by cupping of the optic nerve head and visual-field damage, which may result in irreversible blindness and is the second leading cause of irreversible blindness worldwide.50,51,52 A study using UN World Population Prospects data estimated that by 2040, 3.54% of affected people will be 40–80 years old and 111.8 million will be affected overall.53 Glaucoma can be classified into three types:52 primary glaucoma, which can be divided into open-angle and angle-closure glaucoma, secondary glaucoma, which may result from trauma, certain medications such as corticosteroids, inflammation, tumors, or conditions such as pigment dispersion or pseudoexfoliation, and congenital glaucoma.54 Optic nerve damage is common in patients with glaucoma, which is the main cause of vision loss, while trabecular malformations can exist in the pathology of some types of glaucoma, such as primary open-angle glaucoma (POAG). It has been revealed that different risk factors in glaucoma such as aging, glucocorticoid, ischemic, and harmful mutations, will result in chronic UPR, which can be a conserved characteristic in glaucoma and cause the pathological damage mentioned above. We next discuss the role of ER stress in the development of glaucoma and potential targeted treatment (Fig. 3).

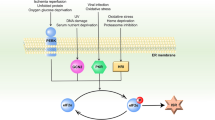
Involvement of ER stress in POAG. POAG can induce aging, aging-related tau, and α-synuclein. Increased ECM, increased TGFβ2, and the accumulation of mutant myocilin can cumulatively lead to ER stress in the trabecular meshwork. Shp2 contributes to ER stress and RGCs loss through the BDNF/TrkB pathway. Following ER stress, the ATF4/CHOP/GADD34 pathway can lead to TM cell apoptosis via Bid/caspase 2/caspase 3, inhibiting autophagy, and stimulating the production of cytokine factors IL-1, IL-8, ERO-1α, and ELAM1. Epigenic modifications like SNHG3/SNAIL2 are involved in the regulation of ECM degradation. OPTN mutation can lead to the accumulation of LCII, which damages autophagy, resulting in ER stress, which induces RGCs death directly. Moreover, mutant OPTN can interact with myocilin accumulation and affect ER stress in TM cells as well. Also, OPTN mutation will facilitate the gliosis which induces cell loss via inflammation. The figure was created with BioRender.com (https://www.biorender.com/). Neurotrophins including P58IPK, MANF and BDNF are involved in the protective effect of ER stress. ECM extracellular matrix, BDNF brain-derived neurotrophic factor, MANF mesencephalic astrocyte-derived neurotrophic factor, RGC retinal ganglion cell, POAG primary open-angle glaucoma, TrkB tropomyosin receptor kinase B, OPTN optineurin, SNHG3 small nucleolar RNA host gene 3, SNAIL2 snail family transcription repressor 2
Involvement of ER stress in primary open-angle glaucoma
Although the pathogenesis of POAG is not fully understood, experts have reached a consensus that high intraocular pressure (IOP) is strongly related to retinal ganglion cells (RGCs) death. The increased IOP results in lamina cribrosa transformation and squeezes the optic nerve head, leading to RGCs death involving ER stress. Trabecular meshwork (TM) cells are an essential part of the maintenance of TM function and normal IOP. TM dysfunction is an important pathogenetic factor of POAG in which ER stress plays a crucial role, and the manifestations include a decreased number of TM cells, stiffness of the TM tissue caused by the correlation of the actin reticulum in TM cells, a conformational change of TM beams, and excess accumulation of extracellular matrix (ECM) (including type I collagen and fibronectin). These changes increase aqueous outflow resistance, causing high IOP, RGCs death, and irreversible vision loss. Meanwhile, a subtype of POAG has normal IOP, and the pathology mainly focuses on RGCs death, which involves ER stress. Therefore, we discuss ER stress involvement in affecting TM cell function, ECM remodeling, and RGCs survival in POAG.
Function of ER stress in TM death
Gene mutations have a close relationship with chronic ER stress in POAG, leading to TM cell death (Table 1). The most common POAG mutations reside within myocilin (MYOC), which is a gene located on chromosome 1 (GLC1A) that encodes the protein myocilin, including N450Y, Y437H, G364V, Q368X, K423E, I477N, and P370L.55,56 Studies have revealed that the MYOC mutation causes about 4% of POAG cases, of which the most common type is juvenile open-angle glaucoma. Disrupting the conformation and production of myocilin by MYOC mutation facilitates mutant myocilin accumulation in TM cells instead of being secreted.57,58 In addition, Ca2+ imbalance in TM cells acts with the myocilin olfactomedin domain, which facilitates the remaining wild-type (WT) myocilin misfolding and accumulating.59 The myocilin accumulation in TM cells and its latter amyloidosis, acting as a key trigger of ER stress in POAG development, induces programmed cell death like apoptosis and impaired autophagy.60,61
ATF4 acts as the most significant upstream regulator of ER stress in TM cells.62 First, it aggravates the myocilin accumulation in TM cells. ATF4 directly acts with misfolded proteins and WT myocilin, which results in reduced secretion of myocilin and aggravates the cytotoxic function in TM cells. Excess ER stress facilitates defects in ERAD, which worsen myocilin accumulation.63 The presence of GRP94 induced by ER stress helps mutant myocilin escape from ERAD, and since mutant myocilin exists as an amyloidogenic protein, stimulating ubiquitin-proteasome degradation is limited.61 Then, ER stress in TM cells induces damage, including oxidative stress, inflammation, impaired autophagy, and mitochondrial dysfunction, and results in a morphological change of human TM cells and disruption of the normal cell cycle, which affects normal TM function. ER stress leads to oxidative stress and inflammation via activation of the ATF4-CHOP-GADD34/ERO-1α pathway, presenting inflammatory cytokines such as interleukin (IL)-1, IL-4, ROS, IL-8, endothelial leukocyte adhesion molecule 1, and cleaved caspase-3.62,64,65 Mitochondrial swelling can be found under mild ER stress during TM cell death.66 The ATF4-CHOP pathway activates impaired autophagy as the downregulated autophagy flux facilitates TM cell death.67,68 Inhibition of mTOR with rapamycin significantly saves TM cells, indicating the harmful function of autophagy in TM cells.55 As discussed above, the resulting TM cell dysfunction will affect the normal ECM. TM cells phagocytose debris from functional TM tissues to keep aqueous humor flowing. TM cells dynamically synthesize and degrade ECM, which provides an architecture to TM tissues and a normal humor aqueous outflow pathway. The overexpression of ATF4 impairs the phagocytic activity of TM cells which contributes to ECM accumulation.62 Also, ECM accumulation (fibronectin and actin) can lead to ER stress in TM cells.58,69,70,71 Transforming growth factor β2 (TGFβ2) increases in the aqueous humor of MYOCY437H mutation patients and facilitates ECM accumulation, which activates ER stress.70,71,72,73,74 In conclusion, protein aggregation in TM tissues, the proliferation of ECM, and changes in cytokines such as TGFβ2 dynamically interact to regulate IOP. Therefore, MYOC mutation leads to ER stress, which affects the lifespan of the TM cells, and ECM remodeling, resulting in glaucoma.60,75,76,77,78
Epigenetic modification is essential for correct myocilin folding and refers to stable and heritable changes in gene expression or cellular phenotype without changes in Watson–Crick DNA base-pairing.79 The core regulates chromatin structure and gene expression by covalently modifying histone proteins and nucleic acids, including DNA methylation, histone modifications, RNA modifications, non-coding RNA, and chromatin remodeling.80 Under OS, long non-coding RNA (lncRNA), small nucleolar RNA host gene 3 (SNHG3), and the snail family transcription repressor 2 (SNAI2) are upregulated in human TM cell culture, in which SNHG3 binds to ELAVL2 to stabilize SNAIL2 mRNA.81 Matrix metalloproteinases (MMPs) are a family of metzincin proteases that cleave the components of the ECM in TM and modulate TM architecture. SNAI2 decreases MMP3 and MMP9 activity, which destroys the balance of synthesis and ECM degradation and increases the aqueous humor outflow resistance.82 SNAI2 activation is dependent on the XBP1 pathway in carcinoma cancer.83 It indicates that SNAI2 in TM cells is responsible for TM cell death, and ECM accumulation is dependent on XBP1 and is under the control of ER stress. Thus, lncRNA and ER stress participate together to facilitate ECM accumulation and TM dysfunction.
N-linked glycosylation, a post-translational modification (PTM), is involved in myocilin synthesis and processing and attaches oligosaccharides to asparagine residues in the N-terminal domain of the protein.67,68 The glycosylated myocilin proteins will form disulfide bonds to achieve the correct spatial conformation. Therefore, inhibition of PTM results in protein accumulation and ER stress in TM cells, which causes TM cell loss and ECM accumulation. Tunicamycin, a broadly used ER stress inducer, acts by inhibiting N-linked glycosylation.
Function of ER stress in RGCs loss
Gene mutations in POAG are significant causes of RGCs loss, such as those in Optineurin (OPTN), TBK1, and WDR36, which are associated with ER stress in RGCs (Table 1). OPTN is an adapter protein mainly expressed in the cytoplasm and the autophagy receptor of the cell,76,84 whose mutation results in autophagy dysfunction, impaired signal transduction, and protein accumulation.85,86 OPTN mutations including E50K, R545Q, M98K, and 691_692insAG can be found in POAG with high IOP and normal IOP.87 TBK1 is associated with the autosomal dominant inheritance of normal glaucoma. Duplications and triplications of TBK1 are related to normal IOP glaucoma.88 The interaction of TBK1 and OPTN determines that the pathology of these two gene mutations is inextricably linked. WDR36 is widely expressed in eye tissues, such as the retina and optic nerve. Although the precise effect of the WDR36 mutation in glaucoma remains unknown, it can result in protein dysfunction and may lead to ER stress.89
OPTN mutation causes optic nerve axon degeneration and then RGCs loss through ER stress. As the optic disk rim loses nerve tissue, the lamina cribrosa recedes and oppresses the optic nerve.90 Neuronal axon transportation is important for neuronal function and RGCs survival, as RGCs deliver nutrients through axons to synapses, and aged organelles or signaling vesicles are retrogradely transported to the soma.91 The crushing and destruction of the optic nerve can recruit microglia and astrocytes, a process called gliosis. Mitochondrial fragments and inflammation cytokines like IL-1α, TNF-α, and C1q released by microglia facilitate the transition of astrocytes to a neurotoxic reactive type, which results in RGCs death.92,93,94 CHOP and BiP are localized in GFAP-positive astrocytes with the OPTNE50K mutation, indicating ER stress involvement in the activation of neurotoxic reactive astrocytes and RGCs loss.95 The morphology of mitochondria changes before the axon in aged OPTNE50K mice, and RGCs degeneration and OPTN mutated cells fail to initiate mitochondria autophagy.96 MAM disturbance activates ER stress and induces RGCs apoptosis.97 OPTN mutations can also affect the RGCs soma through ER stress. RGCs with OPTN mutations tend to possess protein accumulation through blocked autophagy and interaction with TANK-binding kinase 1 (Tbk1). The WT OPTN protein facilitates the transportation of ubiquitin or ubiquitinated aggregates to the autophagosome, whose mutation will cause large molecular protein degradation dysfunction and aggregates formation. TAR DNA-binding protein 43, which is responsible for mRNA processing and trafficking and microRNA biogenesis clearance, is blocked in RGCs, resulting in neurodegeneration like amyotrophic lateral sclerosis (ALS).98 TAR DNA-binding protein 43 accumulation has been proven to be closely related to ER stress and neuronal death.99 OPTN691_692insAG mutation can interact with Tbk1, which leads to LC3-II protein accumulation and directly contributes to cell death via ER stress.100 Furthermore, OPTNE50K enhances the affinity for Tbk1 and increases the insoluble OPTN protein in RGCs.86 As aforementioned, abnormal protein accumulation will induce ER stress, and persistent accumulation will cause RGCs damage. Besides the direct activation of ER stress, OPTNM98K makes RGCs more susceptible to damage caused by ER stress than WT RGCs and enhances apoptosis.101 The RGCs energy metabolism balance is disturbed by the OPTNE50K mutation. Impaired autophagy in OPTN-mutated mice, as aforementioned, will compensatively downregulate mTOR1. AMPK in RGCs, an energy homeostasis regulator and stress sensor, is activated via the downregulation of mTOR1, which is a pathway responsible for neurite growth and stem cell proliferation.102
In other models without gene mutations, ER stress also plays a crucial role in RGCs loss. In a DBA/2J mouse model of chronic glaucoma, neurofilaments in the optic neuronal nerve were lost, and the ER stress marker CHOP was colocalized with the neuronal nerve.103 ER stress decreases expression of Mfn-1 and Ace-tubulin, which contributes to mitochondrial fusion in spinal cord-injured neuron axons. Axon degeneration might further induce mitochondrial dysfunction in RGCs. The mitochondria fusion and rupture increase B-cell lymphoma 2 (Bcl-2) and decrease Bcl2-associated X (BAX) expression, which induces apoptosis. Besides classic intrinsic apoptosis induced by mitochondrial dysfunction, this change can induce a unique Bid-caspase2-caspase3 pathway to induce RGCs death.104 Aging is a risk factor for POAG. CHOP increases with age, and XBP1 decreases in human eye tissue.105,106 In the aged human retina, protein aggregates of non-phosphorylated tau and α-synuclein increase substantially, further supporting the presence of protein misfolding and the resulting ER stress.107 In a micro-bead-injected mouse model and a silicon oil-induced ocular hypertension mouse model, the high IOP and RGCs loss are accompanied by CHOP elevation.108
CHOP KO in vivo or XBP1s overexpression contributes to RGC survival, indicating a complicated and dual function of ER stress in RGCs.109 Protein accumulation in RGCs contributes to the binding of the Sigma-1 receptor (S1R) to IRE1.110,111 The S1R resides on the MAM, described as the “pluripotent modulator,” which only chaperones the ER stress sensor IRE1 to facilitate inter-organelle signaling for survival.112 It can transiently stabilize IRE1 by binding to it. Consequently, the period of conformational IRE1 is prolonged, and the downstream activity of splicing XBP1 increases, which can lead to cell survival.112 Furthermore, under OS, S1R prefers to bind to and phosphorylate BiP, which reduces its ability to refold protein.113 Therefore, XBP1s are upregulated modestly and transiently, whereas CHOP increases dramatically during the injury process. The total effect of ER stress is detrimental to RGCs survival.
The interaction between ER stress and neurotrophic factors plays an important role in glaucoma pathogenesis. p58IPK is an ER-resident chaperone, playing a critical role in facilitating protein folding and protein homeostasis, which protects RGCs from damage like apoptosis under ER stress.23,114,115 Meanwhile, the induction of p58IPK depends on XBP1.23 The mesencephalic astrocyte-derived neurotrophic factor is a member of a newly identified ER-localized neurotrophic factor family and is upregulated during ER stress.116 Upregulated mesencephalic astrocyte-derived neurotrophic factor can protect RGCs from hypoxia-induced apoptosis by inhibiting CHOP.117 p58IPK and mesencephalic astrocyte-derived neurotrophic factor can act together, which provides a protective function for RGC restoration.114 Brain-derived neurotrophic factor (BDNF) facilitates neuron regeneration, whose transcription relies on XBP1 splicing.118 Activated BDNF will form a positive loop for the IRE1-XBP1 pathway.119 Also, BDNF can prevent the upregulation of CHOP in RGCs and reduce RGCs loss.120 Src homology region 2-containing protein tyrosine phosphatase 2 is related to IL-1-induced Ca2+ signaling.121 Its overexpression can dephosphorylate the TrkB receptor, and BDNF/TrkB neuroprotective survival signaling is reduced, resulting in ER stress and RGCs apoptosis.122 Nerve growth factor (NGF) belongs to the neurotrophic factor family and is essential for mature and immature neural cells. In glaucoma, decreased NGF facilitates RGCs apoptosis because NGF ameliorates the expression of the IRE1-JNK-CHOP signaling pathway and reduces Bcl2 and Bad.123 The mechanisms underlying neurotrophic factors and ER stress are still unknown.
Involvement of ER stress in other types of glaucoma
ER stress and acute glaucoma
ER stress, which modulates inflammation and immune response, is involved in acute glaucoma. IRE1α is demonstrated to facilitate translocation of NF-κB to release inflammation cytokines such as IL-6, IL-7, and TNF-α, which results in whole retina inflammation, especially RGCs death.124 Also, IRE1α can induce ROS, which then facilitates NLRP3 binding to mitochondria and activates caspase-2 to lead to mitochondria-related apoptosis.125 In acute glaucoma models, NLRP3 is involved in RGCs pyroptosis, apoptosis, necrosis, ferroptosis and PANoptosis, indicating the potential role of ER stress in mediating RGCs loss through NLRP3 activation.126,127,128,129,130 In acute glaucoma mouse models, the CXC-motif chemokine ligand 10/CXC-motif chemokine receptor 3 (CXCL10/CXCR3) axis is activated via ER stress, which can promote microglial recruitment, inflammation, and mediate leukocytes.131,132,133 The activated CXCL10/CXCR3 axis causes thinning of the retinal ganglion layer, indicating the role of ER stress as the regulator of the retinal immune axis.
Epigenetic modulations, including histone acetylation and methylation, are involved in ER stress and RGCs loss in acute glaucoma. Mice under ischemic/reperfusion (IR) injury present acute IOP elevation and optic neuron injury, which can mimic acute glaucoma. Histone deacetylase (HDAC) 6, an enzyme that deacetylates lysine residues on histones or other proteins in the cytoplasm and nucleus, is upregulated in IR models.134 BiP expression is under the control of the acetylation of histone H3 and histone H4 Arg3 methylation. YY1 recruits P300 (responsible for acetylation) and PRMT1 (responsible for methylation) to the BiP promoter to enhance BiP expression.135 BiP can bind to caspase 12 and block CHOP activation-induced apoptosis. Under IR injury, HDAC elevation decreases BiP expression, which upregulates CHOP and leads to RGC death.136
PTM participates in RGC loss via ER stress as well.137 Peroxiredoxins (Prxs) are a family of peroxidases that can reduce peroxide by oxidizing a specific region of a conserved cysteine residue.138 Acetylation of Prxs will increase their antioxidant ability. HDAC6 targeting Prx2 is specifically increased in glaucoma, which reduces the defensive ability of RGCs under stress.139 In neurons, disruption of Prx4 causes ROS to increase and subsequently induces ER stress.140 Thus, HDAC induces ER stress by deacetylating Prxs and causing RGCs loss.
ER stress and glucocorticoid-induced glaucoma
Glucocorticoids (GCs) are the most widely used medication worldwide. However, persistent use results in secondary glaucoma. Evidence of the involvement of ER stress in GC-induced glaucoma shows a higher level of BiP, GRP94, and CHOP and more phosphorylation level of IRE1α and eIF2α.141 Exerting GCs will result in the overload of MYOC levels and ECM proteins in TM cells, which can induce UPR, cause TM dysfunction, and elevate IOP.69,141 TGFβ2 signaling plays an essential role in glucocorticoid-induced ocular hypertension.142 Under ER stress induced by glucocorticoid, the TGFβ2/SMAD3 pathway is activated, which facilitates ECM deposition and actin accumulation and induces ER stress.141,142 Therefore, glaucomatous characteristics like elevated IOP and RGCs death can result from the simultaneous function of ER stress and TGFβ2.
ER stress and pseudoexfoliation
Pseudoexfoliation (PEX) syndrome is a late-onset disease characterized by the deposition of fibers and ECM accumulation.143 Statistical data reveals that 25% of people with PEX develop glaucoma, called PEX glaucoma which possesses an imbalance in ECM accumulation and subsequent activation of ER stress as BiP and PERK are upregulated.144 In addition, SYVN1, an E3-ubiquitin ligase, is downregulated, indicating decreased proteasome activity, which disturbs protein homeostasis and enhances ER stress in the lens capsule of PEX glaucoma.145 Then, caspase-3, caspase-12, and CHOP levels increase, indicating apoptosis and downstream retinal ER stress.
Therapeutic targets for glaucoma
Currently, the only effective method to treat glaucoma is to lower the intraocular pressure.146 The main treatment goals are slowing disease progression and preserving the quality of life.54 Many medicines aim to reduce intraocular pressure, including prostaglandin analogs, β-adrenergic blockers, α-adrenergic agonists, carbonic anhydrase inhibitors, and cholinergic agonists.147 In particular, the drugs used in POAG are effective only in a few glaucoma cases. Therefore, new drugs aimed at ER stress are required.
Treatment targeting ER stress for repairing damaged TM in glaucoma
ER stress is involved in TM cell dysfunction and loss. Currently, there are chemicals, natural compounds, gene therapies, and stem cell therapies aimed at ER stress.
Grp94 is responsible for protein homeostasis and degradation. In MYOC mutant TM cells, Grp94 recognizes myocilin olfactomedin and facilitates myocilin accumulation, which induces ER stress and TM cell dysfunction or loss.148 Inhibition of Grp94 facilitates mutant myocilin degradation through autophagy rather than ERAD at first and decreases accumulation.63 4-Br-BnIm was proved to be safe and can selectively inhibit Grp94, which facilitates mutant or misfolded myocilin degradation through effective autophagy in TM cells.149 PERK, the upstream component of the UPR branch, can initiate apoptosis and DNA damage in TM cells, which affects the cell cycle, morphology, and function. LDN-0060609, a PERK inhibitor, exists in aqueous solution as ketone, enol, and enolate, and can save TM cells from ER stress.150 4-Phenylbutyric acid (4-PBA), an aromatic short-chain fatty acid, can enhance the outflow of mutant myocilin.63,151 4-PBA can also degrade the ECM by activating MMP9.152 Furthermore, in GC-induced glaucoma, 4-PBA alleviates ER stress, like CHOP expression in the TM tissue, and decreases IOP.141 Astragaloside-IV, once used in renal and cardiac diseases, is effective for preventing myocilin deposition.153 Astragaloside-IV can rescue TGF-β2 induced ocular hypertension by modulating ECM deposition and ER stress in the TM by interacting with the MMP3 and MMP9 systems.153 Regarding the specific MYOCD384N mutation resulting in POAG, trimethylamine N-oxide, a natural osmolyte, can act as an ER chaperone, which alleviates myocilin misfolding and rescues TM cells from ER stress-induced apoptosis.151
Considering the importance of MYOC mutations and ER stress in the pathogenesis of POAG, many researchers have developed new therapies targeting mutant genes. Clustered regularly interspaced short palindromic repeats (CRISPRs) and Cas proteins are broadly expressed in bacteria and archaea.154 The CRISPR-Cas9 system, which is an immune system using RNA-guided nucleases to cleave foreign genetic elements, is commonly used in gene editing. By introducing a single-guide RNA into the CRISPR coding region to achieve specific recognition, target genes can be identified through complementary base pairing. The Cas9 protein can recognize the protospacer-adjacent motif sequence located upstream of target genes.155 Subsequently, cas9 promotes gene editing by inducing DNA double-strand breaks. This technology can be used to insert, replace, and delete target genes by introducing different single-guide RNAs. MYOC mutation results in protein accumulation and later ER stress-induced TM damage and ECM remodeling. MYOCY437H KO by CRISPR-Cas9 assembly Ad5-crMYOC alleviates myocilin accumulation and ER stress, restoring IOP and vision function.77 RNA interference can precisely modulate gene expression in mammalian cells. Small interfering RNA (siRNA) is a component of the RNA interference complex that silences specific genes with complementary sequences.156 After siRNA formation via Dicer or direct introduction into cells, it will form an RNA-induced silencing complex. Subsequently, siRNA binds to the target mRNA through complementary sequences, while an argonaute protein, like endoribonuclease, in the RNA-induced silencing complex cleaves target mRNA to achieve the silence of target genes.157 The introduction of siRNA to reduce mutant MYOC expression contributes to the repopulation of TM cells, prevention of RGCs loss, and recovery of IOP via inhibition of ER stress.75 As ECM remodeling in TM tissue has a close relationship with the overexpression of fibronectin, CRISPR-Cas9 targeting fibronectin successfully downregulates the ECM deposition and reduces ER stress, which protects TM function.69
Stem cells are self-renewable and able to differentiate directionally from functional cells, which can be used in the treatment of a variety of diseases. Human trabecular meshwork stem cells (TMSCs) are extracted from human TM tissue and can differentiate into adipose cells, neuronal cells, and TM cells.158 The pluripotent stem cell marker OCT4 and the neural stem cell marker Nestin can be detected in TMSCs.158,159 Similar to mesenchymal stem cells (MSCs) with directional homing characteristics, TMSCs injected into the anterior chamber can orient homing to TM through the interaction of highly expressed CXCR4 receptors on TMSCs with the SDF1 molecule in TM tissue.160 The highly expressed integrin, α5β1, on the TMSC membrane promotes the anchorage and survival of homing TMSCs in TM tissue by interacting with fibronectin, an ECM component of TM tissue.161 Considering the homing and differentiation functions, transplantation of TMSCs to treat MYOC mutations resulting in TM cell dysfunction is viable. TMSC transplantation can facilitate aqueous humor outflow, recover normal IOP, rescue RGCs function, and reduce RGCs death.162 The potent mechanism is that TMSCs homing TM differentiates into TM cells with phagocytic function, which facilitates relief of ER stress, replication of endogenous TM cells, and the restoration of ECM structure.162 Also, TMSCs are less sensitive and can survive in strong ER stress environments.66 These results indicate the possibility of stem cell therapy by transplantation of TMSCs in the future. Induced pluripotent stem cells (iPSCs) and MSCs can be induced into cells resembling TM cells in vitro.163 MSCs can be distributed to TM and reduce RGCs loss.164 Combining stem cell therapy with superparamagnetic iron oxide nanoparticles is proven to be safe for MSCs and helps stem cells anchor and function in TM tissue accurately by using magnetic field positioning.165 Adipose-derived stem cells can integrate into TM tissue and normalize IOP, whose homing ability is directed by CXCR4/SDF1, which vividly reduces ER stress induced by mutant TM cells.166
Exosomes are thought to be the main mediators of MSCs paracrine effects. MSCs-derived exosomes have low immunogenicity and are relatively safer than stem cell therapy. Direct treatment with MSCs exosomes can achieve a similar therapeutic effect as stem cell transplantation, meaning stem-derived exosomes have become a “cell-free therapy” option for a variety of diseases. Exosomes derived from bone marrow MSCs protect TM cells from oxidative stress and inflammation. The non‐pigmented ciliary epithelium can secrete exosomes rich in miRNA and cytokines, which function on the TM to regulate cell proliferation and ECM. Treating TM cells with NPCE-derived exosomes causes decreased COL3A1 expression. The miR29b component in the exosome is proved to affect autophagy and downregulate molecules involved in ER stress, including CHOP, ATF6, eIF2α, and XBP1.167 miR29b can also downregulate the WNT/β‐catenin pathway, responsible for ECM production.168 In addition, nrf2 enriched in non‐pigmented ciliary epithelium-derived exosomes can protect TM cells from oxidative stress, which can directly inhibit the apoptosis pathway and indirectly reduce TM cell loss by alleviating ER stress.169
Treatment targeting ER stress for rescuing RGCs in glaucoma
RGCs loss is a common reason for vision loss, in which ER stress plays an essential role. Treatments targeting ER stress include chemicals, neurotrophic factors, approved drugs in other fields, and gene therapy.
In acute glaucoma, ischemia resulting in hypoxia and malnutrition causes RGCs to lack ATP, inducing ER stress. Thus, RGCs undergoing ER stress in acute glaucoma are subject to cell death. ATPase Kyoto University Substances (KUSs) are inhibitors of valosin-containing protein (VCP) ATPase, which can be exerted by intravitreal injection.170 KUSs can alleviate CHOP induction by modulating key genes, such as Zfp667, and have been shown to save vision in rats by protecting RGCs, amacrine cells, and photoreceptors.171,172 The PERK-ATF4-CHOP pathway is involved in RGCs death and retinal axon degeneration via apoptosis and other programmed cell death under stress, while the IRE1α-XBP1 pathway is activated in RGCs and protective for RGCs survival. Directly mediating UPR molecules can be a novel strategy for glaucoma. Using adeno-associated viruses (AAV) to deliver proteins and mediate the UPR pathway can be protective for RGCs. Combining CHOP or eIF2α KO by CRISPR/Cas9 with XBP1 activation by AAV-XBP1s provides a synergistic function, that saves both the neuron axon and RGCs soma, and restores vision function.173 AAV-mediated Grp78 injected into the retina can be transported to RGCs and reduces apoptosis by attenuating ER stress through downregulating CHOP, ATF4, and eIF2α.174 KO of CHOP or ATF4 alone by CRISPR/Cas9 relieves UPR downstream DNA damage and facilitates neuron axon regeneration.175 Histamine receptor H1-mediated Ca2+ release and ER stress in RGCs can be blocked by amoxapine, desloratadine, and maprotiline.108 These drugs can inhibit all three UPR pathways and protect RGCs, and they have been approved by the Food and Drug Administration (FDA) to be safe in clinical trials. This protective effect can also be achieved by HR1H deletion in RGCs using CRISPR/Cas9 technology.108 siRNA aimed at ER stress has been verified to protect RGCs. RNA-dependent PKR phosphorylation facilitates eIF2α activation, which leads to RGCs loss. Thus, inhibition of PKR by an imidazolo-oxindole derivative or siRNA can relieve ER stress like CHOP reduction and promote RGCs survival.176 Valdecoxib, a selective COX2 inhibitor that has been used to treat osteoarthritis and arthritis, can reduce RGCs apoptosis in vivo and in vitro via inhibiting ER stress activation like the PERK-ATF4-CHOP pathway.177 p58IPK overexpressed by AAV can elevate RGCs survival rates through refolding proteins and inhibition of ER stress.115 Also, p58IPK might prevent mitochondria-related cell death and provide cells with increased expression of neurotrophins like BDNF. Nervous excitability toxicity and high IOP are correlated with ER stress-induced RGCs death in glaucoma models. Retinoid X receptors, originally highly expressed in ganglion cell layers, are downregulated in glaucoma retinas and induce ER stress. Elevation of retinoid X receptor expression in the retina by resveratrol can reduce ER stress-induced apoptosis and mitochondrial dysfunction.178 It can also repress HDAC1 in RGCs. As aforementioned, HDAC activity is associated with ER stress gene transcription, such as CHOP and BiP, indicating HADC inhibition can be a novel target for inhibition of ER stress in RGCs. Tubacin, an HDAC6 inhibitor, restores prx2 expression, which is a retinal antioxidant, and reduces retinal degeneration.134 Valproate, a bipolar disorder and epilepsy drug, can inhibit HDAC as well and is proven to increase the expression of BiP and decrease CHOP, which disturbs harmful ER stress and protects RGCs.136 Drugs targeting ER stress and autophagy to rescue RGCs with OPTN mutations have been studied. Rapamycin could restore the impaired autophagy in RGCs with OPTN mutations and recover RGCs function. While rapamycin inhibits the mTOR pathway, which is harmful to RGCs proliferation, a new drug, trehalose, can induce autophagy in OPTNE50K retinal organoids independent of the mTOR pathway which decreases OPTN spot accumulation and normal neuron morphology and function compared with WT RGCs.102 Considering the close relationship between ER stress and OS, combining drugs with nanoparticles to deliver inhibitors at the same time can be more effective. A glaucomatous microenvironment-responsive degradable polymer has been designed, characterized by thioketal bonds and a 1,4-dithiane unit in the main chain as well as pendant cholesterol molecules. Thioketal bonds and the 1,4-dithiane unit are responsible for erasing ROS in RGCs while cholesterols are used to target the cell membrane.179 Designing nanoparticle-wrapped drugs to target ER stress could provide innovative therapy.
As aforementioned, S1R can be released and upregulated in glaucoma, which can prolong the protective signal pathway of IRE1-XBP1, while the protective effect of S1R is limited under stress. SR1 ligand (+)-pentazocine can prevent the inhibition effect of S1R on BiP and reduce the phosphorylation of BiP, which reduces ER stress activation as PERK, ATF4, ATF6, IRE1α, and CHOP are downregulated, and saves RGCs in vitro.113 Neurotrophic factors regulate neural system development and function. Utilizing neurotrophins can directly support neuron regeneration, restoring their function.180 Furthermore, considering the reduced content of neurotrophic factors, which are correlated with ER stress, applying BDNF and mesencephalic astrocyte derived neurotrophic factor (MANF) can inhibit the upregulation of CHOP and prevent RGCs apoptosis.117,120 Also, recombinant human NGF prevents RGCs loss and was shown to be safe and effective in a phase 1b randomized controlled study of POAG patients.181
ER stress and diabetic retinopathy
Diabetic retinopathy (DR) is the most common and serious ocular complication of diabetes mellitus.182 DR is the leading cause of vision loss in developed countries.183 The number of diabetes patients worldwide reached 410 million in 2015 and is estimated to reach 640 million in 2040. About 40% of type 2 diabetes patients and 86% of type 1 diabetes patients have diabetes retinopathy. Diabetes retinopathy is classified as non-proliferative or proliferative. Non-proliferative diabetes retinopathy is characterized by the destruction of the blood–retinal barrier (BRB) function. It causes retinal edema, hemorrhage, and exudation. Proliferative diabetes retinopathy manifests as pathological retinal angiogenesis (fibrovascular membrane formation along with the vitreoretinal interface, vitreous hemorrhage, and retinal detachment). Retinal microvascular disease in diabetic patients is often accompanied by retinal neurodegeneration, which is an important reason for decreased vision in diabetic patients. It has been shown that different metabolic pathways are involved in the occurrence of diabetes retinopathy and neuropathy, such as an increase in the polyol pathway, advanced glycation end products, and activation of protein kinase C. It can induce ER stress in retinal cells and cause pathological changes in diabetic patients. We focus on the role of ER in the development of diabetic retinopathy and neuropathy, and describe therapy against it.
Involvement of ER stress in diabetic retinopathy
ER stress mainly functions in DR via BRB breakdown, retinal neovascularization, and neuron damage (Fig. 4). Metabolic changes can facilitate DR and ER stress in DR retinas and can provide metabolism sensors for glucose fluctuations (hypoglycemia and elevated glucose), O-GlcNAcylation, low-density lipoprotein, copper, and advanced glycation end products (AGEs).184,185,186 All three UPR pathways are activated in the DR process.187,188,189,190,191,192,193,194

Involvement of ER stress in DR. Many factors can activate ER stress in a DR model, including hyperglycemia, hypoxia, ROS accumulation, products like LDL, AGE and MGO, and glycemia fluctuation. In pericytes, activated ER stress can induce the ATF4/CHOP pathway and then activate mitochondrial dysfunction, VEGF, and MCP-1, which facilitates leukocyte adhesion and vascular leakage. In DR, macrophages accumulate and facilitate RNV through ER stress or the IL-17A/TXNIP/NLRP3 pathway. Following ER stress, XBP1 facilitates protective anti-inflammatory and anti-neovascularization cytokines. In addition, the ATF4/ CHOP pathway to contributes inflammation, RNV production, and apoptosis of BAX, caspase 3, and PARP. The RPE cells are the most important component of the outer epithelial barrier. ER stress injures the barrier by destroying VE-cadherin and Claudin 5 through O-GlcNAcylation of VE-cadherin/Grp78, MAPK pathway, NF-κB activation and inflammation. ER stress affects the barrier directly through ROS production, mitochondrial membrane potential loss, and PEDF decrease. ATF4/SDF1α leads to RNV in RPE. Impaired autophagy and lncRNA are involved in the development of DR as well. The key enzyme of producing light-sensitive 11-cis-retinol is suppressed, which influences vision. a. retinal ganglion cell; b. amacrine cell; c. bipolar cell; d. horizontal cell; e. Müller cell; f. cone cell; g. rod cell; h. retinal pigment epithelium (RPE). The figure was created with BioRender.com (https://www.biorender.com/). DR diabetic retinopathy, LDL low density lipoprotein, AGE advanced glycation end product, MGO methylglyoxal, VEGF vascular endothelial growth factor, RNV retinal neovascularization, NLRP3 NOD-like receptor protein 3, PEDF pigment epithelium-derived factor, RPE retinal pigment epithelium
Function of ER stress in BRB breakdown in DR
BRB can be divided into inner and outer components, with the inner BRB being formed by tight junctions between neighboring retinal capillary endothelial cells and the outer barrier by tight junctions between retinal pigment epithelium (RPE) cells.195 Besides, Müller cells and pericytes are important for the maintenance of normal inner BRB function. Since ER stress is activated in cells composing the BRB in DR, the importance of BRB destruction in the pathogenesis of DR becomes apparent.192,196
Various candidate gene association studies have revealed an immense association between genetic factors and the development of BRB damage, including the involvement of aldose reductase, which led to the accumulation of sorbitol through the polyol pathway (Table 1).197,198 Aldose reductase contributes to reduced NAD+ levels and inhibits sirtuin protein, leading to an increase in protein acetylation, which may be correlated with O-GlcNAcylation. Also, aldose reductase attributes to Müller glia (MG) activation. Aldose reductase C(-106)T polymorphism is a DR risk.198
PTM and epigenetic modulation, including glycosylation and the lncRNA/miRNA axis, are also involved in BRB damage. Glycosylation of plasma membrane and secretory proteins is a global phenomenon called post-translational modification (PTM).199 The glycosylation process can occur in the cytosol, ER, or Golgi complex. Glycosidases and glycosyltransferases are the essential processors of glycosylation.200 O-GlcNAcylation is a key PTM based on the addition of a single monosaccharide, β-O-D-N-acetylglucosamine (β-O-GlcNAc), ontoserine, or threonine residues of nuclear, cytoplasmic, and mitochondrial proteins.201 ER stress plays an important role in the O-GlcNAcylation of endothelial junction protein, resulting in inner BRB breakdown.202 During high glucose conditions, O-GlcNAcylation is enhanced by upregulating the expression of glutamine-fructose-6-phosphate aminotransferase 2 (GFAT2), which increases the flux from the hexosamine biosynthesis pathway, leading to the formation of uridine 5′-diphosphate-N-acetylglucosamine, the substrate for PTM, loss of retinal endothelial barrier integrity, and transendothelial migration of monocytes.192 Also, under treatment of ER stress inducers in retinal endothelia cells, translocation of GRP78 to the plasma membrane increases O-GlcNAcylation of proteins, including focal adhesion kinase, a known regulator for vascular permeability; cathepsin D, which is responsible for endothelial permeability; and particularly VE-cadherin and β-catenin, which result in defective complex partnering.202 A high-fat diet in mice has been shown to contribute to ER stress in MG and increase expression of O-GlcNAcylation protein.192 ER stress (especially eukaryotic translation in initiation factor 4E, eIF4E) activates the lipotoxicity sensor nuclear receptor subfamily 4 group A member 1 (NR4A1) and GFAT2 to induce O-GlcNAcylation protein.192,203 The O-GlcNAcylation phenomenon in MG might destruct the BRB by upregulating CD40 expression and increasing inflammation.203 The fluctuation of ncRNAs caused by hyperglycemia exists in RPE cells, and their function can be harmful or beneficial for different ncRNAs.188,189 ER stress is associated with ncRNAs in the pathogenetic processing of outer BRB breakdown. lncRNA growth arrest‑specific transcript 5 has multiple cell functions, including promotion of vascular endothelial growth factor (VEGF)-A, apoptosis (decreasing Bcl/BAX ratio), and pyroptosis.204 In ARPE cells, hyperglycemia downregulates expression of growth arrest‑specific transcript 5, which inhibits ER stress by interacting with sarcoplasmic/ER Ca2+ ATPase 2, which leads to inflammation and BRB injury.188,205 miR-204 directly targets and downregulates sirtuin-1 lysine deacetylase in RPE. The activation of the miR-204/sirtuin 1 axis worsens ER stress and leads to apoptosis, presenting as an elevation of cleaved caspase-3, -9, -12, and poly ADP-ribose polymerase.189
ER stress and OS influence each other regarding BRB damage and result in cell death through inflammation, apoptosis, and tight junction protein degradation. Under hyperglycemia, the ATF4/CHOP pathway is activated, and apoptotic-related molecules increase in MG, including BAX, cleaved caspase-3, and poly ADP-ribose polymerase, through the ATF4/CHOP pathway.187,206 One of the mechanisms of ER stress-induced MG apoptosis is that glyceraldehyde 3-phosphate dehydrogenase is transported and localized in the nucleus by binding Siah-1 under triglyceride treatment.207 RPE cells undergoing OS and ER stress have a decreased change in tight junction protein ZO-1 and occlusion.208 Under mild ER stress and chronic proinflammation, a feed-forward loop is formed in the endothelial junction protein, in which TNF-α is exacerbated and visual deficits are caused in the retina.209 Also, following the mitogen-activated protein kinase (MAPK) pathway and NF-κB activation, claudin5 is downregulated among tight junction proteins.210,211 Furthermore, intermittent high glucose may result in the activation of the ATF4-CHOP pathway, which can facilitate the release of MCP-1 in pericytes.
Mitochondrial dysfunction correlates with the metabolic pathway, apoptosis, and inflammation in the pathology of BRB destruction. Stimulator-of-interferon genes (STING) are vital for sensing cytosolic DNA and initiating innate immune responses against microbial infection and tumors and are located in the ER as homodimers. It can be activated by cyclic guanosine monophosphate (GMP)-adenosine monophosphate produced by the key DNA sensor cyclic GMP-adenosine monophosphate synthase and transported to Golgi binding TBK1 and interferon regulatory factor 3. The cGAS-STING axis also activates the NF-κB pathway, resulting in IFN production and inflammation activation, which releases cytokines like IL-1β, TNF-α, and IL-6.212 In human retinal vascular endothelial cells, the IRE1α and ATF6 pathways are upregulated under hyperlipidemia, which facilitates mitochondrial dysfunction and results in mitochondrial DNA leakage.213 As the most abundant and potent STING ligand, mitochondrial DNA stimulates STING activation and enhances the immune response, causing microvascular hyperpermeability.213 ER and mitochondrial coupling is accompanied by elevated mitochondrial calcium ions (Ca2+) and mitochondrial dysfunction in AGE cultured endothelial cells and apoptosis214 via IP3R1-GRP75-VDAC1 which can be inhibited by 4-PBA. In human retinal pericyte death, mitochondria dysfunction is characterized by mitochondrial membrane potential loss and cytokine c release.184,215 The RPE is an important constituent of the outer retinal barrier, and its damage equals the damage to the retina. Accumulation of AGEs and their adduct, methylglyoxal, in RPE can produce ROS, which activates mitochondrial membrane potential loss, intracellular calcium elevation, and an ER stress response to induce RPE cell death.216 Overexpression of mfn2 to induce mitochondrial merging also contributes to RPE death.186 Key isomerases like RPE65, LRAT, and RDH5 that convert light-insensitive all-trans-retinyl ester to light-sensitive 11-cis-retinol for continued visual function in RPE decrease under ER stress.194
The dual function of autophagy in human retinal pericyte survival has been researched. When treated with low-dose low-density lipoprotein, increased autophagy markers like beclin-1 and LC-3 facilitate cell survival, while impaired and exaggerated autophagy is induced under severe oxidative and ER stress. JNK phosphorylation is essential to autophagy induced by low-density lipoprotein and ER stress, which implicates PERK–eIF2a and IRE1–JNK signaling pathways in autophagy. Unlike JNK, which is involved in apoptosis in other cells, it is CHOP that accelerates pericyte apoptosis.184,215
Function of ER stress in retinal neovascularization in diabetic retinopathy
Neovascularization is one of the most important pathogenetic changes in DR, and different cytokines involved in the neovascularization process and the disturbance of normal cell function are related to ER stress.
There is ample evidence that gene polymorphisms play a prominent role in the pathogenesis of DR via ER stress in neovascularization. Gene variation in transcription factor 7 like 2 (TCF7L2) is a susceptible contributor to PDR (Table 1).217 The T allele of TCF7L2 rs7903146 is associated with fibrovascular membrane formation in type 2 diabetes mellitus-PDR patients.218 As part of the Wnt pathway, TCF7L2-regulated pathological neovascularization and VEGFA generation occur in diabetic models via ER stress-dependent pathways.218,219 The distribution of TCF7L2 is mainly in the cell nucleus of the RGCs layer and the inner nuclear layer. TCF7L2 overexpression in retinal progenitor cells (RPCs) affects endothelial cell transcription and leads to microvascular permeability.217 Increased ER stress markers like eIF2α indicate that ER stress elevates the microvascular generation function of rs7903146, and the T risk variation confers additional susceptibility to ER stress. VEGF can induce vascular permeability and retinal neovascularization, ultimately leading to microvascular damage and retinal dysfunction.220 In a survey conducted in a Han Chinese population, insulin-like growth factor 1 gene polymorphisms (rs6218, rs35767, and rs35767) were associated with DR.221 Insulin-like growth factor 1 facilitates the MG to induce neovascularization, whose expression can be upregulated under ER stress.222,223 The single-nucleotide polymorphisms of VEGF, including rs699946, rs833068, rs3025021, and rs10434, have an immense correlation with blinding DR in T1DM and type 2 diabetes mellitus.224 ER stress facilitates VEGF secretion, indicating the correlation between single-nucleotide polymorphisms of VEGF and ER stress in DR pathology. TGFβ1, the encoding protein, can be facilitated via ER stress activation and is involved in the TGF-β/Smad3 signaling pathway in regulating glucose and energy homeostasis.225,226
Epigenetics is defined as “the study of changes in gene function that are mitotically and/or meiotically heritable and that do not entail a change in DNA sequence.”227,228 Epigenetic changes involve both DNA and chromatin molecular modifications that change the expression of genes and genome activity and include DNA methylation of CpG dinucleotide residues, histone modification, most ncRNAs, RNA methylation, and chromatin structure.228 In the last decade, epigenetic modulations have been broadly studied in type 2 diabetes mellitus, but few conclusions have been drawn regarding DR.229 lncRNA metastasis-associated lung adenocarcinoma transcript-1 is found to contribute to inflammation and epigenetic regulation in DR205 and suppresses Grp78 production to regulate ER stress and alleviate inflammation and angiogenesis.205 It also sponges and inhibits miR-125b expression, which can suppress retinal neovascularization characterized by VE-cadherin and VEGF downregulation.230
High glucose results in antioxidant elimination involving SOD, which involves eliminating superoxide radicals; CAT converts harmful H2O2 into H2O and O2, while GR regulates physiological glutathione levels to stabilize ROS levels.231 OS and ROS release can be the early changes in DR and act as upstream regulators of ER stress, and all three UPR branches are involved.232 In particular, ATF4 suppresses antioxidant enzymes to increase ROS and induce OS. Also, ATF4 stabilizes HIF1α and plays a crucial role in generating VEGF. In addition, ER stress is the causal factor for inflammation and angiogenesis, coupled with VEGFA upregulation, overexpressed inflammatory cytokine transcription like TNF-α, IL-6, NF-κB, IL-17A, and ICAM-1,233,234 and inflammation cell infiltration. For example, the proinflammatory cytokine IL-17A is involved in macrophage polarization, which can induce M1 macrophage polarization. In response to hypoxia, the interplay between IL-17A and ER stress contributes to retinal neovascularization via modulation of the TXNIP/NLRP3 signaling pathway.234 TPL2 (tumor progression locus 2) is downstream of proinflammatory cytokines. Sensing AGEs, the TPL2/SDF1α (stromal cell-derived factor-α) axis, which regulates vascular generation, is activated in the RPE, resulting in microvascular dysfunction.235 Therefore, OS, ER stress, and inflammation are correlated and function as a cascade to generate cell death and VEGF production. Notably, retinal neovascularization can trigger retinal inflammation, which forms a loop between ROS, ER stress, inflammation, and VEGF. MG activation is found to be one of the main factors in DR onset and progression.236 Regarding infection or OS (like diabetes), MG is activated and regulates pro-angiogenic factors like pigment epithelium-derived factor (PEDF) and VEGF dependent on the activation of the UPR pathways.237,238,239
Function of ER stress in neuron damage in diabetic retinopathy
When exposed to pathogenic factors of DR, ER stress in the retina is correlated with retinal degeneration through inflammation, apoptosis, and autophagy.
Müller cell abnormalities, including gliosis, activated proliferative and migrative activities, inflammation cytokine release, and dysregulation of neuronal guidance cues, are key events in DR pathogenesis and can result in neuron damage. Semaphorins constitute a large family of endogenous secreted and transmembrane-associated proteins. The role of the secreted protein Sema3A (collapsin-1) is to induce the contraction and collapse of structures on axon growth cones.240 Under high glucose in the early stage, ER stress induces MG secreting Sema3A and is protective for MG resistance to overactivation of ER stress via inhibiting the IRE1α/XBP1 pathway.240 However, in the late stage, Sema3A’s protective function was inadequate, and neuronal degeneration occurred. Inflammation cytokine infiltration in neurons evokes IRE1α, and PERK-eIF2α-ATF4 pathways are involved in DR neurons, which in turn correlates with the generation of inflammatory factors IL-6 and MCP-1.188,190 Meanwhile, RGCs are lost via ER stress-induced apoptosis.241
ER-mitochondria cross-talk plays a vital role in neuronal death. PERK inhibits translation by adding a phosphate group to eIF2α, which stimulates the expression of TXNIP by activating transcription factors ChREBP and ATF5. As a result, TXNIP inhibits the ROS scavenging activity of TRX1 through a disulfide exchange reaction between the redox domains of TRX. Uncombined TXNIP will bind to TRX2 in mitochondria, which can act as key regulators of the NLRP3 inflammasome. This process facilitates mitophagy and dynamin-related protein 1 (Drp1-SNO) interaction with mitochondrial fission 1 protein to trigger the mitochondrial fission process.242
The ubiquitin–proteasome system is responsible for cleaning misfolded and non-functional proteins and is correlated with ERAD through the binding of HRD1 (an E3 ligase) and VCP/p97 (an ATP-driven chaperone governing the ubiquitin–proteasome system).243 Downregulation of deubiquitination enzymes, LCB3 and VCP/p97 in DR illustrates an impaired ubiquitin–proteasome system and low ERAD efficiency, resulting in neuron damage.187
Therapeutic targets for diabetic retinopathy
Treatment targeting ER stress for BRB damage in diabetic retinopathy
ER stress plays an important role in the destruction of the BRB. At present, a variety of natural extracts, chemicals, and gene therapies are used to treat diabetes retinal microvascular disease by alleviating ER stress. Natural chemicals are relatively safe, and attractive study options. Astragalus polysaccharide is a traditional Chinese Medicine and a bioactive polysaccharide extracted from Astragalus membranaceus root (Huang qi), which has been broadly employed clinically for its antitumor and antidiabetic properties.244,245 ER stress is involved in outer BRB damage and the occurrence of macular edema in patients with diabetes. It is associated with the induction of programmed death in RPE cells and the destruction of tight junctions between the cells. It has been demonstrated that astragalus polysaccharide can repair the destroyed diabetic outer BRB by alleviating inflammation and reducing the apoptosis of RPE cells through the miR-204/sirtuin 1 axis.189 Lactucaxanthin is a xanthophyll carotenoid predominantly presenting in lettuce that functions as an antioxidant and antidiabetic substance with anticancer properties dependent on tissues.246,247,248 It can directly reach the retina region and relieve symptoms via ER stress inhibition through potent antioxidant activity by downregulating PC, malonyl dialdehyde, inflammatory markers, OS inhibition, and HIFα induced VEGF reduction.208,233 Furthermore, it can decrease vascular leakage by rescuing the destroyed tight junctions between RPE cells in diabetic models.208 Chrysin is a flavone-type flavonoid that exists in honey, propolis, honeycomb, and passion flowers and exhibits multiple biological effects, including anti-inflammation and neuroprotection. Chrysin can reduce ER stress in RPE cells. It can reverse aberrant production of VEGF, insulin-like growth factor-1, and PEDF in glucose-incubated RPE cells, which contributes to restoring impaired BRB. Furthermore, chrysin could repair the retinoid visual cycle by alleviating ER stress via AGE-RAGE activation in diabetic models.194 Elevated copper levels have been found in the serum of patients with DR. Copper synergistically interacts with high glucose to induce ER stress and inflammation in RPE cells through modulation of mitochondrial function and changing the expression of the mitochondrial fusion protein 2. It eventually causes outer BRB dysfunction. Copper chelation with penicillamine can relieve copper-induced toxicity in RPE. It can reverse outer BRB dysfunction through reduced ER stress and ameliorate mitochondrial fusion protein 2-associated mitochondrial dysfunction in RPE cells under diabetic conditions.186
ER stress is associated with the loss of retinal vascular endothelial cells, pericytes, and Müller cells, which contributes to diabetic inner BRB breakdown. Tauroursodeoxycholic acid (TUDCA) protects cells from DR damage by inhibiting GRP78 translocation, VE-cadherin O-GlcNAcylation, ER-induced apoptosis, and reducing VEGF generation and vascular leakage.204 TUDCA could reverse ER stress-induced damage to vascular endothelial cells via Takeda G protein-coupled receptor 5, suggesting TUDCA is a potential therapeutic candidate for diabetic inner blood barrier damage.249 Nobiletin, a polymethoxylated flavone extracted from citrus explants, can be transported from the blood to retinal tissue. ER stress induces GADPH nuclear translocation, which causes Müller cell death and inner BRB destruction. Meanwhile, the death of Müller cells results in decreased PEDF levels and an imbalanced VEGF/PEDF ratio, which exacerbates inner BRB damage. Nobiletin has been proven to protect Müller cells from HG-induced apoptosis by relieving ER stress in the cells, which facilitates the repair of diabetic-induced iBRB disruption and rebalances the VEGF/PEDF ratio.207 Ghrelin, a gastric-derived acylated peptide, regulates energy homeostasis by transmitting information about peripheral nutritional status to the brain and mainly binds to the growth hormone secretagogue receptor-1a, a seven-transmembrane G protein-coupled receptor.250 It is essential for protecting organisms against famine and is widely distributed in human cells. An advantage of ghrelin is that it can reach ocular tissues through the BRB. Ghrelin protects the inner BRB from high glucose (HG) injury by inhibiting activation of the PERK pathway and reducing ER stress in retinal vascular endothelial cells.250 As aforementioned, chronic hyperglycemia and hyperlipidemia are involved in DR. Hyperlipidemia can induce ER stress and activate the STING signaling pathway in retinal vascular endothelial cells, which is associated with inner BRB breakdown. IRE1α, as an ER stress sensor, can be knocked out by CRISPR-Cas9 technology in retinal vascular endothelial cells, which attenuates the STING signaling pathway and reduces mitochondria leakage by inhibiting IRE1α/XBP1 signaling and alleviating ER stress in the cells. It might provide a novel strategy to treat diabetic inner BRB breakdown.213 RNA interference has been used to treat diabetic retinal microvascular abnormalities by inhibiting ER stress in retinal cells. The transcriptional factor TCF7L2 participates in diabetic damage of the inner BRB. siRNAs targeting TCF7L2 could suppress ATF6 signaling-mediated ER stress and rescue inner BRB breakdown in vascular endothelial cells under high glucose conditions. A 58-kilodalton inhibitor of protein kinase is a member of the heat shock protein 40 family. It is an initiator of eIF2α phosphorylation, which plays a key role in the PERK-induced UPR response.251 siRNA against P58IPK was found to exacerbate diabetic vascular leakage, while overexpression of P58IPK inhibited ER stress-induced CHOP activation and VEGF elevation in retinal vascular endothelial cells.
Treatment targeting ER stress for neurodegeneration in diabetic retinopathy
Diabetic retinal neurodegeneration has been found in patients with diabetes mellitus. It causes vision to decrease even in the absence of diabetic microvascular disease. Neurotrophic factors are used to treat diabetic neuropathy. Neurotrophin-4 (NT-4) is a member of the well-known neurotrophin family that regulates neuronal networks by regulating neuronal survival, differentiation, growth, synaptic development and plasticity, and myelination. NT-4 relieves ER stress in DR and reduces RGCs injury, which can be considered a potential drug for diabetic neuron damage.241 New technologies have developed a complex of NT-4 with dendrimer nanoparticles.252 The NT4-polyamidoamine electrostatic complex can provide a sustained concentration of protein in vitreous and retinal tissues over an extended period after delivery and promote retinal, especially RGCs, recovery from injury.252 Chemical drugs that could directly inhibit ER stress have been investigated to protect retinal neuronal cells from diabetic injury. Liraglutide, a glucagon-like peptide-1 analog, is widely used in the clinic and has a protective effect on neurodegenerative diseases. Liraglutide can also treat diabetic neuropathy through activation of the Erk pathway and regulation of the Trx-ASK1 complex. It subsequently inhibits ER stress and OS in retinal neuron cells. 4-phenylbutyric acid (4-PBA) is a chemical chaperone that mimics endogenous chaperone activity to resolve ER dysfunction in pathological conditions. Administration of 4-PBA could decrease retinal neuron death in the outer nuclear layer and ganglion cell layer by attenuating ER stress in these cells in diabetic models.253 Sulforaphane is widely discovered in cruciferous plants and is a strong antioxidant and activator of nrf2.254 Sulforaphane inhibits ER stress via the AMPK pathway, and it can also reduce inflammation, apoptosis, and OS in retinal cells. It has been demonstrated that sulforaphane can prevent photoreceptor cell death under diabetic stimulus.254 A combination of stem cell therapy with chemical drugs provides a synergetic therapeutic effect on retinal neurodegeneration. Melatonin, a neuroendocrine hormone mainly synthesized in the pineal gland, is involved in pleiotropic biological functions, including control of the circadian rhythm, immune enhancement, and antioxidant, anti-aging, and antitumor effects. Melatonin targeting ER stress function has been revealed recently. It mainly functions in neural cells and is neuroprotective, which mainly inhibits CHOP and then PERK and GRP78/BiP.255 Combining melatonin and adipose-derived MSCs significantly delays the progression of diabetic neuropathy and retinopathy.256
ER stress and age-related macular degeneration
Age-related macular degeneration (AMD) occurs in the macular region and gradually affects central vision.257,258 AMD is the third leading cause of irreversible blindness worldwide, usually affecting people aged >55 years.259 AMD was estimated to affect 196 million people in 2020 and is expected to rise to 288 million by 2040, with the largest number of cases in Asia (113 million).260 Currently, AMD can be classified into two major categories: dry AMD and neovascular AMD (nAMD). nAMD is characterized by a choroidal neovascularization complex, subretinal or intraretinal fluid, hemorrhage, and/or fibrous scar tissue. Dry AMD manifests as the loss of RPE cells overlying photoreceptors and underlying choroidal capillaries.258
Though the concrete pathology of AMD is unclear, it is recognized that physiological and conformational changes happen on photoreceptors, RPE cells, Bruch’s membrane, and the choriocapillaris.261 RPE cells possess many functions for retinal homeostasis, including maintaining the outer BRB and secretion of proteins.262 Proteins secreted by RPE include PEDF, αB crystallin, and prosaposin, which can suppress angiogenesis and reduce drusen.263 First, intracellular debris, including lipofuscin, accumulates in the RPE, which causes RPE injury and ECM accumulation. As the lesion is aggravated, lipid-rich debris is laid down between the basal lamina of the RPE and the inner collagenous layer of Bruch’s membrane, involving RPE debris, lipids, minerals, and immune system-associated elements.264
Risk factors for AMD involve both genetic and environmental factors. Age, race, self-conditions such as basic diseases (diabetes, hypertension, etc.), and complement system activation play important roles in the incidence of AMD.265 Smoking is the strongest environmental risk factor for nAMD. Other elements, including diet, sunlight, glucose deprivation, and complements such as C3 and C3a, accelerate AMD progression.266,267,268 These risk factors can induce ER stress in AMD models.269,270,271 Research has revealed that different metabolic pathways, including the cholesterol pathway, retinoid metabolism, and lipid metabolism, are involved in drusen accumulation, neovascularization, and subretinal fibrosis via ER stress. We elaborate on the role of ER stress in the formation of AMD and its targeted treatment (Fig. 5).

Involvement of ER stress in AMD. Many risk factors contribute to AMD development, including smoke and light. The pathogenesis of AMD is closely associated with RPE death. Complement C3 can be activated and transformed into C3b to induce ER stress. The latter activated ER stress provokes the binding of C3a to C3a receptors, which amplifies the ER stress. ER stress promotes the inflammation in AMD to directly induce inflammatory factors like IL-6 and IL-8. In addition, XBP1 can provoke the protective nrf2 pathway to activate inflammation. Under ER stress, it activates caspase-4, generates ROS, elevates intracellular calcium, and reduces the mitochondrial membrane potential to promote the downstream activation of caspase-3, inducing apoptosis. Mitochondrial damage releases cytochrome c leading to RPE death. Neovascularization and ER stress influence each other through endocrine of VEGF. What’s more, decreased blood in choroid can induce hypoxia and ER stress in RPE cell and macrophages in retina. The polarization of macrophage depends on epigenic modifications of XBP1 gene which facilitates VEGF release and causes neovascularization. The figure was created with BioRender.com (https://www.biorender.com/). VEGF, vascular endothelial growth factor. AMD age-related macular degeneration, nrf2 nuclear factor erythroid2-related factor 2, RPE retinal pigment epithelium, VEGF vascular endothelial growth factor
Involvement of ER stress in age-related macular degeneration
ER stress and Neovascular age-related macular degeneration
Nowadays, genome-wide association studies identify variants associated with AMD (Table 1). The high-temperature requirement A1 (HTRA1) gene (rs11200638) is positive for anti-VEGF therapy.272 HTRA1 belongs to the high-temperature requirement A family and performs dual peptide refolding and degradative chaperone actions, which can be activated to defend against damage under UPR.273 HTRA1 disturbance presents a reduced ability to deal with proteotoxicity and cell apoptosis, indicating the correlation between HTRA1 variants and ER stress in the development of nAMD.273
ER stress-related epigenetic modifications are a vital part of nAMD pathology. ER stress promotes aberrant choroidal neovascularization (CNV) by modulating the expression of angiogenesis-associated genes. In nAMD models, the activated PERK/eIF2α pathway leads to the downregulation of miRNA-106b in vascular endothelial cells. miRNA-106b directly regulates VEGF and HIF-1α expression by targeting its 3′-UTR in vascular endothelial cells. Decreased miRNA-106b promotes the formation of CNV by increasing the proliferation and migration of vascular endothelial cells.209 Reduced blood flow in the choroid and retinal tissue hypoxia is associated with the occurrence of CNV in patients with nAMD. Hypoxia can increase the acetylation and protein stability of XBP1 by enhancing histone acetyltransferase p300, which activates UPR-associated molecules ATF4, GRP94, BiP, and CHOP and triggers UPR in macrophages. Activation of ER stress controls macrophage polarization from M1 to M2, which increases VEGFA. Hence, hypoxia induces p300 activation and regulates UPR and M2 polarization in macrophages, which increases paracrine secretion of VEGFA and promotes the proliferation, migration, and tube formation of choroidal vascular endothelial cells.274
OS interacts with ER stress, leading to AMD lesions. A combination of various stresses can be summarized as the integrated stress response (ISR), in which ER stress usually occurs upstream of the ISR and activated ISR.275,276 Amyloid-β protein precursor (AβPP) is a constituent of drusen, which can accelerate ER stress and ISR.277 ISR facilitates VEGF production and angio-vascularization in endothelial and RPE, in which ATF4 is the main contributor to CNV formation in AMD.274,275 Inflammation might be the mediator between VEGF and ER stress via the MAPK signaling pathway and the P300/XBP1s/Herpud1 axis.278
ER stress and dry age-related macular degeneration
Gene mutations and variants involved in dry AMD include phototransduction, sterol toxicity, and protein-folding dysfunction (Table 1). Abca4, localized on human chromosome 1p21, exists in AMD.279 Variants of Abca4 associated with AMD include D2177N, G1961E, and R1898H.279 The Abca4 protein is an ATP-binding cassette transporter 4 and is responsible for the elimination of metabolic products from phototransduction or lipids. Its mutation results in incomplete RPE phagocytic function and lipid deposition. As Abca4 carries all-trans-retinal out of the photoreceptor disc, its mutation injures retinoid metabolism by releasing free all-trans-retinal which is toxic for RPE and photoreceptors. The toxicity of all-trans-retinal is produced by direct activation of PERK/eIF2α/ATF4/CHOP signaling, which stimulates downstream apoptosis-inducing molecules like p-JNK, p-c-Jun, cleaved caspase-3, and cleaved-PARP.280 The oxysterol-binding protein gene is mainly expressed in the RPE, localized in the macular region, and its protein binds to and transports oxysterols. As the macula ages, oxysterols gradually accumulate and become cytotoxic for RPE, activating ER stress. Nucleotide changes in the oxysterol-binding protein, including c.347A>G, c.450G>T, and c.2351G>A, are found in patients with dry AMD.281 Single-nucleotide polymorphisms in CXCL3 are risk variants for AMD.282 ERp29 deficiency resulting in protein accumulation in CCL2−/−/CX3CR1−/− mouse models might explain the ER stress activation and related AMD.283
ER stress regulated by PTM is involved in dry AMD. Choroidal blood vessel atrophy leads to RPE loss through blood and glucose deprivation. In this circumstance, glycosylation of the VEGFA receptor peptide is incomplete.284 Simultaneously, a perturbation of normal glycosylation leads to VEGFA receptor accumulation, contributing to excessive ER stress, which acts as the connection between PTM and cell loss.
ISR significantly participates in dry AMD development. The whole retina undergoing ER stress produces ROS. Components of the retina, including MG and RPE, react to ER stress and reduce damage. The MG maintains homeostasis in the internal environment, including oxidative redox status and glutamate homeostasis. In response to ER stress, the MG exerts a protective role by upregulating AβPP to defend against apoptosis and recover functional gene transcription.269 Interestingly, AβPP is proven to be harmful in neovascularization AMD, as mentioned before. These data seem to convey that AβPP is a dual regulator of cell survival dependent on VEGF expression level. Under OS disturbance, RPE cells and their tight junctions are damaged, and BRB integrity is damaged. Meanwhile, Erp29, an ER chaperone protein, is upregulated to modulate redox status by increasing nrf2 and inhibiting ER stress in RPE, which enhances BRB integrity.270 When undergoing mild ER stress, activation of XBP1 upregulates the nrf2 expression level, which is protective for RPE.285,286 However, under severe ER stress, these compensatory responses are not sufficient to protect the cells. In the hydrogen-peroxide-induced cell viability loss of RPE, ferroptosis is the main cause of RPE death, which affords new perspectives for AMD pathology.287 Therefore, UPR is protective when the injury is mild but harmful when damage persists.
Since lipid drusen is the most important lesion in AMD, lipid metabolism is involved in ER stress, contributing to dry AMD. Evidence shows that the complement system in combination with ER stress leads to lipid accumulation in the RPE and Bruch’s membrane, in which ER stress is activated by complement C3 and C3a.266,267 Activation of ER stress directly stimulates SREBP and disturbs lipid homeostasis.288
Mitochondrial dysfunction in RPE has a close relationship with ER stress. Mitochondria are important for RPE integrity.289 In a model of AβPP deficiency, ER stress is overexpressed in RPE and results in cell apoptosis through the mitochondrial dysfunction center, which manifests as caspase-4 and caspase-3 upregulation, decreased BCL/BAX ratio, increased release of cytochrome c and glutathione, and mitochondria membrane potential loss.216,290 Under abundant ROS production, mitochondrial fission activates mitophagy to eliminate damaged mitochondria. The morphology changes of mitochondria including swelling and breaking is induced by ER stress, and this dysfunction induces RPE necrosis.291
Autophagy is excessive and impaired under ER stress.283 The normal function of RPE includes phagocytosis and digestion, in which the normal function of autophagy-lysosome-dependent ERAD is crucial to remove accumulated abnormal protein and protein aggregates. PERK-activated autophagy increases over time.292 However, severe and persistent ER stress causes prolonged autophagy and autophagy flux through the PERK pathway and even results in RPE self-death.268 Thus, light injury ER stress is activated and causes dry AMD-like retinal lesions, including drusen, RPE alteration, and photoreceptor degeneration.283 EIF2AK3 downregulation has been discovered in patients with AMD, which lowers the PERK level.
Therapeutic targets for age-related macular degeneration
Zinc, antioxidants such as ascorbic acid and vitamin E, lutein/zeaxanthin, and omega-3 (docosahexaenoic acid) used alone or in combination can delay AMD progression. Inhibition of VEGF is effective in treating neovascular AMD. For example, pegaptanib, bevacizumab, and aflibercept. However, AMD treatment lacks efficacy.
Natural plant extracts can safely and effectively target ER stress in AMD. Grape polyphenols relieve symptoms in both dry AMD and nAMD.293 Curcumin is a yellow pigment found in the rhizome of turmeric. It saves RPE from ER stress, OS, and mitochondrial dysfunction, showing potential for dry AMD treatment.294 Chemicals can also be potential treatments in the future. 4-PBA, the classic ER stress inhibitor, can act on both types of AMD. It suppresses choroidal neovascularization by inhibiting VEGF production and can prevent RPE death in a dry AMD model. Many other drugs can be used in dry AMD, such as paeoniflorin295 and propofol,296 which affect the molecular BAX and apoptosis processes of ER stress. As for nAMD, ISR inhibition can prevent the production of ATF4 and VEGF and act as a treatment target.275
These chemicals can be extracted directly from the human body and have been proven to be effective in dry AMD treatment. Humanin is a 21–24 aa peptide encoded by the mitochondrial MT-RNR2 gene encoding 16S rRNA discovered in surviving neurons in patients with Alzheimer’s disease. Humanin protects RPE cells from oxidative stress-induced cell death and affects not only the ER stress-related pathway but also mitochondrial-ER-associated membranes, which facilitate RPE survival in dry AMD.297 Taurine is one of the most highly expressed amino acids in the eye tissue. In RPE cells, overexpression of Calpain-1 and Calpain-2 can inhibit ER stress and ER stress-induced apoptosis and autophagy.298 Taurine has been proven to protect against starvation-triggered ER stress by upregulating calpain.298
Gene therapy aimed at ER stress has developed rapidly. miRNA has been utilized in in vivo and in vitro experiments and is proven to be anti-vascularization, including MiR-106b.209 Stem cell and stem cell-derived exosomes are proven to act in subretinal fibrosis. The human umbilical cord MSC-derived exosomes secrete several miRNAs that are beneficial for the improvement of nAMD. For example, miR-27b inhibits subretinal fibrosis and increases cell migration, which cannot be achieved with VEGF therapy.299 Stem cell therapy has progressed in the treatment of dry AMD. Injection of human RPCs cocultured into the AMD region releases neurotrophic factors and differentiates into neurons and MG to supply the loss in dry AMD.300 RPE-derived exosomes, including α-Crystallin, can be transported into the eye and protect the RPE from apoptosis.301 However, whether this protein facilitates or inhibits VEGF in nAMD is controversial. Utilizing this exosome in the clinic requires further progress.
In conclusion, these drugs can be used for dry macular degeneration while enhancing the inhibition of angiogenesis by targeting the mechanism of ER stress in nAMD.
ER stress and retinitis pigmentosa
Retinitis pigmentosa (RP) is a hereditary disorder that is the most common form of inherited blindness and is characterized by the degeneration of rod and cone photoreceptors, with a prevalence of 1:3000 worldwide.302,303 The inherent features of RP can be autosomal dominant (30–40% of cases), autosomal recessive (50–60%), or X-linked (5–15%) traits. Although most RP cases are non-syndromic, 20–30% of RP patients develop non-ocular disease. The onset of RP can occur in childhood, teenage, adult, or older individuals. The classic pattern is that it is difficult for RP patients to adapt to dark and night blindness in adolescence and lose their mid-peripheral visual field in young adulthood. As the disease progresses, they will have weak peripheral vision, develop tunnel vision, and eventually lose central sight in their 60s.302 Cone and rod photoreceptors and RPEs are the most affected cells in RP. It has been shown that mutated and misfolded protein accumulation can induce ER stress in RPE, cone, and rod photoreceptors (Fig. 6).

Involvement of ER stress in RP. The major pathogenetic process occurs in retinal cone and rod cells. The death of cone and rod occurs due to an imbalance between autophagy and ERAD. Impaired autophagy occurs through the P53-p38-MAPK-eIF4E cascade and is influenced by ATF4. In addition, ATF4 binds to key autophagy molecules LC3 and P62 to inhibit it. Proteasomes and lysosomes are responsible for clearing mutated protein, which is suppressed in RP. The ratio of autophagy: proteasome is decreased, which facilitates cone and rod cell death. ROS and Ca2+ can induce ER stress in RP. cdk5 is upregulated by ROS and Ca2+ and then stimulates the mekk1/JNK pathway, which promotes apoptosis. The IRE1α/ATF4/CHOP and ATF6 pathways influence caspase cascades and lead to apoptosis. ATF4 also influences p53 and then increases BH3 and Bad, which can lead to apoptosis directly and augment the mitochondrial permeability to facilitate apoptosis. RPE and blood-retinal-barrier (BRB) destruction are involved in the pathological process of RP. PRPF mutations cause the impaired autophagy and activated mTOR, which accelerate the photoreceptor outer segments. It is toxic for RPE. The accumulated PRPF protein accumulated in RPE induces apoptosis of RPE and destruction of BRB. The figure was created with BioRender.com (https://www.biorender.com/). RP retinitis pigmentosa, MAPK Mitogen-activated protein kinase, BRB blood-retinal-barrier, PRPF pre-mRNA processing factor, ERAD ER-associated degradation, ATF4 activating transcription factor 4, ATF6 activating transcription factor 6, eukaryotic translation initiation factor 2α (eIF2α), C/EBP-homologous protein (CHOP), PERK PKR-like ER kinase, IRE1 inositol requiring enzyme 1, XBP1 X-box binding protein 1
Involvement of ER stress in retinitis pigmentosa
Function of ER stress in photoreceptor cell death
Many gene mutations are involved in the development of RP (Table 1), and their pathogenesis is related to ER stress. However, each of these only relates to a small portion of RP. The most common mutation in RP is the rhodopsin gene (RHO) mutation, which directly results in photoreceptor degeneration and contributes to 25% of autosomal dominant retinitis pigmentosa (ADRP). Rhodopsin mutations, such as P23, R135w, Rh1G69D, T17M, and P53R, lead to the translocation of RHO protein from the cell membrane to the ER membrane and acceleration of ER stress.304,305,306,307 Among these, the P23 mutation is the most common.308 The harmful function of the mutant is reflected by protein accumulation and negative effects for WT RHO. The D1080N mutation in interphotoreceptor retinoid-binding protein, a key protein in photoreceptor survival, also prevents the transportation of proteins to the Golgi.309 A phosphodiesterase type 6 (PDE6) gene mutation is one of the main causes of ADRP. PDE6b encodes rod phosphodiesterase (PDE), which is an enzyme hydrolyzing cGMP to GMP in response to light and is only expressed in rod cells. Its mutation disturbs Ca2+ homeostasis and results in ER stress. Variants in interphotoreceptor matrix proteoglycans (IMPG2) have been reported in RP patients. IMPG is the matrix between photoreceptors and the RPE, which supports transduction, cell communication, and photoreceptor differentiation. Thus, IMPG2, resulting in matrix alteration, will cause retinal degeneration. In an IMPG2 KO mouse model, both rod and cone retinal cell degeneration happen at nearly the same time as ER stress activation and cell apoptosis.307
ER stress-induced OS plays a significant role in photoreceptor degeneration. Misfolded and unfolded proteins, including interphotoreceptor retinoid-binding protein and RHO, in photoreceptors can directly induce ER stress. ROS levels and Ca2+ levels increase dramatically, and then Mekk1 is phosphorylated, which induces cdk5 upregulation, and affects the JNK pathway and has a pro-apoptosis function.305,310,311 Rod photoreceptor death and retinal degeneration then occurs.312,313 In a retinal degeneration 10 model that possesses a PDE6 mutation and is widely used for RP studies, intracellular Ca2+ elevation-related ER stress occurs in the whole layer of the retina even before RP symptoms appear.314 As a result, ER chaperone S1R elevates in abundance to antagonize Ca2+.314 In the RGCs layer and cone photoreceptors, this early response can act as a protective response when rod cells are under attack. RNA-binding proteins pTDP-43 accumulates in the whole retina to form RNA stress granules. Over time, these harmful aggregates further activate ER stress, which leads to cell death other than rods, providing a possible mechanism for the death of cones, rod bipolar cells, horizontal cells, and RGCs followed by cone loss.
ER stress leads to mitochondrial dysfunction and photoreceptor mortality. p53 is responsible for the increase in the BH3-only protein BiK/Bad, which can augment the permeability of mitochondria, release apoptosis-inducing factor, and cause apoptosis.315 In RHO-mutated models, ATF4 coincides with p53 and is involved in photoreceptor apoptosis. Furthermore, mitochondrial dysfunction releases ROS and drives NLRP3 inflammasome activation in cone photoreceptors in an R23H model.316 In most cases, the loss of rod function is more severe than the loss of cone sensitivity. This shows cone retinal cell death is dependent on NLRP3 activation and that ER stress modulates the RIP1/RIP3/DRP1 axis of necrosis to induce degeneration.316
When mutated proteins, including RHO and interphotoreceptor retinoid-binding protein, accumulate, two clearance mechanisms, the ubiquitin-proteasome system or the autophagy-lysosome pathway, can be employed to deal with ER stress.317 ERAD is responsible for degrading accumulated protein. The IRE1α pathway is activated under ER stress and relies on functioning proteasomes and lysosomes to degrade the mutated and misfolded rhodopsin.318 Though ERAD is often beneficial for cell survival, it may be too effective and strong, resulting in cell death instead. However, in a RhoP23H/P23H mouse model, the early stage of ERAD activation disturbs RHO homeostasis, which erases almost all RHO in photoreceptors, influences photoreceptor growth, and facilitates photoreceptor death. It can explain why some phenotypes of RP present early-onset photoreceptor degeneration and early vision damage. The ratio of autophagy and proteasome, or “A:P ratio,” influences the clearance of proteins, and the balance is broken when the A:P ratio is overwhelmed in the late stage of ER stress.317 P23H mice experience increased autophagy secondary to ER stress, which leads to proteasome insufficiency and increases retinal degeneration.317 The ATF4/CHOP pathway contributes to impaired autophagy in P23H-accumulated mice through the P53-p38-MAPK-eIF4E cascade and binding to LC3 and p62.313 The downregulation of p62 facilitates the upregulation of keap1 and excessive degradation of antioxidant nrf2. In IMPG2 KO mice, impaired autophagy, gliosis, and apoptotic cell death are induced by ER stress in both rod and cone retinal cells.307 Since activated autophagy is impaired and the autophagy flux is decreased, autophagy functioning in RHO accumulation is harmful to cell survival.319 However, in P10 models, ER stress arouses defective autophagy in the early stage of RP.314 In these cases, autophagy possesses a time-dependent dual function for retinal degeneration. Since ER stress plays a role in facilitating excess autophagy, PERK, independent of the ATF4 pathway, may exert a different role. When inhibiting PERK, RHO accumulation increases and rod retinal cell death is aggravated.308 In addition, in a Drosophila study, PERK inhibited massive autophagy of WT RHO by inhibiting the IRE1/XBP1 pathway,320 indicating that the PERK pathway facilitates photoreceptor survival in the early stage of ER stress.
Function of ER stress in retinal pigment epithelium cell death
Mutation of genes encoding pre-mRNA processing factors (PRPFs), including PRPF3, PRPF4, PRPF6, PRPF8, PRPF31, and SNRNP200, is the second most common cause of ADRP (Table 1).321 The retina is metabolically active tissue, and steady pre-mRNA splicing is required. Mutations in PRPFs may cause damage to retinal cell function by causing global spliceosome dysregulation and influencing important functional gene expression within the retina, including RHO, Abca4, and oxidative stress-related genes, resulting in RP. This explains why patients carrying PRPF mutations possess manifestations such as RPE atrophy and Brunch’s membrane thickening, like AMD pathological changes. In PRPF31A216P/+ mice, mutant PRPF31 protein mainly accumulates in the PRE’s cytoplasm, which can induce ER stress.322 Photoreceptor degeneration is secondary to PRE dysfunction and mortality.
Impaired autophagy, inadequate ERAD, and an activated mTOR pathway contribute to RPE and photoreceptor death in RP models. Ubiquitinations of misfolded proteins are transported to proteasomes for proteolysis. The HSP70 family, a protein chaperone family, is responsible for correcting aggregates folding. When the misfolded or deficient PRPF31 is overloaded, the HSP70 family protein binds to PRPF31 accumulated in the RPE and triggers ubiquitination.323 The photoreceptor outer segment cycle is also dependent on the RPE phagocytosis function, and the dysfunction of RPE will cause their accumulation in the RPE. To deal with inadequate ERAD, autophagy is activated, but the autophagy flux is lower than in normal cells, indicating impaired autophagy. Thus, another protein management system, the mTOR pathway, is activated. With time, the overloaded ubiquitination protein triggers ER stress in the RPE and results in caspase-3-involved apoptosis. This reveals one of the mechanisms behind Bruch’s membrane thickening in retinal PRPF mutations. In addition, the ubiquitination of the tight junctions between RPE accelerates BRB destruction. The NLRP3 inflammasome can be released from the RPE and function on photoreceptors, including cone and rod retinal cells. Therefore, photoreceptor outer segment accumulation, RPE atrophy, and BRB destruction lead to photoreceptors death in RP.
Therapeutic targets for retinitis pigmentosa
There is no effective treatment for RP; only methods that slow its progress are effective; therefore, it is necessary to find effective drugs. There is evidence that oral vitamin A palmitate, vitamin E supplements, and docosahexaenoic acid can improve symptoms.302 Many mechanistically diverse approaches are under investigation, such as gene therapy, mutant genes (gene silencing by siRNA), stem cells, and small-molecule drugs (such as calcium-channel blockers), all of which have been used clinically but with limited influence.
Drugs targeting ER developed in RP are abundant. Many chemicals targeting ER stress can recover whole retinal cell function, including 4-PBA and TUDCA.324 4-PBA used in RP can save the cone cell from necrosis and rod cell death, protect RGCs and prevent vision loss.325,326 In particular, 4-PBA recovers the mitochondrial genesis of rods to defend against cell death induced by mitochondrial dysfunction. Mutant rhodopsin-related cell loss can be suppressed by mTOR inhibition (rapamycin and PP242), AMPK activation (AICAR), and a peIF2α inhibitor (salubrinal).308 mTOR inhibition with rapamycin can restore the A:P ratio in RPE as well. Bilberry extract is antioxidant-rich, and oral administration can ameliorate light injury by suppressing photoreceptor apoptosis and attenuating ER stress in RGCs to reach a neuroprotective function.312 The HSP70 family, like HSPA4L, can colocalize with PRPF31 mutant aggresomes and translocate into nuclear function, which can be protective for RPE. Using a nanoparticle to deliver full-length genomic DNA of the rhodopsin gene can lead to a gain-of-function phenotype.327
Gene therapy offers profound insight into the future treatment of RP since most RP is associated with gene mutations. CRISPR/Cas9 is used in the research of RP mechanisms and treatments because of its high efficiency and accuracy. Since the RHO mutation is a gain-of-function mutation, thorough ablation of the mutation is more effective in ADRP. By selectively erasing the RHO mutation, mutant protein-induced ER stress will be ameliorated and symptoms will be relieved. AAV-CRISPR/Cas9 gene editing and ablation of pathogenic genes have been studied in the S334ter mutation. A study of editing genes using a neonatal rat model of P23H did not affect the survival of individuals and preserved long-term vision sight.328 If proven clinically safe, this technology can improve the quality of life of patients with ADRP from childhood. Silencing mutated RHO mRNA and facilitating WT RHO gene expression by double-stranded siRNA may be effective. For example, a bidirectional gene expression system consisting of a synthetic 75-mer-dsDNA to generate a hairpin loop siRNA and the other expressing the normal gene can be developed for silencing mutant rhodopsin by insertion.329
Stem cells can differentiate into specific cell types, and stem cell therapy can supply the normal cell loss in RP or release neurotrophic factors to support cell survival. RP patient-derived iPSCs with RHO mutations combined with genome editing can correct RP-related mutations.330 In one study, human iPSC-derived RPE cells were transplanted to a pig retina, which developed normal RPE morphology and facilitated photoreceptor survival.331 Transplantation of human embryonic stem cell-derived retinal organoids have been tested in RPE-loss rats and reveals the possibility of host organoid survival and integration of organoid and retina. The RGCs, photoreceptors, and RPE survival rates were improved, and organoids survived for more than 7 months.332 Furthermore, the combination of stem cells and gene editing technology can effectively treat RP, the kind of which begins with RPE cell death. Fibroblasts from RP13 patients who possess PFPR mutations are used as the somatic cell source for producing iPSCs. iPSCs whose P2301S mutation is corrected by CRISPR/Cas9 gene editing can be induced into RPE, which resembles WT RPE and possesses normal functions like phagocytosis.333 Though the injection of iPSC and directional homing ability have not been studied, they provide new ideas for RP treatment. Transplantation of MSCs or MSC-derived RPCs has been studied and proven to be effective in animal models. MSCs mainly express the RPE gene after transplantation into the retina. RPCs can express RGCs, RPE, and the photoreceptor genes and are distributed in different layers of the retina.334 Compared with iPSCs and MSCs transplantation, RPCs protect the whole retina tissue by differentiating and homing to RGCs and photoreceptors and releasing neurotrophic factors like BDNF, GDNF, IGF, CNTF, EG, FGF, and PDGF.335 However, when combining fetal RPE and MSCs, the whole retina can be saved.336
MSCs-derived exosomes can be a new option as cell-free therapy for RP therapy. Injecting MSCs, including bone marrow tissue and adipose tissue-derived exosomes, can suppress TNF-α and IL-1β, inhibit retinal cell apoptosis, and protect RGCs.337,338 RPE can release exosomes to maintain retinal homeostasis. Exosomes from RPEs release anti-inflammatory cytokines, suppress ER stress-induced inflammation and inhibit immune responses in vitro.339 Whether MSCs or RPEs-derived exosomes have a similar therapeutic effect to MSCs or iPSCs and the treatment of RP by inhibiting ER stress needs further study.
Optogenetic therapy has value in providing hope for vision restoration in advanced retinal degeneration.340 In RP, it usually refers to converting non-photosensitive retinal cells, usually bipolar cells or RGCs, into artificial photoreceptors, which is achieved by supplying these cells with a light-sensitive protein. Since the pathology of photoreceptor loss under ER stress is common, the advantage is that it can ignore the mutation type in RP and function in almost all types of RP by recovering light sensitivity. Opsin can be obtained from microbes and animals.341 It has been demonstrated that using a second-generation photoswitch for LiGluR, maleimide-azobenzene-glutamate 0, with peak efficiency at 460 nm, can restore rat vision.342
ER stress and achromatopsia
Achromatopsia (ACHM) is a hereditary dystrophy that affects the central retina and is characterized by cone cell function loss, classically presenting with color blindness, photophobia, nystagmus, and decreased visual potential with a visual acuity of less than 20/200.343 Other symptoms include symmetric nystagmus with high frequency and low amplitude, which can occur in any direction. The actual signs of achromatopsia differ between patients owing to the varying degree of cone loss. It has been found that Ca2+ turbulence and UPR branch damage can result in ER stress, which leads to cone protein accumulation and cone death. We investigated the role of ER stress in the pathology of ACHM and ER stress-targeted therapy.
Involvement of ER stress in achromatopsia
Gene mutations associated with cone cell dysfunction can cause ACHM (Table 1). Cyclic nucleotide-gated (CNG) channel mutations are the most common mutations in ACHM. CNG channel deficiency is characterized by elevated cellular cGMP levels and increased activity of cGMP-dependent protein kinase G, which leads to Ca2+ turbulence, cone degeneration, and impaired cone function, including CNG channel beta 3/ACHM3 and CNG channel alpha 3.343,344 Other gene mutations include PDE6H and PDE6C, which have been discovered in RP sections. Its mutation results in Ca2+ disturbance as well and is attributed to cone proteins, which are supposed to be located in the outer segment but misallocate in the inner segment, including cone proteins M-opsin, S-opsin, and cone phosphodiesterase subunit α' (PDE6C).343 An ATF6 gene mutation is directly related to automatic recessive ACHM, including a homozygous deletion covering exons 8–14, exons 2 and 3, and ATF6 c.970C>T (p.Arg324Cys), which are characterized by early vision loss.345,346 When ATF6 is stimulated, it can be activated but cannot activate its downstream pathways which are related to cell stress and proliferation. It is characterized by the absence of a foveal cone structure in these retinas.347 Currently, there is no effective treatment for achromatopsia. Studying the mechanisms of ACHM and finding new treatments is urgent and necessary.
ER stress is related to OS in cones and results in cone atrophy. Ca2+ turbulence is a contributor to ROS and OS, which can induce ER stress and act as a bridge in ER stress functioning in ACHM. The expression of 84 genes involved in unfolded protein binding, protein folding, ER-associated protein degradation, heat shock proteins, and enzymatic regulation of unfolded glycoproteins increases in a CNG channel deficient mouse model, which reveals the existence of ER stress in ACHM.348 Individuals lacking ATF6 experience severe ER stress-induced cell death and cone degeneration, which demonstrates the protective function of ER stress during the development of the retina in achromatopsia.349 In addition, ATF6 mutation not only influences the transcription activity of ATF6, or transportation of bZip, but also the IRE1 and PERK pathways, indicating an imbalance of ER stress in ACHM,350 leading to mislocalization of cone protein, Ca2+ turbulence, and ER stress-induced apoptosis.351 However, the specific mechanism underlying ER stress in ACHM requires further investigation.
Mitochondrial dysfunction is also a downstream effect of ER stress in ACHM. In retinal organoids generated from patients with ACHM who carried a homozygous ATF6 mutation, mitochondrial morphology changes and mitochondrial respiratory complex gene dysregulation can be identified.352 ER stress correlates with mitochondrial dysfunction, which has been proven in the retina. ATF6 mutation might cause an imbalance in UPR and result in ER stress.
Therapeutic targets for achromatopsia
Owing to the intimate relationship between ER stress and ACHM, we can determine some target treatments aimed at relieving ER stress. In patients with ATF6 mutations, using drugs to increase the expression of this protein may save cone cells in the retina. It has been proven that ATF6 can be activated in two separate pathways: the protein toxicity activation pathway and the lipotoxic activation pathway, including specific sphingolipids, dihydrosphingosine, and dihydroceramide.353 It offers a unique way to treat ACHM with the ATF6 mutation through upregulating dihydroceramide by fenretinide, which facilitates ATF6 only in its normal function but not ER stress. Additionally, the selective ATF6 agonist AA157 can increase transcription in ACHM without effects on other UPR pathways.349 As for mutations in CNG dysfunction and PDE6 defects, recovering Ca2+ homeostasis and cGMP levels are effective ways to relieve ER stress. cGMP/protein kinase G regulates RyR2 expression. Downregulation of RyR2 can weaken cGMP-induced ER stress. For example, the deletion of IP3R1 and inhibition of ryanodine receptors, which control calcium efflux from the ER and are proven to facilitate ER retrotranslocation in Cnga3−/−/Nrl−/− cone cells.347 Inhibition of protein kinase G by Rp-8-Br-cGMPS and KT5823 nearly completely abolishes channel upregulation in CNG channel deficiency.348
Gene therapy for ER stress in mutated cones has great potential. Clinical trials aimed at ACHM Cnga3 and Cngb3 have been successfully conducted, and short-term safety has been evaluated.354 AAV-delivered defect genes or neurotrophic factors can be considered. Subretinal injection of CNG cDNA packaged with AAV recovers cone function and vision.355 In late-stage CNGB3-achromatopsia, AAV-delivered CNGB3 cDNA and neurotrophic factors CNTF by subretinal injection to help restore cone function.356 CRISPR/Cas9 can be used in the ablation of mutated genes such as CNG, PDE6, and ATF6. However, the off-target effects have risks for patients. This can be avoided by using CRISPR/Cas9-mediated gene editing to correct single-nucleotide mutations for PDE6.357
ER stress and cataracts
Cataracts refer to lens opacification and remain the leading cause of blindness in middle- and low-income countries,358 affecting approximately 94 million people worldwide. In 2020, about 15 million people over the age of 50 worldwide will be blind due to cataracts, and about 79 million will have moderate-to-severe visual impairment.359 Currently, there are various classification systems for cataracts. Here, cataracts are classified by the anatomical position of opacity as nuclear cataract, cortical cataract, posterior subcapsular cataract, and mixed cataract, with nuclear and cortical cataracts being the most common. Nuclear cataract lesions present as yellow-brown opacification of the fetus and adults on the lens, causing blurry vision, loss of color, sensitivity, and myopic shift, and the cornea presents as spoke cortical opacity with the same lens fiber shape, which causes astigmatism, monocular diplopia, and glare and halos around lights. Risk factors include smoking, genetic factors (such as KCNAB1 and CRYAA), alcohol consumption, and ultraviolet-B light exposure.358 Studies have shown that different metabolic pathways are involved in the formation of cataract lesions, such as elevation of the coproporphyrinogen pathway, dysregulation of crystal protein synthesis, lactose, etc., and cause pathological damage through yellow-brown opacity formation via ER stress of lens epithelial cells.
Involvement of ER stress in cataracts
Genetic factors account for 35% of the variation in the progression of nuclear cataracts (Table 1). The most frequent transmission is autosomal dominant, whereas autosomal recessive or X-linked transmission can also occur.358 Some Wolfram syndrome patients possess dominant mutations in WSF1, including p.Glu809Lys, p.Glu830Ala, and p.His313Tyr. They can present as neonatal/infancy-onset diabetes, congenital sensorineural deafness, or congenital cataracts. These WSF1 mutations encode unfolded WSF1 proteins and accumulate, which induces ER stress (Table 1). EPHA2 encodes the Eph tyrosine kinase receptor family, which functions in epithelial homeostasis. Mutations and variants of EPHA2, including c.2819C>T, c.2915_2916delTG, rs7543472, and rs11260867, are related to age-related cataracts and congenital cataracts.360,361 In EPHA2 mutated mice, accumulation only results in moderate ER stress and UPR, which are protective, and the opacity is induced by glutathione (GSH) imbalance and fibroblast function turbulence.362 Calnexin is the lens fiber cell gap junction protein, in which connexin 46 and connexin 50 will result in cataracts.363 Mutations in connexin 46 and connexin 50 lead to junction and circulation lens damage, which results in Ca2+ elevation, precipitation, and biomineralization. Lens transparency decreases and can precipitate with proteins. Different connexin50 mutants induce ER stress, which plays different functions for cell survival or death that depend on downstream target genes.364
Epigenetic modifications, including DNA methylation, are regulated by ER stress in the development of cataracts. It is thought that DNA methylation can be inherited in somatic cells at the onset of embryonic development. The failure of DNA methylation can result from passive demethylation by Dnmt1, Dnmt3a, and Dnmt3b following DNA replication or from active enzymatic removal of 5 mC by ten-eleven translocation 1, activation-induced cytidine deaminase, and thymine-DNA glycosylase, independent of replication.365 Under ER stress stimulation, including valproic acid and sodium selenite, or risk factors Hcy for age-related cataracts, enzymes in passive demethylation and active demethylation of lens endothelial cells (LECs) are significantly upregulated.366,367,368,369 Methylglyoxal treatment induces ER stress in LECs, and the promoter DNA methylation status of keap1 is significantly lost, which causes dysregulation of the antioxidant system (nrf2), inflammation, and cell death in LECs.366,368
Cataractogenic stress, including alcohol, lipids, and hypoxia, generates misfolded protein conformations through the interaction of ER stress and OS. Insoluble crystalline is involved in the accumulation of LECs, which results in opacity.362,370 The lens is in a hypoxic environment, and factors such as diabetes will lead to increased anaerobic respiration of lens epithelial cells, which induces ER stress.371 At first, mild ER stress and ATF4 can act with nrf2, and the expression of ARE-dependent phase II antioxidant and detoxification enzymes increases, which reduces OS and ROS generation.372 Then, persistent ER stress induces superfluous ROS by upregulating keap1 and downregulating nrf2 resulting in LEC death and differentiation into fiber cells.372 The neolens fiber cells possess incomplete nrf2 systems and prefer to produce insoluble crystallin and misfolded proteins under ER stress, which can be seen as opacity. Furthermore, Ca2+ levels in the opaque part of cortical cataracts are higher than those in the transparent part, which can result in chronic UPR and is also a consequence of ER stress. Chronic UPR activates ERAD and m-calpain and then degrades sarco/ER Ca2+-ATPases and plasma membrane Ca2+-ATPases (PMCA).365 Thus, Ca2+ in the lens continues to rise and moves toward the lens fiber cells. Excessive ER stress and accompanied OS lead to massive Ca2+ elevation and activate caspase-12 and caspase-3-induced apoptosis.373 In Connexin50D47A mutant mice, gap junction damage between LECs will result in Ca2+ elevation.364 For mild ER stress and UPR for homozygous lenses, only the cell survival pathway PERK/ATF4/Trib3- IRS2 is activated, indicating the effect of ER stress is not static and related to the types and functions of different cataract-related mutations.
LECs go through epithelial–mesenchymal transition (EMT) induced by ER stress. Under ER stress, lens epithelial cells reduce the expression of epithelial genes and transform into fibroblasts in vitro.374 TGFβ facilitates the EMT process and has been proven to have a relationship with ER stress.375 After cataract surgery, remaining lens epithelial cells tend to proliferate excessively and result in fibrotic posterior capsular opacification (PCO). Postoperative stress-induced ER stress strengthens lens epithelial cell migration and EMT, which damages the lens conformation, explaining the development and pathology of PCO.
The interaction of autophagy and ER stress plays an important role in the pathogenesis of PCO. Under sulforaphane treatment in LECs, ER stress elevates ROS and activates the Erk pathway, both of which can contribute to autophagy.376 Combining ER stress and autophagy, lens epithelial cell organelles are impaired, and the recovery process cannot catch up, leading to apoptosis. Considering the role of ER stress in EMT, it can be concluded that the activation of mild ER stress is beneficial for PCO patients since it can inhibit the growth of lens epithelial cells.375 Therefore, ER stress plays a dual function in PCO, dependent on its degree.
Therapeutic targets for cataracts
Surgery is usually performed to treat cataracts. However, the pharmacological prevention of cataracts is an area of future research. Regarding previously described treatments, sulforaphane can promote ER stress and relieve cataract symptoms.376 4-PBA, a protein chaperone, can be used in calnexin mutant congenital cataracts since it can recover the misfolded calnexin and restore the junction between cells. Also, in PCO, 4-PBA has been found to prevent EMT of LECs and reduce fibroplasia and opacity.377 (+)-Pentazocine, a S1R agonist, has been administered to LECs in vitro.378 It can relieve ER stress, OS, and apoptosis, which can make it a potential drug to slow the progression of cataracts. Since oral or injected drugs do not target specific regions in the retina, iontophoresis is an innovative noninvasive strategy to deliver drugs through the anatomic barriers.379
ER stress and uveal disease
ER stress and uveitis
Involvement of ER stress in uveitis
Gene mutations play an important role in uveitis, in which ER stress is associated with pathology. Nucleotide-binding oligomerization domain containing 2 (NOD2) variants, including p.R334W, p.R334Q, p.E383K, p.G481D, p.W490S, p.M513T, p.R587C, and p.N670K, have been found in Chinese populations to be gain-of-function mutations and pathological in uveitis, especially for Blau syndrome. Blau syndrome is an early-onset autoinflammatory and autosomal dominant disorder typically characterized by a triad of uveitis, granulomatous polyarthritis, and skin lesions.380 The incidence is estimated to be 0.05 per 100,000 people per year.381 The NOD2 mutation has been found to directly induce aberrant autoactivation of NF-κB signaling and is attributed to a persistent induction of proinflammatory cytokines, including IFN-γ, IL-6, and IL-17.382 It has been demonstrated in many infection models as a direct ER stress inducer; NOD2 links ER stress with inflammation.383,384 Inflammation induced by IRE1α depends on the activation of NOD2, which activates NF-κB to induce inflammation. Similarly, in posterior uveitis, mutations of NOD-like receptor family genes (NOD2, NLRC4, NLRP3, and NLRP1) are detected to be gain-of-function in uveitis patients and are associated with ER stress and inflammation.385 For example, NLRP3 can be activated under ER stress and then recruit caspase-1, which initiates the pyroptosis pathway, and caspase-3, which induces apoptosis.386 R243L mutation in calpain-5 increases the catalytic activity of calpain.387 Under ER stress at the early stage, mitochondrial calpain-5 is activated first and cleaves caspase-4 to activate cell-programmed death and inflammation.
Behçet syndrome (BS) is an inflammatory disorder characterized by recurrent oral and genital ulcers, skin lesions, and uveitis. A comparison of blood samples from BS patients with those of healthy individuals revealed decreased ER stress-related proteins, such as HSPA8 and GRP78/BiP, but increased CHOP and ATF4, indicating dysregulation of the protein-folding mechanism and ER stress involved in BS pathogenesis.388 In acute anterior uveitis patients, ER stress increases proinflammatory cytokine levels.389
Therapeutic targets in uveitis
Since ER stress interacts with inflammation in uveitis, treatment targeting ER stress can alleviate the inflammation, which is the most significant pathology in uveitis. Using TUDCA or IRE1α inhibitors can relieve the inflammation induced by the IRE1/NOD2 pathway and NLRP3-induced pyroptosis and apoptosis.383,385 Galectin-3, a 31-kDa chimeric lectin characterized by a binding affinity for β-galactose-containing carbohydrates, can ameliorate the intestinal phenotype of BS via downregulation of ER stress and NLRC4 activation in BS.390 The concrete treatment effect on uveitis needs more investigation. Mesalazine is the most broadly utilized medication for mild to moderate ulcerative colitis and has been studied for adverse drug effects and control.391 Mesalazine can inhibit the transcription of proinflammatory and ER stress-associated cytokines and markers in BS.389
ER stress and uveitis melanoma
Uveal melanoma (UM) is the most common intraocular malignancy that arises from melanocytes in the iris, ciliary body, or choroid.392 UM accounts for an estimated 5.5% of primary melanomas.393 Patients can be asymptomatic, and the most common clinical manifestations of primary UM include blurred vision, photopsia, floaters, and visual-field loss.394 Though current optimal treatment or surgery for most UM can be effective, up to 50% of patients possess metastatic symptoms with liver, lung, skin, and bone whose median overall survival is about 13.4 months.395
While patients of cutaneous melanoma harbor mutations in BRAF, RAS, and NF1 related to MAPK activation, few UM patients possess these mutations, but rather mutations in the G-protein α-subunit (Table 1).396 Uveal melanomas display MAP-kinase activation through the GNAQQ209L mutation. GNAQ operates downstream of several G protein-coupled receptors (GPCRs) that are important in melanocyte homeostasis and neoplasia and act as risk links between nevus of Ota and uveal melanoma.397 GNAQ and GNA11 encode an α-subunit of G-protein, which plays an important role in the conversion of GDP to GTP, and their mutation maintains the continuous activation of G-protein, which can activate Gα including activation of the MAPK, PI3K–Akt–mTOR, and Hippo pathways.392 The EIF1AX mutation has been significantly detected in UM, which affects the DNA methylation of cluster 1 tumors, in a multiplatform analysis of 80 UM identifiers.398
Involvement of ER stress in uveal melanoma
ER stress is involved in UM development. By single-cell sequencing of UM, ER stress and the UPR-related genes are shown to be expressed in UM tumor tissue. EIF1AX, which encodes EIF protein, is beneficial for tumors escaping immunological surveillance, and its mutation is attributed to the long latency and low metastatic rate of UM.399 Another study found that EIF1AX can predict the progression of UM and be used as the subtype standard.398,400 Analysis of 80 UM identifiers showed that ATF6 and XBP1 were activated upstream.401 Altogether, ER stress is part of UM’s progress.
Hypoxia broadly exists in the tumor microenvironment as a consequence of rapid cancer cell proliferation. In the UM microenvironment, hypoxia-related genes like BAIAP2L2, CTNNB1, EDNRB, HES6, LGALS1, PPM1K, PROS1, S100A4, and SYNPR can effectively predict the progression of UM.402 For example, HES6 enhances primary UM cell mobility and invasive characteristics.402 Hypoxia would increase the expression of the hypoxia-inducible factor 1 alpha subunit gene and its target gene, VEGF.403 As aforementioned, ER stress can induce the expression of VEGF and contribute to neovascularization. ER stress might be related to the development and prognosis of UM through the HIF1α/VEGF pathway.
Immune cell infiltration can be seen in the tumor microenvironment. It has been demonstrated that among 22 kinds of immune cells, the infiltrating proportions of CD8+T cells, follicular helper T cells, gamma and delta T cells, and activated natural killer (NK) cells were significantly higher in high-risk UM patients, while those of memory resting CD4+T cells, naive B cells, activated mast cells, resting mast cells, monocytes, and resting NK cells were significantly higher in low-risk UM patients.402 In CD8+T cells, ER stress and UPR may be cell-extrinsic regulators of immunity, and ER stress regulates DC cells by transmitting types of polarization and proinflammation, which promote the production of proliferation-deficient T cells.404 This phenomenon may explain the high correlation between CD8+ T cell infiltration and high-risk UM.
ER stress might be potentially related to UM metastasis. MMPs are overexpressed in UM tissues compared to normal tissues, including MMP1, MMP9, MMP10, MMP11, MMP13, MMP14, and MMP17. MMPs can degrade various protein components in the ECM, destroy the histological barrier of tumor cells, and affect tumor migration, invasion, metastasis, and angiogenesis. ER stress can be cytoprotective and drug-resistant in UM.405 MMPs expression can serve as a biomarker of UM prognosis. Meanwhile, in mouse mammary gland tumor models, MMP11 promotes tumor growth through ER stress activation and metabolic rewiring.406 Overexpression of the pro-survival BCL-2 family members has been broadly discovered in cancer and also in primary UM. The effect of BCL-2 inhibitors in UM can be weakened by ER stress.407
Therapeutic targets for UM
Two types of management for localized UM are globe-preserving therapy and enucleation. Globe-preserving therapies can broadly be divided into radiation, surgical, and laser therapy.392 Considering the involvement of neovascularization in tumor metastasis and growth, tumor-targeted approaches, including immunotherapies, are under research. Targeting factor VII, ICON-1 is a structural variant of human factor VII being developed by Iconic Therapeutics and in Phase I study. ICON-1 contains virus-like particles that selectively bind to cancer cells and infrared-activated molecules that destroy tumor membranes when activated by an ophthalmic laser.392 However, current effective therapies for metastasis are limited, and the prognosis is poor. Some studies focus on the MAPK pathway and epigenetic modification with an HDAC inhibitor. Regarding the complicated pathology and poor prognosis of late-stage UM, new, effective approaches are needed.
ER stress has been proven to be a novel target in UM treatment. Direct intervention on ER stress might slow tumor growth and improve prognosis. Though not previously studied in UM, 4-PBA, a selective ER stress inhibitor, has been proven to be effective for BRAF-mutated melanoma.408
ER stress possesses a dual function in UM cell survival. Some research has proved that ER stress plays an anticancer role in UM and contributes to choroidal melanoma cell apoptosis. Current evidence shows that choroidal melanoma is insensitive to many commonly used chemotherapeutic drugs, and properly utilizing ER stress can enhance the chemotherapy effect.409 Cisplatin is a commonly used chemotherapy drug that binds to DNA and results in cell death. The synergistic effect of cisplatin plus pemetrexed contributes to the induction of simultaneous extrinsic and intrinsic apoptosis.410 Intrinsic apoptosis induction is controlled by ER stress through the CHOP/NOXA/Mcl-1 pathway.410 Lithium chloride promotes apoptosis through the CHOP/NOXA/Mcl-1 pathway, which increases NOXA and decreases the anti-apoptosis molecule Mcl-1.411 Using BH3-mimics to inhibit BCL-2 can be considered a treatment strategy for UM. Exerting ABT-263 (Navitoclax) in the treatment of UM is antiproliferative and blocks the PERK pathway while promoting apoptosis, proving the cytoprotective effect of ER stress.407
Immunotherapy can also be enhanced by combining it with ER stress enhancement or reduction. Immunogenic cell death can be evoked by ER stress-induced ROS. Photodynamic therapy and photothermal therapy are commonly used immunotherapies for tumors that rely on ROS in cells. Based on ER stress function, combining targeted ER stress therapy with photodynamic therapy and photothermal therapy enables sustained ROS levels and continuously induces immunogenic cell death.412 An ER-targeting nanosystem consisting of ER-targeting pardaxin peptides modified with indocyanine green conjugated hollow gold nanospheres together with an oxygen-delivering hemoglobin liposome has giant prospects.412 Reversing activated ER stress in CD8+ T cells helps the production of antitumor CD8+ T cells. For example, in human colon cancer, inhibition of stearoyl-CoA desaturase 1 improves antitumor properties by regulating β-catenin signaling in cancer cells and ER stress in T cells.413
ER stress and ocular surface diseases
ER stress and corneal dystrophy
Corneal dystrophies have typically been referred to as a group of inherited disorders that are usually bilateral, symmetric, slowly progressive, and unrelated to environmental or systemic factors. In the ICD3 classification of corneal dystrophy, a modified anatomic classification is proposed consisting of (1) epithelial and subepithelial dystrophies, (2) epithelial–stromal TGFBI dystrophies, (3) stromal dystrophies, and (4) endothelial dystrophies.414 Most corneal dystrophies are characterized by progressive opacification of the cornea and eventually a reduction in visual acuity.
Involvement of ER stress in corneal dystrophy
ER stress has been proven to be involved in various types of corneal dystrophies, including macular corneal dystrophy (MCD), granular corneal dystrophy type 2, congenital stromal corneal dystrophy, Schnyder corneal dystrophy, congenital hereditary endothelial dystrophy, and Fuchs endothelial corneal dystrophy. Corneal dystrophies possess different mutations that code for unfolded proteins, produce accumulations of mutant glycoproteins or proteins, overexpress metabolisms, and result in ER stress (Table 1). Enlarged rough ER can be seen in corneal endothelial cells, which reveals impaired ER function.415,416 In addition, the overload of ECM proteins in the stroma induces aggresomes and ER stress in the corneal endothelium.417 Next, we will discuss the role of ER stress in different coraeal dystrophies.
MCD is a rare autosomal recessive disorder, usually evident in childhood or adolescence, and is clinically characterized by the formation of a diffuse and fine symmetric clouding in the central corneal stroma that extends to the periphery and eventually involves the entire thickness of the cornea, leading to severe bilateral visual disturbance.414,418 Deposition of abnormal glycosaminoglycan material in the stroma can be observed in an irregular position, as an infiltration of the adjacent corneal structures, including Bowman’s layer, Descemet’s membrane, and the endothelium.419 The pathology of MCD has been shown to entail a deficiency in acid mucopolysaccharide metabolism localized in the keratocytes (corneal fibroblasts), whose ER is surrounded by glycosaminoglycan accumulations.415,419 The carbohydrate sulfotransferase gene, encoding an enzyme designated corneal N-acetylglucosamine-6-sulfotransferase (C-GlcNAc6ST), is identified in MCD pathology.420 ER stress markers like GRP78 and CHOP are expressed when ER stress is active.415,421 In cell models transfected with c.463-464del and c.250_272del carbohydrate sulfotransferase gene mutants, ER stress-induced apoptosis was activated.421
Furthermore, the interaction between ECM and ER stress can play a role in the pathogenesis of corneal diseases as well. Congenital stromal corneal dystrophy is a human genetic disease characterized by corneal opacities beginning shortly after birth and is associated with a decorin gene mutation.422 Decorin is a multifunctional small leucine-rich proteoglycan that interacts with collagen fibrils and regulates fibrillogenesis in ECM assembly. Its mutation will cause a deficiency in the correct organization of the corneal stroma. Mutant decorin has an intrinsic deficiency in entering the Golgi apparatus, which causes the accumulation and nonsecretion of decorin in keratocytes.423 The intracellularly ER-retained decorin protein core lacking the C-terminal ear repeat causes ER stress.423 Granular corneal dystrophy type 2 is an autosomal dominant disorder caused by an arginine to histidine substitution at codon 124 (R124H) in the transforming growth factor β-induced protein (TGFBI) gene.424 Mutant-TGFBI protein is an age-related progressive deposition in the corneal epithelia and stroma, and its secretion via the ER/Golgi-dependent secretory pathway is delayed.425,426 TGFBI protein accumulation in fibroblasts results in protein homeostasis disturbance and ER stress.427 Fuchs endothelial corneal dystrophy is characterized by progressive alterations in endothelial cell morphology, excrescences, thickening of the endothelial basement membrane, and cell death, which causes corneal edema and vision loss. The Col8a2Q455K/Q455K mutation results in alpha-2 collagen VIII structural modification, activated UPR (including the IRE and PERK pathways), and intrinsic apoptosis in mouse endothelium.428 Increased TGFβ was discovered in Fuchs endothelial corneal dystrophy which directly increased MMP activity and promoted ECM accumulation and apoptosis.417 The accumulation and overexpression of proteoglycans and ECM, including fibrin and collagen I, help the formation of aggresomes in the endothelium, which act as induction of ER stress followed by cell death.429
Metabolite accumulation or metabolism pathway disturbance in corneal dystrophies is common, which may inhibit normal ERAD systems and affect ER stress. A Slc4a11 protein mutation has been detected in congenital hereditary endothelial dystrophy, where elevation of glutamine is a contributor to mitochondrial ROS and ER stress.416 Schnyder corneal dystrophy is associated with the UbiA prenyltransferase domain-containing protein-1 mutation, which regulates the ER-resident enzyme HMG-CoA reductase in normal condition.430 UbiA prenyltransferase domain-containing protein-1 is responsible for HMGCR ERAD, and its mutation will result in the persistent activation of HMG-CoA reductase and sterol production.430 Thus, the balance between ERAD and autophagy shifts to autophagy.
Therapeutic targets for corneal dystrophy
The current effective treatment for corneal dystrophy is corneal transplantation. Considering the risk of graft rejection and difficulty acquiring corneal donations, finding a new treatment is urgent. As for the important role of ER stress in the onset and development of corneal dystrophy, targeting ER stress can be considered a new method. Inhibiting ER stress with drugs can prevent corneal cell death. Reinforcing ERAD can effectively solve mutant protein accumulation. 4-PBA has been demonstrated to activate ERAD and accelerate TGFBIp degradation.427 Melatonin alleviates the IRE1α/XBP1 pathway and facilitates the activation of ERAD, which can save granular corneal dystrophy type 2 corneal fibroblasts.431 Reduction of ER stress and simultaneous facilitation of protein accumulation by autophagy is beneficial for survival; for example, the use of lithium in Fuchs endothelial corneal dystrophy.432
Appropriately reducing the inducers of ER stress, like the alleviation of OS and facilitating protein folding, can save cells from ER stress-induced death. Glutamine catabolism elevates ROS in Slc4a11−/− cells and leads to mitochondrial dysfunction. Mitochondrial ROS quencher MitoQ can reduce BiP and GADD153 expression in Slc4a11 KO cells and mice.416 It has been successfully demonstrated that the coexpression of R125H and SLC4A11 facilitates mutant protein transportation to the cell membrane without affecting water flux.433 Antioxidative therapy like N-acetylcysteine can relieve ER stress.434 High-throughput assays allow scientists to screen drugs like glafenine and perhaps other non-steroidal anti-inflammatory drugs for helping SLC4A11 properly fold,435 and could also find more potential therapeutics for corneal diseases.
Extracellular vesicles have been broadly investigated in ocular diseases. MSC-derived extracellular vesicles have been verified for their treatment effect in an in vitro model of corneal dystrophy.436 MSC-derived extracellular vesicles contain targeted ER stress miRNAs for ER stress-related genes, including phosphorylated EIF2α, ATF4, and CHOP.436
ER stress and keratoconus
Keratoconus (KCN) is a bilateral and asymmetric disease characterized by restricted cone-like bulging of the cornea, which leads to progressive thinning and steeping of the cornea, leading to irregular astigmatism and decreased visual acuity.437 The prevalence and incidence of KCN vary globally and are estimated to be between 0.2 and 4790 per 100,000 people and 1.5 and 25 per 100,000 people/year, respectively.438,439 Changes in degradation systems and OS can be related to ER stress in KCN pathology.
Genetic factors are essential in the development of KCN, and some of them are found to be related to ER stress in the cornea. Genetic analysis has revealed that ER stress markers and multiple translation initiation and elongation factors (EEF1A1, EEF1B2, EEF1D, EIF3F, EIF3H, and others) downstream of ATF4 are increased in the corneal proteomes of KCN, indicating the involvement of ER stress.440 Mutations in superoxide dismutase 1, including c.169+50 delTAAACAG, have been discovered in KCN patients.441 The protein encoded by SOD 1 can bind to copper and zinc ions, which is an enzyme for radical clearance whose mutation causes OS.442 As aforementioned, OS and ER stress each other, which may cause damage to the cornea. OS will cause peroxynitrite overexpression, causing prolonged oxidative injury. Peroxynitrite can induce ER stress through the depletion of ER-Ca2+ in endothelial cells.443 Increased ROS in cells might cause increased MMP, which can be the mechanism of corneal thinning.444 For example, in fibroblasts, the overexpression of SOD 2 increases the expression of MMP-1, -2, -3, -7, -10, -9, and -11.445 Also, ER stress has been found to elevate MMP expression in neurons, which offers the possibility of an interaction between OS and ER stress to facilitate MMP activation and corneal stromal degradation. TGFB1 is now proven to be involved in KCN, including the G535X.
ER stress and keratitis
Incomplete eyelid closure and blinking cause corneal desiccation and epithelial barrier compromise, which leads to chemosis, erosion, melting, infectious keratitis, and even perforation.445 In the pathogenesis of exposure keratitis, air exposure induces ER stress in corneal epithelial cells and activates autophagy via the PI3K/AKT/mTOR signaling pathway.446 In a keratoconjunctivitis sicca model, ER stress markers, such as GRP78 and sXBP1, are increased, which destroys the ocular surface barrier and facilitates goblet cell death.447 The loss of goblet cell death will result in mucosal decrease and the destruction of the tear film, which is an important eye barrier. Herpes simplex keratitis is an infectious disease caused by herpes simplex virus infection of the cornea. Most herpes simplex viruses (HSVs) infecting the eyes are HSV-1, and some are HSV-2. ER stress is related to immune homeostasis. As aforementioned, STING is crucial for initiating innate immune responses and producing interferon regulatory factor 3 to defend against virus invasion.212 HSV-1 infection results in STING carbonylation and causes inhibition or deactivation of protein function.212 In Mycobacterium bovis, activation of STING/TBK1/interferon regulatory factor 3 is dependent upon ER stress.448 In Brucella abortus, UPR is necessary for STING activation and the production of IFN.182 Therefore, ER stress can be significant in defending against bacteria and viruses through STING/IFN1 activation.
Nowadays, antibiotic abuse results in bacterial resistance in many patients. In an attempt to address antibacterial drug resistance while avoiding toxicity toward mammalian cells, large lipids fraught with small nanoparticles encapsulating antibiotics have been developed and tested against gram-negative bacteria, in which lipids can integrate with bacterial membranes, and nanoparticles are released to kill bacteria.449
ER stress and dry eye syndrome
Oxidative stress correlates with dry eye syndrome (DES) and the core mechanism can be tear-high osmotic pressure during inflammation.450,451 Under high osmotic pressure conditions, increasing ROS levels lead to ER stress in corneal epithelial cells and induce apoptosis.450 Goblet cells, which are integral components of the tear film, are the most important source of mucins and other proteins. In an interferon-γ-induced dry eye model, expression of GRP78 and XBP1s increased, especially in goblet cells, and contributed to cell apoptosis and inflammation.447
In Sjögren’s syndrome, ER stress activates and induces MMP9 activation through the FOX/MAPK pathway, which increases proteolytic activity and results in DES.452 Additionally, MAM functions in this syndrome, during which it can activate ER stress and inflammation.453 In aquaporin 5-deleted mice, the secretion of the lacrimal gland is injured, and there are symptoms of dry eye.454 Moreover, GRP78, CHOP, Caspase12, and BAX levels increase, indicating the occurrence of ER stress.454 High osmotic pressure induces dry eye through GADD34.455
ER stress and myopia
Myopia refers to a condition often developed during childhood and early adulthood where excessive elongation of the eye results in images of distant objects coming into focus in front of the retina, leading to blurred distance vision.456 The global prevalence of myopia in 2010 was about 2 billion individuals, of whom 277 million had high myopia.457 The prevalence is estimated to increase to 4.76 billion individuals for myopia and nearly 1 billion for high myopia by 2050. The most common mechanism is excessive and progressive axial eye growth.458 Besides the elongated axial eye, thinning of the retina and sclera are also important pathophysiological changes in myopia.
Involvement of ER stress in myopia
ER stress is directly involved in axial elongation. The ATF6 and PERK pathways, especially ATF6, can induce axial elongation, albeit with a tendency.459 ER stress plays an important role in scleral remodeling during the progression of myopia. The sclera’s ECM is mainly secreted by fibroblasts. Type I collagen is the major component of scleral ECM, and a decrease will weaken the scleral structural framework. It has been revealed that the decreased synthesis and increased degradation of sclera ECM contribute to sclera remodeling. The HIF-1α signaling pathway is highly activated in myopia, indicating a hypoxic environment.460 One of the mechanisms can be a decrease in choroidal blood perfusion, resulting in hypoxic conditions.461 ER stress signaling eIF2α is activated under HIF-1α inducement, which facilitates myofibroblast transdifferentiation and decreases collagen production (including α1 [COL1A1]).460 Furthermore, under ER stress, Col4a3, Col8a2, Col11a2, and Col15a1 are significantly upregulated, indicating ECM remodeling is regulated by ER stress.458 In an in vitro study, enlarged ER has been observed in fibroblasts, and ER stress is induced under deprivation to induce myopia, which facilitates COL1A1 and TGFβ at the early stage and decreases ECM synthesis at the late stage.462 Therefore, ER stress can be an adaptive response in myopia and compensate for ECM degradation via calreticulin-mediated collagen synthesis.461
Therapeutic targets for myopia
Since hypoxia and ER stress correlate with each other in the development of myopia, they can be novel targets for myopia treatment. Salidroside and formononetin are the active extracts of traditional Chinese medicines that have been proven to resist hypoxia-induced injury separately in heart and retina neovascularization.463,464 By periocular injection of these drugs, the progression of myopia is slowed down, HIF-1α is decreased, and phosphorylation of eIF2α is declined as well, leading to COL1α1 restoration.460 Direct inhibition of ER stress by 4-PBA and TUDCA normalizes the vitreous chamber depth, and retinal thickness shortens in mouse eyes, which tends to shorten myopia progress.459 Gene therapy can be considered since simultaneous inhibition of PERK and ATF6 by CRISPR/Cas9 can slow axial elongation.
Conclusions and prospects
ER is a quality-control organelle for proteostasis. Proteostasis disturbances will result in ER stress, which initiates an adaptive response called the UPR. In recent years, ER stress as a potential modulator in human diseases has been discussed broadly, which can affect ERAD, OS, mitochondrial dysfunction, autophagy, and metabolism. However, the concrete mechanisms of ER stress in different diseases have not been revealed. In most circumstances, ER stress is considered a pro-degenerative process for disease development. However, the different stages of UPR induced by ER stress have opposite functions, indicating the complicated role of ER stress. In this review, we discuss and conclude the molecular mechanisms and treatment targets of ER stress, focusing on ophthalmology, including POAG, AMD, RP, DR, UM, and other ocular diseases.
The notion that ER stress contributes to ocular diseases is commonly researched. The discoveries of mutations in genes that encode the UPR molecule, ER chaperones, misfolded proteins, and metabolisms in all types of ocular diseases indicate the importance of maintaining proteostasis for normal eye function. We point out that ER stress not only affects ocular disease homeostasis through misfolded protein accumulation but also correlates with epigenetic modifications, mitochondrial dysfunction, OS, and impaired autophagy. In concrete terms, ER stress modulates retinal degeneration and neovascularization, leading to pathological changes in glaucoma, AMD, DR, ACHM, and RP. Studies also suggest the involvement of ER stress in lens opacity, epithelial degradation, and ECM remodeling, which have implications for POAG, ocular surface diseases, and cataracts. Additionally, the role of ER stress in regulating the microenvironment underlines the potential effect of ER stress on tumorigenesis and inflammatory diseases like uveitis and keratitis. ER stress is involved in many pathways and plays a key role in many pathogenic processes. Since ER stress plays a complicated role in ocular diseases, further studies are required to elucidate this role.
ER stress is a promising and novel target for the treatment of ocular diseases (Table 2). Recently, several methods, including drugs, nanoparticles, gene therapy, stem cell therapy, and cell-free therapy, have been developed. Drugs, including chemicals, natural extracts, and novel nanoparticles, have been broadly investigated. For example, 4-PBA is an effective ER stress inhibitor that has been utilized in various ocular diseases like glaucoma and DR. Gene therapy has been widely studied, and technologies including CRISPR-Cas9 and ncRNA are innovative and effective. Using gene editing in RP is a hot topic, and some research has been in the clinical trial phase. Stem cell therapy and cell-free therapy provide new insights for treatments targeting ER stress in ocular diseases. As we discussed, TMSCs can differentiate into TM cells and are home to TM tissue, which can survive for months. Treatment targets for ER stress require further research before they can be used clinically. Whether ER stress is involved in ocular diseases and whole-body health remains unclear. Therefore, simply observing aspects of ER stress in ophthalmology is insufficient. Greater efforts are needed for systematic research on the molecular mechanisms of ER stress and the safety of interventions for UPR in ocular diseases. We hope that our review inspires future research into treatment targets for ER stress.
Data availability
Not applicable.
References
Balch, W. E., Morimoto, R. I., Dillin, A. & Kelly, J. W. Adapting proteostasis for disease intervention. Science 319, 916–919 (2008).
Hurtley, S. M. et al. Interactions of misfolded influenza virus hemagglutinin with binding protein (BiP). J. Cell Biol. 108, 2117–2126 (1989).
Kaushik, S. & Cuervo, A. M. Proteostasis and aging. Nat. Med. 21, 1406–1415 (2015).
Smith, M. H., Ploegh, H. L. & Weissman, J. S. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090 (2011).
Travers, K. J. et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 (2000).
Harding, H. P. et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 (2000).
Ron, D. & Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 (2007).
Lin, J. H. et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949 (2007).
Bertolotti, A. et al. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 (2000).
Wang, M. & Kaufman, R. J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335 (2016).
Dombroski, B. A. et al. Gene expression and genetic variation in response to endoplasmic reticulum stress in human cells. Am. J. Hum. Genet. 86, 719–729 (2010).
Ma, K., Vattem, K. M. & Wek, R. C. Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J. Biol. Chem. 277, 18728–18735 (2002).
Han, J. et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490 (2013).
Lee, K. P. et al. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100 (2008).
Credle, J. J. et al. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl Acad. Sci. USA 102, 18773–18784 (2005).
Sidrauski, C. & Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039 (1997).
Zhu, C., Johansen, F. E. & Prywes, R. Interaction of ATF6 and serum response factor. Mol. Cell Biol. 17, 4957–4966 (1997).
Haze, K. et al. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 (1999).
Walter, P. & Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011).
Olzmann, J. A., Kopito, R. R. & Christianson, J. C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 5, a013185 (2013).
Lemberg, M. K. & Strisovsky, K. Maintenance of organellar protein homeostasis by ER-associated degradation and related mechanisms. Mol. Cell 81, 2507–2519 (2021).
Belmont, P. J. et al. Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circ. Res. 106, 307–316 (2010).
Lee, A. H., Iwakoshi, N. N. & Glimcher, L. H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 23, 7448–7459 (2003).
Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 19, 1807–1819 (2007).
Esmaeili, Y. et al. Targeting autophagy, oxidative stress, and ER stress for neurodegenerative disease treatment. J. Control. Release 345, 147–175 (2022).
Marciniak, S. J. et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 (2004).
Mizushima, N. & Komatsu, M. Autophagy: renovation of cells and tissues. Cell 147, 728–741 (2011).
Rashid, H. O., Yadav, R. K., Kim, H. R. & Chae, H. J. ER stress: autophagy induction, inhibition and selection. Autophagy 11, 1956–1977 (2015).
Smith, M. D. et al. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev. Cell 44, 217.e1–232.e1 (2018).
Zielke, S. et al. ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy 17, 2432–2448 (2021).
Liang, J. R. et al. A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 180, 1160.e20–1177.e20 (2020).
Zhang, X. et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 10, 1801–1813 (2014).
B’Chir, W. et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699 (2013).
Sekine, Y., Takeda, K. & Ichijo, H. The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Curr. Mol. Med. 6, 87–97 (2006).
Deegan, S. et al. Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions. Autophagy 10, 1921–1936 (2014).
Giacomello, M. & Pellegrini, L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 23, 1417–1427 (2016).
Pereira, A. C. et al. ER-mitochondria communication is involved in NLRP3 inflammasome activation under stress conditions in the innate immune system. Cell Mol. Life Sci. 79, 213 (2022).
Muñoz, J. P. et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361 (2013).
Wang, H. et al. The XBP1‒MARCH5‒MFN2 axis confers endoplasmic reticulum stress resistance by coordinating mitochondrial fission and mitophagy in melanoma. J. Invest. Dermatol. 141, 2932–2943.e2912 (2021).
McCullough, K. D. et al. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249–1259 (2001).
Puthalakath, H. et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 (2007).
Han, J. & Kaufman, R. J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 57, 1329–1338 (2016).
Ye, J. et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 (2000).
Zeng, L. et al. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 23, 950–958 (2004).
Chen, X. et al. Hepatic ATF6 increases fatty acid oxidation to attenuate hepatic steatosis in mice through peroxisome proliferator-activated receptor α. Diabetes 65, 1904–1915 (2016).
Sriburi, R. et al. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J. Biol. Chem. 282, 7024–7034 (2007).
Lee, A. H., Scapa, E. F., Cohen, D. E. & Glimcher, L. H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496 (2008).
Shao, M. et al. Hepatic IRE1α regulates fasting-induced metabolic adaptive programs through the XBP1s-PPARα axis signalling. Nat. Commun. 5, 3528 (2014).
Oyadomari, S. et al. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 7, 520–532 (2008).
GBD 2019 Blindness and Vision Impairment Collaborators & Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: the Right to Sight: an analysis for the Global Burden of Disease Study. Lancet Glob. Health 9, e144–e160 (2021).
Dandona, L. & Dandona, R. What is the global burden of visual impairment? BMC Med. 4, 6 (2006).
Casson, R. J. et al. Definition of glaucoma: clinical and experimental concepts. Clin. Exp. Ophthalmol. 40, 341–349 (2012).
Tham, Y. C. et al. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 121, 2081–2090 (2014).
Weinreb, R. N., Aung, T. & Medeiros, F. A. The pathophysiology and treatment of glaucoma: a review. JAMA 311, 1901–1911 (2014).
Yan, X. et al. Myocilin gene mutation induced autophagy activation causes dysfunction of trabecular meshwork cells. Front. Cell Dev. Biol. 10, 900777 (2022).
Cheng, Y. et al. Human Pro370Leu mutant myocilin induces the phenotype of open-angle glaucoma in transgenic mice. Cell. Mol. Neurobiol. 43, 2021–2033 (2023).
Allingham, R. R. et al. Gln368STOP myocilin mutation in families with late-onset primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 39, 2288–2295 (1998).
Yang, Y. et al. Cross-talk between MYOC p. Y437H mutation and TGF-β2 in the pathology of glaucoma. Int. J. Med. Sci. 17, 1062–1070 (2020).
Saccuzzo, E. G. et al. Calcium dysregulation potentiates wild-type myocilin misfolding: implications for glaucoma pathogenesis. J. Biol. Inorg. Chem. 27, 553–564 (2022).
Joe, M. K. et al. Accumulation of mutant myocilins in ER leads to ER stress and potential cytotoxicity in human trabecular meshwork cells. Biochem. Biophys. Res. Commun. 312, 592–600 (2003).
Peters, J. C., Bhattacharya, S., Clark, A. F. & Zode, G. S. Increased endoplasmic reticulum stress in human glaucomatous trabecular meshwork cells and tissues. Invest. Ophthalmol. Vis. Sci. 56, 3860–3868 (2015).
Ying, Y. et al. Activation of ATF4 triggers trabecular meshwork cell dysfunction and apoptosis in POAG. Aging 13, 8628–8642 (2021).
Suntharalingam, A. et al. Glucose-regulated protein 94 triage of mutant myocilin through endoplasmic reticulum-associated degradation subverts a more efficient autophagic clearance mechanism. J. Biol. Chem. 287, 40661–40669 (2012).
Kasetti, R. B. et al. ATF4 leads to glaucoma by promoting protein synthesis and ER client protein load. Nat. Commun. 11, 5594 (2020).
Yasuda, M. et al. RNA sequence reveals mouse retinal transcriptome changes early after axonal injury. PLoS ONE 9, e93258 (2014).
Wang, Y. et al. Endoplasmic reticulum stress response of trabecular meshwork stem cells and trabecular meshwork cells and protective effects of activated PERK pathway. Invest. Ophthalmol. Vis. Sci. 60, 265–273 (2019).
Stothert, A. R., Fontaine, S. N., Sabbagh, J. J. & Dickey, C. A. Targeting the ER-autophagy system in the trabecular meshwork to treat glaucoma. Exp. Eye Res. 144, 38–45 (2016).
Kasetti, R. B. et al. Autophagy stimulation reduces ocular hypertension in a murine glaucoma model via autophagic degradation of mutant myocilin. JCI Insight. 6, e143359 (2021).
Kasetti, R. B. et al. Increased synthesis and deposition of extracellular matrix proteins leads to endoplasmic reticulum stress in the trabecular meshwork. Sci. Rep. 7, 14951 (2017).
Kasetti, R. B., Patel, P. D., Maddineni, P. & Zode, G. S. Ex-vivo cultured human corneoscleral segment model to study the effects of glaucoma factors on trabecular meshwork. PLoS ONE 15, e0232111 (2020).
Takai, Y., Tanito, M. & Ohira, A. Multiplex cytokine analysis of aqueous humor in eyes with primary open-angle glaucoma, exfoliation glaucoma, and cataract. Invest. Ophthalmol. Vis. Sci. 53, 241–247 (2012).
Gottanka, J. et al. Effects of TGF-beta2 in perfused human eyes. Invest. Ophthalmol. Vis. Sci. 45, 153–158 (2004).
Kasetti, R. B. et al. Astragaloside IV attenuates ocular hypertension in a mouse model of TGFβ2 induced primary open angle glaucoma. Int. J. Mol. Sci. 22, 12508 (2021).
Shepard, A. R. et al. Adenoviral gene transfer of active human transforming growth factor-{beta}2 elevates intraocular pressure and reduces outflow facility in rodent eyes. Invest. Ophthalmol. Vis. Sci. 51, 2067–2076 (2010).
Li, M., Xu, J., Chen, X. & Sun, X. RNA interference as a gene silencing therapy for mutant MYOC protein in primary open angle glaucoma. Diagn. Pathol. 4, 46 (2009).
Zode, G. S. et al. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J. Clin. Investig. 121, 3542–3553 (2011).
Jain, A. et al. CRISPR-Cas9-based treatment of myocilin-associated glaucoma. Proc. Natl Acad. Sci. USA 114, 11199–11204 (2017).
Kasetti, R. B., Phan, T. N., Millar, J. C. & Zode, G. S. Expression of mutant myocilin induces abnormal intracellular accumulation of selected extracellular matrix proteins in the trabecular meshwork. Invest. Ophthalmol. Vis. Sci. 57, 6058–6069 (2016).
Goldberg, A. D., Allis, C. D. & Bernstein, E. Epigenetics: a landscape takes shape. Cell 128, 635–638 (2007).
Esteve-Puig, R., Bueno-Costa, A. & Esteller, M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett. 474, 127–137 (2020).
Li, S. et al. SNHG3 cooperates with ELAVL2 to modulate cell apoptosis and extracellular matrix accumulation by stabilizing SNAI2 in human trabecular meshwork cells under oxidative stress. Environ. Toxicol. 36, 1070–1079 (2021).
De Groef, L. et al. Aberrant collagen composition of the trabecular meshwork results in reduced aqueous humor drainage and elevated IOP in MMP-9 null mice. Invest. Ophthalmol. Vis. Sci. 57, 5984–5995 (2016).
Cuevas, E. P. et al. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci. Rep. 7, 44988 (2017).
Toth, R. P. & Atkin, J. D. Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Front. Immunol. 9, 1017 (2018).
Sears, N. C., Boese, E. A., Miller, M. A. & Fingert, J. H. Mendelian genes in primary open angle glaucoma. Exp. Eye Res. 186, 107702 (2019).
Minegishi, Y. et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum. Mol. Genet. 22, 3559–3567 (2013).
Rezaie, T. et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295, 1077–1079 (2002).
Quist, T. S., Johnson, C. A., Robin, A. L. & Fingert, J. H. Long-term follow-up of normal tension glaucoma patients with TBK1 gene mutations in one large pedigree. Am. J. Ophthalmol. 214, 52–62 (2020).
Rozpędek-Kamińska, W. et al. The genetic and endoplasmic reticulum-mediated molecular mechanisms of primary open-angle glaucoma. Int. J. Mol. Sci. 21, 4171 (2020).
Quigley, H. A. et al. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am. J. Ophthalmol. 95, 673–691 (1983).
Liu, X. et al. Neuroprotective effects of bone marrow Sca-1(+) cells against age-related retinal degeneration in OPTN E50K mice. Cell Death Dis. 12, 613 (2021).
Guttenplan, K. A. et al. Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Rep. 31, 107776 (2020).
Joshi, A. U. et al. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 22, 1635–1648 (2019).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Gomes, C. et al. Astrocytes modulate neurodegenerative phenotypes associated with glaucoma in OPTN(E50K) human stem cell-derived retinal ganglion cells. Stem Cell Rep. 17, 1636–1649 (2022).
Hou, M. et al. Age-related visual impairments and retinal ganglion cells axonal degeneration in a mouse model harboring OPTN (E50K) mutation. Cell Death Dis. 13, 362 (2022).
Chen, X. et al. Dysfunctional endoplasmic reticulum-mitochondrion coupling is associated with endoplasmic reticulum stress-induced apoptosis and neurological deficits in a rodent model of severe head injury. J. Neurotrauma 39, 560–576 (2022).
Zhang, S. et al. The E50K optineurin mutation impacts autophagy-mediated degradation of TDP-43 and leads to RGC apoptosis in vivo and in vitro. Cell Death Discov. 7, 49 (2021).
Zhao, C., Liao, Y., Rahaman, A. & Kumar, V. Towards understanding the relationship between ER stress and unfolded protein response in amyotrophic lateral sclerosis. Front. Aging Neurosci. 14, 892518 (2022).
Medchalmi, S., Tare, P., Sayyad, Z. & Swarup, G. A glaucoma- and ALS-associated mutant of OPTN induces neuronal cell death dependent on Tbk1 activity, autophagy and ER stress. FEBS J. 288, 4576–4595 (2021).
Sayyad, Z. et al. A glaucoma-associated OPTN polymorphism, M98K sensitizes retinal cells to endoplasmic reticulum stress and tumour necrosis factor α. FEBS J. 290, 3110–3127 (2023).
Huang, K. C. et al. Autophagy disruption reduces mTORC1 activation leading to retinal ganglion cell neurodegeneration associated with glaucoma. Preprint at bioRxiv https://doi.org/10.1101/2023.01.04.522687 (2023).
Ojino, K. et al. Involvement of endoplasmic reticulum stress in optic nerve degeneration after chronic high intraocular pressure in DBA/2J mice. J. Neurosci. Res. 93, 1675–1683 (2015).
Uchibayashi, R. et al. Involvement of Bid and caspase-2 in endoplasmic reticulum stress- and oxidative stress-induced retinal ganglion cell death. J. Neurosci. Res. 89, 1783–1794 (2011).
Naidoo, N. et al. Endoplasmic reticulum stress in wake-active neurons progresses with aging. Aging Cell 10, 640–649 (2011).
McLaughlin, T. et al. Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 13, 16 (2018).
Leger, F. et al. Protein aggregation in the aging retina. J. Neuropathol. Exp. Neurol. 70, 63–68 (2011).
Chen, W. et al. Maprotiline restores ER homeostasis and rescues neurodegeneration via histamine receptor H1 inhibition in retinal ganglion cells. Nat. Commun. 13, 6796 (2022).
Hu, Y. et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron 73, 445–452 (2012).
Mavlyutov, T. A. & Guo, L. W. Peeking into Sigma-1 receptor functions through the retina. Adv. Exp. Med. Biol. 964, 285–297 (2017).
Mysona, B., Kansara, N., Zhao, J. & Bollinger, K. The role of Sigma 1 receptor as a neuroprotective target in glaucoma. Adv. Exp. Med. Biol. 964, 299–307 (2017).
Mori, T., Hayashi, T., Hayashi, E. & Su, T. P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 8, e76941 (2013).
Ha, Y. et al. Sigma receptor 1 modulates endoplasmic reticulum stress in retinal neurons. Invest. Ophthalmol. Vis. Sci. 52, 527–540 (2011).
McLaughlin, T. et al. p58(IPK) is an endogenous neuroprotectant for retinal ganglion cells. Front. Aging Neurosci. 10, 267 (2018).
Boriushkin, E. et al. Identification of p58IPK as a novel neuroprotective factor for retinal neurons. Invest. Ophthalmol. Vis. Sci. 56, 1374–1386 (2015).
Apostolou, A. et al. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp. Cell Res. 314, 2454–2467 (2008).
Gao, F. J. et al. Identification of mesencephalic astrocyte-derived neurotrophic factor as a novel neuroprotective factor for retinal ganglion cells. Front. Mol. Neurosci. 10, 76 (2017).
Hayashi, A. et al. The role of brain-derived neurotrophic factor (BDNF)-induced XBP1 splicing during brain development. J. Biol. Chem. 282, 34525–34534 (2007).
Saito, A. et al. Neuronal activity-dependent local activation of dendritic unfolded protein response promotes expression of brain-derived neurotrophic factor in cell soma. J. Neurochem. 144, 35–49 (2018).
Chen, G. et al. Brain-derived neurotrophic factor suppresses tunicamycin-induced upregulation of CHOP in neurons. J. Neurosci. Res. 85, 1674–1684 (2007).
Wang, Q. et al. Phosphorylation of SHP-2 regulates interactions between the endoplasmic reticulum and focal adhesions to restrict interleukin-1-induced Ca2+ signaling. J. Biol. Chem. 281, 31093–31105 (2006).
Chitranshi, N. et al. Loss of Shp2 rescues BDNF/TrkB signaling and contributes to improved retinal ganglion cell neuroprotection. Mol. Ther. 27, 424–441 (2019).
Lin, B., Zhang, X. & Xu, X. Nerve growth factor protects retinal ganglion cells related to inhibiting endoplasmic reticulum stress by inhibiting IRE1-JNK-CHOP signaling pathway. Ocul. Immunol. Inflamm. 30, 1341–1346 (2022).
Sato, K. et al. CHOP deletion and anti-neuroinflammation treatment with hesperidin synergistically attenuate NMDA retinal injury in mice. Exp. Eye Res. 213, 108826 (2021).
Bronner, D. N. et al. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 43, 451–462 (2015).
Chen, H. et al. NLRP12 collaborates with NLRP3 and NLRC4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol. Neurodegener. 15, 26 (2020).
Chi, W. et al. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-κB pathway in acute glaucoma. J. Neuroinflammation 12, 137 (2015).
Li, L. et al. Airborne particulate matter (PM(2.5)) triggers ocular hypertension and glaucoma through pyroptosis. Part Fibre Toxicol. 18, 10 (2021).
Ye, D. et al. Anti-PANoptosis is involved in neuroprotective effects of melatonin in acute ocular hypertension model. J. Pineal Res. 73, e12828 (2022).
Yao, F. et al. Pathologically high intraocular pressure disturbs normal iron homeostasis and leads to retinal ganglion cell ferroptosis in glaucoma. Cell Death Differ. 30, 69–81 (2023).
Ha, Y. et al. Endoplasmic reticulum stress-regulated CXCR3 pathway mediates inflammation and neuronal injury in acute glaucoma. Cell Death Dis. 6, e1900 (2015).
Rappert, A. et al. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J. Neurosci. 24, 8500–8509 (2004).
Liu, M. et al. GSK872 and necrostatin-1 protect retinal ganglion cells against necroptosis through inhibition of RIP1/RIP3/MLKL pathway in glutamate-induced retinal excitotoxic model of glaucoma. J. Neuroinflammation 19, 262 (2022).
Yuan, H. et al. Involvement of HDAC6 in ischaemia and reperfusion-induced rat retinal injury. BMC Ophthalmol. 18, 300 (2018).
Baumeister, P. et al. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 25, 4529–4540 (2005).
Zhang, Z. et al. Valproate protects the retina from endoplasmic reticulum stress-induced apoptosis after ischemia-reperfusion injury. Neurosci. Lett. 504, 88–92 (2011).
Nashine, S. et al. Role of C/EBP homologous protein in retinal ganglion cell death after ischemia/reperfusion injury. Invest. Ophthalmol. Vis. Sci. 56, 221–231 (2014).
Rhee, S. G. Overview on peroxiredoxin. Mol. Cells 39, 1–5 (2016).
Parmigiani, R. B. et al. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl Acad. Sci. USA 105, 9633–9638 (2008).
Kang, J. H. et al. Peroxiredoxin 4 attenuates glutamate-induced neuronal cell death through inhibition of endoplasmic reticulum stress. Free Radic. Res. 54, 207–220 (2020).
Zode, G. S. et al. Ocular-specific ER stress reduction rescues glaucoma in murine glucocorticoid-induced glaucoma. J. Clin. Investig. 124, 1956–1965 (2014).
Kasetti, R. B. et al. Transforming growth factor β2 (TGFβ2) signaling plays a key role in glucocorticoid-induced ocular hypertension. J. Biol. Chem. 293, 9854–9868 (2018).
Ritch, R. Exfoliation syndrome. Curr. Opin. Ophthalmol. 12, 124–130 (2001).
Tawfik, A. & Smith, S. B. Increased ER stress as a mechanism of retinal neurovasculopathy in mice with severe hyperhomocysteinemia. Austin J. Clin. Ophthalmol. 1, 1023 (2014).
Hayat, B., Padhy, B., Mohanty, P. P. & Alone, D. P. Altered unfolded protein response and proteasome impairment in pseudoexfoliation pathogenesis. Exp. Eye Res. 181, 197–207 (2019).
Boland, M. V. et al. Comparative effectiveness of treatments for open-angle glaucoma: a systematic review for the U.S. Preventive Services Task Force. Ann. Intern. Med. 158, 271–279 (2013).
Gaton, D. D. et al. Increased matrix metalloproteinases 1, 2, and 3 in the monkey uveoscleral outflow pathway after topical prostaglandin F(2 alpha)-isopropyl ester treatment. Arch. Ophthalmol. 119, 1165–1170 (2001).
Stothert, A. R. et al. Exploiting the interaction between Grp94 and aggregated myocilin to treat glaucoma. Hum. Mol. Genet. 23, 6470–6480 (2014).
Stothert, A. R. et al. Isoform-selective Hsp90 inhibition rescues model of hereditary open-angle glaucoma. Sci. Rep. 7, 17951 (2017).
Rozpędek-Kamińska, W. et al. The potential role of small-molecule PERK inhibitor LDN-0060609 in primary open-angle glaucoma treatment. Int. J. Mol. Sci. 22, 4494 (2021).
Jia, L. Y. et al. Correction of the disease phenotype of myocilin-causing glaucoma by a natural osmolyte. Invest. Ophthalmol. Vis. Sci. 50, 3743–3749 (2009).
Maddineni, P. et al. Sodium 4-phenylbutyrate reduces ocular hypertension by degrading extracellular matrix deposition via activation of MMP9. Int. J. Mol. Sci. 22, 10095 (2021).
Dong, Z. et al. Astragaloside-IV alleviates heat-induced inflammation by inhibiting endoplasmic reticulum stress and autophagy. Cell. Physiol. Biochem. 42, 824–837 (2017).
Deveau, H., Garneau, J. E. & Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 64, 475–493 (2010).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Dong, Y., Siegwart, D. J. & Anderson, D. G. Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Deliv. Rev. 144, 133–147 (2019).
Gaynor, J. W., Campbell, B. J. & Cosstick, R. RNA interference: a chemist’s perspective. Chem. Soc. Rev. 39, 4169–4184 (2010).
Du, Y. et al. Multipotent stem cells from trabecular meshwork become phagocytic TM cells. Invest. Ophthalmol. Vis. Sci. 53, 1566–1575 (2012).
McGowan, S. L., Edelhauser, H. F., Pfister, R. R. & Whikehart, D. R. Stem cell markers in the human posterior limbus and corneal endothelium of unwounded and wounded corneas. Mol. Vis. 13, 1984–2000 (2007).
Yun, H. et al. Human stem cells home to and repair laser-damaged trabecular meshwork in a mouse model. Commun. Biol. 1, 216 (2018).
Xiong, S. et al. α5β1 Integrin promotes anchoring and integration of transplanted stem cells to the trabecular meshwork in the eye for regeneration. Stem Cells Dev. 29, 290–300 (2020).
Zhu, W. et al. Restoration of aqueous humor outflow following transplantation of iPSC-derived trabecular meshwork cells in a transgenic mouse model of glaucoma. Invest. Ophthalmol. Vis. Sci. 58, 2054–2062 (2017).
Ding, Q. J. et al. Induction of trabecular meshwork cells from induced pluripotent stem cells. Invest. Ophthalmol. Vis. Sci. 55, 7065–7072 (2014).
Roubeix, C. et al. Intraocular pressure reduction and neuroprotection conferred by bone marrow-derived mesenchymal stem cells in an animal model of glaucoma. Stem Cell Res. Ther. 6, 177 (2015).
Snider, E. J. et al. Improving stem cell delivery to the trabecular meshwork using magnetic nanoparticles. Sci. Rep. 8, 12251 (2018).
Zhou, Y. et al. Adipose-derived stem cells integrate into trabecular meshwork with glaucoma treatment potential. FASEB J. 34, 7160–7177 (2020).
Khater, S. I. et al. β-Cell autophagy pathway and endoplasmic reticulum stress regulating-role of liposomal curcumin in experimental diabetes mellitus: a molecular and morphometric study. Antioxidants 11, 2400 (2022).
Lerner, N., Schreiber-Avissar, S. & Beit-Yannai, E. Extracellular vesicle-mediated crosstalk between NPCE cells and TM cells result in modulation of Wnt signalling pathway and ECM remodelling. J. Cell. Mol. Med. 24, 4646–4658 (2020).
Lerner, N., Chen, I., Schreiber-Avissar, S. & Beit-Yannai, E. Extracellular vesicles mediate anti-oxidative response-in vitro study in the ocular drainage system. Int. J. Mol. Sci. 21, 6105 (2020).
Hata, M. & Ikeda, H. O. Modulation of valosin-containing protein by Kyoto University Substances (KUS) as a novel therapeutic strategy for ischemic neuronal diseases. Neural Regen. Res. 12, 1252–1255 (2017).
Hata, M. et al. KUS121, a VCP modulator, attenuates ischemic retinal cell death via suppressing endoplasmic reticulum stress. Sci. Rep. 7, 44873 (2017).
Hasegawa, T. et al. Effect of VCP modulators on gene expression profiles of retinal ganglion cells in an acute injury mouse model. Sci. Rep. 10, 4251 (2020).
Yang, L. et al. Rescue of glaucomatous neurodegeneration by differentially modulating neuronal endoplasmic reticulum stress molecules. J. Neurosci. 36, 5891–5903 (2016).
Ha, Y. et al. AAV2-mediated GRP78 transfer alleviates retinal neuronal injury by downregulating ER stress and tau oligomer formation. Invest. Ophthalmol. Vis. Sci. 59, 4670–4682 (2018).
Tian, F. et al. Core transcription programs controlling injury-induced neurodegeneration of retinal ganglion cells. Neuron 110, 2607–2624.e2608 (2022).
Shimazawa, M., Ito, Y., Inokuchi, Y. & Hara, H. Involvement of double-stranded RNA-dependent protein kinase in ER stress-induced retinal neuron damage. Invest. Ophthalmol. Vis. Sci. 48, 3729–3736 (2007).
Gao, Z. et al. Valdecoxib protects against cell apoptosis induced by endoplasmic reticulum stress via the inhibition of PERK-ATF4-CHOP pathway in experimental glaucoma. Int. J. Mol. Sci. 23, 12983 (2022).
Dheer, Y. et al. Retinoid x receptor modulation protects against ER stress response and rescues glaucoma phenotypes in adult mice. Exp. Neurol. 314, 111–125 (2019).
Rong, R. et al. Targeting cell membranes, depleting ROS by dithiane and thioketal-containing polymers with pendant cholesterols delivering necrostatin-1 for glaucoma treatment. ACS Nano 16, 21225–21239 (2022).
Huang, E. J. & Reichardt, L. F. Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736 (2001).
Beykin, G. et al. Phase 1b randomized controlled study of short course topical recombinant human nerve growth factor (rhNGF) for neuroenhancement in glaucoma: safety, tolerability, and efficacy measure outcomes. Am. J. Ophthalmol. 234, 223–234 (2022).
Guimarães, E. S. et al. Brucella abortus cyclic dinucleotides trigger STING-dependent unfolded protein response that favors bacterial replication. J. Immunol. 202, 2671–2681 (2019).
Congdon, N. G., Friedman, D. S. & Lietman, T. Important causes of visual impairment in the world today. JAMA 290, 2057–2060 (2003).
Zhong, Y., Wang, J. J. & Zhang, S. X. Intermittent but not constant high glucose induces ER stress and inflammation in human retinal pericytes. Adv. Exp. Med. Biol. 723, 285–292 (2012).
Fresia, D., Cannizzaro, E., Borgo, A. & Roduit, R. GSH-independent induction of ER stress during hypoglycaemia in the retinal cells of mice. J. Clin. Med. 10, 2529 (2021).
Aloysius Dhivya, M., Sulochana, K. N. & Bharathi Devi, S. R. High glucose induced inflammation is inhibited by copper chelation via rescuing mitochondrial fusion protein 2 in retinal pigment epithelial cells. Cell. Signal. 92, 110244 (2022).
Shruthi, K., Reddy, S. S. & Reddy, G. B. Ubiquitin-proteasome system and ER stress in the retina of diabetic rats. Arch. Biochem. Biophys. 627, 10–20 (2017).
Jiang, L., Wang, C. & Shen, X. LncRNA GAS5 suppresses ER stress‑induced apoptosis and inflammation by regulating SERCA2b in HG‑treated retinal epithelial cell. Mol. Med. Rep. 22, 1072–1080 (2020).
Peng, Q. H., Tong, P., Gu, L. M. & Li, W. J. Astragalus polysaccharide attenuates metabolic memory-triggered ER stress and apoptosis via regulation of miR-204/SIRT1 axis in retinal pigment epithelial cells. Biosci. Rep. 40, BSR20192121 (2020).
Wang, Y., Gao, S., Zhu, Y. & Shen, X. Elevated activating transcription factor 4 and glucose-regulated 78 Kda protein levels correlate with inflammatory cytokines in the aqueous humor and vitreous of proliferative diabetic retinopathy. Curr. Eye Res. 42, 1202–1208 (2017).
Yang, J. et al. Loss of X-box binding protein 1 in Müller cells augments retinal inflammation in a mouse model of diabetes. Diabetologia 62, 531–543 (2019).
Dai, W., Toro, A., Dierschke, S. K. & Dennis, M. D. High-fat diet/palmitate–induced ER stress promotes protein O-GlcNAcylation in retina and retinal Muller cells. Diabetes 67, 607-P (2018).
Fu, D. et al. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 55, 3128–3140 (2012).
Kang, M. K. et al. Chrysin ameliorates malfunction of retinoid visual cycle through blocking activation of AGE-RAGE-ER stress in glucose-stimulated retinal pigment epithelial cells and diabetic eyes. Nutrients. 10, 1046 (2018).
Kaur, C., Foulds, W. S. & Ling, E. A. Blood-retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog. Retin. Eye Res. 27, 622–647 (2008).
Tawfik, A., Samra, Y. A., Elsherbiny, N. M. & Al-Shabrawey, M. Implication of hyperhomocysteinemia in blood retinal barrier (BRB) dysfunction. Biomolecules. 10, 1119 (2020).
dos Santos, K. G. et al. The -106CC genotype of the aldose reductase gene is associated with an increased risk of proliferative diabetic retinopathy in Caucasian-Brazilians with type 2 diabetes. Mol. Genet. Metab. 88, 280–284 (2006).
Zhou, M., Zhang, P., Xu, X. & Sun, X. The relationship between aldose reductase C106T polymorphism and diabetic retinopathy: an updated meta-analysis. Invest. Ophthalmol. Vis. Sci. 56, 2279–2289 (2015).
Steentoft, C. et al. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 8, 977–982 (2011).
Kornfeld, R. & Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 (1985).
Saha, A., Bello, D. & Fernández-Tejada, A. Advances in chemical probing of protein O-GlcNAc glycosylation: structural role and molecular mechanisms. Chem. Soc. Rev. 50, 10451–10485 (2021).
Lenin, R., Nagy, P. G., Jha, K. A. & Gangaraju, R. GRP78 translocation to the cell surface and O-GlcNAcylation of VE-Cadherin contribute to ER stress-mediated endothelial permeability. Sci. Rep. 9, 10783 (2019).
Dierschke, S. K. et al. Diabetes enhances translation of Cd40 mRNA in murine retinal Müller glia via a 4E-BP1/2-dependent mechanism. J. Biol. Chem. 295, 10831–10841 (2020).
Xu, Y. et al. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle 19, 3054–3065 (2020).
Wang, Y. et al. Knockdown of MALAT1 attenuates high-glucose-induced angiogenesis and inflammation via endoplasmic reticulum stress in human retinal vascular endothelial cells. Biomed. Pharmacother. 124, 109699 (2020).
Wu, M. et al. Oxidative and endoplasmic reticulum stresses mediate apoptosis induced by modified LDL in human retinal Müller cells. Invest. Ophthalmol. Vis. Sci. 53, 4595–4604 (2012).
Miyata, Y. et al. Regulation of endothelium-reticulum-stress-mediated apoptotic cell death by a polymethoxylated flavone, nobiletin, through the inhibition of nuclear translocation of glyceraldehyde 3-phosphate dehydrogenase in retinal Müller cells. Cells 10, 669 (2021).
Anitha, R. E., Janani, R., Peethambaran, D. & Baskaran, V. Lactucaxanthin protects retinal pigment epithelium from hyperglycemia-regulated hypoxia/ER stress/VEGF pathway mediated angiogenesis in ARPE-19 cell and rat model. Eur. J. Pharmacol. 899, 174014 (2021).
Ménard, C. et al. miR-106b suppresses pathological retinal angiogenesis. Aging 12, 24836–24852 (2020).
Adachi, T. et al. Contribution of p38 MAPK, NF-κB and glucocorticoid signaling pathways to ER stress-induced increase in retinal endothelial permeability. Arch. Biochem. Biophys. 520, 30–35 (2012).
Chung, Y. R., Choi, J. A., Koh, J. Y. & Yoon, Y. H. Ursodeoxycholic acid attenuates endoplasmic reticulum stress-related retinal pericyte loss in streptozotocin-induced diabetic mice. J. Diabetes Res. 2017, 1763292 (2017).
Jia, M. et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 21, 727–735 (2020).
Wen, Z. et al. Hyperlipidemia induces proinflammatory responses by activating STING pathway through IRE1α-XBP1 in retinal endothelial cells. J. Nutr. Biochem. 112, 109213 (2023).
Li, Y. et al. GRP75 modulates endoplasmic reticulum-mitochondria coupling and accelerates Ca(2+)-dependent endothelial cell apoptosis in diabetic retinopathy. Biomolecules 12, 1778 (2022).
Fu, D. et al. Survival or death: a dual role for autophagy in stress-induced pericyte loss in diabetic retinopathy. Diabetologia 59, 2251–2261 (2016).
Chan, C. M. et al. Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction. J. Cell Mol. Med. 20, 1749–1760 (2016).
Wu, K. et al. TCF7L2 promotes ER stress signaling in diabetic retinopathy. Exp. Eye Res. 221, 109142 (2022).
Luo, J. et al. TCF7L2 variation and proliferative diabetic retinopathy. Diabetes 62, 2613–2617 (2013).
Sankrityayan, H. et al. ER stress response mediates diabetic microvascular complications. Drug Discov. Today 24, 2247–2257 (2019).
Cheung, N., Mitchell, P. & Wong, T. Y. Diabetic retinopathy. Lancet 376, 124–136 (2010).
Zhang, J., Chen, X., Zhang, L. & Peng, Y. IGF1 gene polymorphisms associated with diabetic retinopathy risk in Chinese Han population. Oncotarget 8, 88034–88042 (2017).
Liu, Z. et al. Single-cell transcriptome analyses reveal microglia types associated with proliferative retinopathy. JCI Insight 7, e160940 (2022).
Marwarha, G. et al. Palmitate-induced endoplasmic reticulum stress and subsequent C/EBPα homologous protein activation attenuates leptin and Insulin-like growth factor 1 expression in the brain. Cell. Signal. 28, 1789–1805 (2016).
Abhary, S. et al. Common sequence variation in the VEGFA gene predicts risk of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 50, 5552–5558 (2009).
Beránek, M. et al. Polymorphism R25P in the gene encoding transforming growth factor-beta (TGF-beta1) is a newly identified risk factor for proliferative diabetic retinopathy. Am. J. Med. Genet. 109, 278–283 (2002).
Yadav, H. et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. 14, 67–79 (2011).
Wu, C. & Morris, J. R. Genes, genetics, and epigenetics: a correspondence. Science 293, 1103–1105 (2001).
Nilsson, E. E., Sadler-Riggleman, I. & Skinner, M. K. Environmentally induced epigenetic transgenerational inheritance of disease. Environ. Epigenet. 4, dvy016 (2018).
Kwak, S. H. & Park, K. S. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 48, e220 (2016).
Liu, P. et al. LncRNA-MALAT1 promotes neovascularization in diabetic retinopathy through regulating miR-125b/VE-cadherin axis. Biosci. Rep. 39, BSR20181469 (2019).
Nandini, H. S. & Naik, P. R. Antidiabetic, antihyperlipidemic and antioxidant effect of Vincamine, in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 843, 233–239 (2019).
Ghosh, R. et al. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 5, e9575 (2010).
Rani, E. A., Janani, R., Chonche, M. J. & Vallikannan, B. Lactucaxanthin regulates the cascade of retinal oxidative stress, endoplasmic reticulum stress and inflammatory signaling in diabetic rats. Ocul. Immunol. Inflamm. 31, 320–328 (2023).
Wang, Y. et al. Blocking the interaction between interleukin-17A and endoplasmic reticulum stress in macrophage attenuates retinal neovascularization in oxygen-induced retinopathy. Cell Biosci. 11, 82 (2021).
Lai, D. W. et al. TPL2 (therapeutic targeting tumor progression locus-2)/ATF4 (activating transcription factor-4)/SDF1α (chemokine stromal cell-derived factor-α) axis suppresses diabetic retinopathy. Circ. Res. 121, e37–e52 (2017).
Feit-Leichman, R. A. et al. Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Invest. Ophthalmol. Vis. Sci. 46, 4281–4287 (2005).
Ahmad, I., Del Debbio, C. B., Das, A. V. & Parameswaran, S. Müller glia: a promising target for therapeutic regeneration. Invest. Ophthalmol. Vis. Sci. 52, 5758–5764 (2011).
Mills, S. A. et al. Fractalkine-induced microglial vasoregulation occurs within the retina and is altered early in diabetic retinopathy. Proc. Natl Acad. Sci. USA 118, e2112561118 (2021).
Bringmann, A. et al. Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog. Retin. Eye Res. 28, 423–451 (2009).
Li, M. et al. Semaphorin 3A inhibits endoplasmic reticulum stress induced by high glucose in Müller cells. Curr. Eye Res. 48, 70–79 (2023).
Oshitari, T., Yoshida-Hata, N. & Yamamoto, S. Effect of neurotrophin-4 on endoplasmic reticulum stress-related neuronal apoptosis in diabetic and high glucose exposed rat retinas. Neurosci. Lett. 501, 102–106 (2011).
Sharma, I., Yadav, K. S. & Mugale, M. N. Redoxisome and diabetic retinopathy: pathophysiology and therapeutic interventions. Pharm. Res. 182, 106292 (2022).
Kaneko, M. & Nomura, Y. ER signaling in unfolded protein response. Life Sci. 74, 199–205 (2003).
Guo, L., Bai, S. P., Zhao, L. & Wang, X. H. Astragalus polysaccharide injection integrated with vinorelbine and cisplatin for patients with advanced non-small cell lung cancer: effects on quality of life and survival. Med. Oncol. 29, 1656–1662 (2012).
Deng, S., Yang, L., Ma, K. & Bian, W. Astragalus polysaccharide improve the proliferation and insulin secretion of mouse pancreatic β cells induced by high glucose and palmitic acid partially through promoting miR-136-5p and miR-149-5p expression. Bioengineered 12, 9872–9884 (2021).
Khachik, F., Bernstein, P. S. & Garland, D. L. Identification of lutein and zeaxanthin oxidation products in human and monkey retinas. Invest. Ophthalmol. Vis. Sci. 38, 1802–1811 (1997).
Gopal, S. S. et al. Lactucaxanthin - a potential anti-diabetic carotenoid from lettuce (Lactuca sativa) inhibits α-amylase and α-glucosidase activity in vitro and in diabetic rats. Food Funct. 8, 1124–1131 (2017).
Saini, R. K., Moon, S. H., Gansukh, E. & Keum, Y. S. An efficient one-step scheme for the purification of major xanthophyll carotenoids from lettuce, and assessment of their comparative anticancer potential. Food Chem. 266, 56–65 (2018).
Lenin, R. et al. Tauroursodeoxycholic acid alleviates endoplasmic reticulum stress-mediated visual deficits in diabetic tie2-TNF transgenic mice via TGR5 signaling. J. Ocul. Pharm. Ther. 39, 159–174 (2023).
Yanagi, S., Sato, T., Kangawa, K. & Nakazato, M. The homeostatic force of ghrelin. Cell Metab. 27, 786–804 (2018).
Yang, H. et al. Functional characterization of 58-kilodalton inhibitor of protein kinase in protecting against diabetic retinopathy via the endoplasmic reticulum stress pathway. Mol. Vis. 17, 78–84 (2011).
Dąbkowska, M. et al. Electrostatic complex of neurotrophin 4 with dendrimer nanoparticles: controlled release of protein in vitro and in vivo. Int. J. Nanomed. 14, 6117–6131 (2019).
Abdel-Ghaffar, A. et al. Effects of 4-phenylbutyric acid on the development of diabetic retinopathy in diabetic rats: regulation of endoplasmic reticulum stress-oxidative activation. Arch. Physiol. Biochem. 129, 964–974 (2021).
Lv, J. et al. Sulforaphane delays diabetes-induced retinal photoreceptor cell degeneration. Cell Tissue Res. 382, 477–486 (2020).
Yoo, Y. M. & Joo, S. S. Melatonin can modulate neurodegenerative diseases by regulating endoplasmic reticulum stress. Int. J. Mol. Sci. 24, 2381 (2023).
Reda, S., Elsammak, G. A., Elsayed, T. G. & Mostafa, S. A. A comparative study between the possible protective role of melatonin versus its combination with adipose derived-mesenchymal stem cells on experimentally induced diabetic retinopathy in adult male albino rats (histological and immunohistochemical study). Ultrastruct. Pathol. 47, 131–145 (2023).
Lim, L. S. et al. Age-related macular degeneration. Lancet 379, 1728–1738 (2012).
Mitchell, P., Liew, G., Gopinath, B. & Wong, T. Y. Age-related macular degeneration. Lancet 392, 1147–1159 (2018).
Guymer, R. H. & Campbell, T. G. Age-related macular degeneration. Lancet 401, 1459–1472 (2023).
Wong, W. L. et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob. Health 2, e106–e116 (2014).
Karthikeyan, B. et al. Insights on the involvement of (-)-epigallocatechin gallate in ER stress-mediated apoptosis in age-related macular degeneration. Apoptosis 22, 72–85 (2017).
Bok, D. The retinal pigment epithelium: a versatile partner in vision. J. Cell Sci. Suppl. 17, 189–195 (1993).
Paraoan, L. et al. Secretory proteostasis of the retinal pigmented epithelium: impairment links to age-related macular degeneration. Prog. Retin. Eye Res. 79, 100859 (2020).
Johnson, L. V., Leitner, W. P., Staples, M. K. & Anderson, D. H. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp. Eye Res. 73, 887–896 (2001).
Lambert, N. G. et al. Risk factors and biomarkers of age-related macular degeneration. Prog. Retin. Eye Res. 54, 64–102 (2016).
Kunchithapautham, K., Atkinson, C. & Rohrer, B. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J. Biol. Chem. 289, 14534–14546 (2014).
Chen, C. et al. Role of unfolded protein response dysregulation in oxidative injury of retinal pigment epithelial cells. Antioxid. Redox Signal. 20, 2091–2106 (2014).
Song, J. Y. et al. Suppressing endoplasmic reticulum stress-related autophagy attenuates retinal light injury. Aging 12, 16579–16596 (2020).
Chalour, N. et al. AβPP-induced UPR transcriptomic signature of glial cells to oxidative stress as an adaptive mechanism to preserve cell function and survival. Curr. Alzheimer Res. 15, 643–654 (2018).
Ethen, C. M. et al. The proteome of central and peripheral retina with progression of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 47, 2280–2290 (2006).
Decanini, A. et al. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am. J. Ophthalmol. 143, 607–615 (2007).
Wang, Z. et al. Genetic associations of anti-vascular endothelial growth factor therapy response in age-related macular degeneration: a systematic review and meta-analysis. Acta Ophthalmol. 100, e669–e680 (2022).
Gerhardt, M. J. et al. ER stress-induced aggresome trafficking of HtrA1 protects against proteotoxicity. J. Mol. Cell Biol. 9, 516–532 (2017).
Li, W. et al. The P300/XBP1s/Herpud1 axis promotes macrophage M2 polarization and the development of choroidal neovascularization. J. Cell Mol. Med. 25, 6709–6720 (2021).
Yasuda, H. et al. Role of activating transcription factor 4 in murine choroidal neovascularization model. Int. J. Mol. Sci. 22, 8890 (2021).
Zhang, S. X. et al. The unfolded protein response in retinal vascular diseases: implications and therapeutic potential beyond protein folding. Prog. Retin. Eye Res. 45, 111–131 (2015).
Matsui, A. et al. Expression of vascular endothelial growth factor by retinal pigment epithelial cells induced by amyloid-β is depressed by an endoplasmic reticulum stress inhibitor. Ophthalmic Res 55, 37–44 (2015).
Kheitan, S., Minuchehr, Z. & Soheili, Z. S. Exploring the cross talk between ER stress and inflammation in age-related macular degeneration. PLoS ONE 12, e0181667 (2017).
Allikmets, R. et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 277, 1805–1807 (1997).
He, D. et al. eIF2α incites photoreceptor cell and retina damage by all-trans-retinal. J. Biol. Chem. 299, 104686, (2023).
Torrini, M. et al. Mutation analysis of oxisterol-binding-protein gene in patients with age-related macular degeneration. Genet. Test. 11, 421–426 (2007).
Tuo, J. et al. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 18, 1297–1299 (2004).
Tuo, J. et al. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 48, 3827–3836 (2007).
Xu, B. et al. Role of VEGFR2 in mediating endoplasmic reticulum stress under glucose deprivation and determining cell death, oxidative stress, and inflammatory factor expression. Front. Cell Dev. Biol. 9, 631413 (2021).
Chen, C. et al. Regulation of Nrf2 by X box-binding protein 1 in retinal pigment epithelium. Front. Genet. 9, 658 (2018).
Feng, J. et al. Expression of endoplasmic reticulum stress markers GRP78 and CHOP induced by oxidative stress in blue light-mediated damage of A2E-containing retinal pigment epithelium cells. Ophthalmic Res. 52, 224–233 (2014).
Liu, Y. et al. CHAC1 as a novel contributor of ferroptosis in retinal pigment epithelial cells with oxidative damage. Int. J. Mol. Sci. 24, 1582 (2023).
Colgan, S. M., Hashimi, A. A. & Austin, R. C. Endoplasmic reticulum stress and lipid dysregulation. Expert Rev. Mol. Med. 13, e4 (2011).
Tan, L. X., Li, J., Germer, C. J. & Lakkaraju, A. Analysis of mitochondrial dynamics and function in the retinal pigment epithelium by high-speed high-resolution live imaging. Front. Cell Dev. Biol. 10, 1044672 (2022).
Dou, G. et al. Deficiency of αB crystallin augments ER stress-induced apoptosis by enhancing mitochondrial dysfunction. Free Radic. Biol. Med. 53, 1111–1122 (2012).
Baek, A., Son, S., Baek, Y. M. & Kim, D. E. KRT8 (keratin 8) attenuates necrotic cell death by facilitating mitochondrial fission-mediated mitophagy through interaction with PLEC (plectin). Autophagy 17, 3939–3956 (2021).
Saptarshi, N., Porter, L. F. & Paraoan, L. PERK/EIF2AK3 integrates endoplasmic reticulum stress-induced apoptosis, oxidative stress and autophagy responses in immortalised retinal pigment epithelial cells. Sci. Rep. 12, 13324 (2022).
Ha, J. H. et al. Ocular inflammation and endoplasmic reticulum stress are attenuated by supplementation with grape polyphenols in human retinal pigmented epithelium cells and in C57BL/6 mice. J. Nutr. 144, 799–806 (2014).
Lin, Y. H. et al. Retinal protective effect of curcumin metabolite hexahydrocurcumin against blue light-induced RPE damage. Phytomedicine 110, 154606 (2023).
Zhu, X., Wang, K., Zhou, F. & Zhu, L. Paeoniflorin attenuates atRAL-induced oxidative stress, mitochondrial dysfunction and endoplasmic reticulum stress in retinal pigment epithelial cells via triggering Ca(2+)/CaMKII-dependent activation of AMPK. Arch. Pharm. Res. 41, 1009–1018 (2018).
Zhou, X. et al. Propofol decreases endoplasmic reticulum stress-mediated apoptosis in retinal pigment epithelial cells. PLoS ONE 11, e0157590 (2016).
Sreekumar, P. G., Hinton, D. R. & Kannan, R. Endoplasmic reticulum-mitochondrial crosstalk: a novel role for the mitochondrial peptide humanin. Neural Regen. Res. 12, 35–38 (2017).
Zhang, Y. et al. Inhibition of starvation-triggered endoplasmic reticulum stress, autophagy, and apoptosis in ARPE-19 cells by taurine through modulating the expression of Calpain-1 and Calpain-2. Int. J. Mol. Sci. 18, 2146 (2017).
Li, D. et al. Human umbilical cord mesenchymal stem cell-derived exosomal miR-27b attenuates subretinal fibrosis via suppressing epithelial-mesenchymal transition by targeting HOXC6. Stem Cell Res. Ther. 12, 24 (2021).
Yu, J. J. et al. Age-related macular degeneration (AMD) transmitochondrial cybrids protected from cellular damage and death by human retinal progenitor cells (hRPCs). Stem Cells Int. 2021, 6655372 (2021).
Kannan, R., Sreekumar, P. G. & Hinton, D. R. Alpha crystallins in the retinal pigment epithelium and implications for the pathogenesis and treatment of age-related macular degeneration. Biochim. Biophys. Acta 1860, 258–268 (2016).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Megaw, R. et al. Use of induced pluripotent stem-cell technology to understand photoreceptor cytoskeletal dynamics in retinitis pigmentosa. Lancet 385, S69 (2015).
Wang, Y. et al. Variants identified by next-generation sequencing cause endoplasmic reticulum stress in Rhodopsin-associated retinitis pigmentosa. BMC Ophthalmol. 21, 371 (2021).
Palu, R. A. S. & Chow, C. Y. Baldspot/ELOVL6 is a conserved modifier of disease and the ER stress response. PLoS Genet. 14, e1007557 (2018).
Yu, Y. et al. A new rhodopsin R135W mutation induces endoplasmic reticulum stress and apoptosis in retinal pigment epithelial cells. J. Cell. Physiol. 234, 14100–14108 (2019).
Xu, H. et al. Deletion of the Impg2 gene causes the degeneration of rod and cone cells in mice. Hum. Mol. Genet. 29, 1624–1634 (2020).
Athanasiou, D. et al. The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 26, 4896–4905 (2017).
Li, S. et al. Secretory defect and cytotoxicity: the potential disease mechanisms for the retinitis pigmentosa (RP)-associated interphotoreceptor retinoid-binding protein (IRBP). J. Biol. Chem. 288, 11395–11406 (2013).
Kang, M. J., Chung, J. & Ryoo, H. D. CDK5 and MEKK1 mediate pro-apoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nat. Cell Biol. 14, 409–415 (2012).
Jiang, H., Xiong, S. & Xia, X. Retinitis pigmentosa‑associated rhodopsin mutant T17M induces endoplasmic reticulum (ER) stress and sensitizes cells to ER stress-induced cell death. Mol. Med. Rep. 9, 1737–1742 (2014).
Osada, H. et al. Neuroprotective effect of bilberry extract in a murine model of photo-stressed retina. PLoS ONE 12, e0178627 (2017).
Bhootada, Y. et al. Limited ATF4 expression in degenerating retinas with ongoing ER stress promotes photoreceptor survival in a mouse model of autosomal dominant retinitis pigmentosa. PLoS ONE 11, e0154779 (2016).
Yamoah, A. et al. Early alterations of RNA binding protein (RBP) homeostasis and ER stress-mediated autophagy contributes to progressive retinal degeneration in the rd10 mouse model of retinitis pigmentosa (RP). Cells 12, 1094 (2023).
Kunte, M. M. et al. ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest. Ophthalmol. Vis. Sci. 53, 3792–3800 (2012).
Viringipurampeer, I. A. et al. NLRP3 inflammasome activation drives bystander cone photoreceptor cell death in a P23H rhodopsin model of retinal degeneration. Hum. Mol. Genet. 25, 1501–1516 (2016).
Qiu, Y. et al. Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: a novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 10, 547 (2019).
Chiang, W. C. et al. Robust endoplasmic reticulum-associated degradation of rhodopsin precedes retinal degeneration. Mol. Neurobiol. 52, 679–695 (2015).
Yao, J. et al. Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 14, 1226–1238 (2018).
Zhao, N., Li, N. & Wang, T. PERK prevents rhodopsin degradation during retinitis pigmentosa by inhibiting IRE1-induced autophagy. J. Cell Biol. 222, e202208147 (2023).
Wang, J. et al. Landscape of pathogenic variants in six pre-mRNA processing factor genes for retinitis pigmentosa based on large in-house data sets and database comparisons. Acta Ophthalmol. 100, e1412–e1425 (2022).
Valdés-Sánchez, L. et al. Retinal pigment epithelium degeneration caused by aggregation of PRPF31 and the role of HSP70 family of proteins. Mol. Med. 26, 1 (2019).
Georgiou, M. et al. Activation of autophagy reverses progressive and deleterious protein aggregation in PRPF31 patient-induced pluripotent stem cell-derived retinal pigment epithelium cells. Clin. Transl. Med. 12, e759 (2022).
Vang, S., Longley, K., Steer, C. J. & Low, W. C. The unexpected uses of urso- and tauroursodeoxycholic acid in the treatment of non-liver diseases. Glob. Adv. Health Med. 3, 58–69 (2014).
Gorbatyuk, M. S. et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl Acad. Sci. USA 107, 5961–5966 (2010).
Ozawa, Y. et al. Effects of epigenetic modification of PGC-1α by a chemical chaperon on mitochondria biogenesis and visual function in retinitis pigmentosa. Cells 11, 1497 (2022).
Zheng, M., Mitra, R. N., Weiss, E. R. & Han, Z. Rhodopsin genomic loci DNA nanoparticles improve expression and rescue of retinal degeneration in a model for retinitis pigmentosa. Mol. Ther. 28, 523–535 (2020).
Shahin, S. et al. AAV-CRISPR/Cas9 gene editing preserves long-term vision in the P23H rat model of autosomal dominant retinitis pigmentosa. Pharmaceutics 14, 824 (2022).
Shinohara, T., Mulhern, M. L. & Madson, C. J. Silencing gene therapy for mutant membrane, secretory, and lipid proteins in retinitis pigmentosa (RP). Med. Hypotheses 70, 378–380 (2008).
Yoshida, T. et al. The use of induced pluripotent stem cells to reveal pathogenic gene mutations and explore treatments for retinitis pigmentosa. Mol. Brain 7, 45 (2014).
Duarri, A. et al. Transplantation of human induced pluripotent stem cell-derived retinal pigment epithelium in a swine model of geographic atrophy. Int. J. Mol. Sci. 22, 10497 (2021).
Lin, B. et al. Retina organoid transplants develop photoreceptors and improve visual function in RCS rats with RPE dysfunction. Invest. Ophthalmol. Vis. Sci. 61, 34 (2020).
Foltz, L. P., Howden, S. E., Thomson, J. A. & Clegg, D. O. Functional assessment of patient-derived retinal pigment epithelial cells edited by CRISPR/Cas9. Int. J. Mol. Sci. 19, 4127 (2018).
Klassen, H. J. et al. Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behavior. Invest. Ophthalmol. Vis. Sci. 45, 4167–4173 (2004).
Brown, C. et al. Human primitive mesenchymal stem cell-derived retinal progenitor cells improved neuroprotection, neurogenesis, and vision in rd12 mouse model of retinitis pigmentosa. Stem Cell Res. Ther. 13, 148 (2022).
Pan, T. et al. Combined transplantation with human mesenchymal stem cells improves retinal rescue effect of human fetal RPE cells in retinal degeneration mouse model. Invest. Ophthalmol. Vis. Sci. 61, 9 (2020).
Ma, M. et al. Therapeutic effects of mesenchymal stem cell-derived exosomes on retinal detachment. Exp. Eye Res. 191, 107899 (2020).
Dezfuly, A. R. et al. Therapeutic effects of human adipose mesenchymal stem cells and their paracrine agents on sodium iodate induced retinal degeneration in rats. Life Sci. 300, 120570 (2022).
Knickelbein, J. E. et al. Modulation of immune responses by extracellular vesicles from retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 57, 4101–4107 (2016).
Wu, K. Y. et al. Retinitis pigmentosa: novel therapeutic targets and drug development. Pharmaceutics 15, 685 (2023).
Zhang, F. et al. The microbial opsin family of optogenetic tools. Cell 147, 1446–1457 (2011).
Gaub, B. M. et al. Restoration of visual function by expression of a light-gated mammalian ion channel in retinal ganglion cells or ON-bipolar cells. Proc. Natl Acad. Sci. USA 111, E5574–5583 (2014).
Remmer, M. H., Rastogi, N., Ranka, M. P. & Ceisler, E. J. Achromatopsia: a review. Curr. Opin. Ophthalmol. 26, 333–340 (2015).
Wawrocka, A. et al. Five novel CNGB3 gene mutations in Polish patients with achromatopsia. Mol. Vis. 20, 1732–1739 (2014).
Kohl, S. et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 47, 757–765 (2015).
Lee, E. J. et al. Multiexon deletion alleles of ATF6 linked to achromatopsia. JCI Insight 5, e136041 (2020).
Mastey, R. R. et al. Characterization of retinal structure in ATF6-associated achromatopsia. Invest. Ophthalmol. Vis. Sci. 60, 2631–2640 (2019).
Yang, F., Ma, H., Butler, M. R. & Ding, X. Q. Potential contribution of ryanodine receptor 2 upregulation to cGMP/PKG signaling-induced cone degeneration in cyclic nucleotide-gated channel deficiency. FASEB J. 34, 6335–6350 (2020).
Kroeger, H. et al. ATF6 is essential for human cone photoreceptor development. Proc. Natl Acad. Sci. USA 118, e2103196118 (2021).
Chiang, W. C. et al. Achromatopsia mutations target sequential steps of ATF6 activation. Proc. Natl Acad. Sci. USA 114, 400–405 (2017).
Ma, H. et al. Loss of cone cyclic nucleotide-gated channel leads to alterations in light response modulating system and cellular stress response pathways: a gene expression profiling study. Hum. Mol. Genet. 22, 3906–3919 (2013).
Lee, E. J. et al. Mitochondria and endoplasmic reticulum stress in retinal organoids from patients with vision loss. Am. J. Pathol. https://doi.org/10.1016/j.ajpath.2022.12.002 (2022).
Tam, A. B. et al. The UPR activator ATF6 responds to proteotoxic and lipotoxic stress by distinct mechanisms. Dev. Cell 46, 327.e7–343.e7 (2018).
Reichel, F. F. et al. Three-year results of phase I retinal gene therapy trial for CNGA3-mutated achromatopsia: results of a non randomised controlled trial. Br. J. Ophthalmol. 106, 1567–1572 (2022).
Hassall, M. M., Barnard, A. R. & MacLaren, R. E. Gene therapy for color blindness. Yale J. Biol. Med. 90, 543–551 (2017).
Komáromy, A. M. et al. Transient photoreceptor deconstruction by CNTF enhances rAAV-mediated cone functional rescue in late stage CNGB3-achromatopsia. Mol. Ther. 21, 1131–1141 (2013).
Siles, L., Gaudó, P. & Pomares, E. High-efficiency CRISPR/Cas9-mediated correction of a homozygous mutation in achromatopsia-patient-derived iPSCs. Int. J. Mol. Sci. 24, 3655 (2023).
Liu, Y. C. et al. Cataracts. Lancet 390, 600–612 (2017).
Cicinelli, M. V. et al. Cataracts. Lancet 401, 377–389 (2023).
Zhang, H. et al. Association between polymorphisms of OGG1, EPHA2 and age-related cataract risk: a meta-analysis. BMC Ophthalmol. 16, 168 (2016).
Zhang, T. et al. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum. Mutat. 30, E603–611 (2009).
Dave, A. et al. Genotype, age, genetic background, and sex influence Epha2-related cataract development in mice. Invest. Ophthalmol. Vis. Sci. 62, 3 (2021).
Berthoud, V. M. et al. Connexin mutants compromise the lens circulation and cause cataracts through biomineralization. Int. J. Mol. Sci. 21, 5822 (2020).
Berthoud, V. M. et al. The cataract-linked mutant Connexin50D47A causes endoplasmic reticulum stress in mouse lenses. J. Biol. Chem. 291, 17569–17578 (2016).
Periyasamy, P. & Shinohara, T. Age-related cataracts: role of unfolded protein response, Ca(2+) mobilization, epigenetic DNA modifications, and loss of Nrf2/Keap1 dependent cytoprotection. Prog. Retin. Eye Res. 60, 1–19 (2017).
Palsamy, P. et al. Methylglyoxal induces endoplasmic reticulum stress and DNA demethylation in the Keap1 promoter of human lens epithelial cells and age-related cataracts. Free Radic. Biol. Med. 72, 134–148 (2014).
Yang, S. P., Yang, X. Z. & Cao, G. P. Acetyl-l-carnitine prevents homocysteine-induced suppression of Nrf2/Keap1 mediated antioxidation in human lens epithelial cells. Mol. Med. Rep. 12, 1145–1150 (2015).
Palsamy, P., Bidasee, K. R. & Shinohara, T. Valproic acid suppresses Nrf2/Keap1 dependent antioxidant protection through induction of endoplasmic reticulum stress and Keap1 promoter DNA demethylation in human lens epithelial cells. Exp. Eye Res. 121, 26–34 (2014).
Palsamy, P., Bidasee, K. R. & Shinohara, T. Selenite cataracts: activation of endoplasmic reticulum stress and loss of Nrf2/Keap1-dependent stress protection. Biochim. Biophys. Acta 1842, 1794–1805 (2014).
Liu, C. et al. Involvement of increased endoplasmic reticulum stress in the development of cataracts in BALB.NCT-Cpox(nct) mice. Exp. Eye Res. 215, 108905 (2022).
Elanchezhian, R. et al. Low glucose under hypoxic conditions induces unfolded protein response and produces reactive oxygen species in lens epithelial cells. Cell Death Dis. 3, e301 (2012).
Ma, T. J. et al. Nrf2 protects human lens epithelial cells against H(2)O(2)-induced oxidative and ER stress: the ATF4 may be involved. Exp. Eye Res. 169, 28–37 (2018).
Huang, Y. et al. Cataract formation in transgenic HO-1 G143H mutant mice: Involvement of oxidative stress and endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 537, 43–49 (2021).
Zhou, S. et al. Endoplasmic reticulum stress regulates epithelial‑mesenchymal transition in human lens epithelial cells. Mol. Med. Rep. 21, 173–180 (2020).
de Iongh, R. U., Wederell, E., Lovicu, F. J. & McAvoy, J. W. Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells Tissues Organs 179, 43–55 (2005).
Liu, H. et al. Sulforaphane promotes ER stress, autophagy, and cell death: implications for cataract surgery. J. Mol. Med. 95, 553–564 (2017).
Jara, O., Minogue, P. J., Berthoud, V. M. & Beyer, E. C. Chemical chaperone treatment improves levels and distributions of connexins in Cx50D47A mouse lenses. Exp. Eye Res. 175, 192–198 (2018).
Wang, L. et al. Sigma 1 receptor stimulation protects against oxidative damage through suppression of the ER stress responses in the human lens. Mech. Ageing Dev. 133, 665–674 (2012).
Wei, D. et al. Application of iontophoresis in ophthalmic practice: an innovative strategy to deliver drugs into the eye. Drug Deliv. 30, 2165736 (2023).
Sfriso, P. et al. Blau syndrome, clinical and genetic aspects. Autoimmun. Rev. 12, 44–51 (2012).
Milman, N. & Byg, K. E. [Blau syndrome-a chronic granulomatous, genetic disease]. Ugeskr Laeger 168, 3612–3614 (2006).
Ogura, Y. et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 276, 4812–4818 (2001).
Pham, O. H. et al. NOD1/NOD2 and RIP2 regulate endoplasmic reticulum stress-induced inflammation during chlamydia infection. mBio 11, e00979-20 (2020).
Keestra-Gounder, A. M. et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 532, 394–397 (2016).
Li, W. et al. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 104, 6129–6140 (2020).
Lebeaupin, C. et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 6, e1879 (2015).
Wert, K. J. et al. CAPN5 mutation in hereditary uveitis: the R243L mutation increases calpain catalytic activity and triggers intraocular inflammation in a mouse model. Hum. Mol. Genet. 24, 4584–4598 (2015).
Kirectepe Aydin, A. et al. Peripheral blood mononuclear cell proteome profile in Behçet’s syndrome. Rheumatol. Int. 40, 65–74 (2020).
Smatlik, N. et al. Mesalazine suppresses proinflammatory cytokines in patients with acute anterior uveitis independently of HLA-B27. Ocul. Immunol. Inflamm. 30, 1369–1377 (2021).
Lee, H. J. et al. Proteomics-based functional studies reveal that galectin-3 plays a protective role in the pathogenesis of intestinal Behçet’s disease. Sci. Rep. 9, 11716 (2019).
Sehgal, P., Colombel, J. F., Aboubakr, A. & Narula, N. Systematic review: safety of mesalazine in ulcerative colitis. Aliment. Pharmacol. Ther. 47, 1597–1609 (2018).
Krantz, B. A. et al. Uveal melanoma: epidemiology, etiology, and treatment of primary disease. Clin. Ophthalmol. 11, 279–289 (2017).
McLaughlin, C. C. et al. Incidence of noncutaneous melanomas in the U.S. Cancer 103, 1000–1007 (2005).
Damato, E. M. & Damato, B. E. Detection and time to treatment of uveal melanoma in the United Kingdom: an evaluation of 2,384 patients. Ophthalmology 119, 1582–1589 (2012).
Rietschel, P. et al. Variates of survival in metastatic uveal melanoma. J. Clin. Oncol. 23, 8076–8080 (2005).
Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 161, 1681–1696 (2015).
Van Raamsdonk, C. D. et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457, 599–602 (2009).
Robertson, A. G. et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 32, 204.e5–220.e5 (2017).
Durante, M. A. et al. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 11, 496 (2020).
Smit, K. N., Jager, M. J., de Klein, A. & Kiliҫ, E. Uveal melanoma: towards a molecular understanding. Prog. Retin. Eye Res. 75, 100800 (2020).
Pandiani, C. et al. Single-cell RNA sequencing reveals intratumoral heterogeneity in primary uveal melanomas and identifies HES6 as a driver of the metastatic disease. Cell Death Differ. 28, 1990–2000 (2021).
Zhang, X. et al. Construction and verification of a hypoxia-related nine-gene prognostic model in uveal melanoma based on integrated single-cell and bulk RNA sequencing analyses. Exp. Eye Res. 223, 109214 (2022).
Asnaghi, L. et al. Hypoxia promotes uveal melanoma invasion through enhanced Notch and MAPK activation. PLoS ONE 9, e105372 (2014).
Mahadevan, N. R. et al. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8+ T cell priming. PLoS ONE 7, e51845 (2012).
Wang, T. et al. MMP1 and MMP9 are potential prognostic biomarkers and targets for uveal melanoma. BMC Cancer 21, 1068 (2021).
Tan, B. et al. Matrix metalloproteinase-11 promotes early mouse mammary gland tumor growth through metabolic reprogramming and increased IGF1/AKT/FoxO1 signaling pathway, enhanced ER stress and alteration in mitochondrial UPR. Cancers 12, 2357 (2020).
Bellini, L. et al. Endoplasmic reticulum stress mediates resistance to BCL-2 inhibitor in uveal melanoma cells. Cell Death Discov. 6, 22 (2020).
Eigner, K. et al. The unfolded protein response impacts melanoma progression by enhancing FGF expression and can be antagonized by a chemical chaperone. Sci. Rep. 7, 17498 (2017).
Shields, C. L. et al. Choroidal melanoma: clinical features, classification, and top 10 pseudomelanomas. Curr. Opin. Ophthalmol. 25, 177–185 (2014).
Zhao, X., Kong, F., Wang, L. & Zhang, H. c-FLIP and the NOXA/Mcl-1 axis participate in the synergistic effect of pemetrexed plus cisplatin in human choroidal melanoma cells. PLoS ONE 12, e0184135 (2017).
Zhang, Q. et al. LiCl induces apoptosis via CHOP/NOXA/Mcl-1 axis in human choroidal melanoma cells. Cancer Cell Int. 21, 96 (2021).
Li, W. et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat. Commun. 10, 3349 (2019).
Katoh, Y. et al. Inhibition of stearoyl-CoA desaturase 1 (SCD1) enhances the antitumor T cell response through regulating β-catenin signaling in cancer cells and ER stress in T cells and synergizes with anti-PD-1 antibody. J. Immunother. Cancer 10, e004616 (2022).
Weiss, J. S. et al. IC3D classification of corneal dystrophies-edition 2. Cornea 34, 117–159 (2015).
Wang, L. et al. CHST6 mutation screening and endoplasmatic reticulum stress in macular corneal dystrophy. Oncotarget 8, 96301–96312 (2017).
Shyam, R., Ogando, D. G. & Bonanno, J. A. Mitochondrial ROS in Slc4a11 KO corneal endothelial cells lead to ER stress. Front. Cell Dev. Biol. 10, 878395 (2022).
Okumura, N. et al. Activation of TGF-β signaling induces cell death via the unfolded protein response in Fuchs endothelial corneal dystrophy. Sci. Rep. 7, 6801 (2017).
Weiss, J. S. et al. The IC3D classification of the corneal dystrophies. Cornea 27, S1–83 (2008).
Aggarwal, S., Peck, T., Golen, J. & Karcioglu, Z. A. Macular corneal dystrophy: a review. Surv. Ophthalmol. 63, 609–617 (2018).
Akama, T. O. et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat. Genet. 26, 237–241 (2000).
Hao, X. D. et al. Association of macular corneal dystrophy with excessive cell senescence and apoptosis induced by the novel mutant CHST6. Exp. Eye Res. 214, 108862 (2022).
Chen, S. et al. Pathophysiological mechanisms of autosomal dominant congenital stromal corneal dystrophy: C-terminal-truncated decorin results in abnormal matrix assembly and altered expression of small leucine-rich proteoglycans. Am. J. Pathol. 179, 2409–2419 (2011).
Chen, S. et al. Intracellularly-retained decorin lacking the C-terminal ear repeat causes ER stress: a cell-based etiological mechanism for congenital stromal corneal dystrophy. Am. J. Pathol. 183, 247–256 (2013).
Skonier, J. et al. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol. 11, 511–522 (1992).
Klintworth, G. K. Advances in the molecular genetics of corneal dystrophies. Am. J. Ophthalmol. 128, 747–754 (1999).
Choi, S. I. et al. Lysosomal trafficking of TGFBIp via caveolae-mediated endocytosis. PLoS ONE 10, e0119561 (2015).
Choi, S. I. et al. 4-Phenylbutyric acid reduces mutant-TGFBIp levels and ER stress through activation of ERAD pathway in corneal fibroblasts of granular corneal dystrophy type 2. Biochem. Biophys. Res. Commun. 477, 841–846 (2016).
Jun, A. S. et al. An alpha 2 collagen VIII transgenic knock-in mouse model of Fuchs endothelial corneal dystrophy shows early endothelial cell unfolded protein response and apoptosis. Hum. Mol. Genet. 21, 384–393 (2012).
Okumura, N. et al. Sustained activation of the unfolded protein response induces cell death in Fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol. Vis. Sci. 58, 3697–3707 (2017).
Jun, D. J. et al. Schnyder corneal dystrophy-associated UBIAD1 is defective in MK-4 synthesis and resists autophagy-mediated degradation. J. Lipid Res. 61, 746–757 (2020).
Choi, S. I. et al. Melatonin reduces endoplasmic reticulum stress and corneal dystrophy-associated TGFBIp through activation of endoplasmic reticulum-associated protein degradation. J. Pineal Res. https://doi.org/10.1111/jpi.12426 (2017).
Kim, E. C., Meng, H. & Jun, A. S. Lithium treatment increases endothelial cell survival and autophagy in a mouse model of Fuchs endothelial corneal dystrophy. Br. J. Ophthalmol. 97, 1068–1073 (2013).
Loganathan, S. K. & Casey, J. R. Corneal dystrophy-causing SLC4A11 mutants: suitability for folding-correction therapy. Hum. Mutat. 35, 1082–1091 (2014).
Kim, E. C., Meng, H. & Jun, A. S. N-Acetylcysteine increases corneal endothelial cell survival in a mouse model of Fuchs endothelial corneal dystrophy. Exp. Eye Res. 127, 20–25 (2014).
Chiu, A. M. et al. High throughput assay identifies glafenine as a corrector for the folding defect in corneal dystrophy-causing mutants of SLC4A11. Invest. Ophthalmol. Vis. Sci. 56, 7739–7753 (2015).
Buono, L. et al. Mesenchymal stem cell-derived extracellular vesicles protect human corneal endothelial cells from endoplasmic reticulum stress-mediated apoptosis. Int. J. Mol. Sci. 22, 4930 (2021).
Santodomingo-Rubido, J. et al. Keratoconus: an updated review. Cont. Lens Anterior Eye 45, 101559 (2022).
Flockerzi, E. et al. Keratoconus staging by decades: a baseline ABCD classification of 1000 patients in the Homburg Keratoconus Center. Br. J. Ophthalmol. 105, 1069–1075 (2021).
Hwang, S., Lim, D. H. & Chung, T. Y. Prevalence and incidence of keratoconus in South Korea: a nationwide population-based study. Am. J. Ophthalmol. 192, 56–64 (2018).
Shinde, V. et al. Mapping keratoconus molecular substrates by multiplexed high-resolution proteomics of unpooled corneas. Omics 23, 583–597 (2019).
Moschos, M. M. et al. Polymorphism analysis of VSX1 and SOD1 genes in Greek patients with keratoconus. Ophthalmic Genet. 36, 213–217 (2015).
Udar, N. et al. SOD1: a candidate gene for keratoconus. Invest. Ophthalmol. Vis. Sci. 47, 3345–3351 (2006).
Dickhout, J. G. et al. Peroxynitrite causes endoplasmic reticulum stress and apoptosis in human vascular endothelium: implications in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 25, 2623–2629 (2005).
Collier, S. A. Is the corneal degradation in keratoconus caused by matrix-metalloproteinases? Clin. Exp. Ophthalmol. 29, 340–344 (2001).
Nelson, K. K. et al. Elevated sod2 activity augments matrix metalloproteinase expression: evidence for the involvement of endogenous hydrogen peroxide in regulating metastasis. Clin. Cancer Res. 9, 424–432 (2003).
Wang, G. et al. The role of autophagy in the pathogenesis of exposure keratitis. J. Cell Mol. Med. 23, 4217–4228 (2019).
Coursey, T. G. et al. Interferon-γ-induced unfolded protein response in conjunctival goblet cells as a cause of mucin deficiency in Sjögren syndrome. Am. J. Pathol. 186, 1547–1558 (2016).
Cui, Y. et al. Mycobacterium bovis induces endoplasmic reticulum stress mediated-apoptosis by activating IRF3 in a murine macrophage cell line. Front. Cell Infect. Microbiol. 6, 182 (2016).
Fangary, S. et al. Nanoparticle fraught liposomes: a platform for increased antibiotic selectivity in multidrug resistant bacteria. Mol. Pharm. 19, 3163–3177 (2022).
Wang, P. et al. High osmotic pressure increases reactive oxygen species generation in rabbit corneal epithelial cells by endoplasmic reticulum. Am. J. Transl. Res. 8, 860–870 (2016).
Cejka, C. & Cejkova, J. Oxidative stress to the cornea, changes in corneal optical properties, and advances in treatment of corneal oxidative injuries. Oxid. Med. Cell Longev. 2015, 591530 (2015).
Woodward, A. M., Di Zazzo, A., Bonini, S. & Argüeso, P. Endoplasmic reticulum stress promotes inflammation-mediated proteolytic activity at the ocular surface. Sci. Rep. 10, 2216 (2020).
Barrera, M. J. et al. Dysfunctional mitochondria as critical players in the inflammation of autoimmune diseases: potential role in Sjögren’s syndrome. Autoimmun. Rev. 20, 102867 (2021).
Hu, S. et al. Lacrimal gland homeostasis is maintained by the AQP5 pathway by attenuating endoplasmic reticulum stress inflammation in the lacrimal gland of AQP5 knockout mice. Mol. Vis. 27, 679–690 (2021).
Krokowski, D. et al. GADD34 function in protein trafficking promotes adaptation to hyperosmotic stress in human corneal cells. Cell Rep. 21, 2895–2910 (2017).
Baird, P. N. et al. Myopia. Nat. Rev. Dis. Prim. 6, 99 (2020).
Holden, B. A. et al. Global prevalence of myopia and high myopia and temporal trends from 2000 through 2050. Ophthalmology 123, 1036–1042 (2016).
Young, T. L., Metlapally, R. & Shay, A. E. Complex trait genetics of refractive error. Arch. Ophthalmol. 125, 38–48 (2007).
Ikeda, S. I. et al. Scleral PERK and ATF6 as targets of myopic axial elongation of mouse eyes. Nat. Commun. 13, 5859 (2022).
Wu, H. et al. Scleral hypoxia is a target for myopia control. Proc. Natl Acad. Sci. USA 115, E7091–e7100 (2018).
Zhao, F. et al. Scleral HIF-1α is a prominent regulatory candidate for genetic and environmental interactions in human myopia pathogenesis. EBioMedicine 57, 102878 (2020).
Zhu, C. et al. Endoplasmic reticulum stress regulates scleral remodeling in a guinea pig model of form-deprivation myopia. J. Ophthalmol. 2020, 3264525 (2020).
Wu, J. et al. Formononetin, an active compound of Astragalus membranaceus (Fisch) Bunge, inhibits hypoxia-induced retinal neovascularization via the HIF-1α/VEGF signaling pathway. Drug Des. Dev. Ther. 10, 3071–3081 (2016).
Lai, M. C. et al. Protective effect of salidroside on cardiac apoptosis in mice with chronic intermittent hypoxia. Int. J. Cardiol. 174, 565–573 (2014).
Yan, X. et al. Accumulation of Asn450Tyr mutant myocilin in ER promotes apoptosis of human trabecular meshwork cells. Mol. Vis. 26, 563–573 (2020).
Sohn, S. et al. Expression of wild-type and truncated myocilins in trabecular meshwork cells: their subcellular localizations and cytotoxicities. Invest. Ophthalmol. Vis. Sci. 43, 3680–3685 (2002).
Fingert, J. H. et al. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum. Mol. Genet. 8, 899–905 (1999).
Yam, G. H., Gaplovska-Kysela, K., Zuber, C. & Roth, J. Sodium 4-phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Invest. Ophthalmol. Vis. Sci. 48, 1683–1690 (2007).
Mayerl, S. J. et al. Human retinal organoids harboring IMPG2 mutations exhibit a photoreceptor outer segment phenotype that models advanced retinitis pigmentosa. Stem Cell Rep. 17, 2409–2420 (2022).
Yang, J. et al. Differences in unfolded protein response pathway activation in the lenses of three types of cataracts. PLoS ONE 10, e0130705 (2015).
Shiels, A., Bennett, T. M. & Hejtmancik, J. F. Cat-Map: putting cataract on the map. Mol. Vis. 16, 2007–2015 (2010).
Liu, Q. et al. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharm. Res. 152, 104595 (2020).
Shen, D. et al. Therapeutic potential of targeting SHP2 in human developmental disorders and cancers. Eur. J. Med. Chem. 190, 112117 (2020).
Zhu, Y. et al. Allosteric inhibition of SHP2 uncovers aberrant TLR7 trafficking in aggravating psoriasis. EMBO Mol. Med. 14, e14455 (2022).
O’Neil, P. M. et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 392, 637–649 (2018).
Tamborlane, W. V. et al. Liraglutide in children and adolescents with type 2 diabetes. N. Engl. J. Med. 381, 637–646 (2019).
Selvarani, R., Mohammed, S. & Richardson, A. Effect of rapamycin on aging and age-related diseases-past and future. Geroscience 43, 1135–1158 (2021).
Xing, X. et al. PP242 suppresses cell proliferation, metastasis, and angiogenesis of gastric cancer through inhibition of the PI3K/AKT/mTOR pathway. Anticancer Drugs 25, 1129–1140 (2014).
Rashid, M. M., Lee, H. & Jung, B. H. Evaluation of the antitumor effects of PP242 in a colon cancer xenograft mouse model using comprehensive metabolomics and lipidomics. Sci. Rep. 10, 17523 (2020).
Pang, J. et al. Mitochondrial ALDH2 protects against lipopolysaccharide-induced myocardial contractile dysfunction by suppression of ER stress and autophagy. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1627–1641 (2019).
Xu, J., Zhao, H. & Wang, T. Suppression of retinal degeneration by two novel ERAD ubiquitin E3 ligases SORDD1/2 in Drosophila. PLoS Genet. 16, e1009172 (2020).
Acknowledgements
Figures 1–6 are created with BioRender.com. In addition, we have acquired the publication licenses for each figure. We are thankful to many scientists in the field whose seminal works are not cited owing to space limitations. We gratefully acknowledge the help of our supervisors, Professor Xiaobo Xia and Professor Siqi Xiong, who have offered us valuable suggestions in the academic studies. We are thankful to many colleagues and friends who gave professional suggestions to this work. We want to show our appreciation to our beloved parents for their long-lasting love and support.
Funding
This study was financially supported by The National Key Research and Development Program of China (grant number 2020YFC2008205), The National Natural Science Foundation of China (grant numbers 82171058, 81974134, and 81974137), The Key R&D Plan of Hunan Province of China (No. 2020SK2076) and Natural Science Foundation of Hunan Province (No. 2019JJ40507 and 2023JJ30956).
Author information
Authors and Affiliations
Contributions
X.C. and C.S. wrote the manuscript and X.C. contributed to the writing initially. X.C. prepared Figs. 1–6. X.C., C.S., and M.H. collected literature. S.X., X.X., and X.C. conceptualized the manuscript. X.X. and S.X. provided guidance and significant advice throughout the drafting of the manuscript. Corresponding authors X.X. and S.X. provided financial support and supervision. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, X., Shi, C., He, M. et al. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Sig Transduct Target Ther 8, 352 (2023). https://doi.org/10.1038/s41392-023-01570-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01570-w
This article is cited by
-
Construction of an ER stress-related prognostic signature for predicting prognosis and screening the effective anti-tumor drug in osteosarcoma
Journal of Translational Medicine (2024)
-
Integrated proteomics reveals autophagy landscape and an autophagy receptor controlling PKA-RI complex homeostasis in neurons
Nature Communications (2024)
-
Retinoids and EZH2 inhibitors cooperate to orchestrate anti-oncogenic effects on bladder cancer cells
Cancer Gene Therapy (2024)
-
Transcriptional patterns of human retinal pigment epithelial cells under protracted high glucose
Molecular Biology Reports (2024)
-
Reduced Proteolipid Protein 2 promotes endoplasmic reticulum stress-related apoptosis and increases drug sensitivity in acute myeloid leukemia
Molecular Biology Reports (2024)
