
Abstract
Genetic classification helps to disclose molecular heterogeneity and therapeutic implications in diffuse large B-cell lymphoma (DLBCL). Using whole exome/genome sequencing, RNA-sequencing, and fluorescence in situ hybridization in 337 newly diagnosed DLBCL patients, we established a simplified 38-gene algorithm (termed ‘LymphPlex’) based on the information on mutations of 35 genes and rearrangements of three genes (BCL2, BCL6, and MYC), identifying seven distinct genetic subtypes: TP53Mut (TP53 mutations), MCD-like (MYD88, CD79B, PIM1, MPEG1, BTG1, TBL1XR1, PRDM1, IRF4 mutations), BN2-like (BCL6 fusion, NOTCH2, CD70, DTX1, BTG2, TNFAIP3, CCND3 mutations), N1-like (NOTCH1 mutations), EZB-like (BCL2 fusion, EZH2, TNFRSF14, KMT2D, B2M, FAS, CREBBP, ARID1A, EP300, CIITA, STAT6, GNA13 mutations, with or without MYC rearrangement), and ST2-like (SGK1, TET2, SOCS1, DDX3X, ZFP36L1, DUSP2, STAT3, IRF8 mutations). Extended validation of 1001 DLBCL patients revealed clinical relevance and biological signature of each genetic subtype. TP53Mut subtype showed poor prognosis, characterized by p53 signaling dysregulation, immune deficiency, and PI3K activation. MCD-like subtype was associated with poor prognosis, activated B-cell (ABC) origin, BCL2/MYC double-expression, and NF-κB activation. BN2-like subtype showed favorable outcome within ABC-DLBCL and featured with NF-κB activation. N1-like and EZB-like subtypes were predominated by ABC-DLBCL and germinal center B-cell (GCB)-DLBCL, respectively. EZB-like-MYC+ subtype was characterized by an immunosuppressive tumor microenvironment, while EZB-like-MYC- subtype by NOTCH activation. ST2-like subtype showed favorable outcome within GCB-DLBCL and featured with stromal-1 modulation. Genetic subtype-guided targeted agents achieved encouraging clinical response when combined with immunochemotherapy. Collectively, LymphPlex provided high efficacy and feasibility, representing a step forward to the mechanism-based targeted therapy in DLBCL.
Similar content being viewed by others

Introduction
Diffuse large B-cell lymphoma (DLBCL), the most common subtype of non-Hodgkin’s lymphoma, represents a molecularly heterogeneous entity.1,2,3 DLBCL is potentially curable with immunochemotherapy using rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), but recurrent or progressive disease occurs in approximately 40% of the patients.4,5 Attempts to improve the treatment outcome by combining standard immunochemotherapy with promising novel agents targeting specific pathways have encountered difficulties likely due to the biological complexity of DLBCL.6,7 It is thus highly desirable to further reveal molecular heterogeneity of DLBCL by simplified algorithm, that may efficiently be translated for therapeutic use, leading to improved accuracy of outcome prediction and efficacy of the targeted therapy.
As for the classification of DLBCL, a strategy to categorize DLBCL is based upon the detection of rearrangements of MYC, BCL2 and/or BCL6 by fluorescent in-situ hybridization (FISH).2 The most recent revision of the WHO classification of lymphoid neoplasms recognizes the high-grade B-cell double-/triple-hit lymphoma, with MYC rearrangement and BCL2 and/or BCL6 rearrangements, as a particular entity associated with aggressive behavior and inferior outcome.8 Gene expression profiling allows to distinguish three molecular subtypes based on cell of origin (COO), dividing DLBCL cases into activated B-cell like (ABC), germinal center B-cell like (GCB), and unclassified subtypes.9,10 The COO distinction has been proved of prognostic value, as patients with ABC-DLBCL generally show less favorable responses to standard therapy than those with GCB-DLBCL.11 Also, the COO categorization helps to understand the heterogeneous responses of patients with DLBCL to targeted therapies such as ibrutinib, a novel therapeutic agent targeting B-cell receptor (BCR) signaling pathway.12 Since the COO distinction does not solely account for the divergent treatment outcomes of patients with DLBCL following either R-CHOP or targeted therapies, additional genetic complexity remains to be defined.
Genomic studies can illustrate the genetic alterations of DLBCL and facilitate the categorization of genetic subtypes.13,14,15,16,17 The GenClass algorithm first identifies four distinct genetic subtypes, known as MCD (based on co-occurrence of MYD88L265P and CD79B mutations), BN2 (based on BCL6 fusions and NOTCH2 mutations), N1 (based on NOTCH1 mutations), and EZB (based on EZH2 mutations and BCL2 fusions).15 Recently, the LymphGen algorithm has identified additional subtypes, including EZB-MYC (EZB subtype with or without MYC rearrangements), ST2 (based on SGK1 and TET2 mutations), and A53 (based on TP53 mutations and deletions).17 Meanwhile, five molecular clusters related to COO are defined: two ABC-DLBCL groups (one with low risk and possible marginal zone origin (C1) and the other a high-risk group (C5) enriched in cases with mutations in MYD88 and CD79B), two GCB-DLBCL groups (one with a favorable (C4) and the other with a poor (C3) outcome), and an ABC/GCB-independent group (C2) with biallelic inactivation of TP53 and copy number changes.13 Furthermore, using clustering techniques on targeted sequencing data allows characterization of resulting clusters according to the genetic features most enriched in each cluster, for example, identification and characterization of NOTCH2-, MYD88-, BCL2-, TET2/SGK1-, and SOCS1/SGK1-assoicated clusters.14 Interestingly, the above genetic subtypes share partially overlapping classification systems. The MCD subtype (comparable with C5 or MYD88-associated subtype) is associated with ABC-DLBCL, contributing to hyperactivation of NF-κB with poor prognosis.13,14,15,17 The BN2 subtype (comparable with C1 or NOTCH2-associated subtype) includes mutations targeting the BCR-dependent NF-κB pathway.13,14,15,17 The EZB subtype (comparable with C3 or BCL2-associated subtype) is related to GCB-DLBCL, exhibiting frequent mutations in chromatin modifiers with favorable outcome.13,14,15,17 These findings suggest that genetic subtyping helps reveal the diversity of DLBCL in oncogenic pathway engagement, gene expression profile, prognostic value, and therapeutic vulnerabilities.
Despite the progress made, challenges remain in clinical practice regarding genetic subtyping. The high demand of specimen quality for sequencing, the relatively long duration awaiting whole exome sequencing (WES)/whole genome sequencing (WGS) bioinformatics analysis, and the complexity of clustering algorithms limited its broad application.13,14,15,17 To improve the feasibility in clinical use and permit the timely use of targeted agents based on genetic subtypes, there is a great demand to develop simplified but efficient methods for genetic subtyping. Previously, we developed a 20-gene algorithm using the information on mutations of 18 genes and rearrangements of two genes (BCL2 and BCL6) based on the GenClass algorithm,15 and conducted a prospective, phase 2, randomized trial, to explore genetic subtype-guided targeted agents plus R-CHOP (R-CHOP-X) immunochemotherapy in newly diagnosed DLBCL patients (Guidance-1, NCT04025593).18 Here we further developed a 38-gene algorithm (termed ‘LymphPlex’) using information on mutations of 35 genes and rearrangements of three genes (BCL2, BCL6, and MYC) to identify seven genetic subtypes based on the LymphGen algorithm,17 including the TP53 mutated (TP53Mut), MCD-like, BN2-like, N1-like, EZB-MYC+-like, EZB-MYC--like, and ST2-like. We also explore the clinicopathological, prognostic, molecular, and microenvironmental attributes of these genetic subtypes in a large cohort of 1001 patients with newly diagnosed DLBCL. The LymphPlex algorithm may provide a useful tool for genetic subtyping, thereby facilitating the optimization of treatment strategies for DLBCL in the era of precision medicine.
Results
Simplified LymphPlex algorithm established for genetic subtyping in DLBCL
Patient baseline characteristics were summarized in Supplementary Table 1. Among the Ruijin cohort of 1001 patients, 543 (54.2%) were male, 424 (42.4%) were >60 yr, 104 (10.4%) had poor performance status, 467 (46.7%) had elevated serum lactate dehydrogenase (LDH), 457 (45.7%) had advanced Ann Arbor stage, and 266 (26.6%) had multiple extranodal involvement. Accordingly, 516 (51.5%) were at low-risk and 485 (48.5%) were at intermediate- or high-risk, as defined by International Prognostic Index (IPI). Two hundred and forty-three (25.6%) were BCL2/MYC double-expressor lymphoma. As for COO classification, 148 (31.2%) were GCB-DLBCL, 235 (49.5%) were ABC-DLBCL, and 92 (19.4%) were unclassified subtype. Comparing with the training cohort, the validation cohorts14,19 had increased prevalence of elderly patients and poor performance status. Patients with intermediate/high- or high-risk IPI and GCB-DLBCL were also increased.
We first applied the LymphPlex algorithm based on the LymphGen algorithm17 in the training cohort with WES or WGS data (n = 337) to determine the genetic subtype classifier. The following genetic subtypes were identified: mutations in TP53 for TP53Mut; mutations in MYD88, CD79B, PIM1, MPEG1, BTG1, TBL1XR1, PRDM1, and IRF4 for MCD-like; fusion of BCL6 (FBCL6) and mutations in NOTCH2, CD70, DTX1, BTG2, TNFAIP3, and CCND3 for BN2-like; mutations in NOTCH1 for N1-like; fusion of BCL2 (FBCL2) and mutations in EZH2, TNFRSF14, KMT2D, B2M, FAS, CREBBP, ARID1A, EP300, CIITA, STAT6, and GNA13 for EZB-like (with or without MYC fusion); and mutations in SGK1, TET2, SOCS1, DDX3X, ZFP36L1, DUSP2, STAT3, and IRF8 for ST2-like subtype (Fig. 1). Among 337 patients with WES/WGS data, 171 patients (50.7%) were categorized into genetic subtypes, with the remaining 166 patients (49.3%) classified as not otherwise specified (NOS). Of the 171 classified cases, 44 cases (25.7%) were referred as TP53Mut, 37 cases (21.6%) as MCD-like, 39 cases (22.8%) as BN2-like, 12 cases (7.0%) as N1-like, 19 cases (11.1%) as EZB-like, and 20 cases (11.7%) as ST2-like subtype. Within the EZB-like subtype, three (15.8%) had MYC fusion. In terms of our previous 20-gene algorithm based on the GenClass algorithm,15 five genetic subtypes were identified: mutations in TP53 for the TP53Mut; mutations in MYD88, CD79B, PIM1, MPEG1, BTG1, and TBL1XR1 for MCD-like; FBCL6 and mutations in NOTCH2, TNFAIP3, CD70, and DTX1 for BN2-like; mutations in NOTCH1 for N1-like; and FBCL2 and mutations in EZH2, TNFRSF14, CREBBP, EP300, MTOR, and STAT6 for EZB-like (Supplementary Fig. 1a). The newly developed LymphPlex was in principle consistent with our previous algorithm.18 The 17 previously unclassified patients were assigned to specific subtype by using the LymphPlex algorithm: six were assigned to BN2-like with mutations in BTG2 and CCND3, one was assigned to EZB-like with mutations in KMT2D, B2M, FAS, ARID1A, CIITA, and GNA13, and 10 were assigned to the newly identified ST2-like subtype (Supplementary Fig. 1b).

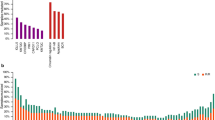
Genetic subtypes based on the LymphPlex algorithm derived from the Ruijin cohort with WES/WGS data (n = 337). Overall landscape of genetic subtypes based on the LymphPlex algorithm derived from the Ruijin cohort with WES/WGS and FISH data (n = 337). Shown on the left panel are genetic features in DLBCL samples that have been classified into each subtype. Shown on the right panel are the mutation rates of corresponding genes. Shown at bottom are MYC rearrangement, COO classification including GCB-DLBCL, ABC-DLBCL, and unclassified subtype, as well as BCL2/MYC double-expression (DE) status. GCB germinal center B-cell, ABC activated B-cell, UNC Unclassified
Evaluation of the LymphPlex algorithm
We applied both the LymphPlex and the LymphGen17 algorithm on the 337 patients with WES/WGS data in the Ruijin cohort. All WES/WGS mutations and copy number variants were used as the input for LymphGen. The LymphGen results were utilized as performance benchmarking. The LymphPlex algorithm assigned a genetic subtype in 50.7% (171/337) cases, while the LymphGen algorithm assigned a genetic subtype in 35.6% (120/337) cases (Fig. 2a). One hundred and six cases were classified into a unique genetic subtype by both algorithms (Fig. 2b). Excluding the TP53Mut cases from LymphPlex and the A53 cases from LymphGen, we calculated the sensitivity, specificity, and precision (positive predictive value) of the LymphPlex algorithm for the subtype assignments in the remaining 87 cases (Fig. 2c). The LymphPlex algorithm performed acceptably, with sensitivity all above 94%, specificity all above 99%, and precision all above 90% (Fig. 2d). The LymphPlex subtypes generally belonged to the corresponding LymphGen subtypes.

Comparison of the LymphPlex and LymphGen algorithms. a Sankey plots showing comparison of the LymphPlex and the LymphGen algorithms in the 337 patients with WES/WGS data including copy number data in the Ruijin cohort or b in the 106 cases for whom a unique genetic subtype was assigned by both algorithms. c Confusion matrixes showing numbers of cases for each subtype assigned by LymphPlex and LymphGen. d Sensitivity, specificity, and precision (positive predictive value (PPV)) of the LymphPlex algorithm for the subtype assignments, as compared to the assignments using the LymphGen algorithm
Genetic and phenotypic attributes of DLBCL genetic subtypes
To evaluate the reproducibility of the LymphPlex algorithm, we analyzed the genetic and phenotypic features of the LymphPlex-assigned subtypes in the Ruijin cohort (n = 1001), BCC cohort (n = 320), and HMRN cohort (n = 928). The distributions of the LymphPlex-assigned subtypes were compared among three cohorts. Comparing with BCC cohort and HMRN cohort, increased incidence of the BN2-like and N1-like subtypes, but decreased incidence of the EZB-like subtype, was observed in Ruijin cohort (Fig. 3a and Supplementary Table 2). Of note, similar distribution pattern of genetic subtypes was observed in both low-risk (IPI 0–1) and intermediate- and high-risk (IPI 2–5) patients (Fig. 3b), indicating that different incidence of genetic subtypes was not resulted from uneven IPI risk group distribution among three cohorts. The clinical characteristics associated with each genetic subtype in the Ruijin cohort were also summarized in Supplementary Table 3. MCD-like had increased, while N1-like had decreased number of elderly patients. TP53Mut and MCD-like had increased, while ST2-like had decreased prevalence of elevated serum LDH. MCD-like had increased, while ST2-like had decreased prevalence of multiple extranodal involvement. Accordingly, patients with intermediate/high- or high- risk IPI were also increased in MCD-like, while decreased in ST2-like.

Genetic and phenotypic attributes of DLBCL genetic subtypes. a Distribution of genetic subtypes based on the LymphPlex algorithm. b Distribution of genetic subtypes according to International Prognostic Index (IPI). c Prevalence of COO subgroups among genetic subtypes. GCB germinal center B-cell, ABC activated B-cell, UNC Unclassified. d Prevalence of BCL2/MYC expression (DE) status among genetic subtypes. e Prevalence of the indicated genetic features among genetic subtypes defined in the BCC and HMRN cohorts, as compared to the Ruijin cohort
Regarding COO classification, the majority of MCD-like patients were ABC-DLBCL, whereas most EZB-like patients were GCB-DLBCL (Fig. 3c), consistent with relatively lower incidence of GCB-DLBCL in Chinese patients.14,19 BCL2/MYC double-expression was frequently observed in the MCD-like subtype (Fig. 3d). The compositions of COO and BCL2/MYC double-expression (Fig. 3c, d) within the genetic subtypes were comparable among three cohorts, with gene mutations and BCL2/BCL6 rearrangements as well (Fig. 3e). To evaluate this genetic coherence, we computed subtype-associated scores using the particular gene sets and compared the scores between gene sets, in which the subtype-defining genetic features were present or absent. In this analysis, we observed significant similarity in the genetic coherence of features defining the MCD-like, BN2-like, EZB-like, and ST2-like subtypes in the training cohort and two validation cohorts (P ≤ 1.3 × 10–9, Supplementary Table 4). The TP53Mut and N1-like subtypes could not be evaluated by this method since TP53Mut and N1-like subtypes were dominated by TP53 mutations and NOTCH1 mutations, respectively.
Prognostic value of DLBCL genetic subtypes
We next examined the prognostic impact of the LymphPlex-assigned genetic subtypes in each cohort. Survival analysis was performed on the patients received R-CHOP. The TP53Mut and MCD-like subtypes presented relatively inferior progression-free survival (PFS), as compared to the BN2-like, EZB-like, and ST2-like subtypes (Fig. 4a). Among GCB-DLBCL, the ST2-like subtype was relatively favorable; among ABC-DLBCL, the BN2-like subtype was relatively favorable (Supplementary Fig. 2). Given these consistent trends in survival, we integrated data from all three cohorts to estimate combined hazard ratios (Fig. 4b). In this model, PFS of the TP53Mut subtype was inferior to the TP53WT patients (P < 0.0001); PFS of the MCD-like subtype was inferior to all non-MCD-like subtypes (P = 0.0038); PFS of the ST2-like subtype was superior to all non-ST2-like subtypes within GCB-DLBCL (P = 0.0005); PFS of the BN2-like subtype was superior to all non-BN2-like subtypes within ABC-DLBCL (P = 0.0233). In the Ruijin cohort, we sub-divided the EZB-like subtype according to MYC rearrangement. Within the EZB-like subtype, PFS of the MYC+ cases were significantly inferior to that of the MYC- cases (P = 0.0003, Fig. 4c). The MYC+ subset presented relatively higher IPI risk and more BCL2/MYC double-expression cases (Supplementary Fig. 3a, b). All cases in the MYC+ subset were GCB-DLBCL, while only 66.7% cases in the MYC- subset were GCB-DLBCL (25.0% were ABC-DLBCL and 8.3% were unclassified subtype, Supplementary Fig. 3c). The gene alterations were similar in the MYC+ and MYC- subsets of the EZB-like subtype (Supplementary Fig. 3d). As for the overall survival (OS), upon R-CHOP treatment, TP53Mut patients showed unfavorable outcome in the Ruijin cohort (P = 0.0153), BCC cohort (P = 0.0042), and HMRN cohort (P = 0.0007), as compared to TP53WT patients (Supplementary Fig. 4a). Furthermore, the OS showed significant differences among genetic subtypes assigned by the LymphPlex algorithm in the BCC cohort and HMRN cohort (P = 0.0005 and P < 0.0001, respectively), but not in the Ruijin cohort, probably because 27.0% of the patients had a follow-up less than 5 years (Supplementary Fig. 4b).

Prognostic attributes of DLBCL genetic subtypes. a Kaplan–Meier plots for PFS in all cases of the indicated DLBCL cohorts. b Hazard ratios (-log2 transformed) for the comparisons between genetic subtypes in the indicated DLBCL cohorts. c Kaplan–Meier plots for PFS in the EZB-like cases of the Ruijin cohort
Given the distinct prognostic value of the TP53Mut subtype, we further investigated the genetic signature of the TP53Mut subtype. Among the 147 TP53Mut patients in the Ruijin cohort, 116 patients (78.9%) possessed mutations in the DNA binding domain (DBD), 16 patients (10.9%) possessed mutations in the non-DBD, and 15 patients (10.2%) possessed multiple TP53 mutations (Supplementary Fig. 5a). Survival analysis was performed on the patients received R-CHOP. No differences on PFS were observed in the patients with DBD and non-DBD TP53 mutations (Supplementary Fig. 5b). To determine the clinical and biological impact of TP53 mutations on DLBCL, we analyzed the molecular alterations accompanied with TP53 mutations (Supplementary Fig. 5c). TP53 mutations were associated with increased frequency of KMT2D mutations in all three cohorts. Additionally, in the Ruijin cohort, TP53Mut patients had significantly increased mutations in EP300 and NOTCH2, but decreased mutations in SOCS1 and CD70. In the BCC cohort, TP53Mut patients had significantly decreased APF36L1 mutations. In the HMRN cohort, TP53Mut patients had significantly increased mutations in EP300 and NOTCH1, but decreased mutations in MYD88, EZH2, and IRF4. With respect to gene functions, in the Ruijin cohort, TP53Mut patients presented decreased mutations associated with response to interferon-γ. In the HMRN cohort, TP53Mut patients had decreased mutations associated with T cell activation, but increased mutations associated with Histone/DNA methylation (Supplementary Fig. 5d and Supplementary Table 5).
Gene expression pattern of DLBCL genetic subtypes
To better understand the biological signatures of genetic subtypes, we analyzed gene expression profile using RNA sequencing (RNA-seq) data of 475 patients in the Ruijin cohort (Fig. 5). The subtypes differed with respect to various biological processes, with the TP53Mut and MCD-like subtypes highly expressing signatures of cell proliferation and MYC oncoprotein, with the TP53Mut and EZB-like-MYC+ subtypes expressing low level signatures of quiescence. Metabolic distinctions among genetic subtypes included highly expressing signatures of glycolytic pathway in the TP53Mut, MCD-like, and BN2-like subtypes, and high expressing signatures of lipid synthesis in the TP53Mut subtype.

Gene expression signature according to DLBCL genetic subtypes. Shown is average normalized expression of signature genes in each subtype versus other DLBCL samples in the Ruijin cohort (n = 475)
The subtypes also appeared to vary from different B cell stage. The MCD-like subtype revealed a predominance of ABC-DLBCL, while the EZB-like and ST2-like subtypes expressed signatures of GCB-DLBCL. The BN2-like subtype was consisted of both GCB- and ABC-DLBCL. The MCD-like and ST2-like subtypes had low expression of plasma cell signatures and memory B cell signatures, respectively. Among transcriptional factors, genes induced by IRF4 were overexpressed in the MCD-like and BN2-like subtypes, genes induced by OCT2 were highly expressed in the MCD-like and EZB-like-MYC+ subtypes, genes repressed by BCL6 were lowest in the TP53Mut and EZB-like-MYC+ subtypes, and genes transactivated by TCF3 were highest in the EZB-like subtype. With respect to oncogenic pathways, NF-κB signaling was activated in the MCD-like and BN2-like subtypes, but not in the EZB-like-MYC+ subtype. The expression of p53-target genes was lowest in the TP53Mut subtype. NOTCH signaling was activated in the BN2-like and EZB-like-MYC- subtypes. PI3K signaling was activated in the TP53Mut subtype. JAK2 signaling was activated in the MCD-like and BN2-like subtypes.
The tumor microenvironment alterations of genetic subtypes were also strikingly discordant: the TP53Mut subtype generally presented low expression of all immune signatures, the MCD-like subtype presented low expression of T cell signatures, the EZB-like-MYC+ subtype had low expressing CD8+T cell signatures, while the EZB-like-MYC– subtype had high expressing GC TFH and CD4+T cell signatures. The stromal-1 signatures, which are prognostically favorable and reflect a fibrotic, macrophage-rich microenvironment, were upregulated in the ST2-like subtype, consistent with their relatively favorable outcomes.
To investigate the potential effect of the LymphPlex algorithm on mechanism-based targeted therapy in DLBCL, we applied R-CHOP in combination with different targeted agents based on the LymphPlex algorithm (R-CHOP-X). Indeed, the TP53Mut patients were treated with intravenous decitabine, the MCD-like, BN2-like, and N1-like patients were treated with BTK inhibitors (ibrutinib, zanubrutinib, or orelabrutinib), the EZB-like, ST2-like, and NOS patients were treated with lenalidomide (Supplementary Fig. 6a). Forty-eight patients were available for response assessment, including 13 TP53Mut, 12 MCD-like, 10 BN2-like, one N1-like, one EZB-like, one ST2-like, and 10 NOS patients (Supplementary Table 6). The overall and complete response rates were 91.7% and 83.3%, respectively (Supplementary Fig. 6b). In terms of genetic subtypes, the overall response rates of the TP53Mut, MCD-like, BN2-like, N1-like, EZB-like, ST2-like, and NOS patients were 92.3% (12/13), 91.7% (11/12), 100% (10/10), 100% (1/1), 100% (1/1), 100% (1/1), and 80.0% (8/10), respectively; the complete response rates of the TP53Mut, MCD-like, BN2-like, N1-like, EZB-like, ST2-like, and NOS patients were 76.9% (10/13), 83.3% (10/12), 90.0% (9/10), 100% (1/1), 100% (1/1), 100% (1/1), and 80.0% (8/10), respectively (Supplementary Fig. 6c).
Discussion
In the present study, we successfully developed a simplified LymphPlex algorithm and defined distinct genetic subtypes, termed as TP53Mut, MCD-like, BN2-like, N1-like, EZB-like-MYC+, EZB-like-MYC-, and ST2-like. The clustering of the LymphPlex algorithm showed a good concordance with that of the LymphGen algorithm. Integrating transcriptomic profiling with these genetic subtypes, we provided further evidence of molecular features and suggested potential targeted approaches in DLBCL.
TP53 mutations are critically involved in tumor progression and indicate poor prognosis in DLBCL.20,21,22 Despite diversity in gene mutations correlated with TP53 mutations among the studied cohorts, we showed similar alterations of response to interferon-γ and T cell activation, indicative of an essential role of TP53 mutations on immune evasion.23 Accordingly, gene expression pattern confirmed dysregulation of p53 signaling and deficiency of anti-tumor immunity. The TP53Mut subtype generally presented low expression of immune signatures including T cells, NK cells, macrophages, and dendritic cells. As reported in our recent phase 1/2 study, decitabine improved clinical efficacy of R-CHOP on TP53Mut DLBCL through targeting tumor immune microenvironment.23,24 Given the above evidence that TP53 mutations had significant biological features and specific response to decitabine treatment, we grouped patients with TP53 mutations as a specific entity of DLBCL, different from previous subtyping as A53 and C2.13,17 Meanwhile, PI3K signaling and lipid synthesis were altered, suggesting that PI3K inhibitors and lipid metabolism reprogramming may also be effective options for the TP53Mut-subtype DLBCL.25,26
The MCD-like subtype was characterized by the co-occurrence of MYD88 and CD79B mutations.13,14,17 We found that the MCD-like subtype was associated with ABC-DLBCL, BCL2/MYC double-expression, and poor prognosis. Subsequent investigation using gene expression data showed strong enrichment in ABC-DLBCL signatures, NF-κB activation, as well as IRF4 and MYC upregulation.17,27 BTK inhibitors targeting BCR and NF-κB signaling are promising agents for the MCD-like subtype.12,28 Recent report from the PHOENIX trial further proved that addition of ibrutinib improves the survival of young DLBCL patients with the MCD subtype.29 The BN2-like subtype was featured by FBCL6 and NOTCH2 mutations and comprised of ABC-, GCB- and unclassified DLBCL.13,14,17 In our cohort, the BN2-like subtype presented favorable outcome among ABC-DLBCL patients, consistent with the results from the NCI study.17 Gene expression profiling also revealed the signatures of BCR-dependent NF-κB activation, indicating the potential response to BTK inhibitors.17 The N1-like subtype was dominated by NOTCH1 mutations, mainly consisted of ABC-DLBCL, and exhibited poor prognosis.14 Limited biological feature and therapeutic targets were disclosed, however, ibrutinib was reported to bring survival benefit in N1 subtype according to the PHOENIX trial.29
The EZB-like subtype was characterized by FBCL2 and EZH2 mutations.13,14,17 This group revealed a predominance of GCB-DLBCL and was generally associated with favorable outcome. According to MYC rearrangement, the EZB-like-MYC+ subtype was distinguished by poor prognosis and CD8+T cell deficiency within tumor microenvironment. Lenalidomide showed survival benefit in ABC-DLBCL and may also had an impact on immunomodulation in the EZB-like-MYC+ subtype.7,30,31 In the EZB-like-MYC- DLBCL, NOTCH pathway was activated, suggesting possible effect of NOTCH inhibitors. In addition, EZH2 inhibitors may be effective in treating the EZB-like DLBCL patients bearing EZH2 mutations via suppressing methyltransferase activity.32 The ST2-like subtype was distinguished by mutations in SGK1, TET2, and SOCS1.13,14,17 Predominantly GCB-DLBCL in origin, the ST2-like subtype was associated with favorable outcome. With respect to gene expression profile, the expression of stromal signatures was increased in the ST2-like subtype, which could be related to stroma modulation induced by SOCS1 mutations and resulted in attenuated tumor growth.33 Moreover, TET2 mutations were reported to be involved in T-cell deficiency. These results may indicate potential efficacy of lenalidomide in ST2-like DLBCL through modulating tumor immune microenvironment.34 To try to translate LymphPlex into clinical practice, we applied R-CHOP in combination with different targeted agents selected by LymphPlex to treat 48 patients with newly diagnosed DLBCL and showed encouraging results. A prospective, phase 3, randomized trial of genetic subtype-guided R-CHOP-X immunochemotherapy in newly diagnosed DLBCL (Guidance-02, NCT05351346) is ongoing.
In conclusion, the simplified LymphPlex algorithm of genetic subtyping displayed high efficacy and clinical practicability in DLBCL. LymphPlex is implemented with a small number of gene alterations revealed by targeted sequencing and FISH and can be applied to paraffin biopsies instead of frozen biopsies, which reveals a good availability for clinical use. Also, the LymphPlex results can be output using targeted sequencing data instead of WES/WGS data, which simplifies the process of data analysis and ensure its application in multicenter clinical trials and timely use of targeted agents based on genetic subtypes. This simple but efficient algorithm facilitated to disclose molecular heterogeneity in DLBCL, thereby contributing to the design of the mechanism-based therapy in the era of precision medicine.
Materials and methods
Patients and procedures
This study was conducted on 1001 patients with newly diagnosed DLBCL (Fig. 6). The patients were included from May 2006 to December 2020, with the last follow-up through September 2021. Histological diagnosis was established according to World Health Organization (WHO) Classification.35 WES and WGS were performed on 228 and 109 patients with available tumor samples, respectively. The remaining 664 cases were analyzed with targeted sequencing data covering 35 lymphoma-associated genes (ARID1A, B2M, BTG1, BTG2, CCND3, CD70, CD79B, CIITA, CREBBP, DDX3X, DTX1, DUSP2, EP300, EZH2, FAS, GNA13, IRF4, IRF8, KMT2D, MPEG1, MYD88, NOTCH1, NOTCH2, PIM1, PRDM1, SGK1, SOCS1, STAT3, STAT6, TBL1XR1, TET2, TNFAIP3, TNFRSF14, TP53, and ZFP36L1), as well as rearrangements in BCL2, BCL6, and MYC. A total of 475 patients were available for RNA-seq, including 207 cases of WES/WGS group and 268 cases of targeted sequencing group.

Flow chart of the study. DLBCL diffuse large B-cell lymphoma, WES whole-exome sequencing, WGS whole-genome sequencing, R-CHOP rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
Clinical parameters included gender, age, Eastern Cooperative Oncology Group performance status, Ann Arbor stage, serum LDH, and number of extranodal involvement. As for BCL2/MYC double-expressor lymphoma, cut-off value of BCL2 and MYC were 50% and 40%, respectively.35 COO classification including GCB-DLBCL, ABC-DLBCL, and unclassified subtype was determined using RNA-seq.36 Rearrangements of BCL2, BCL6, and MYC were assessed by FISH. Survival analysis was performed on 730 patients received R-CHOP immunochemotherapy, excluding those received immunochemotherapy other than R-CHOP (n = 227) or chemo-free regimen (n = 44). The study was approved by the Shanghai Ruijin Hospital Review Board, and informed consent was obtained in accordance with the Declaration of Helsinki.
Sample processing
Genomic DNA (gDNA) was extracted from frozen tumor tissues using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), or from formalin-fixed paraffin-embedded (FFPE) tumor tissues using a GeneRead DNA FFPE Tissue Kit (Qiagen, Hilden, Germany), based on the manufacturer’s guidelines. The concentration of gDNA was measured using Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), and the gDNA was quantified by Qsep100 System (BIOptic, China).
Total RNA was extracted from frozen tumor tissues using Trizol and RNeasy Mini Kit (Qiagen, Hilden, Germany). RNA quantity was assessed on Nanodrop, and the integrity of total RNA was estimated by RNA 6000 Nano Kit on Aligent 2100 Bioanalyzer.
Whole-exome sequencing (WES)/whole-genome sequencing (WGS)
WES was performed on the frozen tumor tissues of 146 patients and the FFPE tumor tissues of 82 patients. Whole exome capture was performed using the SeqCap EZ Human Exome kit (version 3·0), and sequencing was performed on HiSeq 4000 platform with 150 bp paired-end strategy (Shanghai Rightongene Biotechnology Co., Ltd, Shanghai, China). WGS was performed on the frozen tumor tissues of 109 patients. Detailly, library was validated by Agilent 2100 Bioanalyzer, and sequencing was performed on Illumina HiSeq platform with 150 bp paired-end strategy in WuXi NextCODE (Shanghai, China). Also, 42 randomly selected matched peripheral blood samples were submitted to WES (n = 25, divided into five groups) and WGS (n = 17) to build a somatic mutation calling principle, thereafter, excluding the germ-line polymorphisms. The quality control data of 315 samples received WES/WGS were described as previously reported.37 The median depth of the additional samples measured with WES was 158.0X (IQR, 147.1X, 179.6X), with a median mapping ratio of 99.5% (IQR, 98.2%, 99.5%), a median Q30 of 90.6% (IQR, 90.0%, 90.9), and a median 100X coverage rate of 86.2% (IQR, 85.5%, 91.4%).
After sequencing, read pairs were aligned to Human Reference Genome version hg19 (downloaded from UCSC Genome Browser, URLs) by Burrows-Wheeler Aligner38 (BWA) (version 0·7·13-r1126). Samtools39 (version 1.3) was used to generate chromosomal coordinate-sorted bam files and to remove PCR duplications. The reads were then realigned around potential indel regions by Genome Analysis Toolkit40 (GATK) (version 3·4) IndelRealigner with the recommended pipeline. GATK Haplotype Caller and GATK Unified Genotyper were applied to call SNVs (single nucleotide variations) and Indels (Insertion and deletion). Mutation detection and analysis of BAM files were performed using the cancer genome analysis program Mutect2 at the Broad Institute and annotated with ANNOVAR. Noticeably, to reduce systematic error for FFPE samples as the formaldehyde deaminates cytosines and thereby results in C → T transition mutations, the F1R2 and F2R1 annotations were adopted and FilterByOrientationBias was performed to filter the orientation bias.
Next, all the somatic functional mutations, including nonsynonymous SNVs, frameshift or in-frame indels, stopgain or stoploss were obtained. Visual inspection was used to exclude potential false positive results. Homemade pipeline was used to filter SNVs and indels detected by the above software, based on the following criteria: (1) variants with mapping quality >30 were retained; (2) SNVs or Indels with a mutation allele frequency (MAF) <0.001 in databases of 1000 genomes project, 1000 genome East Asian, ExAC all or ExAC East Asian and genomAD were retained; (3) variant allele frequency (VAF) >5% and read depth >10; (4) dbSNP (v147) sites existed COSMIC database (the Catalog of Somatic Mutations in Cancer, version 77) were retained; (5) SNPs or Indels including stopgain, stoploss, frameshift, nonframeshift and splicing sites were retained; (6) duplicate frameshifts found in multiple samples, variants found in repetitive regions, variants found in regions with poor coverage were excluded; (7) germline variants found in control samples or detected by sanger sequencing in paired peripheral blood samples were excluded. A list of variants called was provided in the Supplementary Information (see Supplementary Table 7).
Copy number analyses of WGS/WES samples were conducted with CNVkit41 using recommended pipeline (https://cnvkit.readthedocs.io/en/stable/pipeline.html). GISTIC 2.042 (q < 0.1) was used to verify the.cnv files and determined the copy number profiles.
Targeted sequencing
Targeted sequencing of the lymphoma-related genes was performed on 664 FFPE tumor samples using MultipSeq Custom Panel (Shanghai Rightongene Biotechnology Co., Ltd, China). The capture probes were designed based on ~0.39 Mb genomic regions of the lymphoma-related genes that are frequently mutated in DLBCL, other common lymphoma and hematologic malignancies. In summary, gDNA was fragmented, end repaired, linked with sequencing adapters, purified and went through pre-PCR using Enzyme Plus Library Prep Kit (iGeneTech, Beijing, China). After hybridization and concentration, sequencing was performed on the Novaseq (Illumina) sequencing platform. The mean depth of each sample measured with targeted sequencing was 1261X (IQR, 1085X, 1482X), with a median mapping ratio of 97.0% (IQR, 95.8%, 99.0%), a median Q30 of 93.6% (IQR, 92.9 %, 94.4%), and a median 200X coverage rate of 96.0% (IQR, 93.0%, 97.0%).
After sequencing, the quality of the raw sequencing data was assessed using FastQC software (version 1.11.4). In addition, Trimmomatic (version 3.6) software was used to process raw sequencing data to remove adaptor sequences and low-quality fragments. The data processing process of Broad Institute was used to process all sequence data, and the original sequencing sequence was aligned with the human reference genome hg19 using BWA (0·7·13-r1126). The repeats were eliminated, and the base quality was recalibrated. GATK was used for SNP calling. Mutation detection and analysis of BAM files were performed using the cancer genome analysis program Mutect2 at the Broad Institute and annotated with ANNOVAR. Also, F1R2 and F2R1 annotations were adopted and FilterByOrientationBias was performed to filter the orientation bias. The SNVs and Indels were screened based on the filtering conditions: (1) variants with mapping quality >30 were retained; (2) SNVs or Indels with a mutation allele frequency (MAF) <0.001 in databases of 1000 genomes project, 1000 genome East Asian, ExAC all or ExAC East Asian and genomAD were retained; (2) SNVs or Indels with a VAF ≥5% was retained; (3) dbSNP (v147) sites existed COSMIC database were retained; (4) SNPs or Indels including stopgain, stoploss, frameshift, nonframeshift and splicing sites were retained; (5) missense mutation type require meet the conditions of sift ≤ 0.05, Polyphen2_HVAR_pred ≥0.447 and CADD > 4 were retained. A list of variants called was provided in the Supplementary Information (see Supplementary Table 7).
RNA sequencing
RNA purification, reverse transcription, library construction and sequencing were performed in WuXi NextCODE according to the manufacturer’s instructions (Illumina San Diego, CA, USA). PolyA mRNA was purified from total RNA using oligo-dT-attached magnetic beads and then fragmented by fragmentation buffer. The synthesized cDNA was subjected to end-repair, phosphorylation, and ‘A’ base addition according to Illumina’s library construction protocol. Then Illumina sequencing adapters were added to both size of the cDNA fragments. After PCR amplification for DNA enrichment, the target fragments of 200–300 bp were cleaned up. After library construction, Qubit (Thermo Fisher Scientific) was used to quantify concentration of the resulting sequencing libraries, while the size distribution was analyzed using Agilent BioAnalyzer 2100 (Agilent). After library validation, Illumina cBOT cluster generation system with HiSeq PE Cluster Kits (Illumina) was used to generate clusters. Paired-end sequencing was performed using an Illumina HiSeq system following Illumina-provided protocols for 2 × 150 paired-end sequencing.
Read pairs were aligned to Refseq hg19 by STAR (version 020201). The HTSeq was applied to generate table files containing transcript counts.43 Further analyses were performed by R (v4.0.2). Voom function from R package “limma” (v3.38.3) was used to remove batch effect and normalize raw reads.44 R package “clusterProfiler” (v3.10.1) was used for Gene Ontology enrichment analysis.45
Simplified LymphPlex algorithm
LymphPlex was a novel clustering strategy, based on the LymphGen algorithm17 but designed in a simplified manner. More specifically, a given DLBCL sample was assigned into one of the defined genetic subtypes (TP53Mut, MCD-like, BN2-like, N1-like, EZB-like with or without MYC rearrangements, and ST2-like) by applying the Partitioning Around Medoids (PAM) method with mutation data of 35 genes and rearrangement data of three genes BCL2, BCL6, and MYC.46 These mutated genes (see Supplementary Table 8) were identified from WES and WGS of 337 patients and also were supported by RNA-seq data available for 207 patients (that is, differentially expressed and involved in the functional pathways as revealed by Gene Ontology enrichment analysis). Patients with TP53 mutations were assigned into a particular subtype with a higher priority. The set of features considered for possible association with a class are based on those used in the LymphGen algorithm: TP53Mut (TP53 mutations), MCD-like (MYD88, CD79B, PIM1, MPEG1, BTG1, TBL1XR1, PRDM1, IRF4 mutations), BN2-like (BCL6 fusion, NOTCH2, CD70, DTX1, BTG2, TNFAIP3, CCND3 mutations), N1-like (NOTCH1 mutations), EZB-like (BCL2 fusion, EZH2, TNFRSF14, KMT2D, B2M, FAS, CREBBP, ARID1A, EP300, CIITA, STAT6, GNA13 mutations, with or without MYC rearrangement), and ST2-like (SGK1, TET2, SOCS1, DDX3X, ZFP36L1, DUSP2, STAT3, IRF8 mutations). Computational procedures on the LymphPlex algorithm are shared on https://github.com/difuSJTU/LymphPlex. The online version of the LymphPlex algorithm is provided on https://kylinmu.shinyapps.io/LymphPlexR/.
Validation DLBCL cohorts
To evaluate the LymphPlex, we used two validation cohorts: the BC Cancer (BCC) cohort which consisted of 320 newly diagnosed DLBCL patients received R-CHOP, with available clinical and genomic data from a targeted gene sequencing panel and FISH results,19 and the UK population-based Hematological Malignancy Research Network (HMRN) cohort which consisted of 928 DLBCL patients from a real-world study (617 patients received R-CHOP), with available clinical and panel-based DNA sequencing data.14
Statistical analysis
Fisher’s exact test was used for between-categorical data comparisons. Differences of normalized gene expression in two groups were analyzed using Mann–Whitney U test. PFS was measured from the date of diagnosis to the date when disease progression/relapse was recognized or the date of last follow-up. OS was calculated from the date of diagnosis to the date of death or the date of last follow-up. Survival analysis was performed using the Kaplan–Meier curves and Cox proportional hazards regression models. Statistical significance was defined as two-sided P < 0.05. The above statistical analyses were performed by Statistical Package for the Social Sciences (SPSS) v26.0 (SPSS Inc., Chicago, IL) and R v4.0.2 (R foundation, Vienna, Austria).
Data availability
Genomic and gene expression data have been deposited on https://www.biosino.org/node in project OEP001143. Proposals requesting individual participant data that underlie the results reported in this paper (after de-identification) can be sent to zhao.weili@yahoo.com.
References
Smith, A. et al. Lymphoma incidence, survival and prevalence 2004-2014: sub-type analyses from the UK’s Haematological Malignancy Research Network. Br. J. Cancer 112, 1575–1584 (2015).
Alaggio, R. et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia 36, 1720–1748 (2022).
Teras, L. R. et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 66, 443–459 (2016).
Coiffier, B. et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 235–242 (2002).
Liu, Y. & Barta, S. K. Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 94, 604–616 (2019).
Younes, A. et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J. Clin. Oncol. 37, 1285–1295 (2019).
Nowakowski, G. S. et al. ROBUST: a phase III study of lenalidomide plus R-CHOP versus placebo plus R-CHOP in previously untreated patients with ABC-type diffuse large B-cell lymphoma. J. Clin. Oncol. 39, 1317–1328 (2021).
Rosenwald, A. et al. Prognostic significance of MYC rearrangement and translocation partner in diffuse large B-cell lymphoma: a study by the Lunenburg Lymphoma Biomarker Consortium. J. Clin. Oncol. 37, 3359–3368 (2019).
Alizadeh, A. A. et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000).
Rosenwald, A. et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med 346, 1937–1947 (2002).
Wright, G. et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl Acad. Sci. USA 100, 9991–9996 (2003).
Wilson, W. H. et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med 21, 922–926 (2015).
Chapuy, B. et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med 24, 679–690 (2018).
Lacy, S. E. et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood 135, 1759–1771 (2020).
Schmitz, R. et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med 378, 1396–1407 (2018).
Shaffer, A. L. 3rd, Young, R. M. & Staudt, L. M. Pathogenesis of human B cell lymphomas. Annu Rev. Immunol. 30, 565–610 (2012).
Wright, G. W. et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell 37, 551–568 e514 (2020).
Zhang, M., Xu, P., Wang, L., Cheng, S. & Zhao, W. Genetic subtype guided rituximab‐based immunochemotherapy improves outcome in newly diagnosed diffuse large B‐cell lymphoma: first report of a randomized phase 2 study. Hematol. Oncol. 39, abstr. 026 (2021) https://doi.org/10.1002/hon.26_2879.
Ennishi, D. et al. Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J. Clin. Oncol. 37, 190–201 (2019).
Xu-Monette, Z. Y. et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 120, 3986–3996 (2012).
Chiappella, A. et al. Prognostic impact of TP53 mutation in newly diagnosed diffuse large B-cell lymphoma patients treated in the FIL-DLCL04 trial. Br. J. Haematol. 196, 1184–1193 (2022).
Zenz, T. et al. TP53 mutation and survival in aggressive B cell lymphoma. Int J. Cancer 141, 1381–1388 (2017).
Shouval, R. et al. Impact of TP53 genomic alterations in large B-cell lymphoma treated with CD19-chimeric antigen receptor T-cell therapy. J. Clin. Oncol. 40, 369–381 (2022).
Zhang, M. C. et al. Clinical efficacy and tumour microenvironment influence of decitabine plus R-CHOP in patients with newly diagnosed diffuse large B-Cell lymphoma: Phase 1/2 and biomarker study. Clin. Transl. Med 11, e584 (2021).
O’Brien, S. M. et al. A phase 2 study of idelalisib plus rituximab in treatment-naive older patients with chronic lymphocytic leukemia. Blood 126, 2686–2694 (2015).
Chen, L. L. & Wang, W. J. p53 regulates lipid metabolism in cancer. Int J. Biol. Macromol. 192, 45–54 (2021).
Wang, L. et al. Advances in targeted therapy for malignant lymphoma. Signal Transduct. Target Ther. 5, 15 (2020).
Davis, R. E. et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92 (2010).
Wilson, W. H. et al. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell 39, 1643–1653.e1643 (2021).
Czuczman, M. S. et al. A phase 2/3 multicenter, randomized, open-label study to compare the efficacy and safety of lenalidomide versus investigator’s choice in patients with relapsed or refractory diffuse large B-cell lymphoma. Clin. Cancer Res. 23, 4127–4137 (2017).
Ward, J. P. et al. Phase 1/dose expansion trial of brentuximab vedotin and lenalidomide in relapsed or refractory diffuse large B-cell lymphoma. Blood 139, 1999–2010 (2022).
McCabe, M. T. et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012).
Villalobos-Hernandez, A. et al. SOCS1 inhibits migration and invasion of prostate cancer cells, attenuates tumor growth and modulates the tumor stroma. Prostate Cancer Prostatic Dis. 20, 36–47 (2017).
Zhang, Q. & Casanova, J. L. Human TET2 bridges cancer and immunity. Blood 136, 1018–1019 (2020).
Swerdlow, S. H. et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127, 2375–2390 (2016).
Scott, D. W. et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 123, 1214–1217 (2014).
Shen, R. et al. Influence of oncogenic mutations and tumor microenvironment alterations on extranodal invasion in diffuse large B-cell lymphoma. Clin. Transl. Med. 10, e221 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 12, e1004873 (2016).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Kaufman, L. & Rousseeuw, P. J. in Finding Groups in Data Wiley Series in Probability and Statistics 68–125 (1990).
Acknowledgements
This study was supported, in part, by research funding from the National Key R&D Program of China, National Natural Science Foundation of China (81830007, 82130004, 81670176, and 82070204), Shanghai Municipal Education Commission Gaofeng Clinical Medicine Grant Support (20152206, 20152208, and 202218), Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR1032B, SHDC2022CRD033), and Multicenter Clinical Research Project by Shanghai Jiao Tong University School of Medicine (DLY201601). We appreciate the effort of the physicians for enrolling patients and thank all the patients involved for allowing us to analyze their clinical data.
Author information
Authors and Affiliations
Contributions
W.-L.Z. and P.-P.X. designed and supervised the study. R.S. and D.F. collected the data and performed the analyses. L.D. reviewed the histopathologic diagnoses and gave technical support. M.-C.Z., Q.S., and Z.-Y.S. prepared biological samples. S.C. and L.W. provided executive support and data surveillance. R.S., P.-P.X., W.-L.Z. interpreted the results and wrote the manuscript. P.-P.X., W.-L.Z. verified the underlying data. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, R., Fu, D., Dong, L. et al. Simplified algorithm for genetic subtyping in diffuse large B-cell lymphoma. Sig Transduct Target Ther 8, 145 (2023). https://doi.org/10.1038/s41392-023-01358-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01358-y
This article is cited by
-
Genetic and transcriptomic analyses of diffuse large B-cell lymphoma patients with poor outcomes within two years of diagnosis
Leukemia (2024)
-
Deciphering the Prognostic Significance of MYD88 and CD79B Mutations in Diffuse Large B-Cell Lymphoma: Insights into Treatment Outcomes
Targeted Oncology (2024)
-
Case report: Mutation evolution in a patient with TdT positive high grade B cell lymphoma with MYC and BCL2 rearrangements following the treatment of concurrent follicular lymphoma and diffuse large B-cell lymphoma
Discover Oncology (2024)
-
Combination of acalabrutinib with lenalidomide and rituximab in relapsed/refractory aggressive B-cell non-Hodgkin lymphoma: a single-arm phase II trial
Nature Communications (2024)
