
Abstract
The ageing process is a systemic decline from cellular dysfunction to organ degeneration, with more predisposition to deteriorated disorders. Rejuvenation refers to giving aged cells or organisms more youthful characteristics through various techniques, such as cellular reprogramming and epigenetic regulation. The great leaps in cellular rejuvenation prove that ageing is not a one-way street, and many rejuvenative interventions have emerged to delay and even reverse the ageing process. Defining the mechanism by which roadblocks and signaling inputs influence complex ageing programs is essential for understanding and developing rejuvenative strategies. Here, we discuss the intrinsic and extrinsic factors that counteract cell rejuvenation, and the targeted cells and core mechanisms involved in this process. Then, we critically summarize the latest advances in state-of-art strategies of cellular rejuvenation. Various rejuvenation methods also provide insights for treating specific ageing-related diseases, including cellular reprogramming, the removal of senescence cells (SCs) and suppression of senescence-associated secretory phenotype (SASP), metabolic manipulation, stem cells-associated therapy, dietary restriction, immune rejuvenation and heterochronic transplantation, etc. The potential applications of rejuvenation therapy also extend to cancer treatment. Finally, we analyze in detail the therapeutic opportunities and challenges of rejuvenation technology. Deciphering rejuvenation interventions will provide further insights into anti-ageing and ageing-related disease treatment in clinical settings.
Similar content being viewed by others

Introduction
Ageing is a dynamic and time-varying process, typically manifested by cell damage accumulation, degeneration of tissue and organ structure and function, and increased susceptibility to diseases.1 As the risk factor for human mortality, ageing is closely associated with many chronic diseases, like diabetes, Alzheimer’s disease (AD), chronic kidney diseases (CKD), cardiovascular diseases (CVD), and cancer.2 Therefore, enhancing the knowledge of ageing and the development of rejuvenation interventions are priority targets in biomedical research. Dietary restriction (DR) was found to prolong lifespan in mice and rats in 1934, and there are currently many emerging rejuvenation treatments to enhance health and lengthen lifespan, such as genetic, pharmacological, dietary, and lifestyle modifying approaches (Fig. 1).3 However, these breakthroughs were obtained in short-lived organisms from yeast model to mice model.4 Considering the complex anti-ageing mechanisms, it still takes a long translational phase to implement rejuvenation interventions into clinical applications.

The milestone events for cellular rejuvenation research advances. Starting with the 1934 discovery of the influence of dietary restriction on lifespan extension, important findings on the subject of cellular rejuvenation are emphasized. More recently, the senolytics development and reprogramming technology have been widely applied for cellular rejuvenation. rhEGF recombinant human epidermal growth factor, iPSCs induced pluripotent stem cells, AMPK 5’-AMP-activated protein kinase, NF-κB nuclear factor-κB, STAT3 signal transducer and activator of transcription 3, mTOR mammalian target of rapamycin. Created with BioRender.com
Rejuvenation usually refers to giving aged cells or organisms more “youthful” characteristics through various techniques, such as cellular reprogramming and epigenetic regulation.5 Especially, the technique using induced pluripotent stem cells (iPSCs) in vitro via Yamanaka transcription factors is becoming increasingly proficient in the methodology and applications.6 Different organisms share certain molecular and cellular characteristics that are indicative of ageing, such as cellular senescence, epigenetic changes, telomere attrition, genomic instability, stem cell exhaustion, deregulated nutrient sensing, loss of proteostasis, mitochondrial dysfunction, and altered intercellular communication.7 Targeting these hallmarks of ageing is thus a growingly crucial field of research for the development of innovative cellular rejuvenation strategies. Nevertheless, these interventions have been lacking core criteria of rejuvenation in anti-ageing development and human applications.8 It is necessary to accurately define rejuvenation and characterize the therapeutic effects systemically. To understand whether the ageing can be rejuvenated, it also needs to uncover the common or distinct mechanisms underlying rejuvenation in different cell types that build each organ.9 In addition, a complete framework that describes various rejuvenation strategies should be established, contributing to cellular rejuvenation applications in human diseases. Herein, we provide a systematic and comprehensive discussion of cellular rejuvenation mechanisms and therapeutic interventions. An in-depth understanding of the pivotal roles will provide further insights into cellular rejuvenation in human disease treatment.
Roadblocks and targets for cellular rejuvenation
Intrinsic barriers limiting cell rejuvenation
Epigenetic alterations and genetic instability
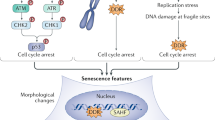
Alterations in physiological and pathological ageing, are frequently caused by disruptions in genetic and epigenetic mechanisms. The universal definition of epigenetics is heritable genomic functionally modifications without DNA sequence variations. Gene expression and chromatin structure are connected with the major epigenetic alterations, which include DNA methylation, histone modifications, and noncoding RNA regulation. Defective transcriptional and chromatin networks have highlighted the contribution to cellular function, stress resistance, and ageing. Thus, epigenetic alterations and genetic instability might affect all cells and tissues in the anti-ageing process, but also provide opportunities for the design of novel rejuvenation treatments (Fig. 2).

The epigenetic states of ageing and rejuvenation. Ageing and rejuvenation can be affected by intrinsic epigenetic alterations and genetic instability, like DNA methylation and chromatin remodeling. Moreover, many extrinsic factors, like microenvironmental cues, intercellular communication, and systemic factors, can also impact the epigenetic states of ageing and rejuvenation. miRNAs microRNAs, lncRNAs long noncoding RNAs, ECM extracellular matrix. Created with BioRender.com
Histone variants and modification
Histone variants serve as non-allelic counterparts of canonical histones and are also a common feature in aged organisms. H2A, H2B, H3, and H4 are four core histones variants with different propensity to diversity, regulating specific chromatin regions and gene transcription programs.10 Numerous research on ageing have demonstrated that histone variations in mammals are associated with a high abundance of macroH2A, H3.3, and H2A.Z.11 These results all suggested that the histone variants could be potential biomarkers for the ageing state.
Histone modifications include acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP ribosylation, deimination, and proline isomerization, depending on the modification process.12 These alterations occur at many sites and have various regulatory effects, including DNA repair, DNA replication, transcriptional control, alternative splicing, and chromosome condensation. Most prominently, histone methylation and histone acetylation play crucial roles in epigenetic alterations during ageing. Transcription is activated by the methylation of H3K4, H3K36, and H3K79, whereas it is repressed by the methylation of H3K9, H3K27, and H4K20.13 Furthermore, numerous investigations have shown that the control of organismal longevity and tissue ageing involves the manipulation of histone methyltransferases or demethylases.14 Therefore, various histone modifications might be important ageing-associated markers with the potential of anti-ageing drug screening targets.
DNA methylation clock
DNA methylation is widely appreciated as the reversible and inheritable epigenetic mark of the genome, participating in gene expression, biological regulation, and developmental processes in various eukaryotes.15 This common DNA modification is the synthesis of 5-methylcytosine (5mC), created when a methyl group is added to cytosine in a context involving a CpG dinucleotide. Long-term alterations in DNA methylation can be influenced by early-life environments and diet, increasing susceptibility to numerous diseases linked to ageing.16
Recent studies have discovered the mechanisms causing ageing-related DNA methylation changes and treated DNA methylation as the most promising biomarker of ageing.17 DNA methylation can directly facilitate transcriptome alterations in cells and tissues and also affect histone modification patterns to regulate gene expression during ageing. Borghesan et al. demonstrated that DNA methylation signatures were the biomarkers of healthy liver ageing and hepatocellular carcinoma progression, and the epigenetic synergism between DNA methylation and histone variant macroH2A1 could control the cancer cell escape from drug-induced senescence.18 Hence, controlling the DNA or histone methylation machinery might contribute to precise drug delivery. Moreover, certain CpG sites spread across the genome can be used to identify the mammalian DNA methylomes in order to determine the organismal biological age.19 DNA methylation-based ageing provides an appealing method of pathology prediction due to its continual readout of molecular changes in development. Many rejuvenation interventions, like calorie restriction, dwarfism, and rapamycin therapy, have been shown to slow down the epigenetic clocks and block some ageing-related changes in DNA methylation.20 Thus, the identification and confirmation of efficient anti-ageing therapies in humans have considerable potential in the DNA methylation.
Nucleosome remodeling
The nucleosome, consisted of pairs of core histones H2A, H2B, H3, and H4 and a DNA strand of 146 base pairs, is the basic unit of chromatin.21 In an ATP-dependent way, nucleosome remodeling controls DNA repair, replication, recombination, transcription, and cell cycle, as well as the expression of genes important for development and cellular functions in anti-ageing processes.22 The highly conserved ATP-dependent chromatin remodelers, referred to as sucrose nonfermenting 2 (SNF2) or switch/ SNF (SWI/SNF)-related enzymes, could utilize the energy released during the hydrolysis of ATP, in order to rebuild the chromatin.23 One particular ATPase might bind with other types of proteins to form distinct remodeling complexes. ATP-dependent nucleosome remodelers have important roles in modulating lifespan in organisms ranging from yeast to humans.
The modifications of histone core residues perform specific recruitment of transcription factors and remodeling complexes, and also shape nucleosome functions themselves.24 With increasing age, the nucleosome positioning and occupancy could be inevitably changed, including the loss of core histones and the substitution of canonical histones with variant histones.25 The availability of DNA for transcription factor binding was proved to depend on different combinations of histone modifications for chromatin remodeling, which also allows the histone code to modify gene expression with ageing.26 In addition, base excision repair (BER) is a vital DNA repair system for eradicating DNA lesions and maintaining the integrity of the genome.27 The deficient BER shows a strong link with ageing in human health. Nucleotide excision repair (NER) defects can also affect nucleosome remodeling, histone ubiquitination, stem cell reprogramming, and transcriptional activation, leading to ageing and developmental abnormalities in mammals.28 Therefore, DNA and histone contents within nucleosomes can go through chemical modifications that change the chromatin conformation and accessibility during ageing process.
Transcriptional signature changes
Numerous transcription factors that control growth, metabolism, and stress resistance are evolutionarily conserved. They are also involved in intricate interactions within various cell types for entire organismal physiologic development and ageing.29 During ageing process, there are many linked genes to prove the senescence phenotype or type.30 These biomarkers are linked with the cell-cycle arrest and SASP, such as increased expression of the cyclin-dependent kinase inhibitors p16 and p21, reduced expression of the nuclear lamina protein LaminB1, increased secretion of many inflammatory cytokines, and others. Many transcriptional responses to oxidative stress and pathogens decrease with ageing, owing to the declining function of the stress-responsive NF-E2-related factor 2 (Nrf2).31 Forkhead box O (FOXO) transcription factors are also revealed as critical mediators in crucial cellular processes in mammals during ageing.32 Thus, ageing was accompanied by both increased transcriptional instability and an accumulation of genetic errors. Besides, RNA N6-methyladenosine (m6A) modification has uncovered a new domain in post-transcriptional epigenetic regulation. In intervertebral discs (IVDs) degeneration, the m6A and the crosstalk between m6A and histone/DNA modifications have been proved to facilitate nucleus pulposus cellular senescence and aggravate cartilage endplate degeneration.33 To determine the diverse biological senescence, the transcriptome signatures associated with senescence and variability of the senescence program contribute to the identification of particular senescence biomarkers in tissues and organisms.
Noncoding RNA profiles
Noncoding RNAs (ncRNAs) are a broad and diverse group that produce non-protein-coding transcripts, including micro RNAs (miRNAs), long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs). Several studies have discussed that the ncRNAs are able to bond to DNA, RNA, and protein, influencing cellular proliferation, quiescence, differentiation, apoptosis, and senescence.34 Specifically, ncRNAs can strongly implicate in controlling senescence at transcriptional, post-transcriptional, and post-translational stages. Especially, miRNAs are the best characterized small ncRNAs influencing ageing and lifespan. Multiple miRNAs, including miRNA-1, miRNA-145, miRNA-140, miRNA-34a, miRNA-106b, and miRNA-449a, are widely considered as critical regulators for cell senescence.35 They impact the SASP phenotype and modulate senescence through the classical p16, p53, and calcium signaling pathways.35 Furthermore, the lncRNAs expressions are known as a result of the disease-triggering stimulation in reduced cardiovascular vigor and cardiovascular ageing. There are increasing lncRNAs as indications of a deteriorating prognosis following cardiac events, such as the lncRNA MIAT, ANRIL, LIPCAR, and MALAT1.36 More importantly, the potential of circRNAs to serve as miRNA sponges and the interacting regulatory network between lncRNAs and miRNAs are all connected to ageing-related changes and the switching of the cell fate.37 In addition, extracellular RNAs in circulation and other bodily fluids also play vital roles within the context of ageing. The pericentromeric ncRNAs can be transported into neighboring cells via extracellular vesicles (EVs), and impaired the DNA binding of the CCCTC-binding factor to modify chromosomal accessibility and trigger an SASP-like inflammatory response.38
Macromolecular damage
The specific complex of cell macromolecules, including telomeres, proteins, and lipids, possesses intrinsically high resistance to modification, contributing to superior longevity in species. Reducing macromolecular damage is associated with an improvement in the majority of ageing-related physiological activities. Thus, the single macromolecular regulator of ageing and the interconnectivity among molecular phenotypes can both induce alterations in ageing-related phenotypes, limiting cell rejuvenation.
Telomere attrition
Telomeres are repetitive DNA sequences at chromosome ends. Telomerase adds repeats to the ends of the chromosomes during genome replication to counteract the loss of telomeric DNA. In most eukaryotes, the telomerase-based mechanism for telomere preservation is crucial for genomic stability and cell viability. The compaction of telomeric chromatin robustly protects the ends by limiting the accessibility of the DNA damage response machinery.39 However, telomeres are gradually shortened with cell division, and the cells enter a replicative senescence state when they cannot effectively guard the ends of the DNA.40 DNA damage accumulation with age also affects the genome randomly. Telomere dysfunction-induced foci and telomere-associated foci are two types of DNA damage that specifically target telomeres.41 Significantly short telomeres or changed telomere architecture can result in dysfunctional telomeres in ageing-associated pathology.42 This telomere dysfunction is closely associated with telomeropathies, telomere biology disorders, and telomere syndromes. In addition, ncRNAs also emerge as key inducers of telomere length maintenance in senescence and ageing-related diseases.43
Loss of proteostasis
Proteome homeostasis is maintained by the proteostasis network (PN), a macromolecular system that coordinates protein synthesis, folding, disaggregation, and degradation for organismal health and longevity. The autophagy-lysosomal system and the ubiquitin-proteasomal system (UPS) are two crucial mechanisms that regulate the turnover of organelles and aggregates.44 Proteasomes are also charged with removing normal and damaged proteins, participating in the evolutionarily conserved ageing mechanism and longevity regulation. However, ageing often shifts the balance between the protein lifecycle in organisms, resulting in pathology. UPS dysregulation occurs in the ageing process and several ageing-related diseases in mammals.45 PN component aggregation during ageing can elicit aberrant transcriptional procedures, reduced folding capacity, and the accumulation of misfolded species.46 This loss of proteostasis might further have profound consequences for ageing progression and age-related disease presentation. In addition, the increased ribosome pausing during ageing can also make the ribosome-associated quality control overloaded, leading to proteostasis impairment and systemic decline.47
Lipid Damage
Lipids are crucial components of all cell types, and perform various biological functions, including energy storage, cell membrane construction, signal transduction, protection, and mitochondrial regulation. There are a variety of bioactive lipids with critical roles in influencing cell age and the progression of several age-associated diseases and metabolic abnormalities.48 Specifically, the lipid assemblies act as scaffolding for the construction and function of signaling complexes and play critical roles in the preservation of proteostasis, thus involving in the ageing process and neurodegenerative disease development.49 Cholesterol, phospholipids, ganglioside GM3, and sphingomyelin are lipid classes that are typically present in cell membranes, playing their respective roles in membrane fluidity and rigidity. The lipid-induced changes in membrane structure and remodeling of lipid composition are the causative agents of ageing phenotypes.50 Moreover, many pathways regulating ageing and longevity are also linked to lipid metabolism and lipid signaling. According to research on the serum and plasma lipidome in centenarians, descendants of centenarians and elderly people without age-related disorders, the lipid signature profile altered with ageing.51 It also showed changes in antioxidant capacity, lipid peroxidation levels, and inflammation along with modifications in lipid metabolic pathways with ageing.
Metabolic imbalance
Age-dependent alterations in the transcriptomes, proteomes, and metabolomes of different organisms and tissues reveal the imbalance of metabolic homeostasis. Remodeling of metabolic signals and metabolites in ageing and the control of lifespan is caused by organelle malfunction, redox imbalance, and changed signaling pathways.52 Both environmental and generated endogenous toxicants by metabolism are major contributors to macromolecular damage and physiological dysregulation during ageing. The metabolic phenotyping of ageing mice revealed the involvement of the adiponectin, growth hormone, and cytokine pathways in autophagy, stress response, genome integrity, mitochondrial biogenesis, energy balance, inflammation, and infection control.53
The main mechanism to foster ageing is the malfunctioning of vital cellular organelles, including the autophagosomal-lysosomal network and mitochondria. With advancing age, there is a reduction in autophagy activity, autophagosome production rate, and lysosome fusion activity.54 Insufficient protective autophagy during ageing might cause damaged cellular components to accumulate and dysfunction of cellular organelles, leading to metabolic imbalance and further ageing. Moreover, defective mitochondrias produce insufficient ATP and frequently produce more ROS to enhance oxidative stress.55 Aged cells commonly develop mitochondria with aberrant characteristics, such as point mutations and mitochondrial DNA (mtDNA) deletions.56 In addition, mitochondrial metabolism also includes carbon metabolism (the tricarboxylic acid (TCA) cycle), the biosynthesis of Fe/S clusters, and the metabolic consequences of mitophagy.57 These metabolic processes are highly dynamic and all influence different facets of ageing.
There are metabolic enzymes and pathways to maintain homeostasis, including acetyl-coenzyme A (acetyl-CoA), pyruvate, 2-oxoglutarate, glycolysis, the TCA cycle, the urea cycle, respiration, and oxidative phosphorylation.58 The dysfunctions of these metabolic enzymes and mechanisms trigger metabolic disorders and restricted lifespan. Many core metabolites are appearing as key regulators of ageing, including nicotinamide adenine dinucleotide (NAD+), reduced nicotinamide dinucleotide phosphate (NADPH), α-ketoglutarate (α-KG), and β-hydroxybutyrate (βHB).59 A change in the NAD+/NADH ratio or the size of the NAD+ pool can cause the biological system to malfunction and result in a variety of metabolic diseases, ageing, and cancer.60 In addition, the telomere shortens and telomerase dysfunction might downregulate peroxisome proliferator-activated receptor gamma coactivator (PGCs) and other metabolically relevant genes, which are linked to hampered mitochondrial biogenesis and function, reduced gluconeogenesis, cardiomyopathy, and elevated ROS.61 It has been demonstrated that altering the insulin/IGF-1 and mammalian target of rapamycin (mTOR) signaling pathways significantly slows down the ageing process in a variety of species.62 Metabolic interventions, like time-restricted feeding, ketone bodies, rapamycin, metformin, resveratrol, NAD boosters, glycolytic inhibition, mitochondrial-derived peptides, and poly (ADP-ribose) polymerase (PARP) activators, all target these conserved pathways and biological ageing mechanisms across species, to boost adaptability, rehabilitation, and postponed ageing.63
Extrinsic factors impacting cellular rejuvenation
Local microenvironmental cues
Extrinsic cues are transmitted by different stromal cell types within their niche and tissues, resulting in an actively responsive microenvironment. The ECM, neighboring cells, and signaling molecules, such as hormones, growth factors, and metabolic products, all play roles in mediating the interaction between the cell and the microenvironment. With time, the microenvironment that maintains multicellular organization is chronically altered, which further remodels the intracellular processes and induces ageing and cancer development.64
MSC lineage shift in ageing is mostly caused by microenvironmental effects. The crucial microenvironmental cues that cause differentiation abnormalities in MSCs are caused by hormonal, immunologic, and metabolic variables. For example, BMSCs in ageing could misdirect the differentiation toward adipocytes to impair osteogenesis, leading to the pathogenesis of osteoporosis.65 Especially, bone-fat reciprocity and the development of mesenchymal progenitors toward an adipogenic fate were mostly caused by the microenvironmental changes that occurred with in vivo ageing.66 Therefore, distinct microenvironmental conditions are deciding on cell fate, including exogenous growth factor stimulation, adjacent cells communication, pH, osmolarity, oxygen concentration, temperature, air pressure, biomechanical and electromagnetical influence.67 These microenvironment changes might increase the difficulty for normal cells to maintain homeostasis and react to damage.
Age-associated changes in ECM structure and composition
As the three-dimensional macromolecular network without cells, the ECM is primarily made of an interconnected system of fibrillar and non-fibrillar collagens, elastic fibers, and glycosaminoglycan-containing non-collagenous glycoproteins (hyaluronan and proteoglycans). Certain enzymes that stimulate ECM destruction, like matrix metalloproteinases (MMPs), mediate the ECM remodeling process.68 ECM maintains tissue integrity, and its dysregulation during ageing leads to various disease disorders by altering its composition, morphology, rigidity, and abundance. Many ECM genes and remodelers can be directly regulated by the mTOR signaling pathway, SIRTs, and numerous longevity-promoting transcription factors, such as KLF4, MYC, and HIF1, which control ECM dynamics during ageing.69
The ECM is necessary for normal tissue repair, but excessive deposition can cause organ malfunction and the onset of fibrotic and degenerative diseases. Especially, the adult dermis quality following complete maturation gradually deteriorates with age, such as atrophy of the elastic network, disintegration of collagen fibers, and alterations modifying proteoglycans.70 Moreover, ageing causes localized flaws and superficial fibrillation of the articular surface to accumulate. For instance, osteoarthritis (OA) might occur due to the altered matrix component composition, declining water content in the tissue, and increased catabolism in the ECM.71 Age-dependent functional deficits of muscle stem cells (MuSCs) are attributed to extensive ECM remodeling during ageing.72 In cancer, the main biochemical, physiological, and mechanical factors associated with ageing ECM promoted invasive and cancer-like activity in both healthy and malignant cells.73
Altered intercellular communication
Intercellular communication networks are essential for the coordination of biological processes in healthy and pathological settings of multicellular organisms. Senescence and ageing are influenced by interferences with intercellular communication caused by metabolic, mechanical, or biochemical triggers. To sustain physiologic function and respond to diseases, the many cell types that form the neurovascular unit (NVU) are in constant contact. The insufficient crosstalk between NVU cells impairs neurovascular coupling and blood-brain barrier dysfunction, thus leading to ageing and related neurological and neurovascular diseases.74 Moreover, there are also many organelle-organelle and organelle-cytosol communications impacting chronological ageing. These communications form an intricate network involving various movements of metabolites between cellular compartments. The process of stem cell ageing and tissue and organ functional declining is attributed to mitochondrial-ER crosstalk.75 Age-related diseases and the ageing process are linked to aberrant EV secretion and disturbance of the mitochondrial-lysosomal axis.76
Senescent cells are extremely proactive and interact with nearby cells through a variety of intercellular channels, including SASP. As the traditional soluble SASP, soluble factors, growth factors, and matrix remodeling enzymes are released. Intercellular communication during senescence via receptor or cell-ECM interaction is referred to as nonclassical SASP, and emerging SASP components include EVs.77 Furthermore, EVs are released into extracellular space and act as a cell-to-cell means of communication. EVs have negative impacts on downstream effectors at the levels of immunology, inflammation, gene expression, and metabolism in the ageing setting and age-related illnesses.78 Aged and senescent cells are proved to release more EVs than young cells.79 In addition, several environmental conditions, including air pollution, ultraviolet light, nutrition, and physical exercise, have been verified to impact the communication network via EVs, further impacting ageing.80
Systemic factors
Alterations in the systemic environment of cells and tissues play a role in the reversible process of ageing. Organ dysfunction with ageing is caused by blood-mediated cell-extrinsic alterations and important molecular mechanisms in the systemic environment. One of the cell types that reacts to young blood exposure is the hematopoietic stem cells (HSCs).81 The hematopoietic and immunological systems can be rejuvenated by the young transcriptional regulation system and cytokine-mediated cell-cell interactions in HSCs. Many agonists and antagonists of specific signaling pathways have the effective capability of resetting tissue stem cells in aged organs into rejuvenating state.82 In addition, systemic obesity, air pollution, exercise, and psychological stress have been clarified to accelerate ageing at molecular and epigenetic levels.83 Some biological techniques, such as heterochronic transplantation and parabiosis, might give cells and substances that are more abundant in young individuals to recover the function of aged tissue. Heterochronic parabiosis is the surgical method of young and aged organisms using a common vascular system, showing the significant impact of the systemic environment on ageing and rejuvenation. Many studies on neurogenesis have reported the pro-neurogenic “youthful” factors in the circulation and “ageing” substances that reduce stem cell activity in heterochronic parabiosis models of young and aged mice.84 Moreover, systemic transplantation of stem cells also displays the therapeutic potential of preventing age-associated degeneration. However, the systemic and hormonal changes with age, including pro-inflammatory cytokine profiles and sex steroid changes, also influence stem cell transplantation effectiveness.85
Rejuvenating-targeted cells for organismal youthful state
Stem cells
The major types of stem cells include adult stem cells, embryonic stem cells (ESCs), and iPSCs created by activating Yamanaka factors from various somatic cells. They have the unique capacity for self-renew and multipotency, and can differentiate into tissue-specific terminal cell types. MSCs are pluripotent cells developed from adult stem cells. Many studies have demonstrated that MSCs produced from various sources, such as bone marrow, adipose tissue, and umbilical cord blood, and MSC-derived compounds slowed ageing process and improved age-related conditions.86 Tissue stem cells are found in particular local tissue microenvironments called “stem cell niches,” which support stem cell maintenance. Tissue stem cells play key roles in facilitating organic tissue renewal and performing regenerative responses to injury.87 ESCs can self-renew and differentiate into multiple cell types of ectoderm, endoderm, and mesoderm lineages.88 More importantly, the iPSCs modified from autologous sources also have ESC-like states and pluripotent potential. iPSCs transform patient-specific samples from early cells into developed target tissues, showing potential for age reversal within the organism.89 These stem cells are excellent rejuvenation targets and all have promising potential in regenerative medicine.
With increasing age, decreased stem cell functionality can lead to diminished organ function and prolonged tissue repair. Targeting the age-related molecular basis of stem cells might reduce the deleterious effects of ageing. There are many rejuvenating approaches based on aged stem cells, such as delayed fasting, gene expression modulation, medicinal intervention, and niche changes.90 However, Ho et al. found that some rejuvenating approaches had no observable renewed effects on aged HSCs and aged bone marrow niches.91 Some rejuvenation techniques might show temporary benefits, but show harmful long-term effects by prematurely diminishing the stem cell pool.92 Thus, it is also vital to strike a balance between the regenerative properties of stem cells and their potential to induce cancer.
Vascular and connective tissue cells
Endothelial cells (ECs) and smooth muscle cells (SMCs) are critical building blocks of blood channels and are negatively impacted by premature or typical ageing processes. In ageing process, dysfunctional ECs and endothelial progenitor cells (EPCs) occur abnormal metabolism, the development into mesenchymal phenotype, vascular detachment, and myofibroblast formation, resulting in fibrosis and organ dysfunction.93 ECs and other neurovascular cells are key cells in maintaining blood-brain barrier function. Dysfunction of pericytes, astrocytes, and endothelial cells increases blood-brain barrier permeability during ageing.94 The intricate biological process of targeted EC regeneration involves migration, survival, proliferation, tube formation, and restoring blood flow to the ischemic organs for tissue homeostasis. EPCs might be obtained from the bloodstream or niches within the vascular wall and restored by the ectopic production of mediators that prevent senescence and the onset of ageing-related traits.95 Thus, targeting the endothelium via regulating the senescence-induced gene expression and other emerging rejuvenation mechanisms is essential for homeostasis and tissue regeneration.
The major stromal cell type is the fibroblast, which regulates tissue morphology by depositing ECM, and promotes cellular and microenvironmental homeostasis by secreting soluble substances and signaling proteins. During the ageing process, fibroblasts lose contractility and exhibit an unbalanced production and degradation of ECM proteins, ultimately leading to reduced connective tissue stiffness and even age-related diseases.96 Activated fibroblasts and senescent fibroblasts secrete inflammatory cytokines with a different ratio, affecting complex reprogramming and wound healing in mice.97 Many studies have insisted that some anti-ageing compounds like triacetylresveratrol and cannabidiol, gene expression, signaling regulation, and cell-cell communication are all effective methods for targeting fibroblasts for rejuvenation, longevity, and health.98 Hence, as a cell type commonly used for iPSC reprogramming, fibroblasts are essential targets for rejuvenation techniques and regenerative medicine.
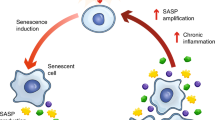
Senescent cells
Senescent cells (SCs) comprise a heterogeneous cell population because of their various cell-autonomous activation pathways and microenvironmental circumstances. Although cell-cycle arrested, SCs are still metabolically active and can perform various functions of the parent cells.99 At present, SCs, as organismal carriers of irreparable damage, are identified by the senescence-associated gene expression, SASP production, DNA damage, and β-galactosidase activity.100 MSC populations with a high number of SCs are found less productive during transplantation.101 The excessive accumulation and activity of SCs are also linked with chronic ageing and age-related illnesses, including atherosclerosis, cardiac and kidney dysfunctions, neurodegeneration, and pulmonary fibrosis.102 SCs are also able to trigger senescence in non-senescent cells. High quantities of SCs secreting chronically SASP are found in aged tissues, causing irreversible reprogramming of their adjacent cells.103 Hence, it also needs to prevent the subsequent development of SCs after the emergence of initial SCs. Partial reprogramming of SCs can reduce the persistent inflammatory state related to ageing and secondary senescence in surrounding cells by inducing the SASP.104 The specific gene expression on the surface of SCs might lead to the advancement of senolysis techniques for selective elimination.105
Immune cells
During ageing, the immune system progressively undergoes disorders of immune cell generation, differentiation, and function, leading to a chronically subclinical inflammatory condition. Several studies have proposed that targeting central immunological processes and specific immune subpopulations can reduce specific age-induced immune changes.106 Neutrophils are abundant immune cell populations in early injury and serve numerous functions in tissue regeneration. Macrophages reside in the bone marrow and are defective in efferocytosis and hyperactivated with ageing. Neutrophils can also function as an anti-inflammatory shift in macrophages by influencing the surrounding microenvironment or controlling the behavior of macrophages during tissue injury.107 The deficiency of immunosurveillance might hamper the SCs clearance and induce a microenvironment of chronic inflammation, leading to pro-tumorigenic events.108 Therefore, enhancing the immune surveillance ability of macrophages is also an effective rejuvenation target, due to the macrophage function of selectively SCs recognition and elimination.
The senescence in immune cells affects innate and adaptive immunity, particularly natural killer (NK) cells, B cell, and T cell function, potently driving age-related changes in solid organs. In vivo reprogramming might be significantly impeded by NK cells, which identify and eliminate partially converted cells in a degranulation-dependent mode.109 T cell generation is decreased because of thymic involution. Some hormones, signaling pathways, cytokines, and growth factors might display T cell reconstitution effects and reduce the negative effects of age-related T cell deficiency.110 Adoptive cell transfer of naive T cells can promote immunological responsiveness to new antigenic stimuli and limit the growth of pathogenic memory T cells.111 However, the quantity of naive T cell reconstitution required to boost immunological defense in ageing organisms still has quantitative constraints.112 Furthermore, B cell production also decreases with age due to the reduction of hematopoietic bone marrow. An in vitro B cell population with youthful characteristics and cellular reactivity to immunological stimulation can be revived after B cell depletion in elderly mice.113
Other somatic cells
Many specialized cells with different sources deserve further research for tissue and organ regeneration and rejuvenation. For example, in the vertebrate retinas, Müller cells serve as the primary supportive and protective glial cells. They can secrete various cytokines and exhibit the potential for self-renew and trans-differentiation into retinal neurons.114 Pathological ageing might impair β-cell function in the pancreas, thus causing the imbalance of glucose homeostasis in the organism. Targeting β-cell and restoration of function is of vital importance for effective therapeutic strategies.115 At present, there are emerging reprogramming strategies conversing differentiated somatic cells into another cell type. For instance, the astrocytes and pancreas exocrine cells can be respectively direct reprogrammed into neuroblasts and β-cells via lineage-specific transcription factors.116 Thus, these cells and related genes and pathways might facilitate the target-based gene delivery and development of effective rejuvenation approaches.
Common or distinct mechanisms of rejuvenation
Signaling pathways
There are various signaling pathways identified in the fields of ageing and rejuvenation, such as nutrient-sensing pathways, DNA damage pathways, ROS and mitochondrial unfolded protein response (UPRmt) pathways, inflammation-related pathways, transforming growth factor-β (TGF-β) pathways and Wnt/β-catenin pathways. The known role of these signaling pathways is complex and mutually connected. Given the prominent association of signaling pathways with rejuvenation and ageing, targeting these signaling systems pharmacologically and therapeutically has great potential for rejuvenation and human health (Fig. 3).

The target signaling pathways for cellular rejuvenation. Target signaling pathways for cellular rejuvenation are listed according to their biological functions. Many interventions, like dietary restriction and drugs, improve metabolism and extend longevity through nutrient-sensing pathways. In addition, targeting the pathways of damage-induced and developmental senescence can regulate the cell cycle and alleviate age-associated phenotypes. Modulating many inflammation pathways also provides an effective route to rejuvenation. IGF-1 insulin-like growth factor 1, PI3K phosphoinositide 3-kinase, AKT protein kinase B, AMPK 5’-AMP-activated protein kinase, NF-κB nuclear factor-κB, STAT3 signal transducer and activator of transcription 3, mTOR mammalian target of rapamycin, SIRTs sirtuins, FOXO forkhead homeobox type protein O, ROS reactive oxygen species, TGF-β transforming growth factor-β, CDKs cyclin-dependent kinase. Created with BioRender.com
Nutrient-sensing pathways
Nutrient availability is crucial in regulating ageing and rejuvenation in mammals. Many growth factors, metabolites, amino acids, and carbohydrates can be recognized by several proteins, such as insulin/IGF-1, mTOR, SIRTs, and AMP-activated protein kinase (AMPK). The insulin/IGF-1 signaling pathway is an evolutionarily conserved glucose sensors mechanism, involving developmental defects and decreased adult functionality with age.117 The insulin/IGF-1 pathway participates in the regulation of many cellular functions, including the metabolism of lipids and carbohydrates, the cellular availability of glucose, gene expression, and cell differentiation, growth, and survival. The stimulation of insulin/IGF-1 receptors can stimulate STAT3 signaling via Janus kinase (JAK) and protein kinase B (AKT)-driven signaling pathways, then forming negative feedback to inhibit insulin/IGF-1 and induce cell immune senescence.118 The insulin/IGF-1 binding to membrane transporters can stimulate the phosphoinositide 3-kinase (PI3K)/AKT signaling pathway, followed by the downstream FOXO1 phosphorylation and mTOR upregulation, hence regulating cellular ageing and rejuvenation.119 Furthermore, the mTOR is also a conserved nutrient-sensing protein kinase that regulates eukaryotic cell growth and metabolism. There also remain AMPK/mTOR and PI3K/AKT/mTOR pathways implicated in the accumulation of unfolded and misfolded proteins and the regulation of the autophagy process during ageing.120 DR and rapamycin have been proved to inhibit the mTOR signaling pathway causing the downregulation of cell growth and lifespan extension.121 Hence, the insulin/IGF-1 and mTOR pathways are promising targets to suppress or delay ageing-associated diseases and extend lifespan.
SIRTs are a family of seven paralogous NAD+-dependent enzymes, that control cell proliferation, energy metabolism, stress tolerance, inflammation, circadian rhythms, neural function, and ageing.122 The activity of SIRTs in many or combinations of tissues might be necessary for lifetime extension and rejuvenation. Meanwhile, the AMPK pathway is a core mediator of energy homeostasis engaged in the pathobiology of ageing and age-linked disorders.123 AMPK is also considered as a crucial integrator of inflammation-controlling signals, including the inflammasome.124 It is well known that among different interventions to promote longevity, like exercise, intermittent fasting and DR, nutrient supply reduction activates AMPK to promote ATP production.125 Metformin can activate AMPK and SIRT1 and downregulates insulin/IGF-1 and mTOR, thus playing beneficial roles in energy metabolism and ageing.126 In general, the above nutrient-sensing pathways and the interaction between SIRT, AMPK, and mTOR all explain a mechanism for rejuvenation.
DNA damaging pathways
The cell functionality can be hampered by persistent DNA damage, which can also accelerate senescence and apoptosis.127 DNA damage response (DDR) pathway can protect against endogenous and exogenous damage and ensures the integrity of the genome.128 In response to stress, the established DDR contributes to the stimulation of p53 and p16 pathways and the depression of cyclin-dependent kinase (CDK) inhibitors to initiate and sustain the arrest of cell cycle.129 The genetic mutations that augment DDR and persistent DNA lesions can continuously activate the DDR. Moreover, the alterations in the integrity and efficacy of mtDNA repair contribute to DNA damage accumulation, illness, and ageing.130
The cytosolic DNA sensor cyclic GMP-AMP Synthase (cGAS) binds to the effector protein stimulator of interferon genes (STING), initiating a DNA sensing signaling pathway for innate immune responses.131 Misplaced cytosolic self-DNA and alteration of mitochondria structure and function can activate cGAS-STING signaling pathway, thus constituting age-related inflammation.132 The DNA mismatch repair system (MMR) is largely conserved across organisms and is essential for maintaining DNA integrity.133 Hence, MMR and related repair proteins are necessary for lifespan extension and rejuvenation targets. There are also a series of systems repairing the DNA, including BER, NER, and double-strand break (DSB).134 Thus, multiple interventions have been developed to lessen the DNA damage accumulation and alleviate age-associated phenotypes, such as lowering destructive molecules, restoring DNA damage, and responding to persistent DNA damage.
ROS and UPRmt pathway
Reactive oxygen species (ROS) are mainly generated from oxidative phosphorylation in mitochondria, and maintain a dynamic balance with antioxidation systems under physiological conditions. Low ROS levels enhance the defensive mechanisms by producing adaptive responses for stress tolerance and longevity, whereas high ROS levels create insufficient adaptive responses that may accelerate the onset and course of ageing.135 The key mediators of the ageing process are ROS and ROS-induced oxidative damage produced by cellular metabolic and respiratory processes. Besides, mtDNA is susceptible to damage by mitochondrial ROS.136 Thus, the ROS causes oxidative stress, damages mitochondria, and induces energetic obstacles that lead to accelerated ageing and various diseases. Peroxiredoxins have been shown to facilitate ROS-based redox signaling and to trigger many cellular stress responses.137 However, these beneficial effects of ROS might be proved to be a sign of toxic adaptation.138 Interventions to ROS pathways are widely proposed as anti-ageing and rejuvenation strategies.
Metabolic stress, hypoxia, protein damage, and mitochondrial ROS all can impair mitochondrial protein homeostasis and functions. The transcriptional activation program of mitochondrial chaperone proteins and proteases is known as the mitochondrial unfolded protein response (UPRmt), which is a mitochondrial response to stress.139 Studies have shown that UPRmt plays a significant role in many physiological processes and that its activation increases longevity and prevents ageing by regulating mitochondrial proteostasis.140 During mitochondrial dysfunction, there are many transcription factors necessary for the activation of UPRmt genes in mammals. Activating transcription factor associated with stress-1 (ATFS-1) participates in the upregulation of genes involved in multiple stress response pathways for organismal survival of acute stressors.141 But, chronic ATFS-1 activation also has a negative effect on longevity. Meanwhile, in the initial development stages of elderly individuals, age-dependent levels of histone 3 methylation partially influence UPRmt activation.142 UPRmt-mediated protective mechanism might be beneficial for rejuvenation mechanisms and therapeutics for diverse metabolic diseases and ageing-related disorders. However, prolonged UPRmt activation might also induce the propagation of mitochondrial damage.143 The mitochondrial permeability transition pore in the inner mitochondrial membrane, can also initiate UPRmt to promote ageing and age-related diseases.144
Inflammation-associated pathways
Multiple signaling cascades whose integration targets the induction of senescence, ageing, and associated disorders can be activated by the cycle of physiological interactions between inflammation and oxidative stress. In mammals, the major pro-inflammatory cytokines, which include interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and IL-1α, significantly remodel the immune system.145 Damage-related stimuli cause the release of danger-associated molecular patterns (DAMPs) with ageing, and DAMPs that activate TLRs or the NLRP3 complex can be involved in sterile inflammation and age-related diseases.146 These age-related increases in chronic and low-level sterile inflammation are known as “inflammageing”, which shows a strong occurrence of various age-related diseases more than with ageing itself.147
The NF-κB signaling pathway is highly linked with the initiation and deterioration of tissue inflammation and ageing process. NF-κB can induce pro-inflammatory mediators, SASP, chemokines, and adhesion molecules, and the crosstalk between upstream signaling elements including MAPK, mTOR, and protein kinase B affects the transcriptional activity of NF-κB.148 Amplification loops for inflammatory processes are created when pro-inflammatory cytokines activate NF-B, which in turn can produce additional cytokines.149 The chronic activation of pro-inflammatory NF-κB/Rel and JAK/STAT signaling pathways contributes to the declined regeneration potential in mouse models.150 The JAK2 gene mutations in hematopoietic ageing can trigger abnormal involvement of downstream signaling pathways and the establishment of an inflammatory environment.151 Overactive JAK signaling is considered as a signature of immune disorders and has a significant impact on inflammation, coagulation, and thrombosis. Therefore, these shared pathways might provide a common route to rejuvenation by modulating inflammation, and some distinct pathways have great potential of targeting improving specific functions.
TGF-β signaling pathway
The transforming growth factor beta (TGF-β) superfamily is a vast protein group, including three TGF-βs (TGF-β1–3), bone morphogenetic proteins (BMPs), and growth differentiation factors (GDFs).152 The TGF-β family play roles through heteromeric combinations of type I and type II receptors, thereby activating many signal transducers, containing SMAD-dependent, SMAD-independent, and non-SMAD signaling pathways.153 These signaling pathways perform multiple functions in the development of embryo, tissue homeostasis and repair, immunological responses, tumor suppression, and metastasis. Many findings on age-related diseases have reported that TGF-β signaling dysfunction or increased levels of TGF-β ligands induce metabolic dysfunction, tissue fibrosis, inflammation, regeneration suppression, and cell degeneration.154 Besides, TGF-β signaling can induce specific epigenetic alterations further promoting senescence and ageing. Meanwhile, ageing also induces many abnormalities at the TGF-β receptor level. TGF-β signaling possesses dual functionality and versatility in some age-related disorders and cancer as a suppressor and a promoter.155 Hence, many strategies targeting TGF-β are mainly focused on inhibition of production, activation, binding to the receptor, and intracellular signaling.
TGF-β signaling can regulate matrix protein synthesis and matrix degradation, and alter cell-cell interaction. TGF-β overexpression results in ECM deposition, epithelial–mesenchymal transition (EMT), and cancer-associated fibroblast (CAF) formation, then leading to fibrosis and cancer.156 More importantly, dysregulation of the TGF-β/SMAD pathway is reported as an essential causative agent in tissue fibrosis, like hepatic, pulmonary, and cardiac fibrosis.157 TGF-β modulates several intracellular signaling cascades to deliver profibrotic effects, and the numerous ways of TGF-β interacting with other profibrotic pathways all offer the potential for therapeutic intervention.158 Furthermore, TGF-β signaling regulates the levels of angiogenesis-related molecules, like VEGF and CTGF, and mediates immune regulation, inflammation, and other pathways.159 In addition, GDF11, one cytokine of the TGF-β superfamily, is identified as a rejuvenating element in neurodegenerative and neurovascular diseases, such as the reversal of senescence and age-related variations, and the modulations of organ regeneration after injury.160
Wnt/β-catenin signaling pathway
Wnt proteins are secreted glycosylated proteins, which are cysteine-rich and can initiate the transcriptional co-activator, β-catenin, leading to the target gene upregulation via the family of T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors.161 By building a stable compound with the cell adhesion molecules of cadherin family, β-catenin, a transcription cofactor with dual roles, participates in cell adhesion.162 The Wnt/β-catenin signaling pathway participates in the modulation of genetic stabilization, cell proliferation, migration, and apoptosis for developmental processes and tissue homeostasis.163 The transcriptional results of Wnt/β-catenin pathway activation change with various cell types. The canonical Wnt/β-catenin pathway as the core mechanism is essential for directing stem cell regeneration and differentiation. For preservation and transition from the pluripotent state during embryo development, stem cells need β-catenin to moderate the response to Wnt signaling.164 Wnt/β-catenin signaling can also regulate the expression of telomerase reverse transcriptase (TERT) and change the telomere length, thereby affecting stem cells, ageing, and cancer.165 Wnt/β-catenin signaling can serve as a promising target to ameliorate the deterioration of stem cell function.
Research findings have verified that Wnt/β-catenin signaling regulates the ageing process of several tissues, performing different changes in different organs. Increased Wnt signaling has been found in aged organisms and excessive levels of Wnt are damaging to organism functionality.166 Wnt/β-catenin pathway is crucial for the growth and development of mineralized tissues, for the regulation of the skeleton in respond to loading and unloading, and for maintaining the viability and fitness of the adult and ageing skeleton.167 More importantly, the dysregulation of Wnt/β-catenin pathway is responsible for the fibrotic tissues associated with ageing, such as kidney, liver, lung, and heart fibrosis.168 Wnt/β-catenin signaling and TGF-β signaling can interact in the fibrosis process. Wnt/β-catenin superfamily members can be activated by TGF-β signaling and vice versa.169 Meanwhile, synaptic assembly, neurotransmission, and synaptic plasticity are all regulated by Wnt ligands, and neurodegenerative diseases are linked to deregulated Wnt signaling.170 Therefore, manipulating Wnt/β-catenin pathway might promote an efficient rejuvenation strategy versus ageing.
Reprogramming-induced rejuvenation
Rejuvenation by induced pluripotent stem cells
iPSC reprogramming is widely defined as the rejuvenation of mature differentiated cells to an embryonic-like fate.171 By forcing the expression of specific transcription factors, induced pluripotency is the potent capacity to differentiate into all cell types. The transcriptome, epigenome, and metabolome of differentiated cells can be significantly altered by the transient expression of Yamanaka factors (OCT4, SOX2, KLF4, and cMYC; OSKM), which can also remodel the cells into iPSCs.172 The most common cell types for reprogramming to iPSCs are fibroblasts, which are greatly implicated in regenerative medicine and rejuvenation strategies. But in distinct subpopulations of fibroblasts, the change of fibroblast component and the level of secreted inflammatory cytokines might affect the in vitro reprogramming effectiveness and in vivo wound healing rate.173 In addition, the generation of iPSCs can realize the rejuvenation of MSCs and the iPSCs differentiation into desired cells via variations in DNA methylation, histone composition, and epigenetic models.174 Therefore, the iPSCs applications can minimize the genetic and epigenetic abnormalities associated with induced pluripotency. The iPSC reprogramming technique has extensive potential for molecular regeneration, disease modeling, and drug discovery.
The somatic cells of elderly donors can be utilized to generate human iPSCs, and cell reprogramming can reverse the key signs of ageing. By lengthening telomeres, reorganizing the mitochondrial network, alleviating oxidative stress, and recovering pluripotency, the reprogramming process transforms aged cells into young condition.175 iPSCs acquire ESC-like features, especially with similar mitochondrial properties, and can modulate mitochondrial or oxidative stress pathways leading to a state of rejuvenation.176 These transformation events can promote an extensive restructuring of mitochondria, including mitochondrial counts, morphology, viability, cellular metabolism, and the complexity of mtDNA. Telomere malfunction and chromosomal fragility can impair the ability of iPSCs to self-renew and the developmental pluripotency to differentiate.177 Telomere rejuvenation is an aspect of epigenetic reprogramming toward pluripotency and reprogramming can maintain or reverse telomere length and chromatin structure. Moreover, in the cells with telomere and mitochondria defects, somatic cell nuclear transfer (SCNT)-mediated reprogramming might be a better technology than current reprogramming factors.178 Therefore, the genetic foundation of ageing and rejuvenation can be modeled using iPSC lines, enabling the identification of new factors that prevent premature ageing or impact cell rejuvenation. In addition, iPSCs can be reprogrammed from patient cells via small molecules, miRNAs, and combinations of reprogramming factors, and be differentiated into somatic cells for drug testing and regenerative medicine.179
However, many studies clarified that ageing also might constitute critical barriers to cell reprogramming due to cellular senescence, inflammation, telomere reduction, and metabolic alterations.6 The aged or pathologic tissues are relatively inefficient at reprogramming and iPSCs derived from these tissue types might lack sufficient pluripotency and differentiation ability.180 Somatic cells produced from iPSCs might undergo premature senescence. The differentiated cells or premature termination of reprogramming can also carry the gene mutation and the full genetic heritage of the patient, which might contribute to long-term risk and tumor formation.181,182 Thus, the application of iPSCs needs efficient reprogramming and differentiation protocols, and the capability to maintain iPSC functionality in ageing microenvironment. Studies have proved that some longevity-promoting compounds and inhibition of age-related pathways enhance reprogramming in regenerative therapy. There are also many methods to boost the safety of iPSCs, such as using suicide genes to eradicate any undifferentiated iPSCs that remain after therapy, choosing younger donors, using appropriate cell sources, improving gene delivery techniques, replacing DNA delivery with proteins, mRNA, or regulatory miRNAs, using small-molecule DNA modifiers, and using low-passage iPSCs.183
Rejuvenation by lineage reprogramming
Lineage reprogramming, also described as direct reprogramming, is the procedure of switching somatic cells from one lineage to another with no transition for intermediate pluripotent states.184 This method of cell reprogramming generates particular cell types by ectopically expressing various lineage-specific transcription factors or miRNAs. For instance, recent research demonstrated that certain pro-neural transcription factors can directly reprogram non-neural somatic cells into neurons, skipping the pluripotent stage.185 Hence, lineage reprogramming techniques can be used to create a variety of cell types, including brain, cardiac, hepatic, and pancreatic cells. Meanwhile, because of the unique advantages of in situ conversion in live organs, lineage reprogramming is efficient and suitable for in vivo tissue repair and rejuvenation. Direct reprogramming in vivo may also benefit from minimizing hazards for genetic changes during prolonged in vitro culture, cancer development associated with de-differentiation, and immunological rejection following transplantation.116
The senescent program induced by ageing process and tissue damage can offer a beneficial microenvironment for in vivo lineage reprogramming. Chiche et al. suggested that tissue damage induced senescence and SASP secretion to promote the plasticity of resident cells, promoting in vivo reprogramming in tissue repair and regeneration.186 However, direct lineage reprogramming might retain epigenetic hallmarks of primary cells, like ageing hallmarks, which makes the reprogrammed cells suitable for modeling ageing-related disease.187 Thus, assigning a new cellular identity to terminally differentiated somatic cells, lineage reprogramming plays key roles in in vivo repair and rejuvenation. This technology can be built to utilize numerous and convenient autologous patient-derived cell types as a source, and is particularly crucial to replicate age-related traits and mimic the onset pathophysiology of diseases.
Rejuvenation by partial reprogramming
Cell reprogramming is a stepwise protocol. Studies have proved that somatic cell reprogramming mediated by OSKM for fewer than 7 days induced transient cellular alterations and reversible dysplasia, but partial reprogramming induction for more than 7 days could lead to tumor formation.188 Accordingly, the partially reprogrammed cells via short-term exposure to Yamanaka factors only partially lose their differentiated identity and undergo molecular rejuvenation without dedifferentiating to pluripotency.189,190 More specifically, partial cell reprogramming indicates that cells gain the ability to multiply and exhibit certain stem cell markers, but do not totally lose the cellular identity or receive all characteristics of pluripotent stem cells.189 Thus, partially reprogrammed cells have no risk of teratoma after transplantation. The cellular re-differentiation to the original phenotype with epigenome rejuvenation and the ability to react to optimum cocktails of certain differentiation factors is the representative feature of partial reprogramming.191
Generally, it is difficult to distinguish between the underlying epigenetic alterations that rejuvenate ageing cells and the changes that regulate the shift in cellular identity. Partial reprogramming is able to restore the common features of cellular ageing without altering the identity or function of the cells.192 Cyclic in vivo short-term induction of OSKM suppresses age-related phenotypes and histological alterations in different organs. Chondronasiou et al. demonstrated that partial and reversible reprogramming could improve the ageing states in cells, increase the ability of old mice to restore tissue damage, and lengthen the lifespan of progeroid mice.193 Potentially reversing the effects of ageing or tissue damage through organ-specific partial reprogramming could lead to the regeneration of the desired organ.194 In addition, partial reprogramming can generate a secretory phenotype that promotes cellular regeneration and improves the chronic inflammatory state linked to ageing and secondary senescence in nearby cells through enhancing SASP.104 Therefore, partial in vivo reprogramming can improve ageing-related traits, like the diminished capability to combat injury and the loss in the capacity of tissues and organs to regenerate during life.
Epigenetic rejuvenation
Reset of ageing molecular signatures by epigenetic rejuvenation
Epigenetic remodeling is associated with biochemical modifications to the genome, leading to an altered response of gene transcription to physiological stimuli. DNA methylation clocks might detect a wide range of ageing-related epigenetic modifications that are indicative of genomic, cell biological, and tissue changes that occur during life.195 Epigenetic clocks can be more accurate than chronological clocks at estimating biological age, which aids in predicting human lifetime via age-reprogramming therapies. Protein-protein interactions can induce allosteric regulatory sites in complicated epigenetic machinery.196 Hence, accessing allosteric sites can assist in the development of epigenetic medicines with improved druggability and pharmacological characteristics. In addition, there are many lifespan-extending conditions, like Prop1df/df dwarfism, calorie restriction, and rapamycin administration, slowing molecular variations linked to the epigenetic clock in mammals.197 Furthermore, reprogramming aged cells to a more youthful status carries the hazard of tumor formation. During reprogramming without de-differentiation, the mobility of heterochromatin protein 1β, an essential epigenetic modifier, has been proved to increase in SCs and promote epigenetic rejuvenation.198 The epigenetic rejuvenation with minimal de-differentiation can be realized by OSKM transduction in partial reprogramming. For epigenetic rejuvenation, distinguishing the rejuvenative features of reprogramming from dedifferentiation is a strong development.
Epigenetic regulation of mitochondria during rejuvenation
Mitochondria role in epigenetic processes mostly involves alterations in DNA methylation, histone modification in nuclear chromatin, and posttranslational gene control by noncoding miRNAs.199 The modulation of mtDNA and mitochondrial proteins by epigenetic and post-translational alterations contributes to the preservation of cellular health and homeostasis. Differential mtDNA methylation is associated with various conditions, including ageing and ageing-related diseases, changed metabolism, alterations in circadian rhythm, and even cancer.200 Thus, removing or counteracting the effects of mtDNA mutations in mitochondria might extend human health and lifespan. Moreover, the conserved histone lysine demethylases JMJD-1.2 and JMJD-3.1 could target the H3K27me2/me3 sites, which is also important for UPRmt induction.201 MET2/ LIN65 histone methyltransferases were proved to mediate the chromatin remodeling and regulate the UPRmt-associated transcriptional networks.202 These findings revealed an epigenetic mechanism for regulating stress signaling and lifespan in response to mitochondrial abnormalities. Besides, all metabolic intermediates that serve as substrates or cofactors for epigenetic alterations originate from the Krebs cycle and other mitochondrial metabolic pathways.203 These metabolites contain acetyl-CoA, α-KG, S-adenosyl methionine (SAM), NAD+, and O-linked β-N-acetylglucosamine (O-GlcNAc) for DNA methylation and histone post-translational modifications, participating in controlling gene transcription and determining cell destiny.
Epigenetic regulation of retro-transposable elements during rejuvenation
Alternate splicing, different promoter or enhancer usage, ncRNAs, and epigenetic changes that impact the structure and function of chromatin all can modulate transcription. Retrotransposon-mediated promoters might also promote gene regulation and expand protein diversity for phenotypic variation and embryo development. During ageing, heterochromatin decay might upregulate the level of silent retrotransposons, leading to promoted mobility of retro-transposable elements (RTEs) within genomes and cellular homeostasis disruption.204 Chromatin of main retrotransposon classes, such as Alu, SVA, and long interspersed nuclear elements (LINEs), become relatively open in SCs and affect the evolutionarily recent elements, leading to increased transcription and ultimately transposition.205 Global hypomethylation of the genome can promote genomic instability and RTE activation, contributing to ageing.206 Hypomethylation of LINEs in cancer cells can restart the recruitment of many variant factors and is connected with an advanced disease stage and poor prognosis.207 DNA methylation can be oxidized by ten-eleven translocation (TET) enzymes as an aspect of the dynamic demethylation mechanism.208 TETs are responsible for LINE-1 demethylation in ESCs, but LINE-1s are negatively regulated by further TET-dependent activities. In nascent RNAs of human cells, m6A actively regulates the expression level of both autonomous LINEs and co-transcribed LINE relics, facilitating the retrotransposition of LINE.209
Epigenetic regulation of inflammation during rejuvenation
The inflammatory response can trigger epigenetic alterations, and epigenetics in turn can interfere with inflammation action. In reaction to severe inflammatory events, transitory activation of NF-κB-related innate immunity and senescence-related inflammatory elements might enhance reparative cellular reprogramming.210 The expression of numerous pro-inflammatory modulators is regulated by epigenetic procedures, which might therefore play a role in the progression of chronic inflammation. DNA methylation and histone acetylation are correlated with TNF-α expression during development and inflammatory disorders.211 Combinations of transcription factors maintain the identity of immune cells by controlling the hypo- and hypermethylation of cell-specific DNA. For instance, Sera et al. found that the X-chromosome-specific enzyme, UTX, maintained the expression of downregulated genes during ageing via demethylase-dependent and -independent epigenetic modulation, contributing to hematopoietic homeostasis and inflammation regulation.212 There are many phytochemicals and short-chain fatty acids regulating DNA methylation and histone modifications, participating in preventing chronic inflammation that worsens neurocognitive and cardiac performance and leads to metabolic disorders.213
Restoration of youthful functions in aged cells by epigenetic rejuvenation
Ageing is unavoidably accompanied by a diminished capacity to maintain tissue integrity and function. Cell or tissue rejuvenation without dedifferentiation is known as epigenetic rejuvenation, and it leads to a more youthful functional state and reversed ageing molecular markers.214 The central epigenetic regulatory mechanisms are based on the enzymes that modulate DNA and histones (methyltransferases, demethylases, acetyltransferases, deacetylases).17 The epigenome reprogramming can initiate ageing plasticity during heterochronic parabiosis, caloric restriction, or cellular reprogramming.215 These epigenetic modifications exhibit a strong capability of youthful function restoration in aged cells. In addition, epigenetic modifications can target several druggable pathways. In addition, senotherapy can increase lifespan, restore the functionality of bone marrow, muscle, and skin progenitor cells, enhance vasomotor function, and decrease the onset of atherosclerosis.216
Metabolic manipulation
Mitochondria-based metabolic remodeling
Mitochondria is responsible for the ATP production required for organisms and apoptosis, autophagy, the creation of iron-sulfur clusters, amino acid synthesis, copper and lipid metabolism.217 The rates of fission and fusion govern the shape, size, and network of the mitochondria, which vary according to both internal and external cues like metabolism and stress.218 The metabolic condition can affect the form and function of mitochondria, consequently influencing organ function. Conversely, the disturbed mitochondrial dynamics, like genetic ablation of mitochondrial fusion and fission components, also cause metabolic changes. Age-related disruption in energy balance and an increased propensity for age-related illnesses may be caused by the reduction in mitochondrial activity. The crosstalk between mitochondria and other organelles like lysosomes might also lead to increased oxidative stress, reduced ATP production, and breakdown of cellular catabolic mechanisms, ultimately inducing metabolic imbalance and ageing.219
The mitochondrial functions in energy homeostasis and metabolism are closely associated with protein quality control factors in disease and age-related disorders, such as PTEN-induced putative kinase 1 (PINK1), Parkin, and TNFR-associated protein 1 (TRAP1).220 SIRTs control the mitochondrial metabolic checkpoint which can control stem cell maintenance and quiescence, and dysregulation of the checkpoint can deteriorate the function of aged stem cells.221 Aiming at the mitochondrial metabolic checkpoint might rejuvenate ageing stem cells and improve the functions of ageing tissue. Mitochondria also segregate many critical metabolic pathways, like the TCA cycle, fatty acid β-oxidation, and the one-carbon cycle. The synthesis of mitochondrial metabolites in these pathways might be involved in additional mechanisms that control stem cell activity and fate decisions.222 Moreover, the mitochondrial UPRmt regulates many genes involved in protein folding, ROS defenses, metabolism, assembly of iron-sulfur clusters, and modulation of the innate immune response.223 Independent of the generation and aggregation of ROS, defective mitochondria also play a significant part in the ageing process. The Mitophagy pathway functions as a crucial mitochondrial switching that guides bioenergetic transition and metabolome remodeling attributes, to eventually define the effectiveness and quality of nuclear reprogramming and stemness transition in somatic cells.224 Mitophagy-induced rejuvenation of mitochondria governs the shift of bioenergetics and metabolome, hence facilitating a change in their capacity for cellular development. Thus, mitochondrial targeting or mitophagy regulation can promote metabolic remodeling, playing potential roles in rejuvenation and regeneration.
Oxidative stress
Deregulation of the redox state causes a rise of peroxides, ROS, and free radicals, which are collectively known as oxidative stress. Adaptive cellular responses to pathogenic challenges in ageing and age-associated disease tolerance, such as ischemia tolerance, can also be greatly benefited by moderate oxidative stress caused by diverse stressors.225 However, an imbalance between the generation of ROS and cellular antioxidant defenses can result in excessive oxidative stress, which accelerates ageing and the pathogenesis of illnesses like cancer.226
It has been demonstrated in numerous human cohorts and animal experiments that oxidative damage and inflammation might promote a state of susceptibility and raise the possibility of unfavorable health outcomes.227 Studies have suggested that the declined ability in response to oxidative stress with ageing is involved with the activated expression of Nrf2/EpRE signaling and its target antioxidant genes.228 Brahma-related gene 1 (BRG1) has been proved to protect cells from oxidative stress harm by encouraging the synthesis of antioxidants and inhibiting the generation of ROS.229 There are also many pathways, including MAPK pathway, PI3K/Akt pathway, heat shock proteins, p53, and NF-κB pathway, playing protective roles in combating oxidative stress for healthful ageing and longevity.230 Thus, these pathways might be effective mediators of oxidative stress for metabolic improvement and rejuvenation.
For oxidative damage regulation, bioactive exosomes have antioxidant effects on reducing the excessive ROS, promoting intracellular anti-oxidative stress defense, immunomodulation by blocking excessive ROS and changing mitochondrial function.231 Antioxidants targeting mitochondria, such as MitoQ and tiron, can penetrate the mitochondrial membrane and neutralize ROS at the core of the origin.232 Many compounds, like dibenzopyrone phenolic derivatives, caused the nuclear accumulation of Nrf2, stimulated Nrf2-governed cytoprotective gene expressions, and enhanced cellular antioxidant capacity.233 In addition, interventions including CR and exercise training targeted at restoring endogenous antioxidant ability and cellular stress reaction can contribute to successful vascular ageing and decreased risk for cardiovascular disease.234 In skeletal muscle, inflammation and oxidative stress are also the primary pathogenic features of ageing, and they are intimately linked to the onset and progression of sarcopenia. There are some promising antioxidant or anti-inflammatory substances, like minerals, vitamins, fatty acids, and antioxidant phytochemicals, to postpone skeletal muscle ageing and the onset of sarcopenia.235
Autophagy modulation
Autophagy is a conserved, physiologic, and self-protective mechanism that supports cellular homeostasis and stress adaption. Autophagosomes with bilayered membrane vesicles can capture the degraded cellular components and subsequently merge with the lysosome to digest long-lived proteins, excess or damaged organelles, and misfolded or aggregation-prone proteins.236 There are three distinct forms of autophagy, namely macroautophagy, microautophagy, and chaperone-mediated autophagy. Depending on the selective autophagic degradation of several organelles, autophagy is subdivided into mitophagy, aggrephagy, pexophagy, reticulophagy, nucleophagy, lysophagy, xenophagy, lipophagy, ferritinophagy, and glycophagy.237 The autophagy process is important for maintaining cellular energetics, cellular reprogramming, organellar remodeling, immunity regulation, metabolism, and cellular survival.
Autophagy serves as one of the central pathways in the protection against functional loss and increased vulnerability to ageing process and age-related disorders. The activity of autophagy and the autophagy gene transcription by specific transcription factors, epigenetic changes, and microRNAs have emerged as crucially conserved pathways for promoting lifespan.238 In autophagy pathways, multiple points with age including autophagosome biogenesis, cargo loading, intracellular transport, and autophagosome-lysosome fusion or acidification have shown ageing or disease-associated deficits. Enhancing the function of the autophagy-lysosome system can eradicate age-related organelle degeneration, which might have regenerative benefits for cellular rejuvenation. The regulator of autophagy is the classical mTOR pathway and some mTOR-independent signaling cascades, such as MAPK-ERK1/2, STAT2, AKT/FOXO3, and CXCR4/GPCR, converge into PI3K signaling node.239 The histone deacetylase SIRT1 also regulates autophagy to influence ageing and age-related disorders.240 Furthermore, autophagy can eliminate the redundant production of ROS and maintain the cell proliferation capacity and regenerative ability.241 In addition, autophagy activation in ageing stem cells by genetic and pharmacological methods can improve cell properties and regenerative functions.242
Activation of circadian clock
Basic molecular factors controlling circadian rhythms
The molecular circadian oscillator consists of transcriptional and translational feedback loops that are interlocked. BMAL1 and CLOCK (or NPAS2) have the ability to heterodimerize and bind to E-box elements of many clock-controlled genes to drive transcription.243 Early in the circadian night, PERIOD (PER) and CRYPTOCHROME (CRY) complexes are formed and suppress the transcription that is mediated by BMAL1 and CLOCK. As PER and CRY degrade over the night, negative regulation of BMAL1 and CLOCK is released, allowing the beginning of a new circadian day. These core clock proteins regulate 24 h oscillations in gene expression and activity, and at protein levels. In addition, the molecular clock controls the rhythmic expression of genes related to numerous cellular processes and nutrient-sensing pathways, which provides feedback to the primary clock system.244 The suprachiasmatic nucleus (SCN) in mammals serves as the body primary circadian clock, coordinating the timing of rhythmic activity with the light/dark cycle. Almost all the major genes implicated in the modulation of the circadian cycle have been found to induce alterations in circadian rhythms in their absence and to have an impact on health status.245 With increasing age, the transition toward morningness involves in epigenetics inside the molecular clock, modifications in the master clock, and downstream oscillator sensitivity to SCN signals.246 Many genes and proteins in circadian expression occur in phase shifts and decreased amplitude.247 The circadian body temperature, melatonin release, sleep-wake periods, patterns of locomotion, and drinking behavior can all vary with age. Thus, improving intracellular synchronization and the synchronization between SCN network and central and peripheral clocks might restore accuracy and stability to the ageing circadian system. Melatonin, produced in the pineal gland and mitochondria, participates in complicated intracellular signaling pathways with anti-ageing, antioxidant, chemopreventive, immunostimulatory, and tumor-inhibitory functions.248 SIRT1 modulation is associated with the activity of clock machinery and might resynchronize the dysfunctional cellular key clock circuits.249 Moreover, many environmental factors, like light exposure, lifestyle, and societal factors, can modify the circadian clock phase.
Circadian clock promotes stem cell rejuvenation
Stem cells contain a functional circadian clock whose rhythmicity contributes to the multipotent cell properties in constant renewal and injury response. Multipotent stem cells from various organs also have distinct clock gene expression profiles with various amplitude ranges.250 Meanwhile, dephased oscillators can provide stem cells the source of heterogeneity, to respond effectively to varied cues.251 Numerous embryonic and adult stem cell-dependent activities, including hematopoietic progenitor cell migration, follicular cycle, osteogenesis, regenerative myogenesis, and neurogenesis, have been linked to rhythmic oscillations and circadian clock regulation.252 The regulation of stem cell division and differentiation by the circadian clock is crucial for adult tissue regeneration. Histone alterations, DNA modifications, non-coding RNAs, huge multisubunit chromatin remodeling complexes, and additional epigenetic changes are also significant points in the circadian modulation of stem cell destiny.250
With ageing, stem cells receive signals from endogenous and external factors operated through circadian rhythms and epigenetic clocks. The circadian output and oscillator system have adapted to the particular homeostatic requirements of the adult stem cell region in the young organism. But the circadian functions of ageing stem cells might switch toward a stress-dominated program.253 Liang et al. found that the overexpression of CLOCK could rejuvenate physiologically and pathologically aged human MSCs.254 Mechanically, nuclear lamina proteins and KAP1 formed complexes with CLOCK, which preserved heterochromatin architecture and stabilized repeated genomic regions. Hence, circadian clock regulation provides opportunities to rejuvenate stem cells for various tissue engineering approaches.
Tissue-specific circadian clocks influence organ rejuvenation
The oscillations of peripheral clocks in numerous peripheral tissues, including the heart, liver, adipose tissue, retina, and multiple brain regions, can be controlled by synchronizing the circadian clock produced by SCN neurons.255 Meanwhile, there is a portion of transcripts expressed cyclically in each peripheral tissue, controlling the function of peripheral tissues.256 These tissue-specific circadian genes govern biological processes necessary for the preservation and dynamic modification of organ functions during the circadian cycle. Cell-autonomous circadian oscillations also have a substantial impact on the physiology and pathology of peripheral organs.257 In addition, the diurnal pattern of the light-dark cycle has a significant effect on the central SCN clock, while the feeding-fasting rhythm constrains the circadian rhythm of peripheral tissues.258
Many studies have suggested that circadian rhythms might be in a noticeably different phase during development in one cell or tissue type compared to another part of the body.259 Circadian programs are tissue-specific and even varied in the same tissue under distinct physiological states.260 During ageing, diminished or asynchronous circadian oscillations can alter signaling networks and tissue-specific gene expression profiles. These alterations in tissue-specific clocks might affect immune hyperactivation with ageing.261 Thus, the tissue-specific targeting of clock subunits by pharmaceutical techniques might aid to the treatment of age-related diseases and recover circadian coherence with a chronically disrupted clock. Besides, tissue susceptibility and reactions to toxicity also change during the circadian cycle, which indicates the development of drug timing administration.262
Designing strategies to promote youthful states in cells and organisms
The molecular mechanisms that mediate cellular and organ ageing provide therapeutic targets for cellular rejuvenation. The increasingly rejuvenative approaches have been developed, and the schematic overview of strategies for cell rejuvenation is shown in Fig. 4. It contains cellular reprogramming, clearance of SCs and SASP inhibitor, metabolic manipulation, the restoration of aged stem cell function, microenvironment remodeling, resetting the circadian clock, immune rejuvenation, and heterochronic parabiosis. Especially, cellular reprogramming can rejuvenate the terminally differentiated cells to the pluripotent state or epigenetically unstable intermediates, and also to another desired cell type for tissue repair. The reprogramming technology mainly includes iPSCs, partial reprogramming, and direct reprogramming (Fig. 5).

Schematic overview of strategies for cell rejuvenation. Various strategies have been developed for cell rejuvenation that leverage intrinsic and extrinsic factors, including epigenetic reprogramming, genetic enhancement, autophagy modulation, and metabolic manipulation. Furthermore, small molecules, growth factors and cytokines, blood factors, iPSC technology, clearance of senescent cells and SASP, microenvironment regulation, and circadian clock modulation can also exert great influences on cell rejuvenation. iPSCs induced pluripotent stem cells, SASP senescence-associated secretory phenotype. Created with BioRender.com

Reprogramming approaches for rejuvenation. The iPSC-mediated cell reprogramming is a protocol that somatic cells are first dedifferentiated into iPSC and then differentiated into the desired somatic cells. Partial reprogramming refers to a short exposure to Yamanaka factors only generates intermediates with high plasticity. Transforming somatic cells from one lineage to another without transitioning through intermediary pluripotent stages is known as direct lineage reprogramming. iPSCs induced pluripotent stem cells, ESCs embryonic stem cells; Oct3/4, Sox2, Klf4, c-Myc, OSKM. Created with BioRender.com
Resetting epigenetic clock in aged somatic cells by reprogramming strategy
Reset epigenetic clocks by reprogramming to a fully pluripotent state
The biological age of our cells, tissues, and organs is determined by an epigenetic clock based on alterations in the DNA methylation profile. A change in permission for future development and a return to a more flexible and pluripotent state are made possible by the removed DNA methylation instruction, and the production of iPSCs can also get around epigenetic constraints and change DNA methylation patterns to support youthful states in cells.263 According to the extensive DNA methylation analysis, the aberrant hyper-methylation spread randomly over the genome during the long-term iPSC reprogramming and then gradually diminished. These cells eventually adapted to closely resemble ESCs.264 Another study that used global DNA methylation and hydroxymethylation analyses demonstrated that DNA demethylation was a miR-29a depletion-mediated process during early reprogramming, and iPSCs produced from miR-29a depletion were epigenetically more similar to ESCs.265 The reprogramming technology to generate iPSCs has matured greatly. iPSCs can be derived from different tissue-specific cell types, such as human endometrial cells, endothelial cells in the placental artery, amnion-derived cells, fibroblast, blood cells, and even cancer cells.264,266 In the terms of differentiation degree, terminally differentiated cells and adult stem cells also could be reprogrammed into iPSCs. Senescent and centenarian cells are revived through pluripotent reprogramming, and the iPSCs produced from these cells have reset telomere length, gene expression profiles, oxidative stress, and mitochondrial metabolism.267 In addition, the fact that these SCs-derived iPSCs can re-differentiate into fully rejuvenated cells further demonstrated that reprogramming is not constrained by cellular senescence and that age-related cellular physiology is reversible.267 There are a variety of strategies to produce iPSCs, including viruses-mediated gene manipulation (e.g., adenoviral gene delivery), non-integrating episomal plasmids, proteins cell membrane-penetrating, the direct transfection of RNA and chemical reprogramming induced by small molecule compounds.268 The evolution of iPSC technology is around the improvement of reprogramming efficiencies and the avoidance of gene risk. Currently, given that each random integration event of a retrovirus poses a possible genetic risk, despite some of these approaches have produced lower efficiency than conventional reprogramming of retroviral administration, they constitute a step closer to therapeutic application.
Reduction in epigenetic age by partial reprogramming
Using partial reprogramming, the epigenetic age of cells can be reduced without losing cell identity, suggesting that full reprogramming of iPSCs is not necessary to reverse ageing of somatic cells. The previous study reported that OSKM + LIN28-mediated partial reprogramming in senescent human fibroblasts could result in the recovery of the high mobility of histone protein 1β, a feature characteristic for young fibroblasts.198 Likewise, multiple cellular features of dermal fibroblasts from middle age donors were rejuvenated following partial reprogramming with forced expression of OSKM, including transcriptome, epigenomes such as histone methylation and DNA methylation, and functional rejuvenation.269
Using a non-integrative partial reprogramming protocol with a cocktail of mRNAs carrying OSKM + LIN28, naturally aged human fibroblasts and endothelial cells exhibited a multifaceted alleviation of cellular ageing, such as resetting the epigenetic clock, reducing inflammatory responses in chondrocytes, and restoring youthful regenerative responses to age, in each case without changing cellular identity.270 Besides, OSKM-mediated partial reprogramming also restored youthful gene expression in adipocytes, MSCs, and myogenic cells, and the identity trajectory mapped by single-cell genomics revealed the temporary suppression of somatic identity programs.271 Partial reprogramming emerges as a powerful tool for the restoration of the cell youthful state.
In vivo reprogramming in ameliorating age-related physiological alternations
In vitro cellular reprogramming has observed alleviation of age-associated phenotypes and the reset of age clock. Due to the physiological complexity of the ageing process, it is necessary to study in vivo reprogramming to gain a deeper understanding of how it can affect cellular and organismal ageing. Ocampo et al. reported that transient cyclic induction of OSKM in vivo ameliorated age-associated hallmarks, extended lifespan in progeroid mice, and promoted tissue repair from streptozotocin-induced pancreatic damage as well as cardiotoxin-caused muscle damage, with no resulting teratoma formation.272 In the physiological ageing mice, the rejuvenating effects of long-term partial reprogramming are manifested in different tissues, including the skin and kidneys, as well as at the organismal level, and such effects were related to a reversion of the epigenetic clock and metabolic and transcriptomic changes, containing the decreased expression of genes mediating inflammation, senescence, and stress response pathways.273 Similar studies in naturally aged mice also reported that a single period of OSKM expression can alter epigenetics, transcriptomes, and metabolomes, resulting in a younger configuration in various tissues and in the serum, such as the reversal of DNA methylation changes and transcriptional changes, and the restoration of some ageing-related serum metabolites and biomarkers to young levels.193 In vivo reprogramming may represent an avenue for facilitating tissue repair. For example, expression of OSKM specifically in hepatocytes can dedifferentiate adult hepatocytes into progenitor cells and promote cell proliferation concurrently, and functionally, short-term in vivo reprogramming increases liver plasticity and promotes regeneration.274 Heart-specific in vivo reprogramming also induces adult cardiomyocytes to a fetal state, conferring regenerative capacity to adult hearts. and the short-term OSKM expression reduces myocardial infarction-induced damage and improves cardiac function.274 In vivo reprogramming might also hold a great promise against age-related diseases. In vivo reprogramming with short-term cyclic expression of OSKM also inhibits the progression of intervertebral disc degeneration (IDD), and significantly improves senescence-associated phenotypes in ageing nucleus pulposus cells (NPCs).275 Exception for OSKM, in vivo cyclic expression of the Forkhead box protein M1 (FOXM1) also ameliorates natural and progeroid ageing phenotypes and increases health span.276 Despite the great progress of in vivo reprogramming for cellular rejuvenation, there are some problems and limitations facing us. The first is teratomas, a type of tumor that could be cancerous, which may be attributable to too much OSKM induced by inappropriate dosage.277 Then, in vivo reprogramming is not enough for systemic rejuvenation, because the reprogramming factors fails to be delivered to certain tissues, which also explains why some study switched their focus to local rejuvenation such as NPCs.275 Finally, several ageing hallmarks such as nuclear and mitochondrial DNA mutations, metabolic aggregates in the cell and extracellular matrix, cannot be reset through in vivo partial reprogramming.
Epigenetic drugs
Currently, “epigenetic drugs” such as those that modulate enzymes that cause epigenetic changes are being considered as potential treatments against ageing. DNA methylation-targeted drugs mainly contain DNA methyltransferase inhibitors, like 5-Azacytidine. The removal of DNA methylation is a prerequisite for epigenetic rejuvenation and the generation of iPSCs.265 5-Azacytidine was used to facilitate the reprogramming of human fibroblast into iPSCs by overcoming epigenetic barriers.268 Besides, brief methylation inhibition with 5-Azacytidine could confer human dermal fibroblasts a relaxed chromatin structure and short plasticity, and the 5-Azacytidine-treated fibroblasts exhibited the upregulation of pluripotency gene expression and differentiation capacity towards other germ layers.278 Bromodomain and extra-terminal inhibitors (BETi) that work to repress transcriptional elongation of acetylated chromatin are also for stem cell rejuvenation. BETi has exhibited the effect to induce keratinocytes dedifferentiation, to enhance the migratory ability in keratinocytes in vitro, and to promote skin wound healing in vivo.279 Then, the high throughput chemical screening discovered I-BET151 can robustly contribute to the expansion of PDX1+NKX6.1+ pancreatic progenitors (PPs), and these expandable PPs (ePPs) maintain pancreatic progenitor cell status in the long term and can efficiently differentiate into functional pancreatic β (ePP-β) cells, which optimize the stem cell therapy in the amelioration of diabetes.280 Histone methylation inhibitors have been reported to re-activate stemness-related genes. The 3-deazaneplanocin A (DZNep) compound, which specifically inhibits H3K27me3 and H4K20me3, is crucial for activating OCT4 during iPSC reprogramming.281 Another intriguing family of anti-ageing drugs that can significantly contribute to the fight against ageing and age-related disorders are histone deacetylase (HDAC) inhibitors. HDAC inhibitors have the ability to reverse the deacetylation of histone tails and trigger the expression of specific genes.282
Rejuvenating strategy by clearance of SCs and decreasing SASP
Elimination of SCs using genetic approaches
Recent advances in our understanding of gene manipulation against age and age-related diseases have prompted several distinct interventional strategies. The first one is the repression of senescence-associated genes to delay or reverse senescence. For example, identifying the driving role of the histone acetyltransferase gene KAT7 in senescence in the Werner syndrome (WS) and Hutchinson-Gilford progeria syndrome (HGPS) models via genome-wide CRISPR-Cas9-based screening. Intravenous administration of lentiviral vectors containing Cas9/sg-KAT7 reduced liver ageing and increased lifespan in progeroid and physiologically old mice.283 Many genes driving senescence and mediating ageing-related disease have been revealed in the past year, and some resource tools also have been developed to facilitate the identification of genes related to cellular senescence and age, including SeneQuest (available at http://Senequest.net) and GenAge: The Ageing Gene Database (available at https://genomics.senescence.info). These ageing-associated genes provide the therapeutic targets for rejuvenation via gene ablation Then, targeting the inhibition of anti‐apoptotic regulators to increase the susceptibility SCs to apoptosis is another approach to eliminating SCs. Analysis of proteomic and transcriptomic datasets revealed senescent cell antiapoptotic pathways (SCAPs), covering Bcl-2 family members, ephrins, PI3K isoforms, p21CIP1, HIF-1α, and plasminogen-activated inhibitors 1 and 2 (PAI-1 and -2), were indeed more highly expressed in senescent than non-senescent cells, which is responsible for the resistance of SCs to apoptosis.284 The small interfering RNAs (siRNAs) against these anti‐apoptotic regulators were effective in killing SCs.285 However, more than one SCAP pathway mediate the resistance to apoptosis, so the gene ablation targeting a single SCAP pathway may be poor. Furthermore, the dominant SCAPs also vary with different cell types, which expands the limitations of genetic silence of SCAPs to remove SCs. Genetic strategy against senescence also can be achieved by overexpression of longevity gene to reset gene expression in SCs. Telomerase reverse transcriptase (TERT) and follistatin (FST) gene therapy using high-capacity cytomegalovirus vector by intranasal inhalation or injection significantly improved phenotypes associated with healthy ageing and extended lifespan in mice without severe side effects.286 Specifically, glucose tolerance and physical performance were significantly enhanced. Further, TERT reduced the telomere shortening associated with ageing, and both therapies halted the degeneration of the mitochondrial structure.286 A clinical trial about TERT gene therapy with adeno-associated virus (AAV) vector to reverse ageing has been approved, and it is also the first clinical trial of genetic editing against ageing that was registered in Clinicaltrials.gov in the world (NCT04133649). Another study reported the gene therapy with three longevity genes (FGF21, sTGF-βR2, and αKlotho) treated multiple age-related diseases.287 In obese and diabetic mice, the findings demonstrated that FGF21 alone totally reversed weight gain and type II diabetes, and that when combined with sTGF-R2, renal fibrosis in mice reduced renal atrophy by 75%. Administration of sTGF-βR2 alone or in combination with two other genes improved cardiac function by 58% in mice with heart failure, indicating that the combination of FGF21 and sTGF-βR2 successfully treated all four diseases with improved health and survival. Importantly, the injected gene did not change the animal’s native DNA, remained separate from its natural genome at all times, or transmit between living things or to subsequent generations.287 These results demonstrate the potential of single or combination gene therapy to treat ageing and ageing-related diseases.
Pharmacological intervention targeting SCs
The association between SCs burden and health span helps to discover chemicals that selectively remove senescent cells. The fact that SCs exhibit resistance to apoptosis supports the idea that these SCs rely on antiapoptotic, pro-survival mechanisms to prevent self-destruction.288 Recent advances have evoked the development of senolytic compounds that work to target the SCAP network nodes and impair the defenses of SCs against apoptosis. Based on bioinformatic analysis, forty-six compounds that target SCAP pathways have been identified as possibly senolytic.284 Senolytic drugs with established safety profiles or FDA approval for additional human purposes were initially and purposefully chosen for further study in order to speed the transition from the bench to the bedside, such as SRC/tyrosine kinase inhibitor dasatinib (D), natural flavonoids quercetin (Q) and fisetin (F). D has the ability to trigger apoptosis brought on by dependency receptors, like as ephrins, in part through blocking Src kinase.289 Senolytic flavonoids primarily induce apoptosis by blocking members of the BCL2 family such BCLxL, as well as HIF-1 and other SCAP network elements.290 Because the different contributions of nodes across the SCAP network in SCs subpopulations, senolytic drugs have cell or tissue-specific sensitivity. For example, senescent human pre-adipocytes or MSCs are susceptible to D but not Q or F, while senescent HUVECs are susceptible to Q or F but not D.284 Likewise, targeting a single SCAP may not be effective in getting rid of SCs due to the heterogeneity of SCAPs among various types of senescent cells. Either D or Q alone has the inability to eliminate senescent mesenchymal embryonic fibroblasts from Ercc1−/− mice and bone marrow mesenchymal progenitors from old mice, but the combination of these agents can do that.284 In vivo experiment revealed that, both in senescent cell-transplanted young mice and naturally aged mice, intermittent oral administration of D + Q alleviated physical dysfunction and reduced mortality hazard to 65%, and meanwhile increased post-treatment survival by 36% while.291 Senolytic agents that focus on a single SCAP node tend to trigger apoptosis in a restricted range of SCs types. Navitoclax (ABT-263), a potent antagonist of BCL-2 and BCL-xL, eliminates senescent myocytes and hematopoietic stem cells but not senescent pre-adipocyte.292 As specific BCL-xL inhibitors, A1331852 and A1155463 have senolytic activity against HUVECs and IMR90 cells. albeit not senescent human pre-adipocytes.290 The discovery of SCAPs promotes the occurrence of more senolytic strategies, and senolytic drugs hold great promise in treating ageing-related diseases.
Immune-mediated SC clearance
Despite the apoptotic SCs induced by senolytic strategies, such apoptotic cells are still finally removed by the immune system. NK cells, macrophages, and cytotoxic T cells are innate and adaptive immune cells that carry out immunosurveillance of SCs and play a crucial role in their elimination when they are young or under physiological conditions. The interaction between the activating NKG2D receptor and its ligands expressed on SCs determines the involvement of NK cells in the elimination of SCs. In cycling human endometrium, decidual senescence is required to support embryonic implantation, and acute decidual senescence can promote the release of IL-15 and attract uterine NKs, further selectively targeting and clearing senescent decidual cells through the interaction of NKG2D and DNAM1 receptors-mediated granule exocytosis.293 SCs can also be eliminated by tissue-resident macrophages or circulating monocytes (MOs). After delivery, MOs are crucial in removing senescent uterine cells to preserve postpartum uterine activity, which helps to maintain the likelihood that a second pregnancy would be successful.294 T cells are also essential players in immune surveillance and healthy longevity. Similar to NKs, CD8 + cytotoxic T cells can function by granular exocytosis and express the same receptor, NKGD2, to identify SCs.295 By identifying MHC II molecules, CD4+ cytotoxic T lymphocytes can directly destroy senescent tumor cells. Accumulating evidences have demonstrated that SCs could be cleared by immune cells, which could be a lever for lowering the burden of SCs. Future researches need to focus on new approaches, such as the use of chimeric antigen receptors (CAR) or modified T-cell receptors to enhance immune cells’ capacity to recognize and eliminate SCs, or explore various approaches including vaccines and small molecule immunomodulators, to enhance the selectivity and efficiency of immune clearance.
Inhibition of SASP
SASP inhibitors, also named senomorphics, target pathways such as p38 MAPK, JAK/STAT pathway, and ILs, further attenuating the SASP of SCs. Suppressing the SASP also has been an alternative strategy for combating cellular senescence-associated phenotypes or diseases. p38MAPK is the MAPK family member and also an important regulator of the SASP, and p38MAPK inhibitors including SB203580, UR-13756, and BIRB 796, potently suppressed SASP expression in SCs.296 JAK inhibitors repress SASP function, alleviate systemic inflammation, and eventually enhance physical function.297 Glucocorticoids act as potent inhibitors of selected pro-inflammatory cytokines (IL-6, IL-1α, and MCP-1).298 As a flavonoid, apigenin (4’,5,7,-trihydroxyflavone) could suppress the level of IL-1α, IL-6, and CXCL10, and attenuate inflammation as well as the related chronic diseases of ageing.299 ROCK inhibitor Y-27632 is also recognized as a senomorphic drug, which has the ability to reduce levels of pro-inflammatory cytokines secreted by senescent normal and dysplastic oral keratinocytes but with no effect on the permanent cell growth arrest.300 Alternatively, senomorphics may be achieved by specific neutralizing antibodies against individual SASP factors. Of note, unlike the intermittent administration of senolytic drugs, most SASP inhibitors are needed to maintain suppression of SASP in a manner of continuous treatment, which raises the likelihood of side effects compared to senolytics taken on an intermittent schedule.
Rejuvenating strategies to restore aged stem cell functions
Improving metabolism in aged stem cells
The metabolic disturbance is known to drive the function decline of adult stem cells with respect to ageing, and the mechanism by which nutrient-sensitive signaling pathways such as the mTOR and AMPK pathways play a central role.301 Targeting nutrient-sensitive signaling pathways to regulate metabolism homeostasis may be the alternative strategy to rejuvenate aged stem cells.
Rapamycin’s suppression of mTORC1 slows the ageing of mouse long-term stem cells (LT-HSCs) by maintaining their ability to self-renew and produce blood cells.302 Rapamycin also rescues adult EpSCs exhaustion and the resulting progressive hair loss in mice.303 In addition, rapamycin improves MuSCs function through induction of autophagy in the Ercc1-/Δ progeria mouse model.304 In the Zmpste24-/- progeria mouse model, rapamycin-treated primary MuSCs could restore differentiation and proliferation potential and reverse senescence.305 Despite the well-established effects of rapamycin on lifespan, it remains debated whether this drug is rejuvenating compound. Long-term (1 year) rapamycin treatment increased memory and learning in both young and old mice, but it also improved some of these traits in young animals, suggesting that rapamycin has beneficial effects that are age-independent.306
Metformin also contributes to metabolic-induced rejuvenation by regulating nutrient-sensitive signaling.306 Metformin has been shown to extend longevity and improve health in mice and C. elegans by activating AMPK in a LKB1-dependent mechanism.307 Metformin also prevents the AGEs-induced inflammatory damage and neuronal dysfunction in human neural progenitor cells through activating AMPK.308 Similarly, metformin rejuvenates ageing oligodendrocyte progenitor cells and restores CNS remyelination capacity by activating the AMPK-mediated mechanism.309 Metformin is also shown to directly inhibit mTORC1 for improving the stemness and alleviating senescence of stem cells, such as the reversal of ageing-associated characteristics in intestinal stem cells.310 Using metformin to improve metabolism for cell rejuvenation and the treatment of age and age-related diseases such as degenerative skeletal diseases is proved to be promising.311
Targeting DNA repair pathways to rescue aged stem cell functions
To maintain genomic integrity and tissue homeostasis, adult tissue-specific stem and progenitor cells have defensive mechanisms that reduce endogenous DNA damage. However, the DNA repair response in stem cells declines with ageing, which results in the loss of stem cell properties and DNA damage-caused cellular senescence and organ atrophy.312 Identifying a suitable target to promote DNA repair may contribute to rescuing the DNA damage-induced function loss in aged stem cells. NAD+, as hydride-accepting coenzyme, is involved in the DNA repair and preservation of genome stability, and the increased NAD+ level reduces DNA damage.313 NAD+ can act as a substrate for PARP and SIRTs and provide adenylate for DNA ligase IV, facilitating nuclear DNA repair.313 NAD+ levels can be increased by supplementation with nicotinamide mononucleotide (NMN), inhibiting NAD+-consuming enzymes, and regulating NAD+ biosynthetic enzymes, in which NMN is widely used. In telomere-dysfunctional animals, administration of the NMN preserves telomere length, inhibits the DNA damage response and p53, enhances mitochondrial activity, and reverses liver fibrosis.314 NMN alleviates retina damage, drops DNA damage by 50%, and eliminates SCs in age-related macular degeneration models.315 NMN protects skin cells such as keratinocytes from UV radiation-caused DNA damage by enhancing DNA repair, contributing to reverse skin ageing.316
Reversing stem cell ageing by improving protein homeostasis
The conducive effect of proteostasis loss on stem cell ageing inspires the strategy of protein homeostasis to protect stem cell function. Chemical/biochemical therapy is an important approach capable of inhibiting the protein aggregation process, and it has received extensive research in “protein misfolding neurodegenerative diseases (PMND)”. Numerous chemical chaperones, including 4-Phenylbutyric Acid (4-PBA) and its derivatives, have been shown to exhibit anti-amyloidogenic activity and to prevent misfolding protein aggregation. By increasing partially folded proteins and stabilizing their more compact native structures, 4-PBA can reduce the formation of unfolded aggregates and increase the pool of folding intermediates. It can also change the structure of Hsps to increase the exposure of hydrophobic surfaces, which improves Hsp chaperone activities.317 In the face of misfolding-induced stress, 4-PBA can elevate the proteostasis ability via activating the heat shock response (HSR) and UPRmt pathway.318 As dual-targeting drugs, chalcone-based derivatives, including nordihydroguaiaretic acid (NDGA), curcumin, resveratrol, quercetin, and isoliquiritigenin, can target both amyloid aggregations and the proteostasis network in vivo.319 The mechanism is partially linked to Hsps-containing protein complexes and ER stress-mediated cellular activity.
Restoring the stem cell pools in ageing tissues
The depletion of adult stem cell pool with age contributes to tissue degeneration, so maintaining tissue homeostasis into old age requires replenishing the stem cell pools. Stem cell transplantation is a therapeutic intervention, and it has been extensively researched as a way for the replenishment of regenerative cells to provide stem cells from an unaffected donor or gene-corrected autologous cells to the recipient. A large number of adult stem cells are available for autograft or allograft therapy, but only HSCs transplantation is widely accepted and used clinically, which has been demonstrated to be successful in treating hematologic disorders such as leukemia and lymphoma.320 Animal experiments revealed replenishing the stem cell pools with stem cell transplantation is also possible for other tissue. For example, EpSCs transplantation can regulate inflammation response, remodel the microenvironment of skin wounds, recapitulate tissue integrity and promote diabetic wound healing.321 MuSCs have the ability to facilitate myofibers regeneration, restore dystrophin expression, and markedly improve contractile function. More importantly, transplanted MuSCs also accessed the satellite cell compartment, replenishing the endogenous stem cell pool and taking part in injury repair.322 Of note, considering the susceptibility of stem cells to ageing, simple cellular replacement therapy with younger stem cells is insufficient. It may be worthwhile to pursue methods for reactivating residual stem cells or combining such procedures with stem cell transplantation. For instance, inhibition of the p38 MAPK signaling pathway in endogenous MuSCs can rescue muscle regeneration ability in ageing animals.323 Triboelectric stimulation boosts the pluripotency and differentiation potential of old BMSCs, enhances their proliferation and reverses senescence phenotypes. The mechanism by which is in a manner of MDM2-dependent p53 degradation.324 Therefore, triboelectric stimulation can be used to rejuvenate endogenously aged BMSCs and replenish the bone stem cell pool, further resulting in the acceleration of bone repair in elderly patients.325 In addition to adult stem cells, reprogramming-derived stem cells can act as alternative cell sources for transplants. A recent clinical trial reported implantation of iPSC-derived dopaminergic progenitors into the putamen (left hemisphere followed by the right hemisphere) of a patient with PD indeed improved clinical symptoms of PD after surgery.326 Reprogramming-derived stem cells are highly amenable to genetic correction and homogeneous than isolated adult stem cells, but there still exist major obstacles limiting the application, including low reprogramming efficacy and potential security concern.
Rejuvenating tissue-specific cells by targeting their microenvironment
Targeting extracellular signals to reverse stem cell ageing
Physiologically, extracellular signaling molecules are cues, such as EVs, neurotransmitters, growth factors, hormones, and cytokines, designed to transmit specific information to target somatic cells or adult stem cells. EVs function as a cutting-edge tool for stem cell rejuvenation because of their ability to transfer genes safely and with systemic effects. For instance, ESC-EVs can directly facilitate pressure ulcer repair in ageing mice through the rejuvenation of tissue-resided senescent endothelial cells.327 Neonatal MSCs-secreted EVs can deposit PCNA to reverse aged BMSCs and slow age-related degeneration.328 In addition, neurotransmitters have been reported to regulate the behavior of nerve-dependent tissue stem cells in wound healing.329 Lgr6+ EpSCs are a regionally restricted population of EpSCs that has the ability to generate all skin lineages and interact with nerves and specialize in wound re-epithelialization, and the activation of Lgr6+ EpSCs rely on neurotransmitters from neuroendocrine signaling and subsequently, promotes wound repair.330 These results suggested that targeting the activation of neuroendocrine signaling might be a promising approach to facilitate wound healing.
Rejuvenating aged cells by environmental preconditioning
Injured tissue has a hostile environment, including inflammation, immune impairment, hypoxic stress, and poor blood supply, which degrades stem cell function, promotes cellular senescence, and results in a low survival rate of transplanted stem cells in vivo. Therefore, it is essential for stem cells to remain viable and maintain their potency before inducing a strong repair response. The optimization of the culture environment, such as hypoxic pretreatment, may achieve preconditioning-induced protection for stem cells.331 The oxygen tensions in natural cell microenvironments appear to be substantially lower. Particularly, ESCs require low oxygen levels to survive, which start at embryonic implantation and last throughout fetal development. The absence of maternal circulation causes a hypoxic environment during embryo implantation, and in the early stages of pregnancy, the oxygen content of the uterine surface is typically only 2%.332 Placental oxygen levels only rise to about 8% even after the embryo forms the connection to the maternal vascular system.333 Relative hypoxia is a feature of the normal physiologic environment of ESCs, and thus, low oxygen concentrations enhance the more efficient growth of ESCs. Adult stem cells such as HSCs and BMSCs, similarly live in hypoxic conditions in vivo.334 For these reasons, hypoxic pretreatment in vitro promotes the long-term expansion of stem cells, delays replicative senescence, and improves the therapeutic potential.
Modulating ECMs for maintaining tissue homeostasis
The extracellular microenvironment’s chemical and mechanical characteristics are sensed by tissue-specific cells, which then produce responses that control a variety of cellular processes, such as expansion, migration, and differentiation, and activation, further maintaining tissue homeostasis. For example, integrin receptors-mediated cell adhesion to the ECM is necessary for cell cycle progression, particularly during G2 to M transition and early mitosis.335 In addition, the structural integrity of the ECM and integrin-associated proteins may also contribute to cellular energy metabolism, because the extracellular matrix proteins regulate nutrient and hormonal flux to balance the energy uptake.336 Changes in the biophysical characteristics of tissue affect how mechanical impulses are received by cells in a variety of disease states, inflammatory conditions, and ageing. Through the process of mechano-transduction, these mechanical cues are transformed into biochemical signals that affect immune cell functions such as cell activation, cytokine generation, metabolism, proliferation, and trafficking.337 As an integral component of all organs, the alternations of biomechanical and biochemical cues exhibit a huge effect on cell behavior and tissue homeostasis. ECM molecules are continuously produced and secreted throughout life, in both physiologically healthy and pathological states, and they regulate a wide range of biological functions, including stem cell differentiation, angiogenesis, innervation, and wound healing.338 Therefore, the modulation of ECM to induce desirable cell-specific responses for cellular rejuvenation is of great importance to tissue homeostasis. Because ECM has a more favorable pro-remodeling host immune response and can offer a natural, instructional microenvironmental habitat for functional tissue remodeling, ECM-based biomaterials have been emerging and exhibit great variability.
Restoring defective intercellular communications to extend healthy lifespan
Intercellular communication has double-edged–sword activities, contributing to tissue homeostasis maintenance but also detrimental in ageing and related diseases, and altered intercellular communication has been the hallmark of ageing.7,77 One of the most prominent and important changes in intercellular signaling that occurs with age is “inflammageing”. The dominant role of inflammation in ageing-related intercellular communication raises the potential of anti-inflammatory agents in lifespan. Aspirin is a common example because it can prolong mouse life and promote good ageing in humans.339 Moreover, age-related changes in the transcriptional landscape of tissues have highlighted the importance of NF-κB-induced inflammatory responses, and the targeted inhibition of this transcription factor may restore the altered intercellular communications for rejuvenation. The conditional expression of an NF-κB inhibitor in transgenic mice’s aged skin rejuvenates the tissue’s phenotype and restores the transcriptional signature associated with youth.340 The novel relationship between inflammation and ageing is that NF-κB-mediated hypothalamic immunity causes neurons to produce less gonadotropin-releasing hormone (GnRH), and this decrease in GnRH can be a factor in several age-related changes, including decreased neurogenesis, muscle weakness, skin atrophy, and bone fragility. Correspondingly, GnRH treatment rejuvenates ageing-impaired neurogenesis and prevents ageing development.341
Restoration of proper circadian clock for anti-ageing
Rejuvenation of the circadian clock by dietary restriction
The use of DR, including time-restricted feeding (TRF) and calorie restriction (CR), has many profound beneficial effects on ageing through the regulation of circadian clock.342 TRF, also known as limiting the time and length of food availability with no calorie decrease, can trigger clock-associated gene expression to drive rhythms in circadian mutant mice. For example, the transcripts, metabolites, and proteins in WT liver exhibit daily rhythms, whose rhythms are lost in clock gene knockout (CRY1-/CRY2-; PER1-/PER2-; BMAL1-) mice, while TRF partially rescues the disturbed rhythms in these mutant mice.343 In addition to the liver, TRF modifies the phase and amplitude of the skin’s circadian clock to rejuvenate ultraviolet radiation-caused skin ageing in old animals.344 For lifespan, TRF even drives a clock-dependent increase in autophagy activity to induce longevity extension.345 CR, a dietary regimen low in energy without malnutrition, can decrease the occurrence of age-related diseases and extends the lifespan via controlling circadian clock. It has been reported that the duration of two months for CR intervention in early life could enhance the amplitude of core clocks in liver.346 From the regulatory site, the central oscillator is impacted by CR, which also entrains the SCN clock. The SCN clockwork’s temporal organization, circadian outputs under the light-dark cycle, and circadian system photic responses are all influenced by CR during the daytime.347 BMAL1-dependent and BMAL1-independent pathways are the primary transcriptional and post-transcriptional mechanisms by which CR modifies the clocks. A functional circadian clock system is necessary for CR-mediated lifespan extension, as evidenced by the fact that CR fails to extend the lifespan of BMAL1 knockout mice.348 Comprehensively, the circadian clock system extends longevity through transmitting the anti-ageing signals brought on by DR.
Genetic regulation of the circadian clock rescues an aged phenotype
Circadian clock-associated genes modulate the extrinsic and intrinsic mechanisms in lifespan modulation and organ ageing. The circadian clock in intervertebral discs (IVDs) is functional and temperature-entrainable, and ageing disrupts the circadian rhythm of IVDs in an inflammation-dependent manner and leads to IDD.349 Restoring the dampened expression of BMAL1 protects against IDD via inhibiting RhoA/ROCK signals.350 The expression of CLOCK is increased in OA cartilage, and by reestablishing autophagic flow, ablation of CLOCK increases antioxidant enzyme activity, lowers ROS generation, and alleviates chondrocyte senescence.120 The downstream gene of CLOCK-BMAL1 complex CRY2 is suppressed in mice with ageing-related OA, which makes the loss of ECM homeostasis in cartilage.351 It also reported the low mRNA and protein expression level of CRY in the old Drosophila melanogaster. The mRNA oscillatory amplitude of multiple genes involved in the clock mechanism was dramatically increased in elderly flies by restoring CRY using the binary GAL4/UAS system, and the flies with higher CRY also displayed a remarkable extension of health span.352
Pharmacological activation of circadian clock for anti-ageing
Numerous parts of human physiology are regulated by the circadian rhythm, opening up windows for interventions that can be made by only giving medications when their targets are at the proper expression level to rescue. There is growing interest in developing small molecules that directly target the circadian system for medicinal benefits. Representative chemicals targeting core clock components include KL001, SR9009/SR9011, and Nobiletin. KL001 specifically interacts with CRY, which prevents ubiquitin-dependent degradation of CRY, leading to extending circadian time.353 KL001 suppressed glucagon-caused gluconeogenesis in primary hepatocytes and improved glucose tolerance in obese mice.353 KL001 also extends the lifespan and modifies the circadian rhythms of Drosophila melanogaster.354 The metabolic regulators SR9009 and SR9011 pharmacologically target REV-ERB receptors. By lowering fat mass and significantly enhancing dyslipidaemia and hyperglycemia, SR9009/SR9011 can change the circadian pattern of expression of a variety of metabolic genes to enhance energy expenditure and lower obesity.355 Nobiletin, a natural polymethoxylated flavone targeting agonizing retinoid acid receptor-related orphan receptors (RORs), is a clock amplitude-enhancing small molecule that has the ability to directly engage circadian cellular clocks to support metabolic health in illness models and healthy ageing in naturally ageing mice.356 Nobiletin is capable of ameliorating steatosis in obesity by restoring aberrant hepatic circadian rhythm,357 and alleviating cognitive deficits as well as the pathological aspects of AD, such as Aβ pathology, hyperphosphorylation of tau, astrogliosis-associated neuroinflammation, and oxidative stress.358 There are other small-molecule modulators with the function to regulate circadian rhythm, which do not directly target core clock genes, such as CKI-7 (CK1 inhibitor), Lithium (GSK3β inhibitor), and OPC-21268 (antagonists for arginine vasopressin receptors V1a).359 The role of these chemical regulators of circadian clock in the rejuvenation of ageing or ageing-related diseases remains being explored.
Organismal rejuvenation via immune system-targeting therapeutic approaches
Therapeutic targeting of immune cells
Immune system is interconnected with all the other systems in the body, and this systemic nature provides the potential opportunity that targeted modifications to a small group of cells (e.g., HSCs or T lymphocytes) for improving the health of various organ systems.
As the origin of blood cell lineages, HSCs are multipotent precursors population that can reconstitute the hematopoietic system and sustain immune homeostasis. Ageing HSCs have less self-renewal activity, fewer cell divisions, decreased homing efficiency and myeloid lineage-biased differentiation as well as reduced output of lymphoid progenitors.360 HSCs play a crucial role in the immune system, hence functional restoration of ageing HSCs has great significance to immune rejuvenation and organismal health. HSCs ageing is accompanied by alterations at the gene level, and genetic modulators by ablation of genes driving ageing or overexpression of rejuvenative genes might be strategies to prevent HSCs dysfunction, such as the deletion of the imprinted gene Grb10 or forced expression of Satb1.90 In addition, pharmacological intervention based on the pathophysiology of HSCs senescence also can promote rejuvenation of the HSC compartment, reconstitute the immune system activity and finally even increase overall lifespan. For example, supplementing elderly mice with a sympathomimetic that specifically targets the adrenoreceptor β3 (β3-AR agonist, BRL37344) greatly improved the in vivo function of aged HSCs.361 In progeroid mice, BRL37344 can decrease premature myeloid and restore the proximal association of HSCs to megakaryocytes.362
Immune lineage-mediated cellular rejuvenation mainly focuses on the restoration of T lymphocytes exhaustion caused by organismal ageing, due to its high susceptibility to ageing-associated changes to the immune system.363 Adoptive transfer of progenitor T (proT)-cells is the merging approach to facilitate T cells function recovery. Preclinical results have shown that proT cells could effectively engraft involuted ageing thymuses, achieve rapid long-term thymic reconstitution and accelerate T cell recovery.364 The process is regulated by receptor activator of NF-B (RANK) and receptor activator of NF-B-ligand (RANKL) interactions, which generate chemokine-rich niches within the cortex and medulla that probably encourage the recruitment of bone marrow-derived thymus seeding progenitors.365 ProT cells are limited to the recipient’s major histocompatibility complex (MHC) and produce host-tolerant T lymphocytes after undergoing positive and negative selection in the host thymus. Therefore, ProT cells avoid the clinical concerns brought on by graft-versus-host disease (GVHD), and it provides an alternative cell-based strategy to rejuvenate T-cell immunity clinically. Surprisingly, immunotherapy with T cell activation, such as the blockage of PD-1/PD-L1, provides a novel alternative to rejuvenate ageing phenotypes.366 In SCs, PD-1/PD-L1 axis inhibition could activate CD8+ T cells and improve senescence surveillance, further resulting in the reduced number of p16+ cells. Besides, anti-PD-1/PD-L1 administration also ameliorated age-associated function decline in vivo, including decreased SASPs and inflammation, and improved alveolar volume, fat accumulation in the liver, grip strength, and athletic ability.366 Clinically, immune checkpoint blockade (ICB) has been widely used, and thus, ICB against ageing is promising.
Other immune-targeting strategies
As the key site of T lymphopoiesis, the thymus orchestrates adaptive immune responses, and ageing-associated thymic atrophy or involution contributes to adaptive immune system deviations. Pharmacologic interventions to increase thymopoiesis have been reported, such as IL‐21, which exhibits beneficial effects on thymic function recovery and T cell output in the aged models.367 The effect is systemic but is also transient and limited. Cellular reprogramming provides an alternative avenue for thymus function restoration and immune rejuvenation. Chemical activation of thymus organogenesis program can direct human ESCs to differentiate into thymic precursor lineage, further promoting functional regeneration of human thymus in vivo.368 First treatment with activin A directs the fate of hESCs towards the definitive endoderm, and the subsequent regulation in signaling pathways of Wnt, TGF‐β, Shh, retinoic acid, BMP4 and FGF facilitates the generation of thymic epithelial progenitors (TEPs).368 hESCs-derived TEPs could yield functional thymic epithelial cells when engrafted into athymic mice. T cells produced in TEPs-recipient mice have functional properties capable of in vitro expansion and in vivo immune responses, and they were detected 10 weeks post-transplantation in the peripheral blood. However, the regenerative efficiency of hESCs-TEPs generated by these protocols is low and there is a progressive decrease in the quantity of T cells produced, which was not sustained over 22 weeks.368 Thymocyte lineage-specific gene FOXN1 manipulation can drive thymus regeneration and restore its function in the age murine.369 iPSCs also can be directed towards thymus fate by ectopic expression of FOXN1.370 FOXN1-mediated iPSC-TEPs contribute to supporting T cell development, and antigen stimulation can cause the produced T cell to activate, which reveals that FOXN1 can promote the functional regeneration of human thymus from iPSCs.370 Similarly, FOXN1 mediates ectopic thymus regeneration from fibroblasts, and the fibroblasts-derived thymus also can revitalize the thymic architecture overall, boost the number of naive T cells, and decrease the number of senescent T cells and inflammatory.371
Targeting systemic signals for organismal youthful state
Heterochronic transplantation and parabiosis for regeneration
In 2005, heterochronic parabiosis was initially demonstrated to reverse the age-dependent loss in stem cell function by restoring the proliferation and regeneration capacity of aged satellite cells and hepatic progenitor cells in naturally aged mice.372 Since then, heterochronic parabiosis has been used as an experimental system to examine potential systemic influences on lifespan, age-related diseases, and the process of organismal ageing. For example, in the central nervous system, heterochronic parabiosis showed young mice exposed to an old systemic environment or plasma from old mice exhibited diminished synaptic plasticity, and worse contextual fear conditioning and spatial learning and memory, while systemic infusion of young blood plasma reversed that process.373 It also revealed that plasma levels of chemokines—including CCL11 were linked to decreased neurogenesis in aged mice. Young mice with elevated peripheral CCL11 chemokine levels in vivo suffered adult neurogenesis loss as well as learning and memory impairments., which provided a new therapeutic target to reverse ageing-caused precipitous decline in neurogenesis.374 Similarly, experiments using heterochronic parabiosis also identify another circulating factor, named β2-microglobulin (B2M), which damages adult hippocampal regeneration function and cognitive performance in an ageing-dependent manner. Aged mice and young heterochronic parabionts have high levels of B2M in their blood and hippocampus, and the ablation of endogenous B2M can alleviate age-related cognitive loss and improve neurogenesis.375 In addition, heterochronic parabiosis discovered the potential therapeutic value of GDF11 in age-related cardiac hypertrophy and age-dependent deterioration of the neurogenic niche,376,377 and the contribution of Ly6G+ plasma extracellular vesicle to improving fracture healing in the elder.378 Heterochronic parabiosis has been a valuable experimental system for addressing some of the fundamental issues surrounding the systemic regulation of cell and tissue ageing, which provide corresponding therapeutic opportunities for organismal rejuvenation. Nevertheless, the key obstacles of parabiotic experimentation facing include the difficulty to control experimental procedures, the influences of shared organs, uncontrollable exercise, and an ambiguous blood-sharing onset.
Blood factors to revitalize aged organs and tissues
The persuasive evidence that blood factors affect organismal ageing has been provided by heterochronic blood exchange (HBE), which has shown that circulating factors not only restore youthful traits to aged tissues but also cause systemic senescence in the young organism. Thus, defining the blood mediators of the rejuvenating effects is of great importance to revitalize aged organs and tissues. The deep proteomic analysis from the large-scale population has confirmed many increased plasma proteins with ageing, like sclerostin, pleiotrophin, and transgelin, while epidermal growth factor receptor (ERBB1) and a2‐antiplasmin were decreased with age.379 Quantification of proteomic alternations revealed ageing protein waves across the lifespan, and for example, structural pathways such as the ECM are downregulated at a young age but increased in middle and old age.379 Metabolomic analysis has identified the blood metabolic profiles that change with ageing or ageing-relevant conditions. A nontargeted, quantitative metabolomics analysis in the blood of 15 young and 15 elderly individuals reported that 14 metabolites showed significantly remarkable age-related alternations.380 The increasing metabolites comprised citrulline, pantothenate and N6-acetyl-lysine, and were associated with declining renal and liver function, while the decreasing counterparts included NAD+, nicotinamide adenine dinucleotide phosphate (NADP+) and UDP-acetyl-glucosamine, which mainly were associated with redox homeostasis. A longitudinal examination of the impacts of ageing on the blood plasma metabolome also identified pathways enriched for age-related metabolites embodied tryptophan, nucleotide, and xenobiotic metabolism.381 HBE also gives clues to the role of circulating EVs in ageing rejuvenation. It was reported that aged mice exposed to young blood showed muscle regeneration, in which serum EVs containing α-Klotho play a key role, and administration of Klotho-enriched EVs into ageing mice could promote muscle regeneration.382 Specifically, identifying blood factors that promote or antagonize ageing can provide therapeutic opportunities for systemic rejuvenation. For example, dilution of old blood plasma with saline that contained 5% albumin to create a “neutral” age blood exchange (NBE), which met or exceeded the rejuvenating effects of promoting muscle repair, lowering liver adiposity and fibrosis, reducing neuroinflammation, and boosting hippocampal neurogenesis in elderly mice.383 These results suggested that blood factors might be the key players in ageing process, and targeting blood factors is expected to yield robust resetting of the systemic signaling to youth.
The importance of cellular rejuvenation in treating human diseases
The accumulation of SCs during ageing accelerates the onset of ageing‐related disorders and chronic diseases, including metabolic disorders, degenerative disorders, inflammation, autoimmune diseases, cancers, etc. Deeper insights into the role of cellular senescence in disease pathogenesis highlight the significance of cellular rejuvenation in treating human disorders, and also provides cues for rejuvenation therapies (Table 1). In addition, a variety of pharmacological treatments with the elimination of SCs or reversal of tissue and organ ageing have been developed for cellular rejuvenation (Table 2). The discussion that follows will thus concentrate on the advances in rejuvenation strategies for alleviating human diseases, and the details of clinical trials in this study were shown in Table 3.
Metabolic disorders
Diabetes
Diabetes, as a metabolic disorder featured by increased blood glucose, result from defective insulin function, impaired insulin secretion, or both, which has increased mortality in recent years. The known etiological basis of diabetes can provide hints for rejuvenation strategy for treatment. SCs contribute to the pathophysiologies of diabetes via their direct impact on pancreatic β-cell function, involvement in adipose tissue malfunction, and SASP-associated tissue inflammation.384 Senolytics for removing SCs and senomorphics capable of attenuating the SASP of SCs can ameliorate diabetes and its complications by alleviating insulin resistance and reducing tissue inflammation.385 Awaking immune cells to kill SCs is conducive to improving blood glucose regulation function in diabetes treatment.386 The increasing exhaustion and senescence of β cells with age is also the driver of diabetes, and the reversal of senescent β cells and enhancing its function contribute to the treatment of diabetes, such as novel drugs for antagonizing β cell senescence,387 regulating metabolism burden to protect β cell function,388 the inhibition of islet inflammation,389 in vivo reprogramming for β cell regeneration390 and epigenetic modifications to rejuvenate senescent β cell, etc.391 Despite various rejuvenation approaches for diabetes treatment, most are still primarily in a developing, preclinical stage. Currently, systemic interventions for diabetes treatment seem to be more likely from bench to bedside, which have been explored in the diabetes population. Intensive lifestyle therapy at the workplace has been conducted on the diabetes population, and it exhibited great beneficial effects on improving the diabetes symptoms, such as significant weight loss and conducive changes in fat mass, glycemic controlling, and multiple organ insulin sensitivity, in which the underlying biological mechanism contains muscle NAD+ production, SIRTs pathways, mitochondrial activity and adipose tissue remodeling.392
Obesity
Obesity is characterized as an abnormal or excessive accumulation of fat that presents a health risk. The signal transduction pathways mediating obesity pathophysiology have been widely explored. Currently, a variety of anti-obesity drugs targeting obesity-related regulation pathways have been explored and even under clinical trials, such as melanocortin-4 receptor (MC4R) agonists, neuropeptide y receptor y5 (NPY5R) antagonist and glucagon-like peptide 1 receptor (GLP-1R) agonist, etc. The mechanisms of these interventions against obesity are mainly related to the control of calory intake, glucose balance and thermogenesis.393 Inflammation and metabolic disturbances associated with obesity could be caused by cellular senescence, and thus elimination of SCs with senolytic interventions may help improve obesity clinically. The previous reported that, in obese mice, the combination of D + Q reduced circulating inflammatory mediators, increased insulin sensitivity, boosted glucose tolerance, and promoted adipogenesis.394 Metabolic disorders in obese are also attributable to adipocyte senescence and unhealthy adipose tissue remodeling, and likewise, senescent adipocyte ablation alleviates adipose tissue inflammation and improves insulin resistance.395 All the results suggest that the emerging strategy with the removal of SCs have the potential to address obesity-associated metabolic dysfunction as well as its complication.
Tissue regeneration
Tissue regeneration refers to the partial regeneration of an organism’s tissue that has been injured by external stimuli, which resulted from the regulation of tissue-residing cells and tissue microenvironment. Cellular rejuvenation for tissue regeneration is devoted to the enhancement of tissue-specific cell function and the regulation microenvironment. The repair of endogenous tissue-specific stem cells was observed in various damaged tissue, like lens stem (LES) cells for lens regeneration after cataract surgery,396 activations of Lgr6+ EpSCs to generate all cell lineage of skin,397 endogenous liver progenitor cell-driven liver regeneration,398 etc. These encourage the development of exogenous activation of tissue-residing stem cells to recapitulate damaged tissue, such as biomaterials-based delivery system featured by controlled and sustainable drugs or other biologically active substances release, in osteochondral regeneration, wound healing, and spinal cord repair, and so on.399 The dysfunction and senescence of differentiated tissue-specific cells and microenvironment imbalance are other key players that impact tissue regeneration. For example, SCs-matrix interaction is responsible for the difficulty in healing in chronic wounds, because the SASP and ROS production from SCs results in increased matrix proteolysis and inflammation, dysfunction in stem cell, impaired vascular endothelial cells and exacerbated inflammatory microenvironment, and in turn, the microenvironment disturbance further accelerates cellular senescence.400 Therefore, the strategies with the elimination of SCs, improvement of tissue cell function, or restoration of microenvironment homeostasis are promising in tissue regeneration. These approaches contain clearance of SCs with senolytics, exogenous stem cell transplantation for replenishing the stem cell pool, EVs and their engineered derivatives for alleviating cellular senescence, and wound inflammatory and chemical compounds for rejuvenating SCs, etc. However, targeting tissue cells and microenvironment fails to achieve the functional regeneration of the large tissue defect or other severe and irreversible tissue damage, such as deep second-degree skin burns, diabetic ulcers, and massive liver defects. Recent advancements in somatic cell reprogramming technology have brought to light the possibility of functional repair of damaged tissue. Impressively, the regeneration of sweat gland cells by epidermal cell, fibroblasts, and MSCs reprogramming not only promotes wound healing in deep second-degree burned skin, but also confers the sweat function, which achieves the functional repair of damaged skin.401,402,403 Ex vivo reprogramming has the disadvantages including the dependence on startling cell sources and potential contamination of in vitro manipulation. Currently, to overcome this obstacle, in situ regeneration based on in vivo reprogramming has developed rapidly, which is applicated to the regeneration of pancreatic β cells, induced cardiomyocyte-like cells, expandable neural stem cells, and sensory hair cells.184 Despite the great leap that has been made, in vivo reprogramming is still in its infancy and lots of bottlenecks need to be overcome.184 Comprehensively, functional tissue regeneration still remains great challenge facing us.
Degenerative disorders
Bone-degenerative disease
The term “bone-degenerative disease” refers to conditions that progressively deteriorate bones and are primarily defined by degeneration, in which osteoporosis is more prevalent. Clearance of SCs has become lucrative methodology for osteoporosis treatment. Age-related osteoporosis has been demonstrated to be improved by genetic elimination of p16,404 and a combination of D and Q also performed well in repairing bone microstructure as seen in radiation-related osteoporosis.405 D + Q for the treatment of osteoporosis is being tested in the clinical trial (NCT04313634). Senomorphics that prevent the production of pro-inflammatory proteins, like the JAK inhibitor ruxolitinib, significantly reduce age-related osteoporosis by possibly suppressing certain factors like IL6, IL8, and PAI1.404
OA is a degenerative disease of the cartilage that develops with ageing and shares a pathogenesis with the SASP and senescence of joint tissue cells. SASP has been linked to cartilage deterioration, and age-related mitochondrial dysfunction and accompanying oxidative stress may cause senescence in joint tissue cells.406 Senolytic agents and senomorphic therapy have potential therapeutic value and implications in OA treatment. Navitoclax and UBX0101 are potential senolytic agents that work to alleviate OA, and the trial of UBX0101 in OA has been completed clinically (NCT03513016). Fisetin is a more selective senolytic candidate with lower hematological toxicity, which can activate SIRT1 to block the inflammatory response in OA chondrocytes,407 and Fisetin is now undergoing a clinical trial as a treatment for OA (NCT04210986). Senomorphic agents targeting MMPs can reduce OA symptom, increase type II collagen and inhibit chondrocyte degeneration in WT mice.408 IL-6 receptor inhibitor tocilizumab is presently being tested in phase III trial for hand OA (NCT02477059).
Neurodegenerative diseases
Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) are three of the neurodegenerative diseases. The accumulating evidences have indicated the contributions of cellular senescence to AD pathophysiology, by which telomerase deficiency and telomere shortening, accumulation of β-amyloid (Aβ), tauopathy, and oxidative stress get involved.409 Removal of SCs pharmacologically can improve memory of AD by reduction of brain Aβ load and tauopathy. The first-generation senolytic ABT263 is able to clear senescent astrocytes and microglia, modulate tau aggregation and inhibit tau-associated cognitive decline.410 Likewise, D + Q senolytic treatment reduced Aβ-related oligodendrocyte senescence, improved cognitive impairments, and decreased Aβ load and neuroinflammation in AD mice.411 In another way, D + Q also decreases cortical Tau‐containing neurofibrillary tangles (NFTs) burden, brain atrophy and neuron damage.412 However, senolytic therapy is rarely reported in treating PD and ALS. Stem cell-based therapies against neurodegenerative diseases hold promise. MSCs can enhance spatial learning and prevent memory impairment in AD via various mechanisms including reducing Aβ plaques and tau hyperphosphorylation, reversing microglial inflammation, and promoting anti-inflammatory response.413 Currently, many clinical trials of MSCs in AD treatment have been approved (e.g., NCT03117738, NCT04040348, and NCT02672306). The transplantation of NSCs with paracrine effect can decrease tau and Aβ levels, alleviate neuroinflammation, improve neurogenesis and enhance cognitive function via releasing neuroprotective or immunomodulatory factors.414 Current stem cell sources that have been implicated in PD include NSCs, human ESCs, iPSCs, as well as directly induced dopamine neurons.415 In addition, some clinical trials for treating ALS with stem cells were completed and it also gained desired results in various countries, including autologous MSC, endogenous MSC mobilization, NSC (NCT05306457), T-cell vaccination combined with autologous MSC and NSC therapy offers proof-of-concept that stem cell grafting as a therapy for ALS is doable and supports a continuous emphasis on perfecting stem cell-based therapeutic techniques to gain maximum benefit in ALS.416 Despite certain results of stem cell therapies that have been achieved neurodegenerative diseases, the disadvantages of this approach including the demand for immunosuppression and a neurosurgery operation, the poor maintenance of stem cell properties, and imperfect cell delivery systems, still need to be solved.
Cardiovascular diseases
The incidence and prevalence of a wide range of cardiovascular diseases (CVD) increase as a function of age. Molecular pathways involved in cellular senescence, oxidative stress, insulin signaling, autophagy, and inflammation may link the cardiovascular homeostasis deterioration.417 Cellular reprogramming is an emerging technology for cardiac regenerative medicine, and it works to treat CVD following three ways: (1) in vitro reprogramming of startling cells such as fibroblasts into cardiomyocytes via initiating cardiac development program, which provides cell source for cardiac regeneration;418 (2) in vivo cardiac reprogramming by lineage transcription factors or microRNA manipulation, in which resident cardiac fibroblasts are converted into cardiomyocytes in situ;419 (3) Partial reprogramming of cardiomyocytes into a fetal state to restore the function of senescent cells via temporarily reactivating the nascent developmental program in mature cardiomyocytes.420 The management of cardiac inflammation, such as targeting inhibition of IL-1 and inflammasome, also work to alleviate CVD, and oral NLRP3 inflammasome inhibitors and IL-1 signaling antagonist including Rilonacept, Canakinumab, and Anakinra have been under the clinical trials.421 Senolytics therapy is another potential approach to attenuate or prevent several CVD, which can improve myocardial function and alleviate atherosclerosis by clearance of senescent endothelial cells, senescent foam cells, and senescent T cells.422
Chronic lung diseases
Chronic lung disease (CLD) refers to a spectrum of persistent lung conditions that impair quality of life and are typically resistant to treatment. The conventional therapy regime for CLD treatment with nasal medications, immune-suppressive drugs, and surgery (in severe cases) only supports the decrease of disease progression rate. Cellular rejuvenation strategies based on stem cell therapy have ushered an alternative in the treatment of CLD. People with CLD may benefit from stem cells in the following ways: (1) lowering airway inflammation and preventing additional harm; (2) creating new, healthy lung tissue to replace any damaged tissue in the lungs; (3) promoting the development of new capillaries with tiny blood channels to enhance lung function.423 The candidate cell sources rejuvenating CLD include HSCs, ESCs, MSCs, and bronchoalveolar stem cells), and desired efficacy in stem cell therapies promotes more related clinical trials to be developed. Based on the differentiation potential of stem cell therapy, ex vivo lung bioengineering also offers exciting new therapeutic approaches for CLD, which contains physical stimuli such as air ventilation and blood perfusion and stem cell to create a functional lung organ.424 In the 3D microenvironment, physical stimuli relevant to lung biology and soluble factors can facilitate the establishment of bioengineered lungs through modulating stem cell fate, which is expected to be a future alternative for transplantation to treat CLD. However, the most important issues to be resolved are how to provide the lung with the ideal conditions of ventilation, perfusion, and oxygenation during the biofabrication process, as well as the best cell types, media, and growth factors (being likely various for the airway and vascular compartments).
Eye-related diseases
Macular degeneration, often known as age-related macular degeneration (AMD), is a serious eye condition underlined by photoreceptor loss and choriocapillaris and retinal pigment epithelium (RPE) degenerating (CC). The accumulating evidence shows that AMD is mainly attributable to oxidative stress, cellular senescence, and inflammation.425 The enhancement of autophagy can resist oxidative damage and mitigate the progression of AMD, and there are potential rejuvenative strategies activating autophagy for the alleviation of AMD, including mTOR inhibitors, hormones, melatonin, and antioxidants in the diet, where the interaction between the NFE2L2, PGC-1, p62, AMPK, and PI3K/Akt/mTOR pathways could be extremely important.425 As a result of the role of cellular senescence in the pathogenesis of AMD, it may be possible to use senolytics that kill senescent cells and stop the bystander effects of SASP to treat AMD. Recently, in a phase I study, Unity Biotechnology Company reports that a single dose of UBX1325 improved wet AMD patients’ visual acuity over the course of 24 weeks.
Glaucoma and age-related cataract (ARC) are also chronic, progressive eye diseases, and regeneration and re-functionalization of damaged trabecular meshwork (TM) or lens with stem cells have been regarded as a therapeutic alternative for these two conditions. A number of stem cells, including trabecular meshwork stem cells (TMSCs), ESCs, iPSCs, and MSCs, have demonstrated efficacy in maintaining intraocular pressure (IOP) equilibrium and restoring TM cellularity.426 These stem cells can home and integrate into the TM tissue, naturally differentiating into functional TM cells as well as secreting factors that facilitate tissue regeneration.427 For ARC treatment, the preservation of endogenous lens epithelial stem/progenitor cells after the extraction of cataractous lens is a new paradigm, which can achieve functional lens regeneration and avoid notable risks of complications caused by the implantation of an artificial intraocular lens.396
Chronic kidney diseases
Chronic kidney disease (CKD) is charactered by persistent kidney function decline, and its progressive nature often results in end-stage renal disease (ESRD). The growing evidences revealed that cellular senescence, stress-induced premature ageing, SASP, oxidative stress, and inflammation-mediated CKD development.428 By the selective elimination of SCs, ABT-263 can improve kidney function and stimulate tubular proliferation to reestablish a regenerative phenotype.429 Subsequently, a clinical trial suggested that senolytics with D + Q improved physical function in CKD patients (NCT02848131). Various drugs targeting SASP, like glucocorticoids, resveratrol and other protease inhibitors have been reported to attenuate CKD, and most of these mainly inhibit NF-κB signals and reduce ROS.430 Stem cell therapy has become the most likely effective therapeutic method to slow and even reverse CKD progression, in which MSC transplantation is widely explored due to their easy collection, low immunogenicity, and high paracrine potential. Both bone marrow MSC and adipose-derived MSC have been shown to have significant renoprotective effects in CKD, including a decrease in fibrosis and glomerulosclerosis as well as an intrarenal inflammatory infiltrate.431 The efficacy and safety of using MSC in CKD treatment have been tested in phase I clinical trials, in which two of them using autologous bone marrow MSCs (NCT02166489 and NCT02195323), and others are adipose-derived MSCs (NCT02266394 and NCT01840540). Trials employing MSCs to treat CKD may encounter a number of difficulties, including choosing the best administration method, assuring flourishing, tracking injected cells, immunological rejection, inadequate homing and engraftment, and low immune acceptance.432
Fibrosis
The term fibrosis describes the deposition of fibrous connective tissue as a reparative response to injury or damage, particularly during chronic inflammatory disorders, and Fibrosis leads to tissue architecture disruption, organ malfunction, and eventually organ failure. Based on that known molecular mechanism, numerous cellular rejuvenation strategies to treat fibrosis have been developed. A key contributor to fibrosis is metabolic reprogramming, which includes increased glycolysis, upregulated glutaminolysis, and accelerated fatty acid oxidation.433,434,435 Targeted inhibition of these metabolic processes can regulate collagen production, reduce ECM accumulation and alleviate progression to fibrosis. Miscommunications of macrophage-fibroblast interactions also result in pathological healing and fibrosis, and the restoration of macrophage and fibroblast crosstalk through IGF1 and platelet-derived growth factor C (PDGFC) can reduce tissue dysfunction.436 Integrin-mediated TGF-β1 activation is a significant pro-fibrogenic signaling pathway, making it a potentially alluring anti-fibrotic target. Pharmacologically blocking TGF-β1 signaling (e.g., SB431542) or inhibiting integrin signaling (e.g., GSK3008348, PLN-74809, and IDL-2965) have become widely accepted antifibrotic therapy.437 Various anti-integrin small molecule agents for idiopathic pulmonary fibrosis (IPF) treatment are under the phase 2 clinical trials (NCT03069989, NCT04072315, and NCT03949530). Strong preclinical studies have identified pro-inflammatory cytokines (e.g., IL-13, CCL2, and CCR2/CCR5) as fibrosis triggers, and likewise, the phase 2 trials of IL-13 inhibitors alone (NCT01266135, NCT00987545, NCT00581997, NCT01872689, and NCT01629667) or IL-4/IL-13 antibodies (NCT02921971 and NCT01529853) have been conducted in the treatment of IPF, skin keloids, and systemic sclerosis. In addition, other clinical trials to treat fibrosis through inhibiting inflammation are also explored, including anti-CCR2/CCR5 (NCT02217475, NCT03028740, NCT03059446, and NCT02330549) for liver fibrosis and non-alcoholic steatohepatitis, anti-IL-1 (NCT01538719) for systemic sclerosis, anti-IL-6 (NCT02453256) for scleroderma, and anti-CCL2 (NCT00786201) for pulmonary fibrosis. During the process of fibrosis, myofibroblasts exhibit remarkable inter-lineage plasticity, which provides opportunities for cellular reprogramming in fibrosis treatment. During cutaneous wound healing, neogenic hair follicles-derived BMP signaling can reprogram myofibroblasts into adipocytes.438 Correspondingly, the BMP pathway activation may be the preventive approach to reduce fibrosis and achieve scarless wound healing. Finally, in vivo reprogramming induces myofibroblasts into other functional desired cells, which can not only attenuate fibrosis and also realize in situ tissue repair.184
Inflammation and autoimmune disorders
The strong evidences link chronic inflammation to autoimmune disease pathogenesis, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), multiple sclerosis (MS), inflammatory bowel disease (IBD), psoriatic arthritis (PsA), primary Sjögren syndrome (PSS) and psoriasis, etc. Inflammatory cytokines (e.g., TNF-ɑ, IL-17, and IL-23), T cell-mediated inflammatory responses, and B cells-mediated antibody production play deleterious roles in the inflammation regulation on autoimmune diseases, which raises the potential for cellular rejuvenation targeted immunotherapy for autoimmune disease.439 Anti-cytokine therapy has revolutionized autoimmune disorders treatment. Targeting TNF-ɑ therapy including monoclonal antibodies (e.g., infliximab) and one receptor-Fc fusion protein (etanercept) is currently availability for RA, psoriasis, and IBD treatment. Inhibition of IL signaling also has been developed against autoimmune diseases, such as IL-1R antagonist (Anakinra), IL-1R1-IgG Fc (Rilonacept), anti-IL-1β mAb (Canakinumab), anti-IL-6 mAb (Sirukumab, Olokizumab, Clazakizumab, and Siltuximab), anti-IL-6R mAb (Tocilizumab, Sarilumab, and Vobarilizumab), anti-IL-17 mAb (Ixekizumab and Secukinumab), anti-IL-17R mAb (Brodalumab), anti-IL17A/F mAb (Bimekizumab), anti-IL-23/p40 mAb (Ustekinumab) and anti-IL-23/p19 mAb (Guselkumab, Risankizumab, Tildrakizumab, and Mirikizumab). Thereinto, monoclonal antibodies targeting IL-6 (Olokizumab: NCT02760407, NCT02760433, and NCT03120949; Clazakizumab: NCT02015520; Vobarilizumab: NCT02518620) have been registered for RA treatment, and other includes anti-IL-23 mAb (Mirikizumab: NCT03482011, NCT03535194, NCT03556202) and anti-IL17A/F mAb (Bimekizumab: NCT03598790 and NCT03766685) for psoriasis treatment, anti-IL-17 mAb (Secukinumab: NCT04181762) for SLE treatment, and anti-IL-23 mAb (Tildrakizumab: NCT03552276, NCT04314544, NCT04314531, NCT04991116) for PsA treatment. Immunosuppression of T cells activation by CTLA4-IgG1 Fc (Abatacept) and anti-CD40 mAb (Iscalimab) in autoimmune diseases also has been studied. Now, Abatacept (NCT02067910 and NCT02915159) and Iscalimab (NCT03905525) have been tested in phase 2 trials for PSS treatment. B cell depletion therapies show great potential for the treatment of autoimmune diseases, and likewise, a large number of monoclonal antibodies targeting B cells function also has been developed, such as anti-CD20 mAb (Rituximab, Ocrelizumab, Ofatumumab, and Ublituximab), anti-CD19 mAb (Inebilizumab), anti-BAFF mAb (Belimumab) and anti-BAFF-R mAb (Ianalumab), in which Ublituximab for MS (NCT03277261, NCT03277248, NCT04130997) and Ianalumab for SLE (NCT05126277) are under the phase 2 trials. Treatment and clinical results for people with autoimmune diseases have been changed by improvements in tailored immunotherapy, but immunosuppression based on decreasing inflammation may lead to serious side effects, such as increasing the risk of infection.440 Therefore, future researches seek to sustain immune defenses while inducing lifelong immunological tolerance.
Other ageing and age-related diseases
Other ageing and age-related diseases are also matters of great concern, including skin ageing, skeletal muscle ageing, reproductive system ageing, and ageing-related hearing loss. Skin ageing is currently a very concerning field, and the reversal or delay of skin ageing has a great market prospect. The process of skin ageing is multifactorial and is influenced by both intrinsic (such as time, genetics, and hormones) and extrinsic (such as UV exposure, and pollution) variables. Senescent skin cells, SASP, oxidative stress, inflammation, and autophagy, all mediate skin ageing pathophysiology. Fu et al. reported the phenomenon that epidermal cells can de-differentiation into stem cells in vivo during wound healing, and this discovery was the first to reveal that adult cells can become adult stem cells, which suggested that the high plasticity of epidermal cells and cellular fate was not one-street.441 The subsequent work of Fu et al. demonstrated that dedifferentiation-derived cells cultivated in vitro shared phenotypic and functional traits with EpSCs.442 Much more importantly, Fu et al. further discovered that aged epidermal cells had the ability to dedifferentiate into EpSCs, a process for which activation of the Wnt/-catenin pathway was crucial.443 Such breakthroughs not only make it possible to reverse skin ageing, but also promote the development of wound repair greatly. Currently, topical cosmetics like sunscreen, retinoids, or resveratrol are the foundation of current strategies for combating skin ageing. However, it is unclear how these products’ effectiveness in preventing skin ageing is related. Anti-ageing drugs such as senolytics or senomorphics may be useful for skin rejuvenation, because such drugs have anti-inflammatory and melanogenic properties that help protect the skin, prevent carcinogenesis, delay ageing, and reduce age-related diseases.444 Application of antioxidants to reduce and neutralize free radicals is an alternative method to slow skin ageing. The administration of natural products, especially polyphenols with known antioxidative properties, show the ability to enhance or remove the undesirable signs of ageing skin. Evidence-based knowledge suggests that the topical or oral use of various polyphenol-rich plants can prevent or reduce, besides others, undesirable conditions of skin ageing.445 Enhancing skin cell function and remodeling elastin networks hold promise for the restoration of skin youthful state. The developed strategies include stem cells and related EVs,446 pharmacological activation of autophagy,447 maintenance of circadian rhythmicity on skin-resided stem cell448, and regulation of metabolism homeostasis to restore mitochondrial integrity and metabolic output.449 Identifying key molecules involved in skin ageing makes great significance to skin rejuvenation. The role of COL17A1 in EpSC competition to orchestrate skin homeostasis and ageing and COL17A1 overexpression against skin ageing also were revealed.450 Therefore, targeting activation of COL17A1 is the potential angle for skin anti-ageing therapeutic intervention. Y27632 and apocynin have been identified as COL17A1-inducing drugs, which show the capacity to facilitate skin regeneration and reduce skin ageing.450 Similarly, the miR-31-CLOCK-ERK pathway is a significant contributor to and therapeutic target for skin ageing. By inhibiting this pathway, either through conditional miR-31 ablation or with clinically proven MAPK/ERK inhibitors, one can safely and effectively delay the ageing process of the skin.451 The molecular mechanisms of skin ageing provide the biological basis for the application of drug-loaded biomaterials to skin rejuvenation. For example, using tetrahedral framework DNA (tFNA) to design a novel bio-switchable miRNA inhibitor delivery system (BiRDS), which can fuse miR inhibitors within the framework and maximize the loading ability. MiR-31 inhibitors-loaded BiRDS have excellent skin penetration and high RNA delivery efficiency, significantly combating skin ageing.452
Utilizing rejuvenation for cancer treatment
Cancer and its relation to rejuvenation
While most studies have focused on the function of senescence in non-cancer cells, it is clear that cancer cells may also establish a senescence response, which makes it possible to exploit senescence to treat cancer. Induction of senescence in tumors includes chemotherapy, radiotherapy, cell cycle inhibition, and telomerase inhibition. However, persistently therapy-induced senescence (TIS) may create a pro-inflammatory microenvironment.453 Meanwhile, SASP can contribute to angiogenesis to facilitate tumor development and an EMT in neighboring cancer cells and induce ECM degradation further resulting in the migration and metastasis of cancer cells.454 To overcome this obstacle, a sequential therapy regimen consisting of TIS followed by a second compound that specifically kills senescent cancer cells (senolytic agent) is highlighted as a one-two-punch method for treating cancer. For example, navitoclax has shown desired results when combined with various TIS, including ionizing radiation, rituximab (CD20 antibody), doxorubicin, paclitaxel, docetaxel, or gemcitabine.455 A phase II study (NCT01087151) demonstrated that, in patients with chronic lymphocytic leukemia, rituximab plus navitoclax exhibit higher effectiveness, prolonged progression-free survival, and well tolerance than that of rituximab monotherapy.456 Other chemotherapeutic agents like etoposide (NCT00878449), irinotecan (NCT01009073), cisplatin (NCT00878449), paclitaxel (NCT00891605), docetaxel (NCT00888108), and gemcitabine (NCT00887757) that are combined with navitoclax also have been tested to treat various cancers. For sequential treatment regimen in cancer, the issue that need to be solved is identifying pro-senescence drugs with high efficacy and selectivity in inducing cancer cell senescence.
Cellular reprogramming strategy converting malignancy to benignity
It is well recognized that cancer cells can develop a malignant transition from benign cells, but the question that whether it is possible to genetically and epigenetically transform malignant cancer cells to benignity is interesting. The discovery of somatic cell reprogramming encourages the idea that cancer cells, featured by genetic and epigenetic plasticity, can rescue benign cell functions by cellular reprogramming theoretically. Cell reprogramming techniques that trigger the switch from malignancy to benignity include transcription factors, chemical cocktails, microRNAs, and exosomes (Fig. 6).457 Transcription factor-mediated cancer cell reprogramming is a pioneering strategy to retrieve benign functions. The conversion of cancer cells to iPSCs with benign phenotypes has been frequently studied. With the induction of pluripotency-associated transcription factors, melanoma cells, hepatoma cells, and colorectal cancer cells, lung adenocarcinoma and gastrointestinal cancer cell can be reprogrammed into iPSCs, and these cancer cells-derived iPSCs maintain benignity without the formation of a visible tumor in vivo.266 In addition, the cancer cells-derived iPSCs can undergo multi-lineage differentiation, such as epithelial and mesenchymal cells as well as neuron. More importantly, these differentiated cancer cells are less aggressive and lack the ability to form tumors than their parental cancer cells. Lineage-specific factors manipulation also has shown proven capabilities to directly convert cancer cells to desired functional cells without the acquisition of pluripotency. For example, overexpression of the transcription factor CCAAT/enhancer-binding protein alpha (C/EBPα) has been utilized to successfully transform human lymphoma and leukemia B cell lines into macrophage-like cells, with approximately 80% efficiency, and the reprogrammed cells exhibited poor tumorigenicity in vivo.458 Likewise, hepatocellular carcinoma can be directly induced into hepatocyte-like cells with normal functions when treated with the combinatorial manipulation of hepatocyte transcription factors.459 Although transcription factor-mediated cellular reprogramming is feasible and ethically acceptable in cancer treatment, such technology has additional challenges including cost and delivery efficiency, and in vivo procedure. The urgent need for developing optional methods is driven by safety and effectiveness concerns brought on by genetic manipulations. The chemical reprogramming technology based on small molecules has some particular advantages, such as affordability, ease of use, versatility that is easily programmable, permeability, and reversibility.460 Small molecule cocktail (SMC) consisted of SB431542 (TGFβ inhibitor), CHIR99021 (GSK3β inhibitor), BIX01294 (H3K9 methyltransferase/G9a inhibitor), and all-trans retinoic acid (ATRA), could give rise to losing malignant phenotype and acquiring hepatocyte properties in various liver cancer cells.461 MicroRNA and exosome delivery are novel anti-cancer alternatives in a manner of cellular reprogramming. Lin et al. reported using microRNA-302s could convert skin cancer cells into iPSCs with decreased tumorigenicity and genomic demethylation. These pluripotent cancer cells displayed more than 86% gene expression similarity to human ESC lines.462 Under the stimulation of lineage-specific factors, such cells could differentiate into functional cells with benignity properties, like neurons and chondrocytes. ESCs-derived exosomes also deliver ESC-related reprogramming factors to convert malignant cells to benignity ones.463 However, the precise and efficient regulation of cellular reprogramming to malignancy conversion with microRNA or exosomes faces obstacles, and few investigations have been conducted in the extending frontiers.

The mechanisms for cellular reprogramming in cancer. Cancer cell reprogramming treatment aims to convert the malignancy to benignity or provide a therapeutic target to inhibit the formation of CSCs. Yamanaka factors-mediated iPSCs technology has been recognized as a common method for the conversion of cancer cells to benign pluripotent cells, and cancer cells-derived iPSCs can re-differentiate into functional cells with less malignancy and free of tumorigenic potential. Cancer cells also can be directly reprogrammed into benign cells via various reprogramming strategies, such as lineage-specific factors, small molecules, microRNAs, and exosomes. Responsive cellular reprogramming based on EMT contributes to CSCs formation, which mediates the initial, progression, metastasis, and post-treatment recurrence. The role of cellular reprogramming in the formation of CSCs suggests that anti-cellular reprogramming strategies may be considered as a therapeutic alternative in cancer treatment. EMT Epithelial–mesenchymal transition. Created with BioRender.com
Anti-cellular reprogramming therapy for cancer treatment
The increasing evidence suggests that cellular reprogramming also contributes to cancer initiation, development, and recurrence (Fig. 6). Specific transcription factors silence- or epigenetic gene mutation-mediated cellular reprogramming initiate cancer initiation. For example, the depletion of PTEN in NSCs could activate Paired Box 7 (PAX7) and induce the generation of glioblastoma stem cell-like cells, which can lead to intracranial tumors in vivo.464 Cellular reprogramming mediates cancer development through cancer cell trans-differentiation or dedifferentiation. In non-small cell lung cancer, Lkb1 deficiency induced the trans-differentiation of adenocarcinoma cells into squamous cell carcinoma.465 In prostate cancer, luminal adenocarcinoma cells may trans-differentiate into neuroendocrine cells as a result of TP53 and PTEN depletion.466 Cancer cells can be reprogrammed by EMT and cell fusion to acquire stemness and differentiate into cancer stem cells (CSCs), which is a crucial step in cancer development. For example, single-cell analysis of organoids derived from CD44+ colorectal CSCs revealed that TWIST1-mediated EMT played an essential role in inducing cancer cell differentiation, and external stimulation with TGF-β1 can initiate EMT and promote the generation of CD44+ CSCs.467 Cellular reprogramming generating CSCs also contributes to therapy resistance and the recurrence of cancers, which may be the biological basis of poor clinical outcomes and post-treatment cancer recurrence. Conventional anti-cancer therapeutics, including chemotherapy and radiotherapy, enable cancer cells to become CSCs. For example, in non-small cell lung cancer, cisplatin-based chemotherapy renders p53 inactive and induces Tribbles Pseudokinase 1 expression, which leads to stemness activation in cancer cells.468 Therefore, cisplatin plus histone deacetylase inhibitor may synergistically suppress non-small cell lung cancer progression. Anti-angiogenesis therapy also has a great deal of potential in treating cancer, and antiangiogenic agents drive CSCs reprogramming by generating tumor hypoxia microenvironment.469 CSCs reprogramming also has been observed in immunotherapy, such as adoptive cell transfer (ACT) therapy. In melanomas, T cell-driven inflammatory stimuli lead to melanomas cells with the transition of the differentiated and dedifferentiated state and confer the ACT resistance.470 Collectively, cellular reprogramming is the key player in cancer initiation, development, recurrence, and therapeutic resistance. Synergistic therapy of conventional therapeutics and anti-cellular reprogramming may represent a promising strategy in cancer treatment. In the future, identifying molecular mechanisms involved in CSCs reprogramming is of great significance to anti-cellular reprogramming therapy.
Summary
The discovery of epidermal cell de-differentiation into stem cells and somatic cell reprogramming into iPSCs revealed that cell fate is not a one-way street. Terminally differentiated cells can reset their genetic and epigenetic properties and acquire the undifferentiated phenotype, and likewise, SCs can also get rid of senescence-related signatures and restore the youthful state. Emerged evidence has indicated the important contribution of cellular senescence in ageing and ageing-related diseases, which encourages the hypothesis that the reversal of ageing may be possible, and targeting cellular senescence may pave the way for that. The development of various cellular rejuvenation strategies provides compelling evidences that the ageing process is not irreversible. Particularly, the effectiveness of stem cell therapy and DR has been tested in the real world, yielding to desired results. Hopefully, there is a great possibility of translation of these rejuvenation strategies to address human ageing, age-associated diseases, and cancers. Therefore, it is reasonable to expect that clinical rejuvenation approaches to treat ageing-related diseases and even to reverse ageing will be boomed within the next two or three decades.
Conclusions and future perspectives
Throughout human history, the quest to extend lifespan or restore youthful state has been relentless. With the enormous progress in medical technology, including a deeper understanding of cell senescence and age-associated pathophysiology, it has become plausible to extend the human lifespan while preserving health. It is extremely essential to gain insight into the basic principles regulating cellular rejuvenation. Although many aged phenotypic and functional characteristics are studied, the formation, maintenance, and functional contribution to the disease process of aged cells still need further exploration. The study of cellular rejuvenation targeting ageing-related mechanisms is promising to develop potential therapeutic interventions for postponing and reversing ageing, and treating ageing-related diseases.
For decades, one of the dominant theories in ageing research has been that ageing results from the accumulation of DNA changes, mainly genetic mutations, which prevent more and more genes from functioning properly over time. These malfunctions, in turn, can cause cells to lose their properties, leading to the breakdown of tissues and organs and ultimately to ageing and diseases. However, the emerging evidences claim that epigenetic information loss over time is the major cause of mammalian ageing, and epigenetic regulation can restore youthful gene expression patterns. Current advances have provided a comprehensive and detailed multi-level epigenetic panorama of ageing, elucidated some common and unique characteristics of physiological ageing and ageing-related diseases, identified novel biomarkers of ageing, and revealed new mechanisms of epigenetic remodeling in cell and organ ageing. It is expected that future research on ageing epigenetic inheritance will further expand the relationship between chromatin three-dimensional structure and function. Especially, with the development of epigenetic editing technology, scientists can make specific perturbations to the epigenome, to distinguish the causal relationship between the three-dimensional structure of chromatin and cell function. Epigenetic changes caused by ageing in more diverse cell types and pathophysiological states should also be extensively explored to discover conditional regulation mechanisms of ageing. For epigenetic rejuvenation, developing safe and stable strategies that modulate the epigenetic landscape of aged cells to a primitive state are important for cells to exert rejuvenating effects without cancer risk. Furthermore, systematic comparisons of epigenetic dynamics during ageing and partial reprogramming will contribute to identifying key checkpoints for reversing the ageing process and inform the design of potential intervention strategies for ageing-related diseases.
Pathological accumulation of SCs is also associated with ageing and a range of diseases, and SCs may be potential pharmacological targets for delaying the ageing process. In respect of targeting SCs, there are still many potential markers, like chromatin dynamics and transcriptional signaling, and pharmacological interventions deserving exploitation, to effectively regulate the secretory phenotype of SCs. SC elimination and SASP inhibition have shown some efficacy in clinical studies of treating functional degeneration and chronic diseases in ageing. Notably, SC populations are heterogeneous in terms of composition, function, and tissue distribution, even among species, which is also a problem in the transition from laboratory to clinic. Therefore, identifying the cell or organ-specific markers of SCs is of great guiding significance for future clinical treatment. Targeting the cell microenvironment and systemic signals makes sense for tissue-specific cell and organism rejuvenation. Stem cells play a crucial role in maintaining tissue homeostasis, and cell microenvironments also regulate stem cell behavior, which together form a regenerative unit. External signals from the ageing microenvironment appear to dominate the intrinsic function of young stem cells. In contrast, signals from the young microenvironment may have a limited effect on the regeneration of aged stem cells. It would be interesting to identify the genes or pathways that make aged stem cells insensitive to external signals in young microenvironment. Although many clinical trials registered for stem cell treatments, an effective and safe stem cell therapy to slow or reverse tissue ageing has not yet been identified. Several obstacles still need to be overcome, including proper differentiation and integration of cells in tissues, maintenance of the youth of stem cells and their progeny in ageing tissues, and prevention of tumorigenesis. It will be important to determine the specific mechanisms, which have the potential to provide better treatment pathways-using stem cell transplantation or utilizing endogenous stem cell banks. Recent advances in single-cell transcriptomics and pedigree tracing techniques provide a systematic understanding of stem cell ageing mechanisms. Developing and utilizing new techniques to track and manipulate stem cells will still be a key field. The systematic identification of gene networks, involved in functional changes, age-dependent changes in RNA and protein and metabolite molecules, and cellular interactions, will contribute to further studies on stem cells in tissue repair and ageing-related diseases. In addition, rejuvenating strong circadian rhythms can optimize organismal physiology and avoid the risk of disease to prolong health. Clinically, circadian rhythms including a series of cyclical life activities, such as diet, sleep and exercise, vary from person to person. It is of great clinical value to develop methods to predict individual circadian rhythms. Machine learning and artificial intelligence methods may help to identify a series of biomarkers to predict circadian rhythms, which has great application prospects for determining the optimal biological clock pattern for each individual.
Despite the great progress in cellular rejuvenation, the potential limitations have led to cellular rejuvenation rarely being tested in human studies. Cellular rejuvenation for reversing ageing and age-related diseases as well as cancers has been extensively studied. While cellular rejuvenation holds great promise, key questions remain to be addressed. (1) Cellular reprogramming strategy can reverse age-related physiological changes and promote tissue regeneration by resetting the epigenetic clock and changing cell fate, but the problems such as relatively low reprogramming efficiency and potential safety concerns, remain the obstacle in the path of its application. (2) The pharmacological delivery system is difficult to express the pluripotency factors with high efficacy. The toxicity might be induced by drug combinations, then reducing the effectiveness of the cocktail and causing side effects in normal cells. (3) Clearance of SCs and decreasing SASP exert a beneficial effect on organ repair and disease treatment, but poor cell selectivity of senolytics may result in the damage of normal tissue, and SASP inhibitors targeting specific secretory factors also have limited therapeutic effects on multiple factors-mediated diseases. Besides, completely senescence-specific markers are still absent. (4) Stem cell therapy as a rejuvenative strategy holds great promise in the reversal of ageing and alleviation of diseases. Despite the advances in many clinical trials of stem cell therapy, optimizing in vitro culture environment, improving the delivery system of stem cells, and reducing immune rejection are still the major challenges to obtain high-quality stem cells and enhance that therapeutic effect. (5) Restoring defective intercellular communications by the inhibition of inflammation can rejuvenate ageing-impaired changes, but long-term inflammation inhibition may lead to immunosuppression.
Collectively, cellular rejuvenation holds great promise for preventing and treating ageing-related diseases from different dimensions. A healthy and rejuvenated state of the organism can maintain stable characteristics and biological functions without excessive ageing-related degeneration or deterioration. Of note, system therapies such as DR and exercise are available rejuvenation strategies mostly closed to bedside, whose mechanism at the cellular level and molecular level has been expounded.471 The role of DR in health and diseases has been discussed above. Exercise greatly activates the immune system, facilitates DNA repair processes, maintains metabolism homeostasis, and is capable of lowering the risk of diabetes, obesity, cancer, osteoporosis, AD, and depression as well as prolonging the lifespan.472 The exploration of diet and exercise programs for different populations is the direction that needs to be paid attention to in the future.
References
Luo, J., Mills, K., le Cessie, S., Noordam, R. & van Heemst, D. Ageing, age-related diseases and oxidative stress: what to do next? Ageing Res. Rev. 57, 100982 (2020).
Kubben, N. & Misteli, T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat. Rev. Mol. Cell Biol. 18, 595–609 (2017).
Dai, X. & Guo, X. Decoding and rejuvenating human ageing genomes: Lessons from mosaic chromosomal alterations. Ageing Res. Rev. 68, 101342 (2021).
Vaiserman, A., De Falco, E., Koliada, A., Maslova, O. & Balistreri, C. R. Anti-ageing gene therapy: Not so far away? Ageing Res. Rev. 56, 100977 (2019).
Gage, F. H., Guarente, L. P. & Wagers, A. J. Aging and rejuvenation: insights from rusty gage, leonard guarente, and amy wagers. Trends Mol. Med. 22, 633–634 (2016).
Soria-Valles, C. & Lopez-Otin, C. iPSCs: on the road to reprogramming aging. Trends Mol. Med. 22, 713–724 (2016).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Zhang, B., Trapp, A., Kerepesi, C. & Gladyshev, V. N. Emerging rejuvenation strategies—reducing the biological age. Aging Cell 21, e13538 (2022).
Sender, R. & Milo, R. The distribution of cellular turnover in the human body. Nat. Med. 27, 45–48 (2021).
Henikoff, S. & Smith, M. M. Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 7, a019364 (2015).
Saul, D. & Kosinsky, R. L. Epigenetics of aging and aging-associated diseases. Int. J. Mol. Sci. 22, 401 (2021).
Wang, Y., Yuan, Q. & Xie, L. Histone modifications in aging: the underlying mechanisms and implications. Curr. Stem Cell Res. Ther. 13, 125–135 (2018).
Greer, E. L. & Shi, Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 (2012).
Braga, D. L., Mousovich-Neto, F., Tonon-da-Silva, G., Salgueiro, W. G. & Mori, M. A. Epigenetic changes during ageing and their underlying mechanisms. Biogerontology 21, 423–443 (2020).
Ciccarone, F., Tagliatesta, S., Caiafa, P. & Zampieri, M. DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech. Ageing Dev. 174, 3–17 (2018).
Lillycrop, K. A., Hoile, S. P., Grenfell, L. & Burdge, G. C. DNA methylation, ageing and the influence of early life nutrition. Proc. Nutr. Soc. 73, 413–421 (2014).
Gabbianelli, R. & Malavolta, M. Epigenetics in ageing and development. Mech. Ageing Dev. 174, 1–2 (2018).
Borghesan, M. et al. DNA hypomethylation and histone variant macroH2A1 synergistically attenuate chemotherapy-induced senescence to promote hepatocellular carcinoma progression. Cancer Res. 76, 594–606 (2016).
Petkovich, D. A. et al. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 25, 954–960.e956 (2017).
Unnikrishnan, A. et al. The role of DNA methylation in epigenetics of aging. Pharm. Ther. 195, 172–185 (2019).
Zhang, Y. & Ren, J. Epigenetics and obesity cardiomyopathy: from pathophysiology to prevention and management. Pharm. Ther. 161, 52–66 (2016).
Watanabe, R., Kanno, S. I., Mohammadi Roushandeh, A., Ui, A. & Yasui, A. Nucleosome remodelling, DNA repair and transcriptional regulation build negative feedback loops in cancer and cellular ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160473 (2017).
Swer, P. B. & Sharma, R. ATP-dependent chromatin remodelers in ageing and age-related disorders. Biogerontology 22, 1–17 (2021).
Tessarz, P. & Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708 (2014).
Kane, A. E. & Sinclair, D. A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem Mol. Biol. 54, 61–83 (2019).
Ilina, A., Khavinson, V., Linkova, N. & Petukhov, M. Neuroepigenetic mechanisms of action of ultrashort peptides in Alzheimer’s disease. Int. J. Mol. Sci. 23, 4259 (2022).
Menoni, H., Di Mascio, P., Cadet, J., Dimitrov, S. & Angelov, D. Chromatin associated mechanisms in base excision repair - nucleosome remodeling and DNA transcription, two key players. Free Radic. Biol. Med. 107, 159–169 (2017).
Kamileri, I., Karakasilioti, I. & Garinis, G. A. Nucleotide excision repair: new tricks with old bricks. Trends Genet. 28, 566–573 (2012).
Martinez Corrales, G. & Alic, N. Evolutionary conservation of transcription factors affecting longevity. Trends Genet. 36, 373–382 (2020).
Loaiza, N. & Demaria, M. Cellular senescence and tumor promotion: Is aging the key? Biochim. Biophys. Acta 1865, 155–167 (2016).
Cheng, Y., Pitoniak, A., Wang, J. & Bohmann, D. Preserving transcriptional stress responses as an anti-aging strategy. Aging Cell 20, e13297 (2021).
Martins, R., Lithgow, G. J. & Link, W. Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15, 196–207 (2016).
Li, G., Zhang, W., Liang, H. & Yang, C. Epigenetic regulation in intervertebral disc degeneration. Trends Mol. Med. 28, 803–805 (2022).
Harries, L. W. Dysregulated RNA processing and metabolism: a new hallmark of ageing and provocation for cellular senescence. FEBS J. 1–14 (2022).
D’Aquila, P., Rose, G., Bellizzi, D. & Passarino, G. Epigenetics and aging. Maturitas 74, 130–136 (2013).
Lozano-Vidal, N., Bink, D. I. & Boon, R. A. Long noncoding RNA in cardiac aging and disease. J. Mol. Cell Biol. 11, 860–867 (2019).
Jusic, A. et al. Noncoding RNAs in age-related cardiovascular diseases. Ageing Res. Rev. 77, 101610 (2022).
Miyata, K. et al. Pericentromeric noncoding RNA changes DNA binding of CTCF and inflammatory gene expression in senescence and cancer. Proc. Natl Acad. Sci. USA 118, e2025647118 (2021).
Bandaria, J. N., Qin, P., Berk, V., Chu, S. & Yildiz, A. Shelterin protects chromosome ends by compacting telomeric chromatin. Cell 164, 735–746 (2016).
Campisi, J. & Vijg, J. Does damage to DNA and other macromolecules play a role in aging? If so, how? J. Gerontol. A Biol. Sci. Med. Sci. 64, 175–178 (2009).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Shay, J. W. Telomeres and aging. Curr. Opin. Cell Biol. 52, 1–7 (2018).
Rossi, M. & Gorospe, M. Noncoding RNAs controlling telomere homeostasis in senescence and aging. Trends Mol. Med. 26, 422–433 (2020).
Fernando, R., Drescher, C., Nowotny, K., Grune, T. & Castro, J. P. Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med. 132, 58–66 (2019).
Korovila, I. et al. Proteostasis, oxidative stress and aging. Redox Biol. 13, 550–567 (2017).
Klaips, C. L., Jayaraj, G. G. & Hartl, F. U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217, 51–63 (2018).
Stein, K. C., Morales-Polanco, F., van der Lienden, J., Rainbolt, T. K. & Frydman, J. Ageing exacerbates ribosome pausing to disrupt cotranslational proteostasis. Nature 601, 637–642 (2022).
Das, U. N. “Cell membrane theory of senescence” and the role of bioactive lipids in aging, and aging associated diseases and their therapeutic implications. Biomolecules 11, 241 (2021).
Roitenberg, N. & Cohen, E. Lipid assemblies at the crossroads of aging, proteostasis, and neurodegeneration. Trends Cell Biol. 29, 954–963 (2019).
Skowronska-Krawczyk, D. & Budin, I. Aging membranes: unexplored functions for lipids in the lifespan of the central nervous system. Exp. Gerontol. 131, 110817 (2020).
Almeida, I., Magalhaes, S. & Nunes, A. Lipids: biomarkers of healthy aging. Biogerontology 22, 273–295 (2021).
Zhu, X. et al. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: the regulation and intervention. Signal Transduct. Target Ther. 6, 245 (2021).
Azzu, V. & Valencak, T. G. Energy metabolism and ageing in the mouse: a mini-review. Gerontology 63, 327–336 (2017).
Rajawat, Y. S., Hilioti, Z. & Bossis, I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res. Rev. 8, 199–213 (2009).
Terman, A., Gustafsson, B. & Brunk, U. T. Mitochondrial damage and intralysosomal degradation in cellular aging. Mol. Asp. Med. 27, 471–482 (2006).
Sreedhar, A., Aguilera-Aguirre, L. & Singh, K. K. Mitochondria in skin health, aging, and disease. Cell Death Dis. 11, 444 (2020).
Breitenbach, M. et al. Mitochondria in ageing: there is metabolism beyond the ROS. FEMS Yeast Res 14, 198–212 (2014).
Suganuma, T. & Workman, J. L. Chromatin and Metabolism. Annu. Rev. Biochem. 87, 27–49 (2018).
Sharma, R. & Ramanathan, A. The aging metabolome-biomarkers to hub metabolites. Proteomics 20, e1800407 (2020).
Amjad, S. et al. Role of NAD(+) in regulating cellular and metabolic signaling pathways. Mol. Metab. 49, 101195 (2021).
Sahin, E. et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470, 359–365 (2011).
Lee, D., Son, H. G., Jung, Y. & Lee, S. V. The role of dietary carbohydrates in organismal aging. Cell Mol. Life Sci. 74, 1793–1803 (2017).
Smith, W. K., Ingram, D. K., de Cabo, R. & Pasquina, P. Metabolic pathways and therapeutics to promote resilience, rehabilitation and delayed aging. Geroscience 43, 1069–1070 (2021).
Castillo, S. P., Keymer, J. E. & Marquet, P. A. Do microenvironmental changes disrupt multicellular organisation with ageing, enacting and favouring the cancer cell phenotype? Bioessays 43, e2000126 (2021).
Li, C. J. et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J. Clin. Invest. 125, 1509–1522 (2015).
Singh, L. et al. Aging alters bone-fat reciprocity by shifting in vivo mesenchymal precursor cell fate towards an adipogenic lineage. Bone 85, 29–36 (2016).
Hass, R. Rejuvenation in distinct cell populations—what does it mean? Exp. Gerontol. 44, 634–638 (2009).
Karamanos, N. K. et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 288, 6850–6912 (2021).
Vidovic, T. & Ewald, C. Y. Longevity-promoting pathways and transcription factors respond to and control extracellular matrix dynamics during aging and disease. Front Aging 3, 935220 (2022).
Haydont, V., Bernard, B. A. & Fortunel, N. O. Age-related evolutions of the dermis: clinical signs, fibroblast and extracellular matrix dynamics. Mech. Ageing Dev. 177, 150–156 (2019).
Rahmati, M., Nalesso, G., Mobasheri, A. & Mozafari, M. Aging and osteoarthritis: central role of the extracellular matrix. Ageing Res. Rev. 40, 20–30 (2017).
Schuler, S. C. et al. Extensive remodeling of the extracellular matrix during aging contributes to age-dependent impairments of muscle stem cell functionality. Cell Rep. 35, 109223 (2021).
Bahcecioglu, G. et al. Aged breast extracellular matrix drives mammary epithelial cells to an invasive and cancer-like phenotype. Adv. Sci. 8, e2100128 (2021).
Costea, L. et al. The blood-brain barrier and its intercellular junctions in age-related brain disorders. Int. J. Mol. Sci. 20, 5472 (2019).
Lin, W. et al. Dynamic regulation of mitochondrial-endoplasmic reticulum crosstalk during stem cell homeostasis and aging. Cell Death Dis. 12, 794 (2021).
Picca, A. et al. Mitochondrial dysfunction and aging: insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 20, 805 (2019).
Fafian-Labora, J. A. & O’Loghlen, A. Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. 30, 628–639 (2020).
Yin, Y., Chen, H., Wang, Y., Zhang, L. & Wang, X. Roles of extracellular vesicles in the aging microenvironment and age-related diseases. J. Extracell. Vesicles 10, e12154 (2021).
Takasugi, M. et al. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 8, 15729 (2017).
Monti, P., Solazzo, G., Ferrari, L. & Bollati, V. Extracellular vesicles: footprints of environmental exposures in the aging process? Curr. Environ. Health Rep. 8, 309–322 (2021).
Ma, S. et al. Heterochronic parabiosis induces stem cell revitalization and systemic rejuvenation across aged tissues. Cell Stem Cell 29, 990–1005.e1010 (2022).
Conboy, I. M., Conboy, M. J. & Rebo, J. Systemic problems: a perspective on stem cell aging and rejuvenation. Aging 7, 754–765 (2015).
Tam, B. T., Morais, J. A. & Santosa, S. Obesity and ageing: two sides of the same coin. Obes. Rev. 21, e12991 (2020).
DeCarolis, N. A., Kirby, E. D., Wyss-Coray, T. & Palmer, T. D. The role of the microenvironmental niche in declining stem-cell functions associated with biological aging. Cold Spring Harb. Perspect. Med. 5, a025874 (2015).
Van Zant, G. & Liang, Y. Concise review: hematopoietic stem cell aging, life span, and transplantation. Stem Cells Transl. Med 1, 651–657 (2012).
Yeo, G. E. C., Ng, M. H., Nordin, F. B. & Law, J. X. Potential of mesenchymal stem cells in the rejuvenation of the aging immune system. Int. J. Mol. Sci. 22, 5479 (2021).
Fuchs, E. & Blau, H. M. Tissue stem cells: architects of their niches. Cell Stem Cell 27, 532–556 (2020).
Deb, K. D., Jayaprakash, A. D., Sharma, V. & Totey, S. Embryonic stem cells: from markers to market. Rejuvenation Res. 11, 19–37 (2008).
Galkin, F., Zhang, B., Dmitriev, S. E. & Gladyshev, V. N. Reversibility of irreversible aging. Ageing Res. Rev. 49, 104–114 (2019).
Li, X. et al. Mechanisms and rejuvenation strategies for aged hematopoietic stem cells. J. Hematol. Oncol. 13, 31 (2020).
Ho, T. T. et al. Aged hematopoietic stem cells are refractory to bloodborne systemic rejuvenation interventions. J. Exp. Med. 218, e20210223 (2021).
Brunet, A., Goodell, M. A. & Rando, T. A. Ageing and rejuvenation of tissue stem cells and their niches. Nat. Rev. Mol. Cell Biol. 24, 45–62 (2022).
Goligorsky, M. S. & Hirschi, K. Stress-induced premature senescence of endothelial and endothelial progenitor cells. Adv. Pharm. 77, 281–306 (2016).
Park, M. H. et al. Vascular and neurogenic rejuvenation in aging mice by modulation of ASM. Neuron 100, 167–182.e9 (2018).
Augustin, H. G. & Kipnis, J. Vascular rejuvenation is geroprotective. Science 373, 490–491 (2021).
Shindyapina, A. V. et al. Mineralization of the connective tissue: a complex molecular process leading to age-related loss of function. Rejuvenation Res. 17, 116–133 (2014).
Mahmoudi, S. et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 574, 553–558 (2019).
Gerasymchuk, M., Robinson, G. I., Kovalchuk, O. & Kovalchuk, I. The effects of nutrient signaling regulators in combination with phytocannabinoids on the senescence-associated phenotype in human dermal fibroblasts. Int. J. Mol. Sci. 23, 8804 (2022).
Roy, A. L. et al. A blueprint for characterizing senescence. Cell 183, 1143–1146 (2020).
Ogrodnik, M., Salmonowicz, H. & Gladyshev, V. N. Integrating cellular senescence with the concept of damage accumulation in aging: Relevance for clearance of senescent cells. Aging Cell 18, e12841 (2019).
Liu, J., Ding, Y., Liu, Z. & Liang, X. Senescence in mesenchymal stem cells: functional alterations, molecular mechanisms, and rejuvenation strategies. Front. Cell Dev. Biol. 8, 258 (2020).
Casella, G. et al. Transcriptome signature of cellular senescence. Nucleic Acids Res. 47, 7294–7305 (2019).
de Keizer, P. L. The fountain of youth by targeting senescent cells? Trends Mol. Med. 23, 6–17 (2017).
Chen, R. & Skutella, T. Synergistic anti-ageing through senescent cells specific reprogramming. Cells 11, 830 (2022).
Krimpenfort, P. & Berns, A. Rejuvenation by therapeutic elimination of senescent. Cells Cell 169, 3–5 (2017).
Borgoni, S., Kudryashova, K. S., Burka, K. & de Magalhaes, J. P. Targeting immune dysfunction in aging. Ageing Res. Rev. 70, 101410 (2021).
Tobin, S. W. et al. Delineating the relationship between immune system aging and myogenesis in muscle repair. Aging Cell 20, e13312 (2021).
Ou, H. L. et al. Cellular senescence in cancer: from mechanisms to detection. Mol. Oncol. 15, 2634–2671 (2021).
Melendez, E. et al. Natural killer cells act as an extrinsic barrier for in vivo reprogramming. Development 149, dev200361 (2022).
Gamez-Garcia, A. & Vazquez, B. N. Nuclear sirtuins and the aging of the immune system. Genes 12, 1856 (2021).
Seledtsov, V. I. & von Delwig, A. A. Immune memory limits human longevity: the role of memory capital ES, CyrillicD4+ T cells in age-related immune abnormalities. Expert Rev. Vaccines 19, 209–215 (2020).
Uhrlaub, J. L. et al. Quantitative restoration of immune defense in old animals determined by naive antigen-specific CD8 T-cell numbers. Aging Cell 21, e13582 (2022).
Avivi, I. et al. Depletion of B cells rejuvenates the peripheral B-cell compartment but is insufficient to restore immune competence in aging. Aging Cell 18, e12959 (2019).
Xu, N. et al. Sphere-induced rejuvenation of swine and human Muller glia is primarily caused by telomere elongation. Stem Cells 35, 1579–1591 (2017).
Bansal, V. S., Raja, C. P., Venkataraman, K. & Vijayalakshmi, M. A. Genes involved in pancreatic islet cell rejuvenation. Indian J. Med. Res. 137, 695–703 (2013).
Taguchi, J. & Yamada, Y. In vivo reprogramming for tissue regeneration and organismal rejuvenation. Curr. Opin. Genet. Dev. 46, 132–140 (2017).
Park, H. H. et al. A PTEN variant uncouples longevity from impaired fitness in Caenorhabditis elegans with reduced insulin/IGF-1 signaling. Nat. Commun. 12, 5631 (2021).
Salminen, A., Kaarniranta, K. & Kauppinen, A. Insulin/IGF-1 signaling promotes immunosuppression via the STAT3 pathway: impact on the aging process and age-related diseases. Inflamm. Res. 70, 1043–1061 (2021).
Hu, F. & Liu, F. Targeting tissue-specific metabolic signaling pathways in aging: the promise and limitations. Protein Cell 5, 21–35 (2014).
Zhong, J. et al. Clock knockdown attenuated reactive oxygen species-mediated senescence of chondrocytes through restoring autophagic flux. Life Sci. 269, 119036 (2021).
Myers, A. & Lithgow, G. J. Drugs that target aging: how do we discover them? Expert Opin. Drug Disco. 14, 541–548 (2019).
Lagunas-Rangel, F. A. SIRT7 in the aging process. Cell Mol. Life Sci. 79, 297 (2022).
Ge, Y., Zhou, M., Chen, C., Wu, X. & Wang, X. Role of AMPK mediated pathways in autophagy and aging. Biochimie 195, 100–113 (2022).
Cordero, M. D., Williams, M. R. & Ryffel, B. AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends Endocrinol. Metab. 29, 8–17 (2018).
Smith, H. J., Sharma, A. & Mair, W. B. Metabolic communication and healthy aging: where should we focus our energy? Dev. Cell 54, 196–211 (2020).
Kulkarni, A. S., Gubbi, S. & Barzilai, N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 32, 15–30 (2020).
Ou, H. L. & Schumacher, B. DNA damage responses and p53 in the aging process. Blood 131, 488–495 (2018).
Pan, M. R., Li, K., Lin, S. Y. & Hung, W. C. Connecting the dots: from dna damage and repair to aging. Int. J. Mol. Sci. 17, 685 (2016).
Shmulevich, R. & Krizhanovsky, V. Cell senescence, DNA damage, and metabolism. Antioxid. Redox Signal 34, 324–334 (2021).
Boesch, P. et al. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim Biophys. Acta 1813, 186–200 (2011).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
Akbari, M., Shanley, D. P., Bohr, V. A. & Rasmussen, L. J. Cytosolic self-DNA—a potential source of chronic inflammation in aging. Cells 10, 3544 (2021).
Conde-Perezprina, J. C., Leon-Galvan, M. A. & Konigsberg, M. DNA mismatch repair system: repercussions in cellular homeostasis and relationship with aging. Oxid. Med. Cell Longev. 2012, 728430 (2012).
Schumacher, B., Garinis, G. A. & Hoeijmakers, J. H. Age to survive: DNA damage and aging. Trends Genet 24, 77–85 (2008).
Davalli, P., Mitic, T., Caporali, A., Lauriola, A. & D’Arca, D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell Longev. 2016, 3565127 (2016).
Quan, Y., Xin, Y., Tian, G., Zhou, J. & Liu, X. Mitochondrial ROS-modulated mtDNA: a potential target for cardiac aging. Oxid. Med. Cell Longev. 2020, 9423593 (2020).
Detienne, G. et al. Beyond ROS clearance: peroxiredoxins in stress signaling and aging. Ageing Res. Rev. 44, 33–48 (2018).
Liochev, S. I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 60, 1–4 (2013).
Zhu, L., Luo, X., Fu, N. & Chen, L. Mitochondrial unfolded protein response: a novel pathway in metabolism and immunity. Pharm. Res. 168, 105603 (2021).
Zhu, L., Zhou, Q., He, L. & Chen, L. Mitochondrial unfolded protein response: an emerging pathway in human diseases. Free Radic. Biol. Med. 163, 125–134 (2021).
Soo, S. K., Traa, A., Rudich, P. D., Mistry, M. & Van Raamsdonk, J. M. Activation of mitochondrial unfolded protein response protects against multiple exogenous stressors. Life Sci. Alliance 4, e202101182 (2021).
Munoz-Carvajal, F. & Sanhueza, M. The mitochondrial unfolded protein response: a hinge between healthy and pathological aging. Front Aging Neurosci. 12, 581849 (2020).
Shpilka, T. & Haynes, C. M. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 19, 109–120 (2018).
Angeli, S. et al. The mitochondrial permeability transition pore activates the mitochondrial unfolded protein response and promotes aging. Elife 10, e63453 (2021).
Rea, I. M. et al. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 9, 586 (2018).
Feldman, N., Rotter-Maskowitz, A. & Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev. 24, 29–39 (2015).
Royce, G. H., Brown-Borg, H. M. & Deepa, S. S. The potential role of necroptosis in inflammaging and aging. Geroscience 41, 795–811 (2019).
Kim, D. H. et al. Anti-aging effects of calorie restriction (CR) and CR mimetics based on the senoinflammation concept. Nutrients 12, 422 (2020).
Libby, P. & Kobold, S. Inflammation: a common contributor to cancer, aging, and cardiovascular diseases-expanding the concept of cardio-oncology. Cardiovasc Res. 115, 824–829 (2019).
Neves, J. & Sousa-Victor, P. Regulation of inflammation as an anti-aging intervention. FEBS J. 287, 43–52 (2020).
Perner, F., Perner, C., Ernst, T. & Heidel, F. H. Roles of JAK2 in aging, inflammation, hematopoiesis and malignant transformation. Cells 8, 854 (2019).
Oshimori, N. & Fuchs, E. The harmonies played by TGF-beta in stem cell biology. Cell Stem Cell 11, 751–764 (2012).
Hata, A. & Chen, Y. G. TGF-beta signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 8, a022061 (2016).
Tominaga, K. & Suzuki, H. I. TGF-beta signaling in cellular senescence and aging-related pathology. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20205002 (2019).
Colak, S. & Ten Dijke, P. Targeting TGF-beta signaling in cancer. Trends Cancer 3, 56–71 (2017).
Peng, D., Fu, M., Wang, M., Wei, Y. & Wei, X. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol. Cancer 21, 104 (2022).
Derynck, R. & Budi, E. H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal 12, eaav5183 (2019).
Gyorfi, A. H., Matei, A. E. & Distler, J. H. W. Targeting TGF-beta signaling for the treatment of fibrosis. Matrix Biol. 68–69, 8–27 (2018).
Wang, K. et al. Emerging roles of transforming growth factor beta signaling in wet age-related macular degeneration. Acta Biochim Biophys. Sin. 51, 1–8 (2019).
Ma, Y. et al. Growth differentiation factor 11: a “rejuvenation factor” involved in regulation of age-related diseases? Aging 13, 12258–12272 (2021).
Gay, A. & Towler, D. A. Wnt signaling in cardiovascular disease: opportunities and challenges. Curr. Opin. Lipido. 28, 387–396 (2017).
Chen, D. et al. Wnt signaling in bone, kidney, intestine, and adipose tissue and interorgan interaction in aging. Ann. N. Y Acad. Sci. 1442, 48–60 (2019).
Maleki Dana, P. et al. Targeting Wnt signaling pathway by polyphenols: implication for aging and age-related diseases. Biogerontology 22, 479–494 (2021).
Ahmadzadeh, A., Norozi, F., Shahrabi, S., Shahjahani, M. & Saki, N. Wnt/beta-catenin signaling in bone marrow niche. Cell Tissue Res. 363, 321–335 (2016).
Hoffmeyer, K. et al. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 336, 1549–1554 (2012).
Lezzerini, M. & Budovskaya, Y. A dual role of the Wnt signaling pathway during aging in Caenorhabditis elegans. Aging Cell 13, 8–18 (2014).
Duan, P. & Bonewald, L. F. The role of the wnt/beta-catenin signaling pathway in formation and maintenance of bone and teeth. Int. J. Biochem. Cell Biol. 77, 23–29 (2016).
Hu, H. H., Cao, G., Wu, X. Q., Vaziri, N. D. & Zhao, Y. Y. Wnt signaling pathway in aging-related tissue fibrosis and therapies. Ageing Res. Rev. 60, 101063 (2020).
Guo, Y., Xiao, L., Sun, L. & Liu, F. Wnt/beta-catenin signaling: a promising new target for fibrosis diseases. Physiol. Res. 61, 337–346 (2012).
Marchetti, B. Nrf2/Wnt resilience orchestrates rejuvenation of glia-neuron dialogue in Parkinson’s disease. Redox Biol. 36, 101664 (2020).
Ohnuki, M. & Takahashi, K. Present and future challenges of induced pluripotent stem cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140367 (2015).
De Vos, J., Bouckenheimer, J., Sansac, C., Lemaitre, J. M. & Assou, S. Human induced pluripotent stem cells: a disruptive innovation. Curr. Res. Transl. Med. 64, 91–96 (2016).
Wehrwein, P. Stem cells: Repeat to fade. Nature 492, S12–S13 (2012).
Wruck, W., Graffmann, N., Spitzhorn, L. S. & Adjaye, J. Human induced pluripotent stem cell-derived mesenchymal stem cells acquire rejuvenation and reduced heterogeneity. Front. Cell Dev. Biol. 9, 717772 (2021).
Rohani, L., Johnson, A. A., Arnold, A. & Stolzing, A. The aging signature: a hallmark of induced pluripotent stem cells? Aging Cell 13, 2–7 (2014).
Prigione, A., Fauler, B., Lurz, R., Lehrach, H. & Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 28, 721–733 (2010).
Wang, F. et al. Molecular insights into the heterogeneity of telomere reprogramming in induced pluripotent stem cells. Cell Res. 22, 757–768 (2012).
Le, R. et al. Enhanced telomere rejuvenation in pluripotent cells reprogrammed via nuclear transfer relative to induced pluripotent stem cells. Cell Stem Cell 14, 27–39 (2014).
Hockemeyer, D. & Jaenisch, R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 18, 573–586 (2016).
Tanaka, N. et al. Development of a high-efficacy reprogramming method for generating human induced pluripotent stem (iPS) cells from pathologic and senescent somatic cells. Int. J. Mol. Sci. 21, 6764 (2020).
Mora, C., Serzanti, M., Consiglio, A., Memo, M. & Dell’Era, P. Clinical potentials of human pluripotent stem cells. Cell Biol. Toxicol. 33, 351–360 (2017).
Gobel, C., Goetzke, R., Eggermann, T. & Wagner, W. Interrupted reprogramming into induced pluripotent stem cells does not rejuvenate human mesenchymal stromal cells. Sci. Rep. 8, 11676 (2018).
Ratajczak, M. Z., Bujko, K. & Wojakowski, W. Stem cells and clinical practice: new advances and challenges at the time of emerging problems with induced pluripotent stem cell therapies. Pol. Arch. Med. Wewn. 126, 879–890 (2016).
Srivastava, D. & DeWitt, N. In vivo cellular reprogramming: the next generation. Cell 166, 1386–1396 (2016).
Yang, S. G., Wang, X. W., Qian, C. & Zhou, F. Q. Reprogramming neurons for regeneration: the fountain of youth. Prog. Neurobiol. 214, 102284 (2022).
Chiche, A. et al. Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell 20, 407–414.e404 (2017).
Wang, H., Yang, Y., Liu, J. & Qian, L. Direct cell reprogramming: approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 22, 410–424 (2021).
Alderton, G. K. Pluripotency: partial reprogramming induces cancer. Nat. Rev. Cancer 14, 216–217 (2014).
Tamanini, S., Comi, G. P. & Corti, S. In vivo transient and partial cell reprogramming to pluripotency as a therapeutic tool for neurodegenerative diseases. Mol. Neurobiol. 55, 6850–6862 (2018).
Lehmann, M. et al. Partial reprogramming as an emerging strategy for safe induced cell generation and rejuvenation. Curr. Gene Ther. 19, 248–254 (2019).
Mendelsohn, A. R., Larrick, J. W. & Lei, J. L. Rejuvenation by partial reprogramming of the epigenome. Rejuvenation Res. 20, 146–150 (2017).
Alle, Q., Le Borgne, E., Milhavet, O. & Lemaitre, J. M. Reprogramming: emerging strategies to rejuvenate aging cells and tissues. Int. J. Mol. Sci. 22, 3990 (2021).
Chondronasiou, D. et al. Multi-omic rejuvenation of naturally aged tissues by a single cycle of transient reprogramming. Aging Cell 21, e13578 (2022).
Marion, R. M. & Blasco, M. A. Long live partial reprogramming. Circ. Res. 120, 1381–1383 (2017).
Bell, C. G. et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 20, 249 (2019).
Ye, F., Huang, J., Wang, H., Luo, C. & Zhao, K. Targeting epigenetic machinery: emerging novel allosteric inhibitors. Pharm. Ther. 204, 107406 (2019).
Wang, T. et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 18, 57 (2017).
Manukyan, M. & Singh, P. B. Epigenome rejuvenation: HP1beta mobility as a measure of pluripotent and senescent chromatin ground states. Sci. Rep. 4, 4789 (2014).
Averbeck, D. & Rodriguez-Lafrasse, C. Role of mitochondria in radiation responses: epigenetic, metabolic, and signaling impacts. Int. J. Mol. Sci. 22, 11047 (2021).
Sharma, N., Pasala, M. S. & Prakash, A. Mitochondrial DNA: epigenetics and environment. Environ. Mol. Mutagen 60, 668–682 (2019).
Merkwirth, C. et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223 (2016).
Tian, Y. et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208 (2016).
Picard, M., McEwen, B. S., Epel, E. S. & Sandi, C. An energetic view of stress: focus on mitochondria. Front Neuroendocrinol. 49, 72–85 (2018).
Pal, S. & Tyler, J. K. Epigenetics and aging. Sci. Adv. 2, e1600584 (2016).
De Cecco, M. et al. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12, 247–256 (2013).
Bacalini, M. G. et al. Present and future of anti-ageing epigenetic diets. Mech. Ageing Dev. 136-137, 101–115 (2014).
Kerachian, M. A. & Kerachian, M. Long interspersed nucleotide element-1 (LINE-1) methylation in colorectal cancer. Clin. Chim. Acta 488, 209–214 (2019).
de la Rica, L. et al. TET-dependent regulation of retrotransposable elements in mouse embryonic stem cells. Genome Biol. 17, 234 (2016).
Xiong, F. et al. RNA m(6)A modification orchestrates a LINE-1-host interaction that facilitates retrotransposition and contributes to long gene vulnerability. Cell Res. 31, 861–885 (2021).
Menendez, J. A. & Alarcon, T. Senescence-inflammatory regulation of reparative cellular reprogramming in aging and cancer. Front. Cell Dev. Biol. 5, 49 (2017).
Sullivan, K. E. et al. Epigenetic regulation of tumor necrosis factor alpha. Mol. Cell Biol. 27, 5147–5160 (2007).
Sera, Y. et al. UTX maintains the functional integrity of the murine hematopoietic system by globally regulating aging-associated genes. Blood 137, 908–922 (2021).
Evans, L. W., Stratton, M. S. & Ferguson, B. S. Dietary natural products as epigenetic modifiers in aging-associated inflammation and disease. Nat. Prod. Rep. 37, 653–676 (2020).
Rando, T. A. & Chang, H. Y. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell 148, 46–57 (2012).
Ocampo, A., Reddy, P. & Belmonte, J. C. I. Anti-aging strategies based on cellular reprogramming. Trends Mol. Med. 22, 725–738 (2016).
Schmitt, R. Senotherapy: growing old and staying young? Pflug. Arch. 469, 1051–1059 (2017).
Osiewacz, H. D. Mitochondrial quality control in aging and lifespan control of the fungal aging model Podospora anserina. Biochem. Soc. Trans. 39, 1488–1492 (2011).
Akbari, M., Kirkwood, T. B. L. & Bohr, V. A. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 54, 100940 (2019).
Terman, A., Kurz, T., Navratil, M., Arriaga, E. A. & Brunk, U. T. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal 12, 503–535 (2010).
Moehle, E. A., Shen, K. & Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 294, 5396–5407 (2019).
Mu, W. C., Ohkubo, R., Widjaja, A. & Chen, D. The mitochondrial metabolic checkpoint in stem cell aging and rejuvenation. Mech. Ageing Dev. 188, 111254 (2020).
Zhang, H., Menzies, K. J. & Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 145, dev143420 (2018).
Sun, N., Youle, R. J. & Finkel, T. The mitochondrial basis of aging. Mol. Cell 61, 654–666 (2016).
Vazquez-Martin, A. et al. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging 8, 1330–1352 (2016).
Yan, L. J. Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol. 2, 165–169 (2014).
Le, H. T. et al. Gap junction intercellular communication mediated by connexin43 in astrocytes is essential for their resistance to oxidative stress. J. Biol. Chem. 289, 1345–1354 (2014).
Alvarez-Satta, M. et al. Relevance of oxidative stress and inflammation in frailty based on human studies and mouse models. Aging 12, 9982–9999 (2020).
Zhang, H., Davies, K. J. A. & Forman, H. J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 88, 314–336 (2015).
You, S. et al. The role of BRG1 in antioxidant and redox signaling. Oxid. Med. Cell Longev. 2020, 6095673 (2020).
Vatner, S. F. et al. Healthful aging mediated by inhibition of oxidative stress. Ageing Res. Rev. 64, 101194 (2020).
Xia, C., Dai, Z., Jin, Y. & Chen, P. Emerging antioxidant paradigm of mesenchymal stem cell-derived exosome therapy. Front. Endocrinol. (Lausanne) 12, 727272 (2021).
Oyewole, A. O. & Birch-Machin, M. A. Mitochondria-targeted antioxidants. FASEB J. 29, 4766–4771 (2015).
Hou, Y., Li, J., Wu, J. C., Wu, Q. X. & Fang, J. Activation of cellular antioxidant defense system by naturally occurring dibenzopyrone derivatives confers neuroprotection against oxidative insults. ACS Chem. Neurosci. 12, 2798–2809 (2021).
El Assar, M., Alvarez-Bustos, A., Sosa, P., Angulo, J. & Rodriguez-Manas, L. Effect of physical activity/exercise on oxidative stress and inflammation in muscle and vascular aging. Int. J. Mol. Sci. 23, 8713 (2022).
Chen, M., Wang, Y., Deng, S., Lian, Z. & Yu, K. Skeletal muscle oxidative stress and inflammation in aging: focus on antioxidant and anti-inflammatory therapy. Front. Cell Dev. Biol. 10, 964130 (2022).
Wang, Y. et al. Insights into autophagy machinery in cells related to skin diseases and strategies for therapeutic modulation. Biomed. Pharmacother. 113, 108775 (2019).
Gatica, D., Lahiri, V. & Klionsky, D. J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242 (2018).
Stavoe, A. K. H. & Holzbaur, E. L. F. Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 35, 477–500 (2019).
Ward, C. et al. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 1861, 269–284 (2016).
Jeong, J. K., Moon, M. H., Lee, Y. J., Seol, J. W. & Park, S. Y. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol. Aging 34, 146–156 (2013).
Li, L., Tan, J., Miao, Y., Lei, P. & Zhang, Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol. Neurobiol. 35, 615–621 (2015).
Vitale, E. et al. Role of chaperone-mediated autophagy in ageing biology and rejuvenation of stem cells. Front. Cell Dev. Biol. 10, 912470 (2022).
Manoogian, E. N. C. & Panda, S. Circadian rhythms, time-restricted feeding, and healthy aging. Ageing Res. Rev. 39, 59–67 (2017).
Acosta-Rodriguez, V. A., Rijo-Ferreira, F., Green, C. B. & Takahashi, J. S. Importance of circadian timing for aging and longevity. Nat. Commun. 12, 2862 (2021).
Hudec, M., Dankova, P., Solc, R., Bettazova, N. & Cerna, M. Epigenetic regulation of circadian rhythm and its possible role in diabetes mellitus. Int. J. Mol. Sci. 21, 3005 (2020).
Hood, S. & Amir, S. The aging clock: circadian rhythms and later life. J. Clin. Invest. 127, 437–446 (2017).
Froy, O. Circadian aspects of energy metabolism and aging. Ageing Res. Rev. 12, 931–940 (2013).
Socaciu, A. I. et al. Melatonin, an ubiquitous metabolic regulator: functions, mechanisms and effects on circadian disruption and degenerative diseases. Rev. Endocr. Metab. Disord. 21, 465–478 (2020).
Jung-Hynes, B., Reiter, R. J. & Ahmad, N. Sirtuins, melatonin and circadian rhythms: building a bridge between aging and cancer. J. Pineal Res. 48, 9–19 (2010).
Gao, W. et al. The circadian clock has roles in mesenchymal stem cell fate decision. Stem Cell Res. Ther. 13, 200 (2022).
Brown, S. A. Circadian clock-mediated control of stem cell division and differentiation: beyond night and day. Development 141, 3105–3111 (2014).
Weger, M., Diotel, N., Dorsemans, A. C., Dickmeis, T. & Weger, B. D. Stem cells and the circadian clock. Dev. Biol. 431, 111–123 (2017).
Benitah, S. A. & Welz, P. S. Circadian regulation of adult stem cell homeostasis and aging. Cell Stem Cell 26, 817–831 (2020).
Liang, C. et al. Stabilization of heterochromatin by CLOCK promotes stem cell rejuvenation and cartilage regeneration. Cell Res. 31, 187–205 (2021).
Kondratova, A. A. & Kondratov, R. V. The circadian clock and pathology of the ageing brain. Nat. Rev. Neurosci. 13, 325–335 (2012).
Blacher, E. et al. Aging disrupts circadian gene regulation and function in macrophages. Nat. Immunol. 23, 229–236 (2022).
Dierickx, P. et al. Circadian networks in human embryonic stem cell-derived cardiomyocytes. EMBO Rep. 18, 1199–1212 (2017).
Kinouchi, K. & Sassone-Corsi, P. Metabolic rivalry: circadian homeostasis and tumorigenesis. Nat. Rev. Cancer 20, 645–661 (2020).
Du Pre, B. C. et al. Circadian rhythms in cell maturation. Physiology 29, 72–83 (2014).
Plikus, M. V. et al. The circadian clock in skin: implications for adult stem cells, tissue regeneration, cancer, aging, and immunity. J. Biol. Rhythms 30, 163–182 (2015).
Lananna, B. V. & Musiek, E. S. The wrinkling of time: Aging, inflammation, oxidative stress, and the circadian clock in neurodegeneration. Neurobiol. Dis. 139, 104832 (2020).
Montaruli, A. et al. Biological rhythm and chronotype: new perspectives in health. Biomolecules 11, 487 (2021).
Dean, W. Pathways of DNA demethylation. Adv. Exp. Med. Biol. 945, 247–274 (2016).
Nishino, K. et al. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 7, e1002085 (2011).
Hysolli, E. et al. Regulation of the DNA methylation landscape in human somatic cell reprogramming by the miR-29 family. Stem Cell Rep. 7, 43–54 (2016).
Shamsian, A. et al. Cancer cells as a new source of induced pluripotent stem cells. Stem Cell Res. Ther. 13, 459 (2022).
Lapasset, L. et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25, 2248–2253 (2011).
Guan, J. et al. Chemical reprogramming of human somatic cells to pluripotent stem cells. Nature 605, 325–331 (2022).
Gill, D. et al. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. Elife 11, e71624 (2022).
Sarkar, T. J. et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 11, 1545 (2020).
Roux, A. E. et al. Diverse partial reprogramming strategies restore youthful gene expression and transiently suppress cell identity. Cell Syst. 13, 574–587.e511 (2022).
Ocampo, A. et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167, 1719–1733.e1712 (2016).
Browder, K. C. et al. In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nat. Aging 2, 243–253 (2022).
Hishida, T. et al. In vivo partial cellular reprogramming enhances liver plasticity and regeneration. Cell Rep. 39, 110730 (2022).
Cheng, F. et al. Partial reprogramming strategy for intervertebral disc rejuvenation by activating energy switch. Aging Cell 21, e13577 (2022).
Ribeiro, R. et al. In vivo cyclic induction of the FOXM1 transcription factor delays natural and progeroid aging phenotypes and extends healthspan. Nat. Aging 2, 397–411 (2022).
Ohnishi, K. et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 156, 663–677 (2014).
Pennarossa, G. et al. Brief demethylation step allows the conversion of adult human skin fibroblasts into insulin-secreting cells. Proc. Natl Acad. Sci. USA 110, 8948–8953 (2013).
Schutzius, G. et al. BET bromodomain inhibitors regulate keratinocyte plasticity. Nat. Chem. Biol. 17, 280–290 (2021).
Ma, X. et al. Human expandable pancreatic progenitor-derived beta cells ameliorate diabetes. Sci. Adv. 8, eabk1826 (2022).
Hou, P. et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341, 651–654 (2013).
Pasyukova, E. G. & Vaiserman, A. M. HDAC inhibitors: a new promising drug class in anti-aging research. Mech. Ageing Dev. 166, 6–15 (2017).
Wang, W. et al. A genome-wide CRISPR-based screen identifies KAT7 as a driver of cellular senescence. Sci. Transl. Med. 13, eabd2655 (2021).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Yosef, R. et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190 (2016).
Jaijyan, D. K. et al. New intranasal and injectable gene therapy for healthy life extension. Proc. Natl Acad. Sci. USA 119, e2121499119 (2022).
Davidsohn, N. et al. A single combination gene therapy treats multiple age-related diseases. Proc. Natl Acad. Sci. USA 116, 23505–23511 (2019).
Chaib, S., Tchkonia, T. & Kirkland, J. L. Cellular senescence and senolytics: the path to the clinic. Nat. Med. 28, 1556–1568 (2022).
Breccia, M. & Alimena, G. Activity and safety of dasatinib as second-line treatment or in newly diagnosed chronic phase chronic myeloid leukemia patients. BioDrugs 25, 147–157 (2011).
Zhu, Y. et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 9, 955–963 (2017).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Brighton, P. J. et al. Clearance of senescent decidual cells by uterine natural killer cells in cycling human endometrium. Elife 6, e31274 (2017).
Egashira, M. et al. F4/80+ macrophages contribute to clearance of senescent cells in the mouse postpartum uterus. Endocrinology 158, 2344–2353 (2017).
Hu, J., Batth, I. S., Xia, X. & Li, S. Regulation of NKG2D(+)CD8(+) T-cell-mediated antitumor immune surveillance: Identification of a novel CD28 activation-mediated, STAT3 phosphorylation-dependent mechanism. Oncoimmunology 5, e1252012 (2016).
Alimbetov, D. et al. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 17, 305–315 (2016).
Xu, M. et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl Acad. Sci. USA 112, E6301–E6310 (2015).
Laberge, R. M. et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 11, 569–578 (2012).
Perrott, K. M., Wiley, C. D., Desprez, P. Y. & Campisi, J. Apigenin suppresses the senescence-associated secretory phenotype and paracrine effects on breast cancer cells. Geroscience 39, 161–173 (2017).
Niklander, S., Bandaru, D., Lambert, D. W. & Hunter, K. D. ROCK inhibition modulates the senescence-associated secretory phenotype (SASP) in oral keratinocytes. FEBS Open Bio. 10, 2740–2749 (2020).
Aiello, A. et al. Nutrient sensing pathways as therapeutic targets for healthy ageing. Expert Opin. Ther. Targets 21, 371–380 (2017).
Chen, C., Liu, Y., Liu, Y. & Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal 2, ra75 (2009).
Castilho, R. M., Squarize, C. H., Chodosh, L. A., Williams, B. O. & Gutkind, J. S. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell 5, 279–289 (2009).
Takayama, K. et al. mTOR signaling plays a critical role in the defects observed in muscle-derived stem/progenitor cells isolated from a murine model of accelerated aging. J. Orthop. Res. 35, 1375–1382 (2017).
Kawakami, Y. et al. Rapamycin rescues age-related changes in muscle-derived stem/progenitor cells from progeroid mice. Mol. Ther. Methods Clin. Dev. 14, 64–76 (2019).
Neff, F. et al. Rapamycin extends murine lifespan but has limited effects on aging. J. Clin. Invest. 123, 3272–3291 (2013).
Martin-Montalvo, A. et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 4, 2192 (2013).
Chung, M. M. et al. Metformin activation of AMPK suppresses AGE-induced inflammatory response in hNSCs. Exp. Cell Res. 352, 75–83 (2017).
Neumann, B. et al. Metformin restores CNS remyelination capacity by rejuvenating aged stem cells. Cell Stem Cell 25, 473–485.e478 (2019).
Na, H. J. et al. Metformin inhibits age-related centrosome amplification in Drosophila midgut stem cells through AKT/TOR pathway. Mech. Ageing Dev. 149, 8–18 (2015).
Kulkarni, A. S. et al. Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell 17, e12723 (2018).
Weeden, C. E. & Asselin-Labat, M. L. Mechanisms of DNA damage repair in adult stem cells and implications for cancer formation. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 89–101 (2018).
Ruszkiewicz, J. A., Burkle, A. & Mangerich, A. Fueling genome maintenance: on the versatile roles of NAD(+) in preserving DNA integrity. J. Biol. Chem. 298, 102037 (2022).
Amano, H. et al. Telomere dysfunction induces sirtuin repression that drives telomere-dependent disease. Cell Metab. 29, 1274–1290.e1279 (2019).
Ren, C. et al. Nicotinamide mononucleotide ameliorates cellular senescence and inflammation caused by sodium iodate in RPE. Oxid. Med. Cell Longev. 2022, 5961123 (2022).
Surjana, D., Halliday, G. M. & Damian, D. L. Nicotinamide enhances repair of ultraviolet radiation-induced DNA damage in human keratinocytes and ex vivo skin. Carcinogenesis 34, 1144–1149 (2013).
Ong, D. S. & Kelly, J. W. Chemical and/or biological therapeutic strategies to ameliorate protein misfolding diseases. Curr. Opin. Cell Biol. 23, 231–238 (2011).
Kolb, P. S. et al. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int. J. Biochem. Cell Biol. 61, 45–52 (2015).
Hekmatimoghaddam, S., Zare-Khormizi, M. R. & Pourrajab, F. Underlying mechanisms and chemical/biochemical therapeutic approaches to ameliorate protein misfolding neurodegenerative diseases. Biofactors 43, 737–759 (2017).
Copelan, E. A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 354, 1813–1826 (2006).
Wang, P. et al. Exosomes derived from epidermal stem cells improve diabetic wound healing. J. Invest. Dermatol. 142, 2508–2517.e2513 (2022).
Cerletti, M. et al. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell 134, 37–47 (2008).
Bernet, J. D. et al. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 20, 265–271 (2014).
Li, G. et al. Rejuvenation of senescent bone marrow mesenchymal stromal cells by pulsed triboelectric stimulation. Adv. Sci. 8, e2100964 (2021).
Wang, B. et al. Bone repairment via mechanosensation of Piezo1 using wearable pulsed triboelectric nanogenerator. Small 18, e2201056 (2022).
Schweitzer, J. S. et al. Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N. Engl. J. Med. 382, 1926–1932 (2020).
Chen, B. et al. Human embryonic stem cell-derived exosomes promote pressure ulcer healing in aged mice by rejuvenating senescent endothelial cells. Stem Cell Res. Ther. 10, 142 (2019).
Lei, Q. et al. Extracellular vesicles deposit PCNA to rejuvenate aged bone marrow-derived mesenchymal stem cells and slow age-related degeneration. Sci. Transl. Med. 13, eaaz8697 (2021).
Shwartz, Y. et al. Cell types promoting goosebumps form a niche to regulate hair follicle stem cells. Cell 182, 578–593.e519 (2020).
Huang, S. et al. Lgr6 marks epidermal stem cells with a nerve-dependent role in wound re-epithelialization. Cell Stem Cell 28, 1582–1596.e1586 (2021).
Pei, M. Environmental preconditioning rejuvenates adult stem cells’ proliferation and chondrogenic potential. Biomaterials 117, 10–23 (2017).
Rodesch, F., Simon, P., Donner, C. & Jauniaux, E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet. Gynecol. 80, 283–285 (1992).
Cowden Dahl, K. D. et al. Hypoxia-inducible factors 1alpha and 2alpha regulate trophoblast differentiation. Mol. Cell Biol. 25, 10479–10491 (2005).
Ma, T., Grayson, W. L., Frohlich, M. & Vunjak-Novakovic, G. Hypoxia and stem cell-based engineering of mesenchymal tissues. Biotechnol. Prog. 25, 32–42 (2009).
Kamranvar, S. A., Rani, B. & Johansson, S. Cell cycle regulation by integrin-mediated adhesion. Cells 11, 2521 (2022).
Draicchio, F. et al. Involvement of the extracellular matrix and integrin signalling proteins in skeletal muscle glucose uptake. J. Physiol. 600, 4393–4408 (2022).
Du, H. et al. Tuning immunity through tissue mechanotransduction. Nat. Rev. Immunol. 1–15 (2022).
Rasouli, M., Rahimi, A., Soleimani, M. & Keshel, S. H. The interplay between extracellular matrix and progenitor/stem cells during wound healing: opportunities and future directions. Acta Histochem. 123, 151785 (2021).
Rothwell, P. M. et al. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 377, 31–41 (2011).
Adler, A. S. et al. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 21, 3244–3257 (2007).
Zhang, G. et al. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 497, 211–216 (2013).
Hodge, B. A. et al. Dietary restriction and the transcription factor clock delay eye aging to extend lifespan in Drosophila Melanogaster. Nat. Commun. 13, 3156 (2022).
Atger, F. et al. Circadian and feeding rhythms differentially affect rhythmic mRNA transcription and translation in mouse liver. Proc. Natl Acad. Sci. USA 112, E6579–E6588 (2015).
Wang, H. et al. Time-restricted feeding shifts the skin circadian clock and alters UVB-induced DNA damage. Cell Rep. 20, 1061–1072 (2017).
Ulgherait, M. et al. Circadian autophagy drives iTRF-mediated longevity. Nature 598, 353–358 (2021).
Sato, S. et al. Circadian reprogramming in the liver identifies metabolic pathways of aging. Cell 170, 664–677.e611 (2017).
Mendoza, J., Drevet, K., Pevet, P. & Challet, E. Daily meal timing is not necessary for resetting the main circadian clock by calorie restriction. J. Neuroendocrinol. 20, 251–260 (2008).
Patel, S. A., Chaudhari, A., Gupta, R., Velingkaar, N. & Kondratov, R. V. Circadian clocks govern calorie restriction-mediated life span extension through BMAL1- and IGF-1-dependent mechanisms. FASEB J. 30, 1634–1642 (2016).
Dudek, M. et al. The intervertebral disc contains intrinsic circadian clocks that are regulated by age and cytokines and linked to degeneration. Ann. Rheum. Dis. 76, 576–584 (2017).
Wang, D. et al. Restoring the dampened expression of the core clock molecule BMAL1 protects against compression-induced intervertebral disc degeneration. Bone Res. 10, 20 (2022).
Bekki, H. et al. Suppression of circadian clock protein cryptochrome 2 promotes osteoarthritis. Osteoarthr. Cartil. 28, 966–976 (2020).
Rakshit, K. & Giebultowicz, J. M. Cryptochrome restores dampened circadian rhythms and promotes healthspan in aging Drosophila. Aging Cell 12, 752–762 (2013).
Hirota, T. et al. Identification of small molecule activators of cryptochrome. Science 337, 1094–1097 (2012).
Solovev, I. A., Shaposhnikov, M. V. & Moskalev, A. A. Chronobiotics KL001 and KS15 extend lifespan and modify circadian rhythms of Drosophila melanogaster. Clocks Sleep. 3, 429–441 (2021).
Solt, L. A. et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 485, 62–68 (2012).
He, B. et al. The small molecule nobiletin targets the molecular oscillator to enhance circadian rhythms and protect against metabolic syndrome. Cell Metab. 23, 610–621 (2016).
Larion, S. et al. The biological clock enhancer nobiletin ameliorates steatosis in obesity by restoring aberrant hepatic circadian rhythm. Am. J. Physiol. Gastrointest. Liver Physiol. 323, G387–G400 (2022).
Wirianto, M. et al. The clock modulator Nobiletin mitigates astrogliosis-associated neuroinflammation and disease hallmarks in an Alzheimer’s disease model. FASEB J. 36, e22186 (2022).
Chen, Z., Yoo, S. H. & Takahashi, J. S. Development and therapeutic potential of small-molecule modulators of circadian systems. Annu. Rev. Pharm. Toxicol. 58, 231–252 (2018).
Yamashita, M. & Iwama, A. Aging and clonal behavior of hematopoietic stem cells. Int. J. Mol. Sci. 23, 1948 (2022).
Maryanovich, M. et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 24, 782–791 (2018).
Ho, Y. H. et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell 25, 407–418.e406 (2019).
Shirakawa, K. & Sano, M. T cell immunosenescence in aging, obesity, and cardiovascular disease. Cells 10, 2435 (2021).
Mohtashami, M., Li, Y. R., Lee, C. R. & Zuniga-Pflucker, J. C. Thymus reconstitution in young and aged mice is facilitated by in vitro-generated progenitor T cells. Front. Immunol. 13, 926773 (2022).
Singh, J., Mohtashami, M., Anderson, G. & Zuniga-Pflucker, J. C. Thymic engraftment by in vitro-derived progenitor T cells in young and aged mice. Front. Immunol. 11, 1850 (2020).
Wang, T. W. et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 611, 358–364 (2022).
Al-Chami, E. et al. Interleukin-21 administration to aged mice rejuvenates their peripheral T-cell pool by triggering de novo thymopoiesis. Aging Cell 15, 349–360 (2016).
Parent, A. V. et al. Generation of functional thymic epithelium from human embryonic stem cells that supports host T cell development. Cell Stem Cell 13, 219–229 (2013).
Sun, L. et al. Declining expression of a single epithelial cell-autonomous gene accelerates age-related thymic involution. Aging Cell 9, 347–357 (2010).
Chhatta, A. R. et al. De novo generation of a functional human thymus from induced pluripotent stem cells. J. Allergy Clin. Immunol. 144, 1416–1419.e1417 (2019).
Oh, J., Wang, W., Thomas, R. & Su, D. M. Thymic rejuvenation via FOXN1-reprogrammed embryonic fibroblasts (FREFs) to counteract age-related inflammation. JCI Insight 5, e140313 (2020).
Conboy, I. M. et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764 (2005).
Villeda, S. A. et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663 (2014).
Villeda, S. A. et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011).
Smith, L. K. et al. beta2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 21, 932–937 (2015).
Loffredo, F. S. et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 153, 828–883 (2013).
Katsimpardi, L. et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344, 630–634 (2014).
Zhang, X. et al. Rejuvenation of neutrophils and their extracellular vesicles is associated with enhanced aged fracture healing. Aging Cell 21, e13651 (2022).
Lehallier, B. et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat. Med. 25, 1843–1850 (2019).
Chaleckis, R., Murakami, I., Takada, J., Kondoh, H. & Yanagida, M. Individual variability in human blood metabolites identifies age-related differences. Proc. Natl Acad. Sci. USA 113, 4252–4259 (2016).
Hoffman, J. M. et al. A longitudinal analysis of the effects of age on the blood plasma metabolome in the common marmoset, Callithrix jacchus. Exp. Gerontol. 76, 17–24 (2016).
Sahu, A. et al. Regulation of aged skeletal muscle regeneration by circulating extracellular vesicles. Nat. Aging 1, 1148–1161 (2021).
Mehdipour, M. et al. Plasma dilution improves cognition and attenuates neuroinflammation in old mice. Geroscience 43, 1–18 (2021).
Palmer, A. K. et al. Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes 64, 2289–2298 (2015).
Palmer, A. K., Tchkonia, T. & Kirkland, J. L. Senolytics: potential for alleviating diabetes and its complications. Endocrinology 162, bqab058 (2021).
Arora, S. et al. Invariant natural killer T cells coordinate removal of senescent cells. Med 2, 938–950 (2021).
Zhong, D. et al. mPGES-2 blockade antagonizes beta-cell senescence to ameliorate diabetes by acting on NR4A1. Nat. Metab. 4, 269–283 (2022).
Ilegems, E. et al. HIF-1alpha inhibitor PX-478 preserves pancreatic beta cell function in diabetes. Sci. Transl. Med. 14, eaba9112 (2022).
Yang, B. et al. RIPK3-mediated inflammation is a conserved beta cell response to ER stress. Sci. Adv. 6, eabd7272 (2020).
Wang, Y. et al. Long-term correction of diabetes in mice by in vivo reprogramming of pancreatic ducts. Mol. Ther. 26, 1327–1342 (2018).
Fu, W. et al. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and beta cells. Elife 3, e04631 (2014).
Yoshino, M. et al. Worksite-based intensive lifestyle therapy has profound cardiometabolic benefits in people with obesity and type 2 diabetes. Cell Metab. 34, 1431–1441.e5 (2022).
Wen, X. et al. Signaling pathways in obesity: mechanisms and therapeutic interventions. Signal Transduct. Target Ther. 7, 298 (2022).
Palmer, A. K. et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18, e12950 (2019).
Lee, G. et al. SREBP1c-PARP1 axis tunes anti-senescence activity of adipocytes and ameliorates metabolic imbalance in obesity. Cell Metab. 34, 702–718.e705 (2022).
Lin, H. et al. Lens regeneration using endogenous stem cells with gain of visual function. Nature 531, 323–328 (2016).
Snippert, H. J. et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 327, 1385–1389 (2010).
So, J., Kim, A., Lee, S. H. & Shin, D. Liver progenitor cell-driven liver regeneration. Exp. Mol. Med. 52, 1230–1238 (2020).
Xia, H. et al. Tissue repair and regeneration with endogenous stem cells. Nat. Rev. Mater. 3, 174–193 (2018).
Wei, X. et al. Senescence in chronic wounds and potential targeted therapies. Burns Trauma 10, tkab045 (2022).
Ji, S. F. et al. Small molecules facilitate single factor-mediated sweat gland cell reprogramming. Mil. Med. Res. 9, 13 (2022).
Sun, X. et al. Sweat gland organoids originating from reprogrammed epidermal keratinocytes functionally recapitulated damaged skin. Adv. Sci. 8, e2103079 (2021).
Sun, S. et al. Targeting ectodysplasin promotor by CRISPR/dCas9-effector effectively induces the reprogramming of human bone marrow-derived mesenchymal stem cells into sweat gland-like cells. Stem Cell Res. Ther. 9, 8 (2018).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079 (2017).
Chandra, A. et al. Targeted reduction of senescent cell burden alleviates focal radiotherapy-related bone loss. J. Bone Min. Res. 35, 1119–1131 (2020).
Coryell, P. R., Diekman, B. O. & Loeser, R. F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 17, 47–57 (2021).
Zheng, W. et al. Fisetin inhibits IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes through activating SIRT1 and attenuates the progression of osteoarthritis in mice. Int. Immunopharmacol. 45, 135–147 (2017).
Wang, M. et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 15, R5 (2013).
Liu, R. M. Aging, cellular senescence, and Alzheimer’s disease. Int. J. Mol. Sci. 23, 1989 (2022).
Bussian, T. J. et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 (2018).
Zhang, P. et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728 (2019).
Musi, N. et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17, e12840 (2018).
Lee, H. J. et al. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 33, 588–602 (2012).
Zhang, Q. et al. Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of Alzheimer’s disease. J. Neurochem 136, 815–825 (2016).
Han, F. et al. Development of stem cell-based therapy for Parkinson’s disease. Transl. Neurodegener. 4, 16 (2015).
Lunn, J. S., Sakowski, S. A. & Feldman, E. L. Concise review: Stem cell therapies for amyotrophic lateral sclerosis: recent advances and prospects for the future. Stem Cells 32, 1099–1109 (2014).
Costantino, S., Paneni, F. & Cosentino, F. Ageing, metabolism and cardiovascular disease. J. Physiol. 594, 2061–2073 (2016).
Cao, N. et al. Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science 352, 1216–1220 (2016).
Sadahiro, T. & Ieda, M. In vivo reprogramming as a new approach to cardiac regenerative therapy. Semin Cell Dev. Biol. 122, 21–27 (2022).
Chen, Y. et al. Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science 373, 1537–1540 (2021).
Abbate, A. et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 126, 1260–1280 (2020).
Dookun, E., Passos, J. F., Arthur, H. M. & Richardson, G. D. Therapeutic potential of senolytics in cardiovascular disease. Cardiovasc Drugs Ther. 36, 187–196 (2022).
Tzouvelekis, A., Ntolios, P. & Bouros, D. Stem cell treatment for chronic lung diseases. Respiration 85, 179–192 (2013).
Nonaka, P. N. et al. Lung bioengineering: physical stimuli and stem/progenitor cell biology interplay towards biofabricating a functional organ. Respir. Res. 17, 161 (2016).
Zhang, Z. Y., Bao, X. L., Cong, Y. Y., Fan, B. & Li, G. Y. Autophagy in age-related macular degeneration: a regulatory mechanism of oxidative stress. Oxid. Med. Cell Longev. 2020, 2896036 (2020).
Coulon, S. J. et al. A novel glaucoma approach: stem cell regeneration of the trabecular meshwork. Prog. Retin Eye Res. 90, 101063 (2022).
Mallick, S., Sharma, M., Kumar, A. & Du, Y. Cell-based therapies for trabecular meshwork regeneration to treat glaucoma. Biomolecules 11, 1258 (2021).
Zhou, B. et al. The emerging role of cellular senescence in renal diseases. J. Cell Mol. Med. 24, 2087–2097 (2020).
Mylonas, K. J. et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci. Transl. Med. 13, eabb0203 (2021).
Wang, W. J., Cai, G. Y. & Chen, X. M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget 8, 64520–64533 (2017).
Peired, A. J., Sisti, A. & Romagnani, P. Mesenchymal stem cell-based therapy for kidney disease: a review of clinical evidence. Stem Cells Int. 2016, 4798639 (2016).
Eirin, A. & Lerman, L. O. Mesenchymal stem cell treatment for chronic renal failure. Stem Cell Res. Ther. 5, 83 (2014).
Wei, Q. et al. Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am. J. Physiol. Ren. Physiol. 316, F1162–F1172 (2019).
Cui, H. et al. Inhibition of glutaminase 1 attenuates experimental pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 61, 492–500 (2019).
Kang, H. M. et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46 (2015).
Shook, B. A. et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 362, eaar2971 (2018).
Zhang, J. et al. A pulsatile release platform based on photo-induced imine-crosslinking hydrogel promotes scarless wound healing. Nat. Commun. 12, 1670 (2021).
Plikus, M. V. et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 355, 748–752 (2017).
Duan, L., Rao, X. & Sigdel, K. R. Regulation of inflammation in autoimmune disease. J. Immunol. Res. 2019, 7403796 (2019).
Jung, S. M. & Kim, W. U. Targeted immunotherapy for autoimmune disease. Immune Netw. 22, e9 (2022).
Fu, X., Sun, X., Li, X. & Sheng, Z. Dedifferentiation of epidermal cells to stem cells in vivo. Lancet 358, 1067–1068 (2001).
Zhang, C. et al. Dedifferentiation derived cells exhibit phenotypic and functional characteristics of epidermal stem cells. J. Cell Mol. Med. 14, 1135–1145 (2010).
Zhang, C. et al. Wnt/beta-catenin signaling is critical for dedifferentiation of aged epidermal cells in vivo and in vitro. Aging Cell 11, 14–23 (2012).
Franco, A. C., Aveleira, C. & Cavadas, C. Skin senescence: mechanisms and impact on whole-body aging. Trends Mol. Med. 28, 97–109 (2022).
Csekes, E. & Rackova, L. Skin aging, cellular senescence and natural polyphenols. Int. J. Mol. Sci. 22, 12641 (2021).
Wu, J. Y. et al. Stem cell-derived exosomes: a new method for reversing skin aging. Tissue Eng. Regen. Med. 19, 961–968 (2022).
Eckhart, L., Tschachler, E. & Gruber, F. Autophagic control of skin aging. Front Cell Dev. Biol. 7, 143 (2019).
Welz, P. S. Clock regulation of skin regeneration in stem cell aging. J. Invest. Dermatol 141, 1024–1030 (2021).
Oblong, J. E. et al. Metabolic dysfunction in human skin: restoration of mitochondrial integrity and metabolic output by nicotinamide (niacinamide) in primary dermal fibroblasts from older aged donors. Aging Cell 19, e13248 (2020).
Liu, N. et al. Stem cell competition orchestrates skin homeostasis and ageing. Nature 568, 344–350 (2019).
Yu, Y. et al. A stress-induced miR-31–CLOCK–ERK pathway is a key driver and therapeutic target for skin aging. Nat. Aging 1, 795–809 (2021).
Li, S. et al. A tetrahedral framework DNA-based bioswitchable miRNA inhibitor delivery system: application to skin anti-aging. Adv. Mater. 34, e2204287 (2022).
Ruhland, M. K. & Alspach, E. Senescence and immunoregulation in the tumor microenvironment. Front. Cell Dev. Biol. 9, 754069 (2021).
Watanabe, S., Kawamoto, S., Ohtani, N. & Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 108, 563–569 (2017).
Paez-Ribes, M., Gonzalez-Gualda, E., Doherty, G. J. & Munoz-Espin, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 11, e10234 (2019).
Kipps, T. J. et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk. Lymphoma 56, 2826–2833 (2015).
Saito, S. et al. Potential application of cell reprogramming techniques for cancer research. Cell Mol. Life Sci. 76, 45–65 (2019).
Rapino, F. et al. C/EBPalpha induces highly efficient macrophage transdifferentiation of B lymphoma and leukemia cell lines and impairs their tumorigenicity. Cell Rep. 3, 1153–1163 (2013).
Cheng, Z. et al. Conversion of hepatoma cells to hepatocyte-like cells by defined hepatocyte nuclear factors. Cell Res. 29, 124–135 (2019).
Ma, X., Kong, L. & Zhu, S. Reprogramming cell fates by small molecules. Protein Cell 8, 328–348 (2017).
Zhang, X. et al. Small molecule-induced differentiation as a potential therapy for liver cancer. Adv. Sci. 9, e2103619 (2022).
Lin, S. L. et al. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA 14, 2115–2124 (2008).
Zhou, S., Abdouh, M., Arena, V., Arena, M. & Arena, G. O. Reprogramming malignant cancer cells toward a benign phenotype following exposure to human embryonic stem cell microenvironment. PLoS ONE 12, e0169899 (2017).
Duan, S. et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 6, 10068 (2015).
Han, X. et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat. Commun. 5, 3261 (2014).
Zou, M. et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Disco. 7, 736–749 (2017).
Nakano, M. et al. Dedifferentiation process driven by TGF-beta signaling enhances stem cell properties in human colorectal cancer. Oncogene 38, 780–793 (2019).
Wang, L. et al. Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell Death Dis. 8, e2746 (2017).
Conley, S. J. et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl Acad. Sci. USA 109, 2784–2789 (2012).
Landsberg, J. et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412–416 (2012).
Sacma, M. & Geiger, H. Exercise generates immune cells in bone. Nature 591, 371–372 (2021).
Duggal, N. A., Niemiro, G., Harridge, S. D. R., Simpson, R. J. & Lord, J. M. Can physical activity ameliorate immunosenescence and thereby reduce age-related multi-morbidity? Nat. Rev. Immunol. 19, 563–572 (2019).
Miyoshi, N. et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc. Natl Acad. Sci. USA 107, 40–45 (2010).
Li, F., Hu, J. & He, T. C. iPSC-based treatment of age-related macular degeneration (AMD): the path to success requires more than blind faith. Genes Dis. 4, 41–42 (2017).
Penney, J., Ralvenius, W. T. & Tsai, L. H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 25, 148–167 (2020).
Nizzardo, M. et al. iPSC-derived LewisX+CXCR4+beta1-integrin+ neural stem cells improve the amyotrophic lateral sclerosis phenotype by preserving motor neurons and muscle innervation in human and rodent models. Hum. Mol. Genet. 25, 3152–3163 (2016).
Kondo, Y., Toyoda, T., Inagaki, N. & Osafune, K. iPSC technology-based regenerative therapy for diabetes. J. Diabetes Investig. 9, 234–243 (2018).
Liu, H. et al. The potential of induced pluripotent stem cells as a tool to study skeletal dysplasias and cartilage-related pathologic conditions. Osteoarthr. Cartil. 25, 616–624 (2017).
Xie, J. et al. Osteogenic differentiation and bone regeneration of iPSC-MSCs supported by a biomimetic nanofibrous scaffold. Acta Biomater. 29, 365–379 (2016).
Hew, M., O’Connor, K., Edel, M. J. & Lucas, M. The possible future roles for iPSC-derived therapy for autoimmune diseases. J. Clin. Med. 4, 1193–1206 (2015).
Wei, R. & Hong, T. Lineage reprogramming: a promising road for pancreatic beta cell regeneration. Trends Endocrinol. Metab. 27, 163–176 (2016).
Gong, L. et al. Cancer cell reprogramming: a promising therapy converting malignancy to benignity. Cancer Commun. 39, 48 (2019).
Wei, Z. D. & Shetty, A. K. Treating Parkinson’s disease by astrocyte reprogramming: progress and challenges. Sci. Adv. 7, eabg3198 (2021).
Xiao, D. et al. Direct reprogramming of fibroblasts into neural stem cells by single non-neural progenitor transcription factor Ptf1a. Nat. Commun. 9, 2865 (2018).
Chen, Y., Yang, Z., Zhao, Z. A. & Shen, Z. Direct reprogramming of fibroblasts into cardiomyocytes. Stem Cell Res. Ther. 8, 118 (2017).
Pearson, R. M., Casey, L. M., Hughes, K. R., Miller, S. D. & Shea, L. D. In vivo reprogramming of immune cells: Technologies for induction of antigen-specific tolerance. Adv. Drug Deliv. Rev. 114, 240–255 (2017).
Banga, A., Akinci, E., Greder, L. V., Dutton, J. R. & Slack, J. M. In vivo reprogramming of Sox9+ cells in the liver to insulin-secreting ducts. Proc. Natl Acad. Sci. USA 109, 15336–15341 (2012).
Kurita, M. et al. In vivo reprogramming of wound-resident cells generates skin epithelial tissue. Nature 561, 243–247 (2018).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Baker, D. J. et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Wang, L., Lankhorst, L. & Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 22, 340–355 (2022).
Li, C., Shen, Y., Huang, L., Liu, C. & Wang, J. Senolytic therapy ameliorates renal fibrosis postacute kidney injury by alleviating renal senescence. FASEB J. 35, e21229 (2021).
Lee, S. et al. A guide to senolytic intervention in neurodegenerative disease. Mech. Ageing Dev. 200, 111585 (2021).
Wu, Y. et al. Senolytics: eliminating senescent cells and alleviating intervertebral disc degeneration. Front Bioeng. Biotechnol. 10, 823945 (2022).
Wang, M. Senolytics for kidney repair. Nat. Rev. Nephrol. 17, 512 (2021).
Szwed, A., Kim, E. & Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 101, 1371–1426 (2021).
Merkt, W., Bueno, M., Mora, A. L. & Lagares, D. Senotherapeutics: targeting senescence in idiopathic pulmonary fibrosis. Semin Cell Dev. Biol. 101, 104–110 (2020).
Krizhanovsky, V. et al. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667 (2008).
Luo, Y. et al. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation 97, 20–29 (2014).
Hong, W., Mo, F., Zhang, Z., Huang, M. & Wei, X. Nicotinamide mononucleotide: a promising molecule for therapy of diverse diseases by targeting NAD+ metabolism. Front. Cell Dev. Biol. 8, 246 (2020).
Squillaro, T., Peluso, G. & Galderisi, U. Clinical trials with mesenchymal stem cells: an update. Cell Transpl. 25, 829–848 (2016).
Wu, Q., Gao, Z. J., Yu, X. & Wang, P. Dietary regulation in health and disease. Signal Transduct. Target Ther. 7, 252 (2022).
Weyand, C. M., Fujii, H., Shao, L. & Goronzy, J. J. Rejuvenating the immune system in rheumatoid arthritis. Nat. Rev. Rheumatol. 5, 583–588 (2009).
Julier, Z., Park, A. J., Briquez, P. S. & Martino, M. M. Promoting tissue regeneration by modulating the immune system. Acta Biomater. 53, 13–28 (2017).
Sharma, H. & Moroni, L. Recent advancements in regenerative approaches for thymus rejuvenation. Adv. Sci. 8, 2100543 (2021).
Baht, G. S. et al. Exposure to a youthful circulaton rejuvenates bone repair through modulation of beta-catenin. Nat. Commun. 6, 7131 (2015).
Iram, T. et al. Young CSF restores oligodendrogenesis and memory in aged mice via Fgf17. Nature 605, 509–515 (2022).
Castellano, J. M. et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 544, 488–492 (2017).
Sahu, A. et al. Age-related declines in alpha-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nat. Commun. 9, 4859 (2018).
Kang, J. S. & Yang, Y. R. Circulating plasma factors involved in rejuvenation. Aging 12, 23394–23408 (2020).
Xu, Q. et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat. Metab. 3, 1706–1726 (2021).
Johmura, Y. et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 371, 265–270 (2021).
He, Y. et al. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell 19, e13117 (2020).
Shan, H. et al. Large-scale chemical screen identifies Gallic acid as a geroprotector for human stem cells. Protein Cell 13, 532–539 (2022).
D’Amico, D. et al. Urolithin A improves mitochondrial health, reduces cartilage degeneration, and alleviates pain in osteoarthritis. Aging Cell 21, e13662 (2022).
Flores, A. et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol. 19, 1017–1026 (2017).
Tamura, H. et al. Long-term melatonin treatment delays ovarian aging. J. Pineal. Res. 62, e12381 (2017).
Petrovski, G., Gurusamy, N. & Das, D. K. Resveratrol in cardiovascular health and disease. Ann. N. Y Acad. Sci. 1215, 22–33 (2011).
de Picciotto, N. E. et al. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15, 522–530 (2016).
Gao, Q. et al. Inhibition of DNA methyltransferase aberrations reinstates antioxidant aging suppressors and ameliorates renal aging. Aging Cell 21, e13526 (2022).
Wong, L. R. et al. Eicosanoid signalling blockade protects middle-aged mice from severe COVID-19. Nature 605, 146–151 (2022).
Lei, Z. et al. Ginsenoside Rb1 improves intestinal aging via regulating the expression of sirtuins in the intestinal epithelium and modulating the gut microbiota of mice. Front. Pharm. 13, 991597 (2022).
Wijaya, Y. T. et al. Ginsenoside Rd ameliorates muscle wasting by suppressing the signal transducer and activator of transcription 3 pathway. J. Cachexia Sarcopenia Muscle 13, 3149–3162 (2022).
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (81871569, 81830064, 81721092, 61803250), the National Key Research and Development Plan (2018YFC1105704, 2017YFC1103304, 2016YFA0101000, 2016YFA0101002), the CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-5-059), the Military Key Basic Research of Foundational Strengthening Program (2020-JCJQ-ZD-256-021), the Military Medical Research and Development Projects (AWS17J005, 2019-126), and the Specific Research Fund of The Innovation Platform for Academicians of Hainan Province.
Author information
Authors and Affiliations
Contributions
S.J., M.X., X.F., and X.S.: the conceptualization and design of this study. S.J. and M.X.: the investigation and methodology of this study. S.J., M.X., H.C., Y.L., L.Z., Y.H., and M.W.: writing—original draft. S.J. and M.X.: Figures and tables of the manuscript. S.J., M.X., C.W., and X.S.: writing—review & editing of the manuscript. X.F. and X.S.: the funding acquisition. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ji, S., Xiong, M., Chen, H. et al. Cellular rejuvenation: molecular mechanisms and potential therapeutic interventions for diseases. Sig Transduct Target Ther 8, 116 (2023). https://doi.org/10.1038/s41392-023-01343-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01343-5
This article is cited by
-
Kdm6a-CNN1 axis orchestrates epigenetic control of trauma-induced spinal cord microvascular endothelial cell senescence to balance neuroinflammation for improved neurological repair
Bone Research (2024)
-
Research progress on the GRP78 gene in the diagnosis, treatment and immunity of cervical cancer
European Journal of Medical Research (2023)
-
Exploring the promising potential of induced pluripotent stem cells in cancer research and therapy
Molecular Cancer (2023)
-
The Prognosis of Cancer Depends on the Interplay of Autophagy, Apoptosis, and Anoikis within the Tumor Microenvironment
Cell Biochemistry and Biophysics (2023)
