
Abstract
Currently, pyroptosis has received more and more attention because of its association with innate immunity and disease. The research scope of pyroptosis has expanded with the discovery of the gasdermin family. A great deal of evidence shows that pyroptosis can affect the development of tumors. The relationship between pyroptosis and tumors is diverse in different tissues and genetic backgrounds. In this review, we provide basic knowledge of pyroptosis, explain the relationship between pyroptosis and tumors, and focus on the significance of pyroptosis in tumor treatment. In addition, we further summarize the possibility of pyroptosis as a potential tumor treatment strategy and describe the side effects of radiotherapy and chemotherapy caused by pyroptosis. In brief, pyroptosis is a double-edged sword for tumors. The rational use of this dual effect will help us further explore the formation and development of tumors, and provide ideas for patients to develop new drugs based on pyroptosis.
Similar content being viewed by others
Introduction
The earliest research on pyroptosis can be traced back to 1986. Friedlander pointed out in the report that treatment of primary mouse macrophages with anthrax lethal toxin (LT) resulted in cell death and rapid release of cell contents.1 Cerretti et al. and Thornberry et al. observed that ICE (interleukin-1β-converting enzyme, caspase-1), discovered for the first time in 1989,2 was an inflammatory caspase, processing precursor IL-1β into mature IL-1β.3,4 In 1992, Zychlinsky et al. discovered pyroptosis for the first time, and they found suicide in the Gram-negative bacterial pathogen Shigella flexneri-infected macrophages.5 In 1996, Chen et al. reported that invasion plasmid antigen B (ipaB) of Shigella flexneri could bind directly to ICE and caused the enzyme to be activated in infected macrophages.6 This form of cell death was originally deemed to be apoptosis because some of its characteristics were similar to apoptosis, such as caspase-dependent, DNA damage, and nuclear condensation. Afterward, this form of death was observed to be different from apoptosis. In 2001, D’Souza et al. pointed out the term of pyroptosis, which comes from the Greek roots pyro (fire/fever) and ptosis (to-sis, falling), to describe pro-inflammatory programmed cell death.7 This is the first definition of pyroptosis, which makes the distinction between pyroptosis and apoptosis (a non-inflammatory program of cell death).7 In 2002, Inflammasome was first considered to activate inflammatory caspases and process pro-IL-1β.8 Afterward, Petr et al. found non-canonical caspase-11 could induce cell death without relying on caspase-1 when the host was infected with Salmonella.9 Pyroptosis was considered to be caspase-1-induced monocyte death for a long time,.10,11 Later, it was reported that caspase-1 or caspase-11/4/5 was activated during this process, and gasdermin D (GSDMD) was cleaved, and the N-terminal domain can oligomerize to form pores in the cell membrane, inducing cell membrane rupture.12 Interestingly, reports in recent years have shown that GSDMD-mediated pyroptosis can be blocked by some factors. Caspase-3/7 cleaved GSDMD at Asp87 site, causing inactivation of the pyroptotic activity of GSDMD.13 In addition, the endosomal sorting complexes required for transport (ESCRT) machinery could eliminate GSDMD pores from the plasma membrane, resulting in the inhibition of GSDMD-mediated pyroptotic cell death and limite the release of IL-1β after the activation of inflammasome.14 Humphries et al. found that the intermediate product of tricarboxylic acid cycle, fumarate, also dampened pyroptosis.15 Both fumarate and dimethyl fumarate (DMF) could prevent GSDMD from being processed and activated by caspases by succinizing the cysteine of GSDMD.15 In 2017, Wang et al. and Rogers et al. showed that chemotherapeutic agents can also induce pyroptosis by activating caspase-3 to shear GSDME.16,17 Subsequently, caspase-8 was indicated to cause pyroptosis and affect inflammasome function.18 In 2020, it was reported that granzyme B (GzmB) can directly cleave GSDME and activate pyroptosis, further activating the antitumor immune response and inhibiting tumor growth.19 In the same year, granzyme A (GzmA) in cytotoxic lymphocytes was found to enter target cells through perforin and induce pyroptosis by hydrolyzing GSDMB at the Lys229/Lys244 site, which renewed our understanding of pyroptosis.20 More recently, pyroptosis has made further progress. Hou et al. found that under hypoxia, activated p-Stat3 promoted nuclear PD-L1 translocation. Nuclear PD-L1 and p-Stat3 synergistically promote the expression of GSDMC, and caspase-8 activated by TNF-α derived from macrophages can cleave GSDMC into N-GSDMC at the D365 site, resulting in pyroptosis ultimately21 (Fig. 1).

The timeline of pyroptosis
Tumors use multiple strategies to avoid or limit the cell death pathway.22 In some cases, tumors can be killed by different ways, such as apoptosis, necrosis, autophagy and pyroptosis.23,24 Apoptosis and other modes of death are important anticancer defense mechanisms that have been widely studied, but the relationship between pyroptosis and cancer is not fully understood at present. Pyroptosis is closely related to nervous system diseases, infectious diseases, autoimmune diseases, cardiovascular diseases, and tumors.25,26,27,28 With further research, the relationship between pyroptosis and tumors is becoming increasingly clear, which provides some strategies for clinical treatments.
We focus on the mechanism, the diverse functions in various tumors and the potential clinical value of pyroptosis. These findings have raised awareness of tumors and identified a number of potential cancer treatments. We look forward to communicating the recent achievements in pyroptosis research and its development in the field of oncology. These findings improve the understanding of tumors and identify a number of potential tumor treatments.
Gasdermin, the executioner of pyroptosis
Pyroptosis was defined as gasdermin-mediated programmed death in 2015.29 The gasdermin superfamily is composed of gasdermin A/B/C/D (GSDMA/B/C/D), gasdermin E (GSDME, also referred to as DFNA5) and DFNB59 (Pejvakin, PJVK) in human (Gsdma1-3, Gsdmc1-4, Gsdmd, Dfna5, and Dfnb59 in mice).30,31,32,33,34 Among these conserved proteins, GSDMD and DFNA5 are most deeply studied in pyroptotic death. Except for Pejvakin, all these members consist of two conserved domains, the N-terminal pore-forming domain and the C-terminal repressor domain (PFD and RD).35,36,37,38 The PFD of most gasdermins could induce pyroptosis, which has not yet been detected for Pejvakin.17,30,39 In general, gasdermins maintain oligomerization through the interaction between PFD and RD, and the cytotoxic effects of PFD could be inhibited by RD.37,38 When the host is stimulated by a variety of exogenous or endogenous factors, gasdermins are cleaved by some caspases or granzymes, and the N-terminal PFD is dissociated from the C-terminal RD, and then the N-terminal PFD oligomerizes and forms pores in the cell membrane, causing the release of inflammatory molecules and cell pyroptotic death.30,40,41 Although a number of gasdermin family proteins have been reported to be linked to human diseases,20,29,39,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63 the specific mechanism and function remain to be studied.
The characteristics of pyroptosis
Pyroptosis is composed of “pyro” and “ptosis”. “Pyro” means fire, indicating the properties of inflammation of pyroptosis, but “ptosis” means falling, which is consistent with other forms of programmed cell death.7 There are some similarities between pyroptosis and apoptosis, such as DNA damage and chromatin condensation.64,65 Interestingly, pyroptotic cells emerged swelling and a lot of bubble-like protrusions appear on the surface of the cellular membrane before its rupture.66 Similarly, membrane blebbing also occurs during apoptosis, and caspase-3 is necessary for this process.67,68,69,70,71,72,73,74,75,76 However, the unique morphological characteristics of pyroptosis are obviously different from those of apoptosis. It is generally believed that apoptosis is a safe form of death, but pyroptosis can cause inflammation, activated by extracellular or intracellular stimulation, such as bacterial, viral, toxin, and chemotherapy drugs.77 In fact, unlike the explosive rupture associated with necrosis, pyroptosis causes flattening of the cytoplasm due to plasma membrane leakage.66 In addition, caspases activation or release of granzymes results in the N-terminal of gasdermin oligomerization and pore formation (1–2 μm in diameter) in the plasma membrane, which allows mature IL-1β/IL-18 with a diameter of 4.5 nm and caspase-1 with a diameter of 7.5 nm to pass through, respectively.30 In the meantime, the water entering through the pores causes cell swelling and osmotic lysis, thus resulting in rupture of the plasma membrane and the release of IL-1β and IL-18.78,79 Thus, the pyroptotic cells are permeable to 7-aminoactinomycin (7-AAD), propidium iodide (PI), and ethidium bromide (EtBr) because of the low molecular weight of these dyes.79 On the contrary, in comparison with pyroptotic cells, apoptotic cells maintain membrane integrity,80 so that these dyes can’t stain them.81,82,83,84 Intriguingly, similar to apoptotic cells, Annexin V also stains pyroptotic cells and the dye binds to phosphatidyl serine (PS).85 Therefore, Annexin V cannot differentiate apoptotic cells from pyroptotic cells. In addition, apoptotic bodies are formed in the process of apoptosis, while pyroptotic bodies are formed in the process of pyroptosis.80 Interestingly, the diameter of pyroptotic bodies is similar to that of apoptotic bodies, and thier size are both 1–5 µm.66
Besides, there is a very special form of DNA damage with dUTP nick-end labeling (TUNEL) staining positive in the early stage of pyroptosis,79,86,87,88,89,90,91,92,93 which is different from that of apoptosis. Compared with apoptosis, the intensity of DNA damage is lower in pyroptotic cells. The DNA fragmentation of pyroptosis is random and the nucleus remains intact,94,95,96,97 while the apoptotic DNA fragment is ordered and the nucleus is fragmented.23,98,99,100 Interestingly, caspase activation is associated with both pyroptosis and apoptosis. Initially, pyroptosis was considered to be caspase-1-related cell death.5,23,101,102 It is worth mentioning that recent studies have shown that other caspases, including caspase-3/4/5/6/8/9/11, also cause pyroptosis in other different cells,16,17,29,39,94,103,104,105 and play major roles in innate immunity and tumorigenesis.106,107,108,109,110,111 Caspase-3 was previously thought to be the executor of apoptosis, but it was suggested that caspase-3 can also induce pyroptosis by cleaving GSDME.16,17,71 More surprisingly, the apoptosis-related protein caspase-8 can also directly cleave GSDMD to induce pyroptosis.103 In addition, the activation of caspase-9 is also involved in pyroptosis by cleaving and activating caspase-3,112 and caspase-6 mediates the cleavage of GSDMC.21 Although current studies have found that caspase-1 and caspase-4/5/11 are only associated with pyroptosis, while caspase-2, caspase-7, and caspase-10 are only related to apoptosis,36,113,114,115,116,117 the connections between caspases and pyroptosis as well as caspases and apoptosis may be reported one after another in the future with the deepening of studies. Apoptosis is affected by the level of ATP, and apoptosis is accompanied by the activation of PARP, causing ATP depletion.118 However, the effector proteins of pyroptosis belong to the gasdermin family23 (Tables 1 and 2).
Molecular mechanism of pyroptosis
Canonical pathway
Canonical pyroptotic death is mediated by inflammasome assembly, which is accompanied by GSDMD cleavage and IL-1β and IL-18 release.10,119,120,121,122,123,124,125,126,127,128 Inflammasomes are multimolecular complexes that are activated when the host is resistant to microbial infection and also facilitate the development of adaptive immune responses. In addition, Inflammasomes are also associated with non-microbial diseases. There is considerable evidence that inflammasomes and their related cytokines play crucial roles in oncogenesis, such as proliferation, metastasis, and invasion.129,130,131,132,133,134,135,136,137 The assembly of inflammasome begins with cytosolic pattern recognition receptors (PRRs, also known as inflammasome sensors), which are capable of recognizing pathogen-associated molecular patterns and danger-associated molecular patterns ((PAMPs and DAMPs).138,139 Activation of PRRs promotes downstream signaling pathway and causes type I interferons generation and pro-inflammatory cytokines release.129,140,141,142,143,144 PRRs assemble with pro-caspase-1 and ASC to form inflammasomes after stimulation of cells by signal molecules such as bacteria and viruses.11,141,145,146,147,148,149,150,151,152
At present, the inflammasome sensors NLRP1, NLRP3, NLRC4, AIM2, and pyrin are able to assemble canonical inflammasomes and have been relatively well studied. Most inflammasomes are constituted by three components: (i) leucine-rich repeat containing proteins (NOD-like receptors, NLRs), (ii) the adapter apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD) (ASC), and (iii) pro-caspase-1. NLRs usually consist of a leucine-rich repeat (LRR), a nucleotide-binding oligomerization domain (NACHT), and a CARD or pyrin domain (PYD),153,154,155,156 divided into NLRPs or NLRCs according to whether their N terminal contain PYD or CARD. The NLRCs have one or more CARD at their N terminal such as NLRC4, but the NLRPs contain PYD such as NLRP1 and NLRP3.101,143,157 Compared with NLRP3, NLRP1 contains extra function-to-find domain (FIIND) and CARD domains at the N terminal.158 Human NLRP1 carry three paralogues in mice: NLRP1a/b/c, and these NLRP1 in mice lack PYD.159 NLRP1b has been studied relatively extensively and responds to the Toxoplasma gondii, Val-boro-Pro, and the Bacillus anthracis anthrax lethal toxin.160,161,162,163 NLRP3 senses a variety of stimulus, such as toxins, pathogens, metabolites, crystalline substances, nucleic acids, and ATP.164The initial recognition of ligands by various NAIP proteins is necessary for NLRC4 inflammasome activation. The human NAIP can directly bind to the flagellin and proteins of the type 3 secretion system (T3SS).165,166,167,168 In contrast to human NAIP, the mouse NAIP1, NAIP2, NAIP5, and NAIP6 recognize the needle of T3SS, the inner rod of the T3SS, flagellin, and flagellin, respectively.165,166,169 Then, NAIPs induce the recruitment and oligomerization of NLRC4 to form the NLRC4 inflammasome complex, leading to the cleavage of caspase-1 and pyroptosis.170 Actually, non-NLRs, such as AIM2 and pyrin can also form inflammasomes. AIM2 consists of an electropositive HIN-200 domain at C terminal and an N-terminal PYD. HIN-200 domain can bind to the electronegative double-stranded DNA (dsDNA),152,171,172,173,174,175,176,177 and PYD can recruit ASC and pro-caspase-1 to assemble the inflammasome.152,171,172,173,174,175,176,177 Another inflammasome sensor, pyrin, is activated after the inactivation of small GTPases of the host RHO family. Rho-inactivating toxins, such as the Clostridium difficile glycosyltransferase TcdB, Vibrio parahaemolyticus VopS, and Clostridium botulinum ADP-ribosylating C3 toxin can induce the assembly of pyrin inflammasome. Besides, YopE (the Yersinia pestis GTPase-activating protein) and YopT (cysteine protease) also trigger the activation of pyrin inflammasome.178,179,180 Specifically, mutations of serine residues, such as S208A and S242R, relieve the phosphorylation, resulting in pyrin activation181,182,183 (Fig. 3).
ASC contains a pyrin domain (PYD) and a caspase activation and recruitment domain (CARD). The CARD is necessary to recruit pro-caspase-1 to form the inflammasome. Some PRRs contain CARD, allowing pro-caspase-1 to also be directly recruited.101 After inflammasome assembly, caspase-1 is activated, and is hydrolyzed into 2 fragments, forming a dimer to become mature cleaved caspase-1.184 On the one hand, caspase-1 cleaves the caustic executor protein GSDMD at the Asp275 site to form the 22 kDa C-terminus (C-GSDMD) and 31 kDa N-terminus (N-GSDMD). N-GSDMD perforates the cell membrane to form nonselective pores with inner diameters of ~10–14 nm, leading to cell swelling and pyroptosis.66,185 On the other hand, caspase-1 also cleaves the precursors of IL-1β and IL-18 to be the mature IL-1β and IL-18, which are released through the pores formed by GSDMD, resulting in pyroptosis.3,4,11,29,31,40 Notably, after inflammasome activation, cells may undergo pyroptosis or release cytokines without cell death, but the specific mechanism is unknown. Some studies have suggested that the Toll-like receptor adapter protein SARM is involved in this regulation186 (Figs. 2 and 3).

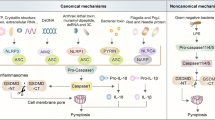
Molecular mechanism of pyroptosis. In the canonical pathway, PAMPs and DAMPs receive intracellular signaling molecule stimulation and assemble with pro-caspase-1 and ASC to form inflammasomes and active caspase-1. Cleaved-caspase-1 cleaves GSDMD and pro-IL-1β/18. N-GSDMD perforates the cell membrane by forming nonselective pores, further causing water influx, lysis, and death. In addition, IL-1β and IL-18 are secreted from the pores formed by N-GSDMD. In the noncanonical pathway, cytosolic LPS activates caspase-4/5 and caspase-11, triggering pyroptosis by cleaving GSDMD. However, oxPAPC competes with LPS to bind caspase-4/1, thus inhibiting pyroptosis. In addition, the cleavage of GSDMD results in efflux of K+, ultimately mediating the assembly of NLRP3 inflammasome, resulting in the cleavage of pro-IL-1β and pro-IL-18. The activated caspase-11 also cleaves Pannexin-1, inducing ATP release and P2X7R-related pyroptotic cell death. In the caspase-3-mediated pathway, active caspase-3 cleaves GSDME to form N-GSDME, inducing pyroptosis. In the caspase-8-mediated pathway, inhibiting TAK1 induces the activation of caspase-8, which cleaves GSDMD, resulting in pyroptosis. In addition, under hypoxia conditions, PD-L1 is transferred to the nucleus and regulates the transcription of GSDMC together with p-Stat3, resulting in the conversion of apoptosis to pyroptosis after TNFα-activated caspase-8. In the granzyme-mediated pathway, CAR T cells rapidly activate caspase-3 in target cells by releasing GzmB, and then GSDME- was activated, causing extensive pyroptosis. In adittion, GzmA and GzmB in cytotoxic lymphocytes enter target cells through perforin and induce pyroptosis. GzmA hydrolyzes GSDMB, and GzmB directly activates GSDME

Multiple assembly mechanisms of canonical inflammasomes. The assembly of canonical inflammasomes occurs in response to PAMPs and DAMPs. Inflammasome sensors have interaction with different target ligand. Bacillus anthracis toxin activates the NLRP1 inflammasome, and NLRP1 recruits pro-caspase-1 into the complex via ASC or direct contact with CARD-CARD interactions. A variety of PAMPs and DAMPs activate NLRP3 inflammasome, which is followed by the recruitment of ASC and pro-caspase-1. Human NAIP senses both bacterial flagellin and proteins of T3SS components. Mouse NAIP1, NAIP2 and NAIP5/6 sense needle, inner rod, and flagellin respectively to assemble and activate the NLRC4 inflammasome. NLRC4 activates caspase-1 in an ASC-dependent or ASC-independent manner. AIM2 inflammasome is assembled when AIM2 senses host- or pathogen-derived dsDNA. The pyrin inflammasome is activated by Rho-modifying proteins. Both AIM2 and pyrin activate caspase-1 in an ASC-dependent manner. Activated caspase-1 can not only cleave GSDMD to form N-GSDMD and induce pyroptosis, but also process the precursors of IL-1β/IL-18 to mature IL-1β/IL-18, which are released through the pores formed by N-GSDMD
Non-canonical pathway
In the non-canonical pyroptosis pathway, the upstream sensory complexes of human caspase-4/5 (mouse orthologs caspase-11) are absent, and these caspases can be activated by directly binding to intracellular lipopolysaccharide (LPS) through the N-terminal CARD.187 It is worth noting that the oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC, a TLR4 agonist) is in competition with LPS to bind caspase-4/11, and reduces the non-canonical inflammasome in macrophages, but not in dendritic cells.188 Activated caspase-4/5/11 can also cleave GSDMD into N-GSDMD, which is oligomerized and transferred to the cell membrane to form plasma membrane pores ultimately.189 However, caspase-4/5/11 cannot cleave pro-IL-1β/pro-IL-18, but they are able to mediate the maturation and secretion of IL-1β/ IL-18 through the NLRP3/caspase-1 pathway in some cells.23 In addition, GSDMD is cleaved by caspase-4/5/11, resulting in the efflux of K+,29 inducing the assembly of NLRP3 inflammasome, eventually leading to pyroptosis.190,191,192 Of note, Yang et al. found that Pannexin-1 is another key protein that mediates pyroptotic cell death in the non-classical pathway induced by caspase-11.193 Under the stimulation of LPS, activated caspase-11 can specifically shear and modify Pannexin-1, causing the release of cellular ATP, thereby inducing pyroptosis mediated by ion channel P2X7 receptor.193 Importantly, murine BMDMs with the deficiency of Pannexin-1 could also cause K+ efflux and NLRP3 inflammasome-mediated caspase-1 cleavage with P2X7 independent,193 which coincides with a published report in 2011.115 Additionally, knockout Pannexin-1 in mice protects against endotoxin shock, suggesting that selective K+ channels modulate non-canonical NLRP3 activation.193 (Fig. 2).
Caspase-3/8-mediated pathway
The members of gasdermin protein family are highly conserved in structure. Except for DFNB59, all gasdermins contain C-terminal and N-terminal domains, and the N-terminus is the pyroptosis executor.30 Previously, apoptosis-related caspases (such as caspase-3/8) were thought to be unable to stimulate gasdermin to induce pyroptosis, while it was shown that chemotherapeutic drugs could induce caspase-3-mediated GSDME cleavage in with high GSDME expression and form N-GSDME termini, which caused pyroptosis of tumor cells.16,17 In addition, it has been found in mouse macrophages that the effector protein YopJ expressed during Yersinia infection can inhibit TGF-β-activated kinase 1 (TAK1) and induce caspase-8-related cleavage of GSDMD,103,104 which further improves and expands the understanding of pyroptosis. Intriguingly, PD-L1 converts TNF-mediated apoptosis into pyroptosis in breast cancer cells. Under hypoxia conditions, p-Stat3 promotes nuclear translocation of PD-L1, and they jointly enhance the transcription of GSDMC. Under the stimulation of TNF-α, Caspase-8 specifically lyses GSDMC to produce N-GSDMC, and forms pores on the cell membrane to induce pyroptosis. Macrophage-derived TNF-α-induced tumor pyroptosis requires nuclear PD-L1, caspase-8, and GSDMC In vivo. In addition, Antibiotic chemotherapy drugs can also trigger caspase-8/GSDMC-mediated pyroptotic death in breast cancer cells21 (Fig. 2).
Granzyme-mediated pathway
In 2020, Liu et al. reported that CAR T cells rapidly activated caspase-3 in target cells by releasing GzmB, and then the caspase-3/GSDME-mediated pyroptotic pathway was activated, causing extensive pyroptosis.194 More recently, researchers found that GzmB directly cleaved GSDME and induced pyroptosis, further activating the antitumor immune response and inhibiting tumor growth.19 Subsequently, it was reported that natural killer cells and cytotoxic T lymphocytes (CTLs) killed GSDMB-positive cells by pyroptosis. The killing effect resulted from GSDMB cleavage at the Lys229/Lys244 site by lymphocyte-derived GzmA. GSDMB is highly expressed in some tissues, especially in the epithelium of the digestive tract, including derived tumors. This study showed for the first time that gasdermin could be hydrolyzed by GzmA at non-aspartic acid sites and form pores, redefining the idea that pyroptosis can only be activated by caspases20 (Fig. 2).
Role of pyroptosis in tumors
Pyroptosis may act as a pivotal part in multiple tumors. With further research, the relationship between pyroptosis and tumors has become increasingly understood and provides some inspiration for clinical treatments.
Pyroptosis and melanoma
Melanoma is a common malignant tumor and is typically associated with BRAF and NRAS mutations.195,196,197,198,199 Previous work by Corey et al. found that the pyroptosis-related protein GSDME from B16-Ova cell lines exerted a tumor suppressive effect.35 In addition, it has been found that melanoma cells with GSDME deficiency generate larger tumors than wild-type melanoma cells; thus, GSDME may have real tumor inhibitory activity.35 In 2020, the combination of BRAF and MEK inhibitors was reported to induce GSDME-dependent pyroptosis in melanoma cells.200,201 In immunocompetent mice, BRAF inhibitor + MEK inhibitor caused an increase in CD4+ T cell and CD8+ T cell infiltration and a decrease in myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs).200 In BRAF inhibitor + MEK inhibitor-resistant tumors, the loss of pyroptosis and GSDME cleavage induced by BRAF inhibitor + MEK inhibitor was associated with a decreased antitumor immune response.201 DHP1808, a new Hsp90/PI3K inhibitor, can effectively induce apoptosis of melanoma cells by interfering with the interaction between Hsp90 and EGFR and inhibiting the downstream MAPK signaling pathway. However, a recent study found that DHP1808 induced less pyroptosis than the combination of Hsp90 and PI3K inhibitors in tumor and intestinal tissue, indicating that DHP1808 is a safe drug and worthy of further development.202 Moreover, z-YVAD-FMK inhibited the ability of the HSV-2 mutant lacking R1 protein kinase activity (ΔPK) to induce melanoma cell death, showing that ΔPK could induce pyroptosis.203 Interestingly, increased etoposide resistance in melanoma cells led to decreased GSDME mRNA levels due to increased cell sensitivity to the activation of caspase-3-dependent signaling pathways, triggering programmed cell death and suggesting that GSDME may be involved in the multimodal mechanism that led to drug resistance in the treatment of malignant melanoma.204 Iron was involved in redox cycle and the activation of ROS,205,206 and ROS was thought to be related to cancer.207,208 Generally, Zhou et al. found that carbonyl cyanide m-chlorophenyl hydrazone (CCCP) activated oxidative stress, and iron significantly enhanced this effect, followed by Tom20 oxidation and oligomerization in melanoma A375 cells.112 Tom20 promoted the recruitment of Bax to mitochondria, then cytochrome c was released into the cytoplasm and caspase-3 was activated, which eventually induced the cleavage of GSDME and caused pyroptosis,112 indicating that iron can enhance the sensitivity of drugs that activate ROS in vivo, and amplify ROS signals to drive pyroptosis. It also reveals a potential iron-based intervention strategy for treatment of melanoma patients.112 In addition, eEF-2K regulated the pathway linking autophagy and pyroptosis.209 Inhibition of eEF-2K makes melanoma cells more sensitive to doxorubicin.209 However, the specific mechanism of eEF-2K regulating GSDME remains to be studied.209 It has been reported that melanoma cells overexpressing GSDMB show obvious characteristics of pyroptosis. The mechanism of GSDMB-induced pyroptosis is not related to the classic caspase pathway but to the protein cleavage mediated by perforin and granzyme, which are produced by cytotoxic particles.20 It was reported that the polymorphism of NLRP1/NLRP3 can affect the susceptibility of malignant melanoma (MM) and the occurrence of nodular melanoma (NM).210 Unlike other NLRPs, the C-terminal domain of NLRP1 contains a CARD-binding motif that interacts with pro-caspase-1 through CARD-CARD.114,211,212 Thus, ASC does not participate in the NLRP1 activation of caspase-1.114,211,212 In 2017, it was found that NLRP1 could not only synergistically promote caspase-1-mediated inflammasome activation, but also inhibit caspase-2/9 related mitochondrial apoptosis of melanoma, thus promoting the tumorigenesis of human melanoma.213 Therefore, NLRP1 may be a new target for the treatment of human melanoma. Additionally, The inhibition of B16F10 lung metastasis in wild type mice was mediated by NK cells,214,215,216,217 and NLRP3 played an important role in this process.218 After intravenous injection of B16F10 melanoma cells, wild-type mice had significantly more lung metastases than NLRP3 knockout mice.218 In addition, the protective effect of NLRP3 deletion on lung metastasis of B16F10 depended on NK cells, but not on CD4+ and CD8+ T cells.218 The absence of NLRP3 increased the number of activated NK cells, secreted more IFN-γ, and killed more tumor cells to reduce B16F10 lung metastasis.218 This may seem strange, since the loss of host NLRP3 inhibited the release of pro-inflammatory cytokines, which contributed to NK cell activation, but the tumor microenvironment was unique. Intervention of NLRP3-dependent immunosuppressive pathway may be a potential therapeutic strategy to enhance host resistance to melanoma metastasis.218 In 2020, Okamoto et al. found that human melanoma cells spontaneously secreted active IL-1β through the structural activation of NLRP3 inflammasome, thus showing the characteristics of autologous inflammatory diseases.219 Interestingly, NLRP3 can also destroy the effect of anti-tumor vaccine. The survival rate of mice with NLRP3 deficient bearing melanoma cell line B16F10 based on dendritic cell vaccination was significantly higher than that of their respective control groups. The increase in survival was attributed to the lower number of MDSCs in NLRP3-deleted tumors, suggesting that NLRP3 played a role in promoting MDSCs-to-TME migration.220 Surprisingly, NLRC4 seemed to play a role different from NLRP1 and NLRP3 in melanoma, which amplified inflammatory signals in macrophages and inhibited the growth of melanoma tumors independent of inflammasome assembly.221 NLRC4 inflammasome consists of NLRC4, ASC, and caspase-1.222,223 Compared with wild-type mice, the tumor volume of NLRC4 gene deficient mice was significantly larger, while the tumor size of mice with knockout inflammasome components ASC and caspase-1 had no significant change, suggesting that NLRC4-mediated regulation of antitumor growth is not associated with NLRC4 inflammasome activation in B16F10 melanoma model.221 In addition, the researchers found that the loss of NLRC4 was related to the inability of CD4+ and CD8+ T cells to produce INF-γ. Although immunotherapy for melanoma that enhances endogenous effector T cell response has yielded promising clinical results,224,225,226,227,228 further studies are needed to determine specifically whether the reduction of chemokine production and the population of CD4+ and CD8+ T cells producing INF-γ directly affect tumor growth. Inflammasome regulates the activation of caspase-1 by ASC, which contains a CARD. However, ASC plays multiple physiological roles in the pathogenesis of melanoma through different regulation of NF-κB and IL-1β.212 On the one hand, ASC appeared to induce metastatic melanoma by activating caspase-1-dependent IL-1β secretion and enhancing its autonomous inflammatory NF-κB activity, suggesting that ASC is a tumor-promoting factor in metastatic melanoma; On the other hand, ASC was highly expressed in primary melanoma, which reduced the phosphorylation of IKKα/β and inhibited the activity of NF-κB, thus playing an anti-tumor role.212 These studies deeply emphasized the cell type and tissue-specific role of inflammasome components in melanoma. In-depth study of ASC and its upstream and downstream molecules or pathways may deepen our comprehension of the molecular mechanism of melanoma and provide new molecular targets for the design of anti-melanoma drugs.212 In particular, pleiotropic cytokine IL-1β/IL-18 also promote the growth, angiogenesis and metastasis of melanoma in an autocrine or paracrine manner.229,230,231 IL-1β-activated hepatic sinusoidal endothelium (HSE) cells release very late antigen-4 stimulating factor to enhance the adhesion of B16 melanoma (B16M) cells to HSE.232 Besides, IL-1β and IL-18 are involved in the process of hepatic metastases of B16M in vivo. The soluble products of B16M cells caused HSE to release TNF-α, IL-1β, and IL-18.233 IL-18 further increased the expression of adhesion molecule-1 (VCAM-1) and promoted the adhesion of melanoma cells.233 PD-1, expressed by activated mature NK cells in lymphoid organs of tumor carriers, can be also upregulated by IL-18.234 Inhibition of IL-18 or IL-18 binding proteins in melanoma was sufficient to stimulate NK cell-dependent immune surveillance in melanoma models,235 indicating that IL-18 can be used as an immunosuppressive cytokine in melanoma, and anti-PD-1 antibody has a new clinical application in human malignant tumors that produce IL-18.
Pyroptosis and breast cancer
Breast cancer is one of the most frequent malignancies.236,237,238,239,240,241 A high level of GSDMB in breast cancers was related to tumor progression, and overexpression of GSDMB indicated a poor response to targeted treatment of HER-2.53 This means that GSDMB could be a novel prognostic marker for tumors. In addition, high levels of GSDMC were associated with poor survival in breast cancer.21 A variety of antibiotics such as doxorubicin, daunorubicin, actinomycin D, and epirubicin could promote the expression of nuclear PD-L1 and GSDMC, and facilitate the activation of caspase-8, resulting in pyroptotic death in breast cancer cells.21 Low expression of GSDME was found in many cancers, and low levels of GSDME were also associated with low survival of breast cancer patients.242 In 2018, it was reported that docosahexaenoic acid exerted anti-tumor effects and induced pyroptosis in breast cancer cells, but this process could be suppressed by caspase-1 inhibitors.243 In addition, p53 induced the expression of GSDME through a specific p53 binding site in GSDME.244 Interestingly, the DAC restored the induction of GSDME by p53 and two other p53 family genes (p63gamma and p73beta), indicating that GSDME may be a transcriptional target of the p53 family.245 Furthermore, the GSDME mRNA level in breast cancer tissues was downregulated because the GSDME promoter was methylated more frequently in tumor tissues than in normal tissues. In addition, GSDME enhanced resistance to triple-negative breast cancer by activating pyroptosis. GSDME methylation was correlated with lymph node metastasis in breast cancer patients, suggesting GSDME promoter methylation is a novel molecular biomarker for human breast cancer.246 P2X7 signaling pathway has been found to be related to tumorigenesis.247,248,249,250,251 Interestingly, a study published in 2015 showed that Ivermectin (an antiparasitic agent approved by FDA)-induced breast cancer pyroptosis depended on the release of ATP and downstream signals of P2X7 receptors, which can be reversed by inhibiting ROS, Ca2+/Calmodulin-dependent protein kinase II (CaMKII) or blocking mitochondrial permeability transition pore (MPTP). Ivermectin induced autophagy and release of ATP and HMGB1.252 Autophagy can not only resist many chemotherapeutic drugs, but also enhance immunogenicity and make cancer cells more sensitive to immune-mediated killing.253,254 The P2X4/P2X7/Pannexin-1 cascade regulated autophagy and ATP release, and participated in the recruitment and activation of many kinds of immune cells, which acted as a positive feedback loop.255,256 Therefore, the anticancer properties of Ivermectin are not only reflected in its direct cytotoxicity. The enhanced P2X4/P2X7 signal can be further associated with the tumor microenvironment rich in ATP, providing clues for purinergic receptors to regulate tumor selectivity and affect the potential of tumor immunotherapy.252 The pyroptosis-related inflammasome and cytokine IL-1β also appeared to be significantly associated with the high recurrence rate of breast cancer, and were very important for the proliferation, angiogenesis, migration, and invasion of breast cancer.229 Compared with the wild type mice, mice with inflammasome deficiency showed higher survival rate.257 After injected orthotopically with PyT8 tumor cells, the tumor growth and metastasis abilities of NLRP3 knockout mice were also lower than that of wild type mice.257 Chemokine (C–C motif) ligand 2 (CCL2), a member of the C–C chemokine family, recruited myeloid cells to inflammatory sites and was shown to promote macrophage infiltration into tumor tissue.258,259,260 Guo et al. found that IL-1β stimulated CCL2 expression in macrophages and tumor cells.257 Importantly, targeting inflammasome/IL-1 signaling pathway inhibited the growth and metastasis of breast cancer in vivo.257 These results suggest that the inhibition of inflammasome/IL-1 signaling pathway in tumor microenvironment may provide a new perspective for the treatment of breast cancer. Pyroptosis and IL-1β produced by pyroptosis were detected in periodontal inflammation (PI) patients, as well as in mouse models.261 PI and IL-1β motivated the expression of CCL5, CXCL12, CCL2, and CXCL5, which recruit MDSC and macrophages to promote breast cancer metastasis.261 Similarly, the use of anti-CCL5 antibodies to target the CCL5 (a prominent marker of poor prognosis in inflammatory breast cancer detected in many clinical specimens of breast cancer262,263,264,265,266) was described as reducing the immunosuppressive activity of MDSCs and decreasing breast cancer metastasis.267 There was a direct relationship between chronic PI and the risk of breast cancer metastasis, and PI induced the recruitment of macrophages, cancer cells, and MDSC.267 As a result, inflammasome inhibitors are suggested to suppress tumor progression.267 Intervention of PI may be an effective way to prevent breast cancer from metastatic to the head and neck. Obesity was related to the elevated risk of ER-positive breast cancer.268,269,270 Generally, tumor microenvironment in obese patients recruited tumor infiltrating myeloid cells with the activation of NLRC4 inflammasome, and causes the production of IL-1β.271 IL-1β promoted the progression of breast cancer through adipocyte-originated angiogenesis and the expression of vascular endothelial growth factor A (VEGFA).271 In 2020, Jiao et al. found NLRP1 inflammasome was involved in breast cancer cell pyroptosis triggged by human umbilical cord MSCs (hUCMSCs)-CM. Surprisedly, in caspase-4-deficient MCF7 cells, hUCMSC-CM mainly induced pyroptosis through classical pathway, while in NLRP1-deficient MCF7 cells, pyroptosis was mainly induced by non-classical pathway.272 These studies facilitate the identification of potential breast cancer treatment targets.
Pyroptosis and colorectal cancer
Colorectal cancer is a heterogeneous disease associated with genetic mutations, age, family history, ethnicity, and lifestyle.273,274,275,276,277,278 The downregulation of GSDMC led to significant decreases in the proliferation and of colorectal cancer cells, while GSDMC overexpression promoted cell proliferation, tumorigenesis suggesting that GSDMC may be a promising therapeutic target in colorectal cancer.54 GSDMD was downregulated in carcinoma cells, while it was slightly overexpressed in normal colorectal epithelial cells.279 In 2019, Ibrahim et al. found that colorectal cancer and normal tissue could be distinguished accurately by a combination of two CpGs regardless of age or stage, indicating that GSDME is a novel diagnostic index for colorectal cancer.280 Lobaplatin-induced ROS/JNK signal transduction, which induced GSDME-mediated pyroptosis in colon cancer cells.281 Interestingly, the lncRNA RP1-85F18.6 is overexpressed in colorectal cancer, playing a key role in tumorigenesis and inhibiting pyroptosis in colorectal cancer cells.279 Pyroptosis-associated inflammasomes have been shown to inhibit tumorigenesis. It was reported that Nlrp1b (−/−), Nlrp3 (−/−), Nlrc4 (−/−), Aim2 (−/−), and pyrin (−/−) mice showed significant increases in inflammation, morbidity, and tumorigenesis compared with those of wild-type animals.282,283,284,285,286,287,288,289 Besides, compared with that of normal tissues, the expression of NALP1 was decreased in colon cancer tissue.290 In addition, DAC could restore the expression of NALP1, thus inhibiting the occurrence of colon cancer, indicating that NALP1 is a potential therapeutic marker for colorectal carcinoma.282,290 Dextran sodium sulfate (DSS) destroys the epithelial barrier, leading to a large amount of inflammation caused by intestinal microflora. Azoxymethane (AOM) is a powerful carcinogen, which causes DNA damage to epithelial cells. Multiple cycles administration of DSS promotes chronic inflammation, thereby accelerating the development of colorectal cancer induced by AOM. Although DSS-treated mice with loss of inflammation are generally more likely to develop induced colitis than wild-type mice, the results may be different on specific NLRP. Some studies have also found that NLRP1b, NLRP3, and pyrin protected against colorectal cancer by facilitating the secretion of IL-18 to promote epithelial barrier regeneration during the early stages of colorectal cancer.283,284,291,292,293,294 Although DSS-treated mice with loss of inflammasome are generally more likely to develop colitis than wild-type mice, the results may be different on specific NLRPs. It has been shown that in the DSS and AOM + DSS model, the inflammasome components possessed protective effects in acute and recurrent colitis and colitis-associated cancer (CAC). Mice with ASC and caspase-1 deficiency showed increased morbidity. The elevated tumor load was associated with decreased IL-1β and IL-18 levels. In addition, acute colitis, recurrent colitis, and CAC were increased in Nlrp3 knockout mice, but disease progression in NLRC4-deletion mice was not significantly different from that in wild-type mice.283 Lack of Nlrp3 inflammasome can also lead to impaired IL-18 signaling on NK cells and aggravate the metastatic growth of colorectal cancer growth in the liver.295 Interestingly, immune surveillance caused by inflammasome was dependent on NK cells but did not require T and B cells.295 Besides, mice with Nlrp3 or caspase-1 deficiency were highly sensitive to inflammation induced by AOM / DSS, and the burden of colon tumor was significantly elevated. This was a consequence of markedly reduced IL-18 levels in mice lacking components of the Nlrp3 inflammasome.284 What’s more, defective inflammasome activation resulted in loss of epithelial integrity, leading to numerous leukocyte infiltration, and an increase in chemokines in the colon. This is due to the reduction of IL-18 in mice, causing higher mortality. Therefore, the Nlrp3 inflammasome plays a vital role in maintaining intestinal homeostasis and preventing colitis.291 These data suggest that the inflammasome can reduce colitis and CAC. However, another report showed that in comparison to wild-type mice, the severity of colitis in NLRP3-deficient mice was less severe after oral administration of DSS.296 It is possible that different experimental methods and inherent differences in variables have caused significant differences between these studies. Interestingly, a report showed that mice with inflammasome component deficiency, such as NLRP3−/− mice, may be characterized by changes in intestinal microflora.297 This suggests that alteration in intestinal flora between different animal facilities may also be responsible for the differences observed in these studies. In addition, mice lacking ASC and caspase-1 are also prone to DSS-induced colitis and colon-related colorectal cancer,283,284,291,294,297 which provides a great deal of evidence for the protective role of inflammasome in colorectal cancer inflammatory models. In addition, IL-18 is also closely related to the development of colorectal cancer. On the one hand, injecting recombinant IL-18 into caspase-1−/− mice reduced disease progression or outcome in response to dextran sulfate sodium DSS and AOM.283 On the other hand, deletion of IL-18 or its receptor IL-18R in intestinal epithelial cells induced resistance to DSS-induced colitis in mice.298 Therefore, local production of IL-18 in intestinal epithelial cells in the early stage could be beneficial to epithelial repair after injury, while excessive production of IL-18 in the late stage during chronic inflammation may promote tumor development, indicating that this pyroptosis-related cytokine could be regarded as a potential candidate for immunotherapy in the treatment of some colorectal cancers.
Pyroptosis and gastric cancer
Gastric cancer is a malignancy that begins in stomach cells and has poor prognosis and high mortality.299,300,301,302,303,304,305 A study showed that GSDMA was a tumor suppressor gene in gastric cancer,306 but GSDMB was overexpressed and in some gastric cancer cells and could act as an oncogene. GSDMB was highly expressed in most cancerous tissue samples, but not in the majority of normal gastric samples, and may be associated with invasion.307 In contrast, GSDMC was downregulated in gastric cancer, indicating that it may function as a tumor-inhibiting factor. Similarly, the expression level of GSDMD was low in gastric cancer cell lines and models.306,308 In addition, the downregulation of GSDMD could activate the signal transducer and activator of STAT3 and PI3K/PKB signaling pathways, and regulate cell cycle-related proteins to accelerate S/G2 phase cell transition, suggesting that GSDMD downregulation may be beneficial for the treatment of gastric cancer.309 It was found that chemotherapeutic drugs induced pyroptosis rather than apoptosis in gastric cancer cells with high expression of GSDME. After treating the gastric cancer cell lines with 5-fluorouracil (5-FU), the cells appeared to exhibit the characteristic of pyroptosis.299 Pyroptosis is mainly caused by microbial infection,11,310 and studies have found that persistent infection of Helicobacter pylori contributes to the progression of multiple gastric and extra-gastric diseases.301,311,312,313,314,315 Importantly, Semper et al. reported that IL-1β produced by Helicobacter pylori in innate immune cells was mainly related to the activation of the NLRP3 inflammasome,316 which was consistent with a study that IL-1β secreted by dendritic cells infected by Helicobacter pylori was associated with the synergism between TLR2/NOD2 and NLRP3 inflammasome.317 NLRP3 played a role in the gastric cancer development in an inflammasome pathway-dependent or independent way.318 The high expression of NLRP3 in gastric cancer accelerated the activation of NLRP3 inflammasome and the release of IL-1β by macrophages.318 Besides, Helicobacter pylori infection significantly promoted the expression of NLRP3, but inhibited the expression of miR-22. miR-22 was an inhibitor of NLRP3, and directly attenuated the carcinogenic activity of NLRP3 in vitro and in vivo to maintain the balance of gastric microenvironment.318 However, the mechanism between the NLRP3 inflammasome and gastric carcinogenesis needs to be further elucidated. Gastric chronic inflammation caused by Helicobacter pylori infection and the release of IL-6, IL-1β, TNF-α can affect the proliferation of gastric cell.319,320 IL-18 produced thrombospondin-1 (a proangiogenic factor) through JNK pathway, which stimulates angiogenesis of gastric cancer cells expressing IL-18R, and affects carcinogenesis and metastasis of gastric cancer.321,322,323,324 IL-1β was also related to an increased risk of spontaneous gastric inflammation and gastric cancer.325 Overexpression of IL-1β in the stomach contributed to the development of gastric cancer by increasing the number of MDSCs.325
Pyroptosis and hepatocellular carcinoma
Hepatocellular carcinoma is the second leading cause of cancer-related death worldwide, and its incidence is expected to further increase globally.326,327,328,329,330,331,332,333 It was reported that DFNA5 was significantly expressed at low levels in hepatocellular carcinoma cells. DFNA5 overexpression resulted in the inhibition of cell proliferation.334 Furthermore, berberine inhibited the viability of HepG2 cells through caspase-1-mediated pyroptosis.335 Interestingly, caspase-1-mediated pyroptosis may also play a role in APOL1-induced cytotoxicity.336 A great deal of evidence has confirmed that NLRP3 inflammasome is associated with liver failure and liver disease.337,338 In a previous study, Wei et al. found that the loss of the NLRP3 inflammasome contributed to the progression of hepatocellular carcinoma. In addition, 17β-estradiol (E2) suppressed the tumor progression of hepatocellular carcinoma by activating the NLRP3 inflammasome.339,340 In 2019, that group demonstrated that E2-induced activation of the NLRP3 inflammasome resulted in caspase 1-mediated pyroptosis.341 Besides, targeting the NLRP3 inflammasome may exert inhibitory effects on proliferation, metastasis, and invasion of hepatocellular carcinoma,342 indicating that it is necessary to understand the exact mechanism of inflammasome in the proliferation, metastasis and aggression of hepatocellular carcinoma. In addition, Wei et al. found that autophagy inhibition enhanced pyroptosis in hepatocellular carcinoma cells,341 suggesting that pyroptosis may be a potential treatment strategy for hepatocellular carcinoma. Hepatic steatosis or cirrhosis is closely related to the occurrence of hepatocellular carcinoma.343,344,345,346 New evidence suggests that inflammasome plays an important role in non-alcoholic fatty liver disease, which is a series of metabolic disorders ranging from steatosis (NAFL) to steatohepatitis (NASH) to liver cirrhosis. Moreover, it has also been reported that inflammasome activity can inhibit hepatic steatosis.347,348,349 The activation of inflammasome and the production of IL-1β induced by endoplasmic reticulum stress produced a positive feedback loop, which amplified the inflammatory response, resulting in hepatic steatosis and injury.347 Similarly, AIM2 was low in hepatocellular carcinoma, the loss of which was significantly associated with mTOR-S6K1 pathway activation and advanced hepatocellular carcinoma progression.350 However, the role of inflammasome in hepatocellular carcinoma has not been deeply explored, and its role in the occurrence and development of hepatocellular carcinoma needs further clinical and experimental research.
Pyroptosis and lung cancer
Lung cancer is one of the primary causes of death and the most common cancer in the world.351,352,353,354,355,356,357,358 GSDMD was found to be upregulated in non-small cell lung cancer (NSCLC).359 In addition, a high level of GSDMD promoted tumor metastasis and suggested a poor prognosis in LUAD.359 Interestingly, in GSDMD‑deficient tumor cells, activation of the pyroptotic signaling pathway (NLRP3/caspase‑1) induced apoptosis but not pyroptosis.359 Moreover, silencing GSDMD inhibited tumor proliferation by attenuating the EGFR/Akt signaling pathway in NSCLC.359 In 2019, Xi et al. demonstrated that GSDMD colocalized with GzmB near immune synapses, and the defect in GSDMD alleviated the cytolytic ability of CD8+ T cells, suggesting that GSDMD is essential in the immune response of tumor cells.360 GSDME is widely expressed in different molecular subtypes of lung cancer. GSDME or caspase-3 depletion significantly weakened GSDME-dependent pyroptosis in A549, PC9 or NCI-H3122 cells.361 Zhang et al. showed that both paclitaxel and cisplatin could obviously induce apoptosis in A549 cells, but some of the dying cells showed a morphology that was highly characteristic of pyroptosis.362 In A549 cells, cisplatin triggered more severe pyroptosis than paclitaxel, indicating that cisplatin may have an additional advantage over paclitaxel in lung cancer with high expression of GSDME.362 Polyphyllin VI (PPVI) induced caspase-1-mediated pyroptosis in NSCLC through the ROS/NF-κB/NLRP3/GSDMD signaling axis, which suggests that PPVI is a potential new therapeutic agent for NSCLC treatment.363 Both AIM2 and NLRP3 inflammasome were highly expressed in NSCLC.364,365 NSCLC cells with high metastatic expressed relatively more inflammasome components and released more interleukin than cells with low metastatic; Besides, cisplatin-sensitive NSCLC cells also possessed higher levels of inflammasome components interleukin release than cisplatin-resistant NSCLC cells; in addition, in resected lung cancer tissues, upregulation of inflammasome components and interleukin release also observed in high-grade adenocarcinoma (ADC) than low-grade ADC, suggesting that inflammasomes may be conducive to chemotherapy sensitivity of lung cancer.364 Taken together, these findings show that the levels of inflammasomes in various lung tumors are dependent on the subtypes, tumor stages, invasive potential, and chemosensitivity of lung cancer, indicating that inflammasomes may be important biomarkers and promising regulators of lung cancer.364 Additional studies have also demonstrated the relationship between inflammasome and lung cancer. For instance, the researchers found that inhibition of NF-κB, a transcription factor associated with the initiation of inflammasome,366,367,368 led to a reduction in the viability and proliferation of lung cancer cells.369,370 LncRNA-XIST was an oncogene in multiple tumors and played a role the regulation of NSCLC cell proliferation, invasion, and metastasis,371,372,373,374,375,376,377,378,379,380,381,382 which significantly highly expressed in NSCLC tissues and cell lines compared with normal tissue or cells,383 regulating the expression level of SOD2 (a mitochondrial antioxidative enzyme can be inhibited by miR-335 in renal senescence384) by targeting miR-335 (a negative tumor regulator that inhibits the progression of NSCLC385,386,387). Interestingly, knocking down LncRNA-XIST increased ROS level and activated NLRP3 inflammasome in A549 cells,383 and the generation of ROS was reported to induce pyroptosis.388,389,390 Besides, ROS scavenger N-acetyl cysteine (NAC) can block pyroptosis in NSCLC cell induced by the knock down of lncRNA-XIST, and the inhibition of cell proliferation can also be reversed by pyroptosis inhibitor necrosulfonamide (NSA).383 Generally, downregulation of lncRNA-XIST inhibits the development of NSCLC by activating miR-335/SOD2/ROS cascade-related pyroptosis.383
Pyroptosis and cervical cancer
Cervical cancer is one of the most common female malignant tumors in worldwide and is one of the primary causes of tumor death among women.391,392,393,394,395,396 The researchers found that HeLa cells with GSDMB overexpression showed obvious characteristics of pyroptosis.20 In addition, the release of GzmB by immune cells cleaved GSDME and promoted HeLa cell pyroptosis, which is an important finding for understanding the interaction between GSDME-mediated cell death and the immune system.19 The results indicate that there is a positive feedback relationship between pyroptosis and the immune response. Indeed, normal cervical epithelial cells released less IL-18 and IL-1β than cervical cancer cells.397 Sirtuin 1 (SIRT1) was overexpressed in HPV-infected cervical cancer cells, and knocking down SIRT1 caused cervical cancer cells to undergo pyroptosis, as well as highly express AIM2 and its downstream genes associated with the inflammasome response. Thus, SIRT1 may be a potential therapeutic target for cervical cancer.398 More recently, Song et al. discovered that human papillomavirus E7 (HPV E7, a sexually transmitted DNA virus) could suppress pyroptotic cell death induced by dsDNA transfection. HPV E7 recruited E3 ligase TRIM21 to cause ubiquitination and degradation of IFI16 inflammasome, resulting in suppression of pyroptosis and self-escape of immune monitoring, revealing a significant immune escape mechanism in HPV infection, which may provide a novel target for the development of immunotherapy strategy for effective restoration of antiviral immunity.399
Pyroptosis and leukemia
Leukemia is characterized by abnormal excessive proliferation of hematopoietic stem cells, which can be divided into acute or chronic leukemia.400,401,402,403,404,405 Tp92 is the only outer membrane protein of T. pallidum which has high homology with the outer membrane proteins of many Gram-negative bacteria.406 Luo et al. found that the recombinant Tp92 protein was able to cause pyroptosis of THP‐1 cells (the human monocytic cell line derived from an acute monocytic leukemia patient) via the caspase‐1 pathway.407 As an effective calcium antagonist, magnesium sulfate (MgSO4) can significantly inhibit the release of calcium-influx dependent histamine, and has a potential anti-inflammatory effect.408,409,410,411 Studies have found that MgSO4 can also inhibit LPS-induced increase of NLRP3 inflammasome, upregulation of IL-1β and pyroptosis in THP-1 cells.412 CARD8, is a protein encoded by the human genome, consisting of a FIIND followed by a CARD.413 The CARD of CARD8 can bind to pro-caspase-1, inducing activation of caspase-1.414,415 In general, anti-cancer drug talabostat (PT-100, Val-boroPro) inhibits DPP8/9 (two intracellular serine dipeptidases Dpp8 and Dpp9),162 and mainly induces pyroptosis dependent on NLRP1b, but not on ASC.162,416,417,418 Surprisingly, talabostat induces CARD8-mediated pyroptosis in THP-1 and other AML cells, which is independent of NLRP1, indicating that CARD8 can also form inflammasome.413,419,420 It has been found that DPP8/9 inhibitors induce pyroptosis in the vast majority of human acute myeloid leukemia (AML) cell lines, and these inhibitors suppress the progression of human AML in mouse models.162,413 What’s more, CARD8 was involved in the pyroptosis of human myeloid cells induced by DPP8/9 inhibitors, which provided a new potential strategy for the treatment of AML.413 Genetic modification enhanced the ability of chimeric antigen receptor (CAR) T cells to kill target tumor cells.421,422,423 Interestingly, CAR T cells activate GSDME-mediated cell death in B leukemic cells by releasing a large amount of perforin and GzmB, resulting in elevated levels of pro-inflammatory cytokines such as IL-1β, IL-6 and increased release of serum amyloid protein A3 (SAA3), leading to cytokine release syndrome (CRS) in CAR T cell-treated patients.194 GSDME can be cleaved by caspase-3 to induce programmed cell death and is considered to be a tumor suppressor gene. However, the high expression of GSDME in B leukemic cells suggests that GSDME may play different role in pore formation of tumor cells.194 It means that transforming target tumor cell death mode from pyroptosis to apoptosis may provide a new perspective for the reduction of CRS caused by CAR T cell therapy. Besides, pyridoxine (vitamin B6), a naturally small molecule with low toxicity, induced GSDME-mediated pyroptosis in THP-1 cells, suggesting that pyridoxine is a potential drug for the treatment of AML.424
Therapeutic implications
Different forms of pyroptosis are closely related to tumors.109 Currently, researchers have been trying to improve the therapeutic efficacy through pyroptosis or combining pyroptosis with a variety of tumor treatment methods.
Gasdermin-related therapy
Gasdermins are the effectors of pyroptosis, and activation of gasdermins might prevent tumor formation. The researchers developed a bioorthogonal chemical system mediated by the probe phenylalanine trifluoroborate (Phe-BF3, a cancer-imaging probe that is significantly and specifically absorbed by tumors) at the cellular and in vivo levels.425 The study revealed that the pyroptosis of a small percentage of tumor cells can effectively regulate the tumor immune microenvironment and activate a strong T cell-mediated antitumor immune response.425 This discovery provides a new strategy for the development of tumor immunotherapy drugs (Fig. 4). Wang et al. and Rogers et al. found that chemotherapy drugs induced the activation of caspase-3 in tumor cells with high GSDME expression and induced pyroptosis.16,17 In tumor cell lines with low expression of GSDME, decitabine can upregulate GSDME expression and increase the sensitivity of tumor cells to chemotherapy drugs, thus making these cells more prone to pyroptosis16,17 (Fig. 4). Lobaplatin-induced ROS led to JNK phosphorylation, and activated JNK promoted the mitochondrial translocation of Bax, which activated GSDME and induced pyroptosis.281 This study further elucidated the relationship between chemotherapeutic drugs and pyroptosis. Chen et al. found that PTX-triggered pyroptosis of A549 cells induced was closely associated with activated caspase-3 and N-GSDME levels. Compared with PTX, cisplatin induced more severe pyroptosis in non-small cell lung carcinoma and esophageal squamous cell carcinoma cells, suggesting that cisplatin may have more advantages than other drugs in treating cancer with high expression of GSDME.362,426 In addition, the expression of GSDME can enhance the tumor cell phagocytosis by tumor-associated macrophages and increase the number and function of tumor-infiltrating natural killer lymphocytes and CD8+ T lymphocytes. Therefore, the tumor suppressor protein GSDME can enhance immune responses to tumors such as triple-negative breast cancer by activating pyroptosis.19 Many different chemotherapy drugs could facilitate nuclear PD-L1 translocation and promote GSDMC expression, but only the antibiotic type drugs was observed to activate caspase-8, and further activated GSDMC activation, resulting in pyroptosis ultimately.21 However, this process was abolished by the PD-L1-NLS/Stat3-Y705F mutations or caspase-8 knockout.21 Thus, therapeutic methods mediated by this type of drug may cause a strong inflammatory response, thereby affecting the antitumor immunity and prognosis of PD-L1+ or GSDMC+ tumors21 (Fig. 4). The combination of BRAF inhibitor and MEK inhibitor led to persistent melanoma regression through immunomodulation.201 The combination of these two inhibitors induced the caspase-3 dependent GSDME lysis and the release of pro-inflammatory factors such as HMGB1.201 HMGB1 activates dendritic cells by interacting with toll-like receptor 4, thereby promoting anti-tumor T cell activity. Besides, during BRAF inhibitor +MEK inhibitor treatment, both TAM and MDSC decreased.201 In addition, immune cells release GzmA and GzmB, which cleave GSDMB and GSDME, respectively, and promote pyroptosis19,20 (Fig. 5). Pyroptosis is accompanied by the release of cytokines, which activate the immune response,23 indicating that there is a positive feedback regulatory relationship between pyroptosis and the immune response (Fig. 5). In view of the strong antitumor activity of this positive feedback relationship, finding the factors that trigger positive feedback or exploring the relevant regulatory pathways may provide new drug targets for antitumor therapy. Gasdermin family proteins are also potential biomarkers of tumor immunotherapy. The next step may be to explore therapeutic strategies to upregulate gasdermin levels, such as using the existing DNA methylation inhibitor decitabine, which has been approved for the treatment of leukemia and myelodysplastic disorders. The agonists of these proteins are likely to become new candidates in antitumor drug research and development, although the efficacy of these new drugs in tumors remains to be determined.

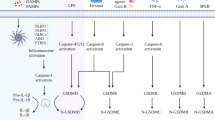
Current status of gasdermin-mediated tumor therapy. The desilylation of Phe-BF3 releases gasdermin from NP-GSDMA3 and induces pyroptosis. A bioorthogonal chemical system was developed in which Phe-BF3 can enter cells through LAT1 (a Phe-BF3 transporter), desilylate and cleave the design linker containing a silyl ether. The purified GSDMA3 (N + C) is conjugated with nanoparticles to become NP-GSDMA3. When Phe-BF3 binds to nanoparticles, the desilylation catalyzed by Phe-BF3 can release active gasdermin from the conjugated nanoparticles and selectively release active gasdermin. Chemotherapeutic drugs induce caspase-3-mediated pyroptosis in tumor cells with high GSDME expression and apoptosis in tumor cells with low expression of GSDME. Decitabine is an inhibitor of DNA methyltransferase, which can demethylate GSDME, changing GSDME expression from low to high, and shift apoptosis to pyroptosis. Almost all chemotherapeutic drugs can induce PD-L1 into the nucleus and promote the expression of GSDMC, but only the antibiotics such as daunorubicin, doxorubicin, epirubicin, and actinomycin D can also activate caspase-8, cause GSDMC cleavage and trigger pyroptosis. Pyroptosis is accompanied by the release of cytokines, which mediate the immune response

Positive feedback loops of pyroptosis and immune response. After the occurrence of pyroptosis, the formed pores release inflammatory factors, such as Il-1β, HMGB1 LDH, and ATP, which act as alarm signals to activate and recruit immune cells and mediate the immune response. BRAF + MEK inhibitors induce GSDME-dependent pyroptosis mediated by caspase-3, and cause an increase in CD4+ T cell and CD8+ T cell infiltration and a decrease in MDSC and TAM. Cytotoxic lymphocytes release perforin and granzyme, and perforin forms pores in tumor cells. Granzyme enters tumor cells through these pores. GzmA cleaves GSDMB, and GzmB cleaves GSDME, further inducing pyroptosis. This forms a positive feedback loop, which means that a small number of cancer cells undergoing pyroptosis can trigger a tumor immune response and expand the death response
Interestingly, the inhibition of gasdermins can also effectively block tumor formation. In 2019, Ángela et al. found a new targeted nanomedicine that can deliver a special anti-GSDMB antibody to HER2-positive breast cancer cells, based on hyaluronic acid–biocompatible nanocapsules. This intracellularly-delivered anti-GSDMB antibody effectively and specifically reduced GSDMB functions, thus providing a novel therapeutic strategy for HER2-positive breast cancers.427 The high expression of GSDMD was associated with tumor invasion and affected tumor size and stage. In addition, the high expression of GSDMD in lung adenocarcinoma indicated a poor prognosis.359 Gsdmd silencing in NSCLC cells decreased EGFR-AKT signaling, enhanced caspase-3 cleavage and apoptosis, and inhibited transplanted tumor growth in NSCLC cells in mice. Gsdmd silencing also reduced EGFR-AKT signaling, enhanced caspase-3 activation and apoptosis, and inhibited transplanted tumor growth in mice.359 These findings indicate that inhibiting gasdermins may also be a potential strategy for tumor therapy.
Inflammasome-related therapy
Inflammasomes are cytoplasmic polyprotein complexes.142 In addition, activated caspase-1 cleaves GSDMD, leading to pyroptosis.428 Inflammasomes take part in diverse hallmarks of tumor formation and progression, promoting and inhibiting tumors.429,430,431
Initially, inflammasomes promote tumorigenesis, and so the inhibition of inflammasomes may provide a promising tumor treatment. The lack of NLRP3 significantly reduced lung metastasis and improved the survival rate of melanoma in response to dendritic cell therapy. The depletion of NLRP3 resulted in a great decrease in the number of MDSCs, thus inhibiting the activity of neighboring immune cells.220 The thymoquinone is the main component extracted from the black seeds of Nigella sativa, and it inhibited the migration of melanoma cells by inhibiting the NLRP3 inflammasome.432 MCC950, an NLRP3 inhibitor, can reduce the number of spheres and colonies formed by SCCHN and suppress the expression of BMI1, ALDH1, and CD44 in SCCHN cells.433 The specific NLRP3 inflammasome blocker β-hydroxybutyrate has also shown promise as a treatment in inflammasome-associated fields.434 In addition, the expression of NLRP3 in tumor-infiltrating macrophages was associated with survival, lymph node invasion, and metastasis in patients with mammary cancer.435 The NLRP3 inflammasome promoted the drug resistance of oral squamous cell carcinoma (OSCC) to 5-fluorouracil (5-FU). Targeting the ROS/NLRP3 inflammasome/IL-1β signaling pathway may be helpful for 5-FU adjuvant chemotherapy in OSCC.436 Bay 11–7082 can inhibit the NLRP3 inflammasome,437 blocking the cell cycle progression of gastric cancer cells, which provides a basis for its clinical application in gastric cancer.438 The expression of ASC protein in metastatic melanoma is lower than that in primary melanoma. In primary melanoma, ASC inhibits tumorigenesis by reducing the phosphorylation of IKKalpha/beta and inhibiting the activity of NF-kappaB.212 ASC has a pro-inflammatory effect on infiltrating cells, which is beneficial to the development of melanoma, but it also limits the proliferation of keratinocytes to respond to harmful stimuli, possibly by activating p53 to inhibit melanoma.439 In addition, antioxidants inhibit the production of ROS, suggesting that they block the expression of NLRP3. N-acetylcysteine (NAC) is a common antioxidant that blocks the activation of the NLRP3 inflammasome in patients.440 NLRP3 and AIM2 were overexpressed in EBV-associated NPC, and the expression level was related to patient survival.441 The exact effects of NLRP3 in different tumors also highlights the therapeutic potential of the inflammasome as a prognostic marker. Knockdown of AIM2 resulted in the inhibition of cell growth and apoptosis in OSCC cells, while B cells activation was downregulated by nuclear factor kappa-light-chain enhancement.442 Furthermore, the NLRC4 inflammasome activation/IL-1a signaling pathway, which is associated with obesity, promoted the progression of breast cancer, which provided a mechanism by which obesity can promote the progression of breast cancer. This result may have a certain reference value in the treatment of obese cancer patients.271 These findings suggest that inhibiting inflammasomes may be beneficial and safe for the prevention and treatment of tumors (Fig. 6).

The status of current NLRP3 inflammasome and caspase-1 inhibitors
In addition, inflammasomes also have antitumor effects, which has been confirmed in a model of colorectal cancer. For example, NLRP1 was significantly dysregulated in colon cancer and attenuated tumorigenesis. Deletion of Nlrp1b significantly increased morbidity and tumorigenesis in mice.282 In addition, Nlrp3 (−/−) mice also showed recurrent colitis and increased colitis-associated cancer.283,284 Besides, the lack of the NLRP6 inflammasome enhanced inflammation-induced CRC development in mice.443 In DSS-induced colitis, pyrin is necessary to activate the inflammasome and mature IL-18.287 Pyrin can also promote the integrity of the intestinal barrier and prevent colitis and tumors.287 In summary, strategies to improve the activity of inflammasomes may be developed to prevent the occurrence of related tumors.
Caspase 1-related therapy
Caspase-1, as an upstream effector, is also crucial in pyroptosis. Thalidomide is an effective anti-inflammatory drug that significantly inhibits the activation and activity of caspase-1, but its application is limited because of its strong teratogenic activity.444 When T cells were depleted, the growth rates of tumors in chimeric mice with adoptive transfer of caspase-1-deficient bone marrow cells were significantly inhibited.445 This antitumor response was reversed by wild-type MDSCs in adoptive metastatic tumor-bearing mice, while the antitumor effect of caspase-1 on MDSCs treated with thalidomide was similar to that in caspase-1-deficient mice.445 Belnacasan (VX-765) is an effective, selective caspase-1 inhibitor. However, its clinical effect has not been optimistic because while it is highly tolerated, preclinical data showed that its curative effect was lower than expected.446 In addition, VX-765 effectively suppressed the production of inflammatory factors, reduced the inflammatory response, and protected the gastric mucosa of mice with acute gastric ulcers induced by cold-restraint stress.447 Ritonavir, once a protease inhibitor used to treat HIV, was later found to effectively reduce the level of IL-18 in mouse pancreatic cancer by suppressing caspase-1.448 Ac-YVAD-CMK, an irreversible and selective inhibitor of caspase-1, effectively blocked the cleavage of pro-IL-1β and pro-IL-18 and reduced the inflammatory response and is expected to be used to inhibit the progression of esophagitis.449 Furthermore, Ac-YVAD-CMK suppressed IFI16- or berberine-induced tumor inhibition in hepatocellular carcinoma 335,450 (Fig. 6).
The negative effects of pyroptosis on tumor therapy
Previous studies have shown that the activation of GSDME is caspase-3-dependent, and the expression level of GSDME is closely associated with the mode of cell death induced by chemotherapeutic drugs. Chemotherapeutic drugs induced pyroptosis in cells with high expression of GSDME, while cells with low or no expression underwent apoptosis. However, the researchers also pointed out that GSDME expression was low in most tumor cell lines due to GSDME gene promoter methylation, while GSDME was widely overexpressed in normal cell lines.16,17 Therefore, chemotherapy could also induce caspase-3-mediated pyroptosis in normal cells with high expression of GSDME, which may be one of the causes of toxicity and side effects associated with chemotherapy. In Gsdme+/+ mice, intraperitoneal injection of cisplatin or 5-FU caused immune cell infiltration and severe small intestinal injury, while the signs of tissue damage decreased significantly in Gsdme−/− mice.451 In addition, Gsdme−/− mice exhibited less inflammation and lung injury under bleomycin or cisplatin treatment.451
Pyroptosis is also involved in the negative effects of radiotherapy. The AIM2 inflammasome played an unexpected role in responding to radiation-induced DNA damage and induced caspase-1-mediated pyroptosis in intestinal epithelial cells and bone marrow cells, which is one of the causes of radiation-induced gastrointestinal and hematological toxicity.452 Inhibition of the AIM2 inflammasome may be a promising treatment target for some patients, such as those exposed to ionizing radiation or cancer patients suffering from hematopoiesis or gastrointestinal toxicity due to radiotherapy or chemotherapy. In addition, radiation also triggered NLRP3 inflammasome-mediated pyroptosis in bone marrow-derived macrophages.453 Targeting the NLRP3 inflammasome may be an effective strategy to reduce radiation damage. In addition, flagellin A N/C inhibited radiation-induced ROS production in intestinal epithelial cells, reduced NLRP3 activity, and suppressed caspase-1-dependent pyroptosis, which may be a factor in protecting intestinal epithelial cells from radiation damage.454 These studies show the negative effects of pyroptosis in chemotherapy and radiotherapy, which offer new insight and hope to tumor treatments.
Conclusion
As an inflammatory and programmed mode of cell death, pyroptosis has been gradually elucidated with further research, but there are still some problems to be solved, such as what other factors regulate pyroptosis and what role other members of the gasdermin family play in pyroptosis.
As described above, increasing evidence has confirmed the important role of pyroptosis in tumors, but few tumor-specific regulatory mechanisms have been identified. Moreover, the conclusion about the relationship between pyroptosis and tumors is not entirely consistent, which indicates the heterogeneity of tumors and the complexity of the immune microenvironment. Pyroptosis can not only inhibit tumor cell proliferation, but also form a microenvironment suitable for tumor cell growth and promote tumor growth. Currently, chronic tumor necrosis caused by pyroptotic cell death of a small number of tumor cells in the central hypoxic area of the tumor inhibits antitumor immunity and accelerates tumor development. The acute inflammation induced by pyroptosis in the tumor microenvironment enhances the immune response and suppresses the progression of the tumor. However, the dual mechanism of pyroptosis related factors promoting and inhibiting tumorgenesis and development remains to be explored. What’s more, the roles of pyroptosis agonists and inhibitors in tumor microenvironment are worthy of further clinical study.
Although there are still many gaps in the knowledge of the role of pyroptosis in tumors, further study on the signaling pathway, regulatory mechanism, and pathological significance of pyroptosis will be helpful in developing new therapeutic options for preventing and treating human tumors.
In addition to the well-studied chemotherapeutic drugs and some small molecular drugs, the therapeutic response to pyroptosis and radiotherapy and the relationship between pyroptosis and specific immunotherapy are not completely clear. Pyroptosis pathway agonists can be used as antitumor activators, and tumor regression can be realized depending on subsequent immune amplification (Fig. 5). The gasdermin proteins do not need to kill all cancer cells but activate tumor immunity by killing a small number of cancer cells. It is difficult to avoid damaging some normal tissue cells. However, since immune cells are able to distinguish normal tissue cells from tumor cells, pyroptosis-mediated immune amplification will selectively occur in tumor cells, greatly reducing the toxic and side effects of drugs on normal tissues. It is possible for researchers to find some small molecules that bind to gasdermin to alleviate the self-inhibition of gasdermin and induce pyroptosis. If such small molecules can be used in clinical treatment by oral or intravenous administration, doctors can accurately control the dose and ensure drug safety. Unfortunately, at present, the regulatory effect of gasdermin family proteins on tumors is not very clear and may be used as tumor-promoting molecules or as tumor suppressors.
We cannot accurately explain whether gasdermin agonists or inhibitors should be used to achieve tumor inhibition. Further elucidation of the important role of gasdermin in the tumor immune response is needed, which will also provide possible strategies for precision treatment of tumor immunity. Specifically, since immune cells can kill cancer cells by inducing cell death,19,20 gasdermin family proteins may be used as biomarkers to divide patients into clinical groups. Patients with high expression of gasdermin family proteins may have improved responses to tumor immunotherapy.
What is the effect of chemotherapeutic drugs on the tumor immune microenvironment by activating GSDME and causing pyroptosis in normal cells with high GSDME expression? How to regulate tumor cell pyroptosis to achieve the desired therapeutic effect and how to avoid normal cell pyroptosis during tumor treatment still need to be explored. It is expected that basic research on pyroptosis and tumors will continue to improve and translate to clinical practice.
References
Friedlander, A. M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 261, 7123–7126 (1986).
Black, R. A., Kronheim, S. R., Merriam, J. E., March, C. J. & Hopp, T. P. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J. Biol. Chem. 264, 5323–5326 (1989).
Thornberry, N. A. et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774 (1992).
Cerretti, D. P. et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science 256, 97–100 (1992).
Zychlinsky, A., Prevost, M. C. & Sansonetti, P. J. Shigella flexneri induces apoptosis in infected macrophages. Nature 358, 167–169 (1992).
Chen, Y., Smith, M. R., Thirumalai, K. & Zychlinsky, A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. 15, 3853–3860 (1996).
D’Souza, C. A. & Heitman, J. Dismantling the Cryptococcus coat. Trends Microbiol 9, 112–113 (2001).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002).
Broz, P. et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291 (2012).
Cookson, B. T. & Brennan, M. A. Pro-inflammatory programmed cell death. Trends Microbiol. 9, 113–114 (2001).
Bergsbaken, T., Fink, S. L. & Cookson, B. T. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109 (2009).
Kovacs, S. B. & Miao, E. A. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 27, 673–684 (2017).
Taabazuing, C. Y., Okondo, M. C. & Bachovchin, D. A. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 24, 507–514.e4 (2017).
Ruhl, S. et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018).
Humphries, F. et al. Succination inactivates gasdermin D and blocks pyroptosis. Science, 369, 1633–1637 (2020).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Rogers, C. et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 8, 14128 (2017).
Newton, K. et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 575, 679–682 (2019).
Zhang, Z. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020).
Zhou, Z. et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368, eaaz7548 (2020).
Hou, J. et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 22, 1264–1275 (2020).
Cerella, C., Teiten, M. H., Radogna, F., Dicato, M. & Diederich, M. From nature to bedside: pro-survival and cell death mechanisms as therapeutic targets in cancer treatment. Biotechnol. Adv. 32, 1111–1122 (2014).
Shi, J., Gao, W. & Shao, F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 42, 245–254 (2017).
Fernandes-Alnemri, T. et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 (2007).
Song, L., Pei, L., Yao, S., Wu, Y. & Shang, Y. NLRP3 inflammasome in neurological diseases, from functions to therapies. Front. Cell Neurosci. 11, 63 (2017).
Shen, H. H. et al. NLRP3: a promising therapeutic target for autoimmune diseases. Autoimmun. Rev. 17, 694–702 (2018).
Jia, C. et al. Role of pyroptosis in cardiovascular diseases. Int. Immunopharmacol. 67, 311–318 (2019).
Pezuk, J. A. Pyroptosis in combinatorial treatment to improve cancer patients’ outcome, is that what we want? EBioMedicine 41, 17–18 (2019).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Ding, J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016).
He, W. T. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 25, 1285–1298 (2015).
Yu, J. et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl Acad. Sci. USA 111, 15514–15519 (2014).
Bergsbaken, T., Fink, S. L., den Hartigh, A. B., Loomis, W. P. & Cookson, B. T. Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine maturation. J. Immunol. 187, 2748–2754 (2011).
Wu, C. et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10, R130 (2009).
Rogers, C. et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 10, 1689 (2019).
Brennan, M. A. & Cookson, B. T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 38, 31–40 (2000).
Kuang, S. et al. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc. Natl Acad. Sci. USA 114, 10642–10647 (2017).
Liu, Z. et al. Crystal structures of the full-length murine and human gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity 51, 43–49.e4 (2019).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Aglietti, R. A. & Dueber, E. C. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 38, 261–271 (2017).
Sollberger, G. et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 3, eaar6689 (2018).
Kambara, H. et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 22, 2924–2936 (2018).
Burgener, S. S. et al. Cathepsin G inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep. 27, 3646–3656.e5 (2019).
Xi, G. et al. GSDMD is required for effector CD8(+) T cell responses to lung cancer cells. Int. Immunopharmacol. 74, 105713 (2019).
Saeki, N. et al. GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF-beta-dependent apoptotic signalling. Oncogene 26, 6488–6498 (2007).
Runkel, F. et al. The dominant alopecia phenotypes Bareskin, Rex-denuded, and Reduced Coat 2 are caused by mutations in gasdermin 3. Genomics 84, 824–835 (2004).
Tanaka, S. et al. A new Gsdma3 mutation affecting anagen phase of first hair cycle. Biochem. Biophys. Res. Commun. 359, 902–907 (2007).
Zhou, Y. et al. Gsdma3 mutation causes bulge stem cell depletion and alopecia mediated by skin inflammation. Am. J. Pathol. 180, 763–774 (2012).
Shi, P. et al. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem J. 468, 325–336 (2015).
Das, S. et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc. Natl Acad. Sci. USA 113, 13132–13137 (2016).
Hergueta-Redondo, M. et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget 7, 56295–56308 (2016).
Hergueta-Redondo, M. et al. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS ONE 9, e90099 (2014).
Miguchi, M. et al. Gasdermin C is upregulated by inactivation of transforming growth factor beta receptor type II in the presence of mutated Apc, promoting colorectal cancer proliferation. PLoS ONE 11, e0166422 (2016).
Kusumaningrum, N., Lee, D. H., Yoon, H. S., Park, C. H. & Chung, J. H. Ultraviolet light-induced gasdermin C expression is mediated via TRPV1/calcium/calcineurin/NFATc1 signaling. Int. J. Mol. Med. 42, 2859–2866 (2018).
Kusumaningrum, N. et al. Gasdermin C is induced by ultraviolet light and contributes to MMP-1 expression via activation of ERK and JNK pathways. J. Dermatol Sci. 90, 180–189 (2018).
Van Laer, L. et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat. Genet. 20, 194–197 (1998).
Booth, K. T. et al. Exonic mutations and exon skipping: lessons learned from DFNA5. Hum. Mutat. 39, 433–440 (2018).
Kim, M. S. et al. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene 27, 3624–3634 (2008).
Croes, L. et al. Large-scale analysis of DFNA5 methylation reveals its potential as biomarker for breast cancer. Clin. Epigenetics 10, 51 (2018).
Delmaghani, S. et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat. Genet. 38, 770–778 (2006).
Schwander, M. et al. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J. Neurosci. 27, 2163–2175 (2007).
Defourny, J. et al. Pejvakin-mediated pexophagy protects auditory hair cells against noise-induced damage. Proc. Natl Acad. Sci. USA 116, 8010–8017 (2019).
Kurokawa, M. & Kornbluth, S. Caspases and kinases in a death grip. Cell 138, 838–854 (2009).
Kerr, J. F., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Chen, X. et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26, 1007–1020 (2016).
Rudel, T. & Bokoch, G. M. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science 276, 1571–1574 (1997).
Poon, I. K., Lucas, C. D., Rossi, A. G. & Ravichandran, K. S. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 14, 166–180 (2014).
Coleman, M. L. et al. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 3, 339–345 (2001).
Sebbagh, M. et al. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 3, 346–352 (2001).
Zheng, T. S. et al. Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc. Natl Acad. Sci. USA 95, 13618–13623 (1998).
Fackler, O. T. & Grosse, R. Cell motility through plasma membrane blebbing. J. Cell Biol. 181, 879–884 (2008).
Torgerson, R. R. & McNiven, M. A. The actin-myosin cytoskeleton mediates reversible agonist-induced membrane blebbing. J. Cell Sci. 111, 2911–2922 (1998).
Janicke, R. U., Ng, P., Sprengart, M. L. & Porter, A. G. Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J. Biol. Chem. 273, 15540–15545 (1998).
Balasubramanian, K., Mirnikjoo, B. & Schroit, A. J. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J. Biol. Chem. 282, 18357–18364 (2007).
Tomiyoshi, G., Horita, Y., Nishita, M., Ohashi, K. & Mizuno, K. Caspase-mediated cleavage and activation of LIM-kinase 1 and its role in apoptotic membrane blebbing. Genes Cells 9, 591–600 (2004).
Tang, R. et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 13, 110 (2020).
Fink, S. L. & Cookson, B. T. Pillars Article: Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006. 8: 1812-1825. J. Immunol. 202, 1913–1926 (2019).
Fink, S. L. & Cookson, B. T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 8, 1812–1825 (2006).
Zhang, Y., Chen, X., Gueydan, C. & Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 28, 9–21 (2018).
Chen, S., Cheng, A. C., Wang, M. S. & Peng, X. Detection of apoptosis induced by new type gosling viral enteritis virus in vitro through fluorescein annexin V-FITC/PI double labeling. World J. Gastroenterol. 14, 2174–2178 (2008).
Zhang, W. & Liang, Z. [Comparison between annexin V-FITC/PI and Hoechst33342/PI double stainings in the detection of apoptosis by flow cytometry]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 30, 1209–1212 (2014).
Chen, Y. et al. Noncovalent proteasome inhibitor PI1840 induces apoptosis and autophagy in osteosarcoma cells. Oncol. Rep. 41, 2803–2817 (2019).
Ray, M., Hostetter, D. R., Loeb, C. R., Simko, J. & Craik, C. S. Inhibition of Granzyme B by PI-9 protects prostate cancer cells from apoptosis. Prostate 72, 846–855 (2012).
Siegel, R. M. Caspases at the crossroads of immune-cell life and death. Nat. Rev. Immunol. 6, 308–317 (2006).
Tajima, T. et al. β-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 95, 1120–1137 (2019).
Li, M. Y. et al. Adrenomedullin alleviates the pyroptosis of Leydig cells by promoting autophagy via the ROS-AMPK-mTOR axis. Cell Death Dis. 10, 489 (2019).
Jia, C. et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 10, 778 (2019).
Ezquerro, S. et al. Ghrelin reduces TNF-α-induced human hepatocyte apoptosis, autophagy, and pyroptosis: role in obesity-associated NAFLD. J. Clin. Endocrinol. Metab. 104, 21–37 (2019).
Liang, J., Wang, Q., Li, J. Q., Guo, T. & Yu, D. Long non-coding RNA MEG3 promotes cerebral ischemia-reperfusion injury through increasing pyroptosis by targeting miR-485/AIM2 axis. Exp. Neurol. 325, 113139 (2020).
Zhao, N., Sun, C., Zheng, M., Liu, S. & Shi, R. Amentoflavone suppresses amyloid β1-42 neurotoxicity in Alzheimer’s disease through the inhibition of pyroptosis. Life Sci. 239, 117043 (2019).
Li, Z. et al. DHA attenuates hepatic ischemia reperfusion injury by inhibiting pyroptosis and activating PI3K/Akt pathway. Eur. J. Pharm. 835, 1–10 (2018).
Zhao, M. W., Yang, P. & Zhao, L. L. Chlorpyrifos activates cell pyroptosis and increases susceptibility on oxidative stress-induced toxicity by miR-181/SIRT1/PGC-1α/Nrf2 signaling pathway in human neuroblastoma SH-SY5Y cells: Implication for association between chlorpyrifos and Parkinson’s disease. Environ. Toxicol. 34, 699–707 (2019).
Jorgensen, I. & Miao, E. A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265, 130–142 (2015).
Bergsbaken, T. & Cookson, B. T. Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 3, e161 (2007).
Mariathasan, S., Weiss, D. S., Dixit, V. M. & Monack, D. M. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049 (2005).
Xu, Y. J., Zheng, L., Hu, Y. W. & Wang, Q. Pyroptosis and its relationship to atherosclerosis. Clin. Chim. Acta 476, 28–37 (2018).
Jaeschke, H., Duan, L., Akakpo, J. Y., Farhood, A. & Ramachandran, A. The role of apoptosis in acetaminophen hepatotoxicity. Food Chem. Toxicol. 118, 709–718 (2018).
Walker, P. R., Leblanc, J., Smith, B., Pandey, S. & Sikorska, M. Detection of DNA fragmentation and endonucleases in apoptosis. Methods 17, 329–338 (1999).
Kijima, M. & Mizuta, R. Histone H1 quantity determines the efficiencies of apoptotic DNA fragmentation and chromatin condensation. Biomed. Res. 40, 51–56 (2019).
Broz, P. & Dixit, V. M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016).
Aachoui, Y., Sagulenko, V., Miao, E. A. & Stacey, K. J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol 16, 319–326 (2013).
Orning, P. et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018).
Sarhan, J. et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl Acad. Sci. USA 115, E10888–E10897 (2018).
Chen, K. W. et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38, e101638 (2019).
Aziz, M., Jacob, A. & Wang, P. Revisiting caspases in sepsis. Cell Death Dis. 5, e1526 (2014).
Vanden, B. T. et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am. J. Respir. Crit. Care Med. 189, 282–291 (2014).
Angosto, D. et al. Evolution of inflammasome functions in vertebrates: Inflammasome and caspase-1 trigger fish macrophage cell death but are dispensable for the processing of IL-1beta. Innate Immun. 18, 815–824 (2012).
Fang, Y. et al. Pyroptosis: a new frontier in cancer. Biomed. Pharmacother. 121, 109595 (2020).
Komada, T. & Muruve, D. A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 15, 501–520 (2019).
Emran, A. A. et al. Do innate killing mechanisms activated by inflammasomes have a role in treating melanoma? Pigment Cell Melanoma Res. 33, 660–670 (2020).
Zhou, B. et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 28, 1171–1185 (2018).
Boise, L. H. & Collins, C. M. Salmonella-induced cell death: apoptosis, necrosis or programmed cell death? Trends Microbiol. 9, 64–67 (2001).
Kolb, R., Liu, G. H., Janowski, A. M., Sutterwala, F. S. & Zhang, W. Inflammasomes in cancer: a double-edged sword. Protein Cell 5, 12–20 (2014).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 (2007).
Li, J. & Yuan, J. Caspases in apoptosis and beyond. Oncogene 27, 6194–6206 (2008).
Ha, H. C. & Snyder, S. H. Poly (ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl Acad. Sci. USA 96, 13978–13982 (1999).
Xia, X. et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis. 10, 650 (2019).
Frank, D. & Vince, J. E. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 26, 99–114 (2019).
Jackson, D. N. & Theiss, A. L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 11, 285–304 (2020).
Man, S. M., Karki, R. & Kanneganti, T. D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 46, 269–280 (2016).
He, Y. & Amer, A. O. Microbial modulation of host apoptosis and pyroptosis. Front. Cell Infect. Microbiol. 4, 83 (2014).
Nunes, T. & de Souza, H. S. Inflammasome in intestinal inflammation and cancer. Mediators Inflamm. 2013, 654963 (2013).
Cervantes, J., Nagata, T., Uchijima, M., Shibata, K. & Koide, Y. Intracytosolic Listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cell Microbiol. 10, 41–52 (2008).
Kelk, P., Johansson, A., Claesson, R., Hanstrom, L. & Kalfas, S. Caspase 1 involvement in human monocyte lysis induced by Actinobacillus actinomycetemcomitans leukotoxin. Infect. Immun. 71, 4448–4455 (2003).
Jesenberger, V., Procyk, K. J., Yuan, J., Reipert, S. & Baccarini, M. Salmonella-induced caspase-2 activation in macrophages: a novel mechanism in pathogen-mediated apoptosis. J. Exp. Med. 192, 1035–1046 (2000).
Li, P. et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80, 401–411 (1995).
He, Y., Hara, H. & Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021 (2016).
Thi, H. & Hong, S. Inflammasome as a therapeutic target for cancer prevention and treatment. J. Cancer Prev. 22, 62–73 (2017).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
McAllister, S. S. & Weinberg, R. A. Tumor-host interactions: a far-reaching relationship. J. Clin. Oncol. 28, 4022–4028 (2010).
Munn, D. H. & Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 39, 1–6 (2016).
Massague, J. & Obenauf, A. C. Metastatic colonization by circulating tumour cells. Nature 529, 298–306 (2016).
Klement, G. L. Eco-evolution of cancer resistance. Sci. Transl. Med. 8, 327fs5 (2016).
Finn, O. J. & Beatty, P. L. Cancer immunoprevention. Curr. Opin. Immunol. 39, 52–58 (2016).
Solinas, G., Marchesi, F., Garlanda, C., Mantovani, A. & Allavena, P. Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev. 29, 243–248 (2010).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Liston, A. & Masters, S. L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 17, 208–214 (2017).
Chen, G. Y. & Nunez, G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 (2010).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Rathinam, V. A. & Fitzgerald, K. A. Inflammasome complexes: emerging mechanisms and effector functions. Cell 165, 792–800 (2016).
Lamkanfi, M. & Dixit, V. M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Place, D. E. & Kanneganti, T. D. Recent advances in inflammasome biology. Curr. Opin. Immunol. 50, 32–38 (2018).
Jorgensen, I., Zhang, Y., Krantz, B. A. & Miao, E. A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 213, 2113–2128 (2016).
Miao, E. A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010).
Minkiewicz, J., de Rivero, V. J. & Keane, R. W. Human astrocytes express a novel NLRP2 inflammasome. Glia 61, 1113–1121 (2013).
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Levy, M. et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443 (2015).
Hara, H. et al. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates Gram-positive pathogen infection. Cell 175, 1651–1664.e14 (2018).
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J. & Alnemri, E. S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Zitvogel, L., Kepp, O., Galluzzi, L. & Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 13, 343–351 (2012).
Kawai, T. & Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 (2011).
Barton, G. M. & Medzhitov, R. Toll-like receptor signaling pathways. Science 300, 1524–1525 (2003).
Lamkanfi, M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 11, 213–220 (2011).
Lamkanfi, M. & Dixit, V. M. Inflammasomes and their roles in health and disease. Annu Rev. Cell Dev. Biol. 28, 137–161 (2012).
Chavarria-Smith, J., Mitchell, P. S., Ho, A. M., Daugherty, M. D. & Vance, R. E. Functional and evolutionary analyses identify proteolysis as a general Mechanism for NLRP1 inflammasome activation. PLoS Pathog. 12, e1006052 (2016).
Boyden, E. D. & Dietrich, W. F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 38, 240–244 (2006).
Chavarria-Smith, J. & Vance, R. E. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 9, e1003452 (2013).
Levinsohn, J. L. et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8, e1002638 (2012).
Okondo, M. C. et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol. 13, 46–53 (2017).
Ewald, S. E., Chavarria-Smith, J. & Boothroyd, J. C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 82, 460–468 (2014).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Kofoed, E. M. & Vance, R. E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Franchi, L. et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582 (2006).
Ren, T., Zamboni, D. S., Roy, C. R., Dietrich, W. F. & Vance, R. E. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18 (2006).
Lightfield, K. L. et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178 (2008).
Duncan, J. A. & Canna, S. W. The NLRC4 Inflammasome. Immunol. Rev. 281, 115–123 (2018).
Roberts, T. L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009).
Jin, T. et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36, 561–571 (2012).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 (2010).
Rathinam, V. A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Pierini, R. et al. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 19, 1709–1721 (2012).
Jones, J. W. et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl Acad. Sci. USA 107, 9771–9776 (2010).
Matyszewski, M., Morrone, S. R. & Sohn, J. Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc. Natl Acad. Sci. USA 115, E1963–E1972 (2018).
Xu, H. et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 (2014).
Chung, L. K. et al. The Yersinia virulence factor YopM hijacks host kinases to inhibit type III effector-triggered activation of the pyrin inflammasome. Cell Host Microbe 20, 296–306 (2016).
Ratner, D. et al. The yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathog. 12, e1006035 (2016).
Park, Y. H., Wood, G., Kastner, D. L. & Chae, J. J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 17, 914–921 (2016).
Masters, S. L. et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl. Med. 8, 332ra45 (2016).
Gao, W., Yang, J., Liu, W., Wang, Y. & Shao, F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl Acad. Sci. USA 113, E4857–E4866 (2016).
Sollberger, G., Strittmatter, G. E., Garstkiewicz, M., Sand, J. & Beer, H. D. Caspase-1: the inflammasome and beyond. Innate Immun. 20, 115–125 (2014).
Sborgi, L. et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778 (2016).
Carty, M. et al. Cell survival and cytokine release after inflammasome activation is regulated by the Toll-IL-1R protein SARM. Immunity 50, 1412–1424.e6 (2019).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Chu, L. H. et al. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat. Commun. 9, 996 (2018).
Aglietti, R. A. et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl Acad. Sci. USA 113, 7858–7863 (2016).
Baker, P. J. et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 45, 2918–2926 (2015).
Ruhl, S. & Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K (+) efflux. Eur. J. Immunol. 45, 2927–2936 (2015).
Schmid-Burgk, J. L. et al. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol. 45, 2911–2917 (2015).
Yang, D., He, Y., Muñoz-Planillo, R., Liu, Q. & Núñez, G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 43, 923–932 (2015).
Liu, Y. et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 5, eaax7969 (2020).
Schadendorf, D. et al. Melanoma. Nat. Rev. Dis. Prim. 1, 15003 (2015).
Ellerhorst, J. A. et al. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin. Cancer Res. 17, 229–235 (2011).
Schadendorf, D. et al. Melanoma. Lancet 392, 971–984 (2018).
O’Neill, C. H. & Scoggins, C. R. Melanoma. J. Surg. Oncol. 120, 873–881 (2019).
Lugovic-Mihic, L. et al. Melanoma development: current knowledge on melanoma pathogenesis. Acta Dermatovenerol. Croat. 27, 163–168 (2019).
Smalley, K. S. M. Two Worlds Collide: unraveling the role of the immune system in BRAF–MEK inhibitor responses. Cancer Discov. 10, 176–178 (2020).
Erkes, D. A. et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 10, 254–269 (2020).
Zhao, Q. et al. Novel HSP90-PI3K dual inhibitor suppresses melanoma cell proliferation by interfering with HSP90-EGFR interaction and downstream signaling pathways. Int. J. Mol. Sci. 21, 1845 (2020).
Bollino, D., Colunga, A., Li, B. & Aurelian, L. ΔPK oncolytic activity includes modulation of the tumour cell milieu. J. Gen. Virol. 97, 496–508 (2016).
Lage, H., Helmbach, H., Grottke, C., Dietel, M. & Schadendorf, D. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 494, 54–59 (2001).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Dixon, S. J. & Stockwell, B. R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 (2014).
Aykin-Burns, N., Ahmad, I. M., Zhu, Y., Oberley, L. W. & Spitz, D. R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 418, 29–37 (2009).
Doskey, C. M. et al. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 10, 274–284 (2016).
Yu, P. et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharm. Sin. 40, 1237–1244 (2019).
Verma, D. et al. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. 25, 506–513 (2012).
Davis, B. K., Wen, H. & Ting, J. P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev. Immunol. 29, 707–735 (2011).
Liu, W. et al. Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J. Invest. Dermatol. 133, 518–527 (2013).
Zhai, Z. et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 36, 3820–3830 (2017).
Street, S. E., Cretney, E. & Smyth, M. J. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 97, 192–197 (2001).
Gilfillan, S. et al. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J. Exp. Med. 205, 2965–2973 (2008).
Werneck, M. B., Lugo-Villarino, G., Hwang, E. S., Cantor, H. & Glimcher, L. H. T-bet plays a key role in NK-mediated control of melanoma metastatic disease. J. Immunol. 180, 8004–8010 (2008).
Takeda, K. et al. IFN-gamma production by lung NK cells is critical for the natural resistance to pulmonary metastasis of B16 melanoma in mice. J. Leukoc. Biol. 90, 777–785 (2011).
Chow, M. T. et al. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 72, 5721–5732 (2012).
Okamoto, M. et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1β. J. Biol. Chem. 285, 6477–6488 (2010).
van Deventer, H. W. et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. 70, 10161–10169 (2010).
Janowski, A. M. et al. NLRC4 suppresses melanoma tumor progression independently of inflammasome activation. J. Clin. Invest. 126, 3917–3928 (2016).
Poyet, J. L. et al. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 276, 28309–28313 (2001).
Zhao, Y. & Shao, F. The NAIP-NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol. Rev. 265, 85–102 (2015).
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996).
Freeman, G. J. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Voronov, E. et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl Acad. Sci. USA 100, 2645–2650 (2003).
Dunn, J. H., Ellis, L. Z. & Fujita, M. Inflammasomes as molecular mediators of inflammation and cancer: potential role in melanoma. Cancer Lett. 314, 24–33 (2012).
Okamoto, M. et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 285, 6477–6488 (2010).
Anasagasti, M. J., Alvarez, A., Martin, J. J., Mendoza, L. & Vidal-Vanaclocha, F. Sinusoidal endothelium release of hydrogen peroxide enhances very late antigen-4-mediated melanoma cell adherence and tumor cytotoxicity during interleukin-1 promotion of hepatic melanoma metastasis in mice. Hepatology 25, 840–846 (1997).
Vidal-Vanaclocha, F. et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc. Natl Acad. Sci. USA 97, 734–739 (2000).
Benson, D. J. et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 116, 2286–2294 (2010).
Terme, M. et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 71, 5393–5399 (2011).
Harbeck, N. et al. Breast cancer. Nat. Rev. Dis. Prim. 5, 66 (2019).
Xiu, B. et al. LINC02273 drives breast cancer metastasis by epigenetically increasing AGR2 transcription. Mol. Cancer 18, 187 (2019).
Mani, S. Microbiota and breast cancer. Prog. Mol. Biol. Transl. Sci. 151, 217–229 (2017).
Pearce, L. Breast cancer. Nurs. Stand 30, 15 (2016).
Woolston, C. Breast cancer. Nature 527, S101 (2015).
Harbeck, N. & Gnant, M. Breast cancer. Lancet 389, 1134–1150 (2017).
Op De Beeck, K., Van Laer, L. & Van Camp, G. DFNA5, a gene involved in hearing loss and cancer: a review. Ann. Otol., Rhinol. Laryngol. 121, 197–207 (2012).
Pizato, N. et al. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci. Rep. 8, 1952 (2018).
Masuda, Y. et al. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J. Hum. Genet. 51, 652–664 (2006).
Fujikane, T. et al. Genomic screening for genes upregulated by demethylation revealed novel targets of epigenetic silencing in breast cancer. Breast Cancer Res. Treat. 122, 699–710 (2010).
Kim, M. S. et al. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem. Biophys. Res. Commun. 370, 38–43 (2008).
Burnstock, G. & Verkhratsky, A. Long-term (trophic) purinergic signalling: purinoceptors control cell proliferation, differentiation and death. Cell Death Dis. 1, e9 (2010).
Fu, W. et al. Activation of P2X7-mediated apoptosis Inhibits DMBA/TPA-induced formation of skin papillomas and cancer in mice. BMC Cancer 9, 114 (2009).
Wen, J., Lv, Z., Ding, H., Fang, X. & Sun, M. Association between PXR polymorphisms and cancer risk: a systematic review and meta-analysis. Biosci. Rep. 38, BSR20171614 (2018).
Di Virgilio, F., Ferrari, D. & Adinolfi, E. P2X7: a growth-promoting receptor—implications for cancer. Purinergic Signal. 5, 251–256 (2009).
Adinolfi, E. et al. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 72, 2957–2969 (2012).
Draganov, D. et al. Modulation of P2X4/P2X7/Pannexin-1 sensitivity to extracellular ATP via Ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci. Rep. 5, 16222 (2015).
Kim, S. et al. Radiation-induced autophagy potentiates immunotherapy of cancer via up-regulation of mannose 6-phosphate receptor on tumor cells in mice. Cancer Immunol. Immunother. 63, 1009–1021 (2014).
Ramakrishnan, R. & Gabrilovich, D. I. The role of mannose-6-phosphate receptor and autophagy in influencing the outcome of combination therapy. Autophagy 9, 615–616 (2013).
Junger, W. G. Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 11, 201–212 (2011).
Ghiringhelli, F. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178 (2009).
Guo, B., Fu, S., Zhang, J., Liu, B. & Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 6, 36107 (2016).
Svensson, S. et al. CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer. Clin. Cancer Res. 21, 3794–3805 (2015).
Lim, S. Y., Yuzhalin, A. E., Gordon-Weeks, A. N. & Muschel, R. J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 7, 28697–28710 (2016).
Qian, B. Z. et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011).
Cheng, R. et al. Periodontal inflammation recruits distant metastatic breast cancer cells by increasing myeloid-derived suppressor cells. Oncogene 39, 1543–1556 (2020).
Yaal-Hahoshen, N. et al. The chemokine CCL5 as a potential prognostic factor predicting disease progression in stage II breast cancer patients. Clin. Cancer Res. 12, 4474–4480 (2006).
Niwa, Y. et al. Correlation of tissue and plasma RANTES levels with disease course in patients with breast or cervical cancer. Clin. Cancer Res. 7, 285–289 (2001).
Eissa, S. A., Zaki, S. A., El-Maghraby, S. M. & Kadry, D. Y. Importance of serum IL-18 and RANTES as markers for breast carcinoma progression. J. Egypt. Natl Canc. Inst. 17, 51–55 (2005).
Bieche, I. et al. Molecular profiling of inflammatory breast cancer: identification of a poor-prognosis gene expression signature. Clin. Cancer Res. 10, 6789–6795 (2004).
Wigler, N. et al. Breast carcinoma: a report on the potential usage of the CC chemokine RANTES as a marker for a progressive disease. Isr. Med. Assoc. J. 4, 940–943 (2002).
Zhang, Y. et al. A novel role of hematopoietic CCL5 in promoting triple-negative mammary tumor progression by regulating generation of myeloid-derived suppressor cells. Cell Res. 23, 394–408 (2013).
Reeves, G. K. et al. Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ 335, 1134 (2007).
Rose, D. P. & Vona-Davis, L. Influence of obesity on breast cancer receptor status and prognosis. Expert Rev. Anticancer Ther. 9, 1091–1101 (2014).
Reeves, G. K., Pirie, K., Green, J., Bull, D. & Beral, V. Comparison of the effects of genetic and environmental risk factors on in situ and invasive ductal breast cancer. Int. J. Cancer 131, 930–937 (2012).
Kolb, R. et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 7, 13007 (2016).
Jiao, Y. et al. The role of caspase-4 and NLRP1 in MCF7 cell pyroptosis induced by hUCMSC-secreted factors. Stem Cells Int. 2020, 1–14 (2020).
Lamichhane, P. et al. Colorectal cancer and probiotics: are bugs really drugs? Cancers (Basel) 12, 1162 (2020).
Shang, A. et al. Exosomal circPACRGL promotes progression of colorectal cancer via the miR-142-3p/miR-506-3p- TGF-β1 axis. Mol. Cancer 19, 117 (2020).
Dekker, E., Tanis, P. J., Vleugels, J., Kasi, P. M. & Wallace, M. B. Colorectal cancer. Lancet 394, 1467–1480 (2019).
Yiu, A. J. & Yiu, C. Y. Biomarkers in colorectal cancer. Anticancer Res. 36, 1093–1102 (2016).
Thanikachalam, K. & Khan, G. Colorectal cancer and nutrition. Nutrients 11, 164 (2019).
Brody, H. Colorectal cancer. Nature 521, S1 (2015).
Ma, Y., Chen, Y., Lin, C. & Hu, G. Biological functions and clinical significance of the newly identified long noncoding RNA RP185F18.6 in colorectal cancer. Oncol. Rep. 40, 2648–2658 (2018).
Ibrahim, J. et al. Methylation analysis of Gasdermin E shows great promise as a biomarker for colorectal cancer. Cancer Med. 8, 2133–2145 (2019).
Yu, J. et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 10, 193 (2019).
Williams, T. M. et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J. Immunol. 194, 3369–3380 (2015).
Allen, I. C. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Zaki, M. H., Vogel, P., Body-Malapel, M., Lamkanfi, M. & Kanneganti, T. D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 185, 4912–4920 (2010).
Hu, B. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl Acad. Sci. USA 107, 21635–21640 (2010).
Wilson, J. E. et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 21, 906–913 (2015).
Sharma, D. et al. Pyrin inflammasome regulates tight junction integrity to restrict colitis and tumorigenesis. Gastroenterology 154, 948–964.e8 (2018).
Zaki, M. H., Lamkanfi, M. & Kanneganti, T. D. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 32, 171–179 (2011).
Chen, G. Y. & Nunez, G. Inflammasomes in intestinal inflammation and cancer. Gastroenterology 141, 1986–1999 (2011).
Chen, C. et al. DAC can restore expression of NALP1 to suppress tumor growth in colon cancer. Cell Death Dis. 6, e1602–e1602 (2015).
Zaki, M. H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Salcedo, R. et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J. Exp. Med. 207, 1625–1636 (2010).
Takagi, H. et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand. J. Gastroenterol. 38, 837–844 (2003).
Dupaul-Chicoine, J. et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378 (2010).
Dupaul-Chicoine, J. et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 43, 751–763 (2015).
Bauer, C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010).
Hirota, S. A. et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 17, 1359–1372 (2011).
Nowarski, R. et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 163, 1444–1456 (2015).
Wang, Y. et al. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem. Biophys. Res. Commun. 495, 1418–1425 (2018).
Wei, L. et al. Noncoding RNAs in gastric cancer: implications for drug resistance. Mol. Cancer 19, 62–17 (2020).
Graham, D. Y. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology 148, 719–31.e3 (2015).
Hu, Y. et al. LncRNA-SNHG1 contributes to gastric cancer cell proliferation by regulating DNMT1. Biochem. Biophys. Res. Commun. 491, 926–931 (2017).
Lordick, F., Lorenzen, S., Yamada, Y. & Ilson, D. Optimal chemotherapy for advanced gastric cancer: is there a global consensus? Gastric Cancer 17, 213–225 (2014).
Wu, W. K. et al. Dysregulation of cellular signaling in gastric cancer. Cancer Lett. 295, 144–153 (2010).
Akino, K. et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 98, 88–95 (2007).
Saeki, N. et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer 48, 261–271 (2009).
Komiyama, H. et al. Alu-derived cis-element regulates tumorigenesis-dependent gastric expression of GASDERMIN B (GSDMB). Genes Genet. Syst. 85, 75–83 (2010).
Qiu, S., Liu, J. & Xing, F. ‘Hints’ in the killer protein gasdermin D: unveiling the secrets of gasdermins driving cell death. Cell Death Differ. 24, 588–596 (2017).
Wang, W. J. et al. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J. Dig. Dis. 19, 74–83 (2018).
Frantz, S. et al. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J. Mol. Cell Cardiol. 35, 685–694 (2003).
Hanada, K. & Graham, D. Y. Helicobacter pylori and the molecular pathogenesis of intestinal-type gastric carcinoma. Expert Rev. Anticancer Ther. 14, 947–954 (2014).
Hardbower, D. M., Peek, R. J. & Wilson, K. T. At the Bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. J. Leukoc. Biol. 96, 201–212 (2014).
Graham, D. Y. History of Helicobacter pylori, duodenal ulcer, gastric ulcer and gastric cancer. World J. Gastroenterol. 20, 5191–5204 (2014).
Salama, N. R., Hartung, M. L. & Muller, A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 11, 385–399 (2013).
Shiotani, A., Cen, P. & Graham, D. Y. Eradication of gastric cancer is now both possible and practical. Semin Cancer Biol. 23, 492–501 (2013).
Semper, R. P. et al. Helicobacter pylori-induced IL-1beta secretion in innate immune cells is regulated by the NLRP3 inflammasome and requires the cag pathogenicity island. J. Immunol. 193, 3566–3576 (2014).
Kim, D. J., Park, J. H., Franchi, L., Backert, S. & Nunez, G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1beta production in Helicobacter pylori infected dendritic cells. Eur. J. Immunol. 43, 2650–2658 (2013).
Li, S. et al. MiR-22 sustains NLRP3 expression and attenuates H. pylori-induced gastric carcinogenesis. Oncogene 37, 884–896 (2018).
Bagheri, V. et al. Cytokine networks and their association withHelicobacter pylori infection in gastric carcinoma. J. Cell. Physiol. 233, 2791–2803 (2018).
Lamb, A. & Chen, L. F. Role of the Helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell Biochem. 114, 491–497 (2013).
Kim, K. E. et al. Expression of ADAM33 is a novel regulatory mechanism in IL-18-secreted process in gastric cancer. J. Immunol. 182, 3548–3555 (2009).
Ye, Z. B., Ma, T., Li, H., Jin, X. L. & Xu, H. M. Expression and significance of intratumoral interleukin-12 and interleukin-18 in human gastric carcinoma. World J. Gastroenterol. 13, 1747–1751 (2007).
Kim, K. E. et al. Interleukin-18 is a critical factor for vascular endothelial growth factor-enhanced migration in human gastric cancer cell lines. Oncogene 26, 1468–1476 (2007).
Kim, J. et al. IL-18 enhances thrombospondin-1 production in human gastric cancer via JNK pathway. Biochem. Biophys. Res. Commun. 344, 1284–1289 (2006).
Tu, S. et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419 (2008).
De Toni, E. N. et al. Age independent survival benefit for patients with hepatocellular carcinoma (HCC) without metastases at diagnosis: a population-based study. Gut 69, 168–176 (2019).
Singal, A. G. & El-Serag, H. B. Hepatocellular carcinoma from epidemiology to prevention: translating knowledge into practice. Clin. Gastroenterol. Hepatol. 13, 2140–2151 (2015).
Liang, Y., Liang, Q., Qiao, L. & Xiao, F. MicroRNAs modulate drug resistance-related mechanisms in hepatocellular carcinoma. Front. Oncol. 10, 920 (2020).
Erratum: Global cancer statistics. 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 70, 313 (2020).
Forner, A., Reig, M. & Bruix, J. Hepatocellular carcinoma. Lancet 391, 1301–1314 (2018).
Zongyi, Y. & Xiaowu, L. Immunotherapy for hepatocellular carcinoma. Cancer Lett. 470, 8–17 (2020).
Ringelhan, M., Pfister, D., O’Connor, T., Pikarsky, E. & Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 19, 222–232 (2018).
Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 380, 1450–1462 (2019).
Wang, C. J. et al. The expression and regulation of DFNA5 in human hepatocellular carcinoma DFNA5 in hepatocellular carcinoma. Mol. Biol. Rep. 40, 6525–6531 (2013).
Chu, Q. et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget 7, 84658 (2016).
Cheng, D. et al. Biogenesis and cytotoxicity of APOL1 renal risk variant proteins in hepatocytes and hepatoma cells. J. Lipid Res. 56, 1583–1593 (2015).
Kim, S. J. & Lee, S. M. NLRP3 inflammasome activation in D-galactosamine and lipopolysaccharide-induced acute liver failure: role of heme oxygenase-1. Free Radic. Biol. Med. 65, 997–1004 (2013).
Ganz, M., Csak, T., Nath, B. & Szabo, G. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J. Gastroenterol. 17, 4772–4778 (2011).
Wei, Q. et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest. 94, 52–62 (2014).
Wei, Q. et al. Estrogen suppresses hepatocellular carcinoma cells through ERbeta-mediated upregulation of the NLRP3 inflammasome. Lab Invest. 95, 804–816 (2015).
Wei, Q., Zhu, R., Zhu, J., Zhao, R. & Li, M. E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC Cells. Oncol. Res. 27, 827–834 (2019).
Fan, S. H. et al. Luteoloside suppresses proliferation and metastasis of hepatocellular carcinoma cells by inhibition of NLRP3 inflammasome. PLoS ONE 9, e89961 (2014).
Musso, G., Cassader, M. & Gambino, R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Disco. 15, 249–274 (2016).
Machado, M. V. & Diehl, A. M. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 150, 1769–1777 (2016).
Pekow, J. R., Bhan, A. K., Zheng, H. & Chung, R. T. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Cancer 109, 2490–2496 (2007).
Lee, Y. B. et al. Association between hepatic steatosis and the development of hepatocellular carcinoma in patients with chronic hepatitis B. Clin. Mol. Hepatol. 25, 52–64 (2019).
Zhang, J., Zhang, K., Li, Z. & Guo, B. ER stress-induced inflammasome activation contributes to hepatic inflammation and steatosis. J. Clin. Cell Immunol. 7, 457 (2016).
Imaeda, A. B. et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Invest. 119, 305–314 (2009).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Ma, X. et al. Loss of AIM2 expression promotes hepatocarcinoma progression through activation of mTOR-S6K1 pathway. Oncotarget 7, 36185–36197 (2016).
Hong, Q. et al. Prevention and management of lung cancer in China. Cancer 121, 3080–3088 (2015).
Sun, R. et al. Long non-coding RNA in drug resistance of non-small cell lung cancer: a mini review. Front. Pharm. 10, 1457 (2019).
Duruisseaux, M. & Esteller, M. Lung cancer epigenetics: from knowledge to applications. Semin. Cancer Biol. 51, 116–128 (2018).
Villalobos, P. & Wistuba, I. I. Lung cancer biomarkers. Hematol. Oncol. Clin. North Am. 31, 13–29 (2017).
Nasim, F., Sabath, B. F. & Eapen, G. A. Lung cancer. Med. Clin. North Am. 103, 463–473 (2019).
Torre, L. A., Siegel, R. L. & Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 893, 1–19 (2016).
Brody, H. Lung cancer. Nature 513, S1 (2014).
Tanoue, L. T., Tanner, N. T., Gould, M. K. & Silvestri, G. A. Lung cancer screening. Am. J. Respir. Crit. Care Med. 191, 19–33 (2015).
Gao, J. et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in non‑small cell lung cancer. Oncol. Rep. 40, 1971–1984 (2018).
Xi, G. et al. GSDMD is required for effector CD8+ T cell responses to lung cancer cells. Int. Immunopharmacol. 74, 105713 (2019).
Lu, H. et al. Molecular targeted therapies elicit concurrent apoptotic and GSDME-dependent pyroptotic tumor cell death. Clin. Cancer Res. 24, 6066–6077 (2018).
Zhang, C. et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 24, 312–325 (2019).
Teng, J. et al. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-κB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers 12, 193 (2020).
Kong, H. et al. Differential expression of inflammasomes in lung cancer cell lines and tissues. Tumor Biol. 36, 7501–7513 (2015).
Zhang, M. et al. AIM2 promotes non-small-cell lung cancer cell growth through inflammasome-dependent pathway. J. Cell Physiol. 234, 20161–20173 (2019).
Zou, J., Yang, Y., Yang, Y. & Liu, X. Polydatin suppresses proliferation and metastasis of non-small cell lung cancer cells by inhibiting NLRP3 inflammasome activation via NF-kappaB pathway. Biomed. Pharmacother. 108, 130–136 (2018).
Li, Q. et al. CPT-11 activates NLRP3 inflammasome through JNK and NF-kappaB signalings. Toxicol. Appl. Pharm. 289, 133–141 (2015).
Harder, J. et al. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J. Immunol. 183, 5823–5829 (2009).
Karin, M. Nuclear factor-κB in cancer development and progression. Nature 441, 431–436 (2006).
Chen, W., Li, Z., Bai, L. & Lin, Y. NF-kappaB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front. Biosci. (Landmark Ed.) 16, 1172–1185 (2011).
Liu, A., Liu, L. & Lu, H. LncRNA XIST facilitates proliferation and epithelial-mesenchymal transition of colorectal cancer cells through targeting miR-486-5p and promoting neuropilin-2. J. Cell Physiol. 234, 13747–13761 (2019).
Chen, Z. et al. Long non-coding RNA XIST promotes the development of esophageal cancer by sponging miR-494 to regulate CDK6 expression. Biomed. Pharmacother. 109, 2228–2236 (2019).
Chen, X. et al. Up-regulated lncRNA XIST contributes to progression of cervical cancer via regulating miR-140-5p and ORC1. Cancer Cell Int. 19, 45 (2019).
Zhang, J. et al. LncRNA XIST facilitates cell growth, migration and invasion via modulating H3 histone methylation of DKK1 in neuroblastoma. Cell Cycle 18, 1882–1892 (2019).
Sun, W., Zu, Y., Fu, X. & Deng, Y. Knockdown of lncRNA-XIST enhances the chemosensitivity of NSCLC cells via suppression of autophagy. Oncol. Rep. 38, 3347–3354 (2017).
Tian, K. et al. Long noncoding RNA X-inactive specific transcript facilitates cellular functions in melanoma via miR-139-5p/ROCK1 pathway. Onco Targets Ther. 13, 1277–1287 (2020).
Wang, C. et al. Upregulation of long non-coding RNA XIST has anticancer effects on epithelial ovarian cancer cells through inverse downregulation of hsa-miR-214-3p. J. Gynecol. Oncol. 29, e99 (2018).
Sun, N., Zhang, G. & Liu, Y. Long non-coding RNA XIST sponges miR-34a to promotes colon cancer progression via Wnt/beta-catenin signaling pathway. Gene 665, 141–148 (2018).
Li, H. et al. Long non-coding RNA XIST serves an oncogenic role in osteosarcoma by sponging miR-137. Exp. Ther. Med. 17, 730–738 (2019).
Zhou, K., Yang, J., Li, X. & Chen, W. Long non-coding RNA XIST promotes cell proliferation and migration through targeting miR-133a in bladder cancer. Exp. Ther. Med. 18, 3475–3483 (2019).
Wang, X. et al. Knockdown of LncRNA-XIST suppresses proliferation and TGF-beta1-Induced EMT in NSCLC through the notch-1 pathway by regulation of miR-137. Genet Test. Mol. Biomark. 22, 333–342 (2018).
Wang, H. et al. The long non-coding RNA XIST controls non-small cell lung cancer proliferation and invasion by modulating miR-186-5p. Cell Physiol. Biochem. 41, 2221–2229 (2017).
Liu, J. et al. Downregulation of LncRNA-XIST inhibited development of non-small cell lung cancer by activating miR-335/SOD2/ROS signal pathway mediated pyroptotic cell death. Aging (Albany NY) 11, 7830–7846 (2019).
Bai, X. Y. et al. miR-335 and miR-34a Promote renal senescence by suppressing mitochondrial antioxidative enzymes. J. Am. Soc. Nephrol. 22, 1252–1261 (2011).
Wang, H. et al. Effect of miR-335 upregulation on the apoptosis and invasion of lung cancer cell A549 and H1299. Tumour Biol. 34, 3101–3109 (2013).
Liu, J. et al. miR-335 inhibited cell proliferation of lung cancer cells by target Tra2beta. Cancer Sci. 109, 289–296 (2018).
Tang, H. et al. CPNE1 is a target of miR-335-5p and plays an important role in the pathogenesis of non-small cell lung cancer. J. Exp. Clin. Cancer Res. 37, 131 (2018).
Cui, J. et al. MST1 suppresses pancreatic cancer progression via ROS-induced pyroptosis. Mol. Cancer Res. 17, 1316–1325 (2019).
Wang, Y. et al. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 11, 1069–1082 (2019).
Sun, W. et al. Ageratina adenophora induces mice hepatotoxicity via ROS-NLRP3-mediated pyroptosis. Sci. Rep. 8, 16032 (2018).
Small, W. J. et al. Cervical cancer: a global health crisis. Cancer 123, 2404–2412 (2017).
Dryden-Peterson, S. et al. HIV infection and survival among women withcervical cancer. J. Clin. Oncol. 34, 3749–3757 (2016).
Rerucha, C. M., Caro, R. J. & Wheeler, V. L. Cervical cancer screening. Am. Fam. Physician 97, 441–448 (2018).
Vu, M., Yu, J., Awolude, O. A. & Chuang, L. Cervical cancer worldwide. Curr. Probl. Cancer 42, 457–465 (2018).
Ngoma, M. & Autier, P. Cancer prevention: cervical cancer. Ecancermedicalscience 13, 952 (2019).
Cohen, P. A., Jhingran, A., Oaknin, A. & Denny, L. Cervical cancer. Lancet 393, 169–182 (2019).
Bostanci, V. et al. Evaluation of IL-1beta, IL-1ra, and IL-10 levels and outcome of periodontal therapy in chronic periodontitis with familial Mediterranean fever. Clin. Oral. Investig. 21, 469–475 (2017).
So, D. et al. Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense. Oncogene 37, 5191–5204 (2018).
Song, Y. et al. HPV E7 inhibits cell pyroptosis by promoting TRIM21-mediated degradation and ubiquitination of the IFI16 inflammasome. Int. J. Biol. Sci. 16, 2924–2937 (2020).
An, Q., Fan, C. H. & Xu, S. M. Recent perspectives of pediatric leukemia—an update. Eur. Rev. Med. Pharm. Sci. 21, 31–36 (2017).
Berg, S. & Nand, S. Neurological complications of the leukemias across the ages. Curr. Neurol. Neurosci. Rep. 17, 13 (2017).
Juliusson, G. & Hough, R. Leukemia. Prog. Tumor Res. 43, 87–100 (2016).
Wayne, A. S., Fitzgerald, D. J., Kreitman, R. J. & Pastan, I. Immunotoxins for leukemia. Blood 123, 2470–2477 (2014).
Gonzalez-Herrero, I. et al. The making of leukemia. Int. J. Mol. Sci. 19, 1494 (2018).
Schiffer, C. A. Leukemia. Curr. Opin. Oncol. 8, 1–2 (1996).
Schroder, N. W., Eckert, J., Stubs, G. & Schumann, R. R. Immune responses induced by spirochetal outer membrane lipoproteins and glycolipids. Immunobiology 213, 329–340 (2008).
Luo, X. et al. The outer membrane protein Tp92 of Treponema pallidum induces human mononuclear cell death and IL-8 secretion. J. Cell Mol. Med. 22, 6039–6054 (2018).
Gao, F. et al. Magnesium sulfate provides neuroprotection in lipopolysaccharide-activated primary microglia by inhibiting NF-kappaB pathway. J. Surg. Res. 184, 944–950 (2013).
Song, W. J. & Chang, Y. S. Magnesium sulfate for acute asthma in adults: a systematic literature review. Asia Pac. Allergy 2, 76–85 (2012).
Suzuki-Kakisaka, H. et al. Magnesium sulfate increases intracellular magnesium reducing inflammatory cytokine release in neonates. Am. J. Reprod. Immunol. 70, 213–220 (2013).
Sugimoto, J. et al. Magnesium decreases inflammatory cytokine production: a novel innate immunomodulatory mechanism. J. Immunol. 188, 6338–6346 (2012).
Chang, Y. Y. et al. Effects of MgSO(4) on inhibiting Nod-like receptor protein 3 inflammasome involve decreasing intracellular calcium. J. Surg. Res. 221, 257–265 (2018).
Johnson, D. C. et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med. 24, 1151–1156 (2018).
Razmara, M. et al. CARD-8 protein, a new CARD family member that regulates caspase-1 activation and apoptosis. J. Biol. Chem. 277, 13952–13958 (2002).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Okondo, M. C. et al. Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem. Biol. 25, 262–267.e5 (2018).
Zhong, F. L. et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem. 293, 18864–18878 (2018).
de Vasconcelos, N. M. et al. DPP8/DPP9 inhibition elicits canonical Nlrp1b inflammasome hallmarks in murine macrophages. Life Sci. Alliance 2, e201900313 (2019).
Chui, A. J. et al. N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85 (2019).
Fenini, G., Karakaya, T., Hennig, P., Di Filippo, M. & Beer, H. D. The NLRP1 inflammasome in human skin and beyond. Int. J. Mol. Sci. 21, 4788 (2020).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Maude, S. L. et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018).
Sadelain, M., Riviere, I. & Riddell, S. Therapeutic T cell engineering. Nature 545, 423–431 (2017).
Yang, W. et al. Pyridoxine induces monocyte-macrophages death as specific treatment of acute myeloid leukemia. Cancer Lett. 492, 96–105 (2020).
Wang, Q. et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426 (2020).
Wu, M. et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine 41, 244–255 (2019).
Molina-Crespo, A. et al. Intracellular delivery of an antibody targeting gasdermin-B reduces HER2 breast cancer aggressiveness. Clin. Cancer Res. 25, 4846–4858 (2019).
Kesavardhana, S., Malireddi, R. & Kanneganti, T. D. Caspases in cell death, inflammation, and pyroptosis. Annu Rev. Immunol. 38, 567–595 (2020).
Karki, R., Man, S. M. & Kanneganti, T. D. Inflammasomes and cancer. Cancer Immunol. Res. 5, 94–99 (2017).
Karki, R. & Kanneganti, T. D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 19, 197–214 (2019).
Christgen, S. & Kanneganti, T. D. Inflammasomes and the fine line between defense and disease. Curr. Opin. Immunol. 62, 39–44 (2020).
Ahmad, I. et al. Thymoquinone suppresses metastasis of melanoma cells by inhibition of NLRP3 inflammasome. Toxicol. Appl Pharm. 270, 70–76 (2013).
Huang, C. F. et al. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 36, 116 (2017).
Youm, Y. H. et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269 (2015).
Weichand, B. et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J. Exp. Med. 214, 2695–2713 (2017).
Feng, X. et al. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 36, 81 (2017).
Juliana, C. et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 285, 9792–9802 (2010).
Chen, L. et al. BAY 11-7082, a nuclear factor-κB inhibitor, induces apoptosis and S phase arrest in gastric cancer cells. J. Gastroenterol. 49, 864–874 (2014).
Drexler, S. K. et al. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc. Natl Acad. Sci. USA 109, 18384–18389 (2012).
Zheng, Q. et al. Reactive oxygen species activated NLRP3 inflammasomes initiate inflammation in hyperosmolarity stressed human corneal epithelial cells and environment-induced dry eye patients. Exp. Eye Res. 134, 133–140 (2015).
Chen, L. C. et al. Tumour inflammasome-derived IL-1beta recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma. EMBO Mol. Med. 4, 1276–1293 (2012).
Kondo, Y. et al. Overexpression of the DNA sensor proteins, absent in melanoma 2 and interferon-inducible 16, contributes to tumorigenesis of oral squamous cell carcinoma with p53 inactivation. Cancer Sci. 103, 782–790 (2012).
Hu, B. et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl Acad. Sci. USA 110, 9862–9867 (2013).
Keller, M., Sollberger, G. & Beer, H. D. Thalidomide inhibits activation of caspase-1. J. Immunol. 183, 5593–5599 (2009).
Zeng, Q. et al. Caspase-1 from human myeloid-derived suppressor cells can promote T cell–independent tumor proliferation. Cancer Immunol. Res. 6, 566–577 (2018).
Wannamaker, W. et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoy l)-pyrrolidine-2-carboxylic acid ((2 R,3 S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J. Pharmacol. Exp. Ther. 321, 509–516 (2007).
Zheng, S. Q., Hong, X. D., Chen, T. S., Luo, P. F. & Xiao, S. C. [Effects of caspase-1 inhibitor VX765 on cold-restraint stress-induced acute gastric ulcer in mice]. Zhonghua Shao Shang Za Zhi 33, 688–693 (2017).
Kast, R. E. Ritonavir and disulfiram may be synergistic in lowering active interleukin-18 levels in acute pancreatitis, and thereby hasten recovery. JOP 9, 350–353 (2008).
Wang, F. et al. Alcohol accumulation promotes esophagitis via pyroptosis activation. Int. J. Biol. Sci. 14, 1245–1255 (2018).
Lin, W. et al. IFI16 restoration in hepatocellular carcinoma induces tumour inhibition via activation of p53 signals and inflammasome. Cell Prolif. 50, e12392 (2017).
Yu, X. & He, S. GSDME as an executioner of chemotherapy-induced cell death. Sci. China Life Sci. 60, 1291–1294 (2017).
Hu, B. et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768 (2016).
Liu, Y. et al. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 8, e2579–e2579 (2017).
Wu, D. et al. Protective effects of flagellin A N/C against radiation-induced NLR pyrin domain containing 3 inflammasome-dependent pyroptosis in intestinal cells. Int J. Radiat. Oncol. Biol. Phys. 101, 107–117 (2018).
Acknowledgements
This work was financially supported by Grant No. 81773341, 82073458, 81830096 from the National Natural Science Foundation; the Major Projects of International Cooperation and Exchanges NSFC Grant No. 81620108024, the Program of Introducing Talents of Discipline to Universities (111 Project, No. B20017), Scientific Research and Innovation Project of postgraduates in Hunan Province Grant No. 1053320192834.
Author information
Authors and Affiliations
Contributions
P.Y. conceived the structure of manuscript and wrote the manuscript; C.P. and X.C. conceived the structure of manuscript; X.Z., N.L., and L.T. collected materials. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, P., Zhang, X., Liu, N. et al. Pyroptosis: mechanisms and diseases. Sig Transduct Target Ther 6, 128 (2021). https://doi.org/10.1038/s41392-021-00507-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00507-5
