
Abstract
Cerebral palsy (CP) is a heterogeneous neurodevelopmental disorder that causes movement and postural disabilities. Recent research studies focused on genetic diagnosis in patients with CP of unknown etiology. The present study was carried out in 20 families with one family member affected with idiopathic CP. Chromosomal microarray and exome sequencing techniques were performed in all patients. Chromosomal microarray analysis did not show any pathological or probable pathological structural variant. However, the next-generation sequencing study showed a high diagnostic yield. We report 11/20 patients (55%) with different pathogenic or potentially pathogenic variants detected by exome sequencing analysis: five patients with mutations in genes related to hereditary spastic paraplegia, two with mutations in genes related to Aicardi–Goutières syndrome, three with mutations in genes related to developmental/epileptic encephalopathies, and one with a mutation in the PGK1 gene. The accurate and precise patients’ selection, the use of a high-throughput genetic platform, the selection of adequate target genes, and the application of rigorous criteria for the clinical interpretation are the most important elements for a good diagnostic performance. Based on our findings, next-generation sequencing should be considered in patients with cryptogenic CP as the first line of genetic workup.
Impact
-
Sequencing techniques in CP of uncertain etiology provides a diagnostic yield of 55%.
-
The appropriate selection of cases optimizes the diagnostic yield.
-
NGS facilitate better understanding of new phenotypes of certain genetic diseases.
Similar content being viewed by others


Introduction
Cerebral palsy (CP) describes a group of permanent disorders of movement and posture development, causing mobility limitation attributed to non-progressive disturbances that occurred in the developing fetal or infant brain. The motor disorders of CP are often accompanied by disturbances of sensation, perception, cognition, communication and behavior, epilepsy, and secondary musculoskeletal problems.1 CP is a heterogeneous condition in terms of etiology as well as in types and severity of impairments. Hypoxic–ischemic encephalopathy, periventricular leukomalacia, brain malformations, intrauterine infections, and neonatal strokes are the most frequent causes of CP; however, up to 20% of patients with CP do not have an etiological diagnosis. This group of patients can be considered to have idiopathic or cryptogenic CP.2 On the other hand, metabolic studies carried out in those patients with unknown etiology have had very low performance.3 Genetic forms of CP account for ~2% in European populations,4 but recent research studies have suggested that genetic variants contribute to CP more than expected. Before the next-generation sequencing (NGS) era, a few studies of single-gene causes of CP involving families with two or more members affected by the disorder were performed. These initial studies led to the identification of mutations in KANK1, AP4M1, and GAD1 genes. Recent studies with high-throughput technologies reported that at least 7% of cases might carry a copy number variation (CNV) of clinical significance and 14% of CP cases had a plausible genetic mutation.5,6 However, similar studies performed by other groups show an obvious disparity in genetic diagnostic yields dependent on the sample sizes and the clinical selection criteria used.7,8,9,10
Genetic and phenotypic heterogeneity may underlie in CP and is difficult for etiological diagnosis. High-throughput technologies such as NGS have made significant advances in the knowledge of genetic causes of CP. In this study, we present the results from our cohort of 20 patients with idiopathic CP after genomic array and next-generation exome sequencing study.
Methods
Subjects
The study was carried out in patients with idiopathic CP. Among 20 families, one family member affected with idiopathic CP was recruited according to the diagnostic criteria established by Rosenbaum et al.1 A brief clinical description of this cohort can be seen in Table 1.
This study was approved by the local ethics committee of the Hospital Universitario y Politécnico La Fe (Valencia, Spain). Informed consent was obtained from all participants.
Clinical features of case series
All patients were evaluated by the same neuropediatrician. Demographic and clinical data were collected: sex, age, details of pregnancy, birth, perinatal and clinical evolution, and magnetic resonance imaging (MRI) study. A physical examination and clinical evaluation were undertaken for each patient, recording the following variables: type of CP (spastic, dyskinetic, ataxic, mixed), functional classification on Gross Motor Function Classification System (GMFS), and the presence of intellectual disability (ID) or epilepsy and other associated conditions such a polyneuropathy or autism.
The inclusion criteria were the presence of clinical findings compatible with CP definition and the absence of the following exclusion criteria: polymalformative syndrome, ataxic CP, progressive encephalopathy, and neuroradiological findings compatible with hypoxic–ischemic encephalopathy, periventricular leukomalacia, cerebral malformation, or leukoencephalopathy. We have excluded ataxic CP because ataxic forms of CP may be hard to distinguish from progressive cerebral disorders.11
Genetic studies
Genomic DNA from peripheral blood samples from individuals and their parents were isolated using the QIAsymphony extractor (Qiagen, Hilden, Germany). DNA purity and concentration were measured using standard procedures in our laboratory.
Chromosomal microarray (CMA) and exome sequencing were performed in all 20 patients. Whole-genome dosage analysis was performed using a genomic array (SNPs Affymetrix CytoScan® 750). Array hybridization and scanning were performed following the manufacturer’s specifications. The data were analyzed using the DNA analytics ChAS software (Chromosome Analysis Suite of Affymetrix). For most patients, a commercial clinical exome (SureSelect Focused Exome from Agilent Technologies) was used for exome sequencing 6110 genes described in different databases as disease-causing genes. Patients 2, 10, 11, 12, and 18 were sequenced using SureSelect Clinical Research Exome to analyze the coding sequences of all known genes in a trio-based study. The libraries were sequenced on an Illumina NextSeq 500 following the manufacturer’s protocol to get a minimum reading depth of 100×. Sequence read alignments, variant detection, and annotation were performed in Unidad de Genómica-IIS La Fe with its own bioinformatics pipeline. To evaluate the clinical impact and to assess the pathogenicity of variants in exome sequencing, we applied the criteria described previously and the standards and guidelines of the Laboratory Quality Assurance Committee (ACMG).12,13 All relevant genetic variants (potential novel and rare variants) detected in the patients as well as the studies in the DNA of their parents were performed by Sanger sequencing (Supplementary Sanger results (online)) (primers and PCR conditions are available on request).
Results
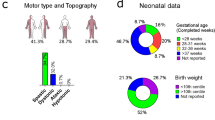
Twenty patients with idiopathic CP have been included, 9 females (45%) and 11 males (55%). None of the patients had a family history of CP or a pre- or perinatal history of interest. The mean age at the time of the study was 8.6 years. Regarding the type of CP, 11 cases (55%) were spastic tetraparesis (ST), 7 (35%) spastic diplegia (SD), and 2 (10%) mixed. Motor assessment using the GMFS scale gave the following results: 5 patients (25%) presented a grade I, 3 (15%) grade II, 1 (5%) grade III, 5 (25%) grade IV, and 6 (30%) grade V. In relation to comorbidities, 8 children (40%) were associated with epilepsy, 5 (25%) presented a severe degree of ID, 5 (25%) a moderate degree, 3 (15%) a mild degree, and 7 (35%) had no ID. Three patients had axonal polyneuropathy and one had autism. Clinical features are summarized in Table 1.
Genomic microarray and exome sequencing were performed in all patients. CMA analysis did not show any pathological or probable pathological copy number variant. On the other hand, the NGS study showed a high diagnostic yield. Pathogenic or probably pathogenic variants were detected by exome sequencing analysis in 55% (11/20) of the patients (Table 2). Our genetic results can be grouped as follows: five patients with mutations in genes related to hereditary spastic paraplegia (HSP), two with mutations in genes related to Aicardi–Goutières syndrome, three with mutations in genes related to developmental/epileptic encephalopathies, and one with mutation in the PGK1 gene, an X-linked recessive condition with a highly variable clinical phenotype where the predominant described feature is not neurological.
Most of the diagnostic variants (7/11) were autosomal dominant and occurred de novo in the patients. In one case, the male patient inherited the X-linked mutation in PGK1 gene from her healthy mother (patient 8). The pathogenic role of the novel PGK1 variant detected was confirmed by an enzymatic study showing low levels of phosphoglycerate kinase. In this patient, the phenotype consisted of SD and normal cognitive level. We believe that diplegia is a neurological manifestation of phosphoglycerate kinase deficiency, not previously described in a disease in which phenotypic variability is the rule. The remaining three cases showed an autosomal-recessive inheritance, where both parents are heterozygous carriers of the pathogenic variants. Although the father of patient 14 was not available for study, only one of the two variants in SPATA5 gene was detected in the mother, so that a paternal inheritance of the other is presumed. In this patient, spasticity was severely affecting his quality of life, requiring the installation of a baclofen pump.
Pathogenic variants have been identified in different genes, except for the ATL1 gene, where three different de novo mutations were found in patients 5, 11, and 18. Patient 5 had typical spastic paraplegia with an early-onset of symptoms, while the other two (cases 11 and 18) had a sensory–motor neuropathy with symptoms associated with an extremely disabling early-onset ST with a GMFS of V. In silico genetic studies suggest that the mutations detected in patients 11 and 18 could impede the tetrameric formation of the active protein, causing considerable loss of function. The in silico study was carried out using the HOPE bioinformatics tool (https://www3.cmbi.umcn.nl/hope/). While the mutation detected in case 5 leads to a decrease in the GTPase activity of the protein and would not affect its function so drastically.14 These different mechanisms illustrate that not all variants in the same gene produce the same phenotype or the same severity in their manifestations.
The 13 different variants are not recurrent and most are missense variants (11/13). None of the dominant or X-linked variants was reported in control databases (GnomAD, Exome Aggregation Consortium), while the recessive variants appear with allele frequencies of 0.1% (p.Ala177Thr in RNASEH2B) or less. Those variants found in patients 2, 11, 14, 15, 17, 18, and 20 were previously reported as likely pathogenic or pathogenic in the ClinVar database, while the variants detected in patients 4 and 5 were reported as pathogenic in the HGMD database. The remaining variants have not been previously described, but the in silico predictions (mutation taster, SIFT, Provean, and Polyphen) classify them as pathogenic.
We have found differences between the group with a genetic diagnosis and the group of idiopathic cerebral palsy with no genetic diagnosis (Table 3). In the first group, spastic tetraparesis is more frequent as a type of CP, has a higher frequency of epilepsy, and greater motor impairment.
Discussion
There are very few studies published to determine the prevalence and characteristics of CNVs and/or point mutations in children with CP of unknown etiology.10,15,16,17 McMichael et al.10 studied children with CP in 50 Caucasian families using two microarray designs. They found 14 CNVs of potential pathogenic relevance (20%), but most were inherited from an unaffected parent (11/14), suggesting another genetic or environmental contributing factor. Segel et al.15 found 19% pathogenic CNVs in patients with cryptogenic CP. The majority of these CNVs were de novo and of large size (median 3 Mb). However, these CNVs were present in patients with dysmorphism and non-motor comorbidities, especially ID. These features are present in many unspecific neurodevelopmental disorders, but none are included in the concept of idiopathic CP. A systematic study carried out by Oskoui et al.5 determined a 7% de novo CNV rate in a CP cohort, but they included an unselected series of children, such as a patient with Wolf–Hirschhorn syndrome. On the other hand, by whole-exome sequencing, McMichael et al.6 found that 14% of the 98 case-parent trios had variants that were putatively disease causing in five known pathogenic genes (KDM5C, SCN8A, TUBA1A, L1CAM, and PAK3) and eight novel candidate genes (TENM1, AGAP1, CD99L2, WIPI2, MAST1, JHDM1D, NAA35, and RFX2).6 In recent years, genetic studies performed by whole-exome sequencing analysis have identified a greater number of patients with CP and genetic mutations. Takezawa et al.16 identified pathogenic/likely pathogenic variants in 9 out of 17 cases (52.9%) and no pathogenic CNVs were identified in a subgroup of CP patients selected by two simple criteria: (i) gestational age of 37 weeks or more and (ii) normal or nonspecific brain MRI findings. In the study of a Greek tertiary care center published in 2019,17 a group of 47 patients with a clinical picture regarded as CP were included. They found the underlying genetic molecular diagnosis in 23 patients (49%). In this regard, our results constitute one of the first publications in a series of idiopathic CP in which no CNV was found through the array technology and with the highest detection rate of NGS studies (11/20 or 55% of cases with a genetic diagnosis).
We identified candidate pathogenic variants of three HSP-related genes in five patients: AP4B1 (case 1), SPAST (case 4), and ATL1 (case 5, 11 and 18), which represents 45% of the total number of cases with a genetic diagnosis and 25% of the total series. HSP is a very heterogeneous group of neurodegenerative disorders characterized by progressive spasticity and weakness of the lower limbs. Traditionally, it is classified as either the pure or the complicated form, depending on the presence of additional neurological features.18 The mean age at onset of symptoms in HSP is 18.9 years old. HSP is genetically very heterogeneous, with at least 77 different loci and 60 associated genes. Our cases were non-progressive and the onset of symptoms was in the infancy. SPAST and ATL1 mutations are the most frequently mutated genes identified in HSP.19
Distinguishing between genetic forms of CP and HSP can be clinically very difficult, especially as new expanding phenotypes of both are continuously being described. This can be seen in AP4M1 mutations, which cause both CP (OMIM#603513) and HSP (OMIM #614066). In the future, boundaries between genetic CP and HSP will probably become more subtle, and therefore the term “CP spectrum,” already described by different authors, could be used more widely. We have identified two patients with mutations in genes previously related to Aicardi–Goutières syndrome, RNASEH2B (case 20) and IFHI (case 2) genes, which correspond to subtypes 2 and 7, respectively, of the genetic classification of this syndrome. The classic form of the syndrome described by Jean Aicardi and Françoise Goutières is characterized by early-onset encephalopathy, progressive course, and basal ganglia calcification.20 However, in recent years, atypical phenotypes have been described, including the absence of basal ganglia calcification21 or late onset.22 The RNASEH2B-mutated patient presented a non-progressive course with white matter abnormalities, but no calcifications. The patient with IFHI mutation apparently presented a progression during the first year of life, but when we assessed him, the patient presented a static picture, with SD and calcifications appeared posteriorly when he was 5 years old. The late onset of calcifications has been described in other cases and is a reason for delayed diagnosis.22 Therefore, both cases were atypical and the contribution of NGS to the diagnosis has been essential.
Patient with GNB1 mutation (case 17) had a phenotype similar to those described in the literature.23 Motor involvement had a mixed component, with dyskinesia in the arms and spasticity in the legs. The patient had severe ID and growth retardation. She did not have epilepsy, despite the fact that up to 50% of patients with mutations in this gene have genitourinary nor gastrointestinal alterations.
In case 15, a pathogenic mutation was found in the GNAO1 gene. Mutations in this gene present two different phenotypes, depending on the type of mutation found: early epileptic encephalopathy (Ohtahara syndrome-like) and movement disorder mainly manifested with chorea or dystonia, but without seizures. In all cases, there is an evident neurodevelopmental delay.24 Although it is true that our patient indeed had seizures, these appeared at 5 years of age and were controlled with antiepileptic treatment; therefore, it cannot be classified as early epileptic encephalopathy. In this case, tetraparesis together with ID were the most prominent features of his phenotype. It is, therefore, a novel phenotypic variant not yet described in GNAO1 mutation. An important aspect when evaluating our results and those of other similar studies is the definition of CP, which is very unspecific. CP is defined as a group of permanent disorders of movement and posture development, causing mobility limitations attributed to non-progressive disturbances that occurred in the developing fetal or infant brain.1 It is, therefore, an umbrella term and includes a heterogeneous group of disorders defined by the clinical description and not by etiology or pathology.
The first problem that we encountered is that the definition of CP is too broad and a heterogeneous number of processes can be included, which hinders the comparison between series. For instance, we have excluded polymalformative syndromes because we have focused our research in patients with unidentified etiology and we think patients with polymalformative syndromes constitute a different scenario. However, a panel of international CP register and surveillance researchers advise the inclusion of these syndromes within the definition of CP, with genetic origin or not, as long as they meet the criteria for this definition.11
Another complicated aspect of the syndromic diagnosis of CP is the exclusion criteria of progressive disease. In this sense, the aforementioned panel of experts recommends including patients who may have experienced a regressive pattern in early ages and since then behave like static encephalopathy. This is the case of the two patients previously discussed with Aicardi–Goutières syndrome who were included in our series.
At this moment, there is a wide debate about the relevance of including patients with a found genetic mutation in the CP definition, taking into account that with the development of NGS technologies, the number of these patients will increase.25 Thus, should we reserve the term CP only for those patients with a perinatal known etiology as hypoxic–ischemic encephalopathy or periventricular leukomalacia, or should we consider the diagnosis of CP to be a syndromic diagnosis and maintained whether or not a genetic diagnosis is established? Different terms have been proposed for those cases of CP in which a genetic diagnosis has been made: CP mimics,26 masqueraders of CP,16 or even some authors propose using the term CP spectrum disorders27 for the whole range of CP regardless of the etiological diagnosis.
The accurate and precise patients’ selection, the use of a high-throughput genetic platform, the selection of adequate target genes, and the application of rigorous criteria for the clinical interpretation of the variants found are the strengths of this study. Based on our findings, NGS should be considered in all patients with cryptogenic CP as the first line of workup at genetic diagnosis.
It has to be noted that our study has several limitations, including the relatively small number of patients and the heterogeneity in the genetic results detected. However, it reflects real-life clinical practice in a large tertiary referral center and in the complex clinical and genetic diagnosis of the cerebral palsy.
References
Rosenbaum, P., Paneth, N., Leviton, A., Goldstein, M. & Bax, M. A report: the definition and classification of cerebral palsy April 2006. Dev. Med. Child Neurol. Suppl. 109, 8–14 (2007).
Majnemer, A. & Shevell, M. Diagnostic yield of the neurologic assessment of the developmentally delayed child. J. Pediatr. 127, 193–199 (1995).
Leonard, J. et al. Should children with cerebral palsy and normal imaging undergo testing for inherited metabolic disorders? Dev. Med. Child Neurol. 53, 226–232 (2011).
Rajab, A. et al. An autosomal recessive form of spastic cerebral palsy with microcephaly and mental retardation. Am. J. Med. Genet. 140, 1504–1510 (2006).
Oskoui, M. et al. Clinically relevant copy number variations detected in cerebral palsy. Nat. Commun. 6, 1–7 (2015).
McMichael, G. et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry 20, 176–182 (2015).
Srivastava, S. et al. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 76, 473–483 (2014).
Parolin Schenkenberg, R. et al. De novo point mutations in patients diagnosed with ataxic cerebral palsy. Brain 138, 1917–1932 (2015).
Zarrei, M. et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet. Med. 20, 172–180 (2017).
McMichael, G. et al. Rare copy number variation in cerebral palsy. Eur. J. Hum. Genet. 22, 40–45 (2014).
Smithers-Sheedy, H. et al. What constitutes cerebral palsy in the twenty-first century? Dev. Med. Child Neurol. 56, 323–328 (2014).
Martinez, F. et al. High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J. Med. Genet. 54, 87–92 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Bian, X. et al. Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl Acad. Sci. USA 108, 3976–3981 (2011).
Segel, R. et al. Copy number variations in cryptogenic cerebral palsy. Neurology 84, 1660–1668 (2015).
Takezawa, Y. et al. Genomic analysis identifies masqueraders of full-term cerebral palsy. Ann. Clin. Transl. Neurol. 5, 538–551 (2018).
Zouvelou, V. et al. The genetic etiology in cerebral palsy mimics: the results from a Greek tertiary care center. Eur. J. Paediatr. Neurol. 23, 427–437 (2019).
Lo Giudice, T., Lombardi, F., Santorelli, F., Kawarai, T. & Orlacchio, A. Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp. Neurol. 261, 518–539 (2014).
Chrestian, N. et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol. Genet. 3, 122 (2016).
Crow, Y. Aicardi-Goutières syndrome. Handb. Clin. Neurol. 113, 1629–1635 (2013).
Tonduti, D. et al. Spontaneous MRI improvement and absence of cerebral calcification in Aicardi-Goutières syndrome: diagnostic and disease-monitoring implications. Mol. Genet. Metab. 126, 489–494 (2019).
Svingen, L. et al. Late diagnosis and atypical brain imaging of Aicardi-Goutières syndrome: are we failing to diagnose Aicardi-Goutières syndrome-2? Dev. Med. Child Neurol. 59, 1307–1311 (2017).
Hemati, P. et al. Refining the phenotype associated with GNB1 mutations: clinical data on 18 newly identified patients and review of the literatura. Am. J. Med. Genet. A 176, 2259–2275 (2018).
Feng, H. et al. Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology 89, 762–770 (2017).
Van Eyk, C., Corbett, M. & Maclennan, A. The emerging genetic landscape of cerebral palsy. Handb. Clin. Neurol. 147, 331–342 (2018).
Pearson, T., Pons, R., Ghaoui, R. & Sue, C. Genetic mimics of cerebral palsy. Mov. Disord. 34, 625–636 (2019).
Moreno-De-Luca, A., Ledbetter, D. & Martin, C. Genetic insights into the causes and clasification of cerebral palsies. Lancet Neurol. 11, 283–292 (2012).
Acknowledgements
This study was supported by grant PI14/00350 (Instituto de Salud Carlos III-Acción Estratégica en Salud 2013-2016; FEDER-Fondo Europeo de Desarrollo Regional). A.C.-L. is supported by a research grant by Fundación Mutua Madrileña (held by S.M.). A.V.M.-H. holds a “Río Ortega” fellowship (reference CM19/00181) funded by the Instituto de Salud Carlos III (ISCIII, Madrid, Spain). We warmly thank all patients and their families for their implication in this study.
Author information
Authors and Affiliations
Contributions
M.R.: responsible for carrying out and interpreting the studies of chromosomal microarray analysis and NGS; substantial contribution to research design, analysis and interpretation of data and drafting the article. A.C.-L.: helped in the carrying out and interpreting the studies of chromosomal microarray analysis and NGS. C.O.: contributed to the research design and analysis and interpretation of data and has revised the paper critically. S.O.: helped in the carrying out and interpreting the studies of chromosomal microarray analysis and NGS. M.A.-A.: revised the paper critically. A.V.M.-H.: contributed to the medical history assessment. S.M.: contributed to the research design and revised the paper. L.P.: helped in carrying out and interpreting the studies of chromosomal microarray analysis and NGS. F.M.: contributed to the research design and analysis, interpretation of data and revised the paper critically. M.T.: responsible for patient selection, as well as for the phenotypic description and medical history assessment; substantial contribution to research design and drafting the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Informed consent
Informed consent was obtained from all participants for the genetic studies. Specific patient consent was not required for the publication of this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Rosello, M., Caro-Llopis, A., Orellana, C. et al. Hidden etiology of cerebral palsy: genetic and clinical heterogeneity and efficient diagnosis by next-generation sequencing. Pediatr Res 90, 284–288 (2021). https://doi.org/10.1038/s41390-020-01250-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-01250-3
This article is cited by
-
Comprehensive whole-genome sequence analyses provide insights into the genomic architecture of cerebral palsy
Nature Genetics (2024)
-
Redefining cerebral palsies as a diverse group of neurodevelopmental disorders with genetic aetiology
Nature Reviews Neurology (2023)
-
Integrative Multi-Omics Research in Cerebral Palsy: Current Progress and Future Prospects
Neurochemical Research (2023)
