
Abstract
The latest study shows that gastric cancer (GC) ranked the fifth most common cancer (5.6%) with over 1 million estimated new cases annually and the fourth most common cause of cancer death (7.7%) globally in 2020. Metastasis is the leading cause of GC treatment failure. Therefore, clarifying the regulatory mechanisms for GC metastatic process is necessary. In the current study, we discovered that calreticulin (CALR) was highly expressed in GC tissues and related to lymph node metastasis and patient’s terrible prognosis. The introduction of CALR dramatically promoted GC cell migration in vitro and in vivo, while the repression of CALR got the opposite effects. Cell migration is a functional consequence of the epithelial-mesenchymal transition (EMT) and is related to adhesion of cells. Additionally, we observed that CALR inhibition or overexpression regulated the expression of EMT markers (E-cadherin, ZO-1, Snail, N-cadherin, and ZEB1) and cellular adhesive moleculars (Fibronectin, integrin β1and MMP2). Mechanistically, our data indicated that CALR could mediate DNA methylation of E-cadherin promoter by interacting with G9a, a major euchromatin methyltransferase responsible for methylation of histone H3 on lysine 9(H3K9me2) and recruiting G9a to the E-cadherin promoter. Knockdown of G9a in CALR overexpressing models restored E-cadherin expression and blocked the stimulatory effects of CALR on GC cell migration. Taken together, these findings not only reveal critical roles of CALR medicated GC metastasis but also provide novel treatment strategies for GC.
Similar content being viewed by others


Introduction
Gastric cancer (GC) ranked the fifth most common cancer (5.6%) with over 1 million estimated new cases annually and the fourth most common cause of cancer death (7.7%) globally in 2020 [1, 2]. In 185 countries, the incidence rates of Eastern Asia and Eastern Europe are highest, and mortality rates remain highest in several South Central Asian countries, including Iran, Afghanistan, Turkmenistan, and Kyrgyzstan, especially in men. Although mortality rates for GC slightly decreased in recent years, clinicians expect to see more GC cases in the future due to aging populations and economic development. In virtue of its frequently advanced stage at diagnosis, high molecular and phenotypical heterogeneity and metastasis as the main causes of GC death, efforts to clarify the molecular mechanisms of GC development and progression and find the potential early detection markers contribute to the precise treatment of GC patients and prolong the overall survival rate.
Calreticulin (CALR), also called CRT, locates in the p13.2–p13.3 region of human chromosome 19 and is a highly conserved chaperone protein that resides primarily in the endoplasmic reticulum [3] and is involved in a spectrum of different physiological and pathological roles, among them, cell adhesion. Additionally, it functions in protein folding quality control and maintenance of calcium homeostasis [4]. CALR is also found in the nucleus, implying that it may have a role in transcriptional activities and gene expression regulation [5]. Because of its multifunction, previous studies have shown a contributing role for CALR in various diseases, including neurodegenerative problems, cancers, autoimmune diseases, and wound healing [6,7,8,9]. In addition, new roles of CALR as possible diagnostic markers in blood or urine have emerged due to its secretion [10,11,12]. In gastric cancer, some researchers found that CALR overexpression was associated with GC invasion, metastasis, lymph node metastasis, angiogenesis, and GC patients’ survival [13]. However, the mechanisms that how CALR mediates GC metastasis remain unclarified, and delineating the mechanism and determining the probable interacting genes and signaling pathways contribute to GC precise targeted therapy. Besides, exploring the diagnostic value of serum CALR is also beneficial to discover novel and specific biomarkers for GC.
Metastasis is the leading cause of GC patients’ death, which involves many processes, including cell adhesion, migration, and invasion. Among them, the EMT process, whose hallmark is the absence of E-cadherin, Integrin β1(ITGB1), and Fibronectin (FN1), plays crucial roles [14,15,16]. Previous studies revealed that CALR enhanced EGF-induced EMT in pancreatic cancer cells via Integrin/EGFR-ERK/MAPK signaling pathway [17] and repressed E-cadherin expression via Snail (SNAI1)/Slug in Madin-Darby canine kidney cells or mouse embryonic stem cells [18, 19].
Intriguingly, in this study, we found that CALR overexpression significantly promoted GC cells migration in vitro and in vivo, while the CALR-mediated effect could be abrogated by blocking G9a (also called EHMT2, euchromatic histone lysine methyltransferase 2) expression in CALR overexpressing model. Mechanistically, we observed that CALR overexpression or suppression significantly regulated the expression of EMT marker (E-cadherin, ZO-1, Snail, N-cadherin and ZEB1) and cellular adhesive molecular (Fibronectin, integrin β1and MMP2). Besides, we demonstrated that CALR interacted with G9a and mediated DNA methylation of E-cadherin promoter, which resulted in suppression of E-cadherin expression. Additionally, we also discovered that serum CALR was better than traditional diagnostic markers for GC detection. Together, our findings expounded the effects of CALR on GC metastasis in detail and provided novel treatment strategies for GC.
Results
CALR is highly expressed in GC tissues and correlated with GC lymph node metastasis and poor prognosis
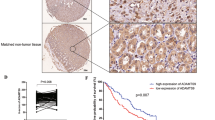
First, our data revealed that CALR protein and mRNA levels were significantly upregulated in GC tissues compared with matched adjacent normal tissues by IHC and qRT-PCR assays. Moreover, the intensity of CALR staining in tumor tissues increased with invasion depth and metastatic lymph node (Fig. 1A–C). These results were consistent with data from the TCGA database (Supplementary Fig. 1A). And then, we analyzed the relationship between CALR expression and the clinical features, and we found that high expression of CALR correlated with GC patients’ TNM stage, distant metastasis, histological differentiation, and tumor size (Fig. 1D, E, Table 1). Furthermore, we also determined that the increased expression of CALR was related to GC grade, postoperative recurrence, and tumor involvement of lymphatic vessels (Supplementary Fig. 1B–E) by analyzing Oncomine data (https://www.oncomine.org/), and overall survival analysis showed a worse survival rate in the GC patients with high CALR expression by Kaplan–Meier Plotter analysis (Fig. 1F) (http://kmplot.com/). These results coincided with Chiung-Nien Chen’s report [13]. Nevertheless, CALR was a secreted protein, so we also examined CALR expression in GC patients’ serum for further exploring its diagnostic values for GC. As a result, receiver operating characteristic (ROC) curve analysis revealed a high AUC value of 0.802 (95% CI, 0.701–0.902) in distinguishing GC from the healthy serums, significantly better than traditional diagnostic markers (CA19-9 and CEA) (Supplementary Fig. 1F). Additionally, we also explored the levels of CALR protein and mRNA in normal gastric mucosal epithelial cells (GES-1) and GC cell lines (SGC-7901, BGC-823, AGS, XN0422, and MGC-803). The results indicated that CALR was highly upregulated in GC cell lines (MGC-803, AGS, and XN0422) and downregulated in SGC-7901 cells compared with GES-1 (Fig. 1G, H). Taken together, the above results demonstrated that CALR expression is significantly increased in GC and is related to GC metastasis. The upregulation signature of CALR expression in GC patients’ serum may be a novel biomarker for the GC diagnosis.

A Representative IHC staining of CALR in GC tissues, normal tissues and metastatic lymph node (scale bar = 50 μm). B RT-qPCR data for CALR expression in 50 paired fresh GC and normal tissues. C Scores analyses of the IHC results; n = 129 vs 129. D, E RT-qPCR data for CALR levels by TNM stage (I + II vs. III + IV) and primary GC tissues with or without lymph node metastasis. F Kaplan–Meier analysis of 876 GC patients implyed that high CALR expression was associated with shorter overall survival. G, H the mRNA and protein level of CALR in normal gastric mucosal epithelial cells (GES-1) and the GC cells (SGC-7901, MGC-803, BGC-823, AGS and XN0422). The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
CALR enhances GC cell migration in vitro
Our clinical findings revealed that CALR expression was correlated with the depth of invasion and lymph node metastasis in GC specimens, implying that CALR might involve in the invasion and metastasis of GC. Based on the results shown in Fig. 1G, H, we chose GC cell lines AGS and MGC-803 for CALR inhibition assays and GC cell line SGC-7901 for establishing CALR stable overexpression model to explore the effects of CALR on GC cell migration; the transfection and infection efficiencies of CALR mRNA and protein level were shown in Supplementary Fig. 2A–E. As shown in Fig. 2A–D, compared with control groups, knockdown of CALR markedly inhibited the migratory capabilities of AGS and MGC-803, while an opposite result was observed in CLAR-overexpressing SGC-7901 cells (Fig. 2E, F). Together, these results indicate that the profiles of CALR influence the migration of GC cells in vitro.

A, B Transwell assays and (C and D) wound healing assays detecting migration abilities of GC cells transfected with CALR siRNAs or control in AGS and MGC-803 cells (scale bar = 100 μm). Wound healing was quantified by measurement of the average linear speed of movement of the wound edges. E, F Representative photomicrographs of transwell assays results for SGC-7901 cells infected with NC vector or CALR vector (scale bar = 100 μm). All of the experiments were performed three times. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
CALR promotes GC cell migration in vivo
Furthermore, to elucidate the role of CALR in GC development, we established the intraperitoneal metastasis models that stable CALR-knockdown MGC-803 cells, CALR overexpressing SGC-7901 cells, and their control cells were respectively injected into the intraperitoneal cavities of nude mice. After 28 days, the mice were sacrificed, and the results revealed that silencing CALR remarkably reduced the formation of metastatic nodules (Fig. 3A–C), whereas CALR overexpression remarkably elevated the number of metastatic nodules compared to the mice implanted with control cells. In summary, these results further support that CALR plays an important role in GC metastasis.

Representative images of the GC abdominal spread model after respectively intraperitoneal injection with GC cells stably knocking down CALR/NC vector (A and B, n = 6 vs 6) and stably overexpressing CALR/NC vector (D, n = 6 vs 6). C, E Evaluation of the number of tumor nodules in mice abdominal cavities. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
CALR promotes migration metastasis of GC cells in association with induction of EMT
To elaborate the mechanism by which the CALR induced GC cells migration, we conducted transcriptome sequencing of CALR stable overexpression model and its control cells. A total of 1522 changed genes (|log2 FoldChange | >0 and padj <0.05) were identified and included 721 upregulated genes and 801 downregulated genes (Supplementary Fig. 3A). KEGG Pathway Analysis revealed that overexpression of CALR remarkably affected cellular focal adhesion and pathways in cancer (Fig. 4A). More importantly, we observed that overexpression of CALR markedly promoted upregulation of EMT markers (Snail and ZEB1) and cell adhesion related molecules (MMP2, Fibronectin and integrin β1) (Fig. 4B). Besides, we still found the expression of CALR was positively related to Snail and negatively Zo-1 by analysis of TCGA data (Supplementary Fig. 3B, C). Increased cell migration is also a functional consequence of EMT [20], so we next examined changes in EMT-related markers by RT-qPCR and Western blot. Indeed, we observed that the inhibition of CALR dramatically decreased the mRNA and protein levels of N-cadherin and Snail and increased E-cadherin and ZO-1 mRNA and protein levels. The reverse results at mRNA and protein levels were observed by overexpression of CALR. The change of ZEB1 protein level was significant, while mRNA level had no difference when knocking down or overexpressing CALR in GC cells. In addition, there were no differences for other EMT markers (Vimentin and Slug) (Fig. 3C, D). Furthermore, according to the results of transcriptome sequencing, we also examined MMP2, Fibronectin, and integrin β1 protein expression, which played important roles in the adhesion of cells [21]. As expected, knockdown of CALR led to inhibition of MMP2, Fibronectin and integrin β1, while CALR overexpression conversely. Taken together, these results suggest that CALR enhances GC metastasis mainly by inducing EMT process and affecting cell adhesion.

A, B KEGG dot analysis of changed genes involved pathways in CALR overexpressing model. C RT-qPCR and (D) western blotting assays verified the results of validated EMT related markers screening by transcriptome sequencing. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
CALR induced EMT requires G9a-mediated H3K9 methylation
As the results above showed, CALR mediated EMT process in GC cells, while the implying mechanism remained unclear. Therefore, proteins that could bind to CALR were examined by IP and MS assays. The list of all potential target proteins for interacting with CALR was provided in Supplementary Table 1. Intriguingly, we noticed that CALR could interact with G9a, which was a euchromatin-associated methyltransferase responsible for mono- and dimethylation of H3K9, and was also recruited to the E-cadherin promoter by Snail and mediated promoter DNA methylation of E-cadherin and the suppression of E-cadherin expression [22]. To validate the interaction of CALR with G9a, we conducted IP assays and immunoprecipitated endogenous CALR in GC cells. In line with this finding of MS analysis, G9a was pulled down by CALR antibody. Besides, CALR was also combined with Snail (Fig. 5A). Similar results were obtained when we used G9a or Snail antibody to immunoprecipitate endogenous CALR (Fig. 5A). Because the interaction between G9a and Snail played a critical role in inducing H3K9me2 and resulting in the DNA methylation of the E-cadherin promoter, we examined methylation levels of H3K9 and E-cadherin promoter by Western Blot assay, DNA Methylation Experiment, and Pyrosequencing. We found that the levels of H3K9me2 and DNA methylation on the E-cadherin promoter were higher in CALR overexpressed SGC-7901 cell lines, while the induction was repressed in the absence of CALR in AGS and MGC-803 cell lines (Fig. 5B–D). To further verify that loss of E-cadherin expression was associated with elevated G9a in GC cells, we blocked G9a expression in CALR overexpressed SGC-7901 cell lines and the efficiency of blocking was shown in (Fig. 5E, F). As expected, knockdown of G9a eliminated the repressed effect of CALR overexpression on E-cadherin expression (Fig. 5G, H). Functionally, consistent result was obtained by transwell assays that knockdown of G9a counteracted acceleration of CALR overexpression on GC cell migration (Fig. 5I, J). These data reveal a cooperative role between CALR and G9a in the suppression of E-cadherin expression and induction of GC metastasis.

A The interactions among CALR, G9a and Snail were confirmed by immunoprecipitation. IgG was used as a negative control. B Western blot analysis of methyl-histone H3 (Lys9) in CALR overexpressing and knocking down models. C, D Methylation position analysis of E-cadherin promoter in CALR overexpressing and knocking down models via pyrosequencing. E–G RT-qPCR and (H) WB analysis the mRNA and protein level of CALR, G9a and E-cadherin in CALR overexpression models transfected with G9a siRNA or siControl. I, J Representative images of G9a siRNA counteracting the acceleration of CALR on GC cells migration in CALR overexpressing models.
Discussion
Over the past years, numerous studies have demonstrated that CALR participated in various cancer cell invasion and migration and was related to patients’ unfavorable clinical prognosis [23,24,25,26]. In GC, researchers found that CALR overexpression enhanced angiogenesis and was associated with microvessel density, metastasis and survival via a independent cohort study of little GC patients [13, 27]. However, the mechanism and function of CALR inducing GC metastasis are obscure. Only Po-Chu Lee et al. [28] found that CALR bound with VEGF-A to stabilize VEGF-A mRNA, thereby promoting the angiogenesis and progression of gastric cancer. MiR-637 could aggravate endoplasmic reticulum stress-induced apoptosis by repressing CALR [29]. In this study, our results indicated that CALR overexpression was indeed relevant to depth of invasion, lymph node metastasis, prognosis, and recurrence in GC specimens via large data and our collected fresh GC samples analysis, which was in concordance with previous studies. More importantly, our results also hinted that serum CALR might serve as a potential biomarker for GC diagnosis. In addition, we established CALR stable overexpression or repression model in GC cells via lentiviral vector and demonstrated that CALR was a key mediator of GC cells migration by cell migration assays and animal assays.
To clarify the mechanism that CALR induced GC metastasis, we performed transcriptome sequencing. Intrestingly, the data showed that the overexpression of CALR dramatically elevated EMT-related markers (ZEB1 and Snail) levels and cellular adhesive molecular (Fibronectin, integrin β1and MMP2) expression. Similar results were obtained by WB assays and big data analysis, which also indicated CALR expression was relative to EMT markers (E-cadherin, ZO-1, and N-cadherin) expression. A cancer-related EMT usually drives early steps of metastatic cascade via transforming epithelial tumor cells into invasive metastatic cancer cells [30, 31]. A previous study by Weiwei Sheng et al. showed that CALR promoted EMT in pancreatic cancer via Integrin/EGFR-ERK/MAPK signaling pathway and mediating Ca2+ dependent acute and chronic endoplasmic reticulum stress [17, 32]. Other research revealed that CALR regulated EMT through modulating Smad signaling and calcium signaling in lung cancer cells [33] and via TGF-β/Smad3/NRP1 pathway in nasopharyngeal carcinoma cells [34]. However, the role of CALR inducing the EMT process in GC is unknown. In this context, with the help of transcriptome sequencing, MS and IP assays, we discovered that CALR could form complexes with G9a and Snail. Although Snail is highly expressed in GC for directly repressing E-cadherin expression [35], the sequential event and other molecular mechanisms leading to DNA methylation at the E-cadherin promoter in GC remain not well characterized [36]. G9a encoded by EHMT2 (euchromatic histone lysine N-methyltransferase 2) is a mammalian histone methyltransferase responsible for histone H3-lysine 9 methylation (H3K9) that contributes to the epigenetic silencing of tumor suppressor genes [37, 38]. Previous studies showed that G9a was upregulated in GC and controlled GC progression via the mTOR pathway [39] or upregulating ITGB3 in a SET domain-independent manner [40]. G9a interacts with Snail, DNA methyltransferases (DNMTs) and forms a complex, which is recruited to the E-cadherin promoter and results in E-cadherin promoter DNA methylation and the suppression of E-cadherin expression in claudin-low breast cancer [41]. However, whether CALR induces GC metastasis depending on G9a remains unclear. Here, our data indicated that CALR expression affected H3K9 dimethylation (H3K9me2) and methylation of E-cadherin promoter by cooperating with Snail and G9a. CALR expression was positively correlated with G9a and Snail. Knockdown of G9a in CALR overexpression model remarkably abrogated the aggravated effect of CALR overexpression on GC cells migration and suppression of E-cadherin mRNA and protein levels. This is the first study to reveal the important role of G9a-Snail complex in the activation of the CALR-mediated EMT process and GC metastasis.
Besides, our data also showed CALR could regulate cellular adhesive molecular (Fibronectin, integrin β1and MMP2) expression. Cell attachment plays an important role in cancer cells migration. Previous studies illustrated that many molecules induced GC metastasis by activating integrin signaling [15, 42,43,44,45]. Matrix metalloprotease-2 (MMP-2) plays a pivotal role in cancer invasion and metastasis. Min Wu et al. [46] found that CALR regulated MMP2 expression via the PI3 pathway. CALR is directly involved in anti-alpha3 integrin antibody-mediated secretion and activation of MMP2 in rhabdomyosarcoma cells [47]. Our results also demonstrate for the first time that CALR is involved in the regulation of Fibronectin, integrin β1and MMP2 expression in GC, which provides a crucial evidence for CALR mediated GC metastasis.
In summary, as displayed in the Fig. 6 schematic, our study demonstrated the expression levels of CALR and regulatory effect of CALR on GC cell migration in vitro and vivo and provided several insights into the regulatory effect of CALR on the methylation status of the E-cadherin promoter in GC, which provided promising therapeutic approaches in the treatment of metastatic GC with high CALR expression. Besides, CALR is a secreted protein, and our study shows it significantly elevated in GC patients’ serum. Therefore, the diagnostic values of the CALR protein level in the blood of GC patients areworth further investigation. In addition, CALR is also found in the nucleus, implying that it may have a role in transcriptional activities and gene expression regulation [5]. Research showed that CALR could be important in gene transcription by interacting with the DNA-binding domain of the glucocorticoid receptor and the synthetic peptide KLGFFKR, and regulating the glucocorticoid receptor and perhaps other members of the superfamily of nuclear receptors [48]. CALR also upregulated neuropilin-1 expression via STAT5A in esophageal cancer cells [49]. Therefore, how CALR is to regulate Snail and G9a expression in GC which deserves further study.

G9a-mediated H3K9me2 can provide a docking site for the Dnmt complex, which results in DNA methylation at the E-cadherin promoter.
Materials and methods
Human specimens
All GC and paired adjacent nontumor tissues (at least 5 cm away from tumor edge) and serum samples in this study were collected from Southwest Hospital of Army Medical University (Chongqing, China). These patients were all confirmed by histopathological analysis and did not undergo chemotherapy, radiotherapy or immunotherapy at the time when tissues or serums samples were collected. Informed consents were obtained from every participants. This research abided by Helsinki Declaration and was performed with the permission of the Ethics Committee of Army Medical University.
Cell culture
GC cell lines (AGS, MGC-803, XN0422, BGC-823, SGC-7901, and GES-1) were purchased from ATCC (Manassas, VA, USA) and were cultured as previously described [50, 51].
Immunohistochemistry (IHC)
IHC staining and scoring were performed as previously described [51, 52]. Differently, the CALR antibody (Sigma-Aldrich, USA) diluted at 1:2000 was used for the primary reaction in this study.
Lentiviral vector construction and infection
Human CALR overexpressing and negative control (NC) lentiviral vectors were purchased from GenePharma (Shanghai, China). SGC-7901 cells were infected with LV18-Puro-CALR lentiviral vector or LV18-Puro-NC vector according to the manufacturer’s protocol. Human CALR knocking down and NC lentiviral vector were purchased from Hanbio (Shanghai, China). MGC-803 and AGS cells were infected with HBLV-Luc-Puro-CALR lentiviral vectors or HBLV-Luc-Puro-NC vectors according to the manufacturer’s protocol. Then stable cell lines were selected with puromycin (1 μg/mL) for 2 weeks.
Oligonucleotides and transfection
CALR, G9a siRNAs, and control siRNAs were synthesized by RIBOBIO (Guangzhou, China). Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used for all transfections in this study according to the manufacturer’s instructions.
RNA isolation and quantitative real-time PCR (RT-qPCR)
Total RNA of GC cells and tissues was extracted via RNAiso reagent (Takara, Dalian, China). PrimeScript™ RT reagent Kit (TaKaRa, Dalian, China) was used for reverse transcription RT-qPCR reactions were conducted by SYBR® Green Real-time PCR Master Mix (TOYOBO, Japan). The primers for GAPDH, Zo-1, Snail, ZEB1, Vimentin, E-cadherin and N-cadherin referred to Yuan Jihang’s [53]. The primers for CALR and G9a were as follows: CALR, (F) 5′-CAGACTCCAAGCCTGAGGAC-3′ and (R) 5′-CCTCCTCTTTGCGTTTCTTG -3′; G9a, (F) 5′-AGGCACCCAAGATTGACC-3′, (R) 5′-GTCTCCCGCTTGAGGATG-3′. All RT-qPCR reactions were performed in triplicate using a Bio-Rad CFX96 real-time PCR system. The gene expression data were normalized to GAPDH. The results were calculated in accordance with the 2−ΔΔCT method.
Transcriptome sequencing
A total amount of 3 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries, transcriptome sequencing, and computational analyses were fulfilled by Novogene (Beijing, China).
Cell migration assays and wound healing assay
GC cells migration ability was evaluated by cell migration assays and wound healing assay whose detailed procedures were described previously [54].
Western blot assay
CALR (1:1000) and Fibronectin (1:1000) polyclonal antibody were obtained from Abcam (Shanghai, China); GAPDH, E-cadherin, N-cadherin, G9a, ZEB1, ZO-1, Snail, Slug, Vimentin, Integrin β1, MMP2 antibodies and methyl-histone H3 (Lys9) antibody sampler kit (1:1000) were purchased from Cell Signaling Technology (Shanghai, China); GAPDH served as an internal reference. Anti-mouse IgG or anti-rabbit horseradish peroxidase (HRP)-conjugated was used as secondary antibody (1:10000; ZhongShan, Beijing, China). Signals were detected by using the SuperSignal West Dura Extended Duration Substrate Kit (Thermo Scientific, Shanghai, China) and the results were analyzed by Image J software (Bio-Rad, Hercules, USA).
Immunoprecipitation and MS analysis
1 mg protein extracted from AGS cells was immunoprecipitated (IP) overnight with anti-CALR antibody (Abcam, Shanghai, China), anti-Snail antibody and anti-G9a antibody (Cell Signaling Technology, USA), or rabbit or normal mouse IgG (Beyotime, Jiangsu, China) at 4 °C and then with protein A + G agarose beads (Beyotime, Jiangsu, China) for at least 2 h. After IP, the potential proteins binding with CALR were analyzed by MS analysis (SANGON BIOTECH, Shanghai, China). To verify the results of MS analysis, the beads were then boiled in 30 μL of 1% SDS loading buffer for western blotting with Snail (Cell Signaling Technology, USA), G9a (Proteintech, USA), or CALR antibodies (Sigma-Aldrich, USA).
Pyrosequencing
Pyrosequencing was performed with the help of Sinotech Genomics (Shanghai, China). Briefly, DNA was isolated from stably overexpressing or repressing CALR GC cells or their controls. Then 300–1000 ng DNA of each sample was used to bisulfite converted using EZ DNA Methylation Kits(Zymo Research, USA), then converted products were put into PCR amplification and Machine sequencing by Sinotech Genomics (Shanghai, China). PCR primers and Pyrosequencing primers were shown in Supplementary Table 2. Finally, The Pyro Q-CPG software of the pyrosequencing device was used to automatically analyze the methylation status of each site. Analysis of the difference between the target sequence and the actual read sequence information.
Intraperitoneal dissemination assay
Intraperitoneal dissemination experiments were conducted as previously described [51]. 1 × 106 stably overexpressing CALR SGC-7901 cells or repressing CALR MGC-803 cells or their controls were respectively injected into the abdominal cavity of female BABL/c nude mice (6–8 weeks old, n = 6 vs 6), which were approved by the Institutional Animal Care and Use Committee of Army Medical University. Four weeks later, the tumor metastasis in the abdominal cavity of the mice was observed by in vivo imaging system and counting the tumor nodules after the mice were sacrificed.
Statistical analyses
Statistical differences were analyzed via GraphPad Prism 5.0 Software (La Jolla, CA, USA) using paired t-tests, Two-tailed Student’s t-tests, and One-way ANOVA as appropriate. The relationships between CALR and Snail/ZO-1/G9a expression levels were analyzed by Pearson’s correlation. Sensitivity and specificity of serum biomarkers were assessed using ROC curve and AUC. The results are presented as mean ± SEM. P ≤ 0.05 was considered statistical significance.
Data availability
The data used and analyzed during the current study are available from the corresponding author on reasonable request.
References
Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635–48.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA cancer J Clinicians. 2021;71:209–49.
Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin: one protein, one gene, many functions. Biochemical J. 1999;344:281–92. Pt 2
Gelebart P, Opas M, Michalak M. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int J Biochem Cell Biol. 2005;37:260–6.
Dey S, Matsunami H. Calreticulin chaperones regulate functional expression of vomeronasal type 2 pheromone receptors. Proc Natl Acad Sci USA. 2011;108:16651–6.
Pronier E, Cifani P, Merlinsky TR, Berman KB, Somasundara AVH, Rampal RK, et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI insight. 2018;3:e122703.
Fucikova J, Spisek R, Kroemer G, Galluzzi L. Calreticulin and cancer. Cell Res. 2021;31:5–16.
Zamanian M, Veerakumarasivam A, Abdullah S, Rosli R. Calreticulin and cancer. Pathol Oncol Res. 2013;19:149–54.
Qiu Y, Michalak M. Transcriptional control of the calreticulin gene in health and disease. Int J Biochem cell Biol. 2009;41:531–8.
Gu VY, Wong MH, Stevenson JL, Crawford KE, Brennecke SP, Gude NM. Calreticulin in human pregnancy and pre-eclampsia. Mol Hum Reprod. 2008;14:309–15.
Lu CM, Lin JJ, Huang HH, Ko YC, Hsu JL, Chen JC, et al. A panel of tumor markers, calreticulin, annexin A2, and annexin A3 in upper tract urothelial carcinoma identified by proteomic and immunological analysis. BMC Cancer. 2014;14:363.
Lin Q, Cao Y, Gao J. Serum calreticulin is a negative biomarker in patients with Alzheimer’s disease. Int J Mol Sci. 2014;15:21740–53.
Chen CN, Chang CC, Su TE, Hsu WM, Jeng YM, Ho MC, et al. Identification of calreticulin as a prognosis marker and angiogenic regulator in human gastric cancer. Ann Surgical Oncol. 2009;16:524–33.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90.
Brakebusch C, Fassler R. beta 1 integrin function in vivo: adhesion, migration and more. Cancer Metastasis Rev. 2005;24:403–11.
Akiyama SK, Olden K, Yamada KM. Fibronectin and integrins in invasion and metastasis. Cancer Metastasis Rev. 1995;14:173–89.
Sheng W, Chen C, Dong M, Wang G, Zhou J, Song H, et al. Calreticulin promotes EGF-induced EMT in pancreatic cancer cells via Integrin/EGFR-ERK/MAPK signaling pathway. Cell Death Dis. 2017;8:e3147.
Karimzadeh F, Opas M. Calreticulin is required for TGF-beta-induced epithelial-to-mesenchymal transition during cardiogenesis in mouse embryonic stem cells. Stem Cell Rep. 2017;8:1299–311.
Hayashida Y, Urata Y, Muroi E, Kono T, Miyata Y, Nomata K, et al. Calreticulin represses E-cadherin gene expression in Madin–Darby canine kidney cells via Slug. J Biol Chem. 2006;281:32469–84.
Diepenbruck M, Tiede S, Saxena M, Ivanek R, Kalathur RKR, Luond F, et al. miR-1199-5p and Zeb1 function in a double-negative feedback loop potentially coordinating EMT and tumour metastasis. Nat Commun. 2017;8:1168.
Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35:347–67.
Collins R, Cheng X. A case study in cross-talk: the histone lysine methyltransferases G9a and GLP. Nucleic Acids Res. 2010;38:3503–11.
Yoon GS, Lee H, Jung Y, Yu E, Moon HB, Song K, et al. Nuclear matrix of calreticulin in hepatocellular carcinoma. Cancer Res. 2000;60:1117–20.
Chiang WF, Hwang TZ, Hour TC, Wang LH, Chiu CC, Chen HR, et al. Calreticulin, an endoplasmic reticulum-resident protein, is highly expressed and essential for cell proliferation and migration in oral squamous cell carcinoma. Oral Oncol. 2013;49:534–41.
Lwin ZM, Guo C, Salim A, Yip GW, Chew FT, Nan J, et al. Clinicopathological significance of calreticulin in breast invasive ductal carcinoma. Mod Pathol. 2010;23:1559–66.
Alfonso P, Nunez A, Madoz-Gurpide J, Lombardia L, Sanchez L, Casal JI. Proteomic expression analysis of colorectal cancer by two-dimensional differential gel electrophoresis. Proteomics. 2005;5:2602–11.
Chen CN, Lin JJ, Lee H, Cheng YM, Chang KJ, Hsieh FJ, et al. Association between color doppler vascularity index, angiogenesis-related molecules, and clinical outcomes in gastric cancer. J Surgical Oncol. 2009;99:402–8.
Lee PC, Chiang JC, Chen CY, Chien YC, Chen WM, Huang CW, et al. Calreticulin regulates vascular endothelial growth factor-A mRNA stability in gastric cancer cells. PloS ONE. 2019;14:e0225107.
Kong Q, Zhang Z, Liang Z. Upregulating miR-637 aggravates endoplasmic reticulum stress-induced apoptosis in gastric cancer cells by suppressing Calreticulin. Anim Cells Syst. 2020;24:267–74.
Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64.
Sheng W, Wang G, Tang J, Shi X, Cao R, Sun J, et al. Calreticulin promotes EMT in pancreatic cancer via mediating Ca(2+) dependent acute and chronic endoplasmic reticulum stress. J Exp Clin Cancer Res. 2020;39:209.
Wu Y, Xu X, Ma L, Yi Q, Sun W, Tang L. Calreticulin regulates TGF-beta1-induced epithelial mesenchymal transition through modulating Smad signaling and calcium signaling. Int J Biochem Cell Biol. 2017;90:103–13.
Ye D, Zhu J, Zhao Q, Ma W, Xiao Y, Xu G, et al. LMP1 up-regulates calreticulin to induce epithelial-mesenchymal transition via TGF-beta/Smad3/NRP1 pathway in nasopharyngeal carcinoma cells. J Cancer. 2020;11:1257–69.
Alves CC, Rosivatz E, Schott C, Hollweck R, Becker I, Sarbia M, et al. Slug is overexpressed in gastric carcinomas and may act synergistically with SIP1 and snail in the down-regulation of E-cadherin. J Pathol. 2007;211:507–15.
Cui H, Hu Y, Guo D, Zhang A, Gu Y, Zhang S, et al. DNA methyltransferase 3A isoform b contributes to repressing E-cadherin through cooperation of DNA methylation and H3K27/H3K9 methylation in EMT-related metastasis of gastric cancer. Oncogene. 2018;37:4358–71.
Lee SH, Kim J, Kim WH, Lee YM. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene. 2009;28:184–94.
Rahman Z, Bazaz MR, Devabattula G, Khan MA, Godugu C. Targeting H3K9 methyltransferase G9a and its related molecule GLP as a potential therapeutic strategy for cancer. J Biochemical Mol Toxicol. 2021;35:e22674.
Yin C, Ke X, Zhang R, Hou J, Dong Z, Wang F, et al. G9a promotes cell proliferation and suppresses autophagy in gastric cancer by directly activating mTOR. FASEB J. 2019;33:14036–50.
Hu L, Zang MD, Wang HX, Zhang BG, Wang ZQ, Fan ZY, et al. G9A promotes gastric cancer metastasis by upregulating ITGB3 in a SET domain-independent manner. Cell Death Dis. 2018;9:278.
Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Investig. 2012;122:1469–86.
Wang H, Chen H, Jiang Z, Lin Y, Wang X, Xiang J, et al. Integrin subunit alpha V promotes growth, migration, and invasion of gastric cancer cells. Pathol, Res Pract. 2019;215:152531.
Yao LW, Wu LL, Zhang LH, Zhou W, Wu L, He K, et al. MFAP2 is overexpressed in gastric cancer and promotes motility via the MFAP2/integrin alpha5beta1/FAK/ERK pathway. Oncogenesis. 2020;9:17.
Izumi D, Ishimoto T, Miyake K, Sugihara H, Eto K, Sawayama H, et al. CXCL12/CXCR4 activation by cancer-associated fibroblasts promotes integrin beta1 clustering and invasiveness in gastric cancer. Int J cancer. 2016;138:1207–19.
Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100.
Wu M, Massaeli H, Durston M, Mesaeli N. Differential expression and activity of matrix metalloproteinase-2 and -9 in the calreticulin deficient cells. Matrix Biol. 2007;26:463–72.
Ito H, Seyama Y, Kubota S. Calreticulin is directly involved in anti-alpha3 integrin antibody-mediated secretion and activation of matrix metalloprotease-2. Biochem Biophys Res Commun. 2001;283:297–302.
Burns K, Duggan B, Atkinson EA, Famulski KS, Nemer M, Bleackley RC, et al. Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nature. 1994;367:476–80.
Shi F, Shang L, Pan BQ, Wang XM, Jiang YY, Hao JJ, et al. Calreticulin promotes migration and invasion of esophageal cancer cells by upregulating neuropilin-1 expression via STAT5A. Clin Cancer Res. 2014;20:6153–62.
Wang B, Liu J, Ma LN, Xiao HL, Wang YZ, Li Y, et al. Chimeric 5/35 adenovirus-mediated Dickkopf-1 overexpression suppressed tumorigenicity of CD44(+) gastric cancer cells via attenuating Wnt signaling. J Gastroenterol. 2013;48:798–808.
Wang L, Yu T, Li W, Li M, Zuo Q, Zou Q, et al. The miR-29c-KIAA1199 axis regulates gastric cancer migration by binding with WBP11 and PTP4A3. Oncogene. 2019;38:3134–50.
Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10:7252–9.
Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666–81.
Wang L, Xiao B, Yu T, Gong L, Wang Y, Zhang X, et al. lncRNA PVT1 promotes the migration of gastric cancer by functioning as ceRNA of miR-30a and regulating Snail. J Cell Physiol. 2021;236:536–48.
Acknowledgements
This work was supported by the National Science Foundation of China (No. 82003109) and Chongqing Natural Science Foundation of General Program(cstc2020jcyj-msxmX0999).
Author information
Authors and Affiliations
Contributions
Study conception, design, and supervision: LW, QMZ, JZ. Manuscript writing: LW, PZ. Experimental analysis: LW, JC, CW, PY, QFZ, TY. Data analysis: LW, CW, HH, JD, CL. Data interpretation: LW, LF, HL. Critical revision and editing of the manuscript: all authors. Reading and approval of the final manuscript: all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Chen, J., Zuo, Q. et al. Calreticulin enhances gastric cancer metastasis by dimethylating H3K9 in the E-cadherin promoter region mediating by G9a. Oncogenesis 11, 29 (2022). https://doi.org/10.1038/s41389-022-00405-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-022-00405-7
This article is cited by
-
Identifying natural products for gastric cancer treatment through pharmacophore creation, 3D QSAR, virtual screening, and molecular dynamics studies
DARU Journal of Pharmaceutical Sciences (2023)
-
Calreticulin as a marker and therapeutic target for cancer
Clinical and Experimental Medicine (2022)

{kind=link}
{kind=link}
{kind=link}