
Abstract
To date, thousands of highly abundant and conserved single-stranded RNA molecules shaped into ring structures (circRNAs) have been identified. CircRNAs are multifunctional molecules that have been shown to regulate gene expression transcriptionally and post-transcriptionally and exhibit distinct tissue- and development-specific expression patterns associated with a variety of normal and disease conditions, including cancer pathogenesis. Over the past years, due to their intrinsic stability and resistance to ribonucleases, particular attention has been drawn to their use as reliable diagnostic and prognostic biomarkers in cancer diagnosis, treatment, and prevention. However, there are some critical caveats to their utility in the clinic. Their circular shape limits their annotation and a complete functional elucidation is lacking. This makes their detection and biomedical application still challenging. Herein, we review the current knowledge of circRNA biogenesis and function, and of their involvement in tumorigenesis and potential utility in cancer-targeted therapy.
Similar content being viewed by others
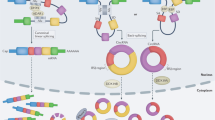
Background: the advent of circRNAs
Despite their recent fame, the discovery of circRNAs dates back over 40 years. CircRNAs were first discovered in the murine respirovirus (Sendai virus) [1] and in plant pathogenic viruses termed viroids [2]. The initial physical evidence of the existence of a circular form of RNA was obtained by electron microscope analysis of the cytoplasmic fraction of eukaryotic (HeLa) cells in 1979 [3] and later in 1986 when they were identified within the hepatitis delta virus (HDV) [4]. Such circular shaped viral genomes possess the distinct property of generating multiple copies of an RNA through the rolling circle replication mechanism that facilitates the spread of infection more efficiently. However, evidence of another type of circRNA as intermediate molecules excised from pre-mRNA started emerging [5,6,7]. In 1991, a group of researchers found the first examples of endogenous spliced circRNAs in humans, transcribed from the DCC gene and end-joined in a scrambled order compared to the canonical linear sequence [8]. In parallel, the mouse Sry gene, encoding a crucial molecule responsible for sex determination during embryogenesis, was found to be expressed in adult mice testes exclusively as a 1.23-kb circular RNA [9]. During the late 1990s and early 2000s, several other studies showed that circRNA-producing genes are widespread in eukaryotic cells from flies to mammals including humans [10,11,12,13,14,15,16,17,18].
Nevertheless, the lack of evidence of their translation into polypeptides left researchers skeptical about the functional significance of such RNAs, and for several decades circRNAs were commonly disregarded as mis-splicing artifacts or by-products of pre-mRNA processing [19]. With the advent of RNA sequencing (RNA-seq) technologies and bioinformatics, the true abundance of circRNAs was revealed. In 2012, an unexpected number of human genes were reported to express “scrambled exons” resulting in circular RNA isoforms [20]. A subsequent analysis of non-polyadenylated libraries prepared from ribosomal RNA-depleted RNAs, revealed >25,000 distinct RNA species containing “non-colinear exons” in human fibroblasts [21] and, at the same time, a new genome-wide in-silico approach identified ~2000 human, 700 nematode and 1900 mouse circRNAs that were more stable than the associated linear mRNAs in vivo [18].
Since their discovery, thousands of circRNAs have been found to be present in multiple organisms and their expression been associated with developmental stages, physiological conditions, and diseases including cancer. This has opened up a new field of study aimed at elucidating their biogenesis and function as an essential part of gene expression programs across eukaryotes (Fig. 1).

Timeline of milestone events leading to the discovery, research and development of circRNAs.
Biogenesis of circRNAs in normal and pathological contexts
General features of circRNAs
Pre-mRNA splicing, facilitated by the spliceosome, is a mechanism that is tightly coupled with transcription [22]. During this process, introns are removed from precursor mRNAs (pre‐mRNAs) and the exons are covalently joined to create linear mRNA molecules that are translated into proteins. By contrast, back-splicing allows a downstream splice donor (5′ splice site) to join backwards to an upstream splice acceptor (3′ splice site), resulting in a closed continuous head-to-tail molecule known as a circRNA (Fig. 2). The resulting molecules are devoid of terminal 5’ caps and 3′ polyadenylated tails [23,24,25], are less accessible to exonucleases and are consequently more stable than linear RNAs [26]. By rearranging the order of genomic information, circRNAs also provide a unique opportunity to further diversify gene expression across eukaryotes.

Starting from the same pre-mRNA molecule, linear splicing (red arrows) and head-to-tail back-splicing (blue arrows) lead to a differential outcome in either processed mRNA/lncRNAs molecules or several types of circRNAs, respectively. Adapted from “RNA Processing in Eukaryotes 2”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates.
Although circRNAs are preferentially back-spliced from hundreds of human genes [20], including neuron-expressed genes [27], back-splicing is typically much less efficient (<1%) than canonical splicing [28] so circRNAs are often expressed at lower levels than their corresponding linear mRNAs [29]. The observed abundance at levels that exceed mRNAs (frequently over tenfold) in a given cell type [21] is largely due to a context- and time-specific accumulation of circRNAs that occurs for example during aging [30,31,32,33] and neurodegenerative [30, 34,35,36] processes. This is primarily attributed to their exceptional stability. Indeed, whilst mRNAs are continually degraded over time, circRNAs persist, thereby increasing the ratio of circRNA to mRNA as time passes. Packaging into exosomes and subsequent release into the extracellular space [37,38,39,40], appears to clear the excessive accumulation of circRNAs avoiding potential toxic effects. In some cases, after being released into the extracellular environment, exosomes can reach recipient cells and deliver circRNAs to trigger functional responses and potentially induce a series of phenotypic changes [41], including the spread of premetastatic niches from cancer-derived exosomes [42, 43]. By contrast, in cancer and highly proliferative cells, circRNAs are maintained at low levels, possibly as a result of dilution effects caused by cell division during the process of cell proliferation [44, 45].
The maturation of circRNAs is a tightly regulated process [46], involving the same spliceosome components engaged in canonical splicing, albeit recruited in a different order [47]. When pre-mRNA processing is inhibited, the intimate connection that exists between canonical splicing and back-splicing [25, 27] results in the splicing machinery facilitating back-splicing and thereby shifting the output of genes toward circRNAs [48, 49]. Additional factors, including epigenetic changes, can impact the biogenesis of circRNA making the expression of mRNA and circRNA at the same locus less predictable. For example, the repression induced by DNA methylation in the main gene body of the parental genes [50, 51] has been found to be one of the causes of altered circRNA expression independent of the linear counterpart [22, 50, 52]. Transcriptional silencing of circRNAs caused by hypermethylation or altered histone modifications at their host gene promoters has also been reported in cancer [53,54,55].
Mechanisms of circRNA biogenesis
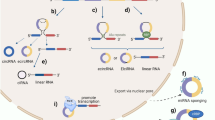
A variety of circRNAs can be generated from a single protein-coding or non-coding genomic locus [18] (Table 1) and can significantly vary in length since not only are entire exons susceptible to circularization, but so are other sequences, including introns, non-coding antisense, 3′ UTR, 5′ UTR, or transcribed intergenic regions. Additional circRNAs can also be assembled from a combination of multiple exons and retained intronic portions [18, 56]. Generally, at least three, often overlapping [57], mechanisms can lead to back-splicing including lariat-driven circularization (exon skipping) [21] (Fig. 3a), intron pairing-driven circularization [21] (Fig. 3b), and RNA binding protein (RBP)‐driven circularization [27] (Fig. 3c).

Three main mechanisms of back-splicing, such as lariat-driven circularization (exon skipping) (A), intron pairing-driven circularization (B), and RNA binding protein (RBP)‐driven circularization (C) lead to the biogenesis of circRNAs. Alternative processes (D) can also lead to the biogenesis of other special classes of circRNAs, including intergenic circRNAs or rt-circRNAs and f-circRNAs, which are frequently associated with pathological conditions. Created with BioRender.com.
In exon skipping a pre-mRNA is spliced into two RNA molecules consisting of an mRNA from which at least one exon is missed, and a lariat containing the skipped exons making circularization possible. The predominant fraction of circRNAs (over 80%) are known to be exon-derived circRNAs (ecircRNAs) [18, 20, 21]; these usually originate from one to five pre-mRNA exons and preferentially localize to the cytoplasm [56], most likely by escaping the nucleus during mitosis [58]. The ATP-dependent RNA helicase DDX39A and the spliceosomal RNA helicase DDX39B are involved in exporting circRNAs from the nucleus to the cytoplasm by sensing the lengths of mature circRNAs by an unclear mechanism [59]. Exonic circularization seems to be strongly favored by longer exon length (generally > 300 nt) [20, 21] flanked by intronic regions of similar length and hosting inverted repeated sequences (e.g., ALU elements) [21, 60, 61]. This appears to occur more frequently through the second mechanism of intron pairing [30] where perfect complementarity between the two introns flanking the exon/exons of the nascent circRNA is required. The successful intron pairing brings the two splice sites into close proximity and the resulting secondary structure facilitates back-splicing [60, 62,63,64]. Although favorable, the presence of intronic repeat sequences is not always associated with exon circularization [65, 66], and excessive stability of intron base pairing can even sometimes prevent circRNA formation [62].
Several RBPs, including quaking (QKI) [67], muscleblind (MBL/MBNL1) [68], and fused-in-sarcoma (FUS) [69] proteins, participate in circRNA biogenesis by tethering specific motifs within adjacent introns of nascent circRNAs and connecting the splice donor and acceptor sites to form a closed intronic-paired RNA. In the cancer context, QKI has been shown to induce the production of up to one-third of the 300 most abundant circRNAs during epithelial-to-mesenchymal transition (EMT) in immortalized human mammary epithelial cells [67]. CircRNA formation via back-splicing is also mediated by heterogeneous nuclear ribonucleoprotein L (HNRNPL) which is required for prostate cancer growth in vitro and is aberrantly expressed in human prostate tumors [70]. In this regard, HNRNPL also facilitates the expression of an oncogenic circRNA from the tumor-suppressor gene ARHGAP35 generally associated with poor survival in cancer patients [71]. Further mechanisms may combine the intron pairing process involving RBPs with the additional involvement of ADAR (RNA editing) proteins that couple A-to-I editing with the unwinding of the dsRNA helical structures [72], preventing the pairing/looping of intron sequences [73, 74]. By contrast, proteins such as NF90 and NF110 [75], can stabilize the intronic RNA pairs at exonic junctions of nascent circRNAs in response to viral infection and favor their production, which is ultimately reduced when the proteins migrate to the cytoplasm and viral infection terminates [76]. Overall, a combination of both cis- and trans-acting factors are likely to provide a more complex set of processes that affect circRNA biogenesis [57].
There is added complexity in the formation of circRNAs. When introns are not excised properly and are retained in the newly generated circRNAs, so-called exon–intron circRNAs (EIciRNAs) are generated [68]. Conversely, if intron lariats that are correctly circularized at the branchpoint 2′–5′ linkage and degraded from the 3′ end up to the branchpoint, somehow escape the usual intron debranching and subsequent degradation, stable circular intronic RNAs (ciRNAs) can also be formed [56, 77]. Specific sequences (a consensus motif containing a 7 nt GU-rich element and a 11 nt C-rich element) near the 5′ splice site and branchpoint close to the branchpoint site can prevent debranching by forming a structure that limits access to the debranching enzyme. Both ciRNAs and EIciRNAs are predominantly located in the nucleus and presumably involved in the regulation of expression of neighboring genes in cis [56, 68] as they have been found to be associated with RNA polymerase II [56, 78, 79]. Screening of the human transcriptome with a bioinformatic tool for circRNA identification has also revealed the existence of a class of non-exonic circRNAs (intergenic circRNAs). These circRNAs originate from intergenic portions of the genome and contain two intronic circRNA fragments flanked by GT-AG (or reverse complementary dinucleotides CT-AC) splicing signals that act as the splice donor and acceptor of the circular junction while forming an integrated circRNA [80]. Other than a weak but significant enrichment of conserved nucleotides between a few ciRNAs and intergenic circRNAs [18], very little is known about the function of intergenic circRNAs (Fig. 3d).
CircRNA biogenesis during pathological processes
Distinct classes of circRNA can also be generated in pathological contexts including cancer (Fig. 3d). For instance, the failure of transcription termination and exceeding transcription into the downstream gene followed by back-splicing can give rise to so-called read-through circRNAs (rt-circRNAs) [49, 81] that may incorporate exons from adjacent and similarly oriented genes and appear to be associated with pathological phenotypes [82]. Uncontrolled gene transcription leading to pervasive transcription read-through is typically associated with cancer. Likewise, fusion-circRNAs (f-circRNAs) have been recently reported as originating from cancer-associated chromosomal translocations and are able to confer resistance to apoptosis-inducing drug therapy, promote transformation and cell survival [83, 84]. For instance, the circRNA generated by the MLL/AF9 fusion gene (f-circM9) in leukemia causes pro-proliferative and pro-oncogenic effects [84]. Similarly, the back-splicing of the non-small cell lung cancer (NSCLC)-associated EML4-ALK fusion variant 3b generates a tumor-promoting circRNA named f-circEA [83] and an additional variant (f-circEA-2a) which enhances cell migration and invasion [85].
Further complexity in circRNA biogenesis
Some circRNAs may also contain modified nucleotides, such as N6-methyl-adenosine patterns (m6A), that can further diversify their biogenesis [86,87,88], as well as their fate, including their degradation and cellular localization [89]. These m6A-modified circRNAs often originate from unmethylated exons of linear mRNAs and they are likely to be methylated during or after circRNA formation [40, 88]. According to recent studies, the efficient depletion of specific enzymes involved in such modifications (e.g., methylation writers, readers, etc.) affects a subset of circRNAs (~20%) without significantly altering their linear isoforms [90]. However, it is unclear whether or not additional factors play a role in the mechanism through which m6A deposition may affect the choice of back-splicing versus canonical splicing.
Alternative explanations of back-splicing should also be contemplated. A proportion of discovered circRNAs might not be functional per se but might offer through their back-splicing a break between transcription of the main gene and translation, allowing post-transcriptional regulatory processes to take place. Offering a more radical perspective, recent studies have also proposed that circRNAs form through pre-mRNA splicing errors and are not able to confer any specific benefit [91].
Biological functions and mechanisms of action of non-coding and translated circRNAs
The number of unique circRNAs produced in human cells (~100,000) [81, 92] largely exceeds the number of protein-coding genes (~20,000) [93,94,95]. Despite their wide prevalence, the majority of circRNAs have not been functionally characterized and the biological role of many remains unclear [15, 16, 91]. A growing body of evidence suggests that those carrying out biological functions are likely to require a specific subcellular localization and that their accumulation in specific disease contexts [18], may indicate a link with the occurrence and development of specific diseases including cancer. Despite the fact that the majority of circRNAs are spliced out from protein-coding pre-mRNAs, they are mainly classified as a special class of non-coding RNAs (ncRNAs) since they are devoid of essential elements for translation such as an open reading frame (ORF) and a 5′ cap and the poly(A) tail, and are characterized by an average length frequently longer than 200, that occasionally (e.g., in the case of ecircRNAs and ciRNAs) can shorten to 100-200 nt [9].
CircRNAs as regulators of transcription
Beyond their regulatory effects on alternative pre-mRNA splicing [27], circRNAs can function in more than a single mechanism (Fig. 4, Table 2), including as regulators of transcription of the same genes from which they are transcribed (parental genes) either alone or in association with RBPs. In this regard, they have been found to either upregulate the expression of specific transcription factors and activate their parental gene’s transcription, or to favor premature transcription termination through the formation of RNA-DNA hybrid (R-loops) with the subsequent upregulation of a truncated, non-functional isoform [96]. CircRNAs can also induce promoter CpG demethylation thereby changing the epigenetic state, and switch on the activity of their parental genes [97, 98]. Through this positive feedback mechanism of inducing DNA hypomethylation in CpG islands of the promoter by recruiting the methylcytosine dioxygenase TET1, circFECR1 activates its parental FL1 (friend leukemia virus integration 1) (onco) gene and favors breast cancer cell metastasis [98]. An interaction with the normal activity of RNA polymerase II and with other components of the transcription machinery proteins has also been reported [56, 68]. CircRNAs that are mainly located in the nucleus, such as EIciRNAs, can bind the small nuclear U1 ribonucleoprotein (U1 snRNP) through RNA-RNA base pairing and then interact with RNA polymerase II at the parental gene promoter thereby enhancing their expression [68]. Similarly, ciRNAs accumulate at their sites of transcription and can increase the transcription rate of parental genes by tethering the elongation Pol lI complex and ultimately regulating its elongation activity [56].

CircRNAs can work as multifunctional devices serving as transcription regulators of their parental genes (1), as microRNA sponges affecting genes post-transcriptionally (2), or as translated short peptides/proteins (3). Additionally, circRNAs can affect the stability of other RNA molecules (mRNAs or lncRNAs) (4), accumulate inside of exosomes and mediate cellular response (5), engage with RBPs and acting as either decoy (6), or scaffold molecules (7), as well as directing RBP cellular localization (8). Adapted from “DNA vs mRNA Transfection”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
CircRNAs as miRNA sponges
Initial examples of functional circRNAs were shown to work as efficient microRNA sponges, post-transcriptionally regulating the activity of their downstream target genes [99], as well as acting as miRNA-reservoirs or miRNA-cargos. This has been widely confirmed by their significant localization inside exosomes [40] and a well reported association with Argonaute proteins [99]. Since the discovery of the antisense transcript cerebellar degeneration-related protein (CDR1as), also known as the circRNA sponge for miR-7 (CiRS-7), and its role as a competing endogenous RNA (ceRNAs) [99] with > 60 miRNA binding sites, more studies have corroborated the ability of circRNAs to act as ceRNA or miRNA sponges [100, 101]. Among several circRNAs associated with altered target gene expression in cancer [102,103,104,105], circHIPK3 regulates cell growth by sponging multiple miRNAs, such as the tumor-suppressor miR-124 [106], whereas circITCH acts as cancer inhibitor by sponging several miRNAs, including miR-7, miR-17, and miR-214, suppressing the Wnt/β-catenin pathway in esophageal squamous cell carcinoma (ESCC) [107].
CircRNAs as modulators of RNA stability
In addition, circRNAs can influence the stability of other RNA molecules, including both lncRNAs and mRNAs [108]. An example is the case of CiRS-7/CDR1as which can stabilize its cognate mRNA by forming an RNA duplex [17]. The stabilization of RNA molecules can also occur in cooperation with proteins; for example, circZNF609 favors the recruitment of the protein ELAV1 (also known as HuR), and its interaction increases the stability and/or translation of a pool of mRNAs, including CKAP5 mRNA expressing a protein that regulates microtubule function and sustains cell-cycle progression in cancer cells [109]. Likewise, circXPO1 promotes lung adenocarcinoma (LUAD) progression by recruiting IGF2BP1 to enhance the stability of CTNNB1 mRNA [110]. A similar mechanism was described a few years earlier in colorectal cancer for circNSUN2 which is able to stabilize HMGA2 mRNA by enhancing its interaction with IGF2BP2 to promote malignant progression [89].
CircRNAs as RBP partners
As well as being required for their back-splicing and gene transcription regulation, circRNAs are also able to engage with RBPs to direct their cellular localization [111]. Given their preferential location inside the cytoplasm, circRNAs can indeed sequester cytoplasmic proteins and prevent their nuclear entry [112], serve as RBP decoys to regulate their function [113] or act as scaffold molecules for complex assembly [55, 114, 115]. For instance, in human cervical carcinoma HeLa cells, circPABPN1, binds the RBP HuR and affects the translation rate of the parental gene PABPN1 by preventing HuR binding to the cognate mRNA [116]. In this regard, a remarkable tumor-suppressor example in breast cancer is represented by the circFOXO3 that binds both p53 and the E3 ubiquitin ligase MDM2, which normally mediates the degradation of the transcription factor Foxo3. This association promotes the degradation of P53 while preventing the degradation of the pro-apoptotic Foxo3 derived from its parental gene [117]. In NSCLC, the inhibitory effects on tumor growth and metastasis are instead caused by the scaffolding action of circNDUFB2 that forms a ternary complex with both TRIM25 and IGF2BPs to facilitate the ubiquitination and degradation of IGF2BPs and ultimately activate an antitumor immune response [118]. Conversely, the successful interaction of the oncogenic circ-Amotl1 with the proto-oncogene c-MYC ensures the retention in the nucleus of c-Myc protein and consequently promotes tumorigenesis [119].
CircRNAs as translated peptides
Although the majority of circRNAs are expected to be non-coding, both in vivo and in vitro experiments have demonstrated their association with ribosomes and translation into proteins [14, 120, 121]. Bioinformatic predictions estimate that only a small proportion of circRNAs host both ORFs and internal ribosome entry site (IRES) elements, or incorporate the m6A RNA modification in their 5′ UTR thereby becoming competent for translation via a cap-independent mechanism. As a result, shortened versions of canonical proteins, acting as modulators of dominant negative protein variants, decoys, or alternative protein complexes are generated [120]. However, according to a recent study, IRES-like short elements that are significantly enriched in endogenous circRNAs, are sufficient to drive extensive circRNA translation [122], suggesting that circRNA translation might be a far more widespread phenomenon than initially estimated [123,124,125]. Besides the better studied translated circZNF609, which harbors IRES elements and undergoes cap-independent translation [120], cancer-associated examples of translated circRNAs include the circular form of the SHPRH gene (circSHPRH) which encodes the novel identified protein termed SHPRH-146aa [126]. Together with circSHPRH, SHPRH-146aa is normally highly expressed in normal human brains but downregulated in glioblastoma, suggesting a potential role as a tumor suppressor [126]. Inhibitory effects on glioma proliferation and cell-cycle acceleration are instead mediated by a 185 amino acid protein encoded from circFBXW7 (FBXW7-185aa) which reduces the half life of c-Myc by antagonizing USP28-induced c-Myc stabilization, suggesting new prognostic implications for glioma patients [127]. By contrast, translation of the circPPP1R12A, (generally up-regulated in colon cancer) into a 73 amino acid small polypeptide (circPPP1R12A-73aa) promotes rapid cancer cell proliferation and metastasis via the Hippo-YAP pathway [128]. A similar oncogenic effect is caused by the E-cadherin variant encoded by the circE-Cad at the CDH1 gene (C-E-Cad- 254aa), involved in the maintenance of the cancer stemness in glioblastoma by interacting with EGFR and activating downstream STAT3 signaling [129]. In hepatocellular carcinoma (HCC), tumor progression, invasion and metastasis, can instead be caused by an exceptionally long (1289aa) oncogenic protein, encoded by circARHGAP35 through alternative m6 A-dependent translation [130] that interacts with the transcription factor TFII-I in the nucleus [71]. Oncogenic virus-derived circRNAs can also be translated into proteins. For example, the highly expressed human papillomavirus-derived circE7, displays oncogenic activity in cervical and head and neck cancers, and has been found to be translated into an E7 oncoprotein [131]. Experimentally validated and functionally characterized peptides encoded by ncRNAs (ncPEPs), including circRNAs, have been recently annotated in the new database FuncPEP [132].
Detection and characterization of circRNAs: challenges and strategies
Clinical applications of circRNAs rely on accurate RNA profiling (Box 1); this includes annotating new RNA species and quantifying their abundance [133]. However, detecting and studying circRNAs poses challenges at several levels due to their circular conformation and sequence overlap with their linear mRNA counterparts.
Methods for the detection of circRNAs
RNA-based high-throughput sequencing technologies (e.g., RNA-seq), have allowed genome-wide annotation and quantification [133] of a number of diverse coding and non-coding RNAs [134, 135] based on the selective isolation of ribosomal RNA (rRNA)-free polyadenylated RNA species using oligo-dT primed reverse transcription. Improved methodologies that employ the preparation of rRNA-depleted libraries and random priming for cDNA synthesis (ribo-depleted total RNA-seq) [135], have enhanced the annotation of RNAs to include large non-polyadenylated transcripts encompassing a compendium of circRNAs [136].
Traditional methods, such as RT-qPCR using sets of “divergently oriented” primers designed to cover the circRNA back-spliced junction, and Northern blotting, which can separate RNA molecules of different size and abundance based on their speed in electrophoresis [25, 30, 34], can help to discern individual circRNAs from their linear counterparts. RNase R treatment-based strategies that preferentially degrade linear over circular RNAs, have been used to validate the presence of and enrichment of circRNAs in a total RNA pool [21, 137]. However, the real efficacy of such techniques remains controversial [45, 138] as demonstrated by one of the best characterized circRNAs, CiRS-7/CDR1as, which is exceptionally sensitive to RNase R treatment [21]. These conventional methods provide useful but limited information and, as in the case of Northern blotting, they are limited by low sensitivity, low throughput, and elaborate steps [139].
The use of microarrays with probes spanning back-splice junctions has also been employed as a suitable screening tool; however, this approach can sometimes lead to the erroneous detection of linear species and can generate data that is not easily consistent between studies [140]. Nonetheless, the identification and quantification of circRNAs requires especially designed bioinformatics pipelines. Over the years, several computational algorithms have been developed to detect non-linear splice events by sequence alignment of reads covering the back-splice site to the reference genome, including algorithms that can reconstruct full-length circular RNAs [141]. Differences in their sensitivity and precision can unfortunately lead to an underestimation and incomplete annotation of circRNAs [136, 142]. A similar challenge applies for both de novo and exon–intron sequence-based prediction tools that can often generate dramatic differences in output and therefore require further validation with other approaches [143]. A comprehensive evaluation of ~100 circRNA bioinformatics tools, including web services, databases, stand-alone programs and pipeline tools based on their performance and limitations, has been recently published [144].
Intracellular visualization, localization, and quantification of RNA molecules are critical for studying their biology and function. Several studies have employed RNA fluorescence in situ hybridization (RNA-FISH) [145] for the quantification and localization of circRNAs, including CiRS-7/CDR1as [146]. RNA-FISH has also been used to confirm the colocalization of circRNA and target miRNAs, such as CiRS-7/CDR1as and miR-135a in bladder cancer [147], circRHOBTB3 and miR-654-3p in gastric cancer cells [148], circFAM114A2 and miR-762 in urothelial bladder carcinoma [149]. However, probe designs that can specifically and uniquely target the back-splice junctions of circRNAs can often be challenging, making the overall technique time-consuming and costly for an efficient signal detection.
Advances in the field of circRNA research have paved the way for a number of novel assays with increased sensitivity and specificity that appear to detect more accurately low abundance circRNAs and hold great promise for their efficient annotation in the future [150]. Among several techniques, the recent development of reverse transcription-droplet digital polymerase chain reaction (RT-ddPCR) demonstrates the ability to provide absolute copy numbers and to detect low abundance circRNAs based on partitioning nucleic acids into nanoliter-sized droplets containing the target sequence for PCR amplification [151]. This approach has been successfully employed to profile circRNA levels in plasma. The method has led to the positive correlation between plasma levels of hsa_circ_0001017 and hsa_circ_0061276 with the overall survival of gastric cancer patients [152]. Particularly suitable for profiling circRNAs is the rolling cycle amplification (RCA) method in which a primer can bind to the junction site on the target circRNA and allow the reverse transcriptase to begin rolling cycle amplification. This produces a single-stranded cDNA of a long-chain of hundreds of repetitive fragments that amplify the signal [153]. A similar strategy based on pairs of stem-loop primers (SLPs) that recognize the junction sequence of circRNAs and form a double stem-loop DNA structure, can induce double exponential amplification (LAMP) and specifically detect circRNAs from linear RNAs [154].
Methods for the functional characterization of circRNAs
Beyond efficient detection, studies aimed at assessing circRNA function require particular attention. Albeit at different levels, circRNAs and linear RNAs transcribed at the same genomic locus are normally co-expressed. This makes gain- and loss-of-function studies particularly challenging since they are based on targeting the original loci and are therefore likely to affect the cognate linear RNAs too. In terms of selective degradation of circRNAs, of particular note is the newly developed RNA-guided, RNA-targeting Cas13 system (CRISPR–Cas13) [155, 156], which has been shown to be far more efficient and specific in circRNA knockdown, compared to standard RNAi approaches [157, 158]. By designing guide RNAs that target sequences spanning the back-splice junction, CRISPR–Cas13 can knock down circRNAs, without any impact on related linear mRNAs. This has enabled the efficient and specific knockdown of the oncogenic circFAM120A, which normally promotes cell proliferation by efficiently favoring the translation of its cognate linear mRNA (FAM120A) by competitively binding to IGF2BP2 (a translation inhibitor) [159]. Strategies based on expression vectors that drive almost exclusively circular and not linear exons [48] by employing flanking introns with base-pairing repeats, have also proven to be, with appropriate controls, hugely advantageous in studying new circRNA functions [62]. A successful genetically engineered mouse model harboring circRNA expression constructs, has recently been employed to study circRNAs in vivo in the context of melanoma [160].
Cutting-edge methodologies, such as the new Oxford Nanopore technology, are particularly suitable for long-read sequencing and may further help in providing information about the entire sequence of circRNAs [161]. Furthermore, the use of NanoString platform offers more accurate detection and quantification of individual circRNAs by using capture and reporter color-coded probes that jointly recognize the back-splice junction without the need for amplification or reverse transcription [162, 163]. For a detailed description of methods used to study and characterize circRNA functions and mechanisms of action, we recommend three excellent recent reviews [164,165,166].
CircRNAs as powerful biomarkers
CircRNAs have been linked to a variety of physiological conditions and cell biology features including stemness and pluripotency [44, 167,168,169,170,171,172] and are therefore potentially implicated in inducing and sustaining cancer development. Moreover, circRNAs have been correlated with some clinical characteristics such as the histological grade, size, metastasis stage and aggressiveness of cancer [163, 173].
Growing evidence suggests that circRNAs can be used as potential biomarkers for early-cancer detection, clinical diagnosis, prognosis and even used in monitoring response to therapy [174,175,176,177] (Fig. 5). Their unique expression patterns, molecular stability, specificity and broad distribution across human body compartments, make circRNAs accessible for relatively easy detection and quantification by liquid biopsy in body fluids, including blood, sputum and urine [81, 178]; the latter are more preferable and effective than tissue biopsy due to the minimal invasiveness and feasibility of repetitive sampling.

An up-to-date summary of circRNAs that show promise as clinical biomarkers (outmost circle) and/or as therapeutic targets (syringe icons pointing towards the chart), associated with different types of cancer. Created with BioRender.com.
CircRNAs as prognostic biomarkers
Some circRNAs have shown a strong prognostic potential including CiRS-7/CDR1as which has been found highly abundant in intratumoral stromal cells, extensively used as prognostic factor in carcinomas of colon [179], lung [180] and breast [181]. A strong potential for predicting gastric cancer prognosis has also been proposed for circERBB2, whose plasma levels in pre-operative gastric cancer patients significantly correlate with the occurrence of lymph node metastasis [177]. Whereas the detection of tumor-suppressor circRNAs such as circLARP4 has been associated with good prognosis in several different cancers [182,183,184,185], the detection of oncogenic circUBAP2 has been linked to unfavorable prognosis in HCC, breast cancer and osteosarcoma through the sponging of different miRNAs [159, 186, 187].
CircRNA panels and/or signatures appear to have a more robust prognostic value than single circRNAs. RNA-seq analysis of frozen tissues collected from post-operation has allowed to profile differential circRNA expression between patients with and without recurrence in four circRNA-based signatures (named circScores), thereby grouping colon cancer patients based on high- or low-risk of recurrence [188]. According to the standard of care for younger patients developed by the Nordic Lymphoma Group, the introduction of cytarabine-containing chemoimmunotherapy followed by autologous stem cells transplant increased mantle-cell lymphoma (MCL) patient’s survival. However, treated MCL patients are likely to experience continuous long-term relapse. Analysis on a cohort of samples from two clinical trials conducted by the Nordic Lymphoma Group, has allowed to profile circRNA expression patterns and help with the identification of high-risk disease patients treated with cytarabine-containing chemoimmunotherapy and autologous stem cells transplant [163].
CircRNAs as diagnostic biomarkers
CircRNA tissue and cell-type specificity allow better correlation with specific pathologies, including the ability to distinguish between cancer subtypes [81, 189,190,191,192] and clinical stages. This is particularly beneficial in early-cancer diagnosis and increases the chances of patient survival. In this regard, a group of researchers have developed a unique circRNA test that detects five circRNAs in urine-derived extracellular vesicles and has the potential to distinguish high-grade prostate cancer from benign prostatic hyperplasia [193]. CircRNA expression profiles could also help with the classification of breast cancer subtypes into HER2-positive, estrogen receptor-positive and triple negative breast cancers [191]. Similarly, the detection of low and high level of expression of circACVR2A and circCCNB1 respectively, has enabled the distinction between adenocarcinoma and squamous cell carcinoma in NSCLC [192]. Plasma-based circRNAs assays with high detection accuracy have also allowed the distinction of HCC patients with hepatitis B virus infection from healthy individuals and patients with chronic hepatitis B and liver cirrhosis [194].
Exosome-derived circRNAs are also useful diagnostic biomarkers depending on relative expression, stability, and exosome coupled targeted delivery pathways [40]. For instance, exosomal circRNAs enriched in serum have shown high potential for the early diagnosis of colorectal cancer [40]. An early diagnosis of the most frequent and deadly human brain cancer, glioblastoma multiforme, could instead be possible by monitoring downregulated levels of exosome-derived circHIPK3 and circSMARCA5 [195].
CircRNAs as both prognostic and diagnostic biomarkers
Some circRNAs could offer a double utility as both prognostic and diagnostic biomarkers. For example, circ0001785 not only shows higher diagnostic efficiency for breast cancer detection than the two most widely investigated biomarkers serum carcinoembryonic antigen (CEA) and cancer antigen 15-3 (CA15-3) [196], but also possesses strong prognostic potential in predicting the histological grade, TNM (Tumor, Node, Metastasis) stage and distant metastasis in breast cancer progression [196]. Similarly, circLDLRAD3 has been proposed as a promising biomarker in both the diagnosis and prognosis of pancreatic cancer since it was found to be elevated in the plasma of patients and strongly correlate with venous and lymphatic invasion [197].
CircRNAs as predictive biomarkers for cancer therapy
The efficacy of cancer therapy is often limited by intrinsic and acquired resistance. CircRNA expression has been proven to vary in response of tumor cells to chemotherapy, radiotherapy and immunotherapy through several mechanisms, thereby serving as a valuable indicator for clinicians to modify cancer patient treatment. For instance, circCRIM1 competitively binds to miR-422a and prevents the inhibitory effects of miR-422a on its target gene FOXQ1, which ultimately leads to metastasis in nasopharyngeal carcinoma, EMT and docetaxel chemoresistance [198]. In endometrial cancer, resistance to paclitaxel, is mediated by a key oncogenic circRNA (circ0007534) which sponges miR-625 and subsequently increases the expression of the miR-625 target gene ZEB2, a master regulator of EMT [199]. Interestingly, prostate cancer therapy with androgen receptors inhibitors such as enzalutamide, dramatically changes the expression of a pool of circRNAs in enzalutamide-resistant cells, opening new scenarios for understanding resistance mechanisms and offering novel opportunities for treating prostate cancer patients [200].
Given their circulating nature, exosomes can also transmit drug resistance between heterogeneous populations of tumor cells. CircRNAs with altered expression in drug-resistant cells can be transferred to drug-sensitive ones. For example, the exosomal circZNF91 functions as an exosomal cargo mediating the signal transmission between hypoxic and normoxic tumor cells in pancreatic cancer and promotes chemoresistance [201]. In a similar vein, the exosome-associated circ0032821 promotes oxaliplatin resistance in gastric cancer (GC) cells by regulating SOX9 via miR-515-5p [202].
Binding of the cell receptor PD-1 to its programmed death-ligand 1 (PD-L1) on tumor cells, activates downstream signaling pathways and inhibits T cell activation. Novel antibody inhibitors, such as anti-PD-1 and anti-PD-L1, are designed to restore the host antitumor immune response effectively, with an overall manageable toxicity compared to chemotherapy and radiotherapy. However, the development of resistance to immune checkpoint inhibitors (ICIs) is one of the major limitations in using novel anti-PD-1 and anti-PD-L1 therapies for treating cancer. CircRNAs play an important role in acquiring resistance features to these therapies by modulating the expression of key cancer pathways and associated immune populations from the tumor microenvironment. For example, in NSCLC circFGFR1 induces resistance to PD-1 antibodies by interacting with miR-381-3p and upregulating its target CXCR4, responsible for NSCLC progression and resistance [203]. In distant metastatic lesions and gastric cancer tissues, overexpressed circDLDG1 sponges miR-141-3p and increases the expression of CXCL12 which in turn promotes EMT, proliferation, metastasis and resistance to anti-PD-1 inhibitors [204]. Another anti-PD-1 resistance-associated circRNA, is circUHRF1 which is generally overexpressed in HCC tissue, cell lines, and linked with poor prognosis. CircUHRF1 is secreted via exosomes by HCC cells into the surrounding microenvironment and by sequestering and preventing miR-449c-5p binding to the checkpoint target gene “T cell immunoglobulin and mucin domain 3” (TIM-3), triggers natural killer (NK) cell dysfunction, immune evasion and resistance to anti-PD-1 immunotherapy [205].
Therapeutic application of circRNAs
The dysregulation of circRNAs can drive tumorigenesis and metastasis in various cancers, making them promising targets for cancer treatment (Fig. 5). Several therapeutic approaches aimed at modifying circRNA expression are currently under investigation. Preclinical studies in animal models are mostly focused on gain- or loss-of-function strategies through circRNA overexpression or knockdown.
Therapeutic applications based on circRNA knockdown
RNA interference (RNAi) accomplished by cytoplasmic delivery of small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) [206] has been used to silence single or multiple circRNAs in vivo. For example, treatment with shRNAs targeting circCUX1, a circRNA generally overexpressed in neuroblastoma (NB) that is able to promote cell proliferation and invasion via sponging miR-16-5p, has been shown to reduce tumor growth in mice efficiently [207]. Repression of tumorigenesis in mice has also been shown upon the silencing of circAGO2 [208], a circRNA that is up-regulated in several cancers and associated with poor prognosis. CircAGO2 interacts with HuR protein to facilitate HuR activation and enrichment on the 3’ UTR of target mRNAs, which reduces the accessibility for AGO2 binding and AGO2/miRNA-mediated silencing to mRNAs associated with cancer progression [208].
Single-stranded DNA antisense oligonucleotides (AONs) have also been employed to inhibit selectively or degrade oncogenic circRNAs. For example, AONs targeting circLONP2, which enhances metastasis and invasiveness of colorectal cancer cells by favoring the maturation and the exosomal dissemination of miR-7, dramatically reduce the extent of metastasis to foreign organs in vivo [209].
The delivery of RNAi molecules can be better accomplished with the use of nanoparticles. Gold nanoparticles (AuNPs), commonly associated with a linker such as PEG or polyethylenimine, can be conjugated with siRNAs, shRNAs or AONs and ensure a more efficient drug delivery in animal studies thanks to their high stability, and easily modifiable surface. Delivery of AuNPs with siRNAs targeting circDNMT1 has been shown to suppress breast tumor growth successfully as well as enhancing the survival in mice [210]. A similar approach with AuNP conjugated with AONs aimed to block the binding sites on circCcnb1 for both Ccnb1 and Cdk1, has also shown to inhibit tumor growth and promote mouse survival [211].
Translated circRNAs with important oncogenic roles in cancer can also be targeted by specific drug inhibitors. This is the case of HER2-103aa which plays a role in tumorigenesis and shares sequence similarity with HER2 CR1 domain (the domain that is targeted by pertuzumab). Treatment with pertuzumab has proven to reduce significantly the tumorigenicity of both circHER2 and its encoded 103 amino acids polypeptide (HER2-103aa) expressing cells in vivo [212].
Therapeutic applications based on circRNA overexpression
CircRNA expression can be increased by direct delivery into cells. This approach is particularly effective in antagonizing oncogenic miRNAs by exploiting the property of circRNAs to act as ceRNAs/miRNA sponges. To obviate possible degradation effects, more efficient systems have adopted cassettes cloned inside lentivirus or adeno-associated virus vectors [213]. However, these vectors may lead to unforeseen adverse effects by producing simultaneously a substantial number of unnecessary cognate mRNA molecules. Alternative delivery of circRNA expression cassettes can be achieved through encapsulation within lipid and/or polymer nanoparticles. To illustrate the latter, a group of researchers ectopically overexpressed circFoxo3 by using plasmid-PEG- AuNPs target to cancer cells and demonstrated that circFoxo3 triggers stress- induced apoptosis, inhibition of tumor xenograft growth in vivo and increases overall survival [117].
Therapeutic strategies based on circRNAs in response to cancer treatment failure
Gain- or loss-of-function-based therapeutic strategies targeting crucial circRNAs associated with cancer therapy resistance have also been investigated to overcome treatment failure. CircRNAs, such as CircRNA-SORE, have been shown to interfere with the activity of tyrosine kinase inhibitors, such as sorafenib. Upregulation of circRNA-SORE in sorafenib-resistant HCC cells ensures the cytoplasmic retention of the oncogenic protein YBX1 and prevents its ubiquitination and degradation by PRP19 in the nucleus and thereby favors resistance to sorafenib. Moreover, circRNA-SORE is loaded into exosomes that are trafficked from sorafenib-resistant cancer cells to sensitive cancer cells, and this helps to spread the resistance phenotype. By treating mice bearing subcutaneous sorafenib-resistant patient-derived xenografts with siRNAs targeting circRNA-SORE, researchers have demonstrated that the responsiveness to sorafenib treatment can be restored [214]. In a similar vein, the 174 amino acids long peptide (AKT3-174aa) encoded by circAKT3 has been shown to have important anti-tumorigenic roles and to be negatively involved in radiotherapy resistance. Mechanistically, AKT3-174aa competes with active phosphorylated PDK1, reduces AKT-thr308 phosphorylation, and acts as a negative regulator modulating PI3K/AKT signal intensity. This reduces cellular proliferation and antagonizes radiotherapy resistance. Downregulation of circAKT3 in glioblastoma causes the subsequent decrease in levels of its associated peptide and the development of a malignant glioblastoma phenotype. The injection of AKT3-174aa in mice models appears to restore glioblastoma cell sensitivity to radiotherapy [215]. Likewise, low levels of circ0025202 affecting the miR-182-5p/FOXO3a axis have been associated with the resistance of one of the most commonly used hormone therapy for hormone receptor (HR)-positive breast cancer patients (tamoxifen). In vivo experiments showed that overexpression of circ0025202 could shrink tumor growth and enhance tamoxifen efficacy [216]. Lastly, studies have revealed that circRNA17 can downregulate the expression of an androgen receptor splice variant 7 via sponging of one of its 3’ UTR targets, miR-181c-5p [217]. CircRNA17 overexpression in in vivo mouse model xenografted with enzalutamide-resistant cells, has shown to restore sensitivity to enzalutamide, used for prostate cancer treatment [217].
As discussed earlier, circUHRF1 plays a crucial role in sustaining anti-PD-1 resistance in HCC [218]. Using an in vivo xenograft model researchers have shown that the therapeutic suppression of circUHRF1 via shRNAs modulates the response to anti-PD-1 treatment and improves overall survival. This may prove an effective method of reversing resistance for ICIs by acting both on tumor cells and on associated dysfunctional immune microenvironment [218]. Immuno-suppression and development of anti-PD-1 therapy resistance in HCC have also been associated with circMET via the miR-30-5p/snail/DPP-4 axis. Treatment with sitagliptin, a dipeptidyl peptidase-4 (DPP-4) inhibitor generally used to treat type 2 diabetes, seems to obviate the resistant effects induced by the circMET. A combination of both sitagliptin and anti-PD-1 molecules has been shown to improve antitumor immunity in immunocompetent mice and is likely to be more effective in treating patients with HCC [219].
Further research is needed before specific circRNAs can be effectively exploited as clinically useful biomarkers or as therapeutic targets for cancer treatment. On-going studies are currently assessing the potential therapeutic benefits for cancer patients by bringing circRNA biology into clinical practice. Table 3 provides a snapshot of the range of projects currently in progress.
Concluding remarks, perspectives and a note of caution
As sequencing approaches improve in terms of depth, accuracy, and read length [220], the annotation of new uncharacterized circRNAs, especially low abundance ones, continues to grow. Beyond their detection, a careful investigation to distinguish what is a splicing artifact from a functional circRNA remains essential. Appropriate controls and accurate validation using multiple approaches remain crucial to rule out false positive circRNAs. To tackle these challenges a series of guidelines in the field of circRNAs research have recently been published [221].
Many open questions regarding their biogenesis and biological functions, remain unanswered. It remains unclear whether or not they are exclusively co- or post-transcriptionally generated, how they are ultimately degraded, to what extent their structures might confer functional differences compared to their linear RNA counterparts and what proportion are translated into proteins. Moreover, many functional aspects of circRNAs might have not been uncovered yet due to the limitations of current approaches.
From a clinical perspective, the advent of circRNA research has opened up new exciting possibilities in cancer research, for better diagnosis and novel therapies. CircRNAs have been associated with a variety of physiological conditions and cell biology features including stemness and pluripotency. It has also been demonstrated that they have potential utility as valid biomarkers for early-cancer detection, diagnosis, prognosis and even prediction of response to therapies. However, how they impact cancer diagnosis and prevention remains debatable and sometimes even contradictory. CircRNAs often participate in more than one molecular mechanism in many tissues and diseases; targeting circRNAs therapeutically might lead to off-target effects in non-cancerous cells and tissue, making their clinical translation particularly challenging. Moreover, the discovery of multiple miRNAs sponged by the same circRNAs in different cancer types suggests that their contribution to a specific cancer phenotype is likely to be context-dependent. Additional related controversies surround the stoichiometry circRNA–miRNA–mRNA and the role of circRNAs as competitive endogenous RNAs [222, 223]. This is poorly understood in physiological conditions suggesting that the number of circRNAs able to effectively contribute to tumorigenesis by sponging miRNAs could be far lower than initially proposed. Measuring correct copy numbers and circRNA/miRNA ratio in cells and tissues remains fundamental in studying circRNAs and a necessary step required to understand how they function in normal development and disease.
The integration of artificial intelligence (AI) and circRNA transcriptome analysis holds great promise for advancing our understanding of this complex biological system, undoubtedly facilitating diagnostics and therapeutics in various pathological contexts including cancer. New approaches based on AI might help to identify shared and unique characteristics in features of circRNAs, such as their expression levels, alternative splicing patterns, co-expression networks, or specific structural motifs, all in different conditions. However, the success of using circRNA transcriptome data combined with AI for building classifiers will rely on high-quality data generation, careful experimental design, and rigorous computational analysis. The interpretability of AI models in this context remains challenging, as circRNA properties and functions are still not fully elucidated.
In summary, there is undoubtedly tremendous potential for circRNAs to have a significant impact on cancer diagnosis and treatment. The initial studies of circRNAs have necessarily focused on their discovery and characterization; what needs to follow in the near future is more detailed biological validation with patient samples in the context of clinical research. These experiments will need to be further corroborated with detailed functional studies in suitable model organisms. Once a more complete understanding of their functional biology is obtained, it will be possible to exploit the full potential of circRNAs in the clinic.
Data availability
All data are included in the published review manuscript.
References
Kolakofsky D. Isolation and characterization of Sendai virus DI-RNAs. Cell 1976;8. https://doi.org/10.1016/0092-8674(76)90223-3.
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single stranded covalently closed circular RNA molecules existing as highly base paired rod like structures. Proc Natl Acad Sci USA. 1976. https://doi.org/10.1073/pnas.73.11.3852.
Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979. https://doi.org/10.1038/280339a0.
Kos A, Dijkema R, Arnberg AC, van der Meide PH, Schellekens H. The hepatitis delta (δ) virus possesses a circular RNA. Nature. 1986. https://doi.org/10.1038/323558a0.
Zaug AJ, Grabowski PJ, Cech TR. Autocatalytic cyclization of an excised intervening sequence RNA is a cleavage-ligation reaction. Nature 1983;301. https://doi.org/10.1038/301578a0.
Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of tetrahymena. Cell. 1982;31. https://doi.org/10.1016/0092-8674(82)90414-7.
Grabowski PJ, Zaug AJ, Cech TR. The intervening sequence of the ribosomal RNA precursor is converted to a circular RNA in isolated nuclei of tetrahymena. Cell 1981;23. https://doi.org/10.1016/0092-8674(81)90142-2.
Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, et al. Scrambled exons. Cell. 1991. https://doi.org/10.1016/0092-8674(91)90244-S.
Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, et al. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell. 1993. https://doi.org/10.1016/0092-8674(93)90279-Y.
Cocquerelle C, Daubersies P, Majerus MA, Kerckaert JP, Bailleul B. Splicing with inverted order of exons occurs proximal to large introns. EMBO J. 1992. https://doi.org/10.1002/j.1460-2075.1992.tb05148.x.
Zaphiropoulos PG. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: correlation with exon skipping. Proc Natl Acad Sci USA. 1996. https://doi.org/10.1073/pnas.93.13.6536.
Zaphiropoulos PG. Exon skipping and circular RNA formation in transcripts of the human cytochrome P-450 2C18 gene in epidermis and of the rat androgen binding protein gene in testis. Mol Cell Biol. 1997. https://doi.org/10.1128/mcb.17.6.2985.
Surono A. Circular dystrophin RNAs consisting of exons that were skipped by alternative splicing. Hum Mol Genet. 1999. https://doi.org/10.1093/hmg/8.3.493.
Li XF, Lytton J. A circularized sodium-calcium exchanger exon 2 transcript. J Biol Chem. 1999. https://doi.org/10.1074/jbc.274.12.8153.
Houseley JM, Garcia-Casado Z, Pascual M, Paricio N, O’Dell KMC, Monckton DG, et al. Noncanonical RNAs from transcripts of the Drosophila muscleblind gene. J Heredity. 2006. https://doi.org/10.1093/jhered/esj037.
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:1–15.
Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, et al. MiRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011. https://doi.org/10.1038/emboj.2011.359.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013. https://doi.org/10.1038/nature11928.
Cocquerelle C, Mascrez B, Hétuin D, Bailleul B. Mis-splicing yields circular RNA molecules. FASEB J. 1993. https://doi.org/10.1096/fasebj.7.1.7678559.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE. 2012. https://doi.org/10.1371/journal.pone.0030733.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013. https://doi.org/10.1261/rna.035667.112.
Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15. https://doi.org/10.1038/nrg3662.
Barrett SP, Wang PL, Salzman J. Circular RNA biogenesis can proceed through an exon-containing lariat precursor. Elife. 2015. https://doi.org/10.7554/eLife.07540.
Schindewolf C, Braun S, Domdey H. In vitro generation of a circular exon from a linear pre-mRNA transcript. Nucleic Acids Res. 1996. https://doi.org/10.1093/nar/24.7.1260.
Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung LH, et al. Exon circularization requires canonical splice signals. Cell Rep. 2015. https://doi.org/10.1016/j.celrep.2014.12.002.
Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J, et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016. https://doi.org/10.1101/gr.202895.115.
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, et al. CircRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014. https://doi.org/10.1016/j.molcel.2014.08.019.
Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, et al. The biogenesis of nascent circular RNAs. Cell Rep. 2016;15. https://doi.org/10.1016/j.celrep.2016.03.058.
Enuka Y, Lauriola M, Feldman ME, Sas-Chen A, Ulitsky I, Yarden Y. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res. 2016. https://doi.org/10.1093/nar/gkv1367.
Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, et al. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014. https://doi.org/10.1016/j.celrep.2014.10.062.
Cortés-López M, Gruner MR, Cooper DA, Gruner HN, Voda AI, van der Linden AM, et al. Global accumulation of circRNAs during aging in Caenorhabditis elegans. BMC Genomics. 2018. https://doi.org/10.1186/s12864-017-4386-y.
Hall H, Medina P, Cooper DA, Escobedo SE, Rounds J, Brennan KJ, et al. Transcriptome profiling of aging Drosophila photoreceptors reveals gene expression trends that correlate with visual senescence. BMC Genomics. 2017. https://doi.org/10.1186/s12864-017-4304-3.
Gruner H, Cortés-López M, Cooper DA, Bauer M, Miura P. CircRNA accumulation in the aging mouse brain. Sci Rep. 2016. https://doi.org/10.1038/srep38907.
Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Hanan M, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2014. https://doi.org/10.1016/j.molcel.2015.03.027.
You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci. 2015. https://doi.org/10.1038/nn.3975.
Venø MT, Hansen TB, Venø ST, Clausen BH, Grebing M, Finsen B, et al. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015. https://doi.org/10.1186/s13059-015-0801-3.
Dou Y, Cha DJ, Franklin JL, Higginbotham JN, Jeppesen DK, Weaver AM, et al. Circular RNAs are down-regulated in KRAS mutant colon cancer cells and can be transferred to exosomes. Sci Rep. 2016;6. https://doi.org/10.1038/srep37982.
Lasda E, Parker R. Circular RNAs co-precipitate with extracellular vesicles: a possible mechanism for circrna clearance. PLoS ONE 2016;11. https://doi.org/10.1371/journal.pone.0148407.
Preußer C, Hung LH, Schneider T, Schreiner S, Hardt M, Moebus A, et al. Selective release of circRNAs in platelet-derived extracellular vesicles. J Extracell Vesicles. 2018;7. https://doi.org/10.1080/20013078.2018.1424473.
Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25. https://doi.org/10.1038/cr.2015.82.
Wang Y, Liu J, Ma J, Sun T, Zhou Q, Wang W, et al. Exosomal circRNAs: Biogenesis, effect and application in human diseases. Mol Cancer. 2019;18. https://doi.org/10.1186/s12943-019-1041-z.
Dai X, Chen C, Yang Q, Xue J, Chen X, Sun B, et al. Exosomal circRNA_100284 from arsenite-transformed cells, via microRNA-217 regulation of EZH2, is involved in the malignant transformation of human hepatic cells by accelerating the cell cycle and promoting cell proliferation. Cell Death Dis. 2018;9:1–14.
Xiao H, Shi J. Exosomal circular RNA_400068 promotes the development of renal cell carcinoma via the miR-210-5p/SOCS1 axis. Mol Med Rep. 2020;22:4810.
Moldovan LI, Hansen TB, Venø MT, Okholm TLH, Andersen TL, Hager H, et al. High-throughput RNA sequencing from paired lesional- and non-lesional skin reveals major alterations in the psoriasis circRNAome. BMC Med Genomics. 2019;12. https://doi.org/10.1186/s12920-019-0616-2.
Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, et al. Correlation of circular RNA abundance with proliferation - exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015;5. https://doi.org/10.1038/srep08057.
Li X, Yang L, Chen LL. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. 2018. https://doi.org/10.1016/j.molcel.2018.06.034.
Wang Y, Wang Z. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015;21. https://doi.org/10.1261/rna.048272.114.
Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S, et al. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 2015;29. https://doi.org/10.1101/gad.270421.115.
Liang D, Tatomer DC, Luo Z, Wu H, Yang L, Chen LL, et al. The output of protein-coding genes shifts to circular RNAs when the Pre-mRNA processing machinery is limiting. Mol Cell. 2017;68. https://doi.org/10.1016/j.molcel.2017.10.034.
Kristensen LS, Okholm TLH, Venø MT, Kjems J. Circular RNAs are abundantly expressed and upregulated during human epidermal stem cell differentiation. RNA Biol. 2018;15. https://doi.org/10.1080/15476286.2017.1409931.
Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, et al. Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell. 2016;19. https://doi.org/10.1016/j.stem.2016.06.020.
Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011. https://doi.org/10.1038/nature10442.
Jakobsen T, Dahl M, Dimopoulos K, Grønbæk K, Kjems J, Kristensen LS. Genome-wide circular rna expression patterns reflect resistance to immunomodulatory drugs in multiple myeloma cells. Cancers. 2021;13. https://doi.org/10.3390/cancers13030365.
Ferreira HJ, Davalos V, de Moura MC, Soler M, Perez-Salvia M, Bueno-Costa A, et al. Circular RNA CpG island hypermethylation-associated silencing in human cancer. Oncotarget. 2018;9. https://doi.org/10.18632/oncotarget.25673.
Hanniford D, Ulloa-Morales A, Karz A, Berzoti-Coelho MG, Moubarak RS, Sánchez-Sendra B, et al. Epigenetic Silencing of CDR1as Drives IGF2BP3-Mediated Melanoma Invasion and Metastasis. Cancer Cell. 2020;37. https://doi.org/10.1016/j.ccell.2019.12.007.
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013. https://doi.org/10.1016/j.molcel.2013.08.017.
Ebbesen KK, Hansen TB, Kjems J. Insights into circular RNA biology. RNA Biol. 2017;14. https://doi.org/10.1080/15476286.2016.1271524.
Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32. https://doi.org/10.1038/nbt.2890.
Huang C, Liang D, Tatomer DC, Wilusz JE. A length-dependent evolutionarily conserved pathway controls nuclear export of circular RNAs. Genes Dev. 2018;32. https://doi.org/10.1101/gad.314856.118.
Zhang XO, Wang H bin, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014. https://doi.org/10.1016/j.cell.2014.09.001.
Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10. https://doi.org/10.1016/j.celrep.2014.12.019.
Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014;28. https://doi.org/10.1101/gad.251926.114.
Pasman Z, Been MD, Garcia-Blanco MA. Exon circularization in mammalian nuclear extracts. RNA. 1996;2.
Dubin RA, Kazmi MA, Ostrer H. Inverted repeats are necessary for circularization of the mouse testis Sry transcript. Gene. 1995;167. https://doi.org/10.1016/0378-1119(95)00639-7.
Wilusz JE. Repetitive elements regulate circular RNA biogenesis. Mob Genet Elements. 2015;5. https://doi.org/10.1080/2159256X.2015.1045682.
Dong R, Ma XK, Chen LL, Yang L. Increased complexity of circRNA expression during species evolution. RNA Biol 2017;14. https://doi.org/10.1080/15476286.2016.1269999.
Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015. https://doi.org/10.1016/j.cell.2015.02.014.
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22. https://doi.org/10.1038/nsmb.2959.
Errichelli L, Dini Modigliani S, Laneve P, Colantoni A, Legnini I, Capauto D et al. FUS affects circular RNA expression in murine embryonic stem cell-derived motor neurons. Nat Commun. 2017;8. https://doi.org/10.1038/ncomms14741.
Fei T, Chen Y, Xiao T, Li W, Cato L, Zhang P, et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc Natl Acad Sci USA. 2017;114:E5207–E5215.
Li Y, Chen B, Zhao J, Li Q, Chen S, Guo T, et al. HNRNPL circularizes ARHGAP35 to produce an oncogenic protein. Adv Sci. 2021;8. https://doi.org/10.1002/advs.202001701.
Koh HR, Xing L, Kleiman L, Myong S. Repetitive RNA unwinding by RNA helicase A facilitates RNA annealing. Nucleic Acids Res. 2014;42. https://doi.org/10.1093/nar/gku523.
Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2. https://doi.org/10.1371/journal.pbio.0020391.
Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22. https://doi.org/10.1038/nbt996.
Wen X, Huang X, Mok BW-Y, Chen Y, Zheng M, Lau S-Y, et al. NF90 Exerts Antiviral Activity through Regulation of PKR Phosphorylation and Stress Granules in Infected Cells. J Immunol. 2014;192. https://doi.org/10.4049/jimmunol.1302813.
Li X, Liu CX, Xue W, Zhang Y, Jiang S, Yin QF, et al. Coordinated circRNA Biogenesis and Function with NF90/NF110 in Viral Infection. Mol Cell. 2017;67. https://doi.org/10.1016/j.molcel.2017.05.023.
Talhouarne GJS, Gall JG. Lariat intronic RNAs in the cytoplasm of Xenopus tropicalis oocytes. RNA. 2014;20. https://doi.org/10.1261/rna.045781.114.
Qian L, Vu MN, Carter M, Wilkinson MF. A spliced intron accumulates as a lariat in the nucleus of T cells. Nucleic Acids Res. 1992;20. https://doi.org/10.1093/nar/20.20.5345.
Kopczynski CC, Muskavitch MAT. Introns excised from the Delta primary transcript are localized near sites of Delta transcription. J Cell Biol. 1992;119. https://doi.org/10.1083/jcb.119.3.503.
Gao Y, Wang J, Zhao F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16:1–16.
Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao L, Zhang Y, et al. The Landscape of Circular RNA in Cancer. Cell 2019;176. https://doi.org/10.1016/j.cell.2018.12.021.
Grosso AR, Leite AP, Carvalho S, Matos MR, Martins FB, Vítor AC, et al. Pervasive transcription read-through promotes aberrant expression of oncogenes and RNA chimeras in renal carcinoma. Elife. 2015;4. https://doi.org/10.7554/eLife.09214.
Tan S, Gou Q, Pu W, Guo C, Yang Y, Wu K, et al. Circular RNA F-circEA produced from EML4-ALK fusion gene as a novel liquid biopsy biomarker for non-small cell lung cancer. Cell Res. 2018;28. https://doi.org/10.1038/s41422-018-0033-7.
Guarnerio J, Bezzi M, Jeong JC, Paffenholz SV, Berry K, Naldini MM, et al. Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell. 2016;165. https://doi.org/10.1016/j.cell.2016.03.020.
Tan S, Sun D, Pu W, Gou Q, Guo C, Gong Y, et al. Circular RNA F-circEA-2a derived from EML4-ALK fusion gene promotes cell migration and invasion in non-small cell lung cancer. Mol Cancer. 2018;17. https://doi.org/10.1186/s12943-018-0887-9.
Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, et al. N6-methyladenosine modification controls circular RNA immunity. Mol Cell. 2019;76. https://doi.org/10.1016/j.molcel.2019.07.016.
Wu J, Guo X, Wen Y, Huang S, Yuan X, Tang L, et al. N6-Methyladenosine modification opens a new chapter in circular RNA biology. Front Cell Dev Biol. 2021;9. https://doi.org/10.3389/fcell.2021.709299.
Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, van Wittenberghe N, et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. 2017;20. https://doi.org/10.1016/j.celrep.2017.08.027.
Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, et al. N 6-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. 2019;10. https://doi.org/10.1038/s41467-019-12651-2.
di Timoteo G, Dattilo D, Centrón-Broco A, Colantoni A, Guarnacci M, Rossi F, et al. Modulation of circRNA Metabolism by m6A Modification. Cell Rep. 2020;31. https://doi.org/10.1016/j.celrep.2020.107641.
Xu C, Zhang J. Mammalian circular RNAs result largely from splicing errors. Cell Rep. 2021;36. https://doi.org/10.1016/j.celrep.2021.109439.
Glažar P, Papavasileiou P, Rajewsky N. CircBase: A database for circular RNAs. RNA. 2014;20. https://doi.org/10.1261/rna.043687.113.
Ji P, Wu W, Chen S, Zheng Y, Zhou L, Zhang J, et al. Expanded Expression Landscape and Prioritization of Circular RNAs in Mammals. Cell Rep. 2019;26. https://doi.org/10.1016/j.celrep.2019.02.078.
Wu W, Ji P, Zhao F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020;21. https://doi.org/10.1186/s13059-020-02018-y.
Dong R, Ma XK, Li GW, Yang L. CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genomics Proteomics Bioinformatics. 2018;16. https://doi.org/10.1016/j.gpb.2018.08.001.
Xu X, Zhang J, Tian Y, Gao Y, Dong X, Chen W, et al. CircRNA inhibits DNA damage repair by interacting with host gene. Mol Cancer. 2020;19. https://doi.org/10.1186/s12943-020-01246-x.
He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333. https://doi.org/10.1126/science.1210944.
Chen N, Zhao G, Yan X, Lv Z, Yin H, Zhang S, et al. A novel FLI1 exonic circular RNA promotes metastasis in breast cancer by coordinately regulating TET1 and DNMT1. Genome Biol. 2018;19. https://doi.org/10.1186/s13059-018-1594-y.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–8.
Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17. https://doi.org/10.1038/nrg.2016.20.
Panda AC. Circular RNAs act as miRNA sponges. In: Advances in experimental medicine and biology; 2018. https://doi.org/10.1007/978-981-13-1426-1_6.
Yu L, Gong X, Sun L, Zhou Q, Lu B, Zhu L. The circular RNA Cdr1as act as an oncogene in hepatocellular carcinoma through targeting miR-7 expression. PLoS ONE. 2016;11. https://doi.org/10.1371/journal.pone.0158347.
Xu XW, Zheng BA, Hu ZM, Qian ZY, Huang CJ, Liu XQ, et al. Circular RNA hsa_circ_000984 promotes colon cancer growth and metastasis by sponging miR-106b. Oncotarget. 2017;8. https://doi.org/10.18632/oncotarget.21748.
Verduci L, Ferraiuolo M, Sacconi A, Ganci F, Vitale J, Colombo T, et al. The oncogenic role of circPVT1 in head and neck squamous cell carcinoma is mediated through the mutant p53/YAP/TEAD transcription-competent complex. Genome Biol. 2017;18. https://doi.org/10.1186/s13059-017-1368-y.
Zheng J, Liu X, Xue Y, Gong W, Ma J, Xi Z, et al. TTBK2 circular RNA promotes glioma malignancy by regulating miR-217/HNF1β/Derlin-1 pathway. J Hematol Oncol. 2017;10. https://doi.org/10.1186/s13045-017-0422-2.
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7. https://doi.org/10.1038/ncomms11215.
Abdelmohsen K, Panda AC, De S, Grammatikakis I, Kim J, Ding J, et al. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/β-catenin pathway. Oncotarget. 2015;5.
Zhao W, Wang S, Qin T, Wang W. Circular RNA (circ-0075804) promotes the proliferation of retinoblastoma via combining heterogeneous nuclear ribonucleoprotein K (HNRNPK) to improve the stability of E2F transcription factor 3 E2F3. J Cell Biochem. 2020;121:3516–25.
Rossi F, Beltran M, Damizia M, Grelloni C, Colantoni A, Setti A, et al. Circular RNA ZNF609/CKAP5 mRNA interaction regulates microtubule dynamics and tumorigenicity. Mol Cell. 2022;82. https://doi.org/10.1016/j.molcel.2021.11.032.
Huang Q, Guo H, Wang S, Ma Y, Chen H, Li H, et al. A novel circular RNA, circXPO1, promotes lung adenocarcinoma progression by interacting with IGF2BP1. Cell Death Dis. 2020;11. https://doi.org/10.1038/S41419-020-03237-8.
Hentze MW, Preiss T. Circular RNAs: splicing’s enigma variations. EMBO J. 2013;32:923–5.
Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z, et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet. 2012;44. https://doi.org/10.1038/ng.2434.
Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7. https://doi.org/10.1038/ncomms12429.
Lou J, Hao Y, Lin K, Lyu Y, Chen M, Wang H, et al. Circular RNA CDR1as disrupts the p53/MDM2 complex to inhibit Gliomagenesis. Mol Cancer. 2020;19. https://doi.org/10.1186/s12943-020-01253-y.
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44. https://doi.org/10.1093/nar/gkw027.
Abdelmohsen K, Panda AC, Munk R, Grammatikakis I, Dudekula DB, De S, et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017;14. https://doi.org/10.1080/15476286.2017.1279788.
Du WW, Fang L, Yang W, Wu N, Awan FM, Yang Z, et al. Induction of tumor apoptosis through a circular RNA enhancing Foxo3 activity. Cell Death Differ 2017;24. https://doi.org/10.1038/cdd.2016.133.
Li B, Zhu L, Lu C, Wang C, Wang H, Jin H, et al. circNDUFB2 inhibits non-small cell lung cancer progression via destabilizing IGF2BPs and activating anti-tumor immunity. Nat Commun. 2021;12. https://doi.org/10.1038/s41467-020-20527-z.
Yang Q, Du WW, Wu N, Yang W, Awan FM, Fang L, et al. A circular RNA promotes tumorigenesis by inducing c-myc nuclear translocation. Cell Death Differ. 2017;24. https://doi.org/10.1038/cdd.2017.86.
Legnini I, di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell. 2017;66:22–37.e9.
He T, Yuan C, Zhao C. Long intragenic non-coding RNA p53-induced transcript (LINC-PINT) as a novel prognosis indicator and therapeutic target in cancer. Biomed Pharmacother. 2021;143. https://doi.org/10.1016/J.BIOPHA.2021.112127.
Fan X, Yang Y, Chen C, Wang Z. Pervasive translation of circular RNAs driven by short IRES-like elements. https://doi.org/10.1038/s41467-022-31327-y.
Stagsted LVW, Nielsen KM, Daugaard I, Hansen TB. Noncoding AUG circRNAs constitute an abundant and conserved subclass of circles. Life Sci Alliance. 2019;2. https://doi.org/10.26508/lsa.201900398.
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, et al. Translation of CircRNAs. Mol Cell. 2017;66. https://doi.org/10.1016/j.molcel.2017.02.021.
Hansen TB. Signal and noise in circRNA translation. Methods. 2021;196. https://doi.org/10.1016/j.ymeth.2021.02.007.
Zhang M, Huang N, Yang X, Luo J, Yan S, Xiao F, et al. A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene. 2018;37. https://doi.org/10.1038/s41388-017-0019-9.
Yang Y, Gao X, Zhang M, Yan S, Sun C, Xiao F, et al. Novel role of FBXW7 circular RNA in repressing glioma tumorigenesis. J Natl Cancer Inst. 2018;110. https://doi.org/10.1093/jnci/djx166.
Zheng X, Chen L, Zhou Y, Wang Q, Zheng Z, Xu B, et al. A novel protein encoded by a circular RNA circPPP1R12A promotes tumor pathogenesis and metastasis of colon cancer via Hippo-YAP signaling. Mol Cancer. 2019;18. https://doi.org/10.1186/s12943-019-1010-6.
Gao X, Xia X, Li F, Zhang M, Zhou H, Wu X, et al. Circular RNA-encoded oncogenic E-cadherin variant promotes glioblastoma tumorigenicity through activation of EGFR–STAT3 signalling. Nat Cell Biol. 2021;23. https://doi.org/10.1038/s41556-021-00639-4.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N 6 -methyladenosine. Cell Res. 2017;27. https://doi.org/10.1038/cr.2017.31.
Zhao J, Lee EE, Kim J, Yang R, Chamseddin B, Ni C, et al. Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat Commun. 2019;10. https://doi.org/10.1038/s41467-019-10246-5.
Dragomir MP, Manyam GC, Ott LF, Berland L, Knutsen E, Ivan C, et al. Funcpep: a database of functional peptides encoded by non-coding rnas. Noncoding RNA. 2020;6. https://doi.org/10.3390/ncrna6040041.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63.
Gerstein MB, Rozowsky J, Yan KK, Wang D, Cheng C, Brown JB, et al. Comparative analysis of the transcriptome across distant species. Nature. 2014;512:445–8.
Mercer TR, Gerhardt DJ, Dinger ME, Crawford J, Trapnell C, Jeddeloh JA, et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol. 2011;30:99–104.
Hansen TB, Venø MT, Damgaard CK, Kjems J. Comparison of circular RNA prediction tools. Nucleic Acids Res. 2016;44. https://doi.org/10.1093/NAR/GKV1458.
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-Type Specific Features of Circular RNA Expression. PLoS Genet. 2013. https://doi.org/10.1371/journal.pgen.1003777.
Vincent HA, Deutscher MP. Substrate recognition and catalysis by the exoribonuclease RNase R. J Biol Chem. 2006;281. https://doi.org/10.1074/jbc.M606744200.
D’Ambra E, Morlando M. Study of circular RNA expression by nonradioactive northern blot procedure. Methods Mol Biol. 2021;2348:371–83.
Li S, Teng S, Xu J, Su G, Zhang Y, Zhao J, et al. Microarray is an efficient tool for circRNA profiling. Brief Bioinform. 2018;20. https://doi.org/10.1093/bib/bby006.
Zheng Y, Ji P, Chen S, Hou L, Zhao F. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med. 2019;11. https://doi.org/10.1186/s13073-019-0614-1.
Zeng X, Lin W, Guo M, Zou Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol. 2017;13. https://doi.org/10.1371/journal.pcbi.1005420.
Szabo L, Salzman J. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet. 2016;17. https://doi.org/10.1038/nrg.2016.114.
Chen L, Wang C, Sun H, Wang J, Liang Y, Wang Y, et al. The bioinformatics toolbox for circRNA discovery and analysis. Brief Bioinform. 2021;22. https://doi.org/10.1093/bib/bbaa001.
Lim AST, Lim TH. Fluorescence in situ hybridization on tissue sections. Methods Mol Biol. 2017;1541. https://doi.org/10.1007/978-1-4939-6703-2_11.
Kocks C, Boltengagen A, Piwecka M, Rybak-Wolf A, Rajewsky N. Single-molecule fluorescence in situ hybridization (FISH) of circular RNA CDR1as. Methods Mol Biol. 2018;1724:77–96.
Li P, Yang X, Yuan W, Yang C, Zhang X, Han J, et al. CircRNA-Cdr1as exerts anti-oncogenic functions in bladder cancer by sponging microRNA-135a. Cell Physiol Biochem. 2018;46:1606–16.
Deng G, Mou T, He J, Chen D, Lv D, Liu H, et al. Circular RNA circRHOBTB3 acts as a sponge for miR-654-3p inhibiting gastric cancer growth. J Exp Clin Cancer Res. 2020;39. https://doi.org/10.1186/S13046-019-1487-2.
Liu T, Lu Q, Liu J, Xie S, Feng B, Zhu W, et al. Circular RNA FAM114A2 suppresses progression of bladder cancer via regulating ∆NP63 by sponging miR-762. Cell Death Dis. 2020;11:1–14.
Mi Z, Zhongqiang C, Caiyun J, Yanan L, Jianhua W, Liang L. Circular RNA detection methods: a minireview. Talanta. 2022;238. https://doi.org/10.1016/J.TALANTA.2021.123066.
Chen DF, Zhang LJ, Tan K, Jing Q. Application of droplet digital PCR in quantitative detection of the cell-free circulating circRNAs. Biotechnol Biotechnol Equip. 2018;32. https://doi.org/10.1080/13102818.2017.1398596.
Li T, Shao Y, Fu L, Xie Y, Zhu L, Sun W, et al. Plasma circular RNA profiling of patients with gastric cancer and their droplet digital RT-PCR detection. J Mol Med. 2018;96. https://doi.org/10.1007/s00109-017-1600-y.
Liu Y, Zhang X, Liu M, Xu F, Zhang Q, Zhang Y, et al. Direct detection of circRNA in real samples using reverse transcription-rolling circle amplification. Anal Chim Acta. 2020;1101. https://doi.org/10.1016/j.aca.2019.12.027.
Zhang P, Gao K, Liang Y, Su F, Wang F, Li Z. Ultrasensitive detection of circular RNA by accurate recognition of the specific junction site using stem-loop primer induced double exponential amplification. Talanta. 2020;217. https://doi.org/10.1016/j.talanta.2020.121021.
Koch L. CRISPR–Cas13 targets circRNAs. Nat Rev Genet. 2020;22:68–68.
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550. https://doi.org/10.1038/nature24049.
Li S, Li X, Xue W, Zhang L, Yang LZ, Cao SM, et al. Screening for functional circular RNAs using the CRISPR–Cas13 system. Nat Methods. 2021;18. https://doi.org/10.1038/s41592-020-01011-4.
Zhang Y, Nguyen TM, Zhang XO, Wang L, Phan T, Clohessy JG, et al. Optimized RNA-targeting CRISPR/Cas13d technology outperforms shRNA in identifying functional circRNAs. Genome Biol. 2021;22. https://doi.org/10.1186/s13059-021-02263-9.
Zhang H, Wang G, Ding C, Liu P, Wang R, Ding W, et al. Increased circular RNA UBAP2 acts as a sponge of miR-143 to promote osteosarcoma progression. Oncotarget. 2017;8. https://doi.org/10.18632/oncotarget.18671.
Mecozzi N, Nenci A, Vera O, Bok I, Falzone A, DeNicola GM, et al. Genetic tools for the stable overexpression of circular RNAs. RNA Biol. 2022;19. https://doi.org/10.1080/15476286.2022.2043041.
Rahimi K, Venø MT, Dupont DM, Kjems J. Nanopore sequencing of brain-derived full-length circRNAs reveals circRNA-specific exon usage, intron retention and microexons. Nat Commun. 2021;12. https://doi.org/10.1038/s41467-021-24975-z.
Dahl M, Daugaard I, Andersen MS, Hansen TB, Grønbæk K, Kjems J, et al. Enzyme-free digital counting of endogenous circular RNA molecules in B-cell malignancies. Lab Investig. 2018;98. https://doi.org/10.1038/s41374-018-0108-6.
Dahl M, Husby S, Eskelund CW, Besenbacher S, Fjelstrup S, Côme C, et al. Expression patterns and prognostic potential of circular RNAs in mantle cell lymphoma: a study of younger patients from the MCL2 and MCL3 clinical trials. Leukemia. 2022;36. https://doi.org/10.1038/s41375-021-01311-4.
Dodbele S, Mutlu N, Wilusz JE. Best practices to ensure robust investigation of circular RNAs: pitfalls and tips. EMBO Rep. 2021;22. https://doi.org/10.15252/embr.202052072.
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20. https://doi.org/10.1038/s41576-019-0158-7.
Chen LL. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol. 2020;21. https://doi.org/10.1038/s41580-020-0243-y.
Xie H, Ren X, Xin S, Lan X, Lu G, Lin Y, et al. Emerging roles of circRNA_001569 targeting miR-145 in the proliferation and invasion of colorectal cancer. Oncotarget. 2016;7. https://doi.org/10.18632/oncotarget.8589.
Xia P, Wang S, Ye B, Du Y, Li C, Xiong Z, et al. A circular RNA protects dormant hematopoietic stem cells from DNA sensor cGAS-Mediated exhaustion. Immunity. 2018;48. https://doi.org/10.1016/j.immuni.2018.03.016.
Zhu P, Zhu X, Wu J, He L, Lu T, Wang Y, et al. IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nat Immunol. 2019;20. https://doi.org/10.1038/s41590-018-0297-6.
Moldovan LI, Tsoi LC, Ranjitha U, Hager H, Weidinger S, Gudjonsson JE, et al. Characterization of circular RNA transcriptomes in psoriasis and atopic dermatitis reveals disease-specific expression profiles. Exp Dermatol. 2021;30. https://doi.org/10.1111/exd.14227.
Gu X, Li X, Jin Y, Zhang Z, Li M, Liu D, et al. CDR1as regulated by hnRNPM maintains stemness of periodontal ligament stem cells via miR-7/KLF4. J Cell Mol Med. 2021;25. https://doi.org/10.1111/jcmm.16541.
Dunn SJ, Martello G, Yordanov B, Emmott S, Smith AG. Defining an essential transcription factor program for naïve pluripotency. Science. 2014;344. https://doi.org/10.1126/science.1248882.
Geng Y, Jiang J, Wu C. Function and clinical significance of circRNAs in solid tumors. J Hematol Oncol. 2018;11. https://doi.org/10.1186/s13045-018-0643-z.
Han YN, Xia SQ, Zhang YY, Zheng JH, Li W. Circular RNAs: A novel type of biomarker and genetic tools in cancer. Oncotarget. 2017;8. https://doi.org/10.18632/oncotarget.18350.
Meng S, Zhou H, Feng Z, Xu Z, Tang Y, Li P, et al. CircRNA: Functions and properties of a novel potential biomarker for cancer. Mol Cancer. 2017;16. https://doi.org/10.1186/s12943-017-0663-2.
Hang D, Zhou J, Qin N, Zhou W, Ma H, Jin G, et al. A novel plasma circular RNA circFARSA is a potential biomarker for non-small cell lung cancer. Cancer Med. 2018;7. https://doi.org/10.1002/cam4.1514.
Nanishi K, Konishi H, Shoda K, Arita T, Kosuga T, Komatsu S, et al. Circulating circERBB2 as a potential prognostic biomarker for gastric cancer: an investigative study. Cancer Sci. 2020;111. https://doi.org/10.1111/cas.14645.
Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, et al. The landscape of MicroRNA, piwi-interacting RNA, and circular RNA in human saliva. Clin Chem. 2015;61. https://doi.org/10.1373/clinchem.2014.230433.
Huijbers A, Tollenaar RAEM, Pelt GWV, Zeestraten ECM, Dutton S, McConkey CC, et al. The proportion of tumor-stroma as a strong prognosticator for stage II and III colon cancer patients: Validation in the victor trial. Ann Oncol. 2013;24. https://doi.org/10.1093/annonc/mds246.
Zhang T, Xu J, Shen H, Dong W, Ni Y, Du J. Tumor-stroma ratio is an independent predictor for survival in NSCLC. Int J Clin Exp Pathol. 2015;8.
Gujam FJA, Edwards J, Mohammed ZMA, Going JJ, McMillan DC. The relationship between the tumour stroma percentage, clinicopathological characteristics and outcome in patients with operable ductal breast cancer. Br J Cancer. 2014;111. https://doi.org/10.1038/bjc.2014.279.
Chen Z, Zuo X, Pu L, Zhang Y, Han G, Zhang L, et al. circLARP4 induces cellular senescence through regulating miR-761/RUNX3/p53/p21 signaling in hepatocellular carcinoma. Cancer Sci. 2019;110. https://doi.org/10.1111/cas.13901.
Zhang J, Liu H, Hou L, Wang G, Zhang R, Huang Y, et al. Circular RNA_LARP4 inhibits cell proliferation and invasion of gastric cancer by sponging miR-424-5p and regulating LATS1 expression. Mol Cancer. 2017;16. https://doi.org/10.1186/s12943-017-0719-3.
Hu Y, Gu J, Shen H, Shao T, Li S, Wang W, et al. Circular RNA LARP4 correlates with decreased Enneking stage, better histological response, and prolonged survival profiles, and it elevates chemosensitivity to cisplatin and doxorubicin via sponging microRNA-424 in osteosarcoma. J Clin Lab Anal. 2020;34. https://doi.org/10.1002/jcla.23045.
Zhang X, Su X, Guo Z, Jiang X, Li X. Circular RNA La-related RNA-binding protein 4 correlates with reduced tumor stage, as well as better prognosis, and promotes chemosensitivity to doxorubicin in breast cancer. J Clin Lab Anal. 2020;34. https://doi.org/10.1002/jcla.23272.
Wang S, Li Q, Wang Y, Li X, Wang R, Kang Y, et al. Upregulation of circ-UBAP2 predicts poor prognosis and promotes triple-negative breast cancer progression through the miR-661/MTA1 pathway. Biochem Biophys Res Commun. 2018;505. https://doi.org/10.1016/j.bbrc.2018.10.026.
Liu B, Tian Y, Chen M, Shen H, Xia J, Nan J, et al. CircUBAP2 Promotes MMP9-Mediated Oncogenic Effect via Sponging miR-194-3p in Hepatocellular Carcinoma. Front Cell Dev Biol. 2021;9. https://doi.org/10.3389/fcell.2021.675043.
Ju H, Zhao Q, Wang F, Lan P, Wang Z, Zuo Z, et al. A circRNA signature predicts postoperative recurrence in stage II/III colon cancer. EMBO Mol Med. 2019;11. https://doi.org/10.15252/emmm.201810168.
Ahmadov U, Bendikas MM, Ebbesen KK, Sehested AM, Kjems J, Broholm H, et al. Distinct circular RNA expression profiles in pediatric ependymomas. Brain Pathol. 2021;31:387–92.
Rickert D, Bartl J, Picard D, Bernardi F, Qin N, Lovino M, et al. Circular RNA profiling distinguishes medulloblastoma groups and shows aberrant RMST overexpression in WNT medulloblastoma. Acta Neuropathol. 2021;141:975–8.
Nair AA, Niu N, Tang X, Thompson KJ, Wang L, Kocher JP, et al. Circular RNAs and their associations with breast cancer subtypes. Oncotarget. 2016;7:80967–79.
Wang C, Tan S, Liu WR, Lei Q, Qiao W, Wu Y, et al. RNA-Seq profiling of circular RNA in human lung adenocarcinoma and squamous cell carcinoma. Mol Cancer. 2019;18:134.
He Y di, Tao W, He T, Wang BY, Tang XM, Zhang LM, et al. A urine extracellular vesicle circRNA classifier for detection of high-grade prostate cancer in patients with prostate-specific antigen 2–10 ng/mL at initial biopsy. Mol Cancer. 2021;20. https://doi.org/10.1186/s12943-021-01388-6.
Yu J, Ding WB, Wang MC, Guo XG, Xu J, Xu QG, et al. Plasma circular RNA panel to diagnose hepatitis B virus-related hepatocellular carcinoma: a large-scale, multicenter study. Int J Cancer. 2020;146. https://doi.org/10.1002/ijc.32647.
Stella M, Falzone L, Caponnetto A, Gattuso G, Barbagallo C, Battaglia R, et al. Serum extracellular vesicle-derived circhipk3 and circsmarca5 are two novel diagnostic biomarkers for glioblastoma multiforme. Pharmaceuticals. 2021;14. https://doi.org/10.3390/ph14070618.
Yin WB, Yan MG, Fang X, Guo JJ, Xiong W, Zhang RP. Circulating circular RNA hsa_circ_0001785 acts as a diagnostic biomarker for breast cancer detection. Clin Chim Acta. 2018;487. https://doi.org/10.1016/j.cca.2017.10.011.
Yang F, Liu DY, Guo JT, Ge N, Zhu P, Liu X, et al. Circular RNA circ-LDLRAD3 as a biomarker in diagnosis of pancreatic cancer. World J Gastroenterol. 2017;23. https://doi.org/10.3748/wjg.v23.i47.8345.
Hong X, Liu N, Liang Y, He Q, Yang X, Lei Y, et al. Circular RNA CRIM1 functions as a ceRNA to promote nasopharyngeal carcinoma metastasis and docetaxel chemoresistance through upregulating FOXQ1. Mol Cancer. 2020;19. https://doi.org/10.1186/s12943-020-01149-x.
Yi H, Han Y, Li S. Oncogenic circular RNA circ_0007534 contributes to paclitaxel resistance in endometrial cancer by sponging miR-625 and promoting ZEB2 expression. Front Oncol. 2022;12. https://doi.org/10.3389/FONC.2022.985470.
Greene J, Baird AM, Casey O, Brady L, Blackshields G, Lim M, et al. Circular RNAs are differentially expressed in prostate cancer and are potentially associated with resistance to enzalutamide. Sci Rep. 2019;9. https://doi.org/10.1038/S41598-019-47189-2.
Zeng Z, Zhao Y, Chen QY, Zhu S, Niu Y, Ye Z, et al. Hypoxic exosomal HIF-1α-stabilizing circZNF91 promotes chemoresistance of normoxic pancreatic cancer cells via enhancing glycolysis. Oncogene. 2021;40:5505–17.
Zhong Y, Wang D, Ding Y, Tian G, Jiang B. Circular RNA circ_0032821 contributes to oxaliplatin (OXA) resistance of gastric cancer cells by regulating SOX9 via miR-515-5p. Biotechnol Lett. 2021;43:339–51.
Zhang PF, Pei X, Li KS, Jin LN, Wang F, Wu J, et al. Circular RNA circFGFR1 promotes progression and anti-PD-1 resistance by sponging miR-381-3p in non-small cell lung cancer cells. Mol Cancer. 2019;18. https://doi.org/10.1186/s12943-019-1111-2.
Chen DL, Sheng H, Zhang DS, Jin Y, Zhao BT, Chen N, et al. The circular RNA circDLG1 promotes gastric cancer progression and anti-PD-1 resistance through the regulation of CXCL12 by sponging miR-141-3p. Mol Cancer. 2021;20. https://doi.org/10.1186/S12943-021-01475-8.
Zhang P-F, Gao C, Huang X-Y, Lu J-C, Guo X-J, Shi G-M, et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19:110.
He AT, Liu J, Li F, Yang BB. Targeting circular RNAs as a therapeutic approach: current strategies and challenges. Signal Transduct Target Ther. 2021;6. https://doi.org/10.1038/S41392-021-00569-5.
Zhang X, Zhang J, Liu Q, Zhao Y, Zhang W, Yang H. Circ-CUX1 accelerates the progression of neuroblastoma via miR-16-5p/DMRT2 axis. Neurochem Res. 2020;45:2840–55.
Chen Y, Yang F, Fang E, Xiao W, Mei H, Li H, et al. Circular RNA circAGO2 drives cancer progression through facilitating HuR-repressed functions of AGO2-miRNA complexes. Cell Death Differ. 2019;26:1346–64.
Han K, Wang FW, Cao CH, Ling H, Chen JW, Chen RX, et al. CircLONP2 enhances colorectal carcinoma invasion and metastasis through modulating the maturation and exosomal dissemination of microRNA-17. Mol Cancer. 2020;19. https://doi.org/10.1186/S12943-020-01184-8.
Du WW, Yang W, Li X, Awan FM, Yang Z, Fang L, et al. A circular RNA circ-DNMT1 enhances breast cancer progression by activating autophagy. Oncogene. 2018;37. https://doi.org/10.1038/s41388-018-0369-y.
Fang L, Du WW, Awan FM, Dong J, Yang BB. The circular RNA circ-Ccnb1 dissociates Ccnb1/Cdk1 complex suppressing cell invasion and tumorigenesis. Cancer Lett. 2019;459. https://doi.org/10.1016/j.canlet.2019.05.036.
Li J, Ma M, Yang X, Zhang M, Luo J, Zhou H, et al. Circular HER2 RNA positive triple negative breast cancer is sensitive to Pertuzumab. Mol Cancer. 2020;19:1–18.
Meganck RM, Borchardt EK, Castellanos Rivera RM, Scalabrino ML, Wilusz JE, Marzluff WF, et al. Tissue-dependent expression and translation of circular RNAs with recombinant AAV vectors in vivo. Mol Ther Nucleic Acids. 2018;13. https://doi.org/10.1016/j.omtn.2018.08.008.
Xu J, Ji L, Liang Y, Wan Z, Zheng W, Song X, et al. CircRNA-SORE mediates sorafenib resistance in hepatocellular carcinoma by stabilizing YBX1. Signal Transduct Target Ther. 2020;5. https://doi.org/10.1038/s41392-020-00375-5.
Xia X, Li X, Li F, Wu X, Zhang M, Zhou H, et al. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol Cancer. 2019;18:1–16.
Sang Y, Chen B, Song X, Li Y, Liang Y, Han D, et al. circRNA_0025202 regulates tamoxifen sensitivity and tumor progression via regulating the miR-182-5p/FOXO3a axis in breast cancer. Mol Ther. 2019;27:1638–52.
Wu G, Sun Y, Xiang Z, Wang K, Liu B, Xiao G, et al. Preclinical study using circular RNA 17 and micro RNA 181c-5p to suppress the enzalutamide-resistant prostate cancer progression. Cell Death Dis. 2019;10. https://doi.org/10.1038/S41419-018-1048-1.
Zhang PF, Zhang PF, Zhang PF, Gao C, Gao C, Huang XY, et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19. https://doi.org/10.1186/S12943-020-01222-5.
Huang XY, Zhang PF, Wei CY, Peng R, Lu JC, Gao C, et al. Circular RNA circMET drives immunosuppression and anti-PD1 therapy resistance in hepatocellular carcinoma via the miR-30-5p/snail/DPP4 axis. Mol Cancer. 2020;19:1–18.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17. https://doi.org/10.1038/nrg.2016.49.
Nielsen AF, Bindereif A, Bozzoni I, Hanan M, Hansen TB, Irimia M, et al. Best practice standards for circular RNA research. Nat Methods. 2022;9. https://doi.org/10.1038/S41592-022-01487-2.
Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell. 2014;54. https://doi.org/10.1016/j.molcel.2014.03.045.
Olesen MTJ, Kristensen LS. Circular RNAs as microRNA sponges: evidence and controversies. Essays Biochem. 2021;65. https://doi.org/10.1042/EBC20200060.
Zhao Y, Zhang C, Tang H, Wu X, Qi Q. Mechanism of RNA circHIPK3 involved in resistance of lung cancer cells to gefitinib. Biomed Res Int. 2022;2022. https://doi.org/10.1155/2022/4541918.
Cheng Z, Yu C, Cui S, Wang H, Jin H, Wang C, et al. circTP63 functions as a ceRNA to promote lung squamous cell carcinoma progression by upregulating FOXM1. Nat Commun. 2019;10. https://doi.org/10.1038/S41467-019-11162-4.
Yu C, Cheng Z, Cui S, Mao X, Li B, Fu Y, et al. circFOXM1 promotes proliferation of non-small cell lung carcinoma cells by acting as a ceRNA to upregulate FAM83D. J Exp Clin Cancer Res. 2020;39. https://doi.org/10.1186/S13046-020-01555-5.
Nie J, Yang R, Zhou R, Deng Y, Li D, Gou D, et al. Circular RNA circFARSA promotes the tumorigenesis of non-small cell lung cancer by elevating B7H3 via sponging miR-15a-5p. Cell Cycle. 2022;21:2575–89.
Wu N, Yuan Z, Du KY, Fang L, Lyu J, Zhang C, et al. Translation of yes-associated protein (YAP) was antagonized by its circular RNA via suppressing the assembly of the translation initiation machinery. Cell Death Differ. 2019;26:2758–73.
Lu W-Y. Cell Cycle Roles of the circular RNA circ-Foxo3 in breast cancer progression. 2017. https://doi.org/10.1080/15384101.2017.1278935.
Du WW, Yang W, Li X, Fang L, Wu N, Li F, et al. The Circular RNA circSKA3 binds integrin β1 to induce invadopodium formation enhancing breast cancer invasion. Mol Ther. 2020;28:1287–98.
Meng L, Liu S, Liu F, Sang M, Ju Y, Fan X, et al. ZEB1-mediated transcriptional upregulation of circwwc3 promotes breast cancer progression through activating ras signaling pathway. Mol Ther Nucleic Acids. 2020;22:124–37.
Xu JZ, Shao CC, Wang XJ, Zhao X, Chen JQ, Ouyang YX, et al. circTADA2As suppress breast cancer progression and metastasis via targeting miR-203a-3p/SOCS3 axis. Cell Death Dis 2019 10:3. 2019;10:1–16.
Zhang X, Wang S, Wang H, Cao J, Huang X, Chen Z, et al. Circular RNA circNRIP1 acts as a microRNA-149-5p sponge to promote gastric cancer progression via the AKT1/mTOR pathway. https://doi.org/10.1186/s12943-018-0935-5.
Li D, Li L, Chen X, Yang W, Cao Y. Circular RNA SERPINE2 promotes development of glioblastoma by regulating the miR-361-3p/miR-324-5p/ BCL2 signaling pathway. Mol Ther Oncolytics. 2021;22:483–94.
Ding L, Zhao Y, Dang S, Wang Y, Li X, Yu X, et al. Circular RNA circ-DONSON facilitates gastric cancer growth and invasion via NURF complex dependent activation of transcription factor SOX4. https://doi.org/10.1186/s12943-019-1006-2.
Zhang J, Hou L, Liang R, Chen X, Zhang R, Chen W, et al. CircDLST promotes the tumorigenesis and metastasis of gastric cancer by sponging miR-502-5p and activating the NRAS/MEK1/ERK1/2 signaling. Mol Cancer. 2019;18. https://doi.org/10.1186/S12943-019-1015-1.
He J, Chen J, Ma B, Jiang L, Zhao G. CircLMTK2 acts as a novel tumor suppressor in gastric cancer. Biosci Rep. 2019;39. https://doi.org/10.1042/BSR20190363.
Jiang T, Xia Y, Lv J, Li B, Li Y, Wang S, et al. A novel protein encoded by circMAPK1 inhibits progression of gastric cancer by suppressing activation of MAPK signaling. Mol Cancer. 2021;20. https://doi.org/10.1186/s12943-021-01358-y.
Morena D, Picca F, Taulli R. CircNT5E/miR-422a: a new circRNA-based ceRNA network in glioblastoma. Transl Cancer Res. 2019;8:S106–S109.
Xu H, Zhang Y, Qi L, Ding L, Jiang H, Yu H. NFIX circular RNA promotes glioma progression by regulating miR-34a-5p via Notch signaling pathway. Front Mol Neurosci. 2018;11. https://doi.org/10.3389/FNMOL.2018.00225.
Yuan DH, Zhao J, Shao GF. Circular RNA TTBK2 promotes the development of human glioma cells via miR-520b/EZH2 axis. Eur Rev Med Pharmacol Sci. 2019;23:10886–98.
Bian A, Wang Y, Liu J, Wang X, Liu D, Jiang J, et al. Circular RNA complement factor H (CFH) Promotes Glioma progression by sponging miR-149 and Regulating AKT1. Med Sci Monit. 2018;24:5704–12.
Barbagallo D, Caponnetto A, Barbagallo C, Battaglia R, Mirabella F, Brex D, et al. The GAUGAA Motif Is Responsible for the Binding between circSMARCA5 and SRSF1 and Related Downstream Effects on Glioblastoma Multiforme Cell Migration and Angiogenic Potential. Int J Mol Sci. 2021;22:1–16.
Shang J, Chen WM, Wang ZH, Wei TN, Chen ZZ, Wu WB. CircPAN3 mediates drug resistance in acute myeloid leukemia through the miR-153-5p/miR-183-5p-XIAP axis. Exp Hematol. 2019;70:42–54.e3.
Wu D-M, Wen X, Han X-R, Wang S, Wang Y-J, Shen M, et al. Role of circular RNA DLEU2 in human acute myeloid leukemia. Mol Cell Biol. 2018;38. https://doi.org/10.1128/MCB.00259-18.
Sheng X, Hong L, Fan L, Zhang Y, Che K, Mu J, et al. Circular RNA PVT1 regulates cell proliferation, migration and apoptosis by stabilizing c-Myc and its downstream target CXCR4 expression in acute myeloid leukemia. Turk J Haematol. 2023. https://doi.org/10.4274/TJH.GALENOS.2023.2022.0435.
Li S, Ma Y, Tan Y, Ma X, Zhao M, Chen B, et al. Profiling and functional analysis of circular RNAs in acute promyelocytic leukemia and their dynamic regulation during all-trans retinoic acid treatment. Cell Death Dis. 2018;9. https://doi.org/10.1038/S41419-018-0699-2.
Liu X, Liu X, Cai M, Luo A, He Y, Liu S, et al. CircRNF220, not its linear cognate gene RNF220, regulates cell growth and is associated with relapse in pediatric acute myeloid leukemia. Mol Cancer. 2021;20. https://doi.org/10.1186/S12943-021-01395-7.
Liu J, Du F, Chen C, Li D, Chen Y, Xiao X, et al. CircRNA ITCH increases bortezomib sensitivity through regulating the miR-615-3p/PRKCD axis in multiple myeloma. Life Sci 2020;262. https://doi.org/10.1016/J.LFS.2020.118506.
Pan Y, Lou J, Wang H, An N, Chen H, Zhang Q, et al. CircBA9.3 supports the survival of leukaemic cells by up-regulating c-ABL1 or BCR-ABL1 protein levels. Blood Cells Mol Dis. 2018;73:38–44.
Li J, Li Z, Jiang P, Peng M, Zhang X, Chen K, et al. Circular RNA IARS (circ-IARS) secreted by pancreatic cancer cells and located within exosomes regulates endothelial monolayer permeability to promote tumor metastasis. J Exp Clin Cancer Res. 2018;37. https://doi.org/10.1186/S13046-018-0822-3.
Li Z, Yanfang W, Li J, Jiang P, Peng T, Chen K, et al. Tumor-released exosomal circular RNA PDE8A promotes invasive growth via the miR-338/MACC1/MET pathway in pancreatic cancer. Cancer Lett. 2018;432:237–50.
Panda AC, De S, Grammatikakis I, Munk R, Yang X, Piao Y, et al. High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res. 2017;45. https://doi.org/10.1093/nar/gkx297.
Acknowledgements
GAC is the Felix L. Haas Endowed Professor in Basic Science. Work in GAC’s laboratory is supported by NCI grants 1R01 CA182905-01 and 1R01CA222007-01A1, NIGMS grant 1R01GM122775-01, DoD Idea Award W81XWH-21-1-0030, a Team DOD grant in Gastric Cancer W81XWH-21-1-0715, a Chronic Lymphocytic Leukemia Moonshot Flagship project, a CLL Global Research Foundation 2019 grant, a CLL Global Research Foundation 2020 grant, a CLL Global Research Foundation 2022 grant, The G. Harold & Leila Y. Mathers Foundation, two grants from Torrey Coast Foundation, and a Development Grant associated with the Brain SPORE 2P50CA127001. THV is the recipient of the Dr. John J. Kopchick Research Fellowship.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conceptual organization and writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
GAC is one of the scientific founders of Ithax Pharmaceuticals, Inc. No conflict of interest to declare for the other authors.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pisignano, G., Michael, D.C., Visal, T.H. et al. Going circular: history, present, and future of circRNAs in cancer. Oncogene 42, 2783–2800 (2023). https://doi.org/10.1038/s41388-023-02780-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-023-02780-w
This article is cited by
-
Non-coding RNAs in disease: from mechanisms to therapeutics
Nature Reviews Genetics (2024)
-
Peptidylprolyl isomerase D circular RNA sensitizes breast cancer to trastuzumab through remodeling HER2 N4-acetylcytidine modification
Journal of Applied Genetics (2024)
-
Biological role and regulation of circular RNA as an emerging biomarker and potential therapeutic target for cancer
Molecular Biology Reports (2024)
-
The role of circular RNA during the urological cancer metastasis: exploring regulatory mechanisms and potential therapeutic targets
Cancer and Metastasis Reviews (2024)
-
A therapeutical insight into the correlation between circRNAs and signaling pathways involved in cancer pathogenesis
Medical Oncology (2024)
