
Abstract
Patients with bipolar disorder (BD) show alterations in both gray matter volume (GMV) and white matter (WM) integrity compared with healthy controls (HC). However, it remains unclear whether the phenotypically distinct BD subtypes (BD-I and BD-II) also exhibit brain structural differences. This study investigated GMV and WM differences between HC, BD-I, and BD-II, along with clinical and genetic associations. N = 73 BD-I, n = 63 BD-II patients and n = 136 matched HC were included. Using voxel-based morphometry and tract-based spatial statistics, main effects of group in GMV and fractional anisotropy (FA) were analyzed. Associations between clinical and genetic features and GMV or FA were calculated using regression models. For FA but not GMV, we found significant differences between groups. BD-I patients showed lower FA compared with BD-II patients (ptfce-FWE = 0.006), primarily in the anterior corpus callosum. Compared with HC, BD-I patients exhibited lower FA in widespread clusters (ptfce-FWE < 0.001), including almost all major projection, association, and commissural fiber tracts. BD-II patients also demonstrated lower FA compared with HC, although less pronounced (ptfce-FWE = 0.049). The results remained unchanged after controlling for clinical and genetic features, for which no independent associations with FA or GMV emerged. Our findings suggest that, at a neurobiological level, BD subtypes may reflect distinct degrees of disease expression, with increasing WM microstructure disruption from BD-II to BD-I. This differential magnitude of microstructural alterations was not clearly linked to clinical and genetic variables. These findings should be considered when discussing the classification of BD subtypes within the spectrum of affective disorders.
Similar content being viewed by others


Introduction
Bipolar disorder (BD) is a heterogeneous mood disorder that is divided into two phenotypically distinct subtypes based on DSM (Diagnostic and Statistical Manual of Mental Disorders) diagnostic criteria [1]. While subtype I (BD-I) is characterized by periods of mania, which may alternate with depressive episodes, subtype II (BD-II) is diagnosed when hypomanic and depressive episodes are present but no episodes of full mania. Beyond diagnostic criteria, differences in the long-term disease course between subtypes have been found, with more (hypo-)manic episodes [2, 3] and hospitalizations [4, 5] in BD-I and more frequent and prolonged depressive episodes [3, 4, 6] in BD-II. Depending on the subtype, different treatment approaches may be more beneficial [4, 7,8,9,10]. Further, differential diagnosis of subtypes is commonly based on phenotypic characteristics and clinical assessment of the degree of impairment, with a clear differentiation only being possible if certain characteristics are present [11]. Therefore, a key challenge for clinicians remains the distinction of BD subtypes [12, 13]. This underlines the importance of investigating other, potentially meaningful markers to enhance subtype diagnosis and treatment response. Studying neuronal [14] and genetic [15, 16] correlates contributes to a more detailed subtype characterization and could provide additional clinical and therapeutic benefits [12, 17].
Previous neuroimaging research showed widespread white and gray matter abnormalities in BD patients compared with healthy controls (HC) [18,19,20,21,22,23]. However, some studies included one subtype only [18, 23,24,25,26] or did not differentiate between subtypes in their analyses [19, 27,28,29,30,31]. Those few studies that directly compared subtypes yielded mainly inconclusive results [20, 21, 32,33,34,35,36,37,38,39,40]: Diffusion tensor imaging (DTI) studies, which usually focus on fractional anisotropy (FA) as a quantitative measure of WM microstructure or integrity, have yielded conflicting results when conducting direct comparisons between subtypes, with some finding reduced FA in the temporal and frontal pathways in BD-I [34, 35, 38] and others in BD-II [32, 39]. Although these studies only allow tentative conclusions due to consistently small samples (mostly n < 30 per group) and varying methods, current evidence rather points to more severe WM microstructural impairments in BD-I compared with BD-II [41]. Regarding gray matter volumes (GMV), some studies found lower volumes in temporal [37, 40, 42], (pre-)frontal [37, 40, 42] and posterior cingulate regions [37] and in the putamen [33] in BD-I compared with BD-II, while others found no GMV differences between subtypes [20, 21, 36, 43, 44]. In conclusion, although there seems to be preliminary data attesting more pronounced WM and GM changes in BD-I compared with BD-II e.g. [35, 37, 38, 42], the evidence is inconsistent and it remains unclear whether existing neuroimaging findings on BD can be generalized to both subtypes.
Going one step further, the issue is whether neurobiological alterations support the categorical classification into clinical subtypes according to DSM, which has so far been based on phenotypic markers only [45]. Even though these phenotypic features are dimensional in nature, they are categorized based on the severity of their expression resulting in the two discrete BD subtypes. Therefore, neurobiological alterations may form a continuum, representing the subtypes as one clinical entity with varying severity. This would be captured by a dimensional diagnostic approach, where neurobiological alterations are associated with dimensional clinical features rather than discrete diagnostic categories [46, 47].
Underlying genetics provide evidence that fit both approaches. Based on the latest genome-wide association study (GWAS), BD subtypes share a large amount of genetic composition, showing a strong between-subtypes correlation of r = 0.85, while correlations with other mental disorders such as schizophrenia (SZ) or MDD were weaker (all r ≤ 0.66) [15]. In contrast, the strength of the GWAS correlations with other mental disorder differed between subtypes, with a greater correlation between BD-II and MDD, and between BD-I and SZ [15, 48, 49]. Moreover, heritability estimates based on twin studies [48, 50] and GWAS-based h2SNP heritability was estimated to be higher for BD-I compared to BD-II [15, 51]. Beyond differences in measures of heritability, identification of distinct loci might be related to subtype-specific symptomatology [15]. BD-I thus might have a stronger genetic component than BD-II.
A higher genetic load, which was also related to a more severe course of BD across subtypes [52], could also be linked to brain structural alterations [53,54,55]. Despite evidence for subtype-specific neuroanatomy and genetics, the relationship between these features in BD subtypes remains uninvestigated. Exploring the effects of genetic factors on brain structure altered in BD subtypes may provide insight into the neural mechanisms by which genetic variation has an impact on the disease at the psychopathological level.
To date, we are not aware of any study investigating subtype-specific differences in GMV and WM within the same sample subdivided by BD type. This study focuses on WM and GMV differences among BD-I, BD-II, and HC, aiming to investigate the neurobiological underpinnings of conventional BD subtype categories. Given previous heterogeneous neuroimaging findings [20, 21, 32,33,34,35,36,37,38,39,40], we employed a whole-brain approach. Rather than adhering to diagnostic categories, we also took a dimensional approach, exploring other potential subtype-specifying factors in relation to neurobiology. Adding this perspective, we aim to contribute to a more nuanced understanding of the neurobiological basis of the subtypes of BD.
First, we expected a decrease in WM integrity and GMV in BD patients, resulting in the following pattern of group differences: BD-I < BD-II < HC (categorical perspective). Second, we exploratively examined potential relationships between white and gray matter, BD polygenic risk scores (PRS) and various clinical characteristics (dimensional perspective).
Materials and methods
Participants
This study included n = 136 patients with BD and n = 136 HC from the Marburg-Münster-Affective-Cohort-Study (MACS; see ref. [56] for a general description). The data in the current study are a subsample of a previously published analysis by our group on WM microstructural differences between healthy, depressed, and bipolar individuals regardless of subtype [22]. Recruitment was conducted via newspaper advertisement and flyers, and in psychiatric hospitals. Inclusion criteria for HC were the absence of any current or lifetime psychiatric disorder, whereas BD patients required a current or lifetime diagnosis of bipolar disorder. The presence or absence of mental disorders was assessed by trained personnel with the Structured Clinical Interview (SCID-I) [57] according to the DSM-IV-TR criteria [58]. Based on this, BD patients were grouped into Bipolar I (BD-I, n = 73; n = 42 female, Mage = 41.77, SDage = 11.51) and Bipolar II (BD-II, n = 63; n = 33 female, Mage = 40.48, SDage = 12.56) subtypes. General exclusion criteria comprised usual magnetic resonance imaging (MRI) contraindications, head trauma, and any history of neurological, cardiovascular, or other severe medical conditions (e.g., cancer, infections, and autoimmune disease). Further exclusion criteria for BD patients were a lifetime diagnosis of alcohol or substance dependence (other than tetrahydrocannabinol dependence), while any current intake of psychotropic medication resulted in exclusion from the study for HC. All participants were aged between 18-65 years and HC and BD patients were matched according to age, sex, and study sites using the MatchIt package in R [59].
During the clinical interview, information about current symptomatology, lifetime course of disease, and psychopharmacological treatment was collected. The 21-item Hamilton Depression Rating Scale [60] and the Young Mania Rating Scale [61] were used to measure current depressive and (hypo-)manic symptoms, respectively. Disease course variables including number and cumulative duration of depressive and (hypo-)manic episodes and psychiatric hospitalizations, time since first symptoms (measured by age minus age of onset), and time since first psychiatric hospitalization were assessed by patients’ self-reports. Current psychopharmacological medication intake was assessed through a previously used composite score, the medication load index [62] (Supplement 1). PRS for bipolar disorder were calculated using genome-wide genotype data and the summary statistics of a recent GWAS of BD (more information in Supplement 2) [15]. Sociodemographic, clinical, and genetic information are provided in Table 1.
The study was approved by the Ethics Committees of the Medical Faculties, University of Marburg (AZ: 07/14) and University of Münster (2014-422-b-S), in accordance with the Declaration of Helsinki. All participants received financial compensation and provided written informed consent prior to participation.
Image acquisition and preprocessing
3 T whole body MRI scanners (Marburg: Tim Trio, Siemens, Erlangen, Germany; Münster: Prisma fit, Siemens, Erlangen, Germany) were used for acquisition of MRI data. All images underwent quality checks according to the quality check protocol of the MACS study [63]. Due to a body-coil change at the Marburg site, we controlled for three different scanner settings (Münster, Marburg body-coil pre, Marburg body-coil post) in all analyses using two dummy-coded variables with Münster as reference category as previously recommended [63].
White matter microstructure (DTI)
DTI data acquisition, preprocessing and quality assurance followed published protocols and were extensively described elsewhere [22, 63]. Preprocessing and analyses were implemented in FSL6.0.1 (http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/) [64,65,66]. For details on DTI acquisition parameters, quality assurance of the data and preprocessing steps see Supplement 3. As the last step, a diffusion tensor model was fitted at each voxel using “DTIFIT” within FMRIB’s Diffusion Toolbox (FDT) [67] and FA, mean diffusivity (MD), radial diffusivity (RD) and axial diffusivity (AD) were calculated for each voxel per participant. FA is a measure of the directionality of water diffusion on a scale from 0 (indicating isotropic diffusion) to 1 (indicating completely anisotropic diffusion) [68]. See Supplement 3 for more information on MD, RD, and AD measures.
Gray matter volumes (GMV)
High-resolution T1-weighted structural images were collected using three-dimensional fast gradient echo sequences (MPRAGE). Details on acquisition of T1 data for GMV analyses are provided in Supplement 4 and have been described elsewhere [69]. Preprocessing of T1-weighted images was conducted using a default pipeline implemented in the CAT12-toolbox (v1720). Steps included bias-correction, tissue classification, realignment and spatial normalization to MNI space using the Geodesic Shooting algorithm. Data were smoothed with an 8 mm full width half maximum Gaussian kernel.
Statistical analyses
Descriptive and clinical variables between BD patients and HC were analyzed in IBM SPSS Statistics 27 (SPSS Inc., Chicago, IL, USA; Table 1).
White matter microstructure (DTI)
Analysis of DTI data was performed using TBSS [70], a technique designed to reduce registration misalignment (Supplement 3). Voxel-wise statistical analyses were performed by using the nonparametric permutation testing implemented in “randomize” from FSL [71] with 5000 permutations. Threshold-Free Cluster Enhancement (TFCE) was applied to obtain cluster-wise statistics corrected for multiple comparisons [72]. Significance was determined using the 95th percentile of the null distribution of permutated input data of the maximum TFCE scores, allowing to correct estimated cluster sizes for family-wise error (FWE) at p < 0.05. The significant effect mask was placed over the raw diffusion metrics maps of each subject and the values of the respective voxels were extracted and averaged, using “fslstats” from FSL. The mean diffusion metric value for each subject from all voxels of the significant cluster was used for visualization in scatterplots.
The results for WM focus on FA. However, to support the interpretation of these results, the same registration steps and analyses were also performed for the other DTI metrics MD, RD, and AD (Supplement 5).
Gray matter volumes (GMV)
Statistical analyses of GMV were performed on the whole-brain level using Statistical Parametric Mapping (SPM12, Wellcome Department of Cognitive Neurology, London, UK, v7771) with an absolute threshold masking of 0.1. TFCE, as implemented in the TFCE-toolbox, with 5000 permutations per test was applied (http://dbm.neuro.uni-jena.de/tfce, Version r210). The significance threshold was set to p < 0.05 FWE corrected.
All DTI and GMV analyses included the covariates age, sex, total intracranial volume (TIV), site, and scanner settings (body-coil pre, body-coil post with Münster as reference category). The following analyses were conducted:
-
1.
To examine brain structural and microstructural differences between the diagnostic groups (HC, BD-I, BD-II), we first performed F-tests. Subsequently, post-hoc pairwise t-contrasts were calculated. Effect sizes were calculated based on the mean t-value of all significant voxels provided by FSL or SPM and respective sample sizes [73].
-
2.
To investigate putative effects of clinical characteristics, we performed additional analyses:
-
a.
In case of significant effects in the contrast BD-II > BD-I in the main analyses (step 1), we repeated the group comparisons by separately including clinical variables (e.g. number of depressive and (hypo-)manic episodes, psychiatric hospitalization, psychopharmacological medication and PRS for BD) potentially related to differences between BD subtypes as additional nuisance variables. An overview of all included clinical variables can be found in Table 1.
-
b.
To further determine which clinical characteristics influence brain structural alterations, associations between the clinical variables mentioned in a) and FA or GMV were calculated for the whole BD sample and per subtype (see Tables S5 and S6) using linear regression models. These analyses were calculated irrespective of significant group differences in step 1. Bonferroni correction for the ten regression analyses was applied, resulting in a significance threshold of p < 0.005.
-
a.
Results
Brain structural differences between HC, BD-I and BD-II
White matter microstructure (DTI)
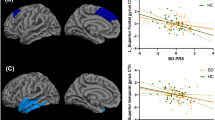
The F-contrast revealed a significant main effect of diagnosis in FA (ptfce-FWE < 0.001, total k = 7028 voxels in seven clusters, Fig. 1, Table S1). Pairwise post-hoc t-contrasts revealed significantly lower FA values in BD-I patients compared with HC in one large bilateral cluster (d = 0.25, ptfce-FWE < 0.001, k = 45712 voxels, Fig. 2A) as well as compared with BD-II patients (d = 0.36, ptfce-FWE = 0.006, k = 6418 voxels in seven clusters, Fig. 2B). Both effects were most probably located in the forceps minor of the corpus callosum (CC), with almost all other major fiber tracts also affected when BD-I and HC were compared (Table S2). BD-II patients also showed significant lower FA values compared with HC in two small clusters in the left body of the CC (d = 0.56, ptfce-FWE = 0.049, k = 27 voxels in two clusters, Fig. 2C). There was also a significant main effect of diagnosis for RD and MD, reflected in RD by significantly higher scores for BD-I compared with HC and BD-II, and in MD by significantly higher scores only for BD-I compared with HC. No effects were found for AD (Supplement 5).

A Mean fractional anisotropy (FA) across healthy controls (HC), bipolar disorder type I (BD-I) and bipolar disorder type II (BD-II). The mean FA value was obtained from FA values of all the voxels that showed a significant main effect of diagnosis (ptfce-FWE < 0.05). Error bars represent 95% confidence intervals. p values were obtained from pairwise post-hoc t-contrasts. B Density estimation plots of FA values showing distributional overlap between HC, BD-I and BD-II.

Differences in fractional anisotropy (FA) between healthy controls (HC), bipolar disorder type I (BD-I), and bipolar disorder type II (BD-II). A Higher FA in HC compared with BD-I. B Higher FA in BD-II compared with BD-I. C Higher FA in HC compared with BD-II. D Differences in FA between the three groups HC, BD-I and BD-II (F-Test). Coordinates are given in MNI space. Highlighted areas represent voxels (using FSL’s ‘fill’ command for better visualization), where significant differences between groups (ptfce-FWE < 0.05) were detected.
Gray matter volumes (GMV)
In the whole-brain analysis no significant main effect of diagnosis was found (ptfce-FWE = 0.509). Exploratory pairwise comparisons between diagnoses on the whole-brain level, uncorrected at p < 0.001 with a cluster threshold of k = 50 voxels pointed towards putative GMV alterations in parietal, frontal and parahippocampal/fusiform regions in the expected pattern of BD-I < BD-II < HC (Table S3).
Additional analyses
Brain structural differences between BD-I and BD-II subtypes correcting for clinical variables and polygenic risk
White matter microstructure (DTI)
Analyses revealed a stably lower FA in BD-I vs. BD-II even when additionally correcting for number of depressive episodes (d = 0.36, ptfce-FWE = 0.004, k = 6832 voxels), number of (hypo-)manic episodes (d = 0.36, ptfce-FWE = 0.005, k = 8061 voxels), number of psychiatric hospitalizations (d = 0.31, ptfce-FWE = 0.003, k = 16683 voxels), time since first symptoms (d = 0.36, ptfce-FWE = 0.007, k = 6689 voxels), time since first psychiatric hospitalization (d = 0.32, ptfce-FWE = 0.007, k = 17,880 voxels), and lifetime psychotic symptoms (d = 0.37, ptfce-FWE = 0.013, k = 5202 voxels). Similarly, the observed pattern of results did not change when correcting the models for PRS for BD (d = 0.38, ptfce-FWE = 0.020, k = 4742 voxels), childhood adversity (d = 0.32, ptfce-FWE = 0.005, k = 17,248 voxels), body mass index (d = 0.53, ptfce-FWE = 0.038, k = 307 voxels) or medication load (d = 0.36, ptfce-FWE = 0.005, k = 6745 voxels, additional analyses for different types of medication are provided in Table S4). Affected tracts consistently included the forceps minor and major of the CC as well as the anterior thalamic radiation (Table S2). The significant increase in RD for BD-I compared with BD-II also proved stable in these additional analyses, except when PRS for BD, body mass index and psychotic symptoms were included as covariates (Supplement 5).
Associations with clinical variables and polygenic risk
White matter microstructure (DTI)
There was a significant positive associations between the time since first psychiatric hospitalization and FA (ptfce-FWE = 0.021), which, however, did not survive Bonferroni correction (Table 2). RD, MD and AD showed a negative association with this variable, which only survived Bonferroni correction in the case of MD (Supplement 5).
Gray matter volumes (GMV)
Regression analyses between clinical variables and GMV revealed no significant associations (all ptfce-FWE > 0.072, Table 3). The results of the additional regression analyses between clinical variables and brain structure in both subtype groups separately can be found in Table S5 (DTI metrics) and Table S6 (GMV).
Discussion
This study examined brain structural differences between patients with BD types I and II and HC, including white and gray matter, PRS for BD and disease course data, attempting to provide a more profound comparison of the subtypes. As the main result, our analyses revealed group differences with the expected pattern of BD-I < BD-II < HC regarding FA as a measure of WM integrity, whereas no group differences were found for GMV. Secondly, the group differences in WM microstructure were not significantly affected by prior disease course or PRS for BD and thus seem to be nearly independent of other clinical factors or underlying genetics. Finally, we found no associations between brain structure and these clinical or genetic parameters in either BD-I or BD-II.
Bringing some clarity to the heterogeneity of previous neuroimaging findings [20, 21, 32, 34, 35, 42], we demonstrated significantly lower FA, along with higher RD, in BD-I patients compared with BD-II. This finding was accompanied by a markedly different extent of impairment when compared with HC: Whereas BD-I patients showed widespread alterations in WM affecting all major WM tracts (including the CC), BD-II patients differed from HC only in a small local cluster in the CC. The FA reduction in the CC in both subtypes compared with HC matches existing reports of reduced FA levels in this tract as one of the most robust effects in BD [19, 22, 74]. As a crucial connecting pathway between brain hemispheres, the CC plays a central role in interhemispheric integration. Meta-analyses [75,76,77,78] have shown reduced WM integrity in the CC across MDD, BD, and SZ, suggesting disruptions in WM interhemispheric connectivity as a common pathophysiological pathway in major mental disorders, being related to deficits in various emotional and cognitive processes, such as executive functions, attention, working and visual memory [75, 77, 79,80,81]. Specifically, alterations in the forceps minor of the CC, composed of fibers extending laterally from the genu of the CC and connecting the cerebral hemispheres anteriorly, primarily indicate impaired interhemispheric communication within the prefrontal cortex, whose involvement in BD has been elaborated in several studies e.g. ref. [82,83,84].
The GMV analyses yielded no significant group effect, refuting our hypothesis. This aligns with findings from two large-cohort studies by the ENIGMA bipolar consortium [20, 21], indicating no subtype-specific (sub-)cortical GMVs differences. In contrast, prior literature found subtype-specific abnormalities [33, 37, 40, 42, 44], but results varied, potentially due to factors such as sample characteristics (e.g. small sample sizes or current mood state) or methodological differences (e.g. voxel-wise comparisons vs. region-specific parcellation). Our study adopted a whole-brain approach to capture this heterogeneity comprehensively, which, given a more stringent statistical threshold, could explain the lack of subtype-specific effects. Accordingly, the uncorrected exploratory analyses (Table S3) suggest the anticipated pattern of BD-I < BD-II < HC in GMVs in parietal, frontal, occipital and parahippocampal/fusiform areas. These regions partly overlap with reported regions in studies reporting subtype-specific differences e.g. [37, 40]. Neurobiological disparities in GMV between BD-I and BD-II may be less distinct than previously thought, necessitating larger, more homogeneous samples for precise investigations and mapping of potential effects. Given this evidence, the brain structural alterations in BD and its subtypes appear to relate less to the volume of structurally independent areas than to impaired fiber connections between these brain regions.
We found significantly lower FA in BD-I compared with BD-II, accompanied by global FA alterations in BD-I compared with HC. The separate diagnosis of BD-II was amended in DSM-IV [85] to acknowledge the disorder’s heterogeneity and to classify less severe symptoms of mania. The significant neurobiological differences between BD-I and BD-II initially support this categorical classification into subtypes [45]. However, the strong distributional overlap in FA reductions between both subtypes challenges a clear-cut classification based on the extent of WM alterations (Fig. 1B). From the latter argument, our data rather suggest a bipolar spectrum with varying severity [46, 47], mirrored by gradual transitions at the clinical level. Only differences in a few criteria and also subjective assessments determine which subtype a patient is diagnosed with [11, 45, 47]. The distinction between bipolar subtypes may be clearer in the presence of critical severity markers, e.g., psychotic symptoms or hospitalization [47], where subtypes differed in our sample (Table 1). However, our dimensional regression analyses revealed no significant linear associations between brain structure and psychotic symptoms, contrasting previous findings [86,87,88]. Subtype differences were largely unaffected by various variables, although single effects lost significance when controlling for body mass index, PRS or psychotic symptomatology, suggesting a more detailed analysis of these variables in future studies. Nevertheless, these factors provide no obvious explanation for the subtype differences found and challenge the notion of neuroprogressive effects in BD [89,90,91,92]. Given this, we can only cautiously interpret our results of distinct subtypes as a neurobiological correlate of overall disease severity in BD-I versus BD-II.
Considering the overlap in genetic compositions and heritability of BD [15], we examined associations between brain structure and PRS for BD. While we observed a significant difference in PRS between subtypes (Table 1), it was not related to gray or white matter features. The PRS employed in this study [15], predominantly derived from BD-I patients, may contribute to the substantial difference observed between subtypes. The debate surrounds the suitability of genome-wide PRS as a genetic tool for investigating psychiatric disorders [93]. While it enhances generalizability by capturing a broad range of common genetic factors for BD [55], its inclusion of alleles with potentially diverse associations with brain structure may mask effects of specific gene sets [93]. This debate is reflected in conflicting findings: On the one hand, there are studies failing to establish significant associations between BD or SZ PRS and brain structure [35, 93]. In contrast, a recent study in bipolar adolescents, benefiting from fewer confounding factors such as illness course or medication, demonstrated higher bipolar PRS scores correlating with both GM structure and WM diffusion [55].
Overall, the inclusion of larger BD-II samples, deep phenotyping, and the investigation of more specific genetic associations may provide relevant insights into the genetic composition of subtypes and putative relationships to brain structural features [94, 95].
This study has several strengths: A large, well-characterized sample of BD patients and comprehensive analyses including clinical, genetic and neuroimaging data. A few limitations are: First, the cross-sectional nature of our analyses entails interpretative difficulties and lower statistical power than longitudinal data. Particularly, the question whether neurobiological abnormalities are a result or precursor for bipolar subtype cannot be addressed. Second, the cross-sectional acquisition of information on previous disease course by self-reports is often biased by inaccuracies and memory deficits [96,97,98]. Therefore, the regression analyses including these clinical variables should be interpreted cautiously. Third, when using a composite score to represent psychopharmacological treatment, we did consider current medication, but without taking medication history into account, so that possible effects of previous medication use or duration of use cannot be excluded. Fourth, while TBSS offers the advantage of fully automated voxel-wise analysis of the whole brain, revealing changes beyond predefined pathways, the addition of newer tractography techniques may be beneficial for hypothesis-driven identification and detailed analysis of specific pathways [70, 99]. Fifth, we have examined only a selection of dimensions of psychopathology relevant to BD subtypes, but the inclusion of other dimensions, such as affective lability [100, 101], emotion regulation deficits [102], or affective temperaments [103], could also provide important insights. Sixth, we decided to include site as a covariate to control for potential scanner effects between Marburg und Münster as suggested by [63]. However, this approach may over- or under-correct for the site effect and only accounts for linear mean differences [104]. Other approaches of data harmonization between different scanners (e.g. ComBat harmonization) could be discussed as an alternative [105].
In summary, BD-I showed widespread alterations in WM connectivity, with no subtype-specific effects on GMV. The results suggest that BD-I phenomenology may be related to brain structural integrity impairment, which seems mainly independent from disease trajectories and genetics. Our findings may improve our understanding of the pathophysiology underlying the clinical and neurobiological spectrum of BD. Thus, microstructural alterations should be included when discussing the categorical or dimensional classification of bipolar subtypes. Similarly, clinicians may consider the degree of impairment in microstructural integrity when classifying BD patients within the spectrum of affective disorders and adjust treatment selection accordingly, although no potential cutoff regarding the extent of WM alterations has yet been established. Future studies should investigate other biological correlates of BD, for example functional connectivity or inflammatory processes, by more consistently taking subtypes into account.
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Arlington VA: American Psychiatric Association; 2013.
Joffe RT, MacQueen GM, Marriott M, Trevor Young L. A prospective, longitudinal study of percentage of time spent ill in patients with bipolar I or bipolar II disorders. Bipolar Disord. 2004;6:62–66.
Judd LL, Akiskal HS, Schettler PJ, Coryell W, Maser J, Rice JA, et al. The comparative clinical phenotype and long term longitudinal episode course of bipolar I and II: a clinical spectrum or distinct disorders? J Affect Disord. 2003;73:19–32.
Tondo L, Miola A, Pinna M, Contu M, Baldessarini RJ. Differences between bipolar disorder types 1 and 2 support the DSM two-syndrome concept. Int J Bipolar Disord. 2022;10:21.
Karanti A, Kardell M, Joas E, Runeson B, Pålsson E, Landén M. Characteristics of bipolar I and II disorder: A study of 8766 individuals. Bipolar Disord. 2020;22:392–400.
Berk M, Dodd S. Bipolar II disorder: a review. Bipolar Disord. 2005;7:11–21.
Vieta E. Bipolar II disorder: frequent, valid, and reliable. Can J Psychiatry. 2019;64:541–3.
Goes FS. Diagnosis and management of bipolar disorders. BMJ. 2023;381:e073591.
National Collaborating Centre for Mental Health (UK). Common Mental Health Disorders: Identification and Pathways to Care. Leicester: British Psychological Society (UK); 2011.
National Collaborating Centre for Mental Health (UK). Bipolar disorder: the NICE guideline on the assessment and management of bipolar disorder in adults, children and young people in primary and secondary care. Leicester: The British Psychological Society and The Royal College of Psychiatrists; 2018.
Parker G, Graham R, Hadzi-Pavlovic D, McCraw S, Hong M, Friend P. Differentiation of bipolar I and II disorders by examining for differences in severity of manic/hypomanic symptoms and the presence or absence of psychosis during that phase. J Affect Disord. 2013;150:941–7.
Phillips ML, Kupfer DJ. Bipolar Disorder 2 - Bipolar disorder diagnosis: challenges and future directions. Lancet. 2013;381:1663–71.
Fritz K, Russell AMT, Allwang C, Kuiper S, Lampe L, Malhi GS. Is a delay in the diagnosis of bipolar disorder inevitable? Bipolar Disord. 2017;19:396–400.
Hozer F, Houenou J. Can neuroimaging disentangle bipolar disorder? J Affect Disord. 2016;195:199–214.
Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JRI, Qiao Z, et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet. 2021;53:817–29.
Menezes IC, von Werne Baes C, Lacchini R, Juruena MF. Genetic biomarkers for differential diagnosis of major depressive disorder and bipolar disorder: a systematic and critical review. Behav Brain Res. 2019;357–358:29–38.
Evans DL. Bipolar disorder: Diagnostic challenges and treatment considerations. J Clin Psychiatry. 2000;61:26–31.
Ambrosi E, Rossi-Espagnet MC, Kotzalidis GD, Comparelli A, Del Casale A, Carducci F, et al. Structural brain alterations in bipolar disorder II: A combined voxel-based morphometry (VBM) and diffusion tensor imaging (DTI) study. J Affect Disord. 2013;150:610–5.
Favre P, Pauling M, Stout J, Hozer F, Sarrazin S, Abé C, et al. Widespread white matter microstructural abnormalities in bipolar disorder: evidence from mega- and meta-analyses across 3033 individuals. Neuropsychopharmacology. 2019;44:2285–93.
Hibar DP, Westlye LT, Van Erp TGM, Rasmussen J, Leonardo CD, Faskowitz J, et al. Subcortical volumetric abnormalities in bipolar disorder. Mol Psychiatry. 2016;21:1710–6.
Hibar DP, Westlye LT, Doan NT, Jahanshad N, Cheung JW, Ching CRK, et al. Cortical abnormalities in bipolar disorder: an MRI analysis of 6503 individuals from the ENIGMA Bipolar Disorder Working Group. Mol Psychiatry. 2018;23:932–42.
Thiel K, Meinert S, Winter A, Lemke H, Waltemate L, Breuer F, et al. Reduced fractional anisotropy in bipolar disorder v. major depressive disorder independent of current symptoms. Psychol Med. 2023;53:4592–602.
Yu H, Meng YJ, Li XJ, Zhang C, Liang S, Li ML, et al. Common and distinct patterns of grey matter alterations in borderline personality disorder and bipolar disorder: Voxel-based meta-analysis. Br J Psychiatry. 2019;215:395–403.
Narita K, Suda M, Takei Y, Aoyama Y, Majima T, Kameyama M, et al. Volume reduction of ventromedial prefrontal cortex in bipolar II patients with rapid cycling: a voxel-based morphometric study. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35:439–45.
Benedetti F, Yeh P-H, Bellani M, Radaelli D, Nicoletti MA, Poletti S, et al. Disruption of white matter integrity in bipolar depression as a possible structural marker of illness. Biol Psychiatry. 2011;69:309–17.
Almeida JRC, Akkal D, Hassel S, Travis MJ, Banihashemi L, Kerr N, et al. Reduced gray matter volume in ventral prefrontal cortex but not amygdala in bipolar disorder: Significant effects of gender and trait anxiety. Psychiatry Res Neuroimaging. 2009;171:54–68.
Abé C, Ekman CJ, Sellgren C, Petrovic P, Ingvar M, Landén M. Manic episodes are related to changes in frontal cortex: a longitudinal neuroimaging study of bipolar disorder 1. Brain. 2015;138:3440–8.
Arnone D, Cavanagh J, Gerber D, Lawrie SM, Ebmeier KP, McIntosh AM. Magnetic resonance imaging studies in bipolar disorder and schizophrenia: Meta-analysis. Br J Psychiatry. 2009;195:194–201.
Kempton MJ, Geddes JR, Ettinger U, Williams SCR, Grasby PM. Meta-analysis, database, and meta-regression of 98 structural imaging studies in bipolar disorder. Arch Gen Psychiatry. 2008;65:1017.
Ganzola R, Duchesne S. Voxel-based morphometry meta-analysis of gray and white matter finds significant areas of differences in bipolar patients from healthy controls. Bipolar Disord. 2017;19:74–83.
Sarıçiçek A, Yalın N, Hıdıroğlu C, Çavuşoğlu B, Taş C, Ceylan D, et al. Neuroanatomical correlates of genetic risk for bipolar disorder: A voxel-based morphometry study in bipolar type I patients and healthy first degree relatives. J Affect Disord. 2015;186:110–8.
Ambrosi E, Chiapponi C, Sani G, Manfredi G, Piras F, Caltagirone C, et al. White matter microstructural characteristics in Bipolar I and Bipolar II Disorder: a diffusion tensor imaging study. J Affect Disord. 2016;189:176–83.
Caseras X, Lawrence NS, Murphy K, Wise RG, Phillips ML. Ventral striatum activity in response to reward: Differences between bipolar i and II disorders. Am J Psychiatry. 2013;170:533–41.
Caseras X, Murphy K, Lawrence NS, Fuentes-Claramonte P, Watts J, Jones DK, et al. Emotion regulation deficits in euthymic bipolar I versus bipolar II disorder: a functional and diffusion-tensor imaging study. Bipolar Disord. 2015;17:461–70.
Foley SF, Bracher-Smith M, Tansey KE, Harrison JR, Parker GD, Caseras X. Fractional anisotropy of the uncinate fasciculus and cingulum in bipolar disorder type I, type II, unaffected siblings and healthy controls. Br J Psychiatry. 2018;213:548–54.
Gutiérrez-Galve L, Bruno S, Wheeler-Kingshott CAM, Summers M, Cipolotti L, Ron MA. IQ and the fronto-temporal cortex in bipolar disorder. J Int Neuropsychol Soc. 2012;18:370–4.
Ha TH, Ha K, Kim JH, Choi JE. Regional brain gray matter abnormalities in patients with bipolar II disorder: A comparison study with bipolar I patients and healthy controls. Neurosci Lett. 2009;456:44–48.
Ha TH, Her JY, Kim JH, Chang JS, Cho HS, Ha K. Similarities and differences of white matter connectivity and water diffusivity in bipolar I and II disorder. Neurosci Lett. 2011;505:150–4.
Liu JX, Chen YS, Hsieh JC, Su TP, Yeh TC, Chen LF. Differences in white matter abnormalities between bipolar i and II disorders. J Affect Disord. 2010;127:309–15.
Maller JJ, Thaveenthiran P, Thomson RH, McQueen S, Fitzgerald PB. Volumetric, cortical thickness and white matter integrity alterations in bipolar disorder type i and II. J Affect Disord. 2014;169:118–27.
Nortje G, Stein DJ, Radua J, Mataix-Cols D, Horn N. Systematic review and voxel-based meta-analysis of diffusion tensor imaging studies in bipolar disorder. J Affect Disord. 2013;150:192–200.
Abé C, Ekman CJ, Sellgren C, Petrovic P, Ingvar M, Landén M. Cortical thickness, volume and surface area in patients with bipolar disorder types I and II. J Psychiatry Neurosci. 2016;41:240–50.
Abé C, Liberg B, Song J, Bergen SE, Petrovic P, Ekman CJ, et al. Longitudinal cortical thickness changes in bipolar disorder and the relationship to genetic risk, mania, and lithium use. Biol Psychiatry. 2020;87:271–81.
Toma S, Islam AH, Metcalfe AWS, Mitchell RHB, Fiksenbaum L, Macintosh BJ, et al. Cortical volume and thickness across bipolar disorder subtypes in adolescents: a preliminary study. J Child Adolesc Psychopharmacol. 2019;29:141–51.
Strakowski SM, Fleck DE, Maj M. Broadening the diagnosis of bipolar disorder: benefits vs. risks. World Psychiatry. 2011;10:181–6.
Guzman-Parra J, Streit F, Forstner AJ, Strohmaier J, González MJ, Gil Flores S, et al. Clinical and genetic differences between bipolar disorder type 1 and 2 in multiplex families. Transl Psychiatry. 2021;11:31.
Parker G, Fletcher K. Differentiating bipolar I and II disorders and the likely contribution of DSM-5 classification to their cleavage. J Affect Disord. 2014;152–154:57–64.
Song J, Kuja-Halkola R, Sjölander A, Bergen SE, Larsson H, Landén M, et al. Specificity in etiology of subtypes of bipolar disorder: evidence from a swedish population-based family study. Biol Psychiatry. 2018;84:810–6.
Charney AW, Ruderfer DM, Stahl EA, Moran JL, Chambert K, Belliveau RA, et al. Evidence for genetic heterogeneity between clinical subtypes of bipolar disorder. Transl Psychiatry. 2017;7:e993.
Edvardsen J, Torgersen S, Røysamb E, Lygren S, Skre I, Onstad S, et al. Heritability of bipolar spectrum disorders. Unity or heterogeneity? J Affect Disord. 2008;106:229–40.
Coleman JRI, Gaspar HA, Bryois J, Breen G, Byrne EM, Forstner AJ, et al. The genetics of the mood disorder spectrum: genome-wide association analyses of more than 185,000 cases and 439,000 controls. Biol Psychiatry. 2020;88:169–84.
Kalman JL, Papiol S, Grigoroiu-Serbanescu M, Adorjan K, Anderson-Schmidt H, Brosch K, et al. Genetic risk for psychiatric illness is associated with the number of hospitalizations of bipolar disorder patients. J Affect Disord. 2022;296:532–40.
Bogdan R, Salmeron BJ, Carey CE, Agrawal A, Calhoun VD, Garavan H, et al. Imaging genetics and genomics in psychiatry: a critical review of progress and potential. Biol Psychiatry. 2017;82:165–75.
Gurung R, Prata DP. What is the impact of genome-wide supported risk variants for schizophrenia and bipolar disorder on brain structure and function? A systematic review. Psychol Med. 2015;45:2461–80.
Jiang X, Zai CC, Kennedy KG, Zou Y, Nikolova YS, Felsky D, et al. Association of polygenic risk for bipolar disorder with grey matter structure and white matter integrity in youth. Transl Psychiatry. 2023;13:322.
Kircher T, Wöhr M, Nenadić I, Schwarting R, Schratt G, Alferink J, et al. Neurobiology of the major psychoses: a translational perspective on brain structure and function—the FOR2107 consortium. Eur Arch Psychiatry Clin Neurosci. 2019;269:949–62.
Wittchen H-U, Wunderlich U, Gruschwitz S, Zaudig M. Strukturiertes Klinisches Interview für DSM-IV. Achse I: Psychische Störungen. In: Interviewheft und Beurteilungsheft. Eine deutschsprachige, erweiterte Bearbeitung der amerikanischen Originalversion des SKID I. Goettingen: Hogrefe; 1997.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV-TR. Washington DC: American Psychiatric Association; 2000.
Ho DE, Imai K, King G, Stuart EA. MatchIt: nonparametric preprocessing for parametric causal inference. J Stat Softw. 2011;42:1–28.
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62.
Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429–35.
Hassel S, Almeida JRC, Kerr N, Nau S, Ladouceur CD, Fissell K, et al. Elevated striatal and decreased dorsolateral prefrontal cortical activity in response to emotional stimuli in euthymic bipolar disorder: no associations with psychotropic medication load. Bipolar Disord. 2008;10:916–27.
Vogelbacher C, Möbius TWD, Sommer J, Schuster V, Dannlowski U, Kircher T, et al. The Marburg-Münster Affective Disorders Cohort Study (MACS): a quality assurance protocol for MR neuroimaging data. Neuroimage. 2018;172:450–60.
Jenkinson M, Beckmann CF, Behrens TEJ, Woolrich MW, Smith SM. FSL. Neuroimage. 2012;62:782–90.
Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TEJ, Johansen-Berg H, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23:S208–19.
Woolrich MW, Jbabdi S, Patenaude B, Chappell M, Makni S, Behrens T, et al. Bayesian analysis of neuroimaging data in FSL. Neuroimage. 2009;45:S173–86.
Behrens TEJ, Woolrich MW, Jenkinson M, Johansen-Berg H, Nunes RG, Clare S, et al. Characterization and propagation of uncertainty in diffusion-weighted MR imaging. Magn Reson Med. 2003;50:1077–88.
Alexander AL, Hurley SA, Samsonov AA, Adluru N, Hosseinbor AP, Mossahebi P, et al. Characterization of cerebral white matter properties using quantitative magnetic resonance imaging stains. Brain Connect. 2011;1:423–46.
Lemke H, Romankiewicz L, Förster K, Meinert S, Waltemate L, Fingas SM, et al. Association of disease course and brain structural alterations in major depressive disorder. Depress Anxiety. 2022;39:441–51.
Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, et al. Tract-based spatial statistics: Voxelwise analysis of multi-subject diffusion data. Neuroimage. 2006;31:1487–505.
Winkler AM, Ridgway GR, Webster MA, Smith SM, Nichols TE. Permutation inference for the general linear model. Neuroimage. 2014;92:381–97.
Smith SM, Nichols TE. Threshold-free cluster enhancement: addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage. 2009;44:83–98.
Borenstein M, Cooper H, Hedges LV, Valentine JC. Effect sizes for continuous data. I: The handbook of research synthesis and meta-analysis. 2nd ed. New York: Russell Sage Foundation; 2009. p. 221–35.
Yang C, Li L, Hu X, Luo Q, Kuang W, Lui S, et al. Psychoradiologic abnormalities of white matter in patients with bipolar disorder: diffusion tensor imaging studies using tract-based spatial statistics. J Psychiatry Neurosci. 2019;44:32–44.
Wise T, Radua J, Via E, Cardoner N, Abe O, Adams TM, et al. Common and distinct patterns of grey-matter volume alteration in major depression and bipolar disorder: evidence from voxel-based meta-analysis. Mol Psychiatry. 2017;22:1455–63.
van Velzen, Kelly LS, Isaev S, Aleman D, Aftanas LI A, Bauer J, et al. White matter disturbances in major depressive disorder: a coordinated analysis across 20 international cohorts in the ENIGMA MDD working group. Mol Psychiatry. 2020;25:1511–25.
Koshiyama D, Fukunaga M, Okada N, Morita K, Nemoto K, Usui K, et al. White matter microstructural alterations across four major psychiatric disorders: mega-analysis study in 2937 individuals. Mol Psychiatry. 2020;25:883–95.
Dong D, Wang Y, Chang X, Jiang Y, Klugah-Brown B, Luo C, et al. Shared abnormality of white matter integrity in schizophrenia and bipolar disorder: a comparative voxel-based meta-analysis. Schizophr Res. 2017;185:41–50.
Meinert S, Nowack N, Grotegerd D, Repple J, Winter NR, Abheiden I, et al. Association of brain white matter microstructure with cognitive performance in major depressive disorder and healthy controls: a diffusion-tensor imaging study. Mol Psychiatry. 2022;27:1103–10.
Videtta G, Squarcina L, Rossetti MG, Brambilla P, Delvecchio G, Bellani M. White matter modifications of corpus callosum in bipolar disorder: A DTI tractography review. J Affect Disord. 2023;338:220–7.
Yamada S, Takahashi S, Ukai S, Tsuji T, Iwatani J, Tsuda K, et al. Microstructural abnormalities in anterior callosal fibers and their relationship with cognitive function in major depressive disorder and bipolar disorder: a tract-specific analysis study. J Affect Disord. 2015;174:542–8.
Phillips ML, Swartz HA. A critical appraisal of neuroimaging studies of bipolar disorder: toward a new conceptualization of underlying neural circuitry and roadmap for future research. Am J Psychiatry. 2014;171:829–43.
Vargas C, López-Jaramillo C, Vieta E. A systematic literature review of resting state network—functional MRI in bipolar disorder. J Affect Disord. 2013;150:727–35.
Wise T, Radua J, Nortje G, Cleare AJ, Young AH, Arnone D. Voxel-based meta-analytical evidence of structural disconnectivity in major depression and bipolar disorder. Biol Psychiatry. 2016;79:293–302.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-IV. Washington DC: American Psychiatric Association; 1994.
Maggioni E, Altamura AC, Brambilla P. Exploring the neuroanatomical bases of psychotic features in bipolar disorder. Epidemiol Psychiatr Sci. 2017;26:358–63.
Lee D-K, Lee H, Ryu V, Kim SW, Ryu S. Different patterns of white matter microstructural alterations between psychotic and non-psychotic bipolar disorder. PLoS ONE. 2022;17:1–13.
Ji A, Godwin D, Rutlin J, Kandala S, Shimony JS, Mamah D. Tract-based analysis of white matter integrity in psychotic and nonpsychotic bipolar disorder. J Affect Disord. 2017;209:124–34.
Abé C, Ching CRK, Liberg B, Lebedev AV, Agartz I, Akudjedu TN, et al. Longitudinal structural brain changes in bipolar disorder: a multicenter neuroimaging study of 1232 individuals by the ENIGMA Bipolar Disorder Working Group. Biol Psychiatry. 2022;91:582–92.
Grewal S, McKinlay S, Kapczinski F, Pfaffenseller B, Wollenhaupt-Aguiar B. Biomarkers of neuroprogression and late staging in bipolar disorder: a systematic review. Aust N Z. J Psychiatry. 2023;57:328–43.
Fries GR, Pfaffenseller B, Stertz L, Paz AVC, Dargél AA, Kunz M, et al. Staging and Neuroprogression in Bipolar Disorder. Curr Psychiatry Rep. 2012;14:667–75.
Förster K, Horstmann RH, Dannlowski U, Houenou J, Kanske P. Progressive grey matter alterations in bipolar disorder across the life span – a systematic review. Bipolar Disord. 2023;25:443–56.
Grama S, Willcocks I, Hubert JJ, Pardiñas AF, Legge SE, Bracher-Smith M, et al. Polygenic risk for schizophrenia and subcortical brain anatomy in the UK Biobank cohort. Transl Psychiatry. 2020;10:309.
O’Connell KS, Coombes BJ. Genetic contributions to bipolar disorder: current status and future directions. Psychol Med. 2021;51:2156–67.
Almeida HS, Mitjans M, Arias B, Vieta E, Ríos J, Benabarre A. Genetic differences between bipolar disorder subtypes: a systematic review focused in bipolar disorder type II. Neurosci Biobehav Rev. 2020;118:623–30.
Mansell W, Lam D. A preliminary study of autobiographical memory in remitted bipolar and unipolar depression and the role of imagery in the specificity of memory. Memory. 2004;12:437–46.
Patten SB, Williams JVA, Lavorato DH, Bulloch AGM, D’Arcy C, Streiner DL. Recall of recent and more remote depressive episodes in a prospective cohort study. Soc Psychiatry Psychiatr Epidemiol. 2012;47:691–6.
Williams JM, Barnhofer T, Crane C, Herman D, Raes F, Watkins E, et al. Autobiographical memory specificity and emotional disorder. Psychol Bull. 2007;133:122–48.
Preti MG, Baglio F, Laganà MM, Griffanti L, Nemni R, Clerici M, et al. Assessing corpus callosum changes in Alzheimer’s disease: comparison between tract-based spatial statistics and atlas-based tractography. PLoS ONE. 2012;7:e35856.
Høegh MC, Melle I, Aminoff SR, Haatveit B, Olsen SH, Huflåtten IB, et al. Characterization of affective lability across subgroups of psychosis spectrum disorders. Int J Bipolar Disord. 2021;9:34.
Bertocci MA, Hanford L, Manelis A, Iyengar S, Youngstrom EA, Gill MK, et al. Clinical, cortical thickness and neural activity predictors of future affective lability in youth at risk for bipolar disorder: initial discovery and independent sample replication. Mol Psychiatry. 2019;24:1856–67.
Kurtz M, Mohring P, Förster K, Bauer M, Kanske P. Deficits in explicit emotion regulation in bipolar disorder: a systematic review. Int J Bipolar Disord. 2021;9:15.
Di Florio A, Hamshere M, Forty L, Green EK, Grozeva D, Jones I, et al. Affective temperaments across the bipolar-unipolar spectrum: Examination of the TEMPS-A in 927 patients and controls. J Affect Disord. 2010;123:42–51.
Bayer JMM, Thompson PM, Ching CRK, Liu M, Chen A, Panzenhagen AC, et al. Site effects how-to and when: An overview of retrospective techniques to accommodate site effects in multi-site neuroimaging analyses. Front Neurol. 2022;13:923988.
Fortin J-P, Parker D, Tunç B, Watanabe T, Elliott MA, Ruparel K, et al. Harmonization of multi-site diffusion tensor imaging data. Neuroimage. 2017;161:149–70.
Wingenfeld K, Spitzer C, Mensebach C, Grabe HJ, Hill A, Gast U, et al. The german version of the Childhood Trauma Questionnaire (CTQ): Preliminary psychometric properties. Psychother Psychosom Med Psychol. 2010;60:442–50.
Acknowledgements
This work is part of the German multicenter consortium “Neurobiology of Affective Disorders. A translational perspective on brain structure and function“. We are deeply indebted to all the study participants, the recruitment sites and their staff. Detailed acknowledgments of the FOR2107 can be found at www.for2107.de/acknowledgements.
Funding
This study was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft DFG; Forschungsgruppe/Research Unit FOR2107): Tilo Kircher (TK, speaker FOR2107; KI588/14-1, KI588/14-2, KI588/15-1, KI588/17-1), Udo Dannlowski (co-speaker FOR2107; DA1151/5-1, DA1151/5-2, DA1151/6-1), Igor Nenadić (NE2254/1-2, NE2254/3-1, NE2254/4-1), Tim Hahn (HA7070/2-2), Andreas Jansen (JA1890/7-1, JA1890/7-2), Stephanie Witt (WI3439/3-1, WI3439/3-2). Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
KT: Drafting the work, Conception and design of the work, Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content HL: Drafting the work, Conception and design of the work, Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content AW: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content KFl: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content LW: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content DG: Acquisition, analysis, or interpretation of data FS: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content KB: Acquisition, analysis, or interpretation of data FO: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content PU: Acquisition, analysis, or interpretation of data LT: Acquisition, analysis, or interpretation of data LB: Critical revision of the paper for important intellectual content KD: Critical revision of the paper for important intellectual content TH: Critical revision of the paper for important intellectual content KFö: Critical revision of the paper for important intellectual content PK: Critical revision of the paper for important intellectual content JR: Critical revision of the paper for important intellectual content NO: Critical revision of the paper for important intellectual content RR: Critical revision of the paper for important intellectual content FD: Critical revision of the paper for important intellectual content AF: Critical revision of the paper for important intellectual content BS: Critical revision of the paper for important intellectual content NA: Critical revision of the paper for important intellectual content HJ: Critical revision of the paper for important intellectual content AJ: Critical revision of the paper for important intellectual content SW: Critical revision of the paper for important intellectual content TA: Critical revision of the paper for important intellectual content AP: Critical revision of the paper for important intellectual content MB: Critical revision of the paper for important intellectual content IN: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content TK: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content SM: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content UD: Acquisition, analysis, or interpretation of data, Critical revision of the paper for important intellectual content. All authors have read and finally approved the submitted version of the paper, and agree to be accountable for all aspects of the work to ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interest
TK received unrestricted educational grants from Servier, Janssen, Recordati, Aristo, Otsuka, neuraxpharm. This funding is not associated with the current work. The corresponding author confirms that no other authors have any potential conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thiel, K., Lemke, H., Winter, A. et al. White and gray matter alterations in bipolar I and bipolar II disorder subtypes compared with healthy controls – exploring associations with disease course and polygenic risk. Neuropsychopharmacol. 49, 814–823 (2024). https://doi.org/10.1038/s41386-024-01812-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-024-01812-7
