
Abstract
Posttraumatic stress disorder can be viewed as a disorder of fear dysregulation. An abundance of research suggests that the prefrontal cortex is central to fear processing—that is, how fears are acquired and strategies to regulate or diminish fear responses. The current review covers foundational research on threat or fear acquisition and extinction in nonhuman animals, healthy humans, and patients with posttraumatic stress disorder, through the lens of the involvement of the prefrontal cortex in these processes. Research harnessing advances in technology to further probe the role of the prefrontal cortex in these processes, such as the use of optogenetics in rodents and brain stimulation in humans, will be highlighted, as well other fear regulation approaches that are relevant to the treatment of posttraumatic stress disorder and involve the prefrontal cortex, namely cognitive regulation and avoidance/active coping. Despite the large body of translational research, many questions remain unanswered and posttraumatic stress disorder remains difficult to treat. We conclude by outlining future research directions related to the role of the prefrontal cortex in fear processing and implications for the treatment of posttraumatic stress disorder.
Similar content being viewed by others
Introduction
Post-traumatic stress disorder (PTSD) is a maladaptive and debilitating psychiatric disorder typically accompanied by an extreme sense of fear at the time of trauma occurrence, with characteristic re-experiencing, avoidance, and hyperarousal symptoms in the months and years following the trauma. PTSD has a prevalence of ~6% but can occur in 25–35% of individuals who have experienced severe psychological trauma, such as combat veterans, refugees, and assault victims [1,2,3]. The differential risk determining those who do versus those who do not develop PTSD is multifactorial [4,5,6,7]. It is in part genetic, with at least 30–40% risk heritability for PTSD following trauma [8,9,10], and in part depends on past personal history, including adult and childhood trauma and psychological factors which may differentially mediate fear and emotion regulation. Additionally, considerable evidence now supports a model in which PTSD can be viewed, in part, as a disorder of fear dysregulation. This is advantageous because the neural circuitry underlying threat and fear-related behaviors in mammals, including the amygdala–hippocampus–medial prefrontal circuit, is among the most well-understood behavioral circuits in neuroscience [11,12,13,14]. Further, the study of threat behavior and its underlying circuitry has led to some of the most rapid progress in understanding learning and memory processes.
Although the amygdala and other subcortical regions are perhaps best understood with relationship to threat processing across species, burgeoning evidence has provided substantial support for the role of different regions of the prefrontal cortex (PFC) in particular in regulating the encoding of threat-related behaviors across species and the emotion of fear in humans. Furthermore, the PFC has a critical role in threat inhibition and extinction, as well as in processes such as emotion regulation and avoidance.
In contrast to the promise of current scientific approaches, in the clinic PTSD remains very difficult to treat [15, 16]. The best current treatments are in the form of exposure-based cognitive-behavioral therapies, which are thought to act on the neurocircuitry of threat extinction, in particular through the PFC. The medication treatments for PTSD are primarily limited to traditional serotonin and norepinephrine reuptake inhibitors, which are used for a broad range of depression and anxiety disorders. Advances in understanding the neural circuit of regulation of threat, fear, and PTSD symptoms may lead to novel and more robust treatment approaches.
This review aims to synthesize our current understanding of the role of the PFC in threat behaviors and threat-related emotional processing, and the role of multiple PFC subregions in PTSD. As acknowledged, this line of research is relevant to the treatment of disorders characterized by fear, such as PTSD. However, in line with the two-system view of fear and anxiety [17] and in order to not make assumptions about emotional states, the term “threat” will be used when referring to the behavioral, psychophysiological, or neural outcomes of conditioning research. The term “fear” will be reserved for describing studies in which the subjective emotion of fear was assessed or discussing the emotion more generally.
Nonhuman animal research on threat processing
The medial prefrontal cortex (mPFC) of the rodent regulates a balance between goal-oriented and habitual behaviors [18, 19]. The mPFC receives massive inputs from subcortical structures, including the amygdala, hippocampus, ventral striatum, hypothalamus, periaqueductal gray, and cerebellum, among others, that allow it to integrate the behavioral state of the animal and adjust behavioral decisions on a moment-to-moment basis. One of the most important mPFC functions is to integrate information about potential threats in the environment with other organismal drives to determine behavioral outputs [20].
Decades of basic research on the mPFC in rodents indicate that it plays a key role in the expression and storage of the Pavlovian threat response and the establishment of threat-related extinction memories [21]. Technological advances have evolved from lesion and pharmacologic studies to experiments utilizing circuit-perturbing and single-cell approaches, which are beginning to provide data at the cell-type resolution for the role of this critical structure in the threat response. Below, we will briefly review the anatomy of the rodent mPFC, the data implicating mPFC circuitry in the threat response and in threat extinction, molecular changes in mPFC cell types with threat acquisition and extinction, and future steps in these lines of research.
Anatomy of rodent mPFC
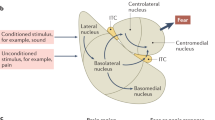
Like most cortical regions, the mPFC is a multi-layered structure of heterogeneous cell types, composed of excitatory pyramidal neurons, inhibitory interneurons, and support cells. Beginning with Brodmann, there have been debates about the existence and location of the mPFC in rodents due to the lack of a prominent granular layer [22; see Preuss and Wise, this issue]. Cross-species comparisons can be more easily made with respect to connectivity patterns [23]. The rodent mPFC is generally considered to consist of the medial precentral area (Fr2), the dorsal anterior cingulate cortex (dACC), prelimbic cortex (PL), and infralimbic cortex (IL) [24] (see Fig. 1a).

a Rodent anatomy highlighting regions involved in threat learning, extinction, avoidance, and the contextual modulation of threat expression; b Human anatomy highlighting regions involved in threat learning, extinction, avoidance, cognitive regulation, and the contextual modulation of threat expression. PL = prelimbic cortex, IL = infralimbic cortex; dACC = dorsal anterior cingulate cortex, vmPFC = ventromedial prefrontal cortex, dlPFC = dorsolateral prefrontal cortex.
For the purposes of this review, we will focus on rodent PL and IL, although recent work has implicated dACC in observational threat pathways, which may be relevant to PTSD from witnessed trauma [25]. Histologically in the mouse, PL and IL differ in the thickness of layer II/III and the prominence of layer separations between superficial II/III and layer V; however, this boundary is not easily demarcated [26, 27]. Both PL and IL receive cortical input, as well as unidirectional projections from the hippocampus, mainly CA1 and subiculum [24]. Projections to the amygdala are bidirectional, although there are differences in the projection patterns of PL and IL to the amygdaloid complex, and there is some controversy about whether PL and IL synapse onto functionally different cell types [28, 29]. Although there is some overlap in projection patterns, IL projects most heavily to lateral septum, bed nucleus of the stria terminalis, amygdala, hypothalamus, and brainstem, while PL sends more projections to insular cortex, nucleus accumbens, thalamus, and raphe nuclei [29]. The differences in these projection patterns suggest diverging functional roles for these adjacent structures.
Evidence for PL/IL distinction
For the past 20 years, there has been an extensive, although debated, literature showing differential roles for PL and IL in threat conditioning and threat extinction [21]. The first study to demonstrate a role for the rodent ventromedial prefrontal cortex (vmPFC) came from Morgan et al., who lesioned the mPFC [30]. A follow-up study demonstrated that more dorsal areas of the mPFC affected threat learning, while more ventral mPFC was required for threat extinction [31]. Quirk et al. supported this result when they [32] performed electrolytic lesions of the rat vmPFC and assessed threat extinction memory. They found that lesions that included caudal IL ablated threat extinction memories, while those that excluded the area had no effect. Pharmacological inactivation of PL and IL with agents such as the GABA agonist muscimol further suggested opposing roles for these structures in threat conditioning and threat extinction, respectively [33]. However, these results have not been universally reproduced [34, 35]. More recent studies from the Quirk laboratory have used optogenetics to drive or inhibit activity in excitatory IL neurons during threat extinction. These data suggest that IL neurons are necessary for encoding threat extinction memories but may not be necessary for threat extinction memory storage or retention [36]. These findings also suggest that the threat extinction memory trace may be represented by different cell populations over time. Indeed, it has been known that the threat memory is likely constituted by a distributed network of cells across a range of brain regions. Inputs to the mPFC likely help to drive evolution of the memory trace over time.
Modulation of mPFC by subcortical structures
Because the mPFC must guide behavior on a moment-to-moment basis, it needs to receive a constant stream of information from subcortical structures and send out a coordinated response. The mPFC receives dense innervation from many subcortical structures, but we will focus here upon three crucial inputs: the hippocampus, amygdala, and thalamus. The canonical role of the hippocampus in threat circuitry is to encode context-specific information of a threat trace, as it is crucial for an organism to be able to distinguish threats as belonging to a particular context. The hippocampus itself appears to have a dorsal-ventral functional gradient, with the dorsal hippocampus encoding context more specifically, while the ventral hippocampus (vHPC) includes affective information as well [37].
The vHPC sends dense direct projections to the mPFC from CA1, but also bidirectional disynaptic indirect connections to the mPFC through the reuniens nucleus of the thalamus and the perirhinal cortex [38]. Lesion studies of the hippocampus suggest a critical role in context processing [39]. Reversible inactivation of the dorsal hippocampus, through either pharmacologic or chemogenetic means, interferes with context-specific information of a threat memory [40, 41]. Inhibition of double-projecting vHPC neurons to the mPFC and basolateral amygdala (BLA) interferes with contextual threat recall [42] and disconnection of the vHPC from the mPFC interferes with renewal of threat memories, a context-dependent process [43]. Activity-tagging coupled with optogenetic inhibition suggests that threat conditioning and extinction memories exist in separate populations of neurons within the hippocampus [44], and the hippocampus may influence mPFC activity through feed-forward inhibition mechanisms through parvalbumin interneurons [45]. In return, the mPFC appears to suppress expression of erroneous contexts in a “top-down” manner through a disynaptic pathway through the reuniens nucleus of the thalamus [46]. In addition, there may be more routes of information flow from the PFC to the hippocampus, including direct routes from the nearby anterior cingulate [47].
The amygdala communicates the salience of the threat cue to the mPFC (see Murray and Fellows, this issue, for further discussion of amygdala-PFC interactions). For thirty years, the amygdala has been implicated in both threat learning [48] and threat extinction [49] processes. The BLA sends bidirectional projections to the mPFC [50]. There is evidence to suggest that there is a dorso-ventral topographic segregation of BLA input to the mPFC; more dorsal projections (to PL) encode threat-stimulating information while more ventral projections (to IL) encode threat extinction-related information [51]. Synaptic connections between PL neurons and BLA inputs also strengthen in response to stress, in part through endocannabinoid-mediated mechanisms [52]. Projection neurons within the BLA exhibit plasticity when conditioned stimulus-unconditioned stimulus pairings occur and convey this information to the mPFC.
Finally, nuclei within the thalamus help bind threat memories to context and facilitate shifts in the mPFC threat memory trace over time. The reuniens nucleus of the thalamus coordinates oscillatory synchrony between the mPFC and the vHPC, which is necessary for proper contextual representation of threat memories [46, 53]. The paraventricular nucleus of the thalamus plays a crucial role in the encoding of threat memories over time [54] and appears to be necessary in shifting the temporal nature of how the mPFC encodes threat memories [55].
Molecular pathways in rodent mPFC
At the molecular level, threat conditioning and extinction are associated with epigenetic, transcriptional, and translational changes that likely modify synaptic weights and cell firing properties that persistently alter circuit function. Introduction of the translational inhibitor anisomycin, either intraventricularly or into the mPFC, causes a failure to retain threat extinction memories. This suggests that translation of new protein is necessary for the formation of a novel threat extinction memory [56]. Threat conditioning and threat recall are associated with unique, cell-type-specific transcriptional changes that persist for weeks after initial training [57]. Threat extinction also requires transcriptional processes within IL: injection of an inhibitor of PARP-1, a gene involved in ADP-ribosylaton that is necessary for transcription, into mPFC impairs contextual threat extinction [58].
The BDNF-TrkB neurotrophic factor pathway has also been extensively studied with regards to mPFC and memory formation in mPFC. Expression of Bdnf in PL is necessary for consolidation of cued threat conditioning [59], while infusion of Bdnf into IL after threat acquisition is sufficient to diminish threat responses in the absence of extinction training [60]. Threat extinction is also associated with epigenetic modification. In the IL, threat extinction is associated with acetylation of histones near the Bdnf locus [61], changes to the p300/CBP complex (PCAF) [62], as well as deposition of DNA-modification marks such as 5-hydroxymethylcytosine and N6-methyl-2’deoxyadenosine (m6dA) near loci of activity-dependent genes such as Bdnf.
Additionally, inhibition of PCAF in IL was shown to interfere with threat extinction [62]. Recently, Li et al. [63], have shown that knockdown of N6amt1, the gene responsible for m6dA deposition, within IL, blocks changes to the m6dA mark at the Bdnf promoter in vivo and impairs threat extinction retention. These findings suggest that alterations in m6dA deposition are necessary for the formation of threat extinction memories within IL [63]. These findings also strongly support the hypothesis that threat extinction memory requires epigenetic changes within IL. Our understanding of the molecular changes that occur within the mPFC during threat-related processes are still in their infancy. Gene expression changes are unique to cell type, and cell-type-specific investigations of mPFC in threat conditioning and extinction are just beginning.
Stress and threat reactions
One potential factor that alters the ability to control emotional responses via altering PFC function is stress (for review, see 64,65, Kalin and Barbas, this issue). Studies in animal models have shown that acute stress leads to changes in neuronal signaling that impair function in the dlPFC [64] and IL cortex [66]. These changes are proposed to be due to the impact of increased catecholamines, in particular noradrenergic and dopaminergic signaling, on PFC neuronal activity with even relatively mild acute stress exposure [64, 65, 67]. Stress also impacts signaling within the amygdala. Noradrenergic signaling from the locus coeruleus to the amygdala was recently shown to be necessary to produce the immediate extinction deficit, an impairment in extinction learning that occurs soon after fear learning and is thought to be related to the stress of the fear learning process [68]. Activity of CRF-expressing neurons within the CeA was also recently shown to contribute to this phenomenon [69]. In rodents, chronic stress also impacts neural activity in both PL and IL cortex [70] and leads to structural changes in IL cortex [71]. One consequence of stress-related PFC impairment is enhanced threat learning and impaired extinction retention in rodent models [66, 70].
In the next section we will explore the role of PFC in human threat processing research, from acquisition and encoding of threat, to its extinction and extinction recall. We will also further integrate additional findings with regards to other threat and avoidance behaviors in response to threat stimuli and the impact of stress on the PFC. Finally, we will examine how these different brain regions and behaviors are dysregulated in threat-related disorders such as PTSD.
Preclinical human threat processing research
Threat learning
Perhaps it is not surprising, given the extensive research with nonhuman animals, that research in humans confirms a role for the amygdala and PFC in threat learning (see Fig. 1b and Fig. 2a). The role of the amygdala was first demonstrated in patients with amygdala damage. Relative to healthy controls, both bilateral [72] and unilateral [73] amygdala damage resulted in impaired conditioned responses, as measured by the skin conductance response (SCR). However, these patients were able to verbally report the contingency between the conditioned stimulus and shock after the procedure, which was impaired in patients with hippocampal damage whose amygdala was intact [72, 74]. These findings suggest that the amygdala is only critical for the implicit, physiological expression of threat learning in humans, with conscious knowledge about the threatening nature of stimuli in the environment remaining intact, despite amygdala lesions. Furthermore, these findings demonstrate that there are additional brain regions that are critical for the expression of the subjective fear and threat responses, including PFC areas that are discussed in more detail below.

a Healthy Threat Circuit. Regions involved in threat learning and the control of threat reactions via extinction, context, avoidance, or cognitive regulation. In healthy individuals the coordination of this circuit enables adaptive threat expression. b PTSD Threat Circuit. The dlPFC, vmPFC/IL, and hippocampus show impaired functioning with PTSD, whereas the amygdala and dACC/PL are enhanced. Disrupted connections between regions are indicated by dashed lines. The disrupted threat circuit with PTSD results in maladaptive threat expression. Prefrontal cortex regions are highlighted within the beige circle. Terms for animal/human homologous regions are in the same circles. PL = prelimbic cortex, IL = infralimbic cortex, dACC = dorsal anterior cingulate cortex, vmPFC = ventromedial prefrontal cortex, dlPFC = dorsolateral prefrontal cortex.
Consistent with these early patient studies, functional magnetic resonance imaging (fMRI) studies soon followed that showed increased blood oxygenation level dependent (BOLD) signal in the amygdala to a conditioned stimulus (relative to stimulus never paired with shock) [75, 76], and the magnitude of this BOLD response was correlated with the strength of the conditioned response [76]. Interestingly, this differential amygdala BOLD response was only apparent in the early stages of threat conditioning. This finding is somewhat surprising given rodent research showing long-lasting changes in the amygdala lateral nucleus with threat learning. However, there is electrophysiological evidence in rodents showing that a subset of lateral nucleus amygdala neurons respond preferentially during initial learning [77], and there is greater responding overall at this time. It may be the case that BOLD changes in the amygdala can only be observed at time windows when there are larger populations of neurons responding, such as initial learning. One major limitation of fMRI for investigations of amygdala function in humans is that it is a relatively coarse measurement. Although the spatial resolution of standard BOLD imaging is generally 3 mm, in practice, with spatial smoothing and group averaging, the actual resolution is greater than 10 mm, which covers a substantial portion of the amygdala (which is slightly more than 1000 mm3 in humans) and makes it very difficult to detect discrete responses in amygdala subnuclei. The challenges of using BOLD imaging to study the human amygdala is reflected in recent meta-analyses of fMRI threat learning studies, which fail to find BOLD changes in the amygdala [78], in spite of its critical role in threat learning in rodent models and patient studies.
In contrast to difficulties in detecting BOLD changes in the amygdala during threat learning, meta-analyses and individual studies reliably show activation in a number of other brain regions, including the insula cortex, which is linked to physiological arousal responses [79], the striatum, and the dACC (e.g., [76, 78, 80]). The dACC is a prefrontal region that is proposed to be the human homolog of the PL cortex in rodents [80]. As discussed earlier, the PL in rodents has been suggested to play a role in the expression of threat learning via projections to the basolateral amygdala, with stimulation of this region increasing conditioned freezing and inactivation reducing it.
In rodents, the PL and IL cortex are located adjacent to one another in the mPFC. In primate models, however, the PL and IL are farther apart. The primate PL cortex is thought to be divided into rostral and caudal regions with different connectivity patterns. The rostral region is thought to be more similar to the PL in rodents, with the dACC being the human homologue for that region [23, 81]. Consistent with this suggestion, Milad et al. [80] found that both cortical thickness, and BOLD response magnitude to a conditioned stimulus in this region, were correlated with the strength of the conditioned response as measured with SCR in humans.
Although the basic circuitry of threat learning seems to be preserved across species, a primary difference between humans and other animals is that humans, more often than not, learn about threats in the environment via social interactions. For example, children learn to fear germs by being told about their existence and observing others engaging in actions attempting to avoid them. This ability is adaptive in that humans do not need to be physically harmed to learn about threats in the environment. It can also be maladaptive in that we can develop robust fears for events that are imagined and anticipated but never actually experienced, contributing greatly to human anxiety and fear-related disorders. To what extent do the brain systems involved in threat learning from direct experience, that have been investigated in rodent models, map onto socially acquired, imagined threats in humans?
To address this question, brain imaging and patient studies have examined threat learning through verbal instruction (e.g., being told a blue square predicts a shock, and then being shown a blue square) or observation (e.g., watching someone else receive a shock paired with a blue square, and then being shown a blue square). Consistent with Pavlovian threat conditioning, fMRI studies of both instructed and observational threat learning show activation in the amygdala, dACC, and insula [82]. For instructed learning, the amygdala BOLD response is left-lateralized [83], and only patients with left, but not right, amygdala damage show impaired physiological evidence of threat learning, perhaps because of the verbally mediated nature of this learning [84]. In contrast, observational learning results in increased bilateral BOLD signal in the amygdala, both when observing someone else receiving a shock paired with a conditioned stimulus (learning), and when viewing the conditioned stimulus afterwards (test). In addition, during observational learning, activation in a rostral mPFC region that has been implicated in mentalizing about others is correlated with the strength of the learned threat response as measured by SCR [85], and learning is stronger with greater empathy for the person being observed [86]. These results suggest that while the social learning of threat may engage unique neural circuitry due to the nature of the learning, it also takes advantage of the phylogenetically older mechanisms of Pavlovian threat conditioning for threat expression.
Threat extinction
Much like threat learning, neuroimaging studies of threat extinction in humans have identified brain regions that parallel those involved in extinction in rodents (see Figs. 1b and 2a). The vmPFC is proposed to be the homologue for the IL in rodents [87] and serves to inhibit threat responses produced by the amygdala. There is consistent evidence of increases in BOLD signal in the vmPFC during extinction learning [88,89,90] and recall [89,90,91,92] (for review, see [93,94,95]). Further, the degree of activation of the vmPFC has been shown to be positively correlated with the degree of extinction, or extinction retention, as measured by SCR [89, 90], consistent with the suggested role of the IL in extinction in rodent models.
Brain morphology studies also point to the human vmPFC being involved in extinction. Milad et al. [96] found vmPFC thickness to be positively correlated with extinction recall. Specifically, greater thickness was associated with smaller SCR to the conditioned stimulus during extinction recall, suggestive of better extinction recall (see also [97]). Subsequently, Winkelman et al. [98] examined the relationship between vmPFC thickness and extinction learning, rather than recall, and found similar results. Greater vmPFC thickness was associated with smaller differential SCR during early extinction learning, suggestive of better extinction learning.
Targeting extinction with neuromodulation, neuroplasticity, and context modulation
One drawback of these MRI studies, however, is that they are correlational in nature. Unlike research conducted in rodents, specific brain regions in humans cannot be lesioned or tagged, nor can regions that are not on the surface of the brain be disrupted. Researchers are able, however, to stimulate or disrupt surface frontal regions of the brain in humans using non-invasive devices. For example, transcranial direct current stimulation (tDCS) applies a low-intensity current through two electrodes attached to the scalp, and transcranial magnetic stimulation (TMS) delivers an electric current through a coiled wire placed on the scalp, creating a magnetic field across the skull. Both of these strategies are thought to modulate neuronal activity in the human brain.
Using these techniques, a few recent brain stimulation studies [99,100,101,102,103] have been conducted in humans to probe the role of the vmPFC in extinction. For example, Dittert et al. [100] administered tDCS via bitemporal electrodes aimed at the vmPFC prior to and during extinction and found that tDCS, relative to sham, stimulation resulted in faster early extinction learning. Similarly, Raij et al. [102] found that TMS, during extinction learning, to an area of the frontal cortex functionally connected to the vmPFC (i.e., the left lateral PFC), but not to an area of the frontal cortex thought to be unconnected to the vmPFC, led to enhanced extinction recall. Although these studies provide some insight into the role of the vmPFC in extinction, given the location of the vmPFC and the fact that tDCS and TMS are applied externally, it is difficult to be certain that the vmPFC in particular was stimulated in these studies.
Consistent with animal models of extinction circuitry, the vmPFC interacts with other regions such as the amygdala and hippocampus to modulate threat responses during extinction. From rodent research showing that intra-amygdala infusion of the NMDA receptor agonist d-cycloserine, which enhances NMDA-dependent plasticity, facilitates extinction learning, and successful translation of this work to humans (see [104] for review), we know that the human amygdala plays a role in extinction learning. Human imaging studies, however, have been less consistent with finding changes in BOLD signal in the amygdala during extinction [76, 88, 90, 95, 105]. Much like with threat acquisition, it may be that the involvement of the amygdala in extinction is more subtle and difficult to detect using standard fMRI techniques [106]. Nonetheless, imaging research does point to changes in the relationship between the PFC and amygdala during extinction. Connectivity analyses have demonstrated functional coupling between the mPFC and amygdala during extinction learning [107], and vmPFC and amygdala during extinction recall [89, 108].
Also consistent with animal models (e.g., [40]), research suggests that the hippocampus is involved in contextual modulation of extinction and works in concert with the PFC during contextual extinction learning. One of the first studies demonstrating hippocampal involvement in extinction showed that patients with damage to the hippocampus failed to show contextually modulated reinstatement of conditioned responses following extinction [74]. Brain imaging studies of the contextual modulation of extinction typically manipulate the visual background during extinction and report hippocampal activation during extinction recall [89, 91, 92]. Importantly, functional connectivity analyses also suggest coupling of the PFC and hippocampus during contextual extinction learning [107] and recall [89].
Sleep is another factor that has been shown to modulate threat control in humans. Sleep has been shown to enhance both threat learning, and the generalization of extinction learning in humans and other animals. The documented role for sleep in memory consolidation is proposed to extend to both threat memories and extinction memories. Which of these competing memory representations is selectively strengthened depends on contextual factors such a recency of learning and replay [109]. Because of evidence for sleep’s modulation of extinction learning across species, it has been suggested that disruptions of sleep following acute trauma, or predating the traumatic experience, may contribute to the etiology or perpetuation of PTSD [110].
Avoidance/active coping
Another method of reducing conditioned threat reactions is through active avoidance or coping. Initial rodent research on the neural circuitry of active avoidance found that while the passive expression of conditioned threat responses engages a pathway from the lateral nucleus to the central nucleus, when the animal engages in an action to avoid the unconditioned stimulus, projections from the lateral nucleus to the basal nucleus to the nucleus accumbens are involved. However, in order for the animal to produce an avoidance action, conditioned freezing must be inhibited which requires the IL cortex, much like in the expression of extinction (see [111] for a review).
One benefit of avoidance learning over extinction for controlling threat reactions is that avoidance learning results in a persistent reduction in the passive conditioned response, even when the avoidance action is no longer available [112]. This is in contrast to extinction in which the conditioned response often returns through spontaneous recovery, renewal, or reinstatement. Both the acquisition of avoidance, and the reduction of the persistent conditioned threat reaction following avoidance learning, are blocked by the injection of protein synthesis inhibitors into the IL. This indicates that plasticity in the IL is critical for the persistent reduction of conditioned responses with avoidance [112]. Mirroring these findings, studies have shown that previous history with escapable shock results in a lasting reduction of the conditioned response, and this effect is eliminated with IL inactivation [113].
In humans, there is evidence that both avoidance learning and history with escapable shock can persistently reduce conditioned threat actions as measured with SCR, even when no avoidance action is available [114, 115]. However, in order to persistently diminish threat conditioned responses in humans, avoidance actions need to be learned through trial and error and there needs to be a subjective sense of control over the unconditioned stimulus during learning [115]. Simply providing the option of an action to avoid the unconditioned stimulus yields no lasting reduction of conditioned responses when the avoidance action is no longer available, and in fact can increase them by preventing extinction learning (called “protection from extinction”, [116]). Consistent with the circuity of avoidance learning detailed in rodent models, trial-by-trial avoidance learning yields increased BOLD activation in the vmPFC and ventral striatum, relative to standard extinction [114], suggesting the brain mechanisms of active coping are preserved across species.
Emotion regulation
Although extinction and active coping can be investigated across species, humans have the unique ability to use cognitive strategies to alter emotional responses, such as responses to fear provoking stimuli (for review, [117]). One common emotion regulation strategy is cognitive reappraisal. This strategy involves reframing thoughts (also called “appraisals”) about a stimulus in order to change the emotional response that that stimulus evokes. Emotion regulation strategies can be employed with the goal of either upregulating (i.e., increasing) or downregulating (i.e., decreasing) emotions. Here, the primary focus is on data related to downregulating negative emotions, fear in particular, as these data are most relevant to PTSD and its treatment. We highlight reappraisal, which is proposed to be similar to cognitive restructuring in clinic. Neuroimaging research has provided insight into the brain regions involved in emotion regulation in humans. Studies to date suggest that emotion regulation strategies aimed at downregulating negative emotions engage cognitive control regions of the PFC, which then modulate the amygdala via various potential pathways to influence negative emotional responses.
The most recent meta-analysis of fMRI studies of emotion regulation [118] found that all strategies aimed at downregulating negative emotions were collectively associated with increased BOLD signal in the following areas: ventrolateral prefrontal cortex (vlPFC), dorsolateral prefrontal cortex (dlPFC), and dorsomedial prefrontal cortex (dmPFC). While these were the largest areas of convergence, activation was also found in other areas (i.e., the bilateral inferior parietal lobule, supplementary motor area, pre-supplementary motor area, left middle temporal gyrus, and posterior cingulate gyrus). These findings were relatively consistent with a prior meta-analysis [119], with the exception that the prior meta-analysis also found decreased BOLD signal in the amygdala and parahippocampal gyrus, consistent with the notion that cognitive control regions of the PFC modulate amygdala activity during emotion regulation. One potential reason for differing results across these two meta-analyses may be differences in the studies examined and proportions of various emotion regulation strategies included. There is some evidence that different emotion regulation strategies may recruit distinct brain regions. For example, Dörfel et al. [120] found that some emotion regulation strategies are associated with reduced activity in the amygdala, whereas others are not.
Nonetheless, the majority of imaging research to date on emotion regulation focuses on the strategy of cognitive reappraisal. Meta-analyses of cognitive reappraisal alone have consistently found increased BOLD signal in the dlPFC, vlPFC, and dmPFC [93, 121, 122] and decreased BOLD signal in the amygdala [93, 121]. The dlPFC is thought to be an important driver of emotion regulation and hypothesized to be involved in the manipulation of appraisals of stimuli in working memory [121,122,123]. The vlPFC is hypothesized to support choosing and inhibiting appraisals of stimuli [121, 124, 125] or potentially may signal salience and the need to reappraise [122]. Finally, the dmPFC is hypothesized to support abstracting affective meaning of stimuli or the processes of self-reflecting and identifying one’s own affective reactions to stimuli [121, 126,127,128,129,130].
Two hypotheses have been proposed for how these cognitive control regions of the PFC (i.e., the dlPFC, vlPFC, and dmPFC) influence the amygdala: (1) that they engage the vmPFC which then modulates the amygdala, similar to the neurocircuitry of extinction [93, 131, 132], and (2) that they modulate lateral temporal areas associated with semantic and perceptual representations, which then indirectly influence the amygdala [121].
In support of the first hypothesis, anatomical research in nonhuman primates shows that connections between the lateral PFC and amygdala are sparse relative to the vmPFC and amygdala (e.g., [133, 134]). Additionally, a study of cognitive reappraisal of threat conditioned stimuli by Delgado et al. [131] demonstrated increased BOLD signal in the dlPFC and decreased BOLD signal in the amygdala, but also changes in vmPFC activity that mirror those that occur during extinction. Specifically, increased BOLD signal in the vmPFC after successful cognitive reappraisal was observed. Further, connectivity analyses [131, 132], a meta-analysis conducted by Diekhof et al. in 2011, and more recent dynamic causal modeling of cognitive reappraisal data [135], also support this hypothesis.
In contrast, two more recent meta-analyses favor the second hypothesis [121, 122]. Specifically, in addition to increased activation of the cognitive control regions of the PFC mentioned above and modulation of the amygdala, Buhle et al. [121] report increased activation of the lateral temporal cortex during cognitive reappraisal. Notably, they did not observe increased activation of the vmPFC. There are many possible reasons for these differences in findings (see [121, 136, 137] for discussion), however, one possibility is that it has to do with the type of cognitive reappraisal procedure used. The study by Delgado etv al. [131] involved cognitive reappraisal of threat conditioned stimuli, whereas most of the studies included in the Buhle et al. [121] meta-analysis involved cognitive reappraisal of negatively-valenced photos. It could also be that the vmPFC is involved in cognitive reappraisal for some individuals more so than others. For example, the nature of vmPFC involvement during cognitive reappraisal has been observed to vary as a function of psychiatric symptoms [138, 139].
To date, the majority of studies on brain regions involved in emotion regulation have examined fMRI data. To our knowledge, only one study has examined brain morphometry in relation to cognitive reappraisal task performance [97] and this study did not find a relationship between success of reappraisal and dlPFC, vlPFC, or vmPFC cortical thickness. Although there do not seem to be structural changes related to the successful cognitive reappraisal, there is one lesion study [140] that supports a role of the dlPFC in cognitive reappraisal. This study examined individuals with dlPFC lesions and found that they showed impaired ability to reappraise threat conditioned stimuli as indexed by poorer subjective fear outcomes, compared to matched controls. Additionally, a few brain stimulation studies have been conducted to probe the role of the PFC in emotion regulation. In this case, given the lateral location of the cognitive control regions of the PFC implicated in emotion regulation, tDCS or TMS can be used to target these regions. However, results of these studies have been inconsistent. While some studies have found that tDCS over the dlPFC or vlPFC enhances cognitive reappraisal [141,142,143], others have not [143, 144].
Stress and threat control
As the discussion above indicates, there are several techniques that can be used to control learned threat reactions in humans when they are no longer adaptive. However, as outlined earlier, stress, both chronic and acute, can impact the function of PFC and subcortical regions implicated in threat control. In humans, experimental studies of chronic stress are not possible due to ethical concerns; however, there is evidence that a history of childhood abuse is correlated with reduced gray matter volume in PFC regions, including the vmPFC and orbital frontal cortex [145]. In addition, mild acute stress in humans impairs the efficacy of previously acquired cognitive reappraisal strategies in reducing conditioned threat and subjective fear [146] and results in enhanced spontaneous recovery following extinction training [147]. These latter findings suggest that even when threat control techniques are successfully learned, relatively mild acute stress may impair the ability to express this learning by impacting the function of PFC inhibitory circuits.
Role of PFC in PTSD
The above evidence suggests that stress, in particular, is associated with altered PFC function and its role in regulating subcortical emotional responses. PTSD is among the most well-understood, prevalent, and medically significant stress-related disorders. A fairly large set of studies now supports a clear role for altered PFC structure and function in PTSD and related disorders (see Fig. 2b).
Structural imaging in PFC and PTSD
Related to stress exposure, independent of PTSD, a number of studies have identified smaller volumes in PFC and decreased structural connectivity between PFC and subcortical areas as a function of violence and trauma exposure. In a prospective study of Israeli soldiers, Admon et al. [148] used diffusion tensor imaging (DTI) pre- and post-military service, reporting that soldiers with decreased hippocampal structural connectivity with the vmPFC had a more maladaptive response to stressful military service. In a small study of victims of urban violence, Rocha-Rego et al. [149] found significant reductions in gray matter volume in the ventral premotor cortex and in the pregenual ACC as a function of civilian violence. Furthermore, in a moderately large sample of post-9/11 veterans, Clausen et al. [150] found that higher combat exposure uniquely related to lower cortical thickness in the left prefrontal lobe; and that, overall, combat exposure, PTSD, and head injuries differentially relate to alterations in cortical thickness.
A number of studies have also found decreased gray matter volumes related to traumatic stress symptoms. In development, with a study of ~50 youth, Keding and Herringa [151] found that those with PTSD had reduced gray matter volume in anterior vmPFC, which inversely correlated with PTSD duration. They suggest that pediatric PTSD is associated with abnormal structure of the vmPFC, possibly related to disrupted extinction and contextual gating of fear. Similarly, in a study of over 100 participants, it was found that maltreated youth with PTSD demonstrated decreased right vmPFC volumes compared to both maltreated youth without PTSD and nonmaltreated healthy controls [152]. Similar findings have been found in adult samples. A study of 85 veterans suggested decreased structural volumes of vmPFC and ACC in those with PTSD compared to controls [153] and a smaller study of 28 veterans found smaller subgenual cingulate volumes compared to controls, in addition to a number of other limbic region structural abnormalities [154].
A number of studies have also examined structural integrity of white matter tracts, including connections to and from the PFC. Koch et al. [155] used DTI to show decreased integrity of the uncinate fasciculus tract, connecting the vmPFC to multiple subcortical, limbic regions including the amygdala, in patients with PTSD. In addition, using DTI, Fani et al. [156] found that civilians with PTSD had decreased structural connectivity via the cingulum bundle, which supports the hippocampus-dACC pathway. They suggest that altered hippocampus-ACC connectivity may represent a highly salient intermediate neural phenotype for PTSD. Further analyses of this cohort found that individuals with the “risk” allele of the FKBP5 genetic biomarker, associated with childhood maltreatment and PTSD risk, also had decreased cingulum structural integrity.
Some of these structural changes may occur quite rapidly in the aftermath of trauma—or may be preexisting and predispose some individuals to a greater risk of PTSD following trauma. Using DTI and structural imaging in the weeks following trauma exposure, research has shown that reduced fractional anisotropy of the uncinate fasciculus at around the time of trauma predicted greater PTSD symptoms (in particular posttraumatic anhedonia) at 12 months post-trauma. Furthermore, as the traumatized participants were followed over time, increased gray matter volume of the vmPFC was also associated with reduced trauma-related symptoms over the 12 months following trauma [157]. In another study recruiting patients after trauma exposure with mild traumatic brain injury, smaller cortical volumes of superior frontal cortex and rostral and caudal cingulate at 2 weeks after trauma exposure contributed to the prediction of increased likelihood of 3-month PTSD diagnosis in multivariable models incorporating other established risk factors [158].
As with many other types of human data, sample size often limits interpretation, as small sample sizes are subject to both false positive and false negative biases. Therefore, as more studies have been performed of PTSD and structural imaging, much larger analyses can be performed via meta-analyses of multiple datasets. A recent meta-analysis confirmed, using voxel-based morphometry, that there were prominent volumetric reductions in the mPFC, including the ACC, when examining over 80 different MRI studies in PTSD compared to depression [159].
In addition, one of the largest meta-analyses to date of structural cortical volumes from the ENIGMA-PGC-PTSD workgroup, compared 1379 PTSD patients to 2192 controls without PTSD. A primary finding was that volumes of left and right lateral orbitofrontal gyri were significantly smaller in PTSD patients than controls and were negatively correlated with symptom severity. Together, these findings indicate that cortical volumes in PTSD patients are smaller in prefrontal regulatory regions, consistent with preclinical work suggesting a critical role for orbital frontal PFC regions in recovery and extinction of threat behaviors [160].
Functional imaging in PFC and PTSD
It is thought that fMRI, including both emotional and cognitive task-based MRI, as well as resting state MRI may be more sensitive to pathology-related functional activity. However, it can also be criticized as there are many processing steps and the data are essentially subtractions and comparisons between different tasks, timepoints, and individuals, making interpretation often more complex than structural MRI. Nonetheless, many of the same themes with regards to the role of PFC in PTSD are found with fMRI, complementing the above structural MRI findings as well as the healthy human and preclinical data.
While many fMRI studies of PTSD have had quite limited sample sizes, some are particularly informative. Script-driven imagery with participants reading or listening to scripts of their prior trauma experiences or other stressful vignettes has been a powerful probe for determining differential brain activity in PTSD. For example, in a case-control twin study with 26 male identical twin pairs (12 PTSD; 14 non-PTSD) discordant for PTSD and combat, script-driven imagery fMRI revealed diminished activation in the mPFC during stressful versus neutral imagery in PTSD patients relative to others [161].
Another powerful method for observing brain activity is using a masked emotional probe. In this design, participants are presented with an emotional stimulus for a very brief period of time (i.e., milliseconds) and then it is replaced by a non-emotional stimulus. This minimizes explicit awareness of the emotional stimulus, while still engaging emotional perception and the associated neutral structures. For example, Killgore et al. [162] used a masked emotional probe task and compared adults with anxiety or PTSD to healthy controls. Patients (all groups combined) showed greater amygdala and reduced vmPFC activation compared to controls during the masked fearful faces. Additionally, in a prospective emergency department study following patients longitudinally, Stevens et al. [163] used an unmasked fearful faces design to demonstrate that dorsal ACC activity during presentation of fearful faces predicts ongoing symptom maintenance in the aftermath of acute trauma in civilians. These findings are consistent with some of the findings of Milad et al. [164,165,166] related to impaired extinction recall, discussed in the next section.
A number of studies have also examined the role of activity within the PFC related to prediction of treatment response. Fonzo et al. [167] reported on fMRI task-based assessment while completing three tasks assessing emotional reactivity and regulation prior to prolonged exposure psychotherapy for PTSD. At baseline, individuals with the greatest symptom improvement with therapy showed, among other findings, greater dorsal PFC and vmPFC activation during emotional conflict regulation. They interpreted these findings to suggest that participants who are most likely to benefit from exposure therapy demonstrate spontaneous activity of PFC when superficially processing threat and adapting to emotional interference. Further analysis of this study suggested that psychotherapy increased lateral frontopolar cortex activity and connectivity with the vmPFC [168]. Additionally, greater increases in frontopolar activation were associated with improvements in hyperarousal symptoms and psychological well-being. Given these findings, the authors argue that frontopolar connectivity with ventromedial regions during emotion regulation is a possible key mechanism of psychotherapeutic improvement in PTSD.
In addition, in a mindfulness-based exposure therapy intervention with a small sample size, King et al. [169] found that posterior cingulate cortex (PCC)-dlPFC functional connectivity was correlated with improvement in PTSD avoidance and hyperarousal symptoms. Overall, they surmised that increased connectivity between PCC-dlPFC brain regions could be related to attentional control and symptom improvement.
It is important to note that the “standard” observation of hyperarousal-related PTSD with increased amygdala activation and decreased mPFC activation during stressful cues seems to be reversed when subjects with PTSD have high levels of dissociative symptoms. Hooper et al. have reported increased mPFC activation in the presence of PTSD with significant dissociative symptoms (e.g., [170]). Dynamic causal modeling has been used to interpret some level of causal and temporal relationship between functionally active brain networks. Nicholson et al. [171] found that PTSD without dissociation was characterized primarily by ‘bottom-up connectivity’ from the amygdala to the vmPFC, whereas PTSD with dissociation had predominant ‘top-down connectivity’ from the vmPFC to the amygdala. The authors suggest that this provides further data supporting a model of enhanced top-down, emotional over-regulation with significant dissociation, in contrast to decreased emotion regulation in the majority of PTSD cases.
Overall, these studies suggest that, in general, decreased vmPFC and dlPFC functional activation to stressful and trauma-related cues, often accompanied by increased dACC activation to such cues, are associated with greater PTSD symptoms and decreased responses to exposure-based therapies. Successful treatment appears to be accompanied by increased activation and functional connectivity of vmPFC/dlPFC regions with other cortical and subcortical areas. Notably, PTSD with significant dissociation seems to have differential connectivity patterns, as recently further demonstrated by Jovanovic et al. [172], and may benefit from distinct approaches to treatment and recovery.
PFC, threat extinction, behavioral inhibition
Work that has done the most to connect neuroimaging findings in PTSD with both healthy human neuroimaging work and preclinical work on threat acquisition and extinction includes neuroimaging studies specifically focused on extinction of threat cues in PTSD as well as inhibitory learning processes. Milad et al. initially demonstrated some of these effects in PTSD through optimization of a within-scanner threat conditioning and extinction protocol. They found that during extinction recall there was decreased activation of vmPFC and greater activation in dACC in PTSD relative to healthy trauma-exposed participants, which was associated with impaired physiological measures of extinction recall [165]. These data suggest that impairments in PTSD recovery may result, in part, from altered PFC regulation of threat extinction recall. Increased dACC activation during threat expression and extinction and decreased vmPFC activation with extinction recall were replicated in another study by this group examining context modulation in PTSD [166]. The dACC also appears to be associated with more PTSD symptoms at rest. Marin et al. [164] found that dACC resting activity positively correlated with PTSD symptom severity and predicted increased dACC activations during extinction recall.
In addition, Helpman et al. [173] examined threat conditioning and extinction in an fMRI task before and after a course of prolonged exposure treatment for PTSD. They found that PTSD patients had pre- to post-treatment reductions in rostral ACC (rACC) activation during extinction recall, and increases in functional coherence between rACC, vmPFC, and sgACC, suggesting these circuits are modified.
In another approach to this issue, Jovanovic et al. [174] found lower vmPFC activation during a simple stop signal (or “Go/No-go”) task related to behavioral inhibition in civilians with PTSD. Inhibition in this task correlated with a physiological measure of threat extinction in a fear-potentiated startle paradigm outside of the scanner. These data suggested that the same circuits involved in behavioral inhibition appear to be involved in fear inhibition processes during differential threat conditioning and extinction. A follow-up study with a larger sample size suggested that the effects of vmPFC/rACC on behavioral inhibition are moderated by childhood maltreatment effects in participants with PTSD [175].
Conclusions and future directions
Considerable progress has been made in understanding the role that the PFC plays in rodent threat learning and threat extinction, in healthy human studies of regulation of threat and fear emotion processing, and in the ways these areas may be dysregulated in threat-related disorders such as PTSD. Still, many questions remain. Much data supports a heuristic in which PL and IL play opposing roles in the rodent threat response, the former driving threat expression, the latter threat extinction. Similarly, in human studies, the dACC and Brodmann 32-related mPFC areas appear to support threat responses and are hyperactive in PTSD associated with hyperarousal and threat emotional dysregulation. Furthermore, human data support subgenual and Brodmann 25-related mPFC areas (i.e., the vmPFC) in regulating/suppressing threat responses, supporting extinction of threat behaviors and fear emotions, and in providing top-down emotional control over amygdala and other subcortical regions.
However, there are other data to suggest that, more generally, PL encodes for the learning of rules, while IL allows for rule reversal [18]. Thus, the precise role for the mPFC is not yet entirely clear. The lens through which the “fear and threat” neuroscientists see these areas needs to become more aligned with how the “appetitive and addiction” and “cognitive control” neuroscientists view them. This will help obtain a more comprehensive overall perspective on these regions, aid in understanding their normal function, and provide better treatment approaches for a large number of disorders associated with their dysfunction. The integration of elegant and powerful circuit tools, from intersectional optogenetic and chemogenetic circuit dissection to cellular and genetically driven in vivo calcium imaging combined with behavior, are providing remarkable cell- and circuit-level appreciation of basic behavioral functions in rodents. In humans, new tools such as fMRI-guided TMS, as one example, are beginning to allow relatively rapid translation of circuit function to targeted, precision-medicine approaches for individually guided care.
Furthermore, as we have learned with subcortical structures, different cell types residing within the same brain structure may have dramatically different, sometimes opposing, functions. The mPFC has immense cellular heterogeneity, and we may expect, functional complexity. Future cell-type specific studies that dissect the circuit- and molecular-adaptations to threat learning within the mPFC will reveal additional layers of complexity to this system, and, hopefully, new therapeutic targets for PTSD. Tools, such as single cell RNA sequencing, are now being used across species from rodent to post-mortem human brain, allowing for molecular dissections to complement the above functional circuit dissections. Their translation across species will support both a scientific understanding of conservation of molecules and circuits across evolution for specific survival-related behaviors, as well as provide much more powerful targets for pharmacological and biological intervention using animal model systems targeting known conserved molecules and cell types for human therapeutic development.
In summary, while much remains to be learned, it is an extraordinarily exciting time for the field in which a great deal of convergence and replication has resulted in a fairly robust understanding of threat processing and its regulation by the PFC. How these areas encode and express threat memories over time is rapidly being dissected and integrating these data into our understanding of pathology in disorders such as PTSD is occurring. Integrating translational research in the coming years promises exciting new discoveries and approaches which may both greatly expand our knowledge of how the brain encodes behavior and also drive development of novel and robust new treatment approaches.
References
Breslau N. The epidemiology of posttraumatic stress disorder: what is the extent of the problem? J Clin Psychiatry. 2001;62:16–22.
Bromet E, Sonnega A, Kessler RC. Risk factors for DSM-III-R posttraumatic stress disorder: findings from the National Comorbidity Survey. Am J Epidemiol. 1998;147:353–61.
McLaughlin KA, Koenen KC, Friedman MJ, Ruscio AM, Karam EG, Shahly V, et al. Subthreshold posttraumatic stress disorder in the world health organization world mental health surveys. Biol Psychiatry. 2015;77:375–84.
Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 2010;167:648–62.
Morgan Iii C, Grillon C, Southwick SM, Davis M, Charney DS. Fear-potentiated startle in posttraumatic stress disorder. Biol Psychiatry. 1995;38:378–85.
Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research—past, present, and future. Biol Psychiatry. 2006;60:376–82.
Yehuda R, LeDoux J. Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron. 2007;56:19–32.
Duncan LE, Ratanatharathorn A, Aiello AE, Almli LM, Amstadter AB, Ashley-Koch AE, et al. Largest GWAS of PTSD (N = 20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry. 2018;23:666–73.
Gelernter J, Sun N, Polimanti R, Pietrzak R, Levey DF, Bryois J, et al. Genome-wide association study of post-traumatic stress disorder reexperiencing symptoms in> 165,000 US veterans. Nat Neurosci. 2019;22:1394–401.
Nievergelt CM, Maihofer AX, Klengel T, Atkinson EG, Chen C-Y, Choi KW, et al. International meta-analysis of PTSD genome-wide association studies identifies sex-and ancestry-specific genetic risk loci. Nat Commun. 2019;10:1–16.
Davis M, Walker DL, Myers KM. Role of the amygdala in fear extinction measured with potentiated startle. Ann NY Acad Sci. 2003;985:218–32.
LeDoux J. Fear and the brain: where have we been, and where are we going? Biol Psychiatry. 1998;44:1229–38.
Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–87.
Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35:169–91.
Abdallah CG, Averill LA, Akiki TJ, Raza M, Averill CL, Gomaa H, et al. The neurobiology and pharmacotherapy of posttraumatic stress disorder. Annu Rev Pharm Toxicol. 2019;59:171–89.
Krystal JH, Davis LL, Neylan TC, Raskind MA, Schnurr PP, Stein MB, et al. It is time to address the crisis in the pharmacotherapy of posttraumatic stress disorder: a consensus statement of the PTSD psychopharmacology working group. Biol Psychiatry. 2017;82:e51–e59.
LeDoux JE, Pine DS. Using neuroscience to help understand fear and anxiety: a two-system framework. Am J Psychiatry. 2016;173:1083–93.
Gourley SL, Taylor JR. Going and stopping: Dichotomies in behavioral control by the prefrontal cortex. Nat Neurosci. 2016;19:656–64.
Woon EP, Sequeira MK, Barbee BR, Gourley SL. Involvement of the rodent prelimbic and medial orbitofrontal cortices in goal-directed action: a brief review. J Neurosci Res. 2020;98:1020–30.
Miller EK. The prefrontal cortex and cognitive control. Nat Rev Neurosci. 2000;1:59–65.
Giustino TF, Maren S. The role of the medial prefrontal cortex in the conditioning and extinction of fear. Front Behav Neurosci. 2015;9:298.
Ongür D, Price JL. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex. 2000;10:206–19.
Heilbronner SR, Rodriguez-Romaguera J, Quirk GJ, Groenewegen HJ, Haber SN. Circuit-based corticostriatal homologies between rat and primate. Biol Psychiatry. 2016;80:509–21.
Heidbreder CA, Groenewegen HJ. The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci Biobehav Rev. 2003;27:555–79.
Allsop SA, Wichmann R, Mills F, Burgos-Robles A, Chang CJ, Felix-Ortiz AC, et al. Corticoamygdala transfer of socially derived information gates observational learning. Cell. 2018;173:1329–42.e18.
Van De Werd HJ, Uylings HB. Comparison of (stereotactic) parcellations in mouse prefrontal cortex. Brain Struct Funct. 2014;219:433–59.
Van De Werd HJ, Rajkowska G, Evers P, Uylings HB. Cytoarchitectonic and chemoarchitectonic characterization of the prefrontal cortical areas in the mouse. Brain Struct Funct. 2010;214:339–53.
Strobel C, Marek R, Gooch HM, Sullivan RKP, Sah P. Prefrontal and auditory input to intercalated neurons of the amygdala. Cell Rep. 2015;10:1435–42.
Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synap. 2004;51:32–58.
Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–13.
Morgan MA, LeDoux JE. Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci. 1995;109:681–8.
Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–31.
Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–38.
Cho JH, Deisseroth K, Bolshakov VY. Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron. 2013;80:1491–507.
Garcia R, Chang CH, Maren S. Electrolytic lesions of the medial prefrontal cortex do not interfere with long-term memory of extinction of conditioned fear. Learn Mem. 2006;13:14–7.
Do-Monte FH, Manzano-Nieves G, Quinones-Laracuente K, Ramos-Medina L, Quirk GJ. Revisiting the role of infralimbic cortex in fear extinction with optogenetics. J Neurosci. 2015;35:3607–15.
Kheirbek MA, Drew LJ, Burghardt NS, Costantini DO, Tannenholz L, Ahmari SE, et al. Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron. 2013;77:955–68.
Eichenbaum H. Prefrontal-hippocampal interactions in episodic memory. Nat Rev Neurosci. 2017;18:547–58.
Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–85.
Corcoran KA, Maren S. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci. 2001;21:1720–6.
Hallock HL, Quillian HM, Maynard KR, Mai Y, Chen HY, et al. Molecularly defined hippocampal inputs regulate population dynamics in the prelimbic cortex to suppress context fear memory retrieval. Biol Psychiatry. 2020;88:554–65.
Kim WB, Cho JH. Synaptic targeting of double-projecting ventral CA1 hippocampal neurons to the medial prefrontal cortex and basal amygdala. J Neurosci. 2017;37:4868–82.
Orsini CA, Kim JH, Knapska E, Maren S. Hippocampal and prefrontal projections to the basal amygdala mediate contextual regulation of fear after extinction. J Neurosci. 2011;31:17269–77.
Lacagnina AF, Brockway ET, Crovetti CR, Shue F, McCarty MJ, Sattler KP, et al. Distinct hippocampal engrams control extinction and relapse of fear memory. Nat Neurosci. 2019;22:753–61.
Marek R, Jin J, Goode TD, Giustino TF, Wang Q, Acca GM, et al. Hippocampus-driven feed-forward inhibition of the prefrontal cortex mediates relapse of extinguished fear. Nat Neurosci. 2018;21:384–92.
Ramanathan KR, Jin J, Giustino TF, Payne MR, Maren S. Prefrontal projections to the thalamic nucleus reuniens mediate fear extinction. Nat Commun. 2018;9:4527.
Rajasethupathy P, Sankaran S, Marshel JH, Kim CK, Ferenczi E, Lee SY, et al. Projections from neocortex mediate top-down control of memory retrieval. Nature. 2015;526:653–9.
LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–29.
Falls WA, Davis M. Lesions of the central nucleus of the amygdala block conditioned excitation, but not conditioned inhibition of fear as measured with the fear-potentiated startle effect. Behav Neurosci. 1995;109:379–87.
Burgos-Robles A, Kimchi EY, Izadmehr EM, Porzenheim MJ, Ramos-Guasp WA, Nieh EH, et al. Amygdala inputs to prefrontal cortex guide behavior amid conflicting cues of reward and punishment. Nat Neurosci. 2017;20:824–35.
Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Gründemann J, et al. Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron. 2014;81:428–37.
Marcus DJ, Bedse G, Gaulden AD, Ryan JD, Kondev V, Winters ND, et al. Endocannabinoid signaling collapse mediates stress-induced amygdalo-cortical strengthening. Neuron. 2020;105:1062–76.e6.
Xu W, Südhof TC. A neural circuit for memory specificity and generalization. Science. 2013;339:1290–5.
Padilla-Coreano N, Do-Monte FH, Quirk GJ. A time-dependent role of midline thalamic nuclei in the retrieval of fear memory. Neuropharmacology. 2012;62:457–63.
Do-Monte FH, Quinones-Laracuente K, Quirk GJ. A temporal shift in the circuits mediating retrieval of fear memory. Nature. 2015;519:460–3.
Santini E, Ge H, Ren K, Pena de Ortiz S, Quirk GJ. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci. 2004;24:5704–10.
Chen MB, Jiang X, Quake SR, Südhof TC. Persistent transcriptional programmes are associated with remote memory. Nature. 2020;587:437–42.
Inaba H, Tsukagoshi A, Kida S. PARP-1 activity is required for the reconsolidation and extinction of contextual fear memory. Mol Brain. 2015;8:63.
Choi DC, Maguschak KA, Ye K, Jang SW, Myers KM, Ressler KJ. Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proc Natl Acad Sci USA. 2010;107:2675–80.
Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–90.
Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–76.
Wei W, Coelho CM, Li X, Marek R, Yan S, Anderson S, et al. p300/CBP-associated factor selectively regulates the extinction of conditioned fear. J Neurosci. 2012;32:11930–41.
Li X, Zhao Q, Wei W, Lin Q, Magnan C, Emami MR, et al. The DNA modification N6-methyl-2’-deoxyadenosine (m6dA) drives activity-induced gene expression and is required for fear extinction. Nat Neurosci. 2019;22:534–44.
Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–22.
Arnsten AF, Raskind MA, Taylor FB, Connor DF. The effects of stress exposure on prefrontal cortex: Translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. 2015;1:89–99.
Izquierdo A, Wellman CL, Holmes A. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci. 2006;26:5733–8.
Fitzgerald PJ, Giustino TF, Seemann JR, Maren S. Noradrenergic blockade stabilizes prefrontal activity and enables fear extinction under stress. Proc Natl Acad Sci USA. 2015;112:E3729–37.
Giustino TF, Ramanathan KR, Totty MS, Miles OW, Maren S. Locus coeruleus norepinephrine drives stress-induced increases in basolateral amygdala firing and impairs extinction learning. J Neurosci. 2020;40:907–16.
Jo YS, Namboodiri VMK, Stuber GD, Zweifel LS. Persistent activation of central amygdala CRF neurons helps drive the immediate fear extinction deficit. Nat Commun. 2020;11:422.
Wilber AA, Walker AG, Southwood CJ, Farrell MR, Lin GL, Rebec GV, et al. Chronic stress alters neural activity in medial prefrontal cortex during retrieval of extinction. Neuroscience. 2011;174:115–31.
Shansky RM, Hamo C, Hof PR, McEwen BS, Morrison JH. Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb Cortex. 2009;19:2479–84.
Bechara A, Tranel D, Damasio H, Adolphs R, Rockland C, Damasio AR. Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science. 1995;269:1115–8.
LaBar KS, LeDoux JE, Spencer DD, Phelps EA. Impaired fear conditioning following unilateral temporal lobectomy in humans. J Neurosci. 1995;15:6846–55.
LaBar KS, Phelps EA. Reinstatement of conditioned fear in humans is context dependent and impaired in amnesia. Behav Neurosci. 2005;119:677.
Büchel C, Morris J, Dolan RJ, Friston KJ. Brain systems mediating aversive conditioning: an event-related fMRI study. Neuron. 1998;20:947–57.
LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–45.
Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE. Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci. 2001;4:724–31.
Fullana M, Harrison B, Soriano-Mas C, Vervliet B, Cardoner N, Àvila-Parcet A, et al. Neural signatures of human fear conditioning: an updated and extended meta-analysis of fMRI studies. Mol Psychiatry. 2016;21:500–08.
MacNamara A, Rabinak CA, Fitzgerald DA, Zhou XJ, Shankman SA, Milad MR, et al. Neural correlates of individual differences in fear learning. Behav Brain Res. 2015;287:34–41.
Milad MR, Quirk GJ, Pitman RK, Orr SP, Fischl B, Rauch SL. A role for the human dorsal anterior cingulate cortex in fear expression. Biol Psychiatry. 2007;62:1191–4.
Roberts AC, Clarke HF. Why we need nonhuman primates to study the role of ventromedial prefrontal cortex in the regulation of threat-and reward-elicited responses. Proc Natl Acad Sci USA. 2019;116:26297–304.
Olsson A, Phelps EA. Social learning of fear. Nat Neurosci. 2007;10:1095–102.
Phelps EA, O’Connor KJ, Gatenby JC, Gore JC, Grillon C, Davis M. Activation of the left amygdala to a cognitive representation of fear. Nat Neurosci. 2001;4:437–41.
Funayama ES, Grillon C, Davis M, Phelps EA. A double dissociation in the affective modulation of startle in humans: effects of unilateral temporal lobectomy. J Cogn Neurosci. 2001;13:721–9.
Olsson A, Nearing KI, Phelps EA. Learning fears by observing others: the neural systems of social fear transmission. Soc Cogn Affect Neurosci. 2007;2:3–11.
Olsson A, McMahon K, Papenberg G, Zaki J, Bolger N, Ochsner KN. Vicarious fear learning depends on empathic appraisals and trait empathy. Psychol Sci. 2016;27:25–33.
Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–51.
Gottfried JA, Dolan RJ. Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci. 2004;7:1144–52.
Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007;62:446–54.
Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905.
Kalisch R, Korenfeld E, Stephan KE, Weiskopf N, Seymour B, Dolan RJ. Context-dependent human extinction memory is mediated by a ventromedial prefrontal and hippocampal network. J Neurosci. 2006;26:9503–11.
Lonsdorf TB, Haaker J, Kalisch R. Long-term expression of human contextual fear and extinction memories involves amygdala, hippocampus and ventromedial prefrontal cortex: a reinstatement study in two independent samples. Soc Cogn Affect Neurosci. 2014;9:1973–83.
Diekhof EK, Geier K, Falkai P, Gruber O. Fear is only as deep as the mind allows: a coordinate-based meta-analysis of neuroimaging studies on the regulation of negative affect. Neuroimage. 2011;58:275–85.
Menz MM, Rihm JS, Büchel C. REM sleep is causal to successful consolidation of dangerous and safety stimuli and reduces return of fear after extinction. J Neurosci. 2016;36:2148–60.
Fullana MA, Albajes-Eizagirre A, Soriano-Mas C, Vervliet B, Cardoner N, Benet O, et al. Fear extinction in the human brain: a meta-analysis of fMRI studies in healthy participants. Neurosci Biobehav Rev. 2018;88:16–25.
Milad MR, Quinn BT, Pitman RK, Orr SP, Fischl B, Rauch SL. Thickness of ventromedial prefrontal cortex in humans is correlated with extinction memory. Proc Natl Acad Sci USA. 2005;102:10706–11.
Hartley CA, Fischl B, Phelps EA. Brain structure correlates of individual differences in the acquisition and inhibition of conditioned fear. Cereb Cortex. 2011;21:1954–62.
Winkelmann T, Grimm O, Pohlack ST, Nees F, Cacciaglia R, Dinu-Biringer R, et al. Brain morphology correlates of interindividual differences in conditioned fear acquisition and extinction learning. Brain Struct Funct. 2016;221:1927–37.
Abend R, Jalon I, Gurevitch G, Sar-El R, Shechner T, Pine D, et al. Modulation of fear extinction processes using transcranial electrical stimulation. Transl Psychiatry. 2016;6:e913–e13.
Dittert N, Hüttner S, Polak T, Herrmann MJ. Augmentation of fear extinction by transcranial direct current stimulation (tDCS). Front Behav Neurosci. 2018;12:76.
Ganho-Ávila A, Gonçalves ÓF, Guiomar R, Boggio PS, Asthana MK, Krypotos A-M, et al. The effect of cathodal tDCS on fear extinction: a cross-measures study. PLoS One. 2019;14:e0221282.
Raij T, Nummenmaa A, Marin M-F, Porter D, Furtak S, Setsompop K, et al. Prefrontal cortex stimulation enhances fear extinction memory in humans. Biol Psychiatry. 2018;84:129–37.
van’t Wout M, Mariano TY, Garnaat SL, Reddy MK, Rasmussen SA, Greenberg BD. Can transcranial direct current stimulation augment extinction of conditioned fear? Brain Stimul. 2016;9:529–36.
Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63:1118–26.
Knight DC, Smith CN, Cheng DT, Stein EA, Helmstetter FJ. Amygdala and hippocampal activity during acquisition and extinction of human fear conditioning. Cogn Affect Behav Neurosci. 2004;4:317–25.
Morriss J, Hoare S, van Reekum CM. It’s time: a commentary on fear extinction in the human brain using fMRI. Neurosci Biobehav Rev. 2018;94:321–2.
Lang S, Kroll A, Lipinski SJ, Wessa M, Ridder S, Christmann C, et al. Context conditioning and extinction in humans: differential contribution of the hippocampus, amygdala and prefrontal cortex. Eur J Neurosci. 2009;29:823–32.
Ganella DE, Barendse ME, Kim JH, Whittle S. Prefrontal-amygdala connectivity and state anxiety during fear extinction recall in adolescents. Front Hum Neurosci. 2017;11:587.
Pace-Schott EF, Germain A, Milad MR. Effects of sleep on memory for conditioned fear and fear extinction. Psychol Bull. 2015;141:835–57.
Pace-Schott EF, Germain A, Milad MR. Sleep and REM sleep disturbance in the pathophysiology of PTSD: the role of extinction memory. Biol Mood Anxiety Disord. 2015;5:3.
LeDoux JE, Moscarello J, Sears R, Campese V. The birth, death and resurrection of avoidance: a reconceptualization of a troubled paradigm. Mol Psychiatry. 2017;22:24–36.
Moscarello JM, LeDoux JE. Active avoidance learning requires prefrontal suppression of amygdala-mediated defensive reactions. J Neurosci. 2013;33:3815–23.
Baratta MV, Lucero TR, Amat J, Watkins LR, Maier SF. Role of the ventral medial prefrontal cortex in mediating behavioral control-induced reduction of later conditioned fear. Learn Mem. 2008;15:84–7.
Boeke EA, Moscarello JM, LeDoux JE, Phelps EA, Hartley CA. Active avoidance: neural mechanisms and attenuation of Pavlovian conditioned responding. J Neurosci. 2017;37:4808–18.
Hartley CA, Gorun A, Reddan MC, Ramirez F, Phelps EA. Stressor controllability modulates fear extinction in humans. Neurobiol Learn Mem. 2014;113:149–56.
Lovibond PF, Davis NR, O’Flaherty AS. Protection from extinction in human fear conditioning. Behav Res Ther. 2000;38:967–83.
Gross JJ. Emotion regulation: current status and future prospects. Psychol Inq. 2015;26:1–26.
Morawetz C, Bode S, Derntl B, Heekeren HR. The effect of strategies, goals and stimulus material on the neural mechanisms of emotion regulation: a meta-analysis of fMRI studies. Neurosci Biobehav Rev. 2017;72:111–28.
Frank D, Dewitt M, Hudgens-Haney M, Schaeffer D, Ball B, Schwarz N, et al. Emotion regulation: quantitative meta-analysis of functional activation and deactivation. Neurosci Biobehav Rev. 2014;45:202–11.
Dörfel D, Lamke J-P, Hummel F, Wagner U, Erk S, Walter H. Common and differential neural networks of emotion regulation by detachment, reinterpretation, distraction, and expressive suppression: a comparative fMRI investigation. Neuroimage 2014;101:298–309.
Buhle JT, Silvers JA, Wager TD, Lopez R, Onyemekwu C, Kober H, et al. Cognitive reappraisal of emotion: a meta-analysis of human neuroimaging studies. Cereb Cortex. 2014;24:2981–90.
Kohn N, Eickhoff SB, Scheller M, Laird AR, Fox PT, Habel U. Neural network of cognitive emotion regulation—an ALE meta-analysis and MACM analysis. Neuroimage. 2014;87:345–55.
Wager TD, Smith EE. Neuroimaging studies of working memory. Cogn Affect Behav Neurosci. 2003;3:255–74.
Robbins TW. Shifting and stopping: fronto-striatal substrates, neurochemical modulation and clinical implications. Philos Trans R Soc Lond Biol Sci. 2007;362:917–32.
Simmonds DJ, Pekar JJ, Mostofsky SH. Meta-analysis of Go/No-go tasks demonstrating that fMRI activation associated with response inhibition is task-dependent. Neuropsychologia. 2008;46:224–32.
Amodio DM, Frith CD. Meeting of minds: the medial frontal cortex and social cognition. Nat Rev Neurosci. 2006;7:268–77.
Binder JR, Desai RH, Graves WW, Conant LL. Where is the semantic system? A critical review and meta-analysis of 120 functional neuroimaging studies. Cereb Cortex. 2009;19:2767–96.
Cato MA, Crosson B, Gökçay D, Soltysik D, Wierenga C, Gopinath K, et al. Processing words with emotional connotation: an FMRI study of time course and laterality in rostral frontal and retrosplenial cortices. J Cogn Neurosci. 2004;16:167–77.
Crosson B, Cato MA, Sadek JR, Gökçay D, Bauer RM, Fischler IS, et al. Semantic monitoring of words with emotional connotation during fMRI: contribution of anterior left frontal cortex. J Int Neuropsychol Soc. 2002;8:607–22.
Olsson A, Ochsner KN. The role of social cognition in emotion. Trends Cogn Sci. 2008;12:65–71.
Delgado MR, Nearing KI, LeDoux JE, Phelps EA. Neural circuitry underlying the regulation of conditioned fear and its relation to extinction. Neuron. 2008;59:829–38.
Schiller D, Delgado MR. Overlapping neural systems mediating extinction, reversal and regulation of fear. Trends Cogn Sci. 2010;14:268–76.
Ghashghaei HT, Barbas H. Pathways for emotion: interactions of prefrontal and anterior temporal pathways in the amygdala of the rhesus monkey. Neuroscience. 2002;115:1261–79.
Ghashghaei H, Hilgetag CC, Barbas H. Sequence of information processing for emotions based on the anatomic dialogue between prefrontal cortex and amygdala. Neuroimage. 2007;34:905–23.
Steward T, Davey CG, Jamieson AJ, Stephanou K, Soriano-Mas C, Felmingham KL, et al. Dynamic neural interactions supporting the cognitive reappraisal of emotion. Cereb Cortex. 2021;31:961–73.
Berboth S, Morawetz C. Amygdala-prefrontal connectivity during emotion regulation: a meta-analysis of psychophysiological interactions. Neuropsychologia 2021;153:107767.
Morawetz C, Riedel MC, Salo T, Berboth S, Eickhoff S, Laird AR, et al. Multiple large-scale neural networks underlying emotion regulation. Neurosci Biobehav Rev. 2020;116:382–95.
Erk S, Mikschl A, Stier S, Ciaramidaro A, Gapp V, Weber B, et al. Acute and sustained effects of cognitive emotion regulation in major depression. J Neurosci. 2010;30:15726–34.
Johnstone T, Van Reekum CM, Urry HL, Kalin NH, Davidson RJ. Failure to regulate: counterproductive recruitment of top-down prefrontal-subcortical circuitry in major depression. J Neurosci. 2007;27:8877–84.
Kroes MC, Dunsmoor JE, Hakimi M, Oosterwaal S, collaboration NP, Meager MR, et al. Patients with dorsolateral prefrontal cortex lesions are capable of discriminatory threat learning but appear impaired in cognitive regulation of subjective fear. Soc Cogn Affect Neurosci. 2019;14:601–12.
Feeser M, Prehn K, Kazzer P, Mungee A, Bajbouj M. Transcranial direct current stimulation enhances cognitive control during emotion regulation. Brain Stimul. 2014;7:105–12.
He Z, Lin Y, Xia L, Liu Z, Zhang D, Elliott R. Critical role of the right VLPFC in emotional regulation of social exclusion: a tDCS study. Soc Cogn Affect Neurosci. 2018;13:357–66.
Marques LM, Morello LY, Boggio PS. Ventrolateral but not dorsolateral prefrontal cortex tDCS effectively impact emotion reappraisal–effects on emotional experience and interbeat interval. Sci Rep. 2018;8:1–12.
Vieira L, Marques D, Melo L, Marques RC, Monte-Silva K, Cantilino A. Transcranial direct current stimulation effects on cognitive reappraisal: an unexpected result? Brain Stimul. 2020;13:650–2.
Gold AL, Sheridan MA, Peverill M, Busso DS, Lambert HK, Alves S, et al. Childhood abuse and reduced cortical thickness in brain regions involved in emotional processing. J Child Psychol Psychiatry. 2016;57:1154–64.
Raio CM, Orederu TA, Palazzolo L, Shurick AA, Phelps EA. Cognitive emotion regulation fails the stress test. Proc Natl Acad Sci USA. 2013;110:15139–44.
Raio CM, Brignoni-Perez E, Goldman R, Phelps EA. Acute stress impairs the retrieval of extinction memory in humans. Neurobiol Learn Mem. 2014;112:212–21.
Admon R, Leykin D, Lubin G, Engert V, Andrews J, Pruessner J, et al. Stress‐induced reduction in hippocampal volume and connectivity with the ventromedial prefrontal cortex are related to maladaptive responses to stressful military service. Hum Brain Mapp. 2013;34:2808–16.
Rocha-Rego V, Pereira MG, Oliveira L, Mendlowicz MV, Fiszman A, Marques-Portella C, et al. Decreased premotor cortex volume in victims of urban violence with posttraumatic stress disorder. PLoS One. 2012;7:e42560.
Clausen AN, Clarke E, Phillips RD, Haswell C, Morey RA. Combat exposure, posttraumatic stress disorder, and head injuries differentially relate to alterations in cortical thickness in military veterans. Neuropsychopharmacology. 2020;45:491–98.
Keding TJ, Herringa RJ. Abnormal structure of fear circuitry in pediatric post-traumatic stress disorder. Neuropsychopharmacology. 2015;40:537–45.
Morey RA, Haswell CC, Hooper SR, De Bellis MD. Amygdala, hippocampus, and ventral medial prefrontal cortex volumes differ in maltreated youth with and without chronic posttraumatic stress disorder. Neuropsychopharmacology. 2016;41:791–801.
Mueller SG, Ng P, Neylan T, Mackin S, Wolkowitz O, Mellon S, et al. Evidence for disrupted gray matter structural connectivity in posttraumatic stress disorder. Psychiatry Res Neuroimaging. 2015;234:194–201.
Herringa R, Phillips M, Almeida J, Insana S, Germain A. Post-traumatic stress symptoms correlate with smaller subgenual cingulate, caudate, and insula volumes in unmedicated combat veterans. Psychiatry Res: Neuroimaging. 2012;203:139–45.
Koch SB, Van Zuiden M, Nawijn L, Frijling JL, Veltman DJ, Olff M. Decreased uncinate fasciculus tract integrity in male and female patients with PTSD: a diffusion tensor imaging study. J Psychiatry Neurosci. 2017;42:331.
Fani N, King TZ, Shin J, Srivastava A, Brewster RC, Jovanovic T, et al. Structural and functional connectivity in posttraumatic stress disorder: associations with FKBP5. Depress Anxiety. 2016;33:300–07.
Harnett NG, Stevens JS, van Rooij SJ, Ely TD, Michopoulos V, Hudak L, et al. Multimodal structural neuroimaging markers of risk and recovery from posttrauma anhedonia: a prospective investigation. Depress Anxiety. 2021;38:79–88.
Stein MB, Yuh E, Jain S, Okonkwo DO, Mac Donald CL, Levin H, et al. Smaller regional brain volumes predict posttraumatic stress disorder at 3 months after mild traumatic brain injury. Biol Psychiatry. 2021;6:352–9.
Bromis K, Calem M, Reinders AA, Williams SC, Kempton MJ. Meta-analysis of 89 structural MRI studies in posttraumatic stress disorder and comparison with major depressive disorder. Am J Psychiatry. 2018;175:989–98.
Zimmermann KS, Li C-C, Rainnie DG, Ressler, et al. Memory retention involves the ventrolateral orbitofrontal cortex: comparison with the basolateral amygdala. Neuropsychopharmacology. 2018;43:373–83.
Dahlgren MK, Laifer LM, VanElzakker MB, Offringa R, Hughes KC, Staples-Bradley LK, et al. Diminished medial prefrontal cortex activation during the recollection of stressful events is an acquired characteristic of PTSD. Psychol Med. 2018;48:1128.
Killgore WD, Britton JC, Schwab ZJ, Price LM, Weiner MR, Gold AL, et al. Cortico‐limbic responses to masked affective faces across ptsd, panic disorder, and specific phobia. Depress Anxiety. 2014;31:150–9.
Stevens JS, Kim YJ, Galatzer-Levy IR, Reddy R, Ely TD, Nemeroff CB, et al. Amygdala reactivity and anterior cingulate habituation predict posttraumatic stress disorder symptom maintenance after acute civilian trauma. Biol Psychiatry. 2017;81:1023–9.
Marin M-F, Song H, VanElzakker MB, Staples-Bradley LK, Linnman C, Pace-Schott EF, et al. Association of resting metabolism in the fear neural network with extinction recall activations and clinical measures in trauma-exposed individuals. Am J Psychiatry. 2016;173:930–8.
Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66:1075–82.
Rougemont‐Bücking A, Linnman C, Zeffiro TA, Zeidan MA, Lebron‐Milad K, Rodriguez‐Romaguera J, et al. Altered processing of contextual information during fear extinction in PTSD: an fMRI study. CNS Neurosci Ther. 2011;17:227–36.
Fonzo GA, Goodkind MS, Oathes DJ, Zaiko YV, Harvey M, Peng KK, et al. PTSD psychotherapy outcome predicted by brain activation during emotional reactivity and regulation. Am J Psychiatry. 2017;174:1163–74.
Fonzo GA, Goodkind MS, Oathes DJ, Zaiko YV, Harvey M, Peng KK, et al. Selective effects of psychotherapy on frontopolar cortical function in PTSD. Am J Psychiatry. 2017;174:1175–84.
King AP, Block SR, Sripada RK, Rauch S, Giardino N, Favorite T, et al. Altered default mode network (DMN) resting state functional connectivity following a mindfulness‐based exposure therapy for posttraumatic stress disorder (PTSD) in combat veterans of Afghanistan and Iraq. Depress Anxiety. 2016;33:289–99.
Hopper JW, Frewen PA, van der Kolk BA, Lanius RA. Neural correlates of reexperiencing, avoidance, and dissociation in PTSD: Symptom dimensions and emotion dysregulation in responses to script‐driven trauma imagery. J Trauma Stress. 2007;20:713–25.
Nicholson AA, Friston KJ, Zeidman P, Harricharan S, McKinnon MC, Densmore M, et al. Dynamic causal modeling in PTSD and its dissociative subtype: Bottom–up versus top–down processing within fear and emotion regulation circuitry. Hum Brain Mapp. 2017;38:5551–61.
Lebois LA, Li M, Baker JT, Wolff JD, Wang D, Lambros AM, et al. Large-scale functional brain network architecture changes associated with trauma-related dissociation. Am J Psychiatry. 2021;178:165–73.
Helpman L, Marin M-F, Papini S, Zhu X, Sullivan GM, Schneier F, et al. Neural changes in extinction recall following prolonged exposure treatment for PTSD: a longitudinal fMRI study. Neuroimage Clin. 2016;12:715–23.
Jovanovic T, Ely T, Fani N, Glover EM, Gutman D, Tone EB, et al. Reduced neural activation during an inhibition task is associated with impaired fear inhibition in a traumatized civilian sample. Cortex. 2013;49:1884–91.
Stevens JS, Ely TD, Sawamura T, Guzman D, Bradley B, Ressler KJ, et al. Childhood maltreatment predicts reduced inhibition‐related activity in the rostral anterior cingulate in PTSD, but not trauma‐exposed controls. Depress Anxiety. 2016;33:614–22.
Funding
KJR has received consulting income from Alkermes and Takeda, research support from NIH, Genomind, and Brainsway, and he is on scientific advisory boards for Janssen and Verily, all of which are unrelated to the present work. This work was supported by NIH (KJR: P50-MH115874, R01-MH108665; EAP: RO1- DA042855), the Frazier Institute at McLean Hospital (KJR), and the James S. McDonnell Foundation (EAP). EAP, MAK, RJF, and ESL have nothing to disclose.
Author information
Authors and Affiliations
Contributions
EAP, RJF, MAK, ESL and KJR conceptualized, wrote, and edited the manuscript. ESL made the figures.
Corresponding authors
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alexandra Kredlow, M., Fenster, R.J., Laurent, E.S. et al. Prefrontal cortex, amygdala, and threat processing: implications for PTSD. Neuropsychopharmacol. 47, 247–259 (2022). https://doi.org/10.1038/s41386-021-01155-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01155-7
This article is cited by
-
Convergent direct and indirect cortical streams shape avoidance decisions in mice via the midline thalamus
Nature Communications (2024)
-
Transcriptional patterns of amygdala functional connectivity in first-episode, drug-naïve major depressive disorder
Translational Psychiatry (2024)
-
Basolateral amygdala neuropeptide Y system modulates binge ethanol consumption
Neuropsychopharmacology (2024)
-
Therapeutic mechanisms of psychedelics and entactogens
Neuropsychopharmacology (2024)
-
Distributed neural representations of conditioned threat in the human brain
Nature Communications (2024)
