
Abstract
Kraepelin, in his early descriptions of schizophrenia (SZ), characterized the illness as having “an orchestra without a conductor.” Kraepelin further speculated that this “conductor” was situated in the frontal lobes. Findings from multiple studies over the following decades have clearly implicated pathology of the dorsolateral prefrontal cortex (DLPFC) as playing a central role in the pathophysiology of SZ, particularly with regard to key cognitive features such as deficits in working memory and cognitive control. Following an overview of the cognitive mechanisms associated with DLPFC function and how they are altered in SZ, we review evidence from an array of neuroscientific approaches addressing how these cognitive impairments may reflect the underlying pathophysiology of the illness. Specifically, we present evidence suggesting that alterations of the DLPFC in SZ are evident across a range of spatial and temporal resolutions: from its cellular and molecular architecture, to its gross structural and functional integrity, and from millisecond to longer timescales. We then present an integrative model based upon how microscale changes in neuronal signaling in the DLPFC can influence synchronized patterns of neural activity to produce macrocircuit-level alterations in DLPFC activation that ultimately influence cognition and behavior. We conclude with a discussion of initial efforts aimed at targeting DLPFC function in SZ, the clinical implications of those efforts, and potential avenues for future development.
Similar content being viewed by others


Introduction
Since the earliest descriptions of the disorder, cognitive deficits have been widely recognized as a core aspect of schizophrenia (SZ) [1]. These cognitive impairments, present prior to the onset of psychosis and persisting through the disease course, have been shown to be a major source of disability in those affected by SZ [2, 3]. Furthermore, impaired cognition in SZ remains largely treatment refractory [3] despite a concerted effort over the past 30 years to find methods for ameliorating this aspect of the illness. As such, understanding the pathophysiology of impaired cognition in SZ and developing effective treatment for cognitive symptoms are major unmet challenges for contemporary biomedical research, particularly when considering the tremendous personal cost to affected individuals and their families and the substantial economic cost ($155 billion in the US alone in 2013) to society [4].
The purpose of this review is to summarize the evidence that the dorsolateral prefrontal cortex (DLPFC) plays a major role in the pathophysiology of higher order cognitive processing in SZ via abnormalities in its cellular architecture and associated circuitry. We first present evidence from clinical cognitive neuroscience that the DLPFC is functionally abnormal in SZ. We then describe how these aberrations may broadly affect cognition, behavioral disorganization symptoms, and everyday functioning in the illness. Next, we review findings suggesting that these functional and behavioral disturbances arise from cellular level alterations in the DLPFC. Finally, we discuss future directions for deepening our understanding of the role of the DLPFC in the disease process of SZ and for developing novel therapeutics to improve cognitive function in people with SZ.
Impairments in cognitive control and working memory: evidence for a prototypical cognitive disturbance in schizophrenia
As conceptualized by Alan Baddeley in the 1970’s, working memory (WM) is the ability to maintain and manipulate information for brief periods of time in order to guide thought or behavior in the absence of ongoing sensory input [5, 6]. Thus, WM is an essential component of the cognitive control processes that guide goal-oriented behaviors based on context. WM includes multiple subprocesses, including the maintenance of relevant information in the absence of sensory stimuli and the suppression of distractor stimuli not required for the task at hand.
WM is perhaps the most extensively studied cognitive domain in SZ. Results from the first systematic investigation of WM in SZ were reported in 1992 by Park and Holtzman [7], who found lower accuracy in SZ relative to psychiatrically unaffected individuals on an oculomotor spatial delayed-response task. Subsequently, deficits in WM have been frequently observed in SZ (e.g., [8, 9], reviewed by Aleman et al. [10]). Indeed, a meta-analysis of 187 studies found that individuals with SZ show robust deficits across a wide range of WM tasks [11], with large effect sizes (mean Cohen’s d = ~1 for each task). Meta-analytic evidence in first episode SZ individuals also suggests that WM deficits are present in the early stages of illness [12]. Interestingly, these deficits likely have a neurodevelopmental component (as they are present prior to illness onset [13]) as well as a genetic component (since they are found in milder form in unaffected siblings [14,15,16,17,18]). Some studies also suggest that larger WM deficits are associated with worse clinical [19, 20] and functional outcomes [2, 21].
A more recent cognitive construct closely related to WM is cognitive control (CC) [22], the ability to use contextual information to guide behavior in the face of conflicting but often habitual (prepotent) responding. CC processes lie at the heart of the “central executive processes” used to support WM task performance, and since CC requires active maintenance of contextual information, WM and CC are strongly related constructs [23]. CC can be measured using a variety of tasks, including the Stroop [24] and AX-Continuous Performance Task (AX-CPT) [25]. Mirroring WM findings, individuals with SZ show robust deficits in CC across all phases of the illness [25,26,27,28,29,30,31,32,33,34,35,36]. Like WM, impaired CC in SZ may also have a developmental and genetic origin as these deficits are present prior to onset of psychosis [37] and have been observed (at a smaller magnitude) in unaffected siblings [38, 39]. Preliminary evidence also suggests that functional impairments in CC may predict lack of clinical improvement after one year of standardized treatment in early psychosis [40, 41].
The role of the dorsolateral prefrontal cortex in working memory and cognitive control
The WM network involves a highly interconnected set of nodes located in multiple cortical and subcortical regions [42]. Among these nodes, the DLPFC is a critical region of convergence that both integrates information processing across nodes in the network and provides top-down control of that processing. The DLPFC is a large frontal brain area comprising lateral portions of Brodmann’s areas 9 and 46. It is evolutionarily relatively recent, being particularly enlarged in non-human primates and humans [43], and therefore may enable the enhanced complex planning and reasoning abilities characteristic of these species. The highly interconnected nature of the DLPFC, with regions such as the parietal, anterior cingulate and sensorimotor cortices and subcortical nuclei [44], is thought to permit integration of information with “on-line” representations to provide flexible internally driven (as opposed to stimulus-driven) control of behavior [examples in 45, 46].
The importance of the DLPFC in WM was first demonstrated in non-human primate studies in which lesions or reversible cooling of the DLPFC impaired WM performance in a delay-dependent manner [47]. In subsequent single-unit recording studies in monkeys, some DLPFC neurons that responded to specific memoranda were active during the delay period of a WM task, appearing to keep the information online [47,48,49]. Particular groups of neurons with delay period activity are therefore thought to encode particular features of the stimulus to be remembered [49] and specific information about the rules of the task at hand [50]. Human neuroimaging work in the early 1990s using positron emission tomography (PET) also found DLPFC recruitment during WM tasks [51], confirming the area as a primary WM hub. Notably, activation of the superficial layers of the DLPFC appears to be particularly important during the maintenance, or “delay” phase of WM, whereas encoding and retrieval processes are associated with activity in the inferior parietal lobule and deeper layers of the DLPFC [52,53,54]. Concordantly, neuronal activity in the DLPFC has been experimentally demonstrated to be predictive of behavioral performance on WM tasks in non-human primates [55], supporting its causal role in the maintenance of WM information. In regard to the WM-related process of CC, based on electrophysiological work in non-human primates [56] and subsequent imaging studies in humans (reviewed by Miller and Cohen [22]), the DLPFC is thought to support this ability by providing a substrate for online maintenance of rules or goals in order to guide responses, including biasing response away from prepotent tendencies [22].
The DLPFC also plays a critical role in resisting interference from distractors [57]. Distractors are defined as extraneous stimuli that occur during the delay period and compete for the neural processing resources required to maintain the task-relevant stimuli in WM. The ability to filter out these distracting stimuli is critical for WM performance and variations in WM capacity in humans reflect the ability to suppress activation of neural activity related to irrelevant stimuli [58]. The central role of the DLPFC in filtering out distracting stimuli during WM tasks is supported by the following lines of evidence. First, WM performance in monkeys with frontal lobe ablations is further impaired when distractor stimuli are presented [59], suggesting that the absence of the DLPFC is associated with an increased susceptibility to the interfering effects of distractors [60]. Second, relative to neurons in the posterior parietal [61, 62], lateral intraparietal [63, 64] and temporal cortices [65], DLPFC neurons are less likely to be activated by distractor stimuli [57]. Third, silencing of DLPFC pyramidal neurons neighboring those activated by a WM memoranda is associated with successful suppression of distractor stimuli [66]. Finally, increases in distractibility are noted with transient inactivation of the DLPFC, but not the parietal cortex, in monkeys [67]. Together, these data support the notion that relative to other cortical regions, neuronal activity in the DLPFC is more resilient to distractor stimuli during a WM task.
Evidence for dorsolateral prefrontal cortical dysfunction during working memory and cognitive control in schizophrenia
Given the extensive behavioral evidence of WM/CC deficits in SZ and the reliance of these constructs on proper DLPFC functioning, it is expected that alterations at the macro- and microscopic levels of resolution in DLPFC circuitry are responsible for these WM/CC deficits. Indeed, in his early descriptions of SZ, the pioneering psychiatrist Kraepelin postulated that “the mind in dementia praecox is like an orchestra without a conductor” [68]. Kraepelin also suggested that “reasoning” deficits may be linked to pathology of the frontal lobes — laying the initial groundwork eventually (nearly a century later) for a line of research linking deficits in executive function to prefrontal cortical pathology in the disorder.
In alignment with this view, a large body of in vivo structural neuroimaging studies of the DLPFC have consistently observed gross macroscopic abnormalities of this region in SZ. These studies, which have been reviewed elsewhere [69,70,71], suggest that the disorder is characterized by smaller DLPFC gray matter volume, thinner gray matter (for which antipsychotic treatment may be a contributing factor [72, 73]), and lower fractional anisotropy of white matter tracts that connect the DLPFC to other brain regions (e.g., the cingulum bundle) [74]. Evidence further indicates that similar pathology is present (albeit less consistently and to a lesser degree) in people at high risk for psychosis, suggesting these differences are not solely the consequence of antipsychotic treatment and may be influenced by genetic risk factors (reviewed by Andreou and Borgwardt [75]).
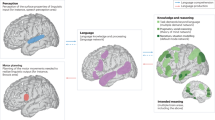
Functional neuroimaging studies also suggest that Kraepelin’s initial supposition of prefrontal pathology was correct (reviewed by Lesh et al. [35, 76]). This supposition was first supported by early studies by Weinberger et al. [77] and Perlstein et al. [78] demonstrating that individuals with SZ show lower cerebral blood flow in the frontal lobes during the Wisconsin Card Sort Task and N-Back WM Task and continuing through more recent studies using the AX-CPT and other tasks [22, 30,31,32,33,34, 79,80,81,82]. Effect sizes may be moderately high for lower DLPFC activation during CC in SZ; one recent study from our group found effect sizes of Cohen’s d = −0.49 at baseline and −0.58 at 12-month follow-up for bilateral DLPFC activation deficits during CC in SZ vs. unaffected comparison individuals [33]. Also mirroring the behavioral findings, deficits in DLPFC activation during WM and CC in SZ are present at the onset of the illness, and even prior to psychosis onset in clinical high-risk states [30, 34, 35, 37, 76, 83] (Fig. 1A).

A DLPFC activation associated with cognitive control during the AX Continuous Performance Task. Note the lower levels of activation in individuals with SZ. Data taken from a previously published sample [33]. B Frequency-time plots of scalp EEG recordings of prefrontal oscillatory activity during the delay period of the Preparing to Overcome Prepotency Task. Warmer colors indicate higher oscillatory power and cooler colors indicate lower power. Note that power in the gamma band (30–80 Hz) is substantially greater in the unaffected comparator group relative to the SZ group. Adapted from [90]. C Schematic representation of neural network synchrony (coordinated intracellular membrane recordings from pyramidal neurons) at gamma frequency in psychiatrically unaffected individuals and the hypothesized alteration of this synchrony in SZ. Adapted from [285]. D Diagram of local neural circuit in cortical layer 3. In individuals without psychiatric illness, it is posited that inhibition from PV basket cells (PVBCs) synchronizes the recurrent excitatory activity of groups of pyramidal neurons at gamma frequency, but lower levels of both excitation (indexed by smaller somal volumes, fewer dendritic spines, and shorter dendritic arbors, as depicted here) and inhibition (indexed by lower levels of GABA markers within PVBCs and by weaker shading here) could contribute to impaired synchrony in schizophrenia, as depicted in panel (C) (right).
It should be noted that, although task fMRI studies show overall reduced activation in SZ [84], some individual studies have reported increased activity in the illness (e.g., [85,86,87,88]). These studies have been interpreted as indicative of an “inverted U” function in the DLPFC, in which increased activity may be seen at low levels of WM demand but decreased activity at higher levels. It is also possible that methodological differences underlie some of these contrasting findings, including small sample sizes and variation in analysis protocols (e.g., matched performance with low performing psychiatrically unaffected participants [86]). For example, a 1999 study that reported increased activation during WM in SZ used a very small sample size (n = 10 psychiatrically unaffected participants, 12 participants with SZ) and included a monetary reward for correct answers [85] not present in most other studies of WM in SZ. A 2003 study that reported locally increased activation during an WM task also had a very small sample size (n = 14 psychiatrically unaffected participants, 12 participants with SZ) and included performance as a nuisance covariate, potentially regressing out effects of interest related to the expected WM deficits in SZ [88]. Overall, evidence supporting the “inverted U” hypothesis is limited, and meta-analytic evidence suggests that the overarching phenotype in SZ is lower DLPFC activation during WM [84].
The temporal dynamics of WM/CC have also been studied in SZ on millisecond-level timescales by electroencephalography (EEG), which has allowed researchers to examine the degree to which synchronous activity at various frequencies are associated with these processes and to what extent neuronal oscillations are disrupted in the disorder. Of the various frequency bands (e.g., theta, beta), the high frequency, gamma band (~30–80 Hz) shows the strongest evidence for alterations in SZ. Gamma band power in the DLPFC has been shown to increase in psychiatrically unaffected individuals during various cognitive operations, including WM and CC [89,90,91]. Furthermore, patterns of gamma band activity may be specific for task-relevant information in WM; a 2012 study demonstrated that when a classifier was trained to distinguish between gamma activations that varied according to WM task-relevant load (i.e., items to remember), it was more likely to encode oscillations during the 3 item + 3 distractor condition as load 3 than as load 6 [92]. Prefrontal gamma oscillations, therefore, may specifically encode task-relevant information without incorporating distractor-associated signals. Recordings in non-human primates have also confirmed that dorsal prefrontal gamma frequency oscillations reflect active maintenance of information during WM [93]. Studies in SZ have generally reported lower gamma oscillatory power relative to controls during the maintenance phase of these tasks (e.g., [90, 94], reviewed by Gonzalez-Burgos [95]) (Fig. 1B). In a recent study, anodal tDCS applied to the DLPFC resulted in transient increases in DLPFC gamma activity along with improved performance on a version of the AX-CPT [96]. Notably, lower DLPFC gamma power may provide a translational bridge between the functional impairments in WM/CC in SZ and histopathological analyses of postmortem brain tissue in the disorder as outlined later in this review (Fig. 1C, D).
In addition to structural and functional MRI studies, measurement of regional levels of glutamate and GABA (the primary excitatory and inhibitory signaling molecules of the brain, respectively) has been undertaken using magnetic resonance spectroscopy (MRS). Since brain MRS was first developed in the 1980s, both GABA and glutamate have been measured extensively in SZ. Most of this previous work has been conducted in the medial prefrontal cortex, for which meta/mega-analyses suggest SZ is associated with lower glutamate [97, 98] but no difference in GABA [99] relative to unaffected participants. For the DLPFC, meta-analyses have not found conclusive evidence for alterations in glutamate [97, 100] or GABA [99] levels in SZ, although the overall number of studies is much smaller relative to the number of medial prefrontal studies and almost all of these previous DLPFC studies have been conducted at 1.5 or 3 T. One study at a higher field strength (7 T) reported lower glutamate in SZ vs. unaffected participants [101], consistent with a recent meta-analysis of 7 T studies of glutamate in the anterior cingulate [102]. Related to WM processes, research suggests that psychiatrically unaffected individuals and individuals with SZ show opposite relationships between DLPFC GABA levels and WM. Specifically, in psychiatrically unaffected individuals, increased GABA predicts better WM [103], whereas in SZ increased GABA predicts worse WM [104]. The reason for this interaction effect is unclear. One speculative possibility is that high GABA concentration represents a homeostatic response to an overall reduction in GABAergic signal. Thus, SZ individuals with higher GABA may actually have more severely disrupted inhibitory circuits (e.g., lower connectivity between GABAergic interneurons and pyramidal cells), resulting in worse WM. The increase in GABA therefore may represent an (unsuccessful) attempt by the brain to restore proper inhibitory function.
A major shortcoming of MRS, however, is its limited spatial resolution. MRS voxel sizes (length/width/height) are typically on the order of centimeters and contain a mix of gray matter, white matter, and CSF. MRS also cannot determine if measured neurotransmitter levels are presynaptic, postsynaptic, and/or involved in metabolic functions not related to neurotransmission. In order to fully understand the circuit level basis for WM deficits in SZ, the DLPFC must be examined at microscale resolutions in postmortem human brain. Following a brief introduction to how specific microcircuits facilitate WM processes in the DLPFC, in the sections below we present a detailed account of postmortem findings that have shaped our understanding of how microstructural aberrations in the DLPFC might lead to circuit-level disruptions during WM in SZ.
Linking dysfunctional dorsolateral prefrontal cortical circuitry to working memory deficits in schizophrenia
Local circuits in the dorsolateral prefrontal cortex and subprocesses of working memory
In order to develop novel therapeutic avenues for targeting DLPFC function in SZ, it is essential to understand how its activation may be fundamentally affected on a circuit level. Within the DLPFC, neurons in the supragranular layers (e.g., layer 3) appear to play a prominent role in WM processes, especially during the delay period of WM tasks [47,48,49]. How might these neurons mediate WM processes? Excitatory pyramidal neurons in layer 3, in addition to furnishing a principal axon which projects through the white matter to other cortical regions in the WM network, also give rise to local axon collaterals that primarily target the dendritic spines of other layer 3 pyramidal neurons [105,106,107]. These connections are thought to provide a local circuit, intrinsic to the DLPFC, for supporting excitatory activity among subsets of DLPFC layer 3 pyramidal neurons during the WM delay period [108, 109]. This interpretation is supported by computational models [110, 111]. In further support of the importance of layer 3 pyramidal neurons in WM function, the dendritic spines of layer 3 pyramidal neurons are enriched in the NR2B subunit of the NMDA receptor which is thought to be critical for delay period activity [109]. In addition, the gamma frequency oscillations that appear to be a neural correlate of WM [112] are primarily generated in layers 2–3 of the DLPFC [93]. These lines of evidence strongly support the role of DLPFC layer 3 pyramidal neurons in the generation of activity necessary for WM.
The activity of layer 3 pyramidal neurons during WM tasks is shaped by inhibitory inputs from different classes of GABA neurons. For example, local administration of a GABAA receptor antagonist in the non-human primate DLPFC impairs WM performance [113] and prevents the activation of neurons that are otherwise engaged by a specific stimulus [114]. Moreover, GABA signaling in non-human primates was found to facilitate interactions between different groups of neurons that are active at discrete time points during a WM task: the presentation of the cue, the delay period, and the response period [115]. Finally, computational models and experimental models of the hippocampus indicates that the gamma frequency activity of large groups of pyramidal neurons is synchronized by inputs from inhibitory interneurons [116].
Among the different subtypes of cortical GABA neurons, parvalbumin (PV)-expressing interneurons have been most extensively studied in relation to WM. The basket cell class of PV interneurons, which are present in high density in layers deep 3 and 4 [117], are a major target of the local axon collaterals of layer 3 pyramidal neurons [118]. These GABA neurons, in turn, provide fast, synaptic inhibition onto the perisomatic region of pyramidal neurons [119]. Based on these targeting properties, PV neurons appear to be positioned to provide strong inhibitory control over the firing of pyramidal neurons. The axons of PV basket cells arborize extensively, enabling them to innervate, and presumably coordinate the activity of, large groups of excitatory pyramidal neurons [120]. Specifically, the strong perisomatic inhibition mediated by PV neurons appears to entrain groups of pyramidal neurons that are responsive to a particular stimulus [121], consistent with experimental data showing that GABAA antagonism can destroy this stimulus-specific responsiveness [114]. Certain lines of evidence suggest that PV neuron activity is also crucial for generating the cortical gamma oscillations that accompany WM tasks. First, in computational models, the excitation of PV-containing basket cells by pyramidal neurons provides strong, coordinated feedback inhibition onto numerous pyramidal neurons [116]. Upon the termination of this inhibition, the targeted pyramidal neurons share a high probability of firing simultaneously and thus in synchrony [122]. Second, the decay kinetics of PV basket cell inhibition are consistent with a resulting firing rate of pyramidal neurons at gamma frequency [123]. Third, in studies of rodent cortex and hippocampus, activation or inhibition of PV cell activity can induce or suppress gamma oscillations, respectively [124,125,126]. Thus, these convergent lines of evidence support a central role for PV neurons in the DLPFC microcircuitry that generates the gamma oscillations associated with WM, although causal evidence for the role of PV neurons in gamma oscillations in the primate cortex is still lacking.
Another class of PV GABA neuron, the chandelier cell, gives rise to vertically arrayed sets of axon terminals (termed cartridges) that synapse on the axon initial segment of pyramidal neurons [127, 128]. Based on the location of these synapses, chandelier cells inputs can exert potent inhibitory regulation over the firing of pyramidal neurons [129]. In contrast, although still controversial, some evidence suggests that chandelier cell inputs can also be depolarizing [130]. Indeed, subsequent studies reported that the state of the cortical circuit can influence the nature of chandelier cell inputs: i.e., in quiescent circuits, these inputs can be depolarizing [131]. These and other findings suggest that unlike PV basket cells, PV chandelier neurons may not participate in the generation of gamma oscillations. In addition, at chandelier neuron inputs onto pyramidal neuron axon initial segments, the postsynaptic GABAA receptors are enriched in α2 subunits which exhibit slower kinetics relative to α1 subunit-enriched GABAA receptors located postsynaptic to PV basket cell inputs [132]. Thus, the available data suggest that PV basket cells, and not PV chandelier cells, likely participate in the generation of gamma oscillatory activity in the DLPFC during the delay period of WM tasks.
Inhibition in the DLPFC could also contribute to distractor resistance, possibly through the activity of a second class of GABA interneurons known as somatostatin (SST)-expressing neurons. SST neurons primarily provide inhibition onto the distal dendrites of pyramidal neurons [133]. This inhibition may play a role in gating excitatory inputs to these neurons, as suggested by studies in the rodent hippocampus [134]. SST neurons in layers 2-superficial 3 also appear to be a major target of local axon collaterals of layer 3 pyramidal neurons [118]. The excitation of layer 3 pyramidal neurons during the delay period of a WM task might activate SST neurons that could, in turn, provide inhibition to pyramidal neurons that are not involved in keeping the WM memoranda online. Indeed, in computational models, cells providing the type of dendritic inhibition characteristic of SST neurons are more efficient in filtering out distractor stimuli than is the perisomatic inhibition provided by PV neurons [121]. Thus, SST neurons might contribute to distractor resistance through their ability to suppress the activity of DLPFC pyramidal neurons [135,136,137] that receive input from task-irrelevant distractor stimuli. This feature of SST neurons might equip the DLPFC to be less sensitive than other cortical regions to the influence of distractors during WM, consistent with the greater abundance of SST neurons in the DLPFC relative to other cortical regions in the non-human primate WM network [138]. While these convergent lines of evidence support a role for SST neurons in distractor resistance during WM, confirmation of this idea requires more evidence from studies in non-human primates.
Overall, the findings reviewed above suggest that the cellular constituents of WM circuitry in the primate DLPFC include (at least in part) layer 3 pyramidal neurons and their interconnected PV and SST interneurons. It is important to note that neuromodulators can also influence the activity of this circuit. For example, dopamine signaling in the DLPFC has long been known to influence WM function [139] and appears to control the firing of neurons exhibiting delay-period activity (see Box 1). In addition, acetylcholine appears to regulate top-down attention crucial for distractor resistance during WM in non-human primates [140]. Other studies have found that acetylcholine influences specific neuronal populations, including SST neurons, in a laminar-specific manner in humans [135, 141]. Therefore, WM impairments in people with SZ likely reflect alterations to one or more of the components of this circuit in the DLPFC. Using results from postmortem tissue studies, we describe the potential contribution(s) of each of these cell types to the WM phenotype in SZ in the following sections.
Alterations in dorsolateral prefrontal layer 3 pyramidal neurons in schizophrenia
Given the important role of layer 3 pyramidal neurons in generating the delay period activity necessary for WM, alterations in this neuronal subtype could be a significant contributor to DLPFC dysfunction in SZ. Indeed, DLPFC layer 3 pyramidal neurons exhibit numerous morphological and molecular anomalies which suggest that SZ is associated with hypoactivity of these neurons. First, pyramidal neurons in layer 3 of the DLPFC of individuals with SZ exhibit smaller somal volumes, shorter dendritic arbors, lower dendritic branching complexity, and lower dendritic spine density relative to matched unaffected comparison individuals [142,143,144,145,146]. These alterations are either not present or present to a smaller degree in pyramidal neurons located in layers 5 and 6 of the DLPFC [147]. Because dendritic spines receive the majority of excitatory inputs to pyramidal neurons [148], the combination of shorter dendrites and lower spine density suggests that layer 3 pyramidal neurons receive a lower level of excitatory input in SZ (Figs. 1D and 2).

Pyramidal neurons in layer 3 of the DLPFC exhibit smaller somal volumes, shorter dendritic branches, and fewer dendritic spines. Two types of parvalbumin (PV) neurons provide inputs onto the perisomatic region of pyramidal neurons: PV basket cells (PVBC) and PV chandelier cells (PVChCs). The available data suggest that PVBC inputs exhibit lower levels of GAD67 protein, and putatively weaker GABA neurotransmission, in SZ. Postsynaptic to PVBC inputs are GABAA receptors enriched in the α1 subunit, and mRNA levels for this subunit are also lower in layer 3 pyramidal neurons in SZ, further contributing to weaker PVBC inhibitory inputs. In contrast, GAD67 levels appear to be unaltered in PVChC inputs, but these terminal cartridges exhibit lower protein levels of the GABA transporter, GAT1, and their synaptic targets, pyramidal neuron axon initial segments, exhibit higher protein levels of postsynaptic α2 subunit-containing GABAA receptors. In concert, these findings suggest that GABA neurotransmission is stronger at PVChC inputs in SZ. Finally, SST neurons, which appear to principally provide inputs onto the dendrites of pyramidal neurons in layers 2–3 of the DLPFC, exhibit lower levels of SST mRNA. Together, these alterations are thought to contribute to the neural substrate of cognitive dysfunction in SZ.
In the absence of increased intrinsic excitability, the lower level of excitatory input is very likely to render layer 3 pyramidal neurons hypoactive in SZ. Indeed, transcriptomic studies of layer 3 pyramidal neurons have reported coordinated downregulation of mitochondrial markers [149, 150], which appears to reflect diminished demand for energy production (rather than an intrinsic dysfunction of mitochondria) in SZ [151]. Further supporting the notion of hypoactive pyramidal neurons in SZ, the expression levels of activity-dependent transcripts are lower in these cells [152,153,154]. It is worth noting that layer 5 pyramidal neurons, which do not appear to exhibit smaller somal sizes or fewer dendritic spines in SZ [143, 147], also exhibit lower expression of mitochondrial-related transcripts [149]. This alteration may reflect lower activity of layer 5 pyramidal neurons (which are a major target of descending axon collaterals from layer 3 pyramidal neurons [105]) secondary to lower excitatory output of layer 3 pyramidal neurons.
Alterations in dorsolateral prefrontal parvalbumin neurons in schizophrenia
As noted above, PV neurons in the middle cortical layers 3–4 are also a major recipient of excitatory inputs from layer 3 pyramidal neurons [118]; thus, hypoactivity of layer 3 pyramidal neurons may subsequently cause activity-dependent alterations to PV neurons in SZ. For example, lower levels of activity-dependent PV mRNA [155,156,157,158] and protein [159, 160] are among the most widely replicated findings in postmortem studies of SZ. Importantly, neither the density nor morphology of PV neurons appears to be altered in SZ [156, 159,160,161,162], supporting the idea that the lower expression of PV is secondary to altered inputs from layer 3 pyramidal neurons.
Consistent with this interpretation, the functional capacity of PV neurons, which can be indexed by measuring cell type-specific expression of transcripts and proteins involved in GABA signaling, appears to be altered in SZ. For example, the expression of GAD67, the principal enzyme responsible for the majority of cortical GABA synthesis, is activity-dependent, and lower levels of DLPFC GAD67 mRNA have been reported in multiple studies of SZ [157, 163,164,165,166,167]. In particular, lower levels of GAD67 mRNA in PV cell bodies [156] and of GAD67 protein in PV axon terminals of PV basket cells have been found in the illness [166, 168] (Fig. 2).
Additional findings lend support to the interpretation that alterations in PV neurons are secondary to weaker excitatory input from layer 3 pyramidal neurons in the DLPFC of SZ. First, levels of KCNS3 mRNA, an activity-dependent potassium channel that is specific to PV neurons in humans [169], are also lower in the DLPFC of SZ [170]. Second, weaker GABA signaling from PV neurons onto pyramidal neurons might be anticipated to result in a homeostatic upregulation of the α1 subunit of the GABAA receptor that is located in pyramidal neurons postsynaptic to PV basket cell inputs. Layer 3 pyramidal neurons in SZ, however, exhibit lower levels of the mRNA encoding the GABAA α1 receptor subunit [171]. Therefore, the concordant downregulation of pre- and post-synaptic markers of inhibition by PV neurons onto layer 3 pyramidal neurons in SZ suggests that the inhibitory strength of PV inputs is reduced to compensate for lower excitatory activity of pyramidal neurons in the circuit. This hypothesis is consistent with findings in experimental systems [134, 172].
In contrast to marked downregulations of GAD67 in the axon terminals of PV basket cells, GAD67 protein levels appears to be unaltered in the axon cartridges of PV chandelier cells [173]. Other studies, however, suggest that levels of the GABA membrane transporter GAT1 protein (which is responsible for the reuptake of GABA) are lower in PV chandelier axon cartridges in SZ [174], whereas levels of the GABAA α2 subunit protein are higher in pyramidal neuron axon initial segments in the illness [175]. In concert, these findings suggest that GABA neurotransmission might be greater in PV chandelier cell inputs to pyramidal neurons in SZ. If as noted above, these inputs are depolarizing in quiescent circuits [130, 176], then the alterations in chandelier cell to pyramidal cell inputs in SZ might reflect a compensatory response to weaker pyramidal neuron activity [177] (Fig. 2).
Alterations in dorsolateral prefrontal somatostatin neurons in schizophrenia
As noted above, the local axon collaterals of layer 3 pyramidal neurons also appear to target SST neurons in layers 2-superficial 3 of the non-human primate DLPFC [133]. Similar to the findings for PV neurons [155, 157] in SZ, multiple studies [155, 157, 178,179,180,181] have reported markedly lower levels of SST mRNA in the DLPFC in a large proportion of individuals with SZ. Unfortunately, postmortem studies of other features of SST neurons in SZ have been limited by several technical challenges. First, SST peptide levels are subject to substantial and rapid postmortem degradation [182], precluding robust proteomic or morphological studies of these neurons [183]. Second, the density of neurons with detectable levels of SST mRNA is ~30% lower in SZ [179]. It is unclear, however, if these findings represent 1) a normal complement of SST neurons in SZ with a subset that fail to express detectable levels of SST mRNA, or 2) a deficit in SST neuron number in SZ. Thus, although SST interneurons are clearly altered in the DLPFC of SZ, the nature of that alteration remains uncertain. One possibility is that like PV neurons, weaker excitatory input to SST neurons from layer 3 pyramidal neurons results in downregulated expression of SST mRNA and possibly GAD67 mRNA within SST neurons. This effect might be strongest in a subset of SST neurons, although a mechanism for such an effect is yet to be discovered.
Possible upstream mechanisms of altered GABA signaling in schizophrenia
As stated above, approximately 50% of the local axon collaterals of layer 3 pyramidal neurons appear to target PV or SST neurons. Therefore, alterations in these two subtypes of GABA neurons in SZ might reflect compensatory responses designed to downregulate inhibition in the face of lower excitatory activity in DLPFC layer 3 pyramidal neurons (Fig. 2). This interpretation is supported by other findings. For example, the μ opioid receptor, which, in rodent hippocampus, is found primarily in SST and PV neurons [184, 185] and mediates hyperpolarization of these neurons [186, 187], is upregulated in SZ [178, 188]; thus, this alteration would also be expected to decrease inhibition from PV and SST neurons. In contrast, other cell types that are not direct targets of layer 3 pyramidal neuron axon collaterals, such as calretinin neurons [118], do not appear to be altered in the DLPFC of individuals with SZ [156, 178, 181, 189]. These findings, in concert with lower levels of markers of GABA neurotransmission, support the idea that convergent transcriptomic alterations take place to downregulate levels of inhibition from GABA neurons to match the lower level of excitation that they receive.
Not all lines of evidence, however, support this hypothesis. For example, although layer 3 pyramidal neurons do not appear to exhibit the same alterations (at least not to the same degree) in the primary visual cortex as they do in the DLPFC in SZ [145], PV and SST mRNAs are significantly lower in the primary visual cortex in SZ [178, 190]. Furthermore, findings of altered ErbB4 splicing [189], and an associated deficit in the density of excitatory inputs onto PV neurons [159], suggest the possibility of a primary pathological process in PV neurons.
Clinical impact: broader contributions of dorsolateral prefrontal cortex pathology on cognitive deficits and clinical symptoms in schizophrenia
To this point in this review, we have emphasized the role of the DLPFC in explaining deficits in WM and CC in SZ. The role of the DLPFC in cognition, however, likely goes well beyond these two cognitive domains. Indeed, a consensus view has emerged that the DLPFC, along with the parietal cortex, insula, and medial prefrontal cortex, belongs to a domain-general CC network in the brain that supports performance across a broad range of tasks [35, 191,192,193,194,195]. CC is engaged in support of these systems under a number of conditions, including distraction, high memory loads, and overcoming prepotency. For this reason, the DLPFC may play an essential role in shifting attentional focus according to task demands and/or maintaining task-related goal/rule information [22] to guide task appropriate responding. In fact, the neural mechanisms in non-human primate DLPFC that underlie attentional focus appear to be similar to those underlying the selection of items available in working memory [196]. A reduction in DLPFC function as observed in SZ would thus be expected to have wide ranging effects on cognitive abilities that extend beyond WM. Indeed, a number of studies have shown DLPFC hypoactivation in SZ under high (but not low) CC demands in cognitive domains such as response selection [197], episodic memory [198], language comprehension [199, 200], and emotion processing [201]. Related to this point, in a 2009 meta-analysis Minzenberg et al. [84] observed significantly lower DLPFC activation in SZ across 41 studies covering a variety of executive function tasks, including oddball tasks, mental arithmetic, word generation, WM, and CC. Similarly, in an activation likelihood estimation-based meta-analysis Ragland et al. [202] showed significantly lower DLPFC activation in SZ across 18 episodic memory studies during both encoding and retrieval of information [195].
Reduced DLPFC activation and impaired performance on tasks related to DLPFC function may also have real-life, functional consequences. To understand how, it may be helpful to first consider the clinical features of SZ. Although the disorder is most well-known for its characteristic positive symptoms (such as hallucinations and delusions), another striking symptom domain is disorganization. The disorganization symptoms of SZ are represented as observable inattention, inappropriate affect, disorganized speech (thought disorder), bizarre behavior, and the inability to initiate or complete complex everyday tasks. Indeed, the pioneering psychiatrist Eugen Bleuler hypothesized that thought disorder, or “loosening of associations,” was close to the primary “organic substrate” of SZ, present over all stages of illness and of principal diagnostic importance [203]. As people with SZ with disorganization symptoms may have difficulties in communication, comprehension, and goal-directed behavior, these symptoms are likely to have deleterious real-life consequences. To this point, a number of studies have reported significant relationships between disorganization and community-based functioning in SZ [204,205,206,207,208].
Conceptually, therefore, it is plausible to hypothesize that disease-related alterations in DLPFC function may contribute to disorganization in the illness. Consistent with this hypothesis, associations have been repeatedly observed between disorganization symptoms and behavioral performance during DLPFC-dependent tasks [27, 31, 208,209,210] (or similarly, communication disturbances and WM [211]) as well between disorganization and reduced DLPFC activation during CC and WM [78, 80, 82, 212]. Related to this point, when individuals with SZ are asked to speak on high stress topics, their speech is more disorganized than when asked to speak on low stress topics [213], possibly due to stress-induced disruption of DLPFC function [214]. Furthermore, in a 2012 study Yoon et al. [215] found that when whole-brain fMRI data during the AX-CPT was used to classify SZ vs. unaffected comparison participants, SZ individuals that were correctly classified showed stronger disorganization symptoms than misclassified individuals with SZ. Relationships between disorganization and task-associated DLPFC activity in the illness were concisely summarized in a 2014 review [210], which reported significant associations between DLPFC response and disorganization in 7 of the 8 studies examined.
Limited evidence suggests abnormal DLPFC function may also play a role in the expression of the negative symptoms of SZ. Early behavioral studies of WM, for example, demonstrated relationships between WM impairment and negative symptom severity (e.g., [216, 217]). Task-based functional imaging studies have observed relationships between DLPFC hypoactivation and negative symptoms as well (e.g., [201, 218].
On a final note, given these symptomatic links, it is important to consider the role of DLPFC dysfunction in SZ given the clinically heterogeneous nature of the illness. As described above, evidence suggests that those individuals with SZ for whom disorganization symptoms are most prominent show the most severe deficits in prefrontal function. On the other hand, people with SZ who do not exhibit severe disorganization are likely to exhibit relatively intact levels of DLPFC function.
Questions for future investigation
In the next section, we briefly consider relevant topics for future investigation, including potential avenues for therapeutic investigation.
Diagnostic specificity
Over the past decade, the field has becoming increasingly interested in transdiagnostic approaches to understanding neuronal dysfunction. This is largely due to the rise of directives such as the NIH Research Domain Criteria (RDoC), which seek to understand the neuronal mechanisms of cognitive dysfunction both within and across Diagnostic and Statistical Manual (DSM)-based diagnostic criteria [219]. Relevant to the present review, therefore, one may ask the question: to what extent are the functional, macrocircuit, and microcircuit level DLPFC abnormalities described for SZ also observed in other psychiatric disorders, particularly in those disorders for which psychosis is a prominent symptom?
At present, approaches towards understanding the diagnostic specificity of DLPFC-related processes (such as WM and CC) have been most extensively applied in studies comparing SZ to bipolar disorder (BD). Behaviorally and functionally, the most common finding appears to be that BD is associated with intermediate performance deficits and prefrontal hypofunction relative to SZ and the unaffected comparison group [31, 32, 220,221,222,223,224,225]. Performance deficits, furthermore, may correlate with disorganization symptoms across diagnoses [31, 32]. As this line of research remains relatively novel, however, these findings require replication with larger sample sizes. Moreover, the majority of this research has been conducted in Type I BD, so it is unclear to what extent these features are present in Type II BD.
Findings at the cellular level have produced mixed results across diagnoses. For example, one study showed lower dendritic spine density on DLPFC layer 3 neurons in SZ relative to both individuals with major depressive disorder (MDD) and individuals without psychiatric illness (with no difference between people with MDD and unaffected comparison individuals) [145]. A later study found lower spine density in people with BD vs. the unaffected comparison group, a trend-level reduction in SZ vs. unaffected individuals, and no difference between SZ and BD individuals [146]. Similarly, PV mRNA levels have been reported to be lower in both SZ and BD relative to individuals who are psychiatrically unaffected, but not in individuals with MDD [153, 226]. Finally, transcriptomic studies in layer 3 pyramidal neurons found that gene expression alterations were much more prominent in the DLPFC of individuals with SZ than those with BD, but that the presence of psychosis in BD individuals was associated with transcriptome alterations similar to SZ [151, 227]. Nonetheless, additional studies to assess diagnostic and phenotypic specificity are needed.
Cortical regional specificity
Although this review focuses on the DLPFC, it would be overly simplistic to claim that reduced WM and CC abilities and associated disorganization in SZ are solely tied to dysfunction in a single brain region. WM and CC are complex processes that not only involve the DLPFC, but also the anterior cingulate, parietal cortex, ventrolateral prefrontal cortex, and inferior frontal cortex. These regions make up an interconnected brain system known as the frontal-cingulate-parietal network [228], which is broadly involved in executive function. Specific components of this network also make distinct contributions to executive processes, e.g., conflict monitoring in the anterior cingulate [229, 230] and attentional orienting in the superior parietal lobule [231]. Furthermore, although less well-studied, microcircuit-level alterations have been detected in one or more of these regions in SZ [178, 232]. Indeed, data suggest that SZ is associated with alterations to SST neurons and PV basket cells not only in the DLPFC but also other regions of the cortical WM network [168, 178, 233]. The latter findings might be consistent with other findings suggesting that abnormal sensory processing streams, e.g., those investigated using early visual N1 event-related potentials, might also contribute to cognitive deficits in SZ [234]. A multisite study of visual processing deficits in SZ, however, found that abnormalities might be explained by attention lapsing (as indexed by catch trials) [235]. This result suggests a role for dysfunctional cognitive control (in the form of attention lapses) in explaining alterations in perceptual task performance in the disorder. Taken together with evidence presented in this review, therefore, substantial data point to abnormalities within the DLPFC that act as primary drivers of WM and CC deficits in the disorder given their fundamental roles in these cognitive processes. The specific contributions of sensory processing streams, nonetheless, remains unresolved and is therefore an important direction for future research.
It is also important to recognize that the neuronal substrate of cognitive deficits in SZ almost certainly extends to other brain areas outside of this frontal network, e.g., the hippocampus for episodic memory [195]. As the nature of functional hippocampal abnormalities is variable, however (with some studies showing hypoactivation and others hyperactivation) [195], the exact extent to which it contributes to the overall neurocognitive phenotype observed in the illness is an important future research direction.
Role of thalamocortical connectivity
The DLPFC is a highly connected brain region. Among its vast array of connecting fibers are prominent reciprocal connections with the mediodorsal nucleus (MDN) of the thalamus. Interest in the role of MDN-DLPFC connectivity in cognition stems from early studies which found that MDN lesions induce similar cognitive deficits to those observed following damage to the prefrontal cortex [236], including deficits in WM function [237]. Postmortem neuroanatomical studies of the MDN in SZ have not shown consistent differences between individuals with SZ relative to those without psychiatric illness in cell number or MDN volume, although sample sizes for these studies are small [238]. Functionally, research in mice suggests the MDN may influence WM by amplifying DLPFC connectivity in order to maintain information during the delay period [239]. Human neuroimaging studies have also found increased activation of the thalamus during WM tasks as well as reduced activation and connectivity between the thalamus and prefrontal cortex in SZ relative to unaffected comparison individuals [240, 241]. Due to limitations of fMRI contrast and resolution, it is unclear if these deficits are specific to the MDN. Combining high field MRI with recent improvements in thalamic parcellation schemes [e.g., 242] may help shed light on the nature of thalamic abnormalities in SZ.
Role of corticostriatal connectivity
In addition to the thalamus, the DLPFC is also connected to the associative striatum, to which it sends efferent projections as part of a cortico-striatal-thalamic feedback loop. The potential role of the striatum in the pathophysiology of SZ has long been appreciated due to its high density of dopaminergic axon terminals and dopamine D2 receptors, as the dopamine system is targeted by all currently FDA-approved antipsychotics. It is now well-established that SZ is associated with dysregulated dopaminergic signaling, and particularly increased dopamine synthesis and release in the striatum [243, 244]. MRI studies further suggest that the illness is associated with disrupted prefrontal-striatal structural and functional connectivity [245,246,247,248,249]. Reduced functional connectivity, furthermore, may be associated with response to antipsychotic treatment [246, 247]. One other possibility suggested by rodent studies is that alterations in prefrontal pyramidal neurons could lead to downstream hyperdopaminergia in the striatum that is sensitive to the effects of antipsychotics [250]. Thus, alterations to the DLPFC in SZ, causing cognitive dysfunction in the disorder, may subsequently lead to hyperdopaminergia and contribute to the emergence of positive symptoms.
In addition to positive symptoms, frontostriatal dysconnectivity may play an important role in cognitive deficits in SZ. For example, aberrant functional connectivity has been observed between the DLPFC and striatum during WM in the illness [251, 252]. Reduced connectivity of this circuit may reflect a relative inability of the DLPFC to regulate the basal ganglia during cognition [252]. Later work in individuals without psychiatric disorders suggests that effective connectivity between the DLPFC and striatum may be particularly important during the encoding phase of WM as it may signal object novelty [253]. Overall, however, despite clear links between striatal dopamine and SZ, how frontostriatal connectivity contributes to the DLPFC-associated cognitive processes in the disorder remains poorly understood.
Antipsychotic effects
Any understanding of DLPFC structure and function in SZ would be incomplete without considering the effects of antipsychotics. Structural imaging studies suggest that people with SZ given antipsychotic treatment have thinner cortices relative to antipsychotic-naive individuals (including a study in a very large (n = 4474) sample) [73, 254, 255], although some evidence suggests cortical thickness is reduced in antipsychotic-naive people with SZ (relative to people without a major psychiatric illness) [256, 257] and may also occur before illness onset (i.e., prior to medication use) [258].
Neuroimaging studies have also reported antipsychotic effects on prefrontal function. A 2017 meta-analysis of longitudinal fMRI studies across various tasks found that the most commonly observed antipsychotic effect was prefrontal normalization (i.e., activation and connectivity patterns that more closely resembled those in psychiatrically unaffected individuals), although the authors of this meta-analysis noted that the included studies were generally naturalistic and therefore did not include placebo groups [259]. Results were also regionally heterogeneous and usually not replicated. A cross-sectional study of CC also found that people with SZ treated with antipsychotics showed levels of DLPFC activation halfway between unmedicated people with SZ and psychiatrically unaffected people [73]. D2 receptor blockade-induced homeostatic increase in prefrontal dopamine release may be a potential mechanism underlying antipsychotic effects [260]. Given that antipsychotic medication is generally ineffective at treating cognitive symptoms, however, it should be noted that these medications are not expected to completely normalize DLPFC activation during WM, CC, or other prefrontal cortex-mediated processes in SZ.
Importantly, the bulk of the cellular level findings described above do not appear to reflect the influence of antipsychotic medications, at least as assessed by comparisons of human participants on or off these medications at time of death and by studies in non-human primates exposed chronically to typical or atypical antipsychotics at doses that produce serum levels associated with therapeutic efficacy in humans. Studies in non-human primates exposed to antipsychotics, however, did find smaller brain tissue volumes [261], suggesting that antipsychotics might contribute to smaller DLPFC volume in SZ.
Potential avenues for therapeutic investigation: psychopharmacology
As described in this review, SZ and WM deficits are associated with disrupted GABAergic signaling. It therefore follows that drugs targeting this receptor may help restore normal function. In a preliminary proof-of-concept study, the subunit selective GABAA receptor agonist MK-0777 improved WM and CC performance as well as increased frontal gamma band power during CC [262]. A larger clinical trial, however, failed to show cognitive benefits of the drug [263]. This apparent lack of clinical benefit might be due to the relatively weak partial agonist activity of MK-0777 or other limitations in study design [264].
As SZ is also associated with reduced glutamatergic signaling, another approach has been to develop agonists for the NMDA receptor. As recently reviewed in a meta-analysis of 25 clinical trials of NMDA receptor-acting agents in SZ [265], a small but significant improvement (standardized mean difference = 0.16) in WM was observed across all trials. It is unclear, however, if this result is clinically meaningful (e.g., improves functional outcomes). In addition, other strategies to augment signaling through NMDARs have been investigated in preclinical models and early stage clinical trials, including agents which inhibit the synthesis of endogenous compounds that block NMDARs [266, 267].
Other neurotransmitter systems, including dopamine signaling in the DLPFC, have been targeted by pharmacologic interventions (Box 1). A limitation inherent in developing compounds that target neurotransmitter systems, however, is that these synthesized molecules are generally less effective at modulating these systems relative to endogenous ligands, for a variety of reasons (e.g., availability, kinetics, affinity, desensitization). It is also unclear if the “normalization” of these systems actually affects the underlying pathology that initiated the cascade of events leading to the disorder. Moreover, these pharmacologic interventions lack regional-specificity contributing to a host of off-region engagements. An alternative approach may be to develop pharmacologic ways of inducing long-lasting microstructural changes that may be more closely tied to disease pathogenesis.
For example, as discussed in this review, SZ is associated with lower dendritic spine density on excitatory pyramidal neurons. One mechanism by which this is thought to occur is chronic inflammation and microglial hyperactivation [268] (Box 2). The clinical features of SZ emerge during late adolescence, a period of brain development during which synaptic pruning is particularly robust. When amplified by an inflammatory stimulus (e.g., environmental stress), microglial activation could be exaggerated during this period, increasing pruning beyond normal levels. Interestingly, a recent finding in neuronal cultures derived from cortical tissue of individuals with SZ found links between abnormalities in microglial-like cells and synapse elimination. Cultures with variants in C4A associated with SZ risk also showed increased neuronal complement disposition and microglial synaptic engulfment. Finally, the antibiotic minocycline prevented pruning, hinting at a potential therapeutic avenue of investigation [269].
If inflammation-induced cortical over-pruning and subsequent loss of excitatory drive is intimately tied to the etiology of SZ, it follows that pharmacologic agents that reverse or prevent this process may have clinical utility. Indeed, significant improvement in visual learning/memory, attention, and/or executive function in SZ has been observed in trials of the anti-inflammatory compounds minocycline and pregnenolone [270]. Notably, however, a 2018 trial of 12 months of minocycline treatment in patients showed no effect of the antibiotic on DLPFC activation associated with WM [271]. The compound also showed no effect on symptoms.
On a final note, a limitation of the majority of clinical trials in SZ is that most do not incorporate functional measures of cognition as biomarkers. For example, effects of treatment on DLPFC function during relevant tasks (e.g., WM) or prefrontal gamma oscillatory activity [262] are rarely included as endpoints. Including such biomarkers might be necessary to determine if treatments are indeed appropriately engaging their intended targets [272].
Potential avenues for therapeutic investigation: neuromodulation
Modulation of DLPFC activity is another potentially exciting avenue for improving cognition in SZ. At present, transcranial direct current stimulation (tDCS) and transcranial magnetic stimulation (TMS) are the two most widely investigated methods for SZ, although the overall body of literature in this area remains small.
tDCS is a well-tolerated, safe, noninvasive technique that applies low current stimulation via scalp electrodes. A number of studies have demonstrated that anodal tDCS over the DLPFC enhances cognitive performance in individuals without a psychiatric illness [273,274,275]. In SZ, overall evidence suggests that anodal tDCS may have modest positive effects on cognition [273, 276, 277] with WM showing the most significant effects among various cognitive domains [277]. Interestingly, however, a meta-analysis in individuals without psychiatric disorders suggests that tDCS effect sizes are larger when administered during the study phase of a cognitive task (compared to the performance phase) [278]. It is possible, therefore, that larger effect sizes in SZ will be observed if the effects are measured following stimulation, as opposed to during stimulation. Indeed, our group found that when anodal prefrontal tDCS is administered during a WM task in SZ, performance and gamma power was enhanced during a CC task that was performed immediately following stimulation [96].
With repetitive transcranial magnetic stimulation (rTMS), brain regions are excited by repeated magnetic pulses. The most recent review of rTMS effects on cognition in SZ revealed mixed findings, with 4 studies showing enhancement and 8 studies reporting no significant effects [279]. A study combined rTMS with fMRI also showed no effect of stimulation on DLPFC activation during WM [280]. Given the higher cost, greater participant discomfort, and increased difficulty of administration for rTMS vs. tDCS, based on these mixed findings we believe that tDCS may ultimately hold more promise as a method for cognitive enhancement in SZ.
Finally, it should be noted that prefrontal stimulation may also improve negative symptoms in SZ, as demonstrated by a 2018 meta-analysis of 24 studies that reported improvement after stimulation with moderate effect sizes (0.64 for rTMS, 0.50 for tDCS) [281].
Potential avenues for therapeutic investigation: cognitive training
As defined by Keshavan et al. [282], cognitive training (CT) is “an intervention that uses specifically designed, behaviorally constrained cognitive or socio-affective learning events delivered in a scalable and reproducible manner to potentially improve neural system operations.” Methodologically, CT involves performing cognitive tasks designed to activate neuronal circuits and enhance their function through neuroplasticity. In relation to “neural system operations” associated with DLPFC function, some evidence suggests potentially promising effects of intensive training in SZ. For example, in a 2014 study Subramaniam et al. [283] found that 1 h of daily computerized auditory/verbal processing and emotional identification exercises improved 2-back WM performance and associated DLPFC BOLD response in SZ relative to a computer games control condition, and a 2019 meta-analysis found that left prefrontal activation was the most commonly observed finding across 16 studies that combined neuroimaging with CT [284]. This meta-analysis, however, also found no significant overlap in the specific brain regions responding to CT, perhaps due to variation in training protocols and tasks. The mechanisms of cognitive enhancement following CT in SZ, therefore, may be variable and remain poorly understood.
Concluding remarks
It has been over a century since Kraepelin posited that SZ was a disorder of the frontal lobes. Since then, anatomical, physiological, and behavioral evidence have converged on a common substrate, localized to the DLPFC, that underlies many of the cognitive deficits observed in the illness. Further, these lines of research are well-interconnected, in that neuroanatomical abnormalities of the DLPFC in SZ can be plausibly linked to aberrant neuronal function to ultimately produce the distinctive neurocognitive phenotypes associated with the disease. Studies of SZ have also helped to identify many potential approaches to novel therapeutic strategies that might be tested for efficacy using well-characterized functional biomarkers that combine neuroimaging and cognitive testing. We anticipate that these therapeutic strategies will eventually make significant inroads towards alleviating the devastating disease burden of SZ, from preempting the emergence of psychosis in at-risk states to improving cognition in chronic illness.
Funding and disclosure
This work was supported by grants MH124329 (SJD), MH043784 (DAL), MH106438 (CSC), MH119546 (CSC), and MH122139 (CSC) from the National Institute of Mental Health. DAL currently receives investigator-initiated research support from Merck. The remaining authors have nothing to disclose.
References
Green MF, Harvey PD. Cognition in schizophrenia: past, present, and future. Schizophr Res Cogn. 2014;1:e1–e9.
Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153:321–30.
Green MF. Impact of cognitive and social cognitive impairment on functional outcomes in patients with schizophrenia. J Clin Psychiatry. 2016;77:8–11. Suppl 2
Cloutier M, Aigbogun MS, Guerin A, Nitulescu R, Ramanakumar AV, Kamat SA, et al. The economic burden of schizophrenia in the United States in 2013. J Clin Psychiatry. 2016;77:764–71.
Baddeley AD, Hitch GJ. Working Memory. In: Bower GA, editor. Recent advances in learning and motivation. New York: Academic; 1974. p. 47–89.
Baddeley AD. Working memory. Philos T R Soc B 1983;302:311–24.
Park S, Holzman PS. Schizophrenics show spatial working memory deficits. Arch Gen Psychiatry. 1992;49:975–82.
Gold JM, Carpenter C, Randolph C, Goldberg TE, Weinberger DR. Auditory working memory and Wisconsin Card Sorting Test performance in schizophrenia. Arch Gen Psychiatry. 1997;54:159–65.
McGurk SR, Coleman T, Harvey PD, Reichenberg A, White L, Friedman J, et al. Working memory performance in poor outcome schizophrenia: relationship to age and executive functioning. J Clin Exp Neuropsychol. 2004;26:153–60.
Aleman A, Hijman R, de Haan EH, Kahn RS. Memory impairment in schizophrenia: a meta-analysis. Am J Psychiatry. 1999;156:1358–66.
Forbes NF, Carrick LA, McIntosh AM, Lawrie SM. Working memory in schizophrenia: a meta-analysis. Psychol Med. 2009;39:889–905.
Mesholam-Gately RI, Giuliano AJ, Goff KP, Faraone SV, Seidman LJ. Neurocognition in first-episode schizophrenia: a meta-analytic review. Neuropsychology 2009;23:315–36.
Bora E, Murray RM. Meta-analysis of cognitive deficits in ultra-high risk to psychosis and first-episode psychosis: do the cognitive deficits progress over, or after, the onset of psychosis? Schizophr Bull. 2014;40:744–55.
Horan WP, Braff DL, Nuechterlein KH, Sugar CA, Cadenhead KS, Calkins ME, et al. Verbal working memory impairments in individuals with schizophrenia and their first-degree relatives: findings from the Consortium on the Genetics of Schizophrenia. Schizophr Res. 2008;103:218–28.
Park S, Holzman PS, Goldman-Rakic PS. Spatial working memory deficits in the relatives of schizophrenic patients. Arch Gen Psychiatry. 1995;52:821–8.
Pirkola T, Tuulio-Henriksson A, Glahn D, Kieseppa T, Haukka J, Kaprio J, et al. Spatial working memory function in twins with schizophrenia and bipolar disorder. Biol Psychiatry. 2005;58:930–6.
Toulopoulou T, Picchioni M, Rijsdijk F, Hua-Hall M, Ettinger U, Sham P, et al. Substantial genetic overlap between neurocognition and schizophrenia: genetic modeling in twin samples. Arch Gen Psychiatry. 2007;64:1348–55.
Seidman LJ, Meyer EC, Giuliano AJ, Breiter HC, Goldstein JM, Kremen WS, et al. Auditory working memory impairments in individuals at familial high risk for schizophrenia. Neuropsychology 2012;26:288–303.
Jenkins LM, Bodapati AS, Sharma RP, Rosen C. Working memory predicts presence of auditory verbal hallucinations in schizophrenia and bipolar disorder with psychosis. J Clin Exp Neuropsychol. 2018;40:84–94.
Bodnar M, Malla A, Joober R, Lepage M. Cognitive markers of short-term clinical outcome in first-episode psychosis. Br J Psychiatry. 2008;193:297–304.
Green MF, Kern RS, Braff DL, Mintz J. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull. 2000;26:119–36.
Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202.
Barch DM, Smith E. The cognitive neuroscience of working memory: relevance to CNTRICS and schizophrenia. Biol Psychiatry. 2008;64:11–7.
Stroop JR. Studies of interference in serial verbal reactions. J Exp Psychol. 1935;18:643–62.
Cohen JD, Barch DM, Carter C, Servan-Schreiber D. Context-processing deficits in schizophrenia: converging evidence from three theoretically motivated cognitive tasks. J Abnorm Psychol. 1999;108:120–33.
Servan-Schreiber D, Cohen JD, Steingard S. Schizophrenic deficits in the processing of context. A test of a theoretical model. Arch Gen Psychiatry. 1996;53:1105–12.
Barch DM, Carter CS, MacDonald AW 3rd, Braver TS, Cohen JD. Context-processing deficits in schizophrenia: diagnostic specificity, 4-week course, and relationships to clinical symptoms. J Abnorm Psychol. 2003;112:132–43.
Stratta P, Daneluzzo E, Bustini M, Casacchia M, Rossi A. Schizophrenic deficits in the processing of context. Arch Gen Psychiatry. 1998;55:186–8.
MacDonald AW 3rd, Carter CS. Event-related FMRI study of context processing in dorsolateral prefrontal cortex of patients with schizophrenia. J Abnorm Psychol. 2003;112:689–97.
Niendam TA, Ray KL, Iosif AM, Lesh TA, Ashby SR, Patel PK, et al. Association of age at onset and longitudinal course of prefrontal function in youth with schizophrenia. JAMA Psychiatry. 2018;75:1252–60.
Smucny J, Lesh TA, Iosif AM, Niendam TA, Tully LM, Carter CS. Longitudinal stability of cognitive control in early psychosis: nondegenerative deficits across diagnoses. J Abnorm Psychol. 2018;127:781–88.
Smucny J, Lesh TA, Newton K, Niendam TA, Ragland JD, Carter CS. Levels of cognitive control: a functional magnetic resonance imaging-based test of an RDoC domain across bipolar disorder and schizophrenia. Neuropsychopharmacology 2018;43:598–606.
Smucny J, Lesh TA, Zarubin VC, Niendam TA, Ragland JD, Tully LM, et al. One-year stability of frontoparietal cognitive control network connectivity in recent onset schizophrenia: a task-related 3T fMRI study. Schizophr Bull. 2020;46:1249–58.
Lesh TA, Westphal AJ, Niendam TA, Yoon JH, Minzenberg MJ, Ragland JD, et al. Proactive and reactive cognitive control and dorsolateral prefrontal cortex dysfunction in first episode schizophrenia. Neuroimage Clin. 2013;2:590–9.
Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36:316–38.
Kang SS, MacDonald AW, Sponheim SR. Dysfunctional neural processes underlying context processing deficits in schizophrenia. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019;4:644–54.
Niendam TA, Lesh TA, Yoon J, Westphal AJ, Hutchison N, Daniel Ragland J, et al. Impaired context processing as a potential marker of psychosis risk state. Psychiatry Res. 2014;221:13–20.
Snitz BE, Macdonald AW 3rd, Carter CS. Cognitive deficits in unaffected first-degree relatives of schizophrenia patients: a meta-analytic review of putative endophenotypes. Schizophr Bull. 2006;32:179–94.
MacDonald AW 3rd, Pogue-Geile MF, Johnson MK, Carter CS. A specific deficit in context processing in the unaffected siblings of patients with schizophrenia. Arch Gen Psychiatry. 2003;60:57–65.
Smucny J, Lesh TA, Carter CS. Baseline frontoparietal task-related BOLD activity as a predictor of improvement in clinical symptoms at 1-year follow-up in recent-onset psychosis. Am J Psychiatry. 2019;176:839–45.
Smucny J, Davidson I, Carter CS. Comparing machine and deep learning-based algorithms for prediction of clinical improvement in psychosis with functional magnetic resonance imaging. Hum Brain Mapp. 2021;42:1197–205.
Christophel TB, Klink PC, Spitzer B, Roelfsema PR, Haynes JD. The distributed nature of working memory. Trends Cogn Sci. 2017;21:111–24.
Semendeferi K, Lu A, Schenker N, Damasio H. Humans and great apes share a large frontal cortex. Nat Neurosci. 2002;5:272–6.
Selemon LD, Goldman-Rakic PS. Common cortical and subcortical targets of the dorsolateral prefrontal and posterior parietal cortices in the rhesus monkey: evidence for a distributed neural network subserving spatially guided behavior. J Neurosci. 1988;8:4049–68.
Zanto TP, Rubens MT, Thangavel A, Gazzaley A. Causal role of the prefrontal cortex in top-down modulation of visual processing and working memory. Nat Neurosci. 2011;14:656–61.
Edin F, Klingberg T, Johansson P, McNab F, Tegner J, Compte A. Mechanism for top-down control of working memory capacity. Proc Natl Acad Sci USA. 2009;106:6802–7.
Fuster JM, Alexander GE. Neuron activity related to short-term memory. Science. 1971;173:652–4.
Kubota K, Niki H. Prefrontal cortical unit activity and delayed alternation performance in monkeys. J Neurophysiol. 1971;34:337–47.
Funahashi S, Bruce CJ, Goldman-Rakic PS. Mnemonic coding of visual space in the monkey’s dorsolateral prefrontal cortex. J Neurophysiol. 1989;61:331–49.
Wallis JD, Anderson KC, Miller EK. Single neurons in prefrontal cortex encode abstract rules. Nature 2001;411:953–6.
Smith EE, Jonides J. Neuroimaging analyses of human working memory. Proc Natl Acad Sci USA. 1998;95:12061–8.
Finn ES, Huber L, Jangraw DC, Molfese PJ, Bandettini PA. Layer-dependent activity in human prefrontal cortex during working memory. Nat Neurosci. 2019;22:1687–95.
Vilberg KL, Rugg MD. Memory retrieval and the parietal cortex: a review of evidence from a dual-process perspective. Neuropsychologia. 2008;46:1787–99.
Wolf RC, Vasic N, Walter H. Differential activation of ventrolateral prefrontal cortex during working memory retrieval. Neuropsychologia. 2006;44:2558–63.
Zhou X, Zhu D, Qi XL, Lees CJ, Bennett AJ, Salinas E, et al. Working memory performance and neural activity in prefrontal cortex of peripubertal monkeys. J Neurophysiol. 2013;110:2648–60.
Asaad WF, Rainer G, Miller EK. Task-specific neural activity in the primate prefrontal cortex. J Neurophysiol. 2000;84:451–9.
Sakai K, Rowe JB, Passingham RE. Active maintenance in prefrontal area 46 creates distractor-resistant memory. Nat Neurosci. 2002;5:479–84.
Vogel EK, McCollough AW, Machizawa MG. Neural measures reveal individual differences in controlling access to working memory. Nature. 2005;438:500–3.
Jacobsen CF. Studies of cerebral function in primates. Comp Psychol Monogr. 1936;13:1–68.
Malmo RB. Interference factors in delayed response in monkeys after removal of frontal lobes. J Neurophysiol. 1942;5:295–308.
Qi XL, Constantinidis C. Lower neuronal variability in the monkey dorsolateral prefrontal than posterior parietal cortex. J Neurophysiol. 2015;114:2194–203.
Qi XL, Elworthy AC, Lambert BC, Constantinidis C. Representation of remembered stimuli and task information in the monkey dorsolateral prefrontal and posterior parietal cortex. J Neurophysiol. 2015;113:44–57.
Powell KD, Goldberg ME. Response of neurons in the lateral intraparietal area to a distractor flashed during the delay period of a memory-guided saccade. J Neurophysiol. 2000;84:301–10.
Bisley JW, Goldberg ME. Neural correlates of attention and distractibility in the lateral intraparietal area. J Neurophysiol. 2006;95:1696–717.
Miller EK, Erickson CA, Desimone R. Neural mechanisms of visual working memory in prefrontal cortex of the macaque. J Neurosci. 1996;16:5154–67.
Lennert T, Martinez-Trujillo J. Strength of response suppression to distracter stimuli determines attentional-filtering performance in primate prefrontal neurons. Neuron. 2011;70:141–52.
Suzuki M, Gottlieb J. Distinct neural mechanisms of distractor suppression in the frontal and parietal lobe. Nat Neurosci. 2013;16:98–104.
Kraepelin E. Ein Lehrbuch fur Studierende und Arzte. Barth: Leipzig; 1913.
Shepherd AM, Laurens KR, Matheson SL, Carr VJ, Green MJ. Systematic meta-review and quality assessment of the structural brain alterations in schizophrenia. Neurosci Biobehav Rev. 2012;36:1342–56.
Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52.
Pearlson GD, Marsh L. Structural brain imaging in schizophrenia: a selective review. Biol Psychiatry. 1999;46:627–49.
van Haren NE, Schnack HG, Cahn W, van den Heuvel MP, Lepage C, Collins L, et al. Changes in cortical thickness during the course of illness in schizophrenia. Arch Gen Psychiatry. 2011;68:871–80.
Lesh TA, Tanase C, Geib BR, Niendam TA, Yoon JH, Minzenberg MJ, et al. A multimodal analysis of antipsychotic effects on brain structure and function in first-episode schizophrenia. JAMA Psychiatry. 2015;72:226–34.
Ellison-Wright I, Bullmore E. Meta-analysis of diffusion tensor imaging studies in schizophrenia. Schizophr Res. 2009;108:3–10.
Andreou C, Borgwardt S. Structural and functional imaging markers for susceptibility to psychosis. Mol Psychiatry. 2020;25:2773–85.
Glahn DC, Ragland JD, Abramoff A, Barrett J, Laird AR, Bearden CE, et al. Beyond hypofrontality: a quantitative meta-analysis of functional neuroimaging studies of working memory in schizophrenia. Hum Brain Mapp. 2005;25:60–9.
Weinberger DR, Berman KF, Zec RF. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch Gen Psychiatry. 1986;43:114–24.
Perlstein WM, Carter CS, Noll DC, Cohen JD. Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry. 2001;158:1105–13.
Barch DM, Carter CS, Braver TS, Sabb FW, MacDonald A 3rd, Noll DC, et al. Selective deficits in prefrontal cortex function in medication-naive patients with schizophrenia. Arch Gen Psychiatry. 2001;58:280–8.
Yoon JH, Minzenberg MJ, Ursu S, Ryan Walter BS, Wendelken C, Ragland JD, et al. Association of dorsolateral prefrontal cortex dysfunction with disrupted coordinated brain activity in schizophrenia: relationship with impaired cognition, behavioral disorganization, and global function. Am J Psychiatry. 2008;165:1006–14.
Snitz BE, MacDonald A 3rd, Cohen JD, Cho RY, Becker T, Carter CS. Lateral and medial hypofrontality in first-episode schizophrenia: functional activity in a medication-naive state and effects of short-term atypical antipsychotic treatment. Am J Psychiatry. 2005;162:2322–9.
MacDonald AW 3rd, Carter CS, Kerns JG, Ursu S, Barch DM, Holmes AJ, et al. Specificity of prefrontal dysfunction and context processing deficits to schizophrenia in never-medicated patients with first-episode psychosis. Am J Psychiatry. 2005;162:475–84.
Zhang R, Picchioni M, Allen P, Toulopoulou T. Working memory in unaffected relatives of patients with schizophrenia: a meta-analysis of functional magnetic resonance imaging studies. Schizophr Bull. 2016;42:1068–77.
Minzenberg MJ, Laird AR, Thelen S, Carter CS, Glahn DC. Meta-analysis of 41 functional neuroimaging studies of executive function in schizophrenia. Arch Gen Psychiatry. 2009;66:811–22.
Manoach DS, Press DZ, Thangaraj V, Searl MM, Goff DC, Halpern E, et al. Schizophrenic subjects activate dorsolateral prefrontal cortex during a working memory task, as measured by fMRI. Biol Psychiatry. 1999;45:1128–37.
Potkin SG, Turner JA, Brown GG, McCarthy G, Greve DN, Glover GH, et al. Working memory and DLPFC inefficiency in schizophrenia: the FBIRN study. Schizophr Bull. 2009;35:19–31.
Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, et al. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex. 2000;10:1078–92.
Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR. Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry. 2003;160:2209–15.
Roux F, Uhlhaas PJ. Working memory and neural oscillations: alpha-gamma versus theta-gamma codes for distinct WM information? Trends Cogn Sci. 2014;18:16–25.
Cho RY, Konecky RO, Carter CS. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci USA. 2006;103:19878–83.
Helfrich RF, Knight RT. Oscillatory dynamics of prefrontal cognitive control. Trends Cogn Sci. 2016;20:916–30.
Roux F, Wibral M, Mohr HM, Singer W, Uhlhaas PJ. Gamma-band activity in human prefrontal cortex codes for the number of relevant items maintained in working memory. J Neurosci. 2012;32:12411–20.
Bastos AM, Loonis R, Kornblith S, Lundqvist M, Miller EK. Laminar recordings in frontal cortex suggest distinct layers for maintenance and control of working memory. Proc Natl Acad Sci USA. 2018;115:1117–22.
Minzenberg MJ, Firl AJ, Yoon JH, Gomes GC, Reinking C, Carter CS. Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology. 2010;35:2590–9.
Gonzalez-Burgos G, Cho RY, Lewis DA. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry. 2015;77:1031–40.
Boudewyn MA, Scangos K, Ranganath C, Carter CS. Using prefrontal transcranial direct current stimulation (tDCS) to enhance proactive cognitive control in schizophrenia. Neuropsychopharmacology. 2020;45:1877–83.
Smucny J, Carter CS, Maddock RJ. Medial prefrontal cortex glutamate is reduced in schizophrenia and moderated by measurement quality: a meta-analysis of 1H-MRS studies. Biol Psychiatry. In Press.
Merritt K, McGuire PK, Egerton A, Investigators HMiS, Aleman A, Block W, et al. Association of age, antipsychotic medication, and symptom severity in schizophrenia with proton magnetic resonance spectroscopy brain glutamate level: a mega-analysis of individual participant-level data. JAMA Psychiatry. 2021. In Press.
Kumar V, Vajawat B, Rao NP. Frontal GABA in schizophrenia: a meta-analysis of (1)H-MRS studies. World J Biol Psychiatry. 2020;22:1–13.
Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK. Nature of glutamate alterations in schizophrenia: a meta-analysis of proton magnetic resonance spectroscopy studies. JAMA Psychiatry. 2016;73:665–74.
Wang AM, Pradhan S, Coughlin JM, Trivedi A, DuBois SL, Crawford JL, et al. Assessing brain metabolism with 7-T proton magnetic resonance spectroscopy in patients with first-episode psychosis. JAMA Psychiatry. 2019;76:314–23.
Sydnor VJ, Roalf DR. A meta-analysis of ultra-high field glutamate, glutamine, GABA and glutathione 1HMRS in psychosis: Implications for studies of psychosis risk. Schizophr Res. 2020;226:61–69.
Yoon JH, Grandelis A, Maddock RJ. Dorsolateral prefrontal cortex GABA concentration in humans predicts working memory load processing capacity. J Neurosci. 2016;36:11788–94.
Ragland JD, Maddock RJ, Hurtado MY, Tanase C, Lesh TA, Niendam TA, et al. Disrupted GABAergic facilitation of working memory performance in people with schizophrenia. Neuroimage Clin. 2020;25:102127.
Kritzer MF, Goldman-Rakic PS. Intrinsic circuit organization of the major layers and sublayers of the dorsolateral prefrontal cortex in the rhesus monkey. J Comp Neurol. 1995;359:131–43.
Melchitzky DS, Gonzalez-Burgos G, Barrionuevo G, Lewis DA. Synaptic targets of the intrinsic axon collaterals of supragranular pyramidal neurons in monkey prefrontal cortex. J Comp Neurol. 2001;430:209–21.
Levitt JB, Lewis DA, Yoshioka T, Lund JS. Topography of pyramidal neuron intrinsic connections in macaque monkey prefrontal cortex (areas 9 and 46). J Comp Neurol. 1993;338:360–76.
Gonzalez-Burgos G, Barrionuevo G, Lewis DA. Horizontal synaptic connections in monkey prefrontal cortex: an in vitro electrophysiological study. Cereb Cortex. 2000;10:82–92.
Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, et al. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron 2013;77:736–49.
Compte A, Brunel N, Goldman-Rakic PS, Wang XJ. Synaptic mechanisms and network dynamics underlying spatial working memory in a cortical network model. Cereb Cortex. 2000;10:910–23.
Camperi M, Wang XJ. A model of visuospatial working memory in prefrontal cortex: recurrent network and cellular bistability. J Comput Neurosci. 1998;5:383–405.
Lundqvist M, Rose J, Herman P, Brincat SL, Buschman TJ, Miller EK. Gamma and beta bursts underlie working memory. Neuron 2016;90:152–64.
Sawaguchi T, Matsumura M, Kubota K. Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res. 1989;75:457–69.
Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–94.
Constantinidis C, Williams GV, Goldman-Rakic PS. A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci. 2002;5:175–80.
Whittington MA, Traub RD, Kopell N, Ermentrout B, Buhl EH. Inhibition-based rhythms: experimental and mathematical observations on network dynamics. Int J Psychophysiol. 2000;38:315–36.
Conde F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA. Local circuit neurons immunoreactive for calretinin, calbindin D-28k or parvalbumin in monkey prefrontal cortex: distribution and morphology. J Comp Neurol. 1994;341:95–116.
Melchitzky DS, Lewis DA. Pyramidal neuron local axon terminals in monkey prefrontal cortex: differential targeting of subclasses of GABA neurons. Cereb Cortex. 2003;13:452–60.
Gonzalez-Burgos G, Krimer LS, Povysheva NV, Barrionuevo G, Lewis DA. Functional properties of fast spiking interneurons and their synaptic connections with pyramidal cells in primate dorsolateral prefrontal cortex. J Neurophysiol. 2005;93:942–53.
Williams SM, Goldman-Rakic PS, Leranth C. The synaptology of parvalbumin-immunoreactive neurons in the primate prefrontal cortex. J Comp Neurol. 1992;320:353–69.
Wang XJ, Tegner J, Constantinidis C, Goldman-Rakic PS. Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. Proc Natl Acad Sci USA. 2004;101:1368–73.
Buzsaki G, Wang XJ. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–25.
Whittington MA, Cunningham MO, LeBeau FE, Racca C, Traub RD. Multiple origins of the cortical gamma rhythm. Dev Neurobiol. 2011;71:92–106.
Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53:591–604.
Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702.
Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–7.
DeFelipe J, Hendry SH, Jones EG, Schmechel D. Variability in the terminations of GABAergic chandelier cell axons on initial segments of pyramidal cell axons in the monkey sensory-motor cortex. J Comp Neurol. 1985;231:364–84.
Szentágothai J. The ‘module-concept’ in cerebral cortex architecture. Brain Res. 1975;95:475–96.
DeFelipe J, Farinas I. The pyramidal neuron of the cerebral cortex: morphological and chemical characteristics of the synaptic inputs. Prog Neurobiol. 1992;39:563–607.
Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–5.
Woodruff AR, McGarry LM, Vogels TP, Inan M, Anderson SA, Yuste R. State-dependent function of neocortical chandelier cells. J Neurosci. 2011;31:17872–86.
Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004;27:569–75.
Melchitzky DS, Lewis DA. Dendritic-targeting GABA neurons in monkey prefrontal cortex: comparison of somatostatin- and calretinin-immunoreactive axon terminals. Synapse. 2008;62:456–65.
Horn ME, Nicoll RA. Somatostatin and parvalbumin inhibitory synapses onto hippocampal pyramidal neurons are regulated by distinct mechanisms. Proc Natl Acad Sci USA. 2018;115:589–94.
Obermayer J, Heistek TS, Kerkhofs A, Goriounova NA, Kroon T, Baayen JC, et al. Lateral inhibition by Martinotti interneurons is facilitated by cholinergic inputs in human and mouse neocortex. Nat Commun. 2018;9:4101.
Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53:735–46.
Zilberter Y, Kaiser KM, Sakmann B. Dendritic GABA release depresses excitatory transmission between layer 2/3 pyramidal and bitufted neurons in rat neocortex. Neuron. 1999;24:979–88.
Dienel SJ, Ciesielski AJ, Bazmi HH, Profozich EA, Fish KN, Lewis DA. Distinct laminar and cellular patterns of GABA neuron transcript expression in monkey prefrontal and visual cortices. Cereb Cortex. 2021;31:2345–63.
Brozoski TJ, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science. 1979;205:929–32.
Sun Y, Yang Y, Galvin VC, Yang S, Arnsten AF, Wang M. Nicotinic alpha4beta2 cholinergic receptor influences on dorsolateral prefrontal cortical neuronal firing during a working memory task. J Neurosci. 2017;37:5366–77.
Obermayer J, Verhoog MB, Luchicchi A, Mansvelder HD. Cholinergic modulation of cortical microcircuits is layer-specific: evidence from rodent, monkey and human brain. Front Neural Circuits. 2017;11:100.
Pierri JN, Volk CL, Auh S, Sampson A, Lewis DA. Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry. 2001;58:466–73.
Rajkowska G, Selemon LD, Goldman-Rakic PS. Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry. 1998;55:215–24.
Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–53.
Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73.
Konopaske GT, Lange N, Coyle JT, Benes FM. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry. 2014;71:1323–31.
Kolluri N, Sun Z, Sampson AR, Lewis DA. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 2005;162:1200–2.
Yuste R. Dendritic spines and distributed circuits. Neuron. 2011;71:772–81.
Arion D, Corradi JP, Tang S, Datta D, Boothe F, He A, et al. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol Psychiatry. 2015;20:1397–405.
Arion D, Huo Z, Enwright JF, Corradi JP, Tseng G, Lewis DA. Transcriptome alterations in prefrontal pyramidal cells distinguish schizophrenia from bipolar and major depressive disorders. Biol Psychiatry. 2017;82:594–600.
Glausier JR, Enwright JF 3rd, Lewis DA. Diagnosis- and cell type-specific mitochondrial functional pathway signatures in schizophrenia and bipolar disorder. Am J Psychiatry. 2020;177:1140–50.
Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN, et al. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci. 2005;25:372–83.
Kimoto S, Zaki MM, Bazmi HH, Lewis DA. Altered markers of cortical gamma-aminobutyric acid neuronal activity in schizophrenia: role of the NARP gene. JAMA Psychiatry. 2015;72:747–56.
Kimoto S, Bazmi HH, Lewis DA. Lower expression of glutamic acid decarboxylase 67 in the prefrontal cortex in schizophrenia: contribution of altered regulation by Zif268. Am J Psychiatry. 2014;171:969–78.
Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, Weickert CS. Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am J Psychiatry. 2010;167:1479–88.
Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–26.
Volk DW, Sampson AR, Zhang Y, Edelson JR, Lewis DA. Cortical GABA markers identify a molecular subtype of psychotic and bipolar disorders. Psychol Med. 2016;46:2501–12.
Chung DW, Chung Y, Bazmi HH, Lewis DA. Altered ErbB4 splicing and cortical parvalbumin interneuron dysfunction in schizophrenia and mood disorders. Neuropsychopharmacology. 2018;43:2478–86.
Chung DW, Fish KN, Lewis DA. Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia. Am J Psychiatry. 2016;173:1131–39.
Enwright JF, Sanapala S, Foglio A, Berry R, Fish KN, Lewis DA. Reduced labeling of parvalbumin neurons and perineuronal nets in the dorsolateral prefrontal cortex of subjects with schizophrenia. Neuropsychopharmacology 2016;41:2206–14.
Woo TU, Miller JL, Lewis DA. Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry. 1997;154:1013–5.
Tooney PA, Chahl LA. Neurons expressing calcium-binding proteins in the prefrontal cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:273–8.
Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase(67) messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57:237–45.
Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE Jr, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–66.
Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci. 2011;31:11088–95.
Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, et al. Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry. 2011;168:921–9.
Duncan CE, Webster MJ, Rothmond DA, Bahn S, Elashoff M, Shannon, et al. Prefrontal GABA(A) receptor alpha-subunit expression in normal postnatal human development and schizophrenia. J Psychiatr Res. 2010;44:673–81.
Fish KN, Rocco BR, DeDionisio AM, Dienel SJ, Sweet RA, Lewis DA. Altered parvalbumin basket cell terminals in the cortical visuospatial working memory network in schizophrenia. Biol Psychiatry. 2021;90:47–57.
Georgiev D, Gonzalez-Burgos G, Kikuchi M, Minabe Y, Lewis DA, Hashimoto T. Selective expression of KCNS3 potassium channel alpha-subunit in parvalbumin-containing GABA neurons in the human prefrontal cortex. PLoS One. 2012;7:e43904.
Georgiev D, Arion D, Enwright JF, Kikuchi M, Minabe Y, Corradi JP, et al. Lower gene expression for KCNS3 potassium channel subunit in parvalbumin-containing neurons in the prefrontal cortex in schizophrenia. Am J Psychiatry. 2014;171:62–71.
Glausier JR, Lewis DA. Selective pyramidal cell reduction of GABA(A) receptor alpha1 subunit messenger RNA expression in schizophrenia. Neuropsychopharmacology 2011;36:2103–10.
Xue M, Atallah BV, Scanziani M. Equalizing excitation-inhibition ratios across visual cortical neurons. Nature. 2014;511:596–600.
Rocco BR, Lewis DA, Fish KN. Markedly lower glutamic acid decarboxylase 67 protein levels in a subset of boutons in schizophrenia. Biol Psychiatry. 2016;79:1006–15.
Woo TU, Whitehead RE, Melchitzky DS, Lewis DA. A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA. 1998;95:5341–6.
Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA. Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex. 2002;12:1063–70.
Woodruff A, Xu Q, Anderson SA, Yuste R. Depolarizing effect of neocortical chandelier neurons. Front Neural Circuits. 2009;3:15.
Lewis DA. The chandelier neuron in schizophrenia. Dev Neurobiol. 2011;71:118–27.
Tsubomoto M, Kawabata R, Zhu X, Minabe Y, Chen K, Lewis DA, et al. Expression of transcripts selective for GABA neuron subpopulations across the cortical visuospatial working memory network in the healthy state and schizophrenia. Cereb Cortex. 2019;29:3540–50.
Morris HM, Hashimoto T, Lewis DA. Alterations in somatostatin mRNA expression in the dorsolateral prefrontal cortex of subjects with schizophrenia or schizoaffective disorder. Cereb Cortex. 2008;18:1575–87.
Hoftman GD, Volk DW, Bazmi HH, Li S, Sampson AR, Lewis DA. Altered cortical expression of GABA-related genes in schizophrenia: illness progression vs developmental disturbance. Schizophr Bull. 2015;41:180–91.
Fung SJ, Fillman SG, Webster MJ, Shannon, Weickert C. Schizophrenia and bipolar disorder show both common and distinct changes in cortical interneuron markers. Schizophr Res. 2014;155:26–30.
Hayes TL, Cameron JL, Fernstrom JD, Lewis DA. A comparative analysis of the distribution of prosomatostatin-derived peptides in human and monkey neocortex. J Comp Neurol. 1991;303:584–99.
Lewis DA. The human brain revisited: opportunities and challenges in postmortem studies of psychiatric disorders. Neuropsychopharmacology. 2002;26:143–54.
Stumm RK, Zhou C, Schulz S, Hollt V. Neuronal types expressing mu- and delta-opioid receptor mRNA in the rat hippocampal formation. J Comp Neurol. 2004;469:107–18.
Drake CT, Milner TA. Mu opioid receptors are in somatodendritic and axonal compartments of GABAergic neurons in rat hippocampal formation. Brain Res. 1999;849:203–15.
Capogna M, Gahwiler BH, Thompson SM. Mechanism of mu-opioid receptor-mediated presynaptic inhibition in the rat hippocampus in vitro. J Physiol. 1993;470:539–58.
Lupica CR. Delta and mu enkephalins inhibit spontaneous GABA-mediated IPSCs via a cyclic AMP-independent mechanism in the rat hippocampus. J Neurosci. 1995;15:737–49. 1 Pt 2
Volk DW, Radchenkova PV, Walker EM, Sengupta EJ, Lewis DA. Cortical opioid markers in schizophrenia and across postnatal development. Cereb Cortex. 2012;22:1215–23.
Chung DW, Volk DW, Arion D, Zhang Y, Sampson AR, Lewis DA. Dysregulated ErbB4 splicing in schizophrenia: selective effects on parvalbumin expression. Am J Psychiatry. 2016;173:60–8.
Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA. Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry. 2008;165:479–89.
Cole MW, Reynolds JR, Power JD, Repovs G, Anticevic A, Braver TS. Multi-task connectivity reveals flexible hubs for adaptive task control. Nat Neurosci. 2013;16:1348–55.
Cole MW, Schneider W. The cognitive control network: Integrated cortical regions with dissociable functions. Neuroimage. 2007;37:343–60.
Niendam TA, Laird AR, Ray KL, Dean YM, Glahn DC, Carter CS. Meta-analytic evidence for a superordinate cognitive control network subserving diverse executive functions. Cogn Affect Behav Neurosci. 2012;12:241–68.
Duncan J. The multiple-demand (MD) system of the primate brain: mental programs for intelligent behaviour. Trends Cogn Sci. 2010;14:172–9.
Guo JY, Ragland JD, Carter CS. Memory and cognition in schizophrenia. Mol Psychiatry. 2019;24:633–42.
Panichello MF, Buschman TJ. Shared mechanisms underlie the control of working memory and attention. Nature. 2021;592:601–05.
Kappenman ES, Luck SJ, Kring AM, Lesh TA, Mangun GR, Niendam T, et al. Electrophysiological evidence for impaired control of motor output in schizophrenia. Cereb Cortex. 2016;26:1891–9.
Ragland JD, Ranganath C, Harms MP, Barch DM, Gold JM, Layher E, et al. Functional and neuroanatomic specificity of episodic memory dysfunction in schizophrenia: a functional magnetic resonance imaging study of the relational and item-specific encoding task. JAMA Psychiatry. 2015;72:909–16.
Swaab TY, Boudewyn MA, Long DL, Luck SJ, Kring AM, Ragland JD, et al. Spared and impaired spoken discourse processing in schizophrenia: effects of local and global language context. J Neurosci. 2013;33:15578–87.
Boudewyn MA, Carter CS, Swaab TY. Cognitive control and discourse comprehension in schizophrenia. Schizophr Res Treat. 2012;2012:484502.
Ursu S, Kring AM, Gard MG, Minzenberg MJ, Yoon JH, Ragland JD, et al. Prefrontal cortical deficits and impaired cognition-emotion interactions in schizophrenia. Am J Psychiatry. 2011;168:276–85.
Ragland JD, Laird AR, Ranganath C, Blumenfeld RS, Gonzales SM, Glahn DC. Prefrontal activation deficits during episodic memory in schizophrenia. Am J Psychiatry. 2009;166:863–74.
Bleuler E. Dementia praecox oder die Gruppe der Schizophrenien. Deuticke: Leipzig, Germany; 1911.
Takahashi H, Iwase M, Nakahachi T, Sekiyama R, Tabushi K, Kajimoto O, et al. Spatial working memory deficit correlates with disorganization symptoms and social functioning in schizophrenia. Psychiatry Clin Neurosci. 2005;59:453–60.
Reed RA, Harrow M, Herbener ES, Martin EM. Executive function in schizophrenia: is it linked to psychosis and poor life functioning? J Nerv Ment Dis. 2002;190:725–32.
Evans JD, Bond GR, Meyer PS, Kim HW, Lysaker PH, Gibson PJ, et al. Cognitive and clinical predictors of success in vocational rehabilitation in schizophrenia. Schizophr Res. 2004;70:331–42.
Smith TE, Hull JW, Huppert JD, Silverstein SM. Recovery from psychosis in schizophrenia and schizoaffective disorder: symptoms and neurocognitive rate-limiters for the development of social behavior skills. Schizophr Res. 2002;55:229–37.
Ventura J, Thames AD, Wood RC, Guzik LH, Hellemann GS. Disorganization and reality distortion in schizophrenia: a meta-analysis of the relationship between positive symptoms and neurocognitive deficits. Schizophr Res. 2010;121:1–14.
O’Leary DS, Flaum M, Kesler ML, Flashman LA, Arndt S, Andreasen NC. Cognitive correlates of the negative, disorganized, and psychotic symptom dimensions of schizophrenia. J Neuropsychiatry Clin Neurosci. 2000;12:4–15.
Goghari VM, Sponheim SR, MacDonald AW 3rd. The functional neuroanatomy of symptom dimensions in schizophrenia: a qualitative and quantitative review of a persistent question. Neurosci Biobehav Rev. 2010;34:468–86.
Docherty NM, Hawkins KA, Hoffman RE, Quinlan DM, Rakfeldt J, Sledge WH. Working memory, attention, and communication disturbances in schizophrenia. J Abnorm Psychol. 1996;105:212–9.
van Veelen NM, Vink M, Ramsey NF, Kahn RS. Left dorsolateral prefrontal cortex dysfunction in medication-naive schizophrenia. Schizophr Res. 2010;123:22–9.
Docherty NM, Evans IM, Sledge WH, Seibyl JP, Krystal JH. Affective reactivity of language in schizophrenia. J Nerv Ment Dis. 1994;182:98–102.
Liston C, McEwen BS, Casey BJ. Psychosocial stress reversibly disrupts prefrontal processing and attentional control. Proc Natl Acad Sci USA. 2009;106:912–7.
Yoon JH, Nguyen DV, McVay LM, Deramo P, Minzenberg MJ, Ragland JD, et al. Automated classification of fMRI during cognitive control identifies more severely disorganized subjects with schizophrenia. Schizophr Res. 2012;135:28–33.
Carter C, Robertson L, Nordahl T, Chaderjian M, Kraft L, O’Shora-Celaya L. Spatial working memory deficits and their relationship to negative symptoms in unmedicated schizophrenia patients. Biol Psychiatry. 1996;40:930–2.
Park S, Puschel J, Sauter BH, Rentsch M, Hell D. Spatial working memory deficits and clinical symptoms in schizophrenia: a 4-month follow-up study. Biol Psychiatry. 1999;46:392–400.
Andreasen NC, Rezai K, Alliger R, Swayze VW 2nd, Flaum M, Kirchner P, et al. Hypofrontality in neuroleptic-naive patients and in patients with chronic schizophrenia. Assessment with xenon 133 single-photon emission computed tomography and the Tower of London. Arch Gen Psychiatry. 1992;49:943–58.
Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167:748–51.
Brambilla P, Macdonald AW 3rd, Sassi RB, Johnson MK, Mallinger AG, Carter CS, et al. Context processing performance in bipolar disorder patients. Bipolar Disord. 2007;9:230–7.
Reilly JL, Hill SK, Gold JM, Keefe RS, Clementz BA, Gershon E, et al. Impaired context processing is attributable to global neuropsychological impairment in schizophrenia and psychotic bipolar disorder. Schizophr Bull. 2017;43:397–406.
Smucny J, Barch DM, Gold JM, Strauss ME, MacDonald AW 3rd, Boudewyn MA, et al. Cross-diagnostic analysis of cognitive control in mental illness: insights from the CNTRACS consortium. Schizophr Res. 2019;208:377–83.
McTeague LM, Huemer J, Carreon DM, Jiang Y, Eickhoff SB, Etkin A. Identification of common neural circuit disruptions in cognitive control across psychiatric disorders. Am J Psychiatry. 2017;174:676–85.
Brandt CL, Eichele T, Melle I, Sundet K, Server A, Agartz I, et al. Working memory networks and activation patterns in schizophrenia and bipolar disorder: comparison with healthy controls. Br J Psychiatry. 2014;204:290–8.
Hamilton LS, Altshuler LL, Townsend J, Bookheimer SY, Phillips OR, Fischer J, et al. Alterations in functional activation in euthymic bipolar disorder and schizophrenia during a working memory task. Hum Brain Mapp. 2009;30:3958–69.
Sibille E, Morris HM, Kota RS, Lewis DA. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol. 2011;14:721–34.
Enwright JF, Lewis DA. Similarities in cortical transcriptome alterations between schizophrenia and bipolar disorder are related to the presence of psychosis. Schizophr Bull. 2021;sbaa195. In press.
Dosenbach NU, Visscher KM, Palmer ED, Miezin FM, Wenger KK, Kang HC, et al. A core system for the implementation of task sets. Neuron. 2006;50:799–812.
Kerns JG, Cohen JD, MacDonald AW 3rd, Cho RY, Stenger VA, Carter CS. Anterior cingulate conflict monitoring and adjustments in control. Science. 2004;303:1023–6.
van Veen V, Cohen JD, Botvinick MM, Stenger VA, Carter CS. Anterior cingulate cortex, conflict monitoring, and levels of processing. Neuroimage 2001;14:1302–8.
Gazzaley A, Nobre AC. Top-down modulation: bridging selective attention and working memory. Trends Cogn Sci. 2012;16:129–35.
Hoftman GD, Dienel SJ, Bazmi HH, Zhang Y, Chen K, Lewis DA. Altered gradients of glutamate and gamma-aminobutyric acid transcripts in the cortical visuospatial working memory network in schizophrenia. Biol Psychiatry. 2018;83:670–79.
Dienel SJ, Lewis DA. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol Dis. 2019;131:104208.
Neuhaus AH, Karl C, Hahn E, Trempler NR, Opgen-Rhein C, Urbanek C, et al. Dissection of early bottom-up and top-down deficits during visual attention in schizophrenia. Clin Neurophysiol. 2011;122:90–8.
Barch DM, Carter CS, Dakin SC, Gold J, Luck SJ, Macdonald A 3rd, et al. The clinical translation of a measure of gain control: the contrast-contrast effect task. Schizophr Bull. 2012;38:135–43.
Mitchell AS, Chakraborty S. What does the mediodorsal thalamus do? Front Syst Neurosci. 2013;7:37.
Parnaudeau S, Bolkan SS, Kellendonk C. The mediodorsal thalamus: an essential partner of the prefrontal cortex for cognition. Biol Psychiatry. 2018;83:648–56.
Dorph-Petersen KA, Lewis DA. Postmortem structural studies of the thalamus in schizophrenia. Schizophr Res. 2017;180:28–35.
Schmitt LI, Wimmer RD, Nakajima M, Happ M, Mofakham S, Halassa MM. Thalamic amplification of cortical connectivity sustains attentional control. Nature 2017;545:219–23.
Woodward ND, Heckers S. Mapping thalamocortical functional connectivity in chronic and early stages of psychotic disorders. Biol Psychiatry. 2016;79:1016–25.
Andrews J, Wang L, Csernansky JG, Gado MH, Barch DM. Abnormalities of thalamic activation and cognition in schizophrenia. Am J Psychiatry. 2006;163:463–9.
Iglesias JE, Insausti R, Lerma-Usabiaga G, Bocchetta M, Van Leemput K, Greve DN, et al. A probabilistic atlas of the human thalamic nuclei combining ex vivo MRI and histology. Neuroimage. 2018;183:314–26.
McCutcheon RA, Abi-Dargham A, Howes OD. Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci. 2019;42:205–20.
Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–9.
Levitt JJ, Nestor PG, Kubicki M, Lyall AE, Zhang F, Riklin-Raviv T, et al. Miswiring of frontostriatal projections in schizophrenia. Schizophr Bull. 2020;46:990–98.
Sarpal DK, Argyelan M, Robinson DG, Szeszko PR, Karlsgodt KH, John M, et al. Baseline striatal functional connectivity as a predictor of response to antipsychotic drug treatment. Am J Psychiatry. 2016;173:69–77.
Sarpal DK, Robinson DG, Lencz T, Argyelan M, Ikuta T, Karlsgodt K, et al. Antipsychotic treatment and functional connectivity of the striatum in first-episode schizophrenia. JAMA Psychiatry. 2015;72:5–13.
Horga G, Cassidy CM, Xu X, Moore H, Slifstein M, Van Snellenberg JX, et al. Dopamine-related disruption of functional topography of striatal connections in unmedicated patients with schizophrenia. JAMA Psychiatry. 2016;73:862–70.
Karcher NR, Rogers BP, Woodward ND. Functional connectivity of the striatum in schizophrenia and psychotic bipolar disorder. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019;4:956–65.
Kim IH, Rossi MA, Aryal DK, Racz B, Kim N, Uezu A, et al. Spine pruning drives antipsychotic-sensitive locomotion via circuit control of striatal dopamine. Nat Neurosci. 2015;18:883–91.
Quide Y, Morris RW, Shepherd AM, Rowland JE, Green MJ. Task-related fronto-striatal functional connectivity during working memory performance in schizophrenia. Schizophr Res. 2013;150:468–75.
Yoon JH, Minzenberg MJ, Raouf S, D’Esposito M, Carter CS. Impaired prefrontal-basal ganglia functional connectivity and substantia nigra hyperactivity in schizophrenia. Biol Psychiatry. 2013;74:122–9.
Geiger LS, Moessnang C, Schafer A, Zang Z, Zangl M, Cao H, et al. Novelty modulates human striatal activation and prefrontal-striatal effective connectivity during working memory encoding. Brain Struct Funct. 2018;223:3121–32.
van Erp TGM, Walton E, Hibar DP, Schmaal L, Jiang W, Glahn DC, et al. Cortical brain abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the Enhancing Neuro Imaging Genetics Through Meta Analysis (ENIGMA) Consortium. Biol Psychiatry. 2018;84:644–54.
Moser DA, Doucet GE, Lee WH, Rasgon A, Krinsky H, Leibu E, et al. Multivariate associations among behavioral, clinical, and multimodal imaging phenotypes in patients with psychosis. JAMA Psychiatry. 2018;75:386–95.
Crespo-Facorro B, Kim J, Andreasen NC, O’Leary DS, Magnotta V. Regional frontal abnormalities in schizophrenia: a quantitative gray matter volume and cortical surface size study. Biol Psychiatry. 2000;48:110–9.
Venkatasubramanian G, Jayakumar PN, Gangadhar BN, Keshavan MS. Automated MRI parcellation study of regional volume and thickness of prefrontal cortex (PFC) in antipsychotic-naive schizophrenia. Acta Psychiatr Scand. 2008;117:420–31.
Jung WH, Kim JS, Jang JH, Choi JS, Jung MH, Park JY, et al. Cortical thickness reduction in individuals at ultra-high-risk for psychosis. Schizophr Bull. 2011;37:839–49.
Kani AS, Shinn AK, Lewandowski KE, Ongur D. Converging effects of diverse treatment modalities on frontal cortex in schizophrenia: a review of longitudinal functional magnetic resonance imaging studies. J Psychiatr Res. 2017;84:256–76.
Ichikawa J, Ishii H, Bonaccorso S, Fowler WL, O’Laughlin IA, Meltzer HY. 5-HT(2A) and D(2) receptor blockade increases cortical DA release via 5-HT(1A) receptor activation: a possible mechanism of atypical antipsychotic-induced cortical dopamine release. J Neurochem. 2001;76:1521–31.
Dorph-Petersen KA, Pierri JN, Perel JM, Sun Z, Sampson AR, Lewis DA. The influence of chronic exposure to antipsychotic medications on brain size before and after tissue fixation: a comparison of haloperidol and olanzapine in macaque monkeys. Neuropsychopharmacology. 2005;30:1649–61.
Lewis DA, Cho RY, Carter CS, Eklund K, Forster S, Kelly MA, et al. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am J Psychiatry. 2008;165:1585–93.
Buchanan RW, Keefe RS, Lieberman JA, Barch DM, Csernansky JG, Goff DC, et al. A randomized clinical trial of MK-0777 for the treatment of cognitive impairments in people with schizophrenia. Biol Psychiatry. 2011;69:442–9.
Lewis DA. Pharmacological enhancement of cognition in individuals with schizophrenia. Biol Psychiatry. 2011;69:397–8.
Chang CH, Lane HY, Tseng PT, Chen SJ, Liu CY, Lin CH. Effect of N-methyl-D-aspartate-receptor-enhancing agents on cognition in patients with schizophrenia: a systematic review and meta-analysis of double-blind randomised controlled trials. J Psychopharmacol. 2019;33:436–48.
Blanco-Ayala T, Sathyasaikumar KV, Uys JD, Perez-de-la-Cruz V, Pidugu LS, Schwarcz R. N-acetylcysteine inhibits kynurenine aminotransferase II. Neuroscience 2020;444:160–69.
Blanco Ayala TB, Ramirez Ortega DR, Ovalle Rodriguez PO, Pineda B, Perez de la Cruz GP, Gonzalez Esquivel DG, et al. Subchronic N-acetylcysteine treatment decreases brain kynurenic acid levels and improves cognitive performance in mice. Antioxidants (Basel). 2021;10:147. https://doi.org/10.3390/antiox10020147.
Cannon TD. How schizophrenia develops: cognitive and brain mechanisms underlying onset of psychosis. Trends Cogn Sci. 2015;19:744–56.
Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci. 2019;22:374–85.
Cho M, Lee TY, Kwak YB, Yoon YB, Kim M, Kwon JS. Adjunctive use of anti-inflammatory drugs for schizophrenia: a meta-analytic investigation of randomized controlled trials. Aust N. Z J Psychiatry. 2019;53:742–59.
Deakin B, Suckling J, Barnes TRE, Byrne K, Chaudhry IB, Dazzan P, et al. The benefit of minocycline on negative symptoms of schizophrenia in patients with recent-onset psychosis (BeneMin): a randomised, double-blind, placebo-controlled trial. Lancet Psychiatry. 2018;5:885–94.
Schoonover KE, Dienel SJ, Lewis DA. Prefrontal cortical alterations of glutamate and GABA neurotransmission in schizophrenia: insights for rational biomarker development. Biomark. Neuropsychiatry. 2020;3:100015.
Chase HW, Boudewyn MA, Carter CS, Phillips ML. Transcranial direct current stimulation: a roadmap for research, from mechanism of action to clinical implementation. Mol Psychiatry. 2020;25:397–407.
Filmer HL, Varghese E, Hawkins GE, Mattingley JB, Dux PE. Improvements in attention and decision-making following combined behavioral training and brain stimulation. Cereb Cortex. 2017;27:3675–82.
Santarnecchi E, Brem AK, Levenbaum E, Thompson T, Kadosh RC, Pascual-Leone A. Enhancing cognition using transcranial electrical stimulation. Curr Opin Behav Sci. 2015;4:171–78.
Yu L, Fang X, Chen Y, Wang Y, Wang D, Zhang C. Efficacy of transcranial direct current stimulation in ameliorating negative symptoms and cognitive impairments in schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2020;224:2–10.
Narita Z, Stickley A, DeVylder J, Yokoi Y, Inagawa T, Yamada Y, et al. Effect of multi-session prefrontal transcranial direct current stimulation on cognition in schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2020;216:367–73.
Simonsmeier BA, Grabner RH, Hein J, Krenz U, Schneider M. Electrical brain stimulation (tES) improves learning more than performance: a meta-analysis. Neurosci Biobehav Rev. 2018;84:171–81.
Sciortino D, Pigoni A, Delvecchio G, Maggioni E, Schiena G, Brambilla P. Role of rTMS in the treatment of cognitive impairments in Bipolar Disorder and Schizophrenia: a review of Randomized Controlled Trials. J Affect Disord. 2021;280:148–55. Pt A
Guse B, Falkai P, Gruber O, Whalley H, Gibson L, Hasan A, et al. The effect of long-term high frequency repetitive transcranial magnetic stimulation on working memory in schizophrenia and healthy controls-a randomized placebo-controlled, double-blind fMRI study. Behav Brain Res. 2013;237:300–7.
Aleman A, Enriquez-Geppert S, Knegtering H, Dlabac-de Lange JJ. Moderate effects of noninvasive brain stimulation of the frontal cortex for improving negative symptoms in schizophrenia: meta-analysis of controlled trials. Neurosci Biobehav Rev. 2018;89:111–18.
Keshavan MS, Vinogradov S, Rumsey J, Sherrill J, Wagner A. Cognitive training in mental disorders: update and future directions. Am J Psychiatry. 2014;171:510–22.
Subramaniam K, Luks TL, Garrett C, Chung C, Fisher M, Nagarajan S, et al. Intensive cognitive training in schizophrenia enhances working memory and associated prefrontal cortical efficiency in a manner that drives long-term functional gains. Neuroimage. 2014;99:281–92.
Mothersill D, Donohoe G. Neural effects of cognitive training in schizophrenia: a systematic review and activation likelihood estimation meta-analysis. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019;4:688–96.
Gonzalez-Burgos G, Lewis DA. GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull. 2008;34:944–61.
Williams SM, Goldman-Rakic PS. Widespread origin of the primate mesofrontal dopamine system. Cereb Cortex. 1998;8:321–45.
Lewis DA, Campbell MJ, Foote SL, Goldstein M, Morrison JH. The distribution of tyrosine hydroxylase-immunoreactive fibers in primate neocortex is widespread but regionally specific. J Neurosci. 1987;7:279–90.
Lewis DA, Melchitzky DS, Sesack SR, Whitehead RE, Auh S, Sampson A. Dopamine transporter immunoreactivity in monkey cerebral cortex: regional, laminar, and ultrastructural localization. J Comp Neurol. 2001;432:119–36.
Lidow MS, Goldman-Rakic PS, Gallager DW, Rakic P. Distribution of dopaminergic receptors in the primate cerebral cortex: quantitative autoradiographic analysis using [3H]raclopride, [3H]spiperone and [3H]SCH23390. Neuroscience. 1991;40:657–71.
Goldman-Rakic PS, Leranth C, Williams SM, Mons N, Geffard M. Dopamine synaptic complex with pyramidal neurons in primate cerebral cortex. Proc Natl Acad Sci USA. 1989;86:9015–9.
Bordelon-Glausier JR, Khan ZU, Muly EC. Quantification of D1 and D5 dopamine receptor localization in layers I, III, and V of Macaca mulatta prefrontal cortical area 9: coexpression in dendritic spines and axon terminals. J Comp Neurol. 2008;508:893–905.
Wang M, Datta D, Enwright J, Galvin V, Yang ST, Paspalas C, et al. A novel dopamine D1 receptor agonist excites delay-dependent working memory-related neuronal firing in primate dorsolateral prefrontal cortex. Neuropharmacology. 2019;150:46–58.
Gamo NJ, Lur G, Higley MJ, Wang M, Paspalas CD, Vijayraghavan S, et al. Stress impairs prefrontal cortical function via D1 dopamine receptor interactions with hyperpolarization-activated cyclic nucleotide-gated channels. Biol Psychiatry. 2015;78:860–70.
Cools R, D’Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry. 2011;69:e113–25.
Arnsten AF, Cai JX, Murphy BL, Goldman-Rakic PS. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology. 1994;116:143–51.
Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22:3708–19.
Abi-Dargham A. Recent evidence for dopamine abnormalities in schizophrenia. Eur Psychiatry. 2002;17:341s–47s. Suppl 4
Akil M, Edgar CL, Pierri JN, Casali S, Lewis DA. Decreased density of tyrosine hydroxylase-immunoreactive axons in the entorhinal cortex of schizophrenic subjects. Biol Psychiatry. 2000;47:361–70.
Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III-the final common pathway. Schizophr Bull. 2009;35:549–62.
Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psychiatry. 2002;51:775–87.
Arnsten AF, Girgis RR, Gray DL, Mailman RB. Novel dopamine therapeutics for cognitive deficits in schizophrenia. Biol Psychiatry. 2017;81:67–77.
Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry. 2015;2:258–70.
Muller N. Inflammation in schizophrenia: pathogenetic aspects and therapeutic considerations. Schizophr Bull. 2018;44:973–82.
Mokhtari R, Lachman HM. The major histocompatibility complex (MHC) in schizophrenia: a review. J Clin Cell Immunol. 2016;7:479.
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83.
Fond G, Lancon C, Korchia T, Auquier P, Boyer L. The role of inflammation in the treatment of schizophrenia. Front Psychiatry. 2020;11:160.
Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. 2013;18:206–14.
Siegel BI, Sengupta EJ, Edelson JR, Lewis DA, Volk DW. Elevated viral restriction factor levels in cortical blood vessels in schizophrenia. Biol Psychiatry. 2014;76:160–7.
Volk DW, Chitrapu A, Edelson JR, Roman KM, Moroco AE, Lewis DA. Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry. 2015;172:1112–21.
Volk DW, Moroco AE, Roman KM, Edelson JR, Lewis DA. The role of the nuclear factor-kappaB transcriptional complex in cortical immune activation in schizophrenia. Biol Psychiatry. 2019;85:25–34.
Plaven-Sigray P, Cervenka S. Meta-analytic studies of the glial cell marker TSPO in psychosis - a question of apples and pears? A commentary on ‘Neuroinflammation in schizophrenia: metaanalysis of in-vivo microglial imaging’ by Marques et al. - ERRATUM. Psychol Med. 2019;49:1583–84.
Kenk M, Selvanathan T, Rao N, Suridjan I, Rusjan P, Remington G, et al. Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr Bull. 2015;41:85–93.
Hafizi S, Tseng HH, Rao N, Selvanathan T, Kenk M, Bazinet RP, et al. Imaging microglial activation in untreated first-episode psychosis: a PET study with [(18)F]FEPPA. Am J Psychiatry. 2017;174:118–24.
Volk DW. Role of microglia disturbances and immune-related marker abnormalities in cortical circuitry dysfunction in schizophrenia. Neurobiol Dis. 2017;99:58–65.
Winter C, Djodari-Irani A, Sohr R, Morgenstern R, Feldon J, Juckel G, et al. Prenatal immune activation leads to multiple changes in basal neurotransmitter levels in the adult brain: implications for brain disorders of neurodevelopmental origin such as schizophrenia. Int J Neuropsychopharmacol. 2009;12:513–24.
Bauman MD, Lesh TA, Rowland DJ, Schumann CM, Smucny J, Kukis DL, et al. Preliminary evidence of increased striatal dopamine in a nonhuman primate model of maternal immune activation. Transl Psychiatry. 2019;9:135.
Meisenzahl EM, Rujescu D, Kirner A, Giegling I, Kathmann N, Leinsinger G, et al. Association of an interleukin-1beta genetic polymorphism with altered brain structure in patients with schizophrenia. Am J Psychiatry. 2001;158:1316–9.
Ellman LM, Deicken RF, Vinogradov S, Kremen WS, Poole JH, Kern DM, et al. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr Res. 2010;121:46–54.
Lesh TA, Careaga M, Rose DR, McAllister AK, Van de Water J, Carter CS, et al. Cytokine alterations in first-episode schizophrenia and bipolar disorder: relationships to brain structure and symptoms. J Neuroinflammation. 2018;15:165.
Keller WR, Kum LM, Wehring HJ, Koola MM, Buchanan RW, Kelly DL. A review of anti-inflammatory agents for symptoms of schizophrenia. J Psychopharmacol. 2013;27:337–42.
Bulzacka E, Boyer L, Schurhoff F, Godin O, Berna F, Brunel L, et al. Chronic peripheral inflammation is associated with cognitive impairment in schizophrenia: results from the multicentric FACE-SZ dataset. Schizophr Bull. 2016;42:1290–302.
Hope S, Hoseth E, Dieset I, Morch RH, Aas M, Aukrust P, et al. Inflammatory markers are associated with general cognitive abilities in schizophrenia and bipolar disorder patients and healthy controls. Schizophr Res. 2015;165:188–94.
Lizano P, Lutz O, Ling G, Lee AM, Eum S, Bishop JR, et al. Association of choroid plexus enlargement with cognitive, inflammatory, and structural phenotypes across the psychosis spectrum. Am J Psychiatry. 2019;176:564–72.
Prasad KM, Chowdari KV, D’Aiuto LA, Iyengar S, Stanley JA, Nimgaonkar VL. Neuropil contraction in relation to Complement C4 gene copy numbers in independent cohorts of adolescent-onset and young adult-onset schizophrenia patients-a pilot study. Transl Psychiatry. 2018;8:134.
Yilmaz M, Yalcin E, Presumey J, Aw E, Ma M, Whelan CW, et al. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat Neurosci. 2021;24:214–24.
Keshavan MS, Anderson S, Pettergrew JW. Is Schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J Psychiatr Res. 1994;28:239–65.
Reichenberg A, Caspi A, Harrington H, Houts R, Keefe RS, Murray RM, et al. Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am J Psychiatry. 2010;167:160–9.
Author information
Authors and Affiliations
Contributions
Authors JS, SJD, DAL, and CSC conceptualized and wrote the paper.
Corresponding authors
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Smucny, J., Dienel, S.J., Lewis, D.A. et al. Mechanisms underlying dorsolateral prefrontal cortex contributions to cognitive dysfunction in schizophrenia. Neuropsychopharmacol. 47, 292–308 (2022). https://doi.org/10.1038/s41386-021-01089-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01089-0
This article is cited by
-
Activation of prefrontal parvalbumin interneurons ameliorates working memory deficit even under clinically comparable antipsychotic treatment in a mouse model of schizophrenia
Neuropsychopharmacology (2024)
-
Modulators of GABAA receptor-mediated inhibition in the treatment of neuropsychiatric disorders: past, present, and future
Neuropsychopharmacology (2024)
-
Longitudinal changes in neural gain and its relationship to cognitive control trajectory in young adults with early psychosis
Translational Psychiatry (2023)
-
Abnormal Structural Network Communication Reflects Cognitive Deficits in Schizophrenia
Brain Topography (2023)
-
Transcranial alternating current stimulation in affecting cognitive impairment in psychiatric disorders: a review
European Archives of Psychiatry and Clinical Neuroscience (2023)
