
Abstract
It has been proposed that binge eating reflects a pathological compulsion driven by the “addictive” properties of foods. Proponents of this argument highlight the large degree of phenomenological and diagnostic overlap between binge eating disorder (BED) and substance use disorders (SUDs), including loss of control over how much is consumed and repeated unsuccessful attempts to abstain from consumption, as well as commonalities in brain structures involved in food and drug craving. To date, very little attention has been given to an additional behavioral symptom that BED shares with SUDs—sleep dysregulation—and the extent to which this may contribute to the pathophysiology of BED. Here, we review studies examining sleep outcomes in patients with BED, which collectively point to a heightened incidence of sleep abnormalities in BED. We identify the orexin (hypocretin) system as a potential neurobiological link between compulsive eating and sleep dysregulation in BED, and provide a comprehensive update on the evidence linking this system to these processes. Finally, drawing on evidence from the SUD literature indicating that the orexin system exhibits significant plasticity in response to drugs of abuse, we hypothesize that chronic palatable food consumption likewise increases orexin system activity, resulting in dysregulated sleep/wake patterns. Poor sleep, in turn, is predicted to exacerbate binge eating, contributing to a cycle of uncontrolled food consumption. By extension, we suggest that pharmacotherapies normalizing orexin signaling, which are currently being trialed for the treatment of SUDs, might also have utility in the clinical management of BED.
Similar content being viewed by others

Introduction
Binge eating disorder (BED) is characterized by repeated episodes of excessive pathological, non-homeostatic food consumption [1]. These binge episodes occur in repeated, discrete time periods, and are accompanied by a perceived loss of control over how much is consumed. BED is highly comorbid with obesity—an estimated 5–15% of obese people have BED, and individuals with BED are 3–6 times more likely to be obese than individuals without an eating disorder [2]. Psychological treatments such as cognitive behavioral therapy and interpersonal therapy are generally considered the first line of treatment for BED [3, 4] although the efficacy of these approaches is limited [5]. Presently, only one medication, lisdexamfetamine (LDX)—a d-amphetamine prodrug—has gained regulatory approval specifically for the treatment of moderate to severe BED in adults [6]. Although daily LDX administration is highly effective at reducing binge frequency, only ~40% of patients report complete binge cessation at 4 weeks, leaving the majority of BED patients with ongoing symptomology [7,8,9], underscoring the need to better understand the phenomenology of BED and associated neurobiological mechanisms.
Recently, there has been significant attention given to the possibility that compulsive overeating, including but not limited to that observed in BED, might reflect the “addictive” properties of foods, particularly those high in sugar and fat content. Proponents of this terminology highlight similarities between the phenomenology of BED and the diagnostic characterization of substance use disorders (SUDs) as outlined in the Diagnostic and Statistical Manual of Mental Disorders (DSM), including frequent and intense cravings (often elicited by exposure to food/drug cues), the consumption of larger amounts than intended, repeated unsuccessful attempts to reduce intake, and continued consumption despite negative consequences [1, 10,11,12,13,14,15]. Moreover, common neural circuits have been demonstrated to underlie food- and drug-seeking and craving [16,17,18,19,20,21], and there is a high (25%) lifetime incidence of SUDs in individuals with BED [22, 23]. In an effort to operationalize food addiction and “addictive eating,” the Yale Food Addiction Scale (YFAS) was developed in 2009, which adapts DSM-IV criteria for substance dependence to eating behavior [10, 24]. Per the YFAS, the majority (57%) of obese BED patients meet the criteria for “food addiction” [25], and one meta-analysis using data from 196,211 predominantly female, overweight/obese individuals indicates that approximately 20% of individuals report experiencing symptoms that align with addictive eating, with an increased rate of addictive eating among those with a current eating disorder diagnosis [10]. These findings have elicited robust discussion regarding whether “food addiction” should be recognized in the DSM as a diagnosis distinct from other eating and SUDs. Notably, several groups oppose the concept of food addiction because of its supposed underdevelopment and oversimplification of complex behavioral phenomena [26, 27], as reviewed elsewhere [13].
By virtue of the YFAS being developed to align with the DSM-IV definition of substance dependence, almost all of the discussion around BED and “food addiction” has focused on the phenomenological parallels with the core diagnostic criteria of SUDs [28]. Consequently, limited attention has been given to examining additional, non-diagnostic, behavioral symptoms of SUDs that may be shared with BED and other forms of compulsive eating, and the extent to which these symptoms may contribute to disease manifestation and severity. Notable in this regard are the strikingly high rates of sleep dysregulation observed in SUDs [29, 30], and the demonstrable link of sleep abnormalities with the initiation, escalation, and relapse of drug use [31, 32]. Moreover, increased sleep quality is associated with reduced drug craving, and total sleep time increases with extended abstinence [33, 34]. Here we examine the available clinical evidence linking sleep dysregulation to the pathophysiology of BED and forms of “addicted” eating, and highlight a paucity of preclinical studies examining the neural mechanisms underlying these phenomena. To this end, we explore the hypothalamic orexin (hypocretin) system as a potential neurobiological link by comprehensively reviewing the separate bodies of literature connecting this system to feeding and sleep/wake processes. Finally, we propose a novel and testable mechanistic framework for sleep dysregulation in BED, whereby binge eating is associated with orexin system plasticity and subsequent hyperactivity. This mechanistic framework for understanding “addicted” eating raises the possibility that orexin-based pharmacotherapies, proven to be highly effective at treating addiction-related outcomes for drugs of abuse [35], may also have some utility as therapeutics for BED due to their ability to concomitantly reduce food craving and normalize sleep.
Sleep dysregulation in binge eating disorder and its relation to night eating syndrome—evidence from clinical populations
“Sleep disorders” is a broad term describing a range of conditions associated with the disturbance of normal sleeping patterns. The International Classification of Sleep Disorders (ICSD) provides a comprehensive classification system of sleep disorders, categorizing disorders into seven diagnostic sections: insomnia, sleep-related breathing disorders, sleep-related movement disorders, central disorders of hypersomnolence, circadian rhythm sleep–wake disorders, parasomnias, and other sleep disorders [36, 37]. The revised edition of the ICSD also includes a separate International Classification of Diseases (ICD) code for substance-induced sleep disorders in its appendices, which reflects clinical evidence indicating that drugs of abuse severely disrupt sleep [38, 39] and that poor sleep is a risk factor for drug taking [40,41,42].
There is a dearth of literature examining whether sleep might also be dysregulated by, or exacerbate the severity of, binge eating. The few studies that have explored this link report a positive association between the phenomena (summarized in Table 1). Briefly, persons with BED are almost three times more likely to report clinically significant insomnia symptoms [43], and consistently score worse on insomnia-related items across several standardized questionnaires that measure sleep quality, compared to controls with no history of an eating disorder [44,45,46,47,48,49]. One study reported that more severe insomnia symptoms predicted more frequent binge eating episodes over the preceding 3 months [43], potentially indicating a causal interaction, although this has not been explored directly. Importantly, these effects do not appear to be mediated by obesity, which is highly comorbid with BED (approximately 36.2% of individuals with BED report lifetime obesity [50]) and is associated with a higher incidence of sleep disorders irrespective of binge eating status [51]. Indeed, significant associations between binge eating and self-reported sleep problems persist when data are adjusted for obesity status [45], and studies that have directly compared patients with obesity and BED, and obese controls with no eating disorders, report an increased incidence and severity of insomnia symptoms in the former population [45, 47]. Notably, one study also reported a higher number of dysregulated sleep symptoms in individuals meeting the criteria for food addiction on the YFAS (independent of BED and obesity) compared to non-food-addicted individuals [52]. We note that, to date, no study has verified self-reported sleep outcomes in patients diagnosed with BED or “food addicted” individuals using polysomnography, an objective measure of sleep that collects several indices (e.g., heart and breathing rate, eye movement, brain dynamics of electroencephalography, etc.) to determine sleep stage/wakefulness status [53]. Moreover, several studies are limited by the small subgroups identified as binge eaters, as well as the lack of a normal weight binge eating comparison group (see Table 1). Nevertheless, these data collectively indicate that compulsive overeating is associated with poor sleep outcomes.
Significant attention has been paid to sleep dysregulation in night eating syndrome (NES), an eating disorder characterized by a circadian delay in food intake and excessive intake (at least 25% of daily caloric intake) after the evening meal and into the night [54,55,56,57,58]. NES is closely tied to sleep disturbances, after being defined by Albert Stunkard in 1955 as a “distinctive syndrome characterized by nocturnal hyperphagia, insomnia and morning anorexia” [59]. BED, described by Stunkard 4 years later as a separate disorder marked by “enormous amounts of food may be consumed in relatively short periods” [60] has a substantial overlap in symptomology with NES [46, 57] and, like BED, NES is associated with a reduction in sleep efficacy and increased sleep disturbance [55, 61]. However, the etiology of NES is different from that of BED—eating behavior in NES is associated with nocturnal anxiety, under-eating relative to unaffected individuals throughout the day, and beliefs about not being able to sleep, or return to sleep, without eating, which is not typically the case with binge eating behavior observed in BED [57, 62]. Furthermore, BED is marked by more frequent, objectively large, eating episodes whereby there is a loss of control over how much is eaten. People with NES, on the other hand, rarely report a loss of control, suggesting that these individuals do not experience true “night binges” [63]. Thus, BED and NES are likely two distinct syndromes mediated, at least in part, by different brain circuits.
What can preclinical models of binge/compulsive eating tell us about sleep dysregulation in BED?
Several rodent models of BED and “addictive” eating effectively recapitulate psychopathological markers of BED, including hyperphagia and the development of food anticipatory activity [17, 18, 64,65,66,67,68,69]. Briefly, binge-like behavioral patterns are often developed through repeated limited access to palatable foods, including sucrose solutions, fat/sucrose mixtures, shortening, and fat presented as solid emulsions [70, 71]. Although periods of food restriction or fasting can be incorporated to mimic dieting patterns often observed in BED (and BN/AN) [66, 72, 73], several models successfully replicate the escalation of intake observed in clinical populations without a food-restriction component, eliminating hunger as a potential confounding variable [74, 75]. Environmental stressors, including noise and overcrowding, further enhance BED-like symptomology [76]. For example, restricted access to food, when combined with a foot shock stressor just prior to palatable food access, induces strong hyperphagia mimicking stress-induced overeating [77]. Although studies have focused primarily on food intake during these discrete binge-like eating sessions, others have examined the expression of “addiction-like” behaviors as a result of binge experience. For example, rats trained to respond for palatable foods on an operant task in brief (1 h) daily sessions exhibit higher motivation for food on a progressive ratio task and continue to consume palatable food in an aversive environment [68, 78]. Interestingly, similar insensitivity to aversive stimuli can also be induced by maintaining rats on palatable high-fat diet ad libitum [18].
Despite the developments in animal models of binge-like eating, remarkably little has been done to examine how a history of binge-like eating disrupts the well-defined neural circuits and mechanisms that mediate sleep and arousal [79,80,81]. To our knowledge, no published works have examined the extent to which sleep disruptions are observed at a phenomenological level in an animal model of BED, let alone the mechanistic underpinnings. This stands in stark contrast to the extensive body of preclinical studies indicating that both acute and chronic drug exposure is associated with significant sleep dysregulation that persists into withdrawal [82, 83]. Some insight can be gleaned, however, from the well-established food entrainment literature, whereby food availability is restricted to a single period scheduled at a fixed time of day, resulting in a temporal decoupling of activity in feeding-related brain regions from the “master clock”, the suprachiasmatic nucleus (SCN), and a subsequent increase in activity aligned with food delivery rather than diurnal light patterns [84]. Although many food entrainment studies restrict access to regular chow [85, 86], a set of studies have reported that intermittent access to palatable foods in otherwise sated animals induces behavioral entrainment, indicating the presence of an endogenous self-sustained neural oscillator entrained specifically by palatable food [87,88,89,90,91,92]. These data are particularly noteworthy given the greater probability of individuals to binge eat in later hours of the day indicated in clinical literature (although see [58]). Early functional analysis studies on binge eating found an increased likelihood of binge eating from 5 p.m. to midnight in all people regardless of binge eating status, and a decreased probability of binge eating in the morning hours from 5 a.m. to 9 a.m. [93]. More recent ecological momentary assessment (EMA) studies, which examine self-reported behavior across several time points throughout the day, indicate heightened binge eating probability at around 1 p.m. and 7–9 p.m. in women with bulimia nervosa [94], and between 6 p.m. and 1 a.m. among women with BED [95]. It is unclear whether these patterns of intake result in behavioral entrainment per se. However, it is possible that regular, timed consumption of palatable food might recruit neural oscillators that drive the search of and craving for palatable food and disrupt normal circadian rhythms [89, 96, 97], thus contributing to uncontrolled food intake.
There is some evidence to indicate that sleep dysregulation itself may contribute altered feeding behavior, possibly via a reduction in “top-down” executive control and increased impulsivity. Corticostriatal projections, which mediate inhibitory control of motivated behavior [98, 99], are selectively weakened in mice following acute (6 h) sleep deprivation [100]. These changes are accompanied by increased sucrose consumption in both an operant and free access task, which is reversed by optogenetic stimulation of corticostriatal projections. Similar effects are observed for drugs of abuse [101], which aligns well with clinical data pointing to increased drug and alcohol use following the emergence of sleep dysregulation in childhood and adolescence [102, 103] and sleep disturbances being a strong risk factor of relapse following protracted abstinence [104]. There is also evidence that drug abuse is associated with a range of cognitive deficits resulting from sleep deprivation [105,106,107], which may compromise attempts to remain abstinent. Although it remains to be tested how sleep deprivation might affect binge eating behavior, a bidirectional relationship likely exists between food intake and sleep dysregulation in BED, whereby dysregulated sleep patterns result in heightened impulsivity and binge-eating behaviors, and binge eating itself produces disruptions to the molecular mechanisms that control sleep.
The hypothalamic orexin system—a common mediator of feeding and sleep
Significant advances have been made to characterize the hypothalamic cell types and circuits involved in reward-seeking and sleep. Perhaps most notable in this regard is the identification in 1998 of the hypothalamic orexin (hypocretin) system, composed of a pair of neuropeptides, orexin-A and orexin-B, synthesized by a relatively small (~4000 in rat, ~70,000 in humans) population of neurons in the caudal hypothalamus [108,109,110,111]. Orexins act on two G-protein coupled receptors, orexin receptor-1 and -2 (Ox1R, Ox2R), with orexin-A having equal affinity for Ox1R and Ox2R, and orexin-B preferentially binding both receptors [108, 109]. The orexin system has manifold roles, simultaneously mediating reward behavior and sleep/wake processes. This dichotomy of function is achieved via broad projections throughout the brain, and to some extent via actions at Ox1R versus Ox2R. For example, orexin acts at Ox1R to modulate the activity of dopamine neurons and promote seeking of food and drug rewards [112,113,114], whereas orexin’s actions at Ox2R in key arousal centers, including tuberomammillary nucleus, appears to underlie its involvement in sleep/wake processes [115,116,117]. Thus, the orexin system is uniquely positioned to serve as a potential common neurobiological substrate underlying aberrant food seeking and sleep dysregulation in BED and other disorders characterized by uncontrolled eating.
Orexin and palatable food intake
A role for orexin in feeding was first identified by Sakurai et al. [108], who demonstrated that intracerebroventricular (i.c.v.) infusions of both orexin-A and orexin-B dose-dependently stimulated chow consumption in freely fed rats. Subsequent studies extended these findings to show that the orexin system acts as a critical link between the peripheral energy balance systems and central nervous system mechanisms that regulate homeostatic feeding behavior, particularly following periods of fasting [118]. Similar to other systems that evolved to promote feeding, the orexin system is readily recruited by highly salient rewards and related stimuli, including palatable foods and drugs of abuse [119]. Indeed, orexin neurons show activation in response to stimuli that predict the availability of food [120] or drugs of abuse [121,122,123]. Moreover, exogenous application of orexin peptides increases both consumption and willingness to work for palatable foods and drugs of abuse; i.c.v. infusions of orexin-A in sated rats increase breakpoint responding for sucrose pellets on a progressive ratio task [124] and local infusions of orexin-A into various reward regions [125,126,127] stimulate binge-like intake of a range of high-sugar and high-fat foods. Similarly, orexin-A peptide infusions increase motivated responding for cocaine [128] and are highly effective at eliciting reinstatement of extinguished drug seeking [129,130,131].
A substantial body of evidence now indicates that orexin mediates reward-seeking and hedonic feeding behavior primarily via actions at Ox1R. Several studies have directly tested the efficacy of Ox1R antagonists in reducing binge-like eating in models of BED. Both selective orexin-1 receptor antagonists (SORA-1s) and dual Ox1R/Ox2R antagonists (DORAs), but not selective Ox2R antagonists (SORA-2s), reduce binge-like eating, often at doses that do not affect homeostatic feeding [132,133,134,135]. Similarly, in low-effort operant schedules of reinforcement, systemic injections of the SORA-1 SB334867 (SB) reduce responding for a variety of palatable foods [136,137,138,139], whereas similar effects are not observed with SORA-2s [140, 141]. SB also reduces responding for palatable foods on operant tasks that require higher effort [124, 136, 138, 142,143,144] (although see [145,146,147]), and also lowers Q0 values (a measure of low-effort intake) and increases alpha values (an inverse measure of motivated, high-effort responding) across a variety of palatable foods on a behavioral economics task [148]. Furthermore, the efficacy of SORA-1s in reducing non-homeostatic intake is enhanced in animals susceptible to diet-induced obesity, indicating that obesity may increase the salience of food-based rewards and induce a propensity for responding to such rewards [149]. Together, these data highlight the possibility of a therapeutic window whereby low dosing of SORA-1s might selectively impair hedonic feeding while leaving homeostatic consumption intact, particularly in those with comorbid obesity. Notably, SORA-1s are also effective at reducing motivated drug seeking across all drugs of abuse tested, often at doses that do not affect homeostatic feeding [150,151,152,153,154,155,156,157,158,159]. SORA-1s are especially effective at reducing craving in response to drug-associated cues and contexts [114, 150, 160,161,162,163,164,165], and thus these compounds may have some utility in reducing binge eating elicited by environmental stimuli. Some of these effects might be driven by improvements in cognitive control, as compounds that block Ox1R signaling improve some (but not all) forms of impulsive responding for food reward [144, 166, 167], and exogenous application of orexin peptides improve cognitive outcomes in aged rats [168]. Currently however, it is unclear what role, if any, the orexin system plays in mediating impulsivity/cognitive control in the context of binge-like eating.
A role for orexin in obesity
Somewhat counterintuitive to the pro-feeding effects of orexin described above, evidence from both clinical populations and animal studies indicate that under some circumstances, orexin serves to enhance metabolism and confers anti-obesogenic effects. Obese (non-BED) patients have depressed orexin plasma levels compared to lean patients, and low orexin levels in obese (non-BED) patients rapidly return to normal levels following bariatric surgery [169, 170]. Moreover, human narcoleptic patients, who are orexin deficient (see below), have been reported to average a higher body mass index and a higher incidence of metabolic syndrome [171]. Consistent with these data is evidence that genetic overexpression of orexin in rodents is associated with increased energy expenditure and a resistance to diet-induced obesity [172,173,174]. These effects are eliminated in OxR2 knockout mice and can be recapitulated by chronic treatment with an OxR2 agonist [172], and endogenous OxR2 levels are higher in obesity-resistant rats compared to obesity-prone rats [174, 175], highlighting a specific role for OxR2 signaling in promoting energy metabolism. In light of these findings, it is notable that chronic exposure to a high-fat diet, which promotes an obesogenic state in rodents, has been routinely demonstrated to result in enhanced hypothalamic orexin mRNA and cell numbers [176,177,178]. It is possible that this increase reflects a compensatory response designed to promote increased metabolism, or it may reveal a differential role for increased orexin levels specifically in obesogenic states induced by overeating (as in BED) compared to genetically driven metabolic dysfunction (as in overexpression studies and narcolepsy). Indeed, given the role for orexins acting at Ox1R in processing salience for palatable foods (reviewed above), it is conceivable that a history of overeating could promote increased signaling at Ox1R in key reward areas, which might then outweigh any protective metabolic effects of orexin signaling at Ox2R.
The orexin system mediates sleep and wakefulness
A role for orexin in sleep stemmed from the discovery that orexin-B has neuroexcitatory properties [109], which subsequently led to an extensive body of work indicating that orexin peptides promote and stabilize wakefulness via broad projections to central arousal centers [179,180,181,182]. Extracellular orexin-A levels exhibit time-dependent changes across 24-h periods, gradually increasing during active periods and rapidly decreasing during inactive periods [183], and Fos immunoreactivity of orexin cells is increased during periods of wakefulness and decreased during non-rapid eye movement (NREM) and rapid eye movement (REM) sleep [184]. Optogenetic and chemogenetic stimulation of orexin neurons increases the number of transitions from both NREM and REM sleep to wakefulness, and increases c-Fos expression in both orexin cells and downstream regions involved in arousal, and inhibition of orexin neurons has the opposite effect [185,186,187,188]. Perhaps the most compelling evidence linking the orexin system to sleep is the demonstration that a deficiency of orexin-producing cells underlies sleep instability in narcolepsy [189, 190]. Individuals with narcolepsy have abnormally low to undetectable orexin-A levels in cerebrospinal fluid and an 85–95% decrease in orexin neuron cell counts [191, 192]. These findings are supported by preclinical studies, including the demonstration that prepro-orexin knockout mice and mice with ablated orexin neurons exhibit fragmented waking patterns and intrusions of REM sleep during active periods [180, 182].
These arousal-promoting effects of orexin are primarily mediated by Ox2R [115,116,117], and thus this receptor subtype has become a target for pharmacological therapeutics to manage sleep disorders, including insomnia [193,194,195,196,197]. Preclinical studies show that administration of SORA-2s or DORAs, but not SORA-1s, improves several sleep parameters, decreasing the latency for persistent sleep and increasing N-REM and REM time [198]. In 2010, Merck’s DORA (MK-4305/suvorexant; subsequently marketed as BelsomraTM), was developed as a treatment for insomnia [199]; following successful preclinical and clinical studies, it was approved by the FDA in 2013. Suvorexant dose dependently promotes sleep and improves sleep architecture [200]. In humans, suvorexant reduces latency to persistent sleep and promotes sleep maintenance, is generally well-tolerated, and has a strong safety profile [201]. Another DORA, lemborexant (marketed by Eisai as DAYVIGO), was granted FDA approval for the treatment of insomnia in 2019 and has also been demonstrated to be safe for use in patients with mild obstructive sleep apnea [202]. Several other DORAs, as well as SORA-2s, are currently in development, with the view to expanding treatment options for insomnia and related sleep disorders [197, 203].
Does the orexin system offer a mechanistic link between binge eating and sleep dysregulation?
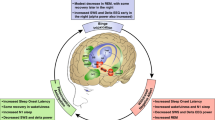
In a 1986 clinical case study describing a patient with nocturnal binge eating behavior, the author speculated that the comorbidity of overeating and dysregulated sleep might be underscored by a common neurobiological substrate—the hypothalamus [204]. Here, with the benefit of several decades of additional research in related fields, we extend this hypothesis to speculate that aberrant hypothalamic orexin system function contributes to the manifestation of both compulsive eating and dysregulated sleep in BED and related disorders. Indeed, we propose that dysregulation of orexin system signaling may contribute to BED pathology in a cyclical manner (Fig. 1), such that binge-eating itself causes orexin system function plasticity, resulting in aberrant sleep patterns that, in turn, promotes further binge eating.

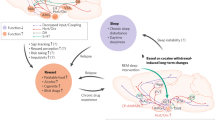
We propose that binge eating results in orexin system plasticity similar to that induced by drugs of abuse, including increased hypothalamic orexin cell numbers, greater orexin input to reward and arousal centers, and greater reactivity to food-related stimuli. This general enhancement of orexin function is associated with the development of a hypermotivated state for food, which in turn increases proclivity to binge eat. Additionally, inappropriately high orexin system function results in hyperarousal and sleep dysregulation, which in turn promotes an increased risk of binge eating due to weakened inhibitory control, as a strategy to combat drowsiness during the daytime, and/or behavioral entrainment. Combined, this creates a self-promoting loop that serves to exacerbate the pathophysiology of BED. Thus, strategies that dampen orexin system signaling, such as orexin receptor antagonists, might be effective at breaking this loop and treating both dysregulated eating and sleep in BED.
Evidence to support this hypothesis comes from a series of recent studies indicating that the number of orexin-producing neurons in hypothalamus is increased in response to repeated exposure to chronic palatable food exposure. For example, increased orexin levels (cell numbers and mRNA) are observed in the hypothalamus of rats and mice exposed to a high-fat diet [176, 177], and this is maintained following a return to a low-fat chow diet [178] (although see [205] and [206]). Similarly, orexin cell numbers and mRNA are increased in monkeys and rats exposed to a high-fat diet during early life [207, 208], further indicating the potential for persistent changes in orexin levels. In addition, antipsychotic medications that are associated with enhanced appetite and weight gain are associated with increased plasma orexin-A levels [209]. Notably, these findings parallel recent demonstrations that rats exposed to several drugs of abuse, including cocaine, alcohol, morphine, and fentanyl, exhibit a robust (~20–25%) upregulation in the number of orexin neurons [122, 152, 210,211,212], which persists well-beyond the cessation of drug use [122]. We predict that chronic, intermittent access to palatable foods (high fat, high sugar or a combination) on a schedule that promotes binge-like eating in rats, would also induce plasticity in the orexin system, resulting in higher orexin gene expression and cell numbers, and potentially increased Ox1R expression in key reward regions, collectively resulting in hyperresponsivity to food-related stimuli. In turn, we predict that binge-induced disruption to the normal orexin levels results in hyperarousal and subsequent sleep dysregulation, which itself would contribute to increased binge-eating behavior via compensatory behaviors (eating to stay awake), reduced inhibitory control, and/or behavioral entrainment. A history of binge eating in conjunction with dieting may also increase the excitability of orexin neurons, as acute food restriction upregulates the number of excitatory glutamatergic inputs onto orexin neurons similar to that seen following cocaine [213,214,215], and the balance of these inputs plays a critical role in regulating the diurnal activity of orexin neurons [216]. Repeated binge-withdrawal cycles might therefore result in hyperarousal and irregular sleep patterns. Significant work is required to test this hypothesis, beginning with experiments to confirm dysregulated sleep in animals with a history of binge-like eating and test whether this is associated with a concomitant increase in orexin levels (central and peripheral). Such correlative studies should be followed by functional studies to show a causal link between these phenomena. For example, orexin system upregulation could be achieved via genetic overexpression [172]; this approach might perturb the diurnal fluctuations of orexin levels in a manner similar to what we propose is observed following chronic binge-like eating, thus affecting both binge eating and sleep outcomes. In contrast, studies focused on ameliorating binge eating might consider reducing orexin signaling at appropriate times of the day/night cycle (i.e. during the inactive period) using approaches that allow for extended periods of cellular inhibition or reduced receptor signaling, including chemogenetics (e.g. [217]) or pharmacology, with a focus on next-day binge outcomes. Similarly, inhibition of orexin neurons using either chemogenetic or pharmacological approaches could be used to test the role of the orexin system in the development versus the expression of these outcomes. Such studies should also consider the circuits through which enhanced orexin signaling might mediate binge eating and/or sleep dysregulation. Orexin neurons project to regions containing monoamine neurotransmitters that exhibit altered signaling in BED, including ventral tegmental area (dopamine) and locus coeruleus (norepinephrine) [218, 219]; it is therefore interesting to consider that binge-induced increases in orexin cell function could result in altered baseline cellular activity or changes in Ox1R vs Ox2R expression in these regions. Moreover, orexin neurons provide excitatory input onto serotoninergic neurons in dorsal raphe nucleus (DRN) [220], and DRN serotonin neurons in turn act to maintain appropriate orexinergic tone by directly inhibiting orexin neuron activity which is critical for the regulation of normal sleep/wake architecture [221, 222]. Thus, effort should be devoted to examining how this orexin–serotonin circuit may be dysregulated in BED and its implications for both sleep and feeding outcomes.
We note that our hypothesis that aberrant eating and sleep in BED are linked with higher orexin activity is seemingly at odds with studies indicating a higher incidence of BED in narcoleptic patients. Indeed, a 1992 case study of a 9-year-old male child with narcolepsy noted, “considerable concern about his voracious appetite with bingeing, which had resulted in excessive weight gain” [223]. Subsequent broader-scale studies have reported that narcolepsy is associated with increased impulsivity and binge-eating behaviors [224,225,226]. A possible explanation for this phenomenon is the use of binge eating as a behavioral intervention to reduce drowsiness and to stave off intrusive sleep episodes [226]. Barson proposes an alternative, developmental stage-based theory, suggesting that postnatal orexin cell loss reduces food intake, while oppositely, adulthood orexin cell loss increases intake [118]. Consistent with this idea, the average age onset of narcolepsy is approximately 24 years [227], and adult orexin cell-knockout mice exhibit an overeating and obesity phenotype, indicating that orexin cell loss in adulthood may lead to binge-like eating and weight gain [228]. In contrast, a second study in which orexin cell loss occurred in mice between weeks 1 and 8 of age reported that cell loss reduced food intake by nearly 30% [182]. Notably however, one study failed to identify heightened rates of binge eating in patients with narcolepsy [229], suggesting such a correlation may be weak, if present at all.
Evidence that orexin signaling is decreased in obese patients and that orexin overexpression confers an obesity-resistant phenotype (discussed above) poses an additional potential challenge to our hypothesis. However, given the dichotomy of roles between OxR1 and OxR2 in mediating food seeking and energy expenditure, respectively, we speculate that binge-induced plasticity to the orexin system results in an imbalance between Ox1R/Ox2R signaling that overall promotes excessive food intake (via Ox1R) which outweighs the metabolic effects of orexin signaling at OxR2. Additionally, orexin’s anti-obesogenic effects are dependent on leptin signaling [172], which is known to decrease in patients with reduced sleep, such as individuals with insomnia [230]. Collectively therefore, we propose that these data do not discount our hypotheses but rather highlight the likely complexity of this relationship when comorbid with other disorders (e.g., diet-induced obesity, metabolic disorder, narcolepsy), which necessitate significant future work to unravel these associations. Relatedly, future studies should also examine the extent to which the orexin system is involved in the delayed circadian rhythm of food intake observed in NES.
Implications for novel pharmacotherapies for BED
The potential involvement of the orexin system as a common mediator of both compulsive eating and sleep dysregulation in BED raises the possibility that more effective clinical management of BED might be achieved via pharmacotherapies designed to reduce orexin signaling. As outlined above, preclinical studies strongly point towards the potential efficacy of SORA-1s to manage food craving and intake in BED. Although there are currently several SORA-1s in the drug development pipeline, no such compounds are yet approved for human use. However, we suggest that a strong rationale exists for the repurposing of already approved DORAs for use in BED patient populations. In addition to reducing food-seeking via actions at Ox1R, these compounds may have the additional benefit of indirectly improving binge eating by normalizing sleep outcomes in BED patients via actions at Ox2R. Indeed, animal studies indicate efficacy of suvorexant and its analogs in reducing drug-seeking and normalizing abstinence-related sleep outcomes [166, 231, 232], and several early-phase clinical studies are currently examining DORA effects in SUD populations [233]. With respect to BED, we are aware of only one study that has reported anti-bingeing effects of a DORA in rats; interestingly, this study also reported that the same DORA had strong hypnotic properties, although bingeing and sleep outcomes were studied in separate groups of rats [132]. Moreover, even though no clinical studies have examined the effect of DORAs on binge eating per se, one report showed efficacy for suvorexant in patients with nighttime eating [234; cited in 235]. Thus, there is a clear need for both preclinical and clinical studies to directly test the efficacy of DORAs on both bingeing and sleep outcomes in BED.
Conclusions
Binge eating and associated disorders, including BED, are characterized by a compulsivity to eat akin to the excessive drive to seek and consume drugs in SUDs, prompting some to propose that these eating disorders reflect an “addiction” to food. Here, we bring together evidence from clinical populations indicating that BED and SUDs share another core, yet non-diagnostic, symptomology: a heightened incidence of sleep disorders, particularly insomnia. Limited evidence from human studies points to an increased incidence of insomnia-related symptoms in BED patients that appears to be independent of any effects of comorbid obesity. Significantly more work is required, both at a clinical and preclinical level, to understand the directionality of the relationship between excessive eating and poor sleep in BED. Regardless, these observations add to a growing body of work supporting the parallels between disordered eating and substance abuse. Animal models of BED, which allow for more invasive interrogation of the neurobiological systems, should be utilized to begin to understand whether excessive eating and dysregulated sleep share common neural substrates, as has been shown in SUD. We propose that a likely candidate system is the hypothalamic orexin system, which is central to the regulation of both hedonic food-seeking and arousal/wakefulness, and shows significant plasticity in response to repeated exposure to salient rewards including palatable food and drugs of abuse. One hypothesis is that chronic binge eating results in hyperactive orexin system function, disrupting the normal diurnal fluctuations in orexin signaling that regulate the sleep/wake cycle. As a consequence, pharmacotherapies that ameliorate orexin signaling might have some therapeutic value in BED; in particular, DORAs might reduce binge eating outcomes by simultaneously reducing food craving via actions at Ox1R and normalizing sleep by reducing signaling at Ox2R. Clearly, significant work is required to test and refine these hypotheses, and it is our hope that this review provides some impetus in this direction.
Funding and disclosure
This work was supported by a Rutgers University Project SUPER award to JBM, an Early Career Fellowship from the National Health and Medical Research Council of Australia (GNT1158276) grant to DM, and a National Institute on Drug Abuse (NIDA) K99/R00 Award (K99 DA045765) to MHJ. The authors declare no competing interests.
References
American Psychiatric A, American Psychiatric A, Force DSMT. Diagnostic and statistical manual of mental disorders: DSM-5. 2013.
McCuen-Wurst C, Ruggieri M, Allison KC. Disordered eating and obesity: associations between binge-eating disorder, night-eating syndrome, and weight-related comorbidities. Ann N Y Acad Sci. 2018;1411:96–105.
Ghaderi A, Odeberg J, Gustafsson S, Råstam M, Brolund A, Pettersson A. et al. Psychological, pharmacological, and combined treatments for binge eating disorder: a systematic review and meta-analysis. PeerJ. 2018;6:e5113
Wilson GT, Wilfley DE, Agras WS, Bryson SW. Psychological treatments of binge eating disorder. Arch Gen Psychiatry. 2010;67:94–101.
Linardon J. Rates of abstinence following psychological or behavioral treatments for binge-eating disorder: meta-analysis. Int J Eat Disord. 2018;51:785–97.
Appolinario JC, Nardi AE, McElroy SL. Investigational drugs for the treatment of binge eating disorder (BED): an update. Expert Opin Invest Drugs. 2019;28:1081–1094.
McElroy SL, Hudson JI, Mitchell JE, Wilfley D, Ferreira-Cornwell MC, Gao J, et al. Efficacy and safety of lisdexamfetamine for treatment of adults with moderate to severe binge-eating disorder: a randomized clinical trial. JAMA Psychiatry. 2015;72:235–46.
McElroy SL, Hudson J, Ferreira-Cornwell MC, Radewonuk J, Whitaker T, Gasior M. Lisdexamfetamine dimesylate for adults with moderate to severe binge eating disorder: results of two pivotal phase 3 randomized controlled trials. Neuropsychopharmacology. 2016;41:1251–60.
Hudson JI, McElroy SL, Ferreira-Cornwell MC, Radewonuk J, Gasior M. Efficacy of lisdexamfetamine in adults with moderate to severe binge-eating disorder: a randomized clinical trial. JAMA Psychiatry. 2017;74:903–10.
Pursey KM, Stanwell P, Gearhardt AN, Collins CE, Burrows TL. The prevalence of food addiction as assessed by the Yale Food Addiction Scale: a systematic review. Nutrients. 2014;6:4552–90.
Innamorati M, Imperatori C, Balsamo M, Tamburello S, Belvederi Murri M, Contardi A, et al. Food Cravings Questionnaire-Trait (FCQ-T) discriminates between obese and overweight patients with and without binge eating tendencies: the Italian version of the FCQ-T. J Pers Assess. 2014;96:632–9.
Schulte EM, Grilo CM, Gearhardt AN. Shared and unique mechanisms underlying binge eating disorder and addictive disorders. Clin Psychol Rev. 2016;44:125–39.
Fletcher PC, Kenny PJ. Food addiction: a valid concept? Neuropsychopharmacology. 2018;43:2506–13.
Meule A, Küppers C, Harms L, Friederich HC, Schmidt U, Blechert J, et al. Food cue-induced craving in individuals with bulimia nervosa and binge-eating disorder. PloS ONE. 2018;13:e0204151.
Wonderlich JA, Breithaupt LE, Crosby RD, Thompson JC, Engel SG, Fischer S. The relation between craving and binge eating: integrating neuroimaging and ecological momentary assessment. Appetite. 2017;117:294–302.
Carlier N, Marshe VS, Cmorejova J, Davis C, Müller DJ. Genetic similarities between compulsive overeating and addiction phenotypes: a case for “food addiction”? Curr Psychiatry Rep. 2015;17:96.
Brown RM, Kupchik YM, Spencer S, Garcia-Keller C, Spanswick DC, Lawrence AJ, et al. Addiction-like synaptic impairments in diet-induced obesity. Biol Psychiatry. 2017;81:797–806.
Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010;13:635–41.
Wiers CE, Zhao J, Manza P, Murani K, Ramirez V, Zehra A, et al. Conscious and unconscious brain responses to food and cocaine cues. Brain Imaging Behav. 2021;15:311–9.
Volkow ND, Wise RA, Baler R. The dopamine motive system: implications for drug and food addiction. Nat Rev Neurosci. 2017;18:741–52.
Volkow ND, Wang GJ, Fowler JS, Tomasi D, Baler R. Food and drug reward: overlapping circuits in human obesity and addiction. Curr Top Behav Neurosci. 2012;11:1–24.
Grilo CM, White MA, Masheb RM. DSM-IV psychiatric disorder comorbidity and its correlates in binge eating disorder. Int J Eat Disord. 2009;42:228–34.
Schreiber LR, Odlaug BL, Grant JE. The overlap between binge eating disorder and substance use disorders: diagnosis and neurobiology. J Behav Addict. 2013;2:191–8.
Gearhardt AN, Corbin WR, Brownell KD. Preliminary validation of the Yale Food Addiction Scale. Appetite 2009;52:430–6.
Gearhardt AN, White MA, Masheb RM, Morgan PT, Crosby RD, Grilo CM. An examination of the food addiction construct in obese patients with binge eating disorder. Int J Eat Disord. 2012;45:657–63.
Hebebrand J, Gearhardt AN. The concept of “food addiction” helps inform the understanding of overeating and obesity: NO. Am J Clin Nutr. 2021;113:268–73.
Finlayson G. Food addiction and obesity: unnecessary medicalization of hedonic overeating. Nat Rev Endocrinol. 2017;13:493–8.
Gearhardt AN, Hebebrand J. The concept of “food addiction” helps inform the understanding of overeating and obesity: YES. Am J Clin Nutr. 2021;113:263–67.
Chakravorty S, Vandrey RG, He S, Stein MD. Sleep management among patients with substance use disorders. Med Clin North Am. 2018;102:733–43.
Roehrs TA, Roth T. Sleep disturbance in substance use disorders. Psychiatr Clin North Am. 2015;38:793–803.
Logan RW, Hasler BP, Forbes EE, Franzen PL, Torregrossa MM, Huang YH, et al. Impact of sleep and circadian rhythms on addiction vulnerability in adolescents. Biol Psychiatry. 2018;83:987–96.
Falcón E, McClung CA. A role for the circadian genes in drug addiction. Neuropharmacology. 2009;56:91–96.
Lydon-Staley DM, Cleveland HH, Huhn AS, Cleveland MJ, Harris J, Stankoski D, et al. Daily sleep quality affects drug craving, partially through indirect associations with positive affect, in patients in treatment for nonmedical use of prescription drugs. Addictive Behav. 2017;65:275–82.
Angarita GA, Canavan SV, Forselius E, Bessette A, Pittman B, Morgan PT. Abstinence-related changes in sleep during treatment for cocaine dependence. Drug Alcohol Depend. 2014;134:343–7.
James MH, Aston-Jones G. Introduction to the Special Issue: “making orexin-based therapies for addiction a reality: what are the steps from here?”. Brain Res. 2020;1731:146665.
American Academy of Sleep M. International Classification of Sleep Disorders—Third Edition (ICSD-3). Diagnostic and coding manual. 2014.
Sateia MJ. International classification of sleep disorders-third edition: highlights and modifications. Chest. 2014;146:1387–94.
Watson R, Bakos L, Compton P, Gawin F. Cocaine use and withdrawal: the effect on sleep and mood. Am J Drug Alcohol Abuse. 1992;18:21–28.
Johanson CE, Roehrs T, Schuh K, Warbasse L. The effects of cocaine on mood and sleep in cocaine-dependent males. Exp Clin Psychopharmacol. 1999;7:338–46.
Young-McCaughan S, Miaskowski C. Measurement of opioid-induced sedation. Pain Manag Nurs. 2001;2:132–49.
Young-McCaughan S, Miaskowski C. Definition of and mechanism for opioid-induced sedation. Pain Manag Nurs. 2001;2:84–97.
Lewis SA, Oswald I, Evans JI, Akindele MO, Tompsett SL. Heroin and human sleep. Electroencephalogr Clin Neurophysiol. 1970;28:374–81.
Kenny TE, Van Wijk M, Singleton C, Carter JC. An examination of the relationship between binge eating disorder and insomnia symptoms. Eur Eat Disord Rev. 2018;26:186–96.
Vardar E, Caliyurt O, Arikan E, Tuglu C. Sleep quality and psychopathological features in obese binge eaters. Stress Health. 2004;20:35–41.
Trace SE, Thornton LM, Runfola CD, Lichtenstein P, Pedersen NL, Bulik CM. Sleep problems are associated with binge eating in women. Int J Eat Disord. 2012;45:695–703.
Yeh SS, Brown RF. Disordered eating partly mediates the relationship between poor sleep quality and high body mass index. Eat Behav. 2014;15:291–7.
Tzischinsky O, Latzer Y. Sleep-wake cycles in obese children with and without binge-eating episodes. J Paediatr Child Health. 2006;42:688–93.
Kim KR, Jung YC, Shin MY, Namkoong K, Kim JK, Lee JH. Sleep disturbance in women with eating disorder: prevalence and clinical characteristics. Psychiatry Res. 2010;176:88–90.
Ulman TF, Von Holle A, Torgersen L, Stoltenberg C, Reichborn-Kjennerud T, Bulik CM. Sleep disturbances and binge eating disorder symptoms during and after pregnancy. Sleep. 2012;35:1403–11.
Kessler RC, Berglund PA, Chiu WT, Deitz AC, Hudson JI, Shahly V, et al. The prevalence and correlates of binge eating disorder in the World Health Organization World Mental Health Surveys. Biol Psychiatry. 2013;73:904–14.
Ogilvie RP, Patel SR. The epidemiology of sleep and obesity. Sleep Health. 2017;3:383–8.
Li JTE, Pursey KM, Duncan MJ, Burrows T. Addictive eating and its relation to physical activity and sleep behavior. Nutrients. 2018;10:10
Marino M, Li Y, Rueschman MN, Winkelman JW, Ellenbogen JM, Solet JM. et al. Measuring sleep: accuracy, sensitivity, and specificity of wrist actigraphy compared to polysomnography. Sleep. 2013;36:1747–55.
Manni R, Ratti MT, Tartara A. Nocturnal eating: prevalence and features in 120 insomniac referrals. Sleep. 1997;20:734–8.
Rogers NL, Dinges DF, Allison KC, Maislin G, Martino N, O'Reardon JP. et al. Assessment of sleep in women with night eating syndrome. Sleep. 2006;29:814–9.
Cleator J, Abbott J, Judd P, Sutton C, Wilding JP. Night eating syndrome: implications for severe obesity. Nutr Diabetes. 2012;2:e44
Allison KC, Lundgren JD, O'reardon JP, Geliebter A, Gluck ME, Vinai P, et al. Proposed diagnostic criteria for night eating syndrome. Int J Eat Disord. 2010;43:241–7.
Kucukgoncu S, Midura M, Tek C. Optimal management of night eating syndrome: challenges and solutions. Neuropsychiatr Dis Treat. 2015;11:751–60.
Stunkard AJ, Grace WJ, Wolff HG. The night-eating syndrome; a pattern of food intake among certain obese patients. Am J Med. 1955;19:78–86.
Stunkard AJ. Eating patterns and obesity. Psychiatr Q. 1959;33:284–95.
Lundgren JD, Allison KC, O’Reardon JP, Stunkard AJ. A descriptive study of non-obese persons with night eating syndrome and a weight-matched comparison group. Eat Behav. 2008;9:343–51.
Sassaroli S, Ruggiero GM, Vinai P, Cardetti S, Carpegna G, Ferrato N, et al. Daily and nightly anxiety among patients affected by night eating syndrome and binge eating disorder. Eat Disord. 2009;17:140–5.
Allison KC, Grilo CM, Masheb RM, Stunkard AJ. Binge eating disorder and night eating syndrome: a comparative study of disordered eating. J Consult Clin Psychol. 2005;73:1107–15.
Corwin RL, Babbs RK. Rodent models of binge eating: are they models of addiction? ILAR J. 2012;53:23–34.
Corwin RL, Avena NM, Boggiano MM. Feeding and reward: perspectives from three rat models of binge eating. Physiol Behav. 2011;104:87–97.
Bello NT, Yeh CY, James MH. Reduced sensory-evoked locus coeruleus-norepinephrine neural activity in female rats with a history of dietary-induced binge eating. Front Psychol. 2019;10:1966.
Moore CF, Leonard MZ, Micovic NM, Miczek KA, Sabino V, Cottone P. Reward sensitivity deficits in a rat model of compulsive eating behavior. Neuropsychopharmacology. 2020;45:589–96.
Velázquez-Sánchez C, Ferragud A, Moore CF, Everitt BJ, Sabino V, Cottone P. High trait impulsivity predicts food addiction-like behavior in the rat. Neuropsychopharmacology. 2014;39:2463–72.
Perello M, Valdivia S, García Romero G, Raingo J. Considerations about rodent models of binge eating episodes. Front Psychol. 2014;5:372.
Berner LA, Bocarsly ME, Hoebel BG, Avena NM. Baclofen suppresses binge eating of pure fat but not a sugar-rich or sweet-fat diet. Behav. Pharmacol. 2009;20:631–4.
Davis JF, Melhorn SJ, Shurdak JD, Heiman JU, Tschöp MH, Clegg DJ, et al. Comparison of hydrogenated vegetable shortening and nutritionally complete high-fat diet on limited access-binge behavior in rats. Physiol Behav. 2007;92:924–30.
Howard CE, Porzelius LK. The role of dieting in binge eating disorder: etiology and treatment implications. Clin Psychol Rev. 1999;19:25–44.
Bello NT, Guarda AS, Terrillion CE, Redgrave GW, Coughlin JW, Moran TH. Repeated binge access to a palatable food alters feeding behavior, hormone profile, and hindbrain c-Fos responses to a test meal in adult male rats. Am J Physiol Regul Integr Comp Physiol. 2009;297:R622–631.
Anversa RG, Campbell EJ, Ch’ng SS, Gogos A, Lawrence AJ, Brown RM. A model of emotional stress-induced binge eating in female mice with no history of food restriction. Genes Brain Behav. 2020;19:e12613.
Corwin RL. Binge-type eating induced by limited access in rats does not require energy restriction on the previous day. Appetite. 2004;42:139–42.
Corwin RL, Buda-Levin A. Behavioral models of binge-type eating. Physiol Behav. 2004;82:123–30.
Hagan MM, Wauford PK, Chandler PC, Jarrett LA, Rybak RJ, Blackburn K. A new animal model of binge eating: key synergistic role of past caloric restriction and stress. Physiol Behav. 2002;77:45–54.
Cottone P, Wang X, Park JW, Valenza M, Blasio A, Kwak J, et al. Antagonism of sigma-1 receptors blocks compulsive-like eating. Neuropsychopharmacology. 2012;37:2593–604.
Venner A, Todd WD, Fraigne J, Bowrey H, Eban-Rothschild A, Kaur S. et al. Newly identified sleep-wake and circadian circuits as potential therapeutic targets. Sleep. 2019;42:5
Scammell TE, Arrigoni E, Lipton JO. Neural circuitry of wakefulness and sleep. Neuron. 2017;93:747–65.
Eban-Rothschild A, Appelbaum L, de Lecea L. Neuronal mechanisms for sleep/wake regulation and modulatory drive. Neuropsychopharmacology. 2018;43:937–52.
Bjorness TE, Greene RW. Interaction between cocaine use and sleep behavior: a comprehensive review of cocaine’s disrupting influence on sleep behavior and sleep disruptions influence on reward seeking. Pharmacol Biochem Behav. 2021;206:173194.
Doyle SE, Feng H, Garber G, Menaker M, Lynch WJ. Effects of circadian disruption on methamphetamine consumption in methamphetamine-exposed rats. Psychopharmacology. 2015;232:2169–79.
Mistlberger RE. Circadian food-anticipatory activity: formal models and physiological mechanisms. Neurosci Biobehav Rev. 1994;18:171–95.
Gooley JJ, Schomer A, Saper CB. The dorsomedial hypothalamic nucleus is critical for the expression of food-entrainable circadian rhythms. Nat Neurosci. 2006;9:398–407.
Mistlberger RE. Food-anticipatory circadian rhythms: concepts and methods. Eur J Neurosci. 2009;30:1718–29.
Mistlberger R, Rusak B. Palatable daily meals entrain anticipatory activity rhythms in free-feeding rats: dependence on meal size and nutrient content. Physiol Behav. 1987;41:219–26.
Mendoza J, Angeles-Castellanos M, Escobar C. Entrainment by a palatable meal induces food-anticipatory activity and c-Fos expression in reward-related areas of the brain. Neuroscience. 2005;133:293–303.
Blancas A, González-Garcí a SD, Rodríguez K, Escobar C. Progressive anticipation in behavior and brain activation of rats exposed to scheduled daily palatable food. Neuroscience. 2014;281:44–53.
Hsu CT, Patton DF, Mistlberger RE, Steele AD. Palatable meal anticipation in mice. PLoS ONE. 2010;5:9.
Flôres DE, Bettilyon CN, Yamazaki S. Period-independent novel circadian oscillators revealed by timed exercise and palatable meals. Sci Rep. 2016;6:21945.
Velázquez-Sánchez C, Santos JW, Smith KL, Ferragud A, Sabino V, Cottone P. Seeking behavior, place conditioning, and resistance to conditioned suppression of feeding in rats intermittently exposed to palatable food. Behav Neurosci. 2015;129:219–24.
Johnson WG, Schlundt DG, Barclay DR, Carr-Nangle RE, Engler LB. A naturalistic functional analysis of binge eating. Behav Ther. 1995;26:101–18.
Smyth JM, Wonderlich SA, Sliwinski MJ, Crosby RD, Engel SG, Mitchell JE, et al. Ecological momentary assessment of affect, stress, and binge-purge behaviors: day of week and time of day effects in the natural environment. Int J Eat Disord. 2009;42:429–36.
Stein RI, Kenardy J, Wiseman CV, Dounchis JZ, Arnow BA, Wilfley DE. What’s driving the binge in binge eating disorder?: A prospective examination of precursors and consequences. Int J Eat Disord. 2007;40:195–203.
Escobar C, Salgado R, Rodriguez K, Blancas Vázquez AS, Angeles-Castellanos M, Buijs RM. Scheduled meals and scheduled palatable snacks synchronize circadian rhythms: consequences for ingestive behavior. Physiol Behav. 2011;104:555–61.
Webb IC, Baltazar RM, Lehman MN, Coolen LM. Bidirectional interactions between the circadian and reward systems: is restricted food access a unique zeitgeber? Eur J Neurosci. 2009;30:1739–48.
Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–13.
Baldo BA, Kelley AE. Discrete neurochemical coding of distinguishable motivational processes: insights from nucleus accumbens control of feeding. Psychopharmacology. 2007;191:439–59.
Liu Z, Wang Y, Cai L, Li Y, Chen B, Dong Y, et al. Prefrontal cortex to accumbens projections in sleep regulation of reward. J Neurosci. 2016;36:7897–910.
Chen B, Wang Y, Liu X, Liu Z, Dong Y, Huang YH. Sleep regulates incubation of cocaine craving. J Neurosci. 2015;35:13300–10.
Roane BM, Taylor DJ. Adolescent insomnia as a risk factor for early adult depression and substance abuse. Sleep. 2008;31:1351–6.
Wong MM, Brower KJ, Nigg JT, Zucker RA. Childhood sleep problems, response inhibition, and alcohol and drug outcomes in adolescence and young adulthood. Alcohol, Clin Exp Res. 2010;34:1033–44.
Brower KJ, Perron BE. Sleep disturbance as a universal risk factor for relapse in addictions to psychoactive substances. Med Hypotheses. 2010;74:928–33.
Morgan PT, Pace-Schott EF, Sahul ZH, Coric V, Stickgold R, Malison RT Sleep, sleep-dependent procedural learning and vigilance in chronic cocaine users: evidence for occult insomnia. Drug alcohol Depend. 2006;82:238–49.
Pace-Schott EF, Stickgold R, Muzur A, Wigren PE, Ward AS, Hart CL. et al. Sleep quality deteriorates over a binge-abstinence cycle in chronic smoked cocaine users. Psychopharmacology. 2005;179:873–83.
Pace-Schott EF, Stickgold R, Muzur A, Wigren PE, Ward AS, Hart CL, et al. Cognitive performance by humans during a smoked cocaine binge-abstinence cycle. Am J Drug Alcohol Abuse. 2005;31:571–91.
Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H. et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:92–585.
de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:95–7.
Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000;27:469–474. https://doi.org/10.1016/S0896-6273(00)00058-1.
Kilduff TS, Peyron C. The hypocretin/orexin ligand-receptor system: implications for sleep and sleep disorders. Trends Neurosci. 2000;23:359–65. https://doi.org/10.1016/s0166-2236(00)01594-0.
Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601.
Thompson JL, Borgland SL. A role for hypocretin/orexin in motivation. Behav Brain Res. 2011;217:446–53.
James MH, Charnley JL, Levi EM, Jones E, Yeoh JW, Smith DW, et al. Orexin-1 receptor signalling within the ventral tegmental area, but not the paraventricular thalamus, is critical to regulating cue-induced reinstatement of cocaine-seeking. Int J Neuropsychopharmacol. 2011;14:684–90.
Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X. et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–76.
Kisanuki Y. The role of orexin receptor type-1 (OX1R) in the regulation of sleep. Sleep. 2000;23:A91
Hondo M, Nagai K, Ohno K, Kisanuki Y, Willie JT, Watanabe T, et al. Histamine-1 receptor is not required as a downstream effector of orexin-2 receptor in maintenance of basal sleep/wake states. Acta Physiol. 2010;198:287–94.
Barson JR. Orexin/hypocretin and dysregulated eating: promotion of foraging behavior. Brain Res. 2020;1731:145915.
Mahler SV, Moorman DE, Smith RJ, James MH, Aston-Jones G. Motivational activation: a unifying hypothesis of orexin/hypocretin function. Nat Neurosci. 2014;17:1298–303.
Campbell EJ, Barker DJ, Nasser HM, Kaganovsky K, Dayas CV, Marchant NJ. Cue-induced food seeking after punishment is associated with increased Fos expression in the lateral hypothalamus and basolateral and medial amygdala. Behav Neurosci. 2017;131:155–67.
Dayas CV, McGranahan TM, Martin-Fardon R, Weiss F. Stimuli linked to ethanol availability activate hypothalamic CART and orexin neurons in a reinstatement model of relapse. Biol Psychiatry. 2008;63:152–7.
James MH, Stopper CM, Zimmer BA, Koll NE, Bowrey HE, Aston-Jones G. Increased number and activity of a lateral subpopulation of hypothalamic orexin/hypocretin neurons underlies the expression of an addicted state in rats. Biol Psychiatry. 2019;85:925–35.
Martin-Fardon R, Cauvi G, Kerr TM, Weiss F. Differential role of hypothalamic orexin/hypocretin neurons in reward seeking motivated by cocaine versus palatable food. Addiction Biol. 2018;23:6–15.
Choi DL, Davis JF, Fitzgerald ME, Benoit SC. The role of orexin-A in food motivation, reward-based feeding behavior and food-induced neuronal activation in rats. Neuroscience. 2010;167:11–20.
Castro DC, Terry RA, Berridge KC. Orexin in rostral hotspot of nucleus accumbens enhances sucrose /‘Liking/' and intake but scopolamine in caudal shell shifts /‘Liking/' toward /‘Disgust/' and /‘Fear/'. Neuropsychopharmacology 2016;41:2101–11.
Terrill SJ, Hyde KM, Kay KE, Greene HE, Maske CB, Knierim AE, et al. Ventral tegmental area orexin 1 receptors promote palatable food intake and oppose postingestive negative feedback. Am J Physiol Regulatory, Integr Comp Physiol. 2016;311:R592–599.
Barson JR, Ho HT, Leibowitz SF. Anterior thalamic paraventricular nucleus is involved in intermittent access ethanol drinking: role of orexin receptor 2. Addict Biol. 2015;20:469–81.
Espana RA, Melchior JR, Roberts DC, Jones SR. Hypocretin 1/orexin A in the ventral tegmental area enhances dopamine responses to cocaine and promotes cocaine self-administration. Psychopharmacology. 2011;214:415–26.
Matzeu A, Kerr TM, Weiss F, Martin-Fardon R. Orexin-A/Hypocretin-1 mediates cocaine-seeking behavior in the posterior paraventricular nucleus of the thalamus via Orexin/Hypocretin Receptor-2. J Pharmacol Exp Ther. 2016;359:273–9.
Wang B, You ZB, Wise RA. Reinstatement of cocaine seeking by hypocretin (orexin) in the ventral tegmental area: independence from the local corticotropin-releasing factor network. Biol Psychiatry. 2009;65:857–62.
Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, et al. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci USA. 2005;102:19168–73.
Piccoli L, Micioni Di Bonaventura MV, Cifani C, Costantini VJ, Massagrande M, Montanari D, et al. Role of orexin-1 receptor mechanisms on compulsive food consumption in a model of binge eating in female rats. Neuropsychopharmacology. 2012;37:1999–2011.
Vickers SP, Hackett D, Murray F, Hutson PH, Heal DJ. Effects of lisdexamfetamine in a rat model of binge-eating. J Psychopharmacol. 2015;29:1290–307.
Alcaraz-Iborra M, Carvajal F, Lerma-Cabrera JM, Valor LM, Cubero I. Binge-like consumption of caloric and non-caloric palatable substances in ad libitum-fed C57BL/6J mice: pharmacological and molecular evidence of orexin involvement. Behav. Brain Res. 2014;272:93–9.
Rorabaugh JM, Stratford JM, Zahniser NR. A relationship between reduced nucleus accumbens shell and enhanced lateral hypothalamic orexin neuronal activation in long-term fructose bingeing behavior. PLoS ONE. 2014;9:e95019.
Cason AM, Aston-Jones G. Attenuation of saccharin-seeking in rats by orexin/hypocretin receptor 1 antagonist. Psychopharmacology. 2013;228:499–507.
Cason AM, Aston-Jones G. Role of orexin/hypocretin in conditioned sucrose-seeking in female rats. Neuropharmacology. 2014;86:97–102.
Jupp B, Krivdic B, Krstew E, Lawrence AJ. The orexin(1) receptor antagonist SB-334867 dissociates the motivational properties of alcohol and sucrose in rats. Brain Res. 2011;1391:54–59.
Nair SG, Golden SA, Shaham Y. Differential effects of the hypocretin 1 receptor antagonist SB 334867 on high-fat food self-administration and reinstatement of food seeking in rats. Br J Pharmacol. 2008;154:406–16.
Brown RM, Khoo SY-S, Lawrence AJ. Central orexin (hypocretin) 2 receptor antagonism reduces ethanol self-administration, but not cue-conditioned ethanol-seeking, in ethanol-preferring rats. Int J Neuropsychopharmacol. 2013;16:2067–79.
Shoblock JR, Welty N, Aluisio L, Fraser I, Motley ST, Morton K. et al. Selective blockade of the orexin-2 receptor attenuates ethanol self-administration, place preference, and reinstatement. Psychopharmacology. 2011;215:191–203.
Borgland SL, Chang SJ, Bowers MS, Thompson JL, Vittoz N, Floresco SB, et al. Orexin A/hypocretin-1 selectively promotes motivation for positive reinforcers. J Neurosci. 2009;29:11215–25.
Espana RA, Oleson EB, Locke JL, Brookshire BR, Roberts DC, Jones SR. The hypocretin-orexin system regulates cocaine self-administration via actions on the mesolimbic dopamine system. Eur J Neurosci. 2010;31:336–48.
Wiskerke J, James MH, Aston-Jones G. The orexin-1 receptor antagonist SB-334867 reduces motivation, but not inhibitory control, in a rat stop signal task. Brain Res. 2020;1731:146222. https://doi.org/10.1016/j.brainres.2019.04.017.
Matzeu A, Martin-Fardon R. Blockade of Orexin receptors in the posterior paraventricular nucleus of the thalamus prevents stress-induced reinstatement of reward-seeking behavior in rats with a history of ethanol dependence. Front Integr Neurosci. 2020;14:599710.
James MH, Yeoh JW, Graham BA, Dayas CV. Insights for developing pharmacological treatments for psychostimulant relapse targeting hypothalamic peptide systems. J Addict Res Ther. 2012;S4:008.
Martin-Fardon R, Weiss F. Blockade of hypocretin receptor-1 preferentially prevents cocaine seeking: comparison with natural reward seeking. Neuroreport. 2014;25:485–8.
Freeman LR, Bentzley BS, James MH, Aston-Jones G. Sex differences in demand for highly palatable foods: role of the Orexin system. Int. J. Neuropsychopharmacol. 2020.
White CL, Ishii Y, Mendoza T, Upton N, Stasi LP, Bray GA. et al. Effect of a selective OX1R antagonist on food intake and body weight in two strains of rats that differ in susceptibility to dietary-induced obesity. Peptides. 2005;26:2331–8.
James MH, Bowrey HE, Stopper CM, Aston-Jones G. Demand elasticity predicts addiction endophenotypes and the therapeutic efficacy of an orexin/hypocretin-1 receptor antagonist in rats. Eur J Neurosci. 2019;50:2602–12.
Fragale JE, Pantazis CB, James MH, Aston-Jones G. The role of orexin-1 receptor signaling in demand for the opioid fentanyl. Neuropsychopharmacology. 2019;44:1690–7.
Fragale JE, James MH, Aston-Jones G. Intermittent self-administration of fentanyl induces a multifaceted addiction state associated with persistent changes in the orexin system. Addiction Biol. 2020;26:e12946.
Mohammadkhani A, Fragale JE, Pantazis CB, Bowrey HE, James MH, Aston-Jones G. Orexin-1 receptor signaling in ventral pallidum regulates motivation for the opioid remifentanil. J Neurosci. 2019;39:9831–40. https://doi.org/10.1523/JNEUROSCI.0255-19.2019.
Mohammadkhani A, James MH, Pantazis CB, Aston-Jones G. Persistent effects of the orexin-1 receptor antagonist SB-334867 on motivation for the fast acting opioid remifentanil. Brain Res. 2019;1731:146461.
LeSage MG, Perry JL, Kotz CM, Shelley D, Corrigall WA. Nicotine self-administration in the rat: effects of hypocretin antagonists and changes in hypocretin mRNA. Psychopharmacology. 2010;209:203–12.
James MH, Fragale JE, O’Connor SL, Zimmer BA, Aston-Jones G. The orexin (hypocretin) neuropeptide system is a target for novel therapeutics to treat cocaine use disorder with alcohol coabuse. Neuropharmacology. 2020;183:108359
Prince CD, Rau AR, Yorgason JT, España RA. Hypocretin/Orexin regulation of dopamine signaling and cocaine self-administration is mediated predominantly by Hypocretin Receptor 1. ACS Chem Neurosci. 2015;6:138–46.
Lawrence AJ, Cowen MS, Yang HJ, Chen F, Oldfield B. The orexin system regulates alcohol-seeking in rats. Br J Pharmacol. 2006;148:752–9.
Hollander JA, Pham D, Fowler CD, Kenny PJ. Hypocretin-1 receptors regulate the reinforcing and reward-enhancing effects of cocaine: pharmacological and behavioral genetics evidence. Front Behav Neurosci. 2012;6:47.
Smith RJ, Tahsili-Fahadan P, Aston-Jones G. Orexin/hypocretin is necessary for context-driven cocaine-seeking. Neuropharmacology. 2010;58:179–84.
Smith RJ, Aston-Jones G. Orexin/hypocretin 1 receptor antagonist reduces heroin self-administration and cue-induced heroin seeking. Eur J Neurosci. 2012;35:798–804.
Matzeu A, Martin-Fardon R. Targeting the Orexin system for prescription opioid use disorder. Brain Sci. 2020;10:4.
Martin-Fardon, R, Weiss, F. N-2-methyl-6-benzoxazolyl)-N’-1,5-naphthyridin-4-yl urea (SB334867), a hypocretin receptor-1 antagonist, preferentially prevents ethanol seeking: comparison with natural reward seeking. Addict Biol. 2014;19:233–6.
Hopf FW. Recent perspectives on orexin/hypocretin promotion of addiction-related behaviors. Neuropharmacology. 2020;168:108013
Kwok C, Lei K, Pedrozo V, Anderson L, Ghotra S, Walsh M, et al. Differential importance of nucleus accumbens Ox1Rs and AMPARs for female and male mouse binge alcohol drinking. Sci Rep. 2021;11:231.
Gentile TA, Simmons SJ, Watson MN, et al. Effects of Suvorexant, a dual orexin/hypocretin receptor antagonist, on impulsive behavior associated with cocaine. Neuropsychopharmacology. 2018;43:1001–9. https://doi.org/10.1038/npp.2017.158.
Muschamp JW, Hollander JA, Thompson JL, Voren G, Hassinger LC, Onvani S, et al. Hypocretin (orexin) facilitates reward by attenuating the antireward effects of its cotransmitter dynorphin in ventral tegmental area. Proc Natl Acad Sci USA. 2014;111:E1648–1655.
Calva CB, Fadel JR. Intranasal administration of orexin peptides: mechanisms and therapeutic potential for age-related cognitive dysfunction. Brain Res. 2020;1731:145921. https://doi.org/10.1016/j.brainres.2018.08.024.
Adam JA, Menheere PP, van Dielen FM, Soeters PB, Buurman WA, Greve JW. Decreased plasma orexin-A levels in obese individuals. Int J Obes Relat Metab Disord. 2002;26:274–6.
Gupta A, Miegueu P, Lapointe M, Poirier P, Martin J, Bastien M, et al. Acute post-bariatric surgery increase in orexin levels associates with preferential lipid profile improvement. PLoS ONE. 2014;9:e84803.
Nishino S. Clinical and neurobiological aspects of narcolepsy. Sleep Med. 2007;8:373–99.
Funato H, Tsai AL, Willie JT, Kisanuki Y, Williams SC, Sakurai T, et al. Enhanced orexin receptor-2 signaling prevents diet-induced obesity and improves leptin sensitivity. Cell Metab. 2009;9:64–76.
Kotz C, Nixon J, Butterick T, Perez-Leighton C, Teske J, Billington C. Brain orexin promotes obesity resistance. Ann N Y Acad Sci. 2012;1264:72–86.
Teske JA, Levine AS, Kuskowski M, Levine JA, Kotz CM. Elevated hypothalamic orexin signaling, sensitivity to orexin A, and spontaneous physical activity in obesity-resistant rats. Am J Physiol Regul Integr Comp Physiol. 2006;291:R889–899.
Kotz C, Nixon J, Butterick T, Perez-Leighton C, Teske J, Billington C. Brain orexin promotes obesity resistance. Ann N Y Acad Sci. 2012;1264:72–86.
Lemus MB, Bayliss JA, Lockie SH, Santos VV, Reichenbach A, Stark R. et al. A stereological analysis of NPY, POMC, Orexin, GFAP astrocyte, and Iba1 microglia cell number and volume in diet-induced obese male mice. Endocrinology. 2015;156:1701–13.
Wortley KE, Chang GQ, Davydova Z, Leibowitz SF. Peptides that regulate food intake: orexin gene expression is increased during states of hypertriglyceridemia. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1454–65.
Morganstern I, Chang GQ, Karatayev O, Leibowitz SF. Increased orexin and melanin-concentrating hormone expression in the perifornical lateral hypothalamus of rats prone to overconsuming a fat-rich diet. Pharmacol, Biochem Behav. 2010;96:413–22.
Xi MC, Morales FR, Chase MH. Effects on sleep and wakefulness of the injection of hypocretin-1 (orexin-A) into the laterodorsal tegmental nucleus of the cat. Brain Res. 2001;901:259–64.
Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C. et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell . 1999;98:437–51.
Fujiki N, Yoshida Y, Ripley B, Mignot E, Nishino S. Effects of IV and ICV hypocretin-1 (orexin A) in hypocretin receptor-2 gene mutated narcoleptic dogs and IV hypocretin-1 replacement therapy in a hypocretin-ligand-deficient narcoleptic dog. Sleep. 2003;26:953–9.
Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM. et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–54.
Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, et al. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14:1075–81.
Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, et al. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–62.
Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–4.
Carter ME, Adamantidis A, Ohtsu H, Deisseroth K, de Lecea L. Sleep homeostasis modulates hypocretin-mediated sleep-to-wake transitions. J Neurosci. 2009;29:29–49.
Sasaki K, Suzuki M, Mieda M, Tsujino N, Roth B, Sakurai T. Pharmacogenetic modulation of orexin neurons alters sleep/wakefulness states in mice. PLoS ONE. 2011;6:e20360.
Tsunematsu T, Kilduff TS, Boyden ES, Takahashi S, Tominaga M, Yamanaka A. Acute optogenetic silencing of orexin/hypocretin neurons induces slow-wave sleep in mice. J Neurosci. 2011;31:10529–39.
Gerhardstein R, Day R, Rosenthal L. Narcolepsy and other causes of excessive daytime sleepiness. Respir Care Clin N Am. 1999;5:427–46, viii-ix.
Slowik JM, Collen JF, Yow AG. Narcolepsy. StatPearls. 2020.
Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40.
Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M. et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:27–74.
Rhyne DN, Anderson SL. Suvorexant in insomnia: efficacy, safety and place in therapy. Ther Adv Drug Saf. 2015;6:189–95.
Norman JL, Anderson SL. Novel class of medications, orexin receptor antagonists, in the treatment of insomnia—critical appraisal of suvorexant. Nat Sci Sleep. 2016;8:239–47.
Janto K, Prichard JR, Pusalavidyasagar S. An update on dual orexin receptor antagonists and their potential role in insomnia therapeutics. J Clin Sleep Med. 2018;14:1399–408.
Muehlan C, Vaillant C, Zenklusen I, Kraehenbuehl S, Dingemanse J. Clinical pharmacology, efficacy, and safety of orexin receptor antagonists for the treatment of insomnia disorders. Expert Opin Drug Metab Toxicol. 2020;16:1063–78.
Recourt K, de Boer P, Zuiker R, Luthringer R, Kent J, van der Ark P, et al. The selective orexin-2 antagonist seltorexant (JNJ-42847922/MIN-202) shows antidepressant and sleep-promoting effects in patients with major depressive disorder. Transl Psychiatry. 2019;9:216.
Dugovic C, Shelton JE, Aluisio LE, Fraser IC, Jiang X, Sutton SW, et al. Blockade of orexin-1 receptors attenuates orexin-2 receptor antagonism-induced sleep promotion in the rat. J Pharmacol Exp therapeutics. 2009;330:142–51.
Cox CD, Breslin MJ, Whitman DB, Schreier JD, McGaughey GB, Bogusky MJ, et al. Discovery of the dual orexin receptor antagonist [(7R)-4-(5-chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H -1,2,3-triazol-2-yl)phenyl]methanone (MK-4305) for the treatment of insomnia. J Med Chem. 2010;53:5320–32.
Winrow CJ, Gotter AL, Cox CD, Doran SM, Tannenbaum PL, Breslin MJ, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25:52–61.
Herring WJ, Snyder E, Budd K, Hutzelmann J, Snavely D, Liu K. et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79:2265–74.
Ardeljan AD, Hurezeanu R. Lemborexant. StatPearls; 2020.
Bonaventure P, Shelton J, Yun S, Nepomuceno D, Sutton S, Aluisio L, et al. Characterization of JNJ-42847922, a selective Orexin-2 receptor antagonist, as a clinical candidate for the treatment of insomnia. J Pharmacol Exp Ther. 2015;354:471–82.
Guirguis WR. Sleepwalking as a symptom of bulimia. Br Med J (Clin Res Ed). 1986;293:587–8.
Iwasa T, Matsuzaki T, Mayila Y, Yano K, Irahara M. Developmental changes in hypothalamic SF-1, POMC, and ERα mRNA expression and their sensitivity to fasting in male and female rats. Endocr J. 2017;64:1157–63.
Tanno S, Terao A, Okamatsu-Ogura Y, Kimura K. Hypothalamic prepro-orexin mRNA level is inversely correlated to the non-rapid eye movement sleep level in high-fat diet-induced obese mice. Obes Res Clin Pract. 2013;7:e251–7.
True C, Arik A, Lindsley S, Kirigiti M, Sullivan E, Kievit P. Early high-fat diet exposure causes dysregulation of the Orexin and Dopamine neuronal populations in nonhuman primates. Front Endocrinol (Lausanne). 2018;9:508.
Beck B, Kozak R, Moar KM, Mercer JG. Hypothalamic orexigenic peptides are overexpressed in young Long-Evans rats after early life exposure to fat-rich diets. Biochem Biophys Res Commun. 2006;342:452–8.
Chen P-Y, Chen C-H, Chang C-K, Kao CF, Lu ML, Lin SK, et al. Orexin-A levels in relation to the risk of metabolic syndrome in patients with schizophrenia taking antipsychotics. Int J Neuropsychopharmacol. 2018;22:28–36.
Thannickal TC, John J, Shan L, Swaab DF, Wu MF, Ramanathan L, et al. Opiates increase the number of hypocretin-producing cells in human and mouse brain and reverse cataplexy in a mouse model of narcolepsy. Sci Transl Med. 2018;10:447.
Collier AD, Halkina V, Min SS, Roberts MY, Campbell SD, Camidge K, et al. Embryonic ethanol exposure affects the early development, migration, and location of hypocretin/orexin neurons in zebrafish. Alcohol Clin Exp Res. 2019;43:1702–13.
Collier AD, Min SS, Campbell SD, Roberts MY, Camidge K, Leibowitz SF. Maternal ethanol consumption before paternal fertilization: stimulation of hypocretin neurogenesis and ethanol intake in zebrafish offspring. Prog Neuropsychopharmacol Biol Psychiatry. 2020;96:109728.
Yeoh JW, James MH, Adams CD, Bains JS, Sakurai T, Aston-Jones G. et al. Activation of lateral hypothalamic group III metabotropic glutamate receptors suppresses cocaine-seeking following abstinence and normalizes drug-associated increases in excitatory drive to orexin/hypocretin cells. Neuropharmacology. 2019;154:22–33.
Yeoh JW, James MH, Graham BA, Dayas CV. Electrophysiological characteristics of paraventricular thalamic (PVT) neurons in response to cocaine and cocaine- and amphetamine-regulated transcript (CART). Front Behav Neurosci. 2014;8:280.
Horvath TL, Gao XB. Input organization and plasticity of hypocretin neurons: possible clues to obesity’s association with insomnia. Cell Metab. 2005;1:279–86.
Laperchia C, Imperatore R, Azeez IA, Del Gallo F, Bertini G, Grassi-Zucconi G, et al. The excitatory/inhibitory input to orexin/hypocretin neuron soma undergoes day/night reorganization. Brain Struct Funct. 2017;222:3847–59.
Bunney PE, Zink AN, Holm AA, Billington CJ, Kotz CM. Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high-fat diet. Physiol Behav. 2017;176:139–48.
Moorman DE, Aston-Jones G. Orexin/hypocretin modulates response of ventral tegmental dopamine neurons to prefrontal activation: diurnal influences. J Neurosci. 2010;30:15585–99.
Horvath TL, Diano S, van den Pol AN. Synaptic interaction between hypocretin (orexin) and neuropeptide Y cells in the rodent and primate hypothalamus: a novel circuit implicated in metabolic and endocrine regulations. J Neurosci. 1999;19:19–1087.
Pollak Dorocic I, Fürth D, Xuan Y, Johansson Y, Pozzi L, Silberberg G. et al. A whole-brain atlas of inputs to serotonergic neurons of the dorsal and median raphe nuclei. Neuron. 2014;83:663–78.
Muraki Y, Yamanaka A, Tsujino N, Kilduff TS, Goto K, Sakurai T. Serotonergic regulation of the orexin/hypocretin neurons through the 5-HT1A receptor. J Neurosci. 2004;24:7159–66.
Saito YC, Tsujino N, Abe M, Yamazaki M, Sakimura K, Sakurai T. Serotonergic input to Orexin neurons plays a role in maintaining wakefulness and REM sleep architecture. Front Neurosci. 2018;12:892.
Allsopp MR, Zaiwalla Z. Narcolepsy. Arch Dis Child. 1992;67:302–6.
Fortuyn HA, Swinkels S, Buitelaar J, Renier WO, Furer JW, Rijnders CA. et al. High prevalence of eating disorders in narcolepsy with cataplexy: a case-control study. Sleep. 2008;31:335–41.
van Holst RJ, van der Cruijsen L, van Mierlo P, Lammers GJ, Cools R, Overeem S. et al. Aberrant food choices after satiation in human Orexin-deficient narcolepsy type 1. Sleep. 2016;39:1951–9.
Dimitrova A, Fronczek R, Van der Ploeg J, Scammell T, Gautam S, Pascual-Leone A, et al. Reward-seeking behavior in human narcolepsy. J Clin Sleep Med. 2011;7:293–300.
Dauvilliers Y, Montplaisir J, Molinari N, Carlander B, Ondze B, Besset A. et al. Age at onset of narcolepsy in two large populations of patients in France and Quebec. Neurology. 2001;57:2029–33.
González JA, Jensen LT, Iordanidou P, Strom M, Fugger L, Burdakov D. Inhibitory interplay between Orexin neurons and eating. Curr Biol. 2016;26:2486–91.
Dahmen N, Becht J, Engel A, Thommes M, Tonn P. Prevalence of eating disorders and eating attacks in narcolepsy. Neuropsychiatr Dis Treat. 2008;4:257–61.
Sharma S, Kavuru M. Sleep and metabolism: an overview. Int J Endocrinol. 2010;2010:270832
O’Connor SL, Fragale JE, James MH, Aston-Jones G. The dual orexin/hypocretin receptor antagonist suvorexant reduces addiction-like behaviors for the opioid fentanyl [Preprint]. 2020. Available from: https://www.biorxiv.org/content/10.1101/2020.04.25.061887v1.
Gamble MC, Katsuki F, McCoy JG, Strecker RE, McKenna JT. The dual orexinergic receptor antagonist DORA-22 improves the sleep disruption and memory impairment produced by a rodent insomnia model. Sleep. 2019;43:3
James MH, Fragale JE, Aurora RN, Cooperman NA, Langleben DD, Aston-Jones G. Repurposing the dual orexin receptor antagonist suvorexant for the treatment of opioid use disorder: why sleep on this any longer? Neuropsychopharmacology. 2020;45:717–19.
Kotorii N. Treatment strategies when multiple sleep disturbances coexist. The abstract of Japanese society of sleep research meeting 192; Japan, 2015.
Ono H, Kanbayashi T, Iwaki S, et al. Clinical experience with a dual orexin receptor antagonist, Suvorexant (Belsomra) in Japan. J Sleep Disord Med Care. 2018;1:1–5.
Bastien CH, Vallières A, Morin CM. Validation of the Insomnia Severity Index as an outcome measure for insomnia research. Sleep Med. 2001;2:297–307. https://doi.org/10.1016/s1389-9457(00)00065-4.
Acknowledgements
Figure was prepared using BioRender.
Author information
Authors and Affiliations
Contributions
JBM, DM, HEB, and MHJ all contributed to draft versions of the manuscript and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mehr, J.B., Mitchison, D., Bowrey, H.E. et al. Sleep dysregulation in binge eating disorder and “food addiction”: the orexin (hypocretin) system as a potential neurobiological link. Neuropsychopharmacol. 46, 2051–2061 (2021). https://doi.org/10.1038/s41386-021-01052-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01052-z
This article is cited by
-
Sleep-mediated regulation of reward circuits: implications in substance use disorders
Neuropsychopharmacology (2023)
-
The abuse potential of lemborexant, a dual orexin receptor antagonist, according to the 8 factors of the Controlled Substances Act
Psychopharmacology (2023)
-
Sleep disruption as a potential contributor to the worsening of eating disorder pathology during the COVID-19-pandemic
Journal of Eating Disorders (2022)
-
Orexin-1 receptor signaling in ventral tegmental area mediates cue-driven demand for cocaine
Neuropsychopharmacology (2022)
