
Abstract
Our modern society suffers from both pervasive sleep loss and substance abuse—what may be the indications for sleep on substance use disorders (SUDs), and could sleep contribute to the individual variations in SUDs? Decades of research in sleep as well as in motivated behaviors have laid the foundation for us to begin to answer these questions. This review is intended to critically summarize the circuit, cellular, and molecular mechanisms by which sleep influences reward function, and to reveal critical challenges for future studies. The review also suggests that improving sleep quality may serve as complementary therapeutics for treating SUDs, and that formulating sleep metrics may be useful for predicting individual susceptibility to SUDs and other reward-associated psychiatric diseases.
Similar content being viewed by others
Introduction
Sleep, as “the main course in life’s feast, and the most nourishing” (—William Shakespeare, Macbeth), powerfully influences our emotional well-being and motivational states. Why is this the case and how does sleep do so? Decades of research in sleep as well as in motivated behaviors have laid the foundation for us to begin to understand the relationship between the two. Studies at circuit, cellular, and molecular levels have lent us increasing insights into their intricate interactions, which have profound implications for questions such as: How does loss of sleep alter motivated behaviors? How does acute sleep loss differ from chronic sleep disturbance? And for our modern society suffering from both pervasive sleep loss and substance abuse—what may be the indications for sleep loss on the disease process of substance use disorders (SUDs) and the individual variations? We will primarily focus on sleep-cocaine interactions to demonstrate the relationships and potential underlying mechanisms, then extend to other substances. Reviews on sleep-opioid or sleep-cannabis interactions can be found elsewhere [1,2,3].
Sleep composition, function, and association with psychiatric disorders
Sleep is a rapidly reversable and quiescent state characterized by specific sleep postures, reduced response to stimuli, and increased arousal threshold. During sleep, animals cannot forage, reproduce, and are vulnerable to predators. Despite the potential maladaptive nature and disadvantages associated with the prolonged immobility, sleep has been preserved through evolution [4, 5]. Vertebrates such as mammals, birds, and reptiles sleep, and a sleep-like state also exists in invertebrates [6,7,8]. Sleep can be broadly divided into two main states: non-rapid-eye-movement (NREM) sleep and rapid-eye-movement (REM) sleep (Box 1), which are also preserved across mammalian and avian species [9]. Following acute sleep deprivations (SD), both NREM and REM sleep show rebounds in the duration or intensity during recovery sleep, suggesting that they are under homeostatic regulations [10, 11].
Sleep is important for maintaining various vital physiological functions, including survival [12, 13], restoration of body and mind [4, 14, 15], energy conservation [16], immune functions [17, 18], brain development [19], brain metabolism and waste cleaning [20,21,22], learning and memory [23], and regulation of emotion and motivation [24, 25]. Good sleep is associated with positive affect and psychophysiological well-being [26], while poor sleep quality is often linked to negative valence and impaired regulation of emotion [27].
Sleep disturbance is a common comorbidity in almost all psychiatric disorders [28, 29]. Insomnia is often observed among patients with mood disorders, anxiety, and SUDs, and thus, chronic sleep disturbance often serves as a diagnostic checkmark for these disorders [30]. In the context of SUDs, sleep problems have been associated with the use or abuse of many substances, including alcohol, nicotine, cannabis, opioids, cocaine, amphetamines, and caffeine (reviewed in [31,32,33]). Psychostimulants, such as cocaine, nicotine, and amphetamine, may cause sleep loss acutely [33, 34]; narcotics such as opioids can both increase sleepiness and impair sleep quality acutely, and the complex effects with the specific contexts are reviewed elsewhere [1, 2, 35]. Chronic substance uses often lead to persistent sleep disturbances, including difficulty falling asleep, sleep fragmentation, frequent awakenings, reduction of sleep time, poor sleep quality, daytime sleepiness, and abnormalities or shifts in the timing of the cyclic sleep architecture [3, 33, 34]. Following withdrawal or detoxification from alcohol, psychostimulants, or narcotics, sleep disturbances are prevalent [3, 33], with some notable differences. For example, patients with alcohol or cannabis use disorder have higher incidence of sleep-onset insomnia, whereas patients with cocaine or heroin use disorder frequently experience sleep-maintenance insomnia [36]. The extent of sleep disturbances further predicts subsequent use of illicit drugs and alcohol [37]. There have been animal models that recapitulate some of the sleep abnormalities in SUDs, including intermittent ethanol vaper exposure [38], opioid self-administration (SA) [39], and cocaine SA in rodents [40] (Box 2).
Sleep-mediated regulation of reward-seeking behaviors
In generally healthy populations, acute sleep loss is often associated with an increase in reward-seeking behaviors. In human adolescents, shorter sleep duration is associated with more snacking and over-consumption of high-calorie food [41]. Pregnant women with poor sleep quality are more likely to have stronger and more frequent food cravings, together with higher hedonic hunger [42]. In young adult males, one night of SD increases the desire for high-calorie foods [43]. Other than food reward, overnight SD increases smoking in healthy cigarette smokers [44], shifts economic preferences toward higher gains [45], and promotes risk taking for higher gains [46]. Similar phenomena have also been observed in animals. Sleep disturbance animal models (Table 1) are often used to examine the impact on a variety of reward-seeking behaviors, including natural and drug reward. In male mice, acute SD (zeitgeber time: ZT0-6, gentle handling) increases the seeking and consumption of sucrose reward but not lab chow [47]. Chronic sleep restriction in male rats (ZT2-6, gentle handling, 4 h/day × 7 days) increases voluntary alcohol consumption [48]. Acute SD (ZT0-4/8, novel object exploration) in male rats increases the rate and efficiency of cocaine infusion during a motivational test, without changing the perceived value of cocaine [49]. Chronic sleep restriction (EEG-based disk-treadmill method, ~25% reduction in baseline sleep over 8 days) in male rats increases the perceived value of cocaine selectively in high drug-taking rats [50]. Chronic REM SD (flowerpot-over-water) in rats lowers the threshold for intra-cranial self-stimulation for reward sensation [51]. While increasing reward seeking during sleep loss may have adaptive values such as consuming calories to sustain energy expenditure, it can also be maladaptive—increasing chances for developing obesity [52, 53], risk taking, and substance use. Meanwhile, the widespread increase in reward-seeking behaviors across species and reward modalities following sleep disturbance also suggests potential common underlying neural substrates.
The brain reward circuitry—composition and functional interaction with sleep
The mesolimbic reward pathway is an interconnected neurocircuit that regulates reward-cue encoding, reward evaluation, and execution of reward seeking. The nucleus accumbens (NAc) resides in the ventral striatum and serves as a limbic-motor interface that integrates and prioritizes emotional and motivational inputs for motor outputs [54,55,56,57]. The NAc receives convergent glutamatergic inputs from the medial prefrontal cortex (mPFC), hippocampus, amygdala, among other regions, which carry various information on reward-associated cues, context, and executive controls. Moreover, the NAc is an important target of the mesolimbic dopamine (DA) projection, which carries information on reward-cue salience and reward prediction error [58, 59]. In addition, NAc neurons express a rich repertoire of neuropeptide receptors, including opioid, hypocretin/orexin (Hcrt), and melanin-concentrating hormone (MCH) receptors and many more, relaying information from the hypothalamus, thalamus, midbrain, brain stem, etc. to influence various reward-associated behaviors [60]. Thus, the NAc is a converging hub where top-down controls from the cortex interact with a variety of bottom-up emotional and motivational drives (Fig. 1a). Increasing evidence suggests that the reward circuit presents multi-layered targets for sleep and sleep disturbance to regulate reward-associated emotional and motivational responses.

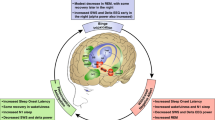
a Acute sleep loss often results in reduced top-down controls, contrasting with increased bottom-up drives. Following acute SD, PFC shows reduced coupling with the NAc and amygdala (AMG), possibly through adenosine build-up and reduced glutamatergic transmission efficacy; some subregional increase in activity is also observed. Hippocampus (Hipp) shows deficits in glutamatergic signaling and synaptic plasticity. NAc shows increased reactivity to reward or risk taking, possibly through decreased dopamine D2/3 receptor signaling, thus biasing toward D1-receptor signaling. The VTA and AMG show increased reactivity to emotional stimuli. The AMG also shows increased reactivity to food reward-associated cues. The hypocretin (Hcrt)/orexin (Orx) system based in the LH may represent a regulatory hub. It shows enhanced activity following sleep disturbances through increase in the activity of Hcrt neurons, increase in prepro-orexin synthesis and orexin release, and increase in OX1R and OX2R receptor expressions in the target regions. Hcrt signaling may orchestrate various neurotransmitter and modulator systems, including glutamate, GABA, acetylcholine, DA, norepinephrine, and 5-HT, and promote reward-associated sign-tracking (PVT-NAc), attention (basal forebrain-PFC), impulsivity and risk taking (PFC, NAc), and reward perception (NAc). These effects may, together, lead to typical increases in reward-seeking behaviors, including palatable food wanting and consumption, alcohol intake, cigarette smoking, and illicit drug use. DR: dorsal raphe nucleus (a main source of 5-HT neurons). Details see Tables 2 and 3. b Withdrawal from chronic drug use affects not only the reward circuit, but also the sleep-regulatory mechanisms, often resulting in persistent sleep disturbances at night and excessive sleepiness during the day, which may, in turn, facilitate drug relapse. In rats trained to self-administer cocaine, drug withdrawal induces excessive activation of Hcrt neurons in the LH, which may contribute to sleep instability as well as increase in drug seeking. Moreover, cocaine withdrawal dampens MCH neuron intrinsic membrane excitability and impairs glutamatergic transmissions, which may compromise MCH neuron’s role in REM sleep regulations. Hypofunction of MCH neurons may result in dis-inhibition of downstream targets, worsening cocaine-induced hyperactivity of Hcrt neurons in LH and exacerbating CP-AMPAR accumulation at NAc principal neuron synapses, which together facilitate incubation of cocaine craving. REM sleep interventions, including REM sleep restriction-rebound and sleep-warming, engage MCH neuron activities in sleep, and promote REM sleep through downstream targets such as medial septum (MS), tuberomammillary nucleus (TMN), and dorsal raphe (DR), among others. MCH neuron activities in REM sleep may also produce anti-relapse effects hypothetically through MCH-Hcrt neuron interactions in the LH as well as downstream mutual targets including the NAc, hippocampus, amygdala, and VTA.
The NAc-interconnected reward circuit has substantial anatomical overlap with the sleep-regulatory network (Table 2; details reviewed in [1, 61]). Some regions regulate both reward and sleep, including the PFC, NAc, ventral tegmental area (VTA), habenula (Hb), and lateral hypothalamus (LH); most of them are known to be affected by sleep disturbance in various ways (Table 2). How these regional changes induced by sleep disturbances may orchestrate to produce behavioral outcome is not fully understood. One example comes from human functional MRI studies. Following acute SD, the medial frontal cortex shows reduced coupling with the amygdala and NAc in response to pleasure-evoking stimuli, suggesting reduced top-down controls, whereas amygdala, NAc, and VTA activities show increase to various reward stimuli, suggesting increased bottom-up drives [62]. Thus, the overall compromised top-down controls combined with increased bottom-up drives may synergize to result in biased reward processing favoring risk taking and reward seeking after acute SD [63]. This notion would be consistent with (1) increased impulsivity and decreased inhibitory control sometimes observed after acute SD [64,65,66,67]; and (2) increased subjective value of the reward or reward-associated cues following acute sleep disturbances. In rats and mice, acute SD increases the preference to contextual cues associated with cocaine or amphetamine experience [68, 69]. In humans, preference for the stimulant methylphenidate is driven by sleepiness [70]; and higher perception of cocaine strength is reported after 24 h of SD [71]. Consistent with these changes in top-down versus bottom-up drives, SD can shift the decision-making strategy from loss-defending toward gain-seeking [45, 72].
It should be noted that the above results in humans are mostly obtained from adults or young adults, and there may be important differences in the adolescent brain regarding the sleep-reward responding relationship. In early through late pubertal adolescents, there is less activation in the caudate during both reward anticipation and reward outcome in individuals with shorter sleep/later onset and earlier offset and lower sleep quality [73]. However, there have been limited sleep manipulation studies in adolescents—it is not clear whether acute SD in adolescents may similarly blunt subcortical reward responding, and whether reward “insensitivity” in this context may also contribute to risk taking and high-reward seeking.
Compared to acute SD, less is known about the impact of chronic sleep disturbance on the reward circuit. Recently, using a low-stress sleep fragmentation paradigm in mice, it was shown that the medial Hb (mHb) exhibits increased spontaneous pace-making activity following 5–7 days of chronic REM sleep fragmentation [74]. This is postulated to promote mHb-mediated anxiety and anhedonia [75,76,77,78,79] following chronic sleep disturbance. The potential molecular and cellular mechanisms mediating these sleep disturbance-induced changes will be discussed in the next section.
Molecular and cellular mechanisms that contribute to sleep-mediated regulation of reward function
Neurotransmitter and neuromodulator systems
Our insights into sleep-mediated regulation of neurotransmitter/modulator systems predominantly come from acute SD studies, with limited results from chronic sleep manipulations. Acute SD has broad impact across brain regions and over many neurotransmitter systems, including SD-induced extracellular accumulation of metabolites (e.g., adenosine, ceremide) that promote sleep, and changes in pro-wakefulness neurotransmissions (e.g., DA, Hcrt). When these changes occur in the reward circuit, they may result in acute changes in reward processing and thus impact associated behaviors. Summarized in Table 3 are details on the sleep manipulation paradigms, impacted neurotransmitter/modulator systems, brain regions, cellular effects, and outcome or expected outcome on reward-associated behaviors. A few are highlighted below.
Hcrt
Hcrt signaling not only sustains wakefulness and promotes arousal [80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100], but also facilitates reward seeking including natural and drug rewards [68, 98, 101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124]. Acute SD and chronic sleep restriction both enhance Hcrt signaling through increasing prepro-orexin synthesis, orexin-A release, and/or OX1 and OX2 receptor expressions (Table 3). Sleep disturbance-induced increase in the activities of Hcrt system is thought to critically contribute to dysregulated reward seeking ranging from binge eating disorder to SUDs (reviewed in [123,122,125]). However, direct assessment of such causal relationships has been limited. Recently, it was shown in male mice that acute SD (4 h starting at ZT0-ZT2) enhances cocaine conditioned place preference (CPP), and that systemic administration of an orexin 1 receptor antagonist attenuates the SD effect [68], suggesting that SD-induced increase in Hcrt signaling can indeed enhance drug-reward seeking. The Hcrt system may regulate diverse aspects of reward-seeking behaviors through its wide projection targets and versatile cellular effects. For example, Hcrt through receptor signaling in the paraventricular thalamus (PVT) critically regulates reward-associated sign-tracking conditioned responses [126]; Hcrt signaling in the VTA increases glutamate release and transmission efficacy onto DA neurons, facilitates DA release (reviewed in [127]), and promotes impulsive behaviors [128]; in the basal forebrain, Hcrt increases cholinergic cell activity, resulting in robust acetylcholine release in the PFC and sustained attention [129]; in the NAc, Hcrt increases GABAergic transmission onto the principal neurons (tested in dissociated neurons [130]), and increases hedonic “liking” and/or “wanting” of palatable food reward [131]. In addition, Hcrt also regulates serotonergic (5-HT) system in dorsal raphe, norepinephrinergic system in the locus coeruleus, and histaminergic system in the tuberomammillary nucleus (TMN) (reviewed in [1]). Thus, the Hcrt system orchestrates a full collection of neurotransmitter and modulator systems to promote motivational activation in various circumstances (reviewed in [132]). Their up-regulation in response to sleep disturbances may tip the top-down (e.g. attention) versus bottom-up balance (e.g. reward-cue salience, hedonic values etc.), resulting in reward dysfunction.
DA
There are mixed results regarding DA release following sleep disturbance. Acute SD (6 h, gentle handling) in rats increases extracellular DA metabolites in the basal forebrain [133]; acute SD (4 h, gentle handling) in hamsters increases hypothalamic DA and its metabolites [134]; chronic REM SD (flowerpot-over-water; 96 h) in rats increases DA metabolites in the striatum but not cortex [135]; yet REM SD for 16 h in rats does not appear to modulate overall DA release in the NAc assessed by microdialysis [136], suggesting procedure-dependent, and brain region-specific regulations. Furthermore, acute SD decreases the availability of DA D2/3 receptors in the NAc in humans [136]. This is thought to result in a D1-D2/3 imbalance, which may increase the tendency for impulsivity and risk taking [137, 138].
Adenosine
The accumulation of extracellular adenosine upon acute SD suppresses neurotransmitter release, and, when occurs in the cortex, would “tune down” the top-down control. On the other hand, adenosine modulates postsynaptic mGluR5-homer1a-mTOR signaling as shown in the cortex, producing anti-depressive effects (Table 3).
Ceramide
A recent study identified a lipid product ceramide, which accumulates during acute SD. It promotes sleep through direct inhibition of thalamic reticular neuron firing [139]. Interestingly, the source of ceramide is thought to be microglia [139], the CNS immune cells that play active roles in synaptic pruning during sleep [140, 141]. Related to reward and drug seeking, a decrease in ceramide in dorsal hippocampus is associated with faster extinction of learned appetitive behaviors in rats [142]. Moreover, inhibition of ceramide biosynthesis attenuates the development of tolerance to morphine [143]. Whether and how ceramide accumulation from SD may impact reward function is not known.
In summary, changes in neurotransmitter systems underlie the reduced top-down controls and increased bottom-up drives, which may together increase reward motivation and seeking (Fig. 1a). This will have important implications for SUDs (see below).
Synaptic plasticity
In addition to acute modulation of neurotransmissions, sleep can also impose enduring effects by inducing long-term synaptic plasticity. A great deal of insight was obtained from learning-and-memory studies in the visual, somatosensory, and motor cortices as well as the hippocampal circuits, where diverse mechanisms are engaged in sleep to produce long-term synaptic potentiation or depression as well as homeostatic synaptic scaling [23, 144,145,146,147,148,149,150,151,152,153]. On the one hand, slow waves during NREM sleep and theta waves in REM sleep sort different neuronal ensembles across the peaks and throughs, enabling spike timing-dependent synaptic potentiation or depression [149, 154, 155]; on the other hand, NREM and REM sleep states provide dramatic fluctuations in neuromodulators such as increase in acetylcholine and MCH in REM sleep, decrease in 5-HT, and almost complete silence of norepinephrine transmission in REM sleep, which further modulate synaptic plasticity [145, 153, 155,156,157,158,159,160,161,162,163]. Furthermore, imposed upon these background fluctuations are the “memory replays” that occur both during NREM and REM sleep, which combine with the synchronous population activity and fluctuation of neuromodulators to influence synapses and neurocircuits. These sleep effects are postulated to mediate distinct aspects of learning, memory, as well as creativity [151].
Whereas most of sleep-assisted synaptic plasticity is studied in the context of sensory cortex development, cognition, or motor learning and memory, similar cellular processes may occur in the limbic circuit to regulate emotion and motivation. Many “signature” sleep waves occur in the reward circuit. Slow-wave activity in NREM sleep, the most prominent sleep EEG feature, is frequently generated in the PFC-orbital frontal cortices and propagates as a wave [164]; theta waves as a prominent feature in REM sleep are found in the hippocampus, amygdala, and cortex (reviewed in [165]). These waves could provide opportunities for spike timing-dependent plasticity to take place in large scales in the reward circuit. Moreover, “memory replays” are often observed in hippocampal “place” cells during sleep [166,167,168]. In mice acquired cocaine CPP, the hippocampal “place” cells are coupled to NAc principal neurons in wake and sleep through theta oscillations, which is thought to underlie the potentiation of hippocampal-NAc coupling and the increase in NAc principal neuron firing in the cocaine zone [169]. In addition to promoting synaptic strengthening, sleep may also facilitate learning-associated synaptic weakening. In the mouse frontal association cortex, auditory-cued fear conditioning induces spine eliminations. This process is dependent on REM sleep through a calcium-dependent mechanism [170]. Finally, opposite to promoting “memory”, sleep may also enable “forgetfulness”. For example, in the mouse hippocampus, MCH neuron terminal activity suppresses pyramidal neuron firing, and MCH neuron activities during REM sleep promote the elimination of contextual fear memory [171]. Thus, REM sleep may serve to modify contextual memory, which together with reducing the affective tone of emotional memory [25, 63], may provide protections against the development of post-traumatic stress disorder phenotypes in rodents and humans [172,173,174]. In summary, sleep is integral to synaptic plasticity across cortex, hippocampus, and the interconnected circuit. It is fully capable of playing diverse roles in the formation and modification of emotional memories. However, direct demonstrations of sleep-specific synaptic plasticity that regulates natural or drug reward-seeking behaviors have been limited.
Sleep-mediated regulation of gene expression
Another avenue for sleep to impose long-term changes is through regulation of gene expressions. This is reflected in sleep disturbance-induced changes in the transcriptome [175,176,177] or epigenome. The latter is just beginning to be understood [178]. Changes in gene expression and regulation could be the result of neuronal and glial ensemble activity in sleep or sleep disturbance combined with neurochemical changes discussed above, which may, in turn, feedback to sustained regulation of neural activity. Indeed, comparing mouse cortical transcriptome between sleep, 6-h-SD, and 4-h-SD + 2-h-sleep recovery conditions, more than half of the SD-altered genes continue to be differentially expressed after recovery sleep, suggesting sustained changes induced by SD [175]. Although acute SD profoundly alters the cortical transcriptomes [175, 177], whether and how these changes alter subsequent reward processing is poorly understood. Notably, a few of the SD-sensitive transcriptional hubs in the cortex or basal forebrain are similarly affected by repeated drug exposures, including MEF2C, PER2, JUNB, NFκB, FOSB, and EGR1 etc., some of which have been examined for potential implications in SUDs (Table 4). Thus, sleep disturbance may interact with drug experience at the level of transcription regulation to exert long-term impact on cellular functions and behaviors, including predisposition to drug use, drug experience, and drug withdrawal. Nonetheless, the transcriptomic data from sleep studies are mostly focused on the cortex, whereas data from SUD patients or addiction animal models are overwhelmingly focused on the NAc [179]. Thus, future studies need to bridge the two by examining overlapping brain regions shared by sleep and reward regulations.
Chronic sleep disturbance may affect the reward circuit and cellular processes in ways distinct from acute SD effects. Research in this area has been limited, and mostly relies on high-stress sleep disturbance models (Table 1; [180]). Using disk-over-water method for chronic sleep restriction in rats, it was shown that long-term sleep restriction alters cortical transcriptome qualitatively different from that following acute SD [176]. In a separate study, rats were sleep-restricted using a periodically rotating wheel for 18 h daily, and adrenergic receptors of different subtypes in the basal forebrain and anterior cingulate cortex show differential expressions following 1 day versus 3–5 days of sleep restriction [181], consistent with the notion that sleep undergoes allostasis as sleep debt accumulates [182, 183]. However, it is not known how the specific changes may affect cortical function or reward processing. Using a customized treadmill system for chronic selective REM sleep fragmentation with minimum stress, it was shown that the mHb cholinergic neurons in mice increase tonic firing through reduced activity of an acid-sensing potassium channel KCNK9 [74]. The behavioral consequences of this effect are not known, although increase in mHb activity may modulate a variety of affect/reward functions ranging from aversion, anxiety, anhedonia, to nicotine and alcohol abuse, as well as reinstatement of cocaine CPP [77,78,79, 184,185,186,187]. Understanding the impact of chronic sleep disturbance on the reward circuit is of high clinical significance. This will be greatly facilitated by the development of robust, automated, and noninvasive chronic sleep disturbance models that introduce minimum procedural stress.
Implications in SUDs
Initial drug use
In humans, initiation of drug use often occurs during adolescence [188,189,190]. The median ages for first opportunity and first use of illicit drugs are 13 years and 14 years, respectively, in the US as of 2011 [191]. In 2019, 1.8 million among 12–17-year olds had first time use of alcohol, 1 million for marijuana, 385,000 for cigarette smoking, and 82,000 for cocaine [192]. Moreover, based on a 2010 survey in a nationally representative sample (N = 2524) of 10th graders in the US, 8% were “predominant polysubstance users” [193]. This is greatly concerning because of the association between younger initiation and poorer outcome in developing SUDs [188,189,190]. There are many reasons that set adolescence a vulnerable period for initiating drug use [194], and compromised sleep is an unequivocal one. Although 8–10 h of sleep per night is needed for optimal function in adolescents, only 29% of US high school students reach this amount, and 44% of US high schoolers sleep for less than 6 h per night [195]. Acute sleep loss weakens top-down controls and increases bottom-up drives for reward, which may result in biases toward reward-associated sign-tracking (speculated above), increases in impulsivity, risk taking, and reward responding [46, 196] (Fig. 1a). All these aspects, combined with the fast-developing mesolimbic system and slowly maturing prefrontal cortex in the adolescent brain [194], may facilitate drug seeking and taking behaviors.
Compared to acute SD, implications of chronic sleep disturbance in SUDs are less explored. A longitudinal study shows that over 70% of individuals with insomnia or hypersomnia develop some forms of psychiatric problems, including major depression and SUDs, in their lifetime [197]. Insomnia is also a commonly reported cause for self-medication with alcohol, which often precedes the development of alcohol use disorder [198]. It is not clear, however, whether it is chronic sleep problems that lead to substance use as self-medication, or there could be a third causal factor—e.g., genetic-environmental predisposition—that leads to both sleep problems and SUDs, with sleep problems manifesting first.
Relapse to drug use after withdrawal—preclinical studies targeting sleep improvement
Sleep-drug use interactions take place since the initial drug use and persist through long-term withdrawal [2, 3, 199]. On the one hand, sleep dysregulation persists, indicating drug withdrawal-induced prolonged neurophysiological changes; and conversely, chronic sleep disturbance is thought to precipitate relapse to drug use [2, 49, 50, 199,200,201,202,203,204,205]. The relationship between sleep disturbance and SUDs is thus thought to be bi-directional, which forms a vicious cycle that drives continued substance use and relapse [200]. Sleep disturbance may tilt the balance between top-down controls and bottom-up drives that contribute to drug craving and relapse after withdrawal. However, the bottom-up drive at this stage is likely to be more about alleviating negative affect than obtaining reward [206].
Sleep disturbance resulting from drug withdrawal could be pathophysiologically different from externally imposed SD. For example, withdrawal-induced sleep disturbance may reflect sleep allostasis, and experimentally induced sleep disturbance in either naïve or drug-experienced individuals is a deviation from homeostasis. Considering this difference, it is important to use animal models that recapitulate the long-term sleep disturbances following substance use and withdrawal, and examine whether increasing or improving sleep may be beneficial.
Increasing sleep is not the mathematical opposite of SD, as sleep is not simply a lack of wakefulness, but rather an active process of similar scales. This is clearly demonstrated in transcriptomic studies, which show a large number of sleep-specific gene expressions qualitatively distinct from those during wakefulness or SD [177]. In this regard, it is equally important to complement sleep disturbance studies by addressing whether increasing sleep time or improving sleep quality after drug withdrawal affects relapse.
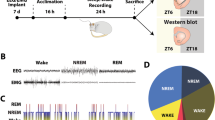
To tackle this problem, we have used a rat cocaine SA model. Following SA training (0.75 mg/kg/infusion, 1 overnight + 5 day × 2 h/day) and withdrawal, rats experience sustained sleep loss and fragmentation [40], qualitatively recapitulating the human situations [3]. Moreover, this cocaine procedure induces “incubation of cocaine craving”—a progressive intensification of cue-induced cocaine seeking after withdrawal, which indicates increased propensity for drug relapse (Box 2). Additionally, this cocaine procedure induces a persistent synaptic accumulation of calcium-permeable AMPA receptors (CP-AMPARs) after withdrawal in NAc principal neurons [40, 207,208,209], which are key synaptic substrates for cocaine incubation [210,211,212,213,214]. These features allow for behavioral and electrophysiological assessment of the effects of sleep interventions after cocaine experience.
First, using a sleep restriction-rebound strategy to increase and consolidate light-phase (inactive phase) sleep in rats, we found that selective REM sleep restriction-rebound during withdrawal day 22–42 leads to decreased incubation of cocaine craving tested on withdrawal day 45. This is accompanied by decreased CP-AMPARs at NAc principal neuron synapses [40]. Next, focusing on the role of REM sleep in this regulation, we used environmental warming to selectively increase REM sleep. Warming to near thermoneutrality preferentially increases REM sleep in drug naïve animals [215,216]. In rats after cocaine withdrawal, warming leads to a selective increase in total REM sleep time and decrease in REM sleep fragmentation, and the REM sleep effects are accompanied by reduced incubation of cocaine craving as well as decrease in NAc CP-AMPARs [207]. Together, these results suggest a potential causal relationship between REM sleep and relapse to cocaine use.
LH MCH system comes into play—contrasting with Hcrt system
LH MCH neurons critically regulate REM sleep onsets and/or bout durations [217,218,219]. Both REM sleep restriction-induced rebound in REM sleep and warming-induced increase of REM sleep engage the activity of LH MCH neurons [216, 220]. These neurons are predominantly REM sleep-active [221], with greater population activities during long-bout versus short-bout REM sleep episodes [207]. Cocaine withdrawal-induced REM sleep fragmentation concurs with persistent decrease in the membrane excitability of these neurons, as well as impairment in glutamatergic transmission efficacy [222]. To counteract cocaine-induced deficits in MCH neurons, enhancing MCH neuron activities in sleep by environmental warming or direct chemo- or optogenetic stimulations similarly decreases cocaine incubation [207]. Moreover, locally infusing MCH peptide into NAc decreases cocaine-induced synaptic accumulation of CP-AMPARs and reduces incubation [207], suggesting that MCH-to-NAc projections could play an important role. In the NAc, cocaine-induced CP-AMPARs are enriched in GluA1 subunits [210, 211, 223, 224]. MCH receptor activation in the NAc dephosphorylates GluA1 at Serine 845 and facilitates their removal from synapses [225]. Thus, consolidated REM sleep may engage MCH neurons to regulate cocaine seeking after withdrawal in part through regulating NAc CP-AMPARs (Fig. 1b).
In contrast to the hypoactivity of MCH neurons, the LH Hcrt neurons show enhanced functionality following cocaine experience. After rats learn to self-administer cocaine with intermittent access for 14 d (0.2 mg/infusion, 5 min/30 min × 12 epochs/d), they undergo withdrawal for >150 d, followed by a re-exposure to the SA environment. The rats show an increase in the number of Hcrt neurons in LH and increase in Hcrt neuron activities, as measured by the proportion of Hcrt neurons that show Fos immunoreactivity, which further show a significant correlation with incubation of cocaine craving [101]. Excessive activation of Hcrt neurons may promote drug seeking through diverse projection targets as discussed above. In addition, Hcrt neurons may drive sleep instability after cocaine withdrawal, as they do in the aging brain [100].
In summary, repeated cocaine exposure and withdrawal can lead to reduced activities of MCH neurons and enhanced activities of Hcrt neurons in the LH, which may synergistically impair sleep and promote drug seeking (Fig. 1b).
Targeting sleep for SUDs
Through diverse neurotransmitter and modulator systems, all substance uses result in sleep changes, and persistent sleep disturbances are a common complaint following drug and alcohol withdrawal [3, 33, 34, 36, 226]. Sleep problems are thought to fuel drug relapse, and could indeed predict the outcome of drug and alcohol relapse in human patients [37, 227]. This may also be directly relevant to polysubstance uses, where patients may intend to self-medicate and resort to alcohol, marijuana, or opioids to mend impaired sleep from various drug withdrawal; or alternatively, use psychostimulants to promote wakefulness to counteract the withdrawal-induced excessive sleepiness during the day [228]. Polysubstance use is increasingly gaining attention [229,230], for which animal models are beginning to be developed (reviewed in [230]). In summary, targeting sleep may be applicable to both mono- and poly-SUDs, and may benefit drug relapse far beyond sleep improvement per se.
The Hcrt system represents a prominent candidate for this goal. Excessive activation of Hcrt system is commonly observed during withdrawal from a variety of substances, including cocaine, opioids, nicotine, and alcohol [1, 123, 124, 231], suggesting the possibility of targeting Hcrt system in general either for individual substance use or polysubstance use (e.g. cocaine + alcohol [232]). Recent studies focused on suvorexant, an FDA-approved, insomnia-treating dual Hcrt/orexin receptor OX1R/OX2R antagonist, show promising results for improving withdrawal symptoms in various preclinical models, raising the possibility that the reward circuit can be targeted through its shared components with the sleep-regulatory machinery [233]. A few other sleep-promoting medications have been proposed to be tested for treating SUDs, including topiramate, tiagabine, gaboxadol, vigabatrin, and lisuride [3]. The reported sleep effects are either a selective increase in slow-wave sleep time, or a reduction in REM sleep. While some have safety issues as GABA enhancers, none are shown to be particularly effective yet in reducing drug relapse [3]. More recently, modafinil, a mild psychostimulant acting on DA transporters, shows promising results in reducing cocaine relapse. In the study, modafinil is taken in the morning to increase alertness during the day, resulting in improved nighttime sleep. Patients with this modafinil regimen exhibit reduced rate of cocaine relapse, with the improvement positively correlated with the amount of N3 stage NREM sleep; and the positive correlation persists even after correction for modafinil treatment [234]. However, modafinil does not alter REM sleep architecture in this study (but see [235]).
Could REM sleep-MCH neuron activity benefit SUDs in general? The notion of enhancing REM sleep to achieve benefits may appear counter-intuitive, as many antidepressants that increase brain monoaminergic signaling suppress or delay REM sleep [236]. However, REM sleep may have both pro-relapse (i.e. sleep fragmentation) and anti-relapse components, and MCH neuron activity in REM sleep may represent a key to the anti-relapse effects [207]. MCH neurons project throughout the brain, and many limbic structures important for reward processing have moderate to high levels of MCH receptor expressions, including NAc, hippocampus, VTA, PFC, amygdala, and LH [237, 238]. Many of these regions also express Hcrt receptors and receive Hcrt fiber projections [239,240,241,242]. Thus, in the LH, MCH neurons inhibit Hcrt neurons through local inhibitory circuits, which has been detailed in sleep and energy expenditure regulations [243, 244]. In ventral hippocampus, MCH neuron terminal activities and MCH receptor signaling regulate impulsivity [245]; stimulating MCH neuron axon terminals suppresses pyramidal neuron firing, and REM sleep-active MCH neuron activity facilitates the erasure of contextual fear memory [171]. This may be in contrast with the role of Hcrt in facilitating hippocampus-dependent memory formation [246]. Other brain regions receiving MCH and Hcrt dual innervations include PFC, amygdala, NAc, VTA, among others [107, 237,238,239,240,241,242, 247, 248]. Therefore, it is possible that MCH neuron activities modulate Hcrt effects from both the source and the target regions, shaping the reward network through synaptic and cellular plasticity mechanisms in REM sleep (Fig. 1b). It will be meaningful to test whether REM sleep-enhancing medications, particularly those that can increase MCH neuron activity or MCH signaling during REM sleep and prolong REM sleep episodes without causing REM sleep fragmentation, alleviate withdrawal symptoms and reduce relapse to drug use.
Individual variations
Both sleep phenotypes and SUD development exhibit substantial individual variations. Sleep time, architecture, and waveforms are influenced by genetic and environmental factors [249]. SUD development is also complex, involving personality and physiological traits as well as individual experiences [250]. Genetic factors may strongly influence personality and physiological traits such as impulsivity, risk taking, and reward/stress responsivity, which tap into the vulnerability to SUDs [250]. Environmental factors include drug availability, peer influence, stress, lifestyle, and others, to which circadian misalignment and sleep disturbance could also be contributors [194]. Specifically, how may sleep interact with genetics to produce diverse individual variations in SUDs?
An inspiring example comes from rodent studies of sign-tracking versus goal-tracking behaviors. These are naturally occurring behaviors through Pavlovian learning, in which reward-associated cues acquire incentive salience in sign-tracking animals, whereas in goal-tracking ones the cues obtain predictive value that predominates over incentive salience [251]. It is thought that sign-trackers may be more vulnerable to addiction, as their reaction to reward-associated cues is more relying on subcortical drive rather than cortical executive control [252]. Mechanistically, sign-tracking relies on PVT Hcrt receptor signaling [126] and (presumed downstream) phasic DA release in the NAc core [58]. In response to reward-associated cues, both c-Fos activities in LH-PVT and PVT-NAc projection neurons and phasic DA release in the NAc are more prominent in sign-trackers than in goal trackers [58, 253]. Thus, comparing the top-down versus bottom-up dynamics, sign-trackers may have stronger bottom-up drive than goal trackers at baseline, which may render sign-trackers higher susceptibility to acute SD-induced weakening in top-down controls (reviewed in [61]).
Although sign-tracking may not be directly applied to humans, certain genetic polymorphisms affecting Hcrt or DA systems are associated with SUDs and/or SD susceptibility. For example, in a genome-wide association study, a Hcrt receptor 2 (HCRTR2) gene polymorphism shows association with nicotine dependence as well as the age for initial methamphetamine use [254]. HCRTR2 is also a candidate gene in sertraline- (a selective serotonin reuptake inhibitor antidepressant) associated insomnia in depressed patients [255]. Regarding the DA system, a variable number tandem repeat polymorphism in the human PER2 gene is associated with lower availability of striatal D2 receptors and cocaine abuse [256]. Moreover, a nine-repeat DA transporter (DAT) allele in human is linked to higher phasic DA activity and increased striatal responsivity to reward anticipation following SD, and a ten-repeat DAT allele is linked to lower phasic DA activity and increased striatal responsivity to punishment following SD [257]. These examples highlight the interaction between sleep and genotypes in producing individual variations.
Based on the sleep-reward interactions at circuit, cellular, and molecular levels discussed above, we suggest that future efforts be focused on the following three aspects to further dissect sleep-based individual variations in SUDs: (1) to explore the potential correlation between the sensitivity to acute SD in reward-associated tasks and susceptibility to drug-taking and seeking tests at the level of individual subjects; (2) to explore the potential correlation between key sleep parameters (e.g., REM sleep fragmentation and microstructures) and scores in SUD tests; (3) to explore the potential correlation between sleep-associated genetic polymorphisms and SUD phenotypes.
Conclusions and future directions
By summarizing recent results, this review discusses how sleep influences reward processing through circuit, cellular, and molecular substrates. This sleep-mediated regulation and the underlying mechanisms offer a conceptual possibility not only to improve the wellbeing of healthy populations and safe-guard adolescence development, but also develop treatment for SUDs. Considering sleep disruption both as a comorbidity to SUDs and a potential causal factor for drug relapse, manipulating sleep quality may serve as complementary therapeutics treating SUDs and mood disorders. Moreover, by integrating key sleep parameters, a sleep metrics may be formulated to predict individual susceptibility to SUDs and other reward-associated psychiatric diseases.
References
Valentino RJ, Volkow ND. Drugs, sleep, and the addicted brain. Neuropsychopharmacology. 2020;45:3–5.
Eacret D, Veasey SC, Blendy JA. Bidirectional relationship between opioids and disrupted sleep: putative mechanisms. Mol Pharmacol. 2020;98:445–53.
Angarita GA, Emadi N, Hodges S, Morgan PT. Sleep abnormalities associated with alcohol, cannabis, cocaine, and opiate use: a comprehensive review. Addict Sci Clin Pract. 2016;11:9.
Anafi RC, Kayser MS, Raizen DM. Exploring phylogeny to find the function of sleep. Nat Rev Neurosci. 2019;20:109–16.
Siegel JM. Do all animals sleep? Trends Neurosci. 2008;31:208–13.
Sauer S, Kinkelin M, Herrmann E, Kaiser W. The dynamics of sleep-like behaviour in honey bees. J Comp Physiol A. 2003;189:599–607.
Cirelli C, Bushey D. Sleep and wakefulness in Drosophila melanogaster. Ann N Y Acad Sci. 2008;1129:323–29.
Kanaya HJ, Park S, Kim J-H, Kusumi J, Krenenou S, Sawatari E, et al. A sleep-like state in Hydra unravels conserved sleep mechanisms during the evolutionary development of the central nervous system. Sci Adv. 2020;6:eabb9415.
Yamazaki R, Toda H, Libourel PA, Hayashi Y, Vogt KE, Sakurai T. Evolutionary origin of distinct NREM and REM sleep. Front Psychol. 2020;11:567618.
Rechtschaffen A, Bergmann BM, Gilliland MA, Bauer K. Effects of method, duration, and sleep stage on rebounds from sleep deprivation in the rat. Sleep. 1999;22:11–31.
Beersma D, Dijk D, Blok C, Everhardus I. REM sleep deprivation during 5 h leads to an immediate REM sleep rebound and to suppression of non-REM sleep intensity. Electroencephalogr Clin Neurophysiol. 1990;76:114–22.
Everson CA, Bergmann BM, Rechtschaffen A. Sleep deprivation in the rat: III. Total sleep deprivation. Sleep 1989;12:13–21.
Kushida CA, Bergmann BM, Rechtschaffen A. Sleep deprivation in the rat: IV. Paradoxical sleep deprivation. Sleep 1989;12:22–30.
Fulda S, Schulz H. Cognitive dysfunction in sleep disorders. Sleep Med Rev. 2001;5:423–45.
Walker MP. Cognitive consequences of sleep and sleep loss. Sleep Med. 2008;9:S29–S34.
Berger RJ, Phillips NH. Energy conservation and sleep. Behav Brain Res. 1995;69:65–73.
Besedovsky L, Lange T, Born J. Sleep and immune function. Pflügers Arch-Eur J Physiol. 2012;463:121–37.
Zager A, Andersen ML, Ruiz FS, Antunes IB, Tufik S. Effects of acute and chronic sleep loss on immune modulation of rats. Am J Physiol Reguly Integr Comp Physiol. 2007;293:R504–R09.
Graven S. Sleep and brain development. Clin Perinatol. 2006;33:693–706.
Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–60.
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7.
Benveniste H, Heerdt PM, Fontes M, Rothman DL, Volkow ND. Glymphatic system function in relation to anesthesia and sleep states. Anesth Analg. 2019;128:747–58.
Wang G, Grone B, Colas D, Appelbaum L, Mourrain P. Synaptic plasticity in sleep: learning, homeostasis and disease. Trends Neurosci. 2011;34:452–63.
Palmer CA, Alfano CA. Sleep and emotion regulation: an organizing, integrative review. Sleep Med Rev. 2017;31:6–16.
Walker MP. Sleep, memory and emotion. Prog Brain Res. 2010;185:49–68.
Steptoe A, O’Donnell K, Marmot M, Wardle J. Positive affect, psychological well-being, and good sleep. J Psychosom Res. 2008;64:409–15.
Fairholme CP, Manber R. Sleep, emotions, and emotion regulation: an overview. Elsevier Science: EBook; 2015; p. 45–61.
Krystal AD. Psychiatric disorders and sleep. Neurologic Clin. 2012;30:1389–413.
Benca RM. Sleep in psychiatric disorders. Neurologic Clin. 1996;14:739–64.
American Psychiatric Association D, Association AP. Diagnostic and statistical manual of mental disorders: DSM-5. Washington, DC: American Psychiatric Association; 2013.
Conroy DA, Arnedt JT. Sleep and substance use disorders: an update. Curr Psychiatry Rep. 2014;16:1–9.
Freeman D, Sheaves B, Waite F, Harvey AG, Harrison PJ. Sleep disturbance and psychiatric disorders. The Lancet. Lancet Psychiatry. 2020;7:628–37.
Basu A, Anand N, Das M. Sleep and substance-use disorder. Sleep and Neuropsychiatric Disorders. Singapore: Springer; 2022. p. 435–64.
Cruickshank CC, Dyer KR. A review of the clinical pharmacology of methamphetamine. Addiction. 2009;104:1085–99.
Cutrufello NJ, Ianus VD, Rowley JA. Opioids and sleep. Curr Opin Pulm Med. 2020;26:634–41.
Grau-Lopez L, Grau-Lopez L, Daigre C, Palma-Alvarez RF, Martinez-Luna N, Ros-Cucurull E, et al. Insomnia symptoms in patients with substance use disorders during detoxification and associated clinical features. Front Psychiatry. 2020;11:540022.
Fortuna LR, Cook B, Porche MV, Wang Y, Amaris AM, Alegria M. Sleep disturbance as a predictor of time to drug and alcohol use treatment in primary care. Sleep Med. 2018;42:31–37.
Ehlers CL, Sanchez-Alavez M, Wills D. Effect of gabapentin on sleep and delta and theta EEG power in adult rats exposed to chronic intermittent ethanol vapor and protracted withdrawal during adolescence. Psychopharmacology 2018;235:1783–91.
Steinfels G, Young G, Khazan N. Opioid self-administration and REM sleep EEG power spectra. Neuropharmacology. 1980;19:69–74.
Chen B, Wang Y, Liu X, Liu Z, Dong Y, Huang YH. Sleep regulates incubation of cocaine craving. J Neurosci. 2015;35:13300–10.
Weiss A, Xu F, Storfer-Isser A, Thomas A, Ievers-Landis CE, Redline S. The association of sleep duration with adolescents’ fat and carbohydrate consumption. Sleep. 2010;33:1201–09.
Betts GM, Lipsky LM, Temmen CD, Siega-Riz AM, Faith MS, Nansel TR. Poorer mental health and sleep quality are associated with greater self-reported reward-related eating during pregnancy and postpartum: an observational cohort study. Int J Behav Nutr Phys Act. 2021;18:1–9.
Greer SM, Goldstein AN, Walker MP. The impact of sleep deprivation on food desire in the human brain. Nat Commun. 2013;4:2259.
Hamidovic A, de Wit H. Sleep deprivation increases cigarette smoking. Pharmacol Biochem Behav. 2009;93:263–69.
Venkatraman V, Huettel SA, Chuah LY, Payne JW, Chee MW. Sleep deprivation biases the neural mechanisms underlying economic preferences. J Neurosci. 2011;31:3712–8.
McKenna BS, Dickinson DL, Orff HJ, Drummond SP. The effects of one night of sleep deprivation on known-risk and ambiguous-risk decisions. J Sleep Res. 2007;16:245–52.
Liu Z, Wang Y, Cai L, Li Y, Chen B, Dong Y, et al. Prefrontal cortex to accumbens projections in sleep regulation of reward. J Neurosci. 2016;36:7897–910.
Garcia-Garcia F, Priego-Fernandez S, Lopez-Mucino LA, Acosta-Hernandez ME, Pena-Escudero C. Increased alcohol consumption in sleep-restricted rats is mediated by delta FosB induction. Alcohol. 2021;93:63–70.
Puhl MD, Fang J, Grigson PS. Acute sleep deprivation increases the rate and efficiency of cocaine self-administration, but not the perceived value of cocaine reward in rats. Pharmacol Biochem Behav. 2009;94:262–70.
Puhl MD, Boisvert M, Guan Z, Fang J, Grigson PS. A novel model of chronic sleep restriction reveals an increase in the perceived incentive reward value of cocaine in high drug-taking rats. Pharmacol Biochem Behav. 2013;109:8–15.
Steiner SS, Ellman SJ. Relation between REM sleep and intracranial self-stimulation. Science 1972;177:1122–4.
Cooper CB, Neufeld EV, Dolezal BA, Martin JL. Sleep deprivation and obesity in adults: a brief narrative review. BMJ Open Sport Exerc Med. 2018;4:e000392.
Beccuti G, Pannain S. Sleep and obesity. Curr Opin Clin Nutr Metab Care. 2011;14:402–12.
Pickens CL, Airavaara M, Theberge F, Fanous S, Hope BT, Shaham Y. Neurobiology of the incubation of drug craving. Trends Neurosci. 2011;34:411–20.
Kelley AE. Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev. 2004;27:765–76.
Mogenson GJ, Jones DL, Yim CY. From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol. 1980;14:69–97.
Robbins TW, Everitt BJ. Neurobehavioural mechanisms of reward and motivation. Curr Opin Neurobiol. 1996;6:228–36.
Flagel SB, Clark JJ, Robinson TE, Mayo L, Czuj A, Willuhn I, et al. A selective role for dopamine in stimulus-reward learning. Nature. 2011;469:53–7.
Schultz W. Dopamine signals for reward value and risk: basic and recent data. Behav Brain Funct. 2010;6:24.
Castro DC, Bruchas MR. A motivational and neuropeptidergic hub: anatomical and functional diversity within the nucleus accumbens shell. Neuron. 2019;102:529–52.
Ahrens AM, Ahmed OJ. Neural circuits linking sleep and addiction: animal models to understand why select individuals are more vulnerable to substance use disorders after sleep deprivation. Neurosci Biobehav Rev. 2020;108:435–44.
Gujar N, Yoo SS, Hu P, Walker MP. Sleep deprivation amplifies reactivity of brain reward networks, biasing the appraisal of positive emotional experiences. J Neurosci. 2011;31:4466–74.
Krause AJ, Simon EB, Mander BA, Greer SM, Saletin JM, Goldstein-Piekarski AN, et al. The sleep-deprived human brain. Nat Rev Neurosci. 2017;18:404–18.
Saksvik-Lehouillier I, Saksvik SB, Dahlberg J, Tanum TK, Ringen H, Karlsen HR, et al. Mild to moderate partial sleep deprivation is associated with increased impulsivity and decreased positive affect in young adults. Sleep. 2020;43:zsaa078.
Pezzato F, Silveira D, Novais D, Hoshino K. Assessment of impulsivity in REM-sleep deprived rats. Sleep Sci. 2012;5:79–83.
Zhao R, Zhang X, Fei N, Zhu Y, Sun J, Liu P, et al. Decreased cortical and subcortical response to inhibition control after sleep deprivation. Brain Imaging Behav. 2019;13:638–50.
Rossa KR, Smith SS, Allan AC, Sullivan KA. The effects of sleep restriction on executive inhibitory control and affect in young adults. J Adolesc Health. 2014;55:287–92.
Bjorness TE, Greene RW. Sleep deprivation enhances cocaine conditioned place preference in an orexin receptor-modulated manner. eNeuro. 2020;7. https://doi.org/10.1523/ENEURO.0283-20.2020.
Berro LF, Tufik SB, Frussa-Filho R, Andersen ML, Tufik S. Sleep deprivation precipitates the development of amphetamine-induced conditioned place preference in rats. Neurosci Lett. 2018;671:29–32.
Roehrs T, Papineau K, Rosenthal L, Roth T. Sleepiness and the reinforcing and subjective effects of methylphenidate. Exp Clin Psychopharmacol. 1999;7:145.
Fischman MW, Schuster CR. Cocaine effects in sleep-deprived humans. Psychopharmacology. 1980;72:1–8.
Killgore WD, Balkin TJ, Wesensten NJ. Impaired decision making following 49 h of sleep deprivation. J Sleep Res. 2006;15:7–13.
Holm SM, Forbes EE, Ryan ND, Phillips ML, Tarr JA, Dahl RE. Reward-related brain function and sleep in pre/early pubertal and mid/late pubertal adolescents. J Adolesc Health. 2009;45:326–34.
Ge F, Mu P, Guo R, Cai L, Liu Z, Dong Y, et al. Chronic sleep fragmentation enhances habenula cholinergic neural activity. Mol Psychiatry. 2021;26:941–54.
Han S, Yang SH, Kim JY, Mo S, Yang E, Song KM, et al. Down-regulation of cholinergic signaling in the habenula induces anhedonia-like behavior. Sci Rep. 2017;7:900.
Salas R, Sturm R, Boulter J, De, Biasi M. Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci. 2009;29:3014–8.
Yamaguchi T, Danjo T, Pastan I, Hikida T, Nakanishi S. Distinct roles of segregated transmission of the septo-habenular pathway in anxiety and fear. Neuron. 2013;78:537–44.
Zhang J, Tan L, Ren Y, Liang J, Lin R, Feng Q, et al. Presynaptic excitation via GABAB receptors in habenula cholinergic neurons regulates fear memory expression. Cell. 2016;166:716–28.
Lopez AJ, Jia Y, White AO, Kwapis JL, Espinoza M, Hwang P, et al. Medial habenula cholinergic signaling regulates cocaine-associated relapse-like behavior. Addict Biol. 2019;24:403–13.
Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, et al. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–62.
Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, et al. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci USA. 1999;96:10911–6.
Eggermann E, Serafin M, Bayer L, Machard D, Saint-Mleux B, Jones BE, et al. Orexins/hypocretins excite basal forebrain cholinergic neurones. Neuroscience. 2001;108:177–81.
Xi MC, Morales FR, Chase MH. Effects on sleep and wakefulness of the injection of hypocretin-1 (orexin-A) into the laterodorsal tegmental nucleus of the cat. Brain Res. 2001;901:259–64.
Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, Watanabe T, et al. Arousal effect of orexin A depends on activation of the histaminergic system. Proc Natl Acad Sci USA. 2001;98:9965–70.
Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, et al. Distinct narcolepsy syndromes in Orexin receptor-2 and Orexin null mice: molecular genetic dissection of Non-REM and REM sleep regulatory processes. Neuron 2003;38:715–30.
Mieda M, Willie JT, Hara J, Sinton CM, Sakurai T, Yanagisawa M. Orexin peptides prevent cataplexy and improve wakefulness in an orexin neuron-ablated model of narcolepsy in mice. Proc Natl Acad Sci USA. 2004;101:4649–54.
Yamanaka A, Tsujino N, Funahashi H, Honda K, Guan JL, Wang QP, et al. Orexins activate histaminergic neurons via the orexin 2 receptor. Biochem Biophys Res Commun. 2002;290:1237–45.
Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–98.
Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–20.
Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–54.
Hara J, Yanagisawa M, Sakurai T. Difference in obesity phenotype between orexin-knockout mice and orexin neuron-deficient mice with same genetic background and environmental conditions. Neurosci Lett. 2005;380:239–42.
Wu MF, John J, Maidment N, Lam HA, Siegel JM. Hypocretin release in normal and narcoleptic dogs after food and sleep deprivation, eating, and movement. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1079–86.
Yukitake H, Fujimoto T, Ishikawa T, Suzuki A, Shimizu Y, Rikimaru K, et al. TAK-925, an orexin 2 receptor-selective agonist, shows robust wake-promoting effects in mice. Pharmacol Biochem Behav. 2019;187:172794.
Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–81.
Chieffi S, Carotenuto M, Monda V, Valenzano A, Villano I, Precenzano F, et al. Orexin system: the key for a healthy life. Front Physiol. 2017;8:357.
Gotter AL, Webber AL, Coleman PJ, Renger JJ, Winrow CJ. International Union of Basic and Clinical Pharmacology. LXXXVI. Orexin receptor function, nomenclature and pharmacology. Pharmacol Rev. 2012;64:389–420.
Willie JT, Chemelli RM, Sinton CM, Yanagisawa M. To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci. 2001;24:429–58.
Mahler SV, Smith RJ, Moorman DE, Sartor GC, Aston-Jones G. Multiple roles for orexin/hypocretin in addiction. Prog Brain Res. 2012;198:79–121.
Adamantidis A, de Lecea L. The hypocretins as sensors for metabolism and arousal. J Physiol. 2009;587:33–40.
Li SB, Damonte VM, Chen C, Wang GX, Kebschull JM, Yamaguchi H, et al. Hyperexcitable arousal circuits drive sleep instability during aging. Science. 2022;375:eabh3021.
James MH, Stopper CM, Zimmer BA, Koll NE, Bowrey HE, Aston-Jones G. Increased number and activity of a lateral subpopulation of hypothalamic orexin/hypocretin neurons underlies the expression of an addicted state in rats. Biol Psychiatry. 2019;85:925–35.
Muschamp JW, Hollander JA, Thompson JL, Voren G, Hassinger LC, Onvani S, et al. Hypocretin (orexin) facilitates reward by attenuating the antireward effects of its cotransmitter dynorphin in ventral tegmental area. Proc Natl Acad Sci USA. 2014;111:E1648–55.
Olney JJ, Navarro M, Thiele TE. The role of orexin signaling in the ventral tegmental area and central amygdala in modulating binge-like ethanol drinking behavior. Alcohol Clin Exp Res. 2017;41:551–61.
Fatahi Z, Assar N, Mahmoudi D, Pahlevani P, Moradi M, Haghparast A. Functional interaction between the orexin-1 and CB1 receptors within the nucleus accumbens in the conditioned place preference induced by the lateral hypothalamus stimulation. Pharmacol Biochem Behav. 2015;132:42–48.
Narita M, Nagumo Y, Hashimoto S, Narita M, Khotib J, Miyatake M, et al. Direct involvement of orexinergic systems in the activation of the mesolimbic dopamine pathway and related behaviors induced by morphine. J Neurosci. 2006;26:398–405.
Guo SJ, Cui Y, Huang ZZ, Liu H, Zhang XQ, Jiang JX, et al. Orexin A-mediated AKT signaling in the dentate gyrus contributes to the acquisition, expression and reinstatement of morphine-induced conditioned place preference. Addict Biol. 2016;21:547–59.
Farzinpour Z, Taslimi Z, Azizbeigi R, Karimi-Haghighi S, Haghparast A. Involvement of orexinergic receptors in the nucleus accumbens, in the effect of forced swim stress on the reinstatement of morphine seeking behaviors. Behav Brain Res. 2019;356:279–87.
Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601.
James MH, Charnley JL, Levi EM, Jones E, Yeoh JW, Smith DW, et al. Orexin-1 receptor signalling within the ventral tegmental area, but not the paraventricular thalamus, is critical to regulating cue-induced reinstatement of cocaine-seeking. Int J Neuropsychopharmacol. 2011;14:684–90.
Prince CD, Rau AR, Yorgason JT, Espana RA. Hypocretin/Orexin regulation of dopamine signaling and cocaine self-administration is mediated predominantly by hypocretin receptor 1. ACS Chem Neurosci. 2015;6:138–46.
Korotkova TM, Sergeeva OA, Eriksson KS, Haas HL, Brown RE. Excitation of ventral tegmental area dopaminergic and nondopaminergic neurons by orexins/hypocretins. J Neurosci. 2003;23:7–11.
Tung LW, Lu GL, Lee YH, Yu L, Lee HJ, Leishman E, et al. Orexins contribute to restraint stress-induced cocaine relapse by endocannabinoid-mediated disinhibition of dopaminergic neurons. Nat Commun. 2016;7:12199.
Haynes AC, Jackson B, Chapman H, Tadayyon M, Johns A, Porter RA, et al. A selective orexin-1 receptor antagonist reduces food consumption in male and female rats. Regul Pept. 2000;96:45–51.
Yamada H, Okumura T, Motomura W, Kobayashi Y, Kohgo Y. Inhibition of food intake by central injection of anti-orexin antibody in fasted rats. Biochem Biophys Res Commun. 2000;267:527–31.
Haynes AC, Chapman H, Taylor C, Moore GB, Cawthorne MA, Tadayyon M, et al. Anorectic, thermogenic and anti-obesity activity of a selective orexin-1 receptor antagonist in ob/ob mice. Regul Pept. 2002;104:153–9.
Thorpe AJ, Kotz CM. Orexin A in the nucleus accumbens stimulates feeding and locomotor activity. Brain Res. 2005;1050:156–62.
Shoblock JR, Welty N, Aluisio L, Fraser I, Motley ST, Morton K, et al. Selective blockade of the orexin-2 receptor attenuates ethanol self-administration, place preference, and reinstatement. Psychopharmacology. 2011;215:191–203.
Alizamini MM, Farzinpour Z, Ezzatpanah S, Haghparast A. Role of intra-accumbal orexin receptors in the acquisition of morphine-induced conditioned place preference in the rats. Neurosci Lett. 2017;660:1–5.
Khosrowabadi E, Karimi-Haghighi S, Jamali S, Haghparast A. Differential roles of intra-accumbal orexin receptors in acquisition and expression of methamphetamine-induced conditioned place preference in the rats. Neurochem Res. 2020;45:2230–41.
Fartootzadeh R, Alaei H, Reisi P. Mutual assistance of nucleus accumbens cannabinoid receptor-1 and orexin receptor-2 in response to nicotine: a single-unit study. Res Pharm Sci. 2021;16:173–81.
Sahafzadeh M, Karimi-Haghighi S, Mousavi Z, Haghparast A. Role of the orexin receptors within the nucleus accumbens in the drug priming-induced reinstatement of morphine seeking in the food deprived rats. Brain Res Bull. 2018;137:217–24.
Assar N, Mahmoudi D, Mousavi Z, Zarrabian S, Haghparast A. Role of orexin-1 and −2 receptors within the nucleus accumbens in the acquisition of sensitization to morphine in rats. Behav Brain Res. 2019;373:112090.
James MH, Mahler SV, Moorman DE, Aston-Jones G. A decade of orexin/hypocretin and addiction: where are we now? Curr Top Behav Neurosci. 2017;33:247–81.
Mehr JB, Bilotti MM, James MH. Orexin (hypocretin) and addiction. Trends Neurosci. 2021;44:852–55.
Mehr JB, Mitchison D, Bowrey HE, James MH. Sleep dysregulation in binge eating disorder and “food addiction”: the orexin (hypocretin) system as a potential neurobiological link. Neuropsychopharmacology. 2021;46:2051–61.
Iglesias AG, Flagel SB. The paraventricular thalamus as a critical node of motivated behavior via the hypothalamic-thalamic-striatal circuit. Front Integr Neurosci. 2021;15:706713.
Baimel C, Borgland SL. Hypocretin modulation of drug-induced synaptic plasticity. Prog Brain Res. 2012;198:123–31.
Gentile TA, Simmons SJ, Watson MN, Connelly KL, Brailoiu E, Zhang Y, et al. Effects of suvorexant, a dual orexin/hypocretin receptor antagonist, on impulsive behavior associated with cocaine. Neuropsychopharmacology. 2018;43:1001–09.
Fadel J, Burk JA. Orexin/hypocretin modulation of the basal forebrain cholinergic system: role in attention. Brain Res. 2010;1314:112–23.
Martin G, Fabre V, Siggins GR, de Lecea L. Interaction of the hypocretins with neurotransmitters in the nucleus accumbens. Regul Pept. 2002;104:111–7.
Castro DC, Terry RA, Berridge KC. Orexin in rostral hotspot of nucleus accumbens enhances sucrose ‘liking’ and intake but scopolamine in caudal shell shifts ‘liking’ toward ‘disgust’ and ‘fear’. Neuropsychopharmacology. 2016;41:2101–11.
Mahler SV, Moorman DE, Smith RJ, James MH, Aston-Jones G. Motivational activation: a unifying hypothesis of orexin/hypocretin function. Nat Neurosci. 2014;17:1298–303.
Zant JC, Leenaars CH, Kostin A, Van Someren EJ, Porkka-Heiskanen T. Increases in extracellular serotonin and dopamine metabolite levels in the basal forebrain during sleep deprivation. Brain Res. 2011;1399:40–8.
Asikainen M, Deboer T, Porkka-Heiskanen T, Stenberg D, Tobler I. Sleep deprivation increases brain serotonin turnover in the Djungarian hamster. Neurosci Lett. 1995;198:21–4.
Farooqui SM, Brock JW, Zhou J. Changes in monoamines and their metabolite concentrations in REM sleep-deprived rat forebrain nuclei. Pharmacol Biochem Behav. 1996;54:385–91.
Volkow ND, Tomasi D, Wang GJ, Telang F, Fowler JS, Logan J, et al. Evidence that sleep deprivation downregulates dopamine D2R in ventral striatum in the human brain. J Neurosci. 2012;32:6711–7.
Dalley JW, Fryer TD, Brichard L, Robinson ES, Theobald DE, Laane K, et al. Nucleus accumbens D2/3 receptors predict trait impulsivity and cocaine reinforcement. Science. 2007;315:1267–70.
Linnet J, Moller A, Peterson E, Gjedde A, Doudet D. Inverse association between dopaminergic neurotransmission and Iowa Gambling Task performance in pathological gamblers and healthy controls. Scand J Psychol. 2011;52:28–34.
Liu H, Wang X, Chen L, Chen L, Tsirka SE, Ge S, et al. Microglia modulate stable wakefulness via the thalamic reticular nucleus in mice. Nat Commun. 2021;12:4646.
Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–34.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–8.
Huston JP, Kornhuber J, Muhle C, Japtok L, Komorowski M, Mattern C, et al. A sphingolipid mechanism for behavioral extinction. J Neurochem. 2016;137:589–603.
Ndengele MM, Cuzzocrea S, Masini E, Vinci MC, Esposito E, Muscoli C, et al. Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J Pharmacol Exp Ther. 2009;329:64–75.
Gronli J, Soule J, Bramham CR. Sleep and protein synthesis-dependent synaptic plasticity: impacts of sleep loss and stress. Front Behav Neurosci. 2013;7:224.
Poe GR. Sleep Is for forgetting. J Neurosci. 2017;37:464–73.
Seibt J, Frank MG. Primed to sleep: the dynamics of synaptic plasticity across brain states. Front Syst Neurosci. 2019;13:2.
Frank MG. Sleep and synaptic plasticity in the developing and adult brain. Curr Top Behav Neurosci. 2015;25:123–49.
Kuhn M, Wolf E, Maier JG, Mainberger F, Feige B, Schmid H, et al. Sleep recalibrates homeostatic and associative synaptic plasticity in the human cortex. Nat Commun. 2016;7:12455.
Puentes-Mestril C, Roach J, Niethard N, Zochowski M, Aton SJ. How rhythms of the sleeping brain tune memory and synaptic plasticity. Sleep. 2019;42:zsz095.
Frank MG, Cantera R. Sleep, clocks, and synaptic plasticity. Trends Neurosci. 2014;37:491–501.
Lewis PA, Knoblich G, Poe G. How memory replay in sleep boosts creative problem-solving. Trends Cogn Sci. 2018;22:491–503.
Abel T, Havekes R, Saletin JM, Walker MP. Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol. 2013;23:R774–88.
Gonzalez-Rueda A, Pedrosa V, Feord RC, Clopath C, Paulsen O. Activity-dependent downscaling of subthreshold synaptic inputs during slow-wave-sleep-like activity in vivo. Neuron. 2018;97:1244–52.
Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–48.
Miyamoto D, Hirai D, Murayama M. The roles of cortical slow waves in synaptic plasticity and memory consolidation. Front Neural Circuits. 2017;11:92.
Brzosko Z, Mierau SB, Paulsen O. Neuromodulation of spike-timing-dependent plasticity: past, present, and future. Neuron. 2019;103:563–81.
Samanta A, Alonso A, Genzel L. Memory reactivations and consolidation: considering neuromodulators across wake and sleep. Curr Opin Physiol. 2020;15:120–27.
Tully K, Bolshakov VY. Emotional enhancement of memory: how norepinephrine enables synaptic plasticity. Mol Brain. 2010;3:15.
Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76:116–29.
Pachoud B, Adamantidis A, Ravassard P, Luppi PH, Grisar T, Lakaye B, et al. Major impairments of glutamatergic transmission and long-term synaptic plasticity in the hippocampus of mice lacking the melanin-concentrating hormone receptor-1. J Neurophysiol. 2010;104:1417–25.
Crispino M, Volpicelli F, Perrone-Capano C. Role of the serotonin receptor 7 in brain plasticity: from development to disease. Int J Mol Sci. 2020;21:505.
Meunier CN, Chameau P, Fossier PM. Modulation of synaptic plasticity in the cortex needs to understand all the players. Front Synaptic Neurosci. 2017;9:2.
Lesch KP, Waider J. Serotonin in the modulation of neural plasticity and networks: implications for neurodevelopmental disorders. Neuron. 2012;76:175–91.
Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G. The sleep slow oscillation as a traveling wave. J Neurosci. 2004;24:6862–70.
Hutchison IC, Rathore S. The role of REM sleep theta activity in emotional memory. Front Psychol. 2015;6:1439.
Lee AK, Wilson MA. Memory of sequential experience in the hippocampus during slow wave sleep. Neuron. 2002;36:1183–94.
Nadasdy Z, Hirase H, Czurko A, Csicsvari J, Buzsaki G. Replay and time compression of recurring spike sequences in the hippocampus. J Neurosci. 1999;19:9497–507.
Skaggs WE, McNaughton BL. Replay of neuronal firing sequences in rat hippocampus during sleep following spatial experience. Science. 1996;271:1870–3.
Sjulson L, Peyrache A, Cumpelik A, Cassataro D, Buzsaki G. Cocaine place conditioning strengthens location-specific hippocampal coupling to the nucleus accumbens. Neuron. 2018;98:926–34 e5.
Zhou Y, Lai CSW, Bai Y, Li W, Zhao R, Yang G, et al. REM sleep promotes experience-dependent dendritic spine elimination in the mouse cortex. Nat Commun. 2020;11:4819.
Izawa S, Chowdhury S, Miyazaki T, Mukai Y, Ono D, Inoue R, et al. REM sleep-active MCH neurons are involved in forgetting hippocampus-dependent memories. Science. 2019;365:1308–13.
Davis CJ, Vanderheyden WM. Optogenetic sleep enhancement improves fear-associated memory processing following trauma exposure in rats. Sci Rep. 2020;10:18025.
Harrington MO, Ashton JE, Sankarasubramanian S, Anderson MC, Cairney SA. Losing control: sleep deprivation impairs the suppression of unwanted thoughts. Clin Psychol Sci. 2021;9:97–113.
Germain A, Buysse DJ, Shear MK, Fayyad R, Austin C. Clinical correlates of poor sleep quality in posttraumatic stress disorder. J Trauma Stress. 2004;17:477–84.
Bjorness TE, Kulkarni A, Rybalchenko V, Suzuki A, Bridges C, Harrington AJ, et al. An essential role for MEF2C in the cortical response to loss of sleep in mice. Elife. 2020;9:e58331.
Cirelli C, Faraguna U, Tononi G. Changes in brain gene expression after long-term sleep deprivation. J Neurochem. 2006;98:1632–45.
Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41:35–43.
Gaine ME, Chatterjee S, Abel T. Sleep deprivation and the epigenome. Front Neural Circuits. 2018;12:14.
Nestler EJ. Transcriptional mechanisms of drug addiction. Clin Psychopharmacol Neurosci. 2012;10:136–43.
Reynolds AC, Banks S. Total sleep deprivation, chronic sleep restriction and sleep disruption. Prog Brain Res. 2010;185:91–103.
Kim Y, Chen L, McCarley RW, Strecker RE. Sleep allostasis in chronic sleep restriction: the role of the norepinephrine system. Brain Res. 2013;1531:9–16.
Kim Y, Laposky AD, Bergmann BM, Turek FW. Repeated sleep restriction in rats leads to homeostatic and allostatic responses during recovery sleep. Proc Natl Acad Sci USA. 2007;104:10697–702.
McEwen BS. Sleep deprivation as a neurobiologic and physiologic stressor: allostasis and allostatic load. Metabolism 2006;55:S20–3.
Kobayashi Y, Sano Y, Vannoni E, Goto H, Suzuki H, Oba A, et al. Genetic dissection of medial habenula-interpeduncular nucleus pathway function in mice. Front Behav Neurosci. 2013;7:17.
Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601.
Frahm S, Slimak MA, Ferrarese L, Santos-Torres J, Antolin-Fontes B, Auer S, et al. Aversion to nicotine is regulated by the balanced activity of beta4 and alpha5 nicotinic receptor subunits in the medial habenula. Neuron. 2011;70:522–35.
McLaughlin I, Dani JA, De, Biasi M. The medial habenula and interpeduncular nucleus circuitry is critical in addiction, anxiety, and mood regulation. J Neurochem. 2017;142:130–43.
Volkow ND, Han B, Einstein EB, Compton WM. Prevalence of substance use disorders by time since first substance use among young people in the US. JAMA Pediatr. 2021;175:640–43.
Sharma A, Morrow JD. Neurobiology of adolescent substance use disorders. Child Adolesc Psychiatr Clin N Am. 2016;25:367–75.
Chen CY, Storr CL, Anthony JC. Early-onset drug use and risk for drug dependence problems. Addict Behav. 2009;34:319–22.
Swendsen J, Burstein M, Case B, Conway KP, Dierker L, He J, et al. Use and abuse of alcohol and illicit drugs in US adolescents: results of the National Comorbidity Survey-Adolescent Supplement. Arch Gen Psychiatry. 2012;69:390–8.
SAMHSA. Key substance use and mental health indicators in the United States: results from the 2020 National Survey on Drug Use and Health. Rockville, MD: SAMHSA (Substance Abuse and Mental Health Services Administration); 2021.
Conway KP, Vullo GC, Nichter B, Wang J, Compton WM, Iannotti RJ, et al. Prevalence and patterns of polysubstance use in a nationally representative sample of 10th graders in the United States. J Adolesc Health. 2013;52:716–23.
Logan RW, Hasler BP, Forbes EE, Franzen PL, Torregrossa MM, Huang YH, et al. Impact of sleep and circadian rhythms on addiction vulnerability in adolescents. Biol Psychiatry. 2018;83:987–96.
Basch CE, Basch CH, Ruggles KV, Rajan S. Prevalence of sleep duration on an average school night among 4 nationally representative successive samples of American high school students, 2007-2013. Prev Chronic Dis. 2014;11:E216.
Venkatraman V, Chuah YM, Huettel SA, Chee MW. Sleep deprivation elevates expectation of gains and attenuates response to losses following risky decisions. Sleep. 2007;30:603–9.
Breslau N, Roth T, Rosenthal L, Andreski P. Sleep disturbance and psychiatric disorders: a longitudinal epidemiological study of young adults. Biol Psychiatry. 1996;39:411–18.
Brower KJ, Aldrich MS, Robinson EA, Zucker RA, Greden JF. Insomnia, self-medication, and relapse to alcoholism. Am J Psychiatry. 2001;158:399–404.
Bjorness TE, Greene RW. Interaction between cocaine use and sleep behavior: a comprehensive review of cocaine’s disrupting influence on sleep behavior and sleep disruptions influence on reward seeking. Pharmacol Biochem Behav. 2021;206:173194.
Ara A, Jacobs W, Bhat IA, McCall WV. Sleep disturbances and substance use disorders: a bi-directional relationship. Psychiatr Ann. 2016;46:408–12.
Brower KJ, Perron BE. Sleep disturbance as a universal risk factor for relapse in addictions to psychoactive substances. Med Hypotheses. 2010;74:928–33.
Gillin J. Are sleep disturbances risk factors for anxiety, depressive and addictive disorders? Acta Psychiatr Scand. 1998;98:39–43.
Malcolm R, Myrick LH, Veatch LM, Boyle E, Randall PK. Self-reported sleep, sleepiness, and repeated alcohol withdrawals: a randomized, double blind, controlled comparison of lorazepam vs gabapentin. J Clin Sleep Med. 2007;3:24–32.
Roehrs T, Johanson CE, Meixner R, Turner L, Roth T. Reinforcing and subjective effects of methylphenidate: dose and time in bed. Exp Clin Psychopharmacol. 2004;12:180–9.
Teplin D, Raz B, Daiter J, Varenbut M, Tyrrell M. Screening for substance use patterns among patients referred for a variety of sleep complaints. Am J drug alcohol Abus. 2006;32:111–20.
Koob GF, Schulkin J. Addiction and stress: an allostatic view. Neurosci Biobehav Rev. 2019;106:245–62.
Guo R, Wang Y, Yan R, Chen B, Ding W, Gorczyca MT, et al. REM sleep engages MCH neurons to reduce cocaine seeking. Biological Psychiatry. (in press).
Ma YY, Lee BR, Wang X, Guo C, Liu L, Cui R, et al. Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron. 2014;83:1453–67.
Lee BR, Ma YY, Huang YH, Wang X, Otaka M, Ishikawa M, et al. Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat Neurosci. 2013;16:1644–51.
Loweth JA, Tseng KY, Wolf ME. Adaptations in AMPA receptor transmission in the nucleus accumbens contributing to incubation of cocaine craving. Neuropharmacology. 2014;76:287–300.
Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–21.
McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M. Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J Neurosci. 2011;31:5737–43.
Terrier J, Luscher C, Pascoli V. Cell-type specific insertion of GluA2-lacking AMPARs with cocaine exposure leading to sensitization, cue-induced seeking, and incubation of craving. Neuropsychopharmacology. 2016;41:1779–89.
Zinsmaier AK, Dong Y, Huang YH. Cocaine-induced projection-specific and cell type-specific adaptations in the nucleus accumbens. Mol Psychiatry. 2022;27:669–86.
Szymusiak R, Satinoff E. Maximal REM sleep time defines a narrower thermoneutral zone than does minimal metabolic rate. Physiol Behav. 1981;26:687–90.
Komagata N, Latifi B, Rusterholz T, Bassetti CLA, Adamantidis A, Schmidt MH. Dynamic REM sleep modulation by ambient temperature and the critical role of the melanin-concentrating hormone system. Curr Biol. 2019;29:1976–87 e4.
Tsunematsu T, Ueno T, Tabuchi S, Inutsuka A, Tanaka KF, Hasuwa H, et al. Optogenetic manipulation of activity and temporally controlled cell-specific ablation reveal a role for MCH neurons in sleep/wake regulation. J Neurosci. 2014;34:6896–909.
Konadhode RR, Pelluru D, Blanco-Centurion C, Zayachkivsky A, Liu M, Uhde T, et al. Optogenetic stimulation of MCH neurons increases sleep. J Neurosci. 2013;33:10257–63.
Jego S, Glasgow SD, Herrera CG, Ekstrand M, Reed SJ, Boyce R, et al. Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat Neurosci. 2013;16:1637–43.
Modirrousta M, Mainville L, Jones BE. Orexin and MCH neurons express c-Fos differently after sleep deprivation vs. recovery and bear different adrenergic receptors. Eur J Neurosci. 2005;21:2807–16.
Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci USA. 2009;106:2418–22.
Wang Y, Guo R, Chen B, Rahman T, Cai L, Li Y, et al. Cocaine-induced neural adaptations in the lateral hypothalamic melanin-concentrating hormone neurons and the role in regulating rapid eye movement sleep after withdrawal. Mol Psychiatry. 2021;26:3152–68.
Wolf ME, Tseng KY. Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front Mol Neurosci. 2012;5:72.
Ferrario CR, Loweth JA, Milovanovic M, Ford KA, Galinanes GL, Heng LJ, et al. Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca(2)(+)-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacology. 2011;61:1141–51.
Sears RM, Liu RJ, Narayanan NS, Sharf R, Yeckel MF, Laubach M, et al. Regulation of nucleus accumbens activity by the hypothalamic neuropeptide melanin-concentrating hormone. J Neurosci. 2010;30:8263–73.
Chakravorty S, Vandrey RG, He S, Stein MD. Sleep management among patients with substance use disorders. Med Clin N Am. 2018;102:733–43.
Gann H, Feige B, Hohagen F, van Calker D, Geiss D, Dieter R. Sleep and the cholinergic rapid eye movement sleep induction test in patients with primary alcohol dependence. Biol Psychiatry. 2001;50:383–90.
Volkow ND. Connections between sleep and substance use disorders. 2020. https://nida.nih.gov/about-nida/noras-blog/2020/03/connections-between-sleep-substance-use-disorders.
Liu Y, Williamson V, Setlow B, Cottler LB, Knackstedt LA. The importance of considering polysubstance use: lessons from cocaine research. Drug Alcohol Depend. 2018;192:16–28.
Crummy EA, O’Neal TJ, Baskin BM, Ferguson SM. One is not enough: understanding and modeling polysubstance use. Front Neurosci. 2020;14:569.
Walker LC, Lawrence AJ. The role of orexins/hypocretins in alcohol use and abuse. Curr Top Behav Neurosci. 2017;33:221–46.
James MH, Fragale JE, O’Connor SL, Zimmer BA, Aston-Jones G. The orexin (hypocretin) neuropeptide system is a target for novel therapeutics to treat cocaine use disorder with alcohol coabuse. Neuropharmacology. 2021;183:108359.
James MH, Fragale JE, Aurora RN, Cooperman NA, Langleben DD, Aston-Jones G. Repurposing the dual orexin receptor antagonist suvorexant for the treatment of opioid use disorder: why sleep on this any longer? Neuropsychopharmacology. 2020;45:717–19.
Morgan PT, Angarita GA, Canavan S, Pittman B, Oberleitner L, Malison RT, et al. Modafinil and sleep architecture in an inpatient-outpatient treatment study of cocaine dependence. Drug Alcohol Depend. 2016;160:49–56.
Morgan PT, Pace-Schott E, Pittman B, Stickgold R, Malison RT. Normalizing effects of modafinil on sleep in chronic cocaine users. Am J Psychiatry. 2010;167:331–40.
Vogel GW, Buffenstein A, Minter K, Hennessey A. Drug effects on REM sleep and on endogenous depression. Neurosci Biobehav Rev. 1990;14:49–63.
Hervieu GJ, Cluderay JE, Harrison D, Meakin J, Maycox P, Nasir S, et al. The distribution of the mRNA and protein products of the melanin-concentrating hormone (MCH) receptor gene, slc-1, in the central nervous system of the rat. Eur J Neurosci. 2000;12:1194–216.
Saito Y, Cheng M, Leslie FM, Civelli O. Expression of the melanin-concentrating hormone (MCH) receptor mRNA in the rat brain. J Comp Neurol. 2001;435:26–40.
Ikeno T, Yan L. A comparison of the orexin receptor distribution in the brain between diurnal Nile grass rats (Arvicanthis niloticus) and nocturnal mice (Mus musculus). Brain Res. 2018;1690:89–95.
Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, et al. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25.
Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015.
Nixon JP, Smale L. A comparative analysis of the distribution of immunoreactive orexin A and B in the brains of nocturnal and diurnal rodents. Behav Brain Funct. 2007;3:28.
Konadhode RR, Pelluru D, Shiromani PJ. Neurons containing orexin or melanin concentrating hormone reciprocally regulate wake and sleep. Front Syst Neurosci. 2014;8:244.
Rao Y, Lu M, Ge F, Marsh DJ, Qian S, Wang AH, et al. Regulation of synaptic efficacy in hypocretin/orexin-containing neurons by melanin concentrating hormone in the lateral hypothalamus. J Neurosci. 2008;28:9101–10.
Noble EE, Wang Z, Liu CM, Davis EA, Suarez AN, Stein LM, et al. Hypothalamus-hippocampus circuitry regulates impulsivity via melanin-concentrating hormone. Nat Commun. 2019;10:4923.
Yang L, Zou B, Xiong X, Pascual C, Xie J, Malik A, et al. Hypocretin/orexin neurons contribute to hippocampus-dependent social memory and synaptic plasticity in mice. J Neurosci. 2013;33:5275–84.
Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, et al. The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J Comp Neurol. 1992;319:218–45.
Steininger TL, Kilduff TS, Behan M, Benca RM, Landry CF. Comparison of hypocretin/orexin and melanin-concentrating hormone neurons and axonal projections in the embryonic and postnatal rat brain. J Chem Neuroanat. 2004;27:165–81.
Van Dongen HP, Vitellaro KM, Dinges DF. Individual differences in adult human sleep and wakefulness: Leitmotif for a research agenda. Sleep. 2005;28:479–96.
Kreek MJ, Nielsen DA, Butelman ER, LaForge KS. Genetic influences on impulsivity, risk taking, stress responsivity and vulnerability to drug abuse and addiction. Nat Neurosci. 2005;8:1450–7.
Flagel SB, Akil H, Robinson TE. Individual differences in the attribution of incentive salience to reward-related cues: implications for addiction. Neuropharmacology. 2009;56 Suppl 1:139–48.
Flagel SB, Robinson TE. Neurobiological basis of individual variation in stimulus-reward learning. Curr Opin Behav Sci. 2017;13:178–85.
Haight JL, Fuller ZL, Fraser KM, Flagel SB. A food-predictive cue attributed with incentive salience engages subcortical afferents and efferents of the paraventricular nucleus of the thalamus. Neuroscience. 2017;340:135–52.
Nishizawa D, Kasai S, Hasegawa J, Sato N, Yamada H, Tanioka F, et al. Associations between the orexin (hypocretin) receptor 2 gene polymorphism Val308Ile and nicotine dependence in genome-wide and subsequent association studies. Mol Brain. 2015;8:50.
Firouzabadi N, Navabzadeh N, Moghimi-Sarani E, Haghnegahdar M. Orexin/hypocretin type 2 receptor (HCRTR2) gene as a candidate gene in sertraline-associated insomnia in depressed patients. Neuropsychiatr Dis Treat. 2020;16:1121–28.
Shumay E, Fowler JS, Wang GJ, Logan J, Alia-Klein N, Goldstein RZ, et al. Repeat variation in the human PER2 gene as a new genetic marker associated with cocaine addiction and brain dopamine D2 receptor availability. Transl Psychiatry. 2012;2:e86.
Greer SM, Goldstein AN, Knutson B, Walker MP. A genetic polymorphism of the human dopamine transporter determines the impact of sleep deprivation on brain responses to rewards and punishments. J Cogn Neurosci. 2016;28:803–10.
Raven F, Meerlo P, Van der Zee EA, Abel T, Havekes R. A brief period of sleep deprivation causes spine loss in the dentate gyrus of mice. Neurobiol Learn Mem. 2019;160:83–90.
Mang GM, Nicod J, Emmenegger Y, Donohue KD, O’Hara BF, Franken P. Evaluation of a piezoelectric system as an alternative to electroencephalogram/electromyogram recordings in mouse sleep studies. Sleep. 2014;37:1383–92.
Fenzl T, Romanowski CP, Flachskamm C, Honsberg K, Boll E, Hoehne A, et al. Fully automated sleep deprivation in mice as a tool in sleep research. J Neurosci Methods. 2007;166:229–35.
Mendelson WB, Guthrie RD, Frederick G, Wyatt RJ. The flower pot technique of rapid eye movement (REM) sleep deprivation. Pharmacol Biochem Behav. 1974;2:553–56.
Machado RB, Hipólide DC, Benedito-Silva AA, Tufik S. Sleep deprivation induced by the modified multiple platform technique: quantification of sleep loss and recovery. Brain Res. 2004;1004:45–51.
Rechtschaffen A, Bergmann BM. Sleep deprivation in the rat by the disk-over-water method. Behav Brain Res. 1995;69:55–63.
Van Luijtelaar E, Kaiser J, Coenen A. Deprivation of paradoxical sleep and intracranial self-stimulation. Sleep. 1982;5:284–89.
Van Hulzen Z, Coenen A. The pendulum technique for paradoxical sleep deprivation in rats. Physiol Behav. 1980;25:807–11.
Amici R, Cerri M, Ocampo-Garcés A, Baracchi F, Dentico D, Jones CA, et al. Cold exposure and sleep in the rat: REM sleep homeostasis and body size. Sleep. 2008;31:708–15.
NOR NSM, NAWI A, AHMAD WANW, NOORDIN L. Sleep deprivation models in rodents. J Sains Kesihat Malays. 2021;19:1–6.
Lieberman MD, Straccia MA, Meyer ML, Du M, Tan KM. Social, self, (situational), and affective processes in medial prefrontal cortex (MPFC): causal, multivariate, and reverse inference evidence. Neurosci Biobehav Rev. 2019;99:311–28.
Donnelly NA, Holtzman T, Rich PD, Nevado-Holgado AJ, Fernando AB, Van Dijck G, et al. Oscillatory activity in the medial prefrontal cortex and nucleus accumbens correlates with impulsivity and reward outcome. PLoS ONE. 2014;9:e111300.
Haber SN. Anatomy and connectivity of the reward circuit. Decision neuroscience: An integrative perspective. San Diego, CA, US: Elsevier Academic Press; 2017. p. 3–19.
Mander BA, Rao V, Lu B, Saletin JM, Lindquist JR, Ancoli-Israel S, et al. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat Neurosci. 2013;16:357–64.
Nir Y, Staba RJ, Andrillon T, Vyazovskiy VV, Cirelli C, Fried I, et al. Regional slow waves and spindles in human sleep. Neuron. 2011;70:153–69.
LeGates TA, Kvarta MD, Tooley JR, Francis TC, Lobo MK, Creed MC, et al. Reward behaviour is regulated by the strength of hippocampus-nucleus accumbens synapses. Nature. 2018;564:258–62.
Prince TM, Abel T. The impact of sleep loss on hippocampal function. Learn Mem. 2013;20:558–69.
Tang W, Jadhav SP. Sharp-wave ripples as a signature of hippocampal-prefrontal reactivation for memory during sleep and waking states. Neurobiol Learn Mem. 2019;160:11–20.
Squire LR, Genzel L, Wixted JT, Morris RG. Memory consolidation. Cold Spring Harb Perspect Biol. 2015;7:a021766.
Mullin BC, Phillips ML, Siegle GJ, Buysse DJ, Forbes EE, Franzen PL. Sleep deprivation amplifies striatal activation to monetary reward. Psychol Med. 2013;43:2215–25.
Wang Y, Liu Z, Cai L, Guo R, Dong Y, Huang YH. A critical role of basolateral amygdala-to-nucleus accumbens projection in sleep regulation of reward seeking. Biol Psychiatry. 2020;87:954–66.
Oishi Y, Xu Q, Wang L, Zhang BJ, Takahashi K, Takata Y, et al. Slow-wave sleep is controlled by a subset of nucleus accumbens core neurons in mice. Nat Commun. 2017;8:734.
McCullough KM, Missig G, Robble MA, Foilb AR, Wells AM, Hartmann J, et al. Nucleus Accumbens Medium Spiny Neuron Subtypes Differentially Regulate Stress-Associated Alterations in Sleep Architecture. Biol Psychiatry. 2021;89:1138–49.
Lazarus M, Shen HY, Cherasse Y, Qu WM, Huang ZL, Bass CE, et al. Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. J Neurosci. 2011;31:10067–75.
Luo YJ, Li YD, Wang L, Yang SR, Yuan XS, Wang J, et al. Nucleus accumbens controls wakefulness by a subpopulation of neurons expressing dopamine D1 receptors. Nat Commun. 2018;9:1576.
Ranaldi R. Dopamine and reward seeking: the role of ventral tegmental area. Rev Neurosci. 2014;25:621–30.
Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR, de Lecea L. VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat Neurosci. 2016;19:1356–66.
Baxter MG, Murray EA. The amygdala and reward. Nat Rev Neurosci. 2002;3:563–73.
Hikosaka O. The habenula: from stress evasion to value-based decision-making. Nat Rev Neurosci. 2010;11:503–13.
Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 2007;447:1111–5.
Tyree SM, de Lecea L. Lateral hypothalamic control of the ventral tegmental area: reward evaluation and the driving of motivated behavior. Front Syst Neurosci. 2017;11:50.
Olsson M, Arlig J, Hedner J, Blennow K, Zetterberg H. Sleep deprivation and cerebrospinal fluid biomarkers for Alzheimer’s disease. Sleep. 2018;41. https://doi.org/10.1093/sleep/zsy025.
Mehta R, Khanday MA, Mallick BN. REM sleep loss associated changes in orexin-A levels in discrete brain areas in rats. Neurosci Lett. 2015;590:62–7.
Xu A, Sakurai E, Kuramasu A, Zhang J, Li J, Okamura N, et al. Roles of hypothalamic subgroup histamine and orexin neurons on behavioral responses to sleep deprivation induced by the treadmill method in adolescent rats. J Pharm Sci. 2010;114:444–53.
Pedrazzoli M, D’Almeida V, Martins PJ, Machado RB, Ling L, Nishino S, et al. Increased hypocretin-1 levels in cerebrospinal fluid after REM sleep deprivation. Brain Res. 2004;995:1–6.
Ono D, Yamanaka A. Hypothalamic regulation of the sleep/wake cycle. Neurosci Res. 2017;118:74–81.
Nakamura K. The role of the dorsal raphe nucleus in reward-seeking behavior. Front Integr Neurosci. 2013;7:60.
Luo M, Zhou J, Liu Z. Reward processing by the dorsal raphe nucleus: 5-HT and beyond. Learn Mem. 2015;22:452–60.
Gardner JP, Fornal CA, Jacobs BL. Effects of sleep deprivation on serotonergic neuronal activity in the dorsal raphe nucleus of the freely moving cat. Neuropsychopharmacology. 1997;17:72–81.
Monti JM. The role of dorsal raphe nucleus serotonergic and non-serotonergic neurons, and of their receptors, in regulating waking and rapid eye movement (REM) sleep. Sleep Med Rev. 2010;14:319–27.
Hefti K, Holst SC, Sovago J, Bachmann V, Buck A, Ametamey SM, et al. Increased metabotropic glutamate receptor subtype 5 availability in human brain after one night without sleep. Biol Psychiatry. 2013;73:161–8.
Holst SC, Sousek A, Hefti K, Saberi-Moghadam S, Buck A, Ametamey SM, et al. Cerebral mGluR5 availability contributes to elevated sleep need and behavioral adjustment after sleep deprivation. Elife. 2017;6:e28751.
Holz A, Mulsch F, Schwarz MK, Hollmann M, Dobrossy MD, Coenen VA, et al. Enhanced mGlu5 signaling in excitatory neurons promotes rapid antidepressant effects via AMPA receptor activation. Neuron. 2019;104:338–52.
Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, et al. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci USA. 2007;104:20090–5.
Zhu J, Hafycz J, Keenan BT, Guo X, Pack A, Naidoo N. Acute sleep loss upregulates the synaptic scaffolding protein, Homer1a, in non-canonical sleep/wake brain regions, claustrum, piriform and cingulate cortices. Front Neurosci. 2020;14:188.
Kammermeier PJ, Worley PF. Homer 1a uncouples metabotropic glutamate receptor 5 from postsynaptic effectors. Proc Natl Acad Sci USA 2007;104:6055–60.
Ango F, Prezeau L, Muller T, Tu JC, Xiao B, Worley PF, et al. Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature. 2001;411:962–5.
Serchov T, Clement HW, Schwarz MK, Iasevoli F, Tosh DK, Idzko M, et al. Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of Homer1a. Neuron 2015;87:549–62.
Wagner KV, Hartmann J, Labermaier C, Hausl AS, Zhao G, Harbich D, et al. Homer1/mGluR5 activity moderates vulnerability to chronic social stress. Neuropsychopharmacology. 2015;40:1222–33.
Martinez D, Broft A, Foltin RW, Slifstein M, Hwang DR, Huang Y, et al. Cocaine dependence and d2 receptor availability in the functional subdivisions of the striatum: relationship with cocaine-seeking behavior. Neuropsychopharmacology. 2004;29:1190–202.
Volkow ND, Fowler JS, Wang GJ, Hitzemann R, Logan J, Schlyer DJ, et al. Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse. 1993;14:169–77.
Wiers CE, Shumay E, Cabrera E, Shokri-Kojori E, Gladwin TE, Skarda E, et al. Reduced sleep duration mediates decreases in striatal D2/D3 receptor availability in cocaine abusers. Transl Psychiatry. 2016;6:e752.
Elmenhorst D, Kroll T, Matusch A, Bauer A. Sleep deprivation increases cerebral serotonin 2A receptor binding in humans. Sleep. 2012;35:1615–23.
Zhao X, Ozols AB, Meyers KT, Campbell J, McBride A, Marballi KK, et al. Acute sleep deprivation upregulates serotonin 2A receptors in the frontal cortex of mice via the immediate early gene Egr3. Mol Psychiatry. 2022;27:1599–1610.
Asikainen M, Toppila J, Alanko L, Ward DJ, Stenberg D, Porkka-Heiskanen T. Sleep deprivation increases brain serotonin turnover in the rat. Neuroreport. 1997;8:1577–82.
Bjorvatn B, Gronli J, Hamre F, Sorensen E, Fiske E, Bjorkum AA, et al. Effects of sleep deprivation on extracellular serotonin in hippocampus and frontal cortex of the rat. Neuroscience. 2002;113:323–30.
Albert PR, Benkelfat C, Descarries L. The neurobiology of depression-revisiting the serotonin hypothesis. I. Cellular and molecular mechanisms. Philos Trans R Soc Lond B Biol Sci. 2012;367:2378–81.
Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, et al. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14:1075–81.
Briggs C, Bowes SC, Semba K, Hirasawa M. Sleep deprivation-induced pre- and postsynaptic modulation of orexin neurons. Neuropharmacology. 2019;154:50–60.
Wang L, Gu Y, Zhang J, Gong L. Effects of sleep deprivation (SD) on rats via ERK1/2 signaling pathway. Med Sci Monit. 2019;25:2886–95.
Martins PJ, Marques MS, Tufik S, D’Almeida V. Orexin activation precedes increased NPY expression, hyperphagia, and metabolic changes in response to sleep deprivation. Am J Physiol Endocrinol Metab. 2010;298:E726–34.
Elmenhorst D, Basheer R, McCarley RW, Bauer A. Sleep deprivation increases A(1) adenosine receptor density in the rat brain. Brain Res. 2009;1258:53–8.
Basheer R, Bauer A, Elmenhorst D, Ramesh V, McCarley RW. Sleep deprivation upregulates A1 adenosine receptors in the rat basal forebrain. Neuroreport. 2007;18:1895–9.
Elmenhorst D, Meyer PT, Winz OH, Matusch A, Ermert J, Coenen HH, et al. Sleep deprivation increases A1 adenosine receptor binding in the human brain: a positron emission tomography study. J Neurosci. 2007;27:2410–5.
Greene RW, Bjorness TE, Suzuki A. The adenosine-mediated, neuronal-glial, homeostatic sleep response. Curr Opin Neurobiol. 2017;44:236–42.
Pasquini S, Contri C, Merighi S, Gessi S, Borea PA, Varani K, et al. Adenosine receptors in neuropsychiatric disorders: fine regulators of neurotransmission and potential therapeutic targets. Int J Mol Sci. 2022;23:1219.
Hines DJ, Schmitt LI, Hines RM, Moss SJ, Haydon PG. Antidepressant effects of sleep deprivation require astrocyte-dependent adenosine mediated signaling. Transl Psychiatry. 2013;3:e212.
Hines DJ, Haydon PG. Astrocytic adenosine: from synapses to psychiatric disorders. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130594.
Riddle EL, Rau KS, Topham MK, Hanson GR, Fleckenstein AE. Ceramide-induced alterations in dopamine transporter function. Eur J Pharmacol. 2003;458:31–6.
Host L, Dietrich JB, Carouge D, Aunis D, Zwiller J. Cocaine self-administration alters the expression of chromatin-remodelling proteins; modulation by histone deacetylase inhibition. J Psychopharmacol. 2011;25:222–9.
Dietrich JB, Takemori H, Grosch-Dirrig S, Bertorello A, Zwiller J. Cocaine induces the expression of MEF2C transcription factor in rat striatum through activation of SIK1 and phosphorylation of the histone deacetylase HDAC5. Synapse. 2012;66:61–70.
Franken P, Dijk DJ. Circadian clock genes and sleep homeostasis. Eur J Neurosci. 2009;29:1820–9.
Falcon E, McClung CA. A role for the circadian genes in drug addiction. Neuropharmacology. 2009;56 Suppl 1:91–6.
Kim M, Custodio RJ, Botanas CJ, de la Pena JB, Sayson LV, Abiero A, et al. The circadian gene, Per2, influences methamphetamine sensitization and reward through the dopaminergic system in the striatum of mice. Addict Biol. 2019;24:946–57.
Ribeiro EA, Scarpa JR, Garamszegi SP, Kasarskis A, Mash DC, Nestler EJ. Gene network dysregulation in dorsolateral prefrontal cortex neurons of humans with cocaine use disorder. Sci Rep. 2017;7:5412.
Huggett SB, Stallings MC. Genetic architecture and molecular neuropathology of human cocaine addiction. J Neurosci. 2020;40:5300–13.
Chen Z, Gardi J, Kushikata T, Fang J, Krueger JM. Nuclear factor-kappaB-like activity increases in murine cerebral cortex after sleep deprivation. Am J Physiol. 1999;276:R1812–8.
Seney ML, Kim SM, Glausier JR, Hildebrand MA, Xue X, Zong W, et al. Transcriptional alterations in dorsolateral prefrontal cortex and nucleus accumbens implicate neuroinflammation and synaptic remodeling in opioid use disorder. Biol Psychiatry. 2021;90:550–62.
Terao A, Greco MA, Davis RW, Heller HC, Kilduff TS. Region-specific changes in immediate early gene expression in response to sleep deprivation and recovery sleep in the mouse brain. Neuroscience. 2003;120:1115–24.
O’Donovan KJ, Tourtellotte WG, Millbrandt J, Baraban JM. The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci. 1999;22:167–73.
Steiner H, Gerfen CR. Dynorphin opioid inhibition of cocaine-induced, D1 dopamine receptor-mediated immediate-early gene expression in the striatum. J Comp Neurol. 1995;353:200–12.
Moratalla R, Robertson HA, Graybiel AM. Dynamic regulation of NGFI-A (zif268, egr1) gene expression in the striatum. J Neurosci. 1992;12:2609–22.
Cole AJ, Bhat RV, Patt C, Worley PF, Baraban JM. D1 dopamine receptor activation of multiple transcription factor genes in rat striatum. J Neurochem. 1992;58:1420–6.
Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–28.
Chellappa SL, Schmidt C, Cajochen C. Neurophysiology of sleep and wakefulness. Sleepiness and human impact assessment. Milano: Springer; 2014. p. 23–41.
Carskadon MA, Dement WC. Normal human sleep: an overview. Princ Pract sleep Med. 2005;4:13–23.
Waterhouse J, Fukuda Y, Morita T. Daily rhythms of the sleep-wake cycle. J Physiol Anthropol. 2012;31:1–14.
Lesku JA, Roth TC II, Amlaner CJ, Lima SL. A phylogenetic analysis of sleep architecture in mammals: the integration of anatomy, physiology, and ecology. Am Nat. 2006;168:441–53.
Grimm JW, Hope BT, Wise RA, Shaham Y. Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature. 2001;412:141–2.
Tran-Nguyen LT, Fuchs RA, Coffey GP, Baker DA, O’Dell LE, Neisewander JL. Time-dependent changes in cocaine-seeking behavior and extracellular dopamine levels in the amygdala during cocaine withdrawal. Neuropsychopharmacology. 1998;19:48–59.
Acknowledgements
We thank Dr. Yan Dong for helpful discussions.
Funding
This work was supported by the National Institutes of Health under Award Numbers DA043826 (YH), DA046491 (YH), AA028145 (YH), DA046346 (YH).
Author information
Authors and Affiliations
Contributions
YH designed the framework; RG, DV, AA, and YH collected the literature and wrote the review. We agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guo, R., Vaughan, D.T., Rojo, A.L.A. et al. Sleep-mediated regulation of reward circuits: implications in substance use disorders. Neuropsychopharmacol. 48, 61–78 (2023). https://doi.org/10.1038/s41386-022-01356-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-022-01356-8
This article is cited by
-
Perceived Educational Needs of Substance Use Peer Support Specialists: A Qualitative Study
Community Mental Health Journal (2024)
-
Sleep disturbance in substance use disorders: the orexin (hypocretin) system as an emerging pharmacological target
Neuropsychopharmacology (2023)
