
Abstract
Most cigarette smokers who wish to quit too often relapse within the first few days of abstinence, primarily due to the aversive aspects of the nicotine withdrawal syndrome (NWS), which remains poorly understood. Considerable research has suggested that the dorsal anterior cingulate cortex (dACC) plays a key role in nicotine dependence, with its functional connections between other brain regions altered as a function of trait addiction and state withdrawal. The flow of information between dACC and fronto-striatal regions is secured through different pathways, the vast majority of which are glutamatergic. As such, we investigated dACC activity using resting state functional connectivity (rsFC) with functional magnetic resonance imaging (fMRI) and glutamate (Glu) concentration with magnetic resonance spectroscopy (MRS). We also investigated the changes in adenosine levels in plasma during withdrawal as a surrogate for brain adenosine, which plays a role in fine-tuning synaptic glutamate transmission. Using a double-blind, placebo-controlled, randomized crossover design, nontreatment seeking smoking participants (N = 30) completed two imaging sessions, one while nicotine sated and another after 36 h nicotine abstinence. We observed reduced dACC Glu (P = 0.029) along with a significant reduction in plasma adenosine (P = 0.03) and adenosine monophosphate (AMP; P < 0.0001) concentrations during nicotine withdrawal in comparison with nicotine sated state. This withdrawal state manipulation also led to an increase in rsFC strength (P < 0.05) between dACC and several frontal cortical regions, including left superior frontal gyrus (LSFG), and right middle frontal gyrus (RMFG). Moreover, the state-trait changes in dACC Glu and rsFC strength between the dACC and both SFG and MFG were positively correlated (P = 0.012, and P = 0.007, respectively). Finally, the change in circuit strength between dACC and LSFG was negatively correlated with the change in withdrawal symptom manifestations as measured by the Wisconsin Smoking Withdrawal Scale (P = 0.04) and Tobacco Craving Questionnaire (P = 0.014). These multimodal imaging-behavioral findings reveal the complex cascade of changes induced by acute nicotine deprivation and call for further investigation into the potential utility of adenosine- and glutamate-signaling as novel therapeutic targets to treat the NWS.
Similar content being viewed by others
Introduction
Acute smoking abstinence induces a rapid nicotine craving state characterized by increased irritability, sadness, and reduced ability to focus, concentrate and get refreshing sleep that peaks about 24–48 h following nicotine abstinence. These symptoms are known collectively as the nicotine withdrawal syndrome (NWS) [1, 2]. Because of the intensity of symptoms, most quit attempts fail within days of smoking cessation, even with the best available pharmacological and behavioral treatment options, making it a priority to identify novel therapeutic targets to attenuate the NWS severity [3]. As such, new insights into how the brain signals the nicotine deprivation state and in turn, the NWS at the circuit and neurochemical levels are needed to inform novel treatment interventions to improve long-term abstinence rates.
Several lines of evidence suggest that the anterior cingulate cortex (ACC) is a key brain region that influences and is, in turn, influenced by different stages of nicotinic addiction [4,5,6,7,8,9,10,11,12,13,14,15]. As a central hub integrating the flow of information between various limbic and cortical networks, the ACC is strategically positioned to regulate emotions and monitor performance errors and behavioral conflict and plays a major role in reward-based decision-making, all of which are dysregulated during acute nicotine abstinence [16, 17]. Functionally, the ACC can be parsed mainly into an ‘affective’ rostral-ventral (rACC), and a ‘cognitive’ dorsal (dACC) subregion [18,19,20]. The dACC plays an important role in cognitive self-control processes critical for initiating and maintaining abstinence. Indeed, high levels of cognitive control contribute to smoking resistance [21], while higher impulsivity and lower cognitive control predicts a higher likelihood of taking up smoking in adolescence and a lower likelihood of quitting in adulthood [22]. Similarly, adult smokers with poor cognitive control are more likely to relapse in the short term [23]. Moreover, during smoking abstinence (vs satiety), smoking cue reactivity engenders increased ACC activity [24,25,26,27], while the degree of cue-induced activation in ACC predicts abstinence duration [26, 28]. The vast majority of ACC neurons are glutamatergic and dACC glutamatergic alterations across different stages of addiction have been well documented (Reviewed in [17, 29,30,31]).
Hence, there is a growing interest in integrating glutamatergic neurotransmission within the dACC and its network functional connectivity to better understand the mechanistic underpinnings of NWS. Specifically, abstinence induced changes in these metrics linked to alterations in withdrawal symptom may serve as a potential marker for severity of the nicotine deprivation state and subsequent relapse risk. However, measuring subtle changes in dACC Glu in humans and then linking such changes to behavioral symptoms tends to be challenging.
Proton magnetic resonance spectroscopy (1H-MRS) is a non-invasive tool that can quantify Glu and other brain metabolites [32], with the caveat that MRS reflects total Glu concentration and not specifically synaptic Glu neurotransmitter levels. At magnetic field strengths of 3 T or above, the spectra for structurally similar Glu and glutamine (Gln) can be separately resolved [33, 34]. Using this modality, smokers who relapsed while undergoing nicotine replacement therapy (NRT, aka, the patch) were found to have lower baseline dACC Glu concentration compared to those who remained abstinent for 8 weeks [7]. However, other research has found no difference in Glu concentration based on smoking status [35].
Several factors may be responsible for the inconsistency in these studies, including the acute state of the individual during measurement (i.e., nicotine sated or withdrawn). For example, our group has previously reported increased rsFC strength between dACC and posterior insula during abstinence versus smoking satiety [13] that correlated with withdrawal symptom severity. In contrast, dACC connectivity strength with the ventral striatum correlates with the degree of nicotine dependence (trait), but is not modified with NRT after abstinence (i.e., state), which we thus termed an addiction trait circuit [11, 14, 36]. These state- and trait-based changes in FC suggest a mechanistic role for Glu in modulating network connectivity under different homeostatic conditions.
Moreover, quantifying Glu in isolation may not provide adequate information of dACC functionality. Glu concentration levels, which have previously been associated with task-induced BOLD signal change [37, 38], have been simultaneously measured in abstinent smokers seeking treatment during cognitive task performance [39] and in sated and experimentally stressed smokers [40, 41]. Collectively, these studies show that in nicotine dependent subjects, changes in Glu concentrations are associated with fMRI measures of task activation and behavior. Specifically, one study reported a significant decrease in dACC Glx (Glutamate + Glutamine) associated with reduction in rACC BOLD activation induced by the Stroop color-naming task before and after smoking cessation with varenicline [39]. Janes et al. showed significant positive association between dACC Glu and Default Mode Network (DMN) reactivity to smoking cues relative to neutral cues in nicotine-dependent participants [40], while Woodcock et al. reported that pharmacologically induced stress attenuated the increase in dlPFC Glu during a working memory task [41].
Taken together, these studies attempt to resolve the relationship between relatively static Glu measures that require several minutes to acquire and the more dynamic (seconds) cognitive task-based changes in the BOLD signal. In an effort to combat this challenge, we acquired both MRS glutamate (Glu) and resting state BOLD signal (averaged over many minutes and more similar to the time for MRS acquisition) instead of task-related activation in smokers during both smoking satiety and acute nicotine withdrawal. We hypothesized that acute withdrawal will be associated with altered dACC Glu concentration and dACC-based rsFC circuit strength and that these changes will correlate with specific clinical manifestations of nicotine use (trait) and NWS (state).
Methods
Participants
Thirty healthy nicotine cigarette smokers (18 M/12 F) completed all experimental procedures. Participants were right-handed, 18–60 years of age, free of current alcohol or drug use disorders except for nicotine, reported no lifetime psychiatric diagnoses (DSM-5) or neurological disorders and had no contraindications for MRI. Written informed consent was obtained in accordance with the National Institute on Drug Abuse-Intramural Research Program Institutional Review Board. Detailed history, physical examination, laboratory assessments (complete blood count, thyroid, liver and kidney functions), drug use history, IQ (Wechsler Adult Intelligence Scale; WAIS; Wechsler 1958), depressive (Beck Depression Inventory; BDI) [42], and anxiety symptoms (Beck Anxiety Inventory; BAI) [43] were obtained from each participant. See consort flow diagram (Supplementary Fig. 1) for detailed description of enrollment, randomization, and excluded subjects.
Experimental design
Each participant completed two MR scanning sessions, one during nicotine-withdrawal and one during nicotine satiety. To achieve these two states in a double-blind, cross-over design, participants were required to abstain from smoking for 36 h prior to scanning and were given a smoking dose-matched nicotine (satiety) or placebo (nicotine-withdrawal) patch after 12 h of abstinence and a second patch 1–2 h prior to the scan session. Nicotine abstinence was biochemically verified by urine nicotine, cotinine, and exhaled carbon monoxide concentrations (Vitalograph, Lexington, KS) at time of scanning. The mean (±SEM) interval between the two scans was 47 days (±15) with the order of nicotine-satiety and withdrawal days counterbalanced across subjects. Participants were required to refrain from drinking caffeinated beverages for 12 h and from the use of alcohol or other drugs for at least 24 h before sessions, which was confirmed by a urine drug screen and a breath alcohol test. Each imaging day consisted of two scan sessions. Resting state fMRI and EEG data were collected during the morning session, while several fMRI tasks were performed on both morning and afternoon sessions (data not reported here). Blood for genetic testing, nicotine, cotinine, and adenosine assay was collected in the afternoon scan session prior to an unpredictable shock task [44] 1H-MRS was acquired about 30 min after the shock task at the end of the afternoon session.
Biological and subjective withdrawal measures
Prior to each scanning session, participants completed a detailed assessment battery of withdrawal related symptoms including: the Wisconsin Smoking Withdrawal Scale (WSWS), a 28-item self-report test comprising of seven subscales (anger, anxiety, concentration, craving, hunger, sadness, sleep) [45]; State-Trait Anxiety Inventory-State (STAI-S) consisting of 20 items designed to quantify temporary anxiety conditions such as feelings of apprehension, tension, nervousness, and worry [46]; Tobacco Craving Questionnaire (TCQ) short (12 item) version to assess current feelings related to smoking and craving and which loads on four factors: (1) emotionality: smoking in anticipation of relief from withdrawal or negative mood; (2) expectancy: anticipation of positive outcomes from smoking; (3) compulsivity: an inability to control tobacco use; and (4) purposefulness: intention and planning to smoke for positive outcome [47, 48]; Tobacco Craving Scale (TCS) consists of 5 self-report items that are rated on a 10-point scales. These item pertain to desire and urge for a cigarette (1) how strong is your desire for a cigarette right now, (2) how strong was your desire for a cigarette during the last 24 h, (3) how often you had the urge to smoke during the past 24 h, (4) how strong your urges have been for a cigarette when something in the environment reminded you of it over the past 24 h, and (5) imagine yourself in the environment in which you previously used drugs and/or alcohol. If you were in this environment right now, what is the likelihood that you would smoke? Participants’ affective state was rated using the Positive And Negative Affective State (PANAS), a 20-item scale composed of 10 items describing negative affect and 10 items describing positive affect [49]; the Perceived Stress Scale, a brief 10-item instrument measures the degree of perceived stress during the past month [50] and the Snaith Hamilton Anhedonia Scale (SHAPS) designed to measure hedonic tone [51].
Plasma assays
Liquid chromatography/atmospheric pressure chemical ionization-tandem mass spectrometry was used to assay plasma levels of nicotine, cotinine, adenosine, adenosine monophosphate (AMP), and adenosine triphosphate (ATP) according to previously published methods [52, 53].
Resting state fMRI
Resting-state BOLD images were acquired (eyes open with participants asked to focus on a fixation cross) on a Siemens Tim Trio 3T MRI scanner (Erlangen, Germany) using a single shot echo planer imaging sequence (TE/TR = 27/2000 ms, image resolution: 3.4 × 3.4 × 4.0 mm3, 39 oblique-axial slices, 30° to AC-PC, 300 volumes, total resting-state acquisition time = 8 min). T1-weighted anatomical scans were collected using magnetization-prepared rapid gradient echo (MPRAGE) sequence (TE/TR = 3.51/1900 ms, image resolution: 1 mm3, oblique-axial), which covered the whole brain.
1H-MRS
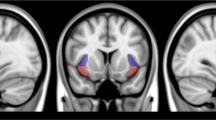
High resolution T1-weighted anatomical images were used to position two MRS voxels (15 × 20 × 30 mm3) that encompassed the dACC and posterior cingulate cortex (PCC) (shown in Supplementary Fig. 2B). The PCC voxel served as a negative control region, with no expected changes in metabolites between satiety and withdrawal. Shimming of the magnetic field within the voxels was performed using an automated shimming routine. MRS was acquired using a point-resolved spectroscopy sequence, TE/TR = 30/3000 ms, and 128 averages.
MRS data were quantified using LCModel version 6.3 [54]. The basis set included the following metabolites: alanine (Ala), aspartate (Asp), creatine (Cre), γ-aminobutyric acid (GABA), Gln, Glu, glycerophosphocholine (GPC), phosphocholine (PCh), myo-inositol (mI), and N-acetylaspartate (NAA). A subset of this basis set of macromolecules (MM) and lipids was incorporated into the LCModel analysis to take into account the impact of MM/lipids on spectral fitting.
LCModel reported both estimated metabolite concentrations in institutional units (i.u.), using the unsuppressed water signal as the scaling reference, and the ratio of metabolite concentration to the concentration of creatine + Phosphocreatine (Cr + PCr). We used metabolites as ratioed to Cr + PCr to correct for partial volume effects assuming the stability of creatine [32, 55]. Only metabolite concentration estimations with a Cramer-Rao Lower Bound (CRLB) of less than 20% were included in the final data analysis. These metabolites and their CRLB values for dACC voxel were as follows: Gln: 9–20%, Glu: 5–10%, GPC: 2–5%, mI: 3–6%, NAA: 2–4%, GPC + PCh: 2–5%, NAA + NAAG: 2–4%, Cr + PCr: 2–4%, Glu + Gln: 4–6% and for the PCC voxel: Gln: 10–20%, Glu: 4–7%, GPC: 3–5%, mI: 3–6%, NAA: 2–4%, GPC + PCh: 3–5%, NAA + NAAG: 2–4%, Cr+PCr: 2–3%, Glu+Gln: 4–7%. The volume fractions of different tissue types were calculated from segmented T1 images (SPM 12.0, http://www.fil.ion.ucl.ac.uk/spm). Tissue-corrected metabolite concentrations were then calculated by dividing metabolite concentrations by the total fraction of GM and WM in that voxel [56]. A representative spectrum for dACC is shown in Supplementary Fig. 2A and for PCC in Supplementary Fig. 2C.
Statistical analysis
Clinical plasma markers and MRS data are presented as mean (±SEM). The differences in mean scores of biological and clinical withdrawal manifestations and in Glu/Cr + PCr ratios between nicotine satiety and nicotine withdrawal states were compared with paired t tests. Pearson correlation analyses were performed to evaluate associations between dACC Glu/Cr + PCr, rsFC, and clinical variables. Analyses were performed using GraphPad Prism V7 software (La Jolla, CA). Results are considered significant at P < 0.05.
Resting BOLD data were analyzed using the AFNI software package [57] and SPM12. Data were slice-time and motion-corrected, quadratically detrended, and spatially normalized to MNI space via non-linear registration. Following motion correction, motion censoring was performed on any two consecutive time points with Euclidean distance derivative values >0.35 mm. Individual white matter (WM), and cerebrospinal fluid (CSF) masks were obtained by anatomical image segmentation in SPM and were used to extract signals from non-neuronal sources. Nuisance covariates, including six-motion parameters and signals in the first four principal components of WM/CSF were regressed out [58], followed by temporal band-pass filtering (0.01–0.1 Hz) and Gaussian spatial smoothing (full-width at half-maximum = 8 mm). Functional connectivity maps were created for the withdrawal (placebo patch) and satiety (nicotine patch) conditions using the merged dACC MRS voxels (with at least 80% overlap) as a seed in 22 participants, whose MRS data survived data quality criteria (Fig. 2, top panel). The correlation coefficients were converted to z-scores using the Fisher’s r-to-z transformation. Differences between abstinent vs. sated rsFC-maps [ΔFC-maps] were calculated within participants and a one-sample t-test was carried out for group analysis. Results from group analysis were corrected for multiple comparisons using 3dClustSim, based on Monte Carlo simulations [59] using the non-Gaussian spatial autocorrelation function [60]; statistical significance was set to Puncorrected < 0.001 and Pcorrected < 0.05. Finally, we performed a regression analysis for the significant Δ(withdrawal (-) satiety) rsFC, ΔGlu/Cr+PCr and Δwithdrawal manifestations that showed significant smoking state related changes (ΔWSWS, ΔTCQ, ΔPANAS positive subscale, ΔSTAI-S, and ΔTCS). Results of the regression analyses were Bonferroni corrected for multiple comparisons.
Results
Clinical characterization
Participants started smoking at about age 16 (±0.7) and smoked 17.7(±1.4) cigarettes/day for 19.8 (±1.9) years. The severity of nicotine addiction averaged 5.3 ± 0.3 and ranged between low and severe (Fagerstrom Test for Nicotine Dependence: 2–8). Participants scored moderately high in impulsivity [BIS; 62(±1.7)] and alexithymia [TAS-20; subscales: difficulty identifying feelings 10 (±0.8), difficulty describing feelings 7.8(±0.7) and externally oriented thinking 18.2(±0.7)]. A total of 29/30 participants completed both scanning sessions (see Table 1).
Biological and clinical manifestations of acute nicotine withdrawal
Abstinence from smoking prior to each imaging session was verified by measuring expired air CO, plasma nicotine, and cotinine concentrations (Table 2). The change (Δ) in plasma nicotine concentrations (withdrawal-satiety) showed significant positive correlations with ΔPANAS positive subscale (r = 0.393, P = 0.03, Fig. 1a) and ΔSATI-S (r = −0.506, P = 0.019, Fig. 1b). Paired t-tests revealed significant reductions between satiety and withdrawal in plasma adenosine [0.114(±0.04) vs. 0.019(±0.00), P = 0.03] and AMP [0.146(±0.02) vs. 0.013(±0.00), P < 0.0001], but not ATP [0.013(±0.00) vs. 0.008(±0.0), P = 0.3] concentrations. Moreover, these physiological measures were consistent with clinical assessments of withdrawal: WSWS total score, anxiety, concentration, and sadness subscales all were significantly elevated in abstinence compared to satiety. Similarly, during the withdrawal state, participants reported heightened anxiety (STAI-S) and tobacco cravings (TCS and TCQ total score, factor 1 (emotionality) and factor 3 (compulsivity)) and a significant drop in positive affect (PANAS positive subscale). However, the changes observed in negative affective state, perceived stress, and anhedonia scales (PANAS negative subscale, PSS, and SHAPS) did not reach significance.

Correlation analysis between the change (Δ) in plasma nicotine concentrations (Withdrawal-satiety) and a ΔPANAS positive subscale; r = 0.393, P = 0.03), and b with ΔSATI-S, r = −0.506 m P = 0.019).
The effect of acute nicotine withdrawal on dACC glutamate and functional connectivity
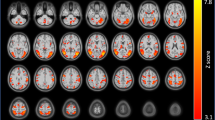
There was a significant reduction in dACC Glu/Cr + PCr ratio during withdrawal vs. satiety: 1.54(±0.029) vs. 1.50(±0.03), P = 0.029, (Supplementary Fig. 3, and Supplementary Table 1). We next investigated whether this reduction in dACC glutamate was related to dACC rsFC. We used the merged dACC MRS voxels with ≥80% overlap between subjects and scans as the seed in a whole brain connectivity analysis (Fig. 2a, b) contrasting rsFC strength (z-values) between satiety and withdrawal scans and identified significant (Pcorrected < 0.05) clusters in the left superior frontal gyrus (LSFG, Fig. 2c; z-values are shown in Fig. 2d) and right middle frontal gyrus (RMFG, Fig. 2f; z-values are shown in Figure 4G). PCC Glu, on the other hand, did not significantly change between withdrawal and satiety: 1.31(±0.016) vs. 1.37(±0.046), P = 0.4.

a Merged dACC MRS voxel in the sagittal, axial and coronal planes. Color spectrum from green to red denotes the degree of overlap. b The ≥80% voxel overlap used as seed for whole brain resting state functional connectivity analysis. c Voxel-wise resting state one-sample T-test analysis when contrasting withdrawal and satiety conditions. Significant cluster (Pcorrected < 0.05) at the left superior frontal gyrus (LSFG) shown in the three planes. d rsFC z-values in both satiety and withdrawal conditions and e shows the correlation between Δ-dACC Glu and Δ-rsFC (dACC-LSFG) F(1,20) = 7.497, P = 0.012 by linear regression. f Voxel-wise resting state one-sample T-test analysis when contrasting withdrawal and satiety conditions. Significant cluster (Pcorrected < 0.05) at the right middle frontal gyrus (RMFG) in the three planes. g rsFC z-values in both satiety and withdrawal and h the correlation between Δ-dACC Glu and Δ-rsFC (dACC-RMFG) F(1,20) = 8.864, P = 0.007 by linear regression.
We also found that the ΔdACC Glu/Cr + PCr was significantly correlated with both the ΔrsFC between dACC and LIFG (Pcorrected = 0.024, Fig. 3e) and between dACC and RMFG (Pcorrected =0.014; Fig. 3h). Finally, there was a significant association between ΔdACC-LSFG rsFC and ΔWSWS total score (P = 0.04, Fig. 3a) and ΔTCQ total score (P = 0.014, Fig. 3b). However, as we also tested for relationships between dACC Glu, PANAS and STAI-S, neither connectivity relationship survived Bonferroni correction.

The change (withdrawal-satiety) in rsFC between dACC and LSFG correlates with a change in WSWS total score F(1,20) = 4.824, P = 0.04 and b with the change in total TCS: F(1,20) = 7.231, P = 0.014 by linear regression.
Exploratory analyses
We examined whether dACC Cr impacts dACC Glu concentration or rsFC and found no significant effects. Furthermore, there were no significant effects of dACC Cr, age, sex or smoking history on ΔdACC Glu and Δplasma adenosine concentration (See Supplementary material for additional details).
Discussion
To the best of our knowledge, this is the first study that combines resting state fMRI with 1H-MRS along with plasma adenosine to investigate neurobiological mechanisms of short-term nicotine withdrawal. Our results show that acute nicotine abstinence is associated with a significant reduction in dACC Glu that is linked with a marked increase in FC strength between the same dACC region and two specific subregions of the frontal cortex (LSFG and RMFG). Moreover, the changes in Glu and functional connectivity between nicotine satiety and abstinence state (ΔdACC Glu and ΔrsFC dACC-LSFG and ΔdACC-RMFG) were significantly positively correlated.
We focused our investigation on the dACC as it is a key component of the Salience Network (SN) [61] and has previously been linked with nicotine dependence [62]. Moreover, the tripartite network hypothesis of neuropsychiatric diseases [63], and more specifically as it applies to drug addiction [64], links the dACC (along with the anterior insula), a key component of the SN [65], with the medial frontal cortex, part of the DMN [61, 66] and the dorsolateral prefrontal cortex and posterior parietal cortex, which are key components of the Executive Control Network (ECN). It has been hypothesized that homeostatically relevant information is constantly exchanged between these three networks in order to switch attention and cognitive resources (via SN) between externally salient stimuli (ECN) and internal, mind-wandering/ruminations, (processed within the DMN) [67]. Moreover, this important communication is secured through anatomical glutamatergic projections [61, 68] that are likely functionally synchronized between the networks [69, 70]. It has been shown that perturbations in Glu (e.g., following ketamine infusion) negatively impact the efficiency of rsFC in healthy subjects [71, 72] and in patients with depression [71]. Furthermore, a recent study showed that baseline dACC Glu concentration was associated with nicotine cue-induced activation in the frontal lobe [40].
Our results strengthen such current evidence and reveal a strong dynamic association between ΔdACC Glu and ΔrsFC when a smoker transitions from nicotine satiety to abstinence. Interestingly, the reduction in dACC Glu was associated with an increase in dACC rsFC strength with distal frontal cortical regions. Here we speculate that the increase in connectivity strength is related to increased subjective withdrawal (ΔWSWS) and craving (ΔTCS) symptoms, i.e., as the connectivity strength decreased, withdrawal symptoms increased. We speculate that acute nicotine abstinence caused the reduction in dACC Glu, which in turn increased FC strength. However, the flow of information is hampered, as evident by the increase in withdrawal symptoms. During nicotine deprivation, Glu is reduced across certain brain regions such as dACC, which allows only limited dynamic changes at rest and hence more synchronization, which is reflected as greater rsFC strength and manifests clinically as nicotine withdrawal and craving. This presumptive model, albeit simplistic, is in agreement with a recent model proposed by Moeller et al. where the authors argue that corticolimbic rsFC can provide an intermediate phenotype to explain associations between addiction-relevant glutamatergic alterations and addiction symptomatology (e.g., craving, drug-seeking, engagement with treatment) [31]. This schema is also consistent with previous reports showing a reduction in dACC Glx in smokers after 48 h of nicotine deprivation [73] and significant reduction after 12 weeks of abstinence on varenicline treatment [39]. Of course, the data presented here are correlational and interventional studies are required to demonstrate causation.
The roles that the two frontal subregions (SFG and MFG) identified as part of our functional circuits play in nicotine withdrawal remains under investigation. The SFG is involved in a variety of functions ranging from cognitive control [74] and switching between distinct cognitive tasks [67], to working memory [75], humor appreciation [76], and overall cognitive wellbeing [77]. In contrast, the MFG is strategically positioned in the junction between top-down, goal-directed and bottom-up, stimulus-driven visual attention control streams [78]. With this unique nodal location, the MFG integrates and computes “on-line” storage and processing of spatial information [79] to reorient attention from exogenous to endogenous attentional control [78]. These cognitive processes are modulated by nicotine. Specifically, nicotine enhances reorienting of attention in visuospatial tasks [80,81,82,83,84] while nicotine withdrawal disrupts spatial memory in preclinical model [85].
Given that smokers experience decrements in cognitive performance during withdrawal [13, 36, 86,87,88], it is plausible that information processing in this frontal subregion is compromised and may in turn contribute to processing of smoking-associated stimuli. In support of this conjuncture, McClernon et al. reported greater SFG and MFG activation to smoking cues in dependent smokers following overnight abstinence with abstinence-induced changes in craving positively correlated with changes in bilateral MFG amplitude [25]. Similarly, Due et al. observed smoking cue-activation in MFG [89]. We found herein significant correlation between changes in withdrawal and craving (ΔWSWS and ΔTCQ total scores) and rsFC between dACC and LSFG. Further studies are needed to explore the specific role of these frontal subregions in mediating cognitive domains associated with the NWS and whether, e.g., non-invasive brain stimulation (i.e. TMS, tDCS, etc.) could be utilized to enhance cognitive function during early abstinence.
The effect of nicotine withdrawal on glutamatergic signaling seems to depend on several factors. For instance, in preclinical models nicotine withdrawal leads to downregulation of metabotropic glutamatergic 2/3 receptors (mGluR2/3) [90] and astrocytic glutamate transporter (GLT1) expression in limbic regions [91]. Since mGluR2/3 are presynaptic and function to inhibit glutamate release, its downregulation is expected to cause an increase in synaptic glutamate. Similarly, as GLT1 removes glutamate from the synaptic region, its inhibition also could lead to an increase in synaptic glutamate concentration. However, without quick replenishment, increased release and reduced uptake of synaptic glutamate eventually reduces the astrocytic glutamate–glutamine shuttle and leads to reduced glutamate concentration [92]. Furthermore, glutamatergic signaling is involved in mediating different aspects of nicotine use disorder, including the motivation to smoke [93]. Mechanistically, presynaptic nAChRs positively regulate the release of various neurotransmitters, including glutamate [94, 95]. As most cortical projecting efferent fibers utilize glutamate as their transmitter, enhanced glutamate release modifies the motivation to self-administer nicotine in preclinical models [96]. Taken together, it is possible that glutamate concentration changes over the course of withdrawal, with initial increases followed by normalization and then an overall reduction. However, this explanation remains speculative and requires further testing in preclinical models.
We investigated the changes in adenosine levels during withdrawal because of its important role in fine-tuning synaptic transmission [97]. It is well accepted that to ensure maintenance of synaptic transmission, adenosine can inhibit [98,99,100] or increase [101, 102] glutamate release and uptake [103] in specific brain regions depending on the state of neuronal activity. We observed marked reduction in plasma adenosine and AMP concentrations (Table 2) while ATP remained unchanged during withdrawal.
Adenosine is synthesized in neuronal and glial cells mostly through the hydrolysis of ATP [104] and is then released into the synaptic space through equilibrative nucleoside transporter (ENT) to exert its action by activating multiple G-protein coupled adenosine receptor subtypes, i.e., A1, A2A, A2B, and A3 [104]. ENT also transports adenosine back into astrocytes where it undergoes rapid degradation into AMP by adenosine kinase or into inosine by adenosine deaminase [104, 105]. Brain-synthesized adenosine is transported across the blood brain barrier [106], while plasma adenosine is sequestered by the brain microvasculature after transport and is rapidly converted to adenosine metabolites [106]. Nicotine increases the activity of ATPase [107, 108] and the concentration of extracellular adenosine [109] and cyclic AMP [110]. As such, our results suggest that during nicotine deprivation, the synthesis of ATP is not affected, while the conversion of ATP into adenosine and then into AMP is inhibited, leading to reduced adenosine and AMP levels. It is important here to note that we measured peripheral and not brain adenosine and it is not entirely clear whether changes in plasma adenosine reflect corresponding changes in brain adenosine. However, one study showed rapid rise in plasma adenosine levels following acute brain insults such as transient ischemic attacks and strokes [111]. Taking this into consideration, it is plausible to suggest that acute nicotine withdrawal caused reduction in brain adenosine concentration that is reflected in low plasma adenosine levels. This hypothesis requires testing in preclinical models. Given the well documented influence of adenosine on glutamatergic terminals, if nicotine withdrawal state indeed reduces brain adenosine concentration, this could bring new insight into the mechanisms through which nicotine modulates glutamatergic neurotransmission.
Interestingly, we did not find any correlation between levels of adenosine, ATP or AMP and clinical NWS manifestations. These negative results are in line with a previous report that showed that the severity of mecamylamine-induced nicotine withdrawal was not different between wild-type and A2AR knockout mice, which suggest that A2AR do not participate in the expression of the somatic component of nicotine withdrawal [112, 113]. However, a second study reported that either increasing or decreasing the activity at A2AR was found to prevent the aversive motivational response to withdrawal from chronic, but not acute nicotine administration [114]. It is relevant here to note that A1 receptors are widely distributed in the cortex, hippocampus, and cerebellum, while A2A receptors are localized mainly in the striatum and olfactory bulb (A2B and A3 receptors are found at low levels of expression) [115]. A2A receptors are found in receptor clusters with dopamine and glutamate receptors [116] and A2A-D2-mGlu5 receptor mosaics have been discovered [117]. Striatal A2A receptors colocalize with dopamine D2 receptor to form A2A-D2 heteromers, which modulate dopamine-dependent striatal functions such as reward-oriented behavior and learning of stimulus-reward and reward-response associations [118, 119]. In addition, there is a high density of A2A receptors that regulate cortico-accumbens glutamatergic transmission, among the main circuits involved in compulsive drug seeking and relapse [120]. As such, adenosine is a major modulator for dopaminergic and glutamatergic neurotransmitter systems involved in nicotine reward and withdrawal [112]. Moreover, adenosine enhances nicotine-induced locomotor sensitization possibly through functional interaction between nicotinic and adenosine A2A receptors in striatal dopaminergic terminals [121]. Potential therapeutic benefits of A2A receptor agonists or antagonists is still a matter of debate. While nicotine-induced conditioned place preference was suppressed in A2A knockout mice [113], treatment with A2A receptor agonists reduced locomotor sensitization and conditioned locomotion to nicotine, which can help counteract the abuse actions of nicotine [122].
In contrast, caffeine, a non-selective A2AR antagonist [123] has been reported to produce an anxiolytic effect in nicotine-dependent rats, and enhance sensitivity to nicotine during nicotine withdrawal [124]. However, whether drinking caffeinated beverages during smoking cessation would help smokers stay abstinent remains largely unknown. These mixed results, along with ours and recent data showing a pivotal role of adenosine in mediating nicotine-induced upregulation of α7 nAChRs without affecting α4β2 receptor upregulation [125], reflect the complex relationship between nicotine and adenosine and call for further studies to examine the potential utility of adenosine signaling pathway (and potentially its relationship with dACC Glu) as a novel therapeutic target to attenuate withdrawal manifestations.
Surprisingly, our results did not show any effect for age, sex, duration of smoking or cigarette packs smoked over lifetime on ΔdACC Glu or Δadenosine concentrations (See Supplementary materials). Durazzo et al. reported negative correlation between ACC Glu and age among sated smokers (n = 35) but not among non-smokers (n = 30) [4]. Others have reported age-related differences in glutamate concentration in other brain regions in healthy controls [126]. We did not find significant correlation between age and sated dACC Glu or between age and ΔdACC Glu. This discrepancy could be related to subtle differences in voxel placement (i.e., perigenual ACC in Durazzo et al. and dACC in this study), sample size, or in subjects sex distribution. Durazzo’s cohort consisted predominantly of males (31/4) while our sample consisted of both males and females (18/12).
The results of this study should be viewed in light of its strength and limitations. First, we compared nicotine satiety to deprivation by using a nicotine and matching placebo patch to ensure blinding of nicotine status. Nicotine delivery through the patch has different pharmacokinetics from smoking, so our results may differ in deprived vs. active smoking individuals. Second, we chose to use a cross-over study design where every participant acts as his or her own control to nullify variations between groups, control for scan order and to enhance statistical power compared to cross-sectional designs [127]. In addition, we did not include a non-smoking control group, so our study does not provide insights related to the impact of smoking per se on dACC Glu levels and rsFC. Third, participants in this study were non-treatment seeking smokers who had only to consent to two acute abstinent days prior to scanning. The study was not designed to understand smoking cessation or test for an intervention. As smokers often take brief smoking holidays due to travel, illness, or other factors, we do not believe that motivation to quit interacted with this short-term experimental induced abstinence. They did report on average 7.1 ± 1.7 quit attempts during their lifetime. Fourth, our estimates of Glu may not be completely resolved from Gln as the presence of macromolecular resonances complicate quantification given the current acquisition parameters. That said, the changes we observed in the Glx peak region is most likely attributed to Glu. It should also be noted that Glu measured by 1H-MRS reflects total intracellular and extracellular glutamatergic pools and is not specific for synaptic glutamate [128]. However, given that the concentration of Glu is 20-fold higher in synaptic vesicles than in the cytoplasm [129] and assuming the stability of the TCA cycle rate, observed changes in measured Glu can likely be attributed to changes in synaptic glutamate concentration. This assumption requires further testing through 13C-spectroscopy studies [130].
Finally, we used spectroscopy metabolites expressed as ratios to creatine to correct for partial volume effects assuming the stability of creatine. This method is widely accepted by many [32, 55], but not all [131] spectroscopy researchers.
Despite these limitations, the results of this study improve our understanding of the complex brain changes underlying the NWS and the intriguing role of glutamatergic signaling in initiating the cascade of events that translate into clinical manifestations and highlight several potential novel therapeutic targets for further investigation. Equally important, our results highlight the association between glutamatergic neurotransmitter on network functional connectivity strength and raise an important point that increased connectivity strength does not necessarily mean better functionality.
Funding and disclosure
No pharmaceutical company or other funding agents has financially supported the study. The authors have no conflict of interest to declare.
Data availability
Data files are available upon request.
References
Piasecki TM. Relapse to smoking. Clin Psychol Rev. 2006;26:196–215.
Hughes JR. Effects of abstinence from tobacco: etiology, animal models, epidemiology, and significance: a subjective review. Nicotine Tob Res. 2007;9:329–39.
Henningfield JE, Shiffman S, Ferguson SG, Gritz ER. Tobacco dependence and withdrawal: science base, challenges and opportunities for pharmacotherapy. Pharm Ther. 2009;123:1–16.
Durazzo TC, Meyerhoff DJ, Mon A, Abe C, Gazdzinski S, Murray DE. Chronic cigarette smoking in healthy middle-aged individuals is associated with decreased regional brain N-acetylaspartate and glutamate levels. Biol Psychiatry. 2016;79:481–8.
Gutzeit A, Froehlich JM, Hergan K, Graf N, Binkert CA, Meier D, et al. Insula-specific H magnetic resonance spectroscopy reactions in heavy smokers under acute nicotine withdrawal and after oral nicotine substitution. Eur Addict Res. 2013;19:184–93.
O’Neill J, Tobias MC, Hudkins M, Oh EY, Hellemann GS, Nurmi EL, et al. Thalamic glutamate decreases with cigarette smoking. Psychopharmacology. 2014;231:2717–24.
Mashhoon Y, Janes AC, Jensen JE, Prescot AP, Pachas G, Renshaw PF, et al. Anterior cingulate proton spectroscopy glutamate levels differ as a function of smoking cessation outcome. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1709–13.
Mason GF, Petrakis IL, de Graaf RA, Gueorguieva R, Guidone E, Coric V, et al. Cortical gamma-aminobutyric acid levels and the recovery from ethanol dependence: preliminary evidence of modification by cigarette smoking. Biol Psychiatry. 2006;59:85–93.
Ding X, Lee SW. Changes of functional and effective connectivity in smoking replenishment on deprived heavy smokers: a resting-state FMRI study. PLoS ONE. 2013;8:e59331.
Lerman C, Gu H, Loughead J, Ruparel K, Yang Y, Stein EA. Large-scale brain network coupling predicts acute nicotine abstinence effects on craving and cognitive function. JAMA Psychiatry. 2014;71:523–30.
Sutherland MT, Carroll AJ, Salmeron BJ, Ross TJ, Hong LE, Stein EA. Down-regulation of amygdala and insula functional circuits by varenicline and nicotine in abstinent cigarette smokers. Biol Psychiatry. 2013;74:538–46.
Wang K, Yang J, Zhang S, Wei D, Hao X, Tu S, et al. The neural mechanisms underlying the acute effect of cigarette smoking on chronic smokers. PLoS ONE. 2014;9:e102828.
Fedota JR, Ding X, Matous AL, Salmeron BJ, McKenna MR, Gu H, et al. Nicotine abstinence influences the calculation of salience in discrete insular circuits. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3:150–59.
Hong LE, Gu H, Yang Y, Ross TJ, Salmeron BJ, Buchholz B, et al. Association of nicotine addiction and nicotine’s actions with separate cingulate cortex functional circuits. Arch Gen Psychiatry. 2009;66:431–41.
Sutherland MT, Ray KL, Riedel MC, Yanes JA, Stein EA, Laird AR. Neurobiological impact of nicotinic acetylcholine receptor agonists: an activation likelihood estimation meta-analysis of pharmacologic neuroimaging studies. Biol Psychiatry. 2015;78:711–20.
Peoples LL. Neuroscience. Will, anterior cingulate cortex, and addiction. Science. 2002;296:1623–4.
Ashare RL, Falcone M, Lerman C. Cognitive function during nicotine withdrawal: Implications for nicotine dependence treatment. Neuropharmacology. 2014;76:581–91.
Bush G, Luu P, Posner MI. Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn Sci. 2000;4:215–22.
Bush G, Vogt BA, Holmes J, Dale AM, Greve D, Jenike MA, et al. Dorsal anterior cingulate cortex: a role in reward-based decision making. Proc Natl Acad Sci USA. 2002;99:523–8.
Vogt BA, Finch DM, Olson CR. Functional heterogeneity in cingulate cortex: the anterior executive and posterior evaluative regions. Cereb Cortex. 1992;2:435–43.
Daly M, Egan M, Quigley J, Delaney L, Baumeister RF. Childhood self-control predicts smoking throughout life: Evidence from 21,000 cohort study participants. Health Psychol. 2016;35:1254–63.
Daly M, Egan M. Childhood cognitive ability and smoking initiation, relapse and cessation throughout adulthood: evidence from two British cohort studies. Addiction. 2017;112:651–59.
Waters AJ, Shiffman S, Sayette MA, Paty JA, Gwaltney CJ, Balabanis MH. Attentional bias predicts outcome in smoking cessation. Health Psychol. 2003;22:378–87.
Kuhn S, Gallinat J. Common biology of craving across legal and illegal drugs—a quantitative meta-analysis of cue-reactivity brain response. Eur J Neurosci. 2011;33:1318–26.
McClernon FJ, Hiott FB, Huettel SA, Rose JE. Abstinence-induced changes in self-report craving correlate with event-related FMRI responses to smoking cues. Neuropsychopharmacology. 2005;30:1940–7.
Allenby C, Falcone M, Wileyto EP, Cao W, Bernardo L, Ashare RL, et al. Neural cue reactivity during acute abstinence predicts short-term smoking relapse. Addict Biol. 2020;25:e12733.
Wilson SJ, Sayette MA, Fiez JA. Prefrontal responses to drug cues: a neurocognitive analysis. Nat Neurosci. 2004;7:211–4.
Owens MM, MacKillop J, Gray JC, Beach SRH, Stein MD, Niaura RS, et al. Neural correlates of tobacco cue reactivity predict duration to lapse and continuous abstinence in smoking cessation treatment. Addict Biol. 2018;23:1189–99.
Alasmari F, Al-Rejaie SS, AlSharari SD, Sari Y. Targeting glutamate homeostasis for potential treatment of nicotine dependence. Brain Res Bull. 2016;121:1–8.
Spencer S, Scofield M, Kalivas PW. The good and bad news about glutamate in drug addiction. J Psychopharmacol. 2016;30:1095–98.
Moeller SJ, London ED, Northoff G. Neuroimaging markers of glutamatergic and GABAergic systems in drug addiction: Relationships to resting-state functional connectivity. Neurosci Biobehav Rev. 2016;61:35–52.
Mason GF. Magnetic resonance spectroscopy for studies of neurotransmission in vivo. Psychopharmacol Bull. 2003;37:26–40.
Hancu I. Optimized glutamate detection at 3T. J Magn Reson Imaging. 2009;30:1155–62.
Mullins PG, Chen H, Xu J, Caprihan A, Gasparovic C. Comparative reliability of proton spectroscopy techniques designed to improve detection of J-coupled metabolites. Magn Reson Med. 2008;60:964–9.
Gallinat J, Schubert F. Regional cerebral glutamate concentrations and chronic tobacco consumption. Pharmacopsychiatry. 2007;40:64–7.
Lesage E, Aronson SE, Sutherland MT, Ross TJ, Salmeron BJ, Stein EA. Neural signatures of cognitive flexibility and reward sensitivity following nicotinic receptor stimulation in dependent smokers: a randomized trial. JAMA Psychiatry. 2017;74:632–40.
Ip IB, Berrington A, Hess AT, Parker AJ, Emir UE, Bridge H. Combined fMRI-MRS acquires simultaneous glutamate and BOLD-fMRI signals in the human brain. Neuroimage. 2017;155:113–19.
Gu H, Hu Y, Chen X, He Y, Yang Y. Regional excitation-inhibition balance predicts default-mode network deactivation via functional connectivity. Neuroimage. 2019;185:388–97.
Wheelock MD, Reid MA, To H, White DM, Cropsey KL, Lahti AC. Open label smoking cessation with varenicline is associated with decreased glutamate levels and functional changes in anterior cingulate cortex: preliminary findings. Front Pharmacol. 2014;5:158.
Janes AC, Betts J, Jensen JE, Lukas SE. Dorsal anterior cingulate glutamate is associated with engagement of the default mode network during exposure to smoking cues. Drug Alcohol Depend. 2016;167:75–81.
Woodcock EA, Stanley JA, Diwadkar VA, Khatib D, Greenwald MK. A neurobiological correlate of stress-induced nicotine-seeking behavior among cigarette smokers. Addict Biol. 2020;25:e12819.
Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–71.
Beck AT, Epstein N, Brown G, Steer RA. An inventory for measuring clinical anxiety: psychometric properties. J Consult Clin Psychol. 1988;56:893–7.
Schmitz A, Grillon C. Assessing fear and anxiety in humans using the threat of predictable and unpredictable aversive events (the NPU-threat test). Nat Protoc. 2012;7:527–32.
Welsch SK, Smith SS, Wetter DW, Jorenby DE, Fiore MC, Baker TB. Development and validation of the Wisconsin Smoking Withdrawal Scale. Exp Clin Psychopharmacol. 1999;7:354–61.
Spielberg CD, Gorsuch RL, Lushen RE. STAI Manual for the State-Trait Anxiety Inventory. Palo Alto, CA: Consulting Psychologist Press; 1970.
Heishman SJ, Singleton EG, Pickworth WB. Reliability and validity of a Short Form of the Tobacco Craving Questionnaire. Nicotine Tob Res. 2008;10:643–51.
Singleton EG, Anderson LM, Heishman SJ. Reliability and validity of the Tobacco Craving Questionnaire and validation of a craving-induction procedure using multiple measures of craving and mood. Addiction. 2003;98:1537–46.
Watson D, Clark LA, Tellegen A. Development and validation of brief measures of positive and negative affect: the PANAS scales. J Pers Soc Psychol. 1988;54:1063–70.
Cohen S, Kamarck T, Mermelstein R. A global measure of perceived stress. J Health Soc Behav. 1983;24:385–96.
Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiatry. 1995;167:99–103.
Van Dycke A, Verstraete A, Pil K, Raedt R, Vonck K, Boison D, et al. Quantitative analysis of adenosine using liquid chromatography/atmospheric pressure chemical ionization-tandem mass spectrometry (LC/APCI-MS/MS). J Chromatogr B Anal Technol Biomed Life Sci. 2010;878:1493–8.
Taylor PJ, Forrest KK, Landsberg PG, Mitchell C, Pillans PI. The measurement of nicotine in human plasma by high-performance liquid chromatography-electrospray-tandem mass spectrometry. Ther Drug Monit. 2004;26:563–8.
Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30:672–9.
Hellem T, Shi X, Latendresse G, Renshaw PF. The utility of magnetic resonance spectroscopy for understanding substance use disorders: a systematic review of the literature. J Am Psychiatr Nurses Assoc. 2015;21:244–75.
Hu Y, Chen X, Gu H, Yang Y. Resting-state glutamate and GABA concentrations predict task-induced deactivation in the default mode network. J Neurosci. 2013;33:18566–73.
Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 1996;29:162–73.
Behzadi Y, Restom K, Liau J, Liu TT. A component based noise correction method (CompCor) for BOLD and perfusion based fMRI. Neuroimage. 2007;37:90–101.
Ward BD. Simultaneous inference for FMRI data. 2000; http://afni.nimh.nih.gov/pub/dist/doc/manual/AlphaSim.pdf.
Cox RW, Chen G, Glen DR, Reynolds RC, Taylor PA. FMRI clustering in AFNI: false-positive rates redux. Brain Connect. 2017;7:152–71.
Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci USA. 2003;100:253–8.
Fedota JR, Stein EA. Resting-state functional connectivity and nicotine addiction: prospects for biomarker development. Ann N Y Acad Sci. 2015;1349:64–82.
Uddin LQ, Supekar K, Menon V. Typical and atypical development of functional human brain networks: insights from resting-state FMRI. Front Syst Neurosci. 2010;4:21.
Sutherland MT, McHugh MJ, Pariyadath V, Stein EA. Resting state functional connectivity in addiction: Lessons learned and a road ahead. Neuroimage. 2012;62:2281–95.
Seeley WW. The salience network: a neural system for perceiving and responding to homeostatic demands. J Neurosci. 2019;39:9878–82.
Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci USA. 2001;98:676–82.
Cutini S, Scatturin P, Menon E, Bisiacchi PS, Gamberini L, Zorzi M, et al. Selective activation of the superior frontal gyrus in task-switching: an event-related fNIRS study. Neuroimage. 2008;42:945–55.
Beckmann M, Johansen-Berg H, Rushworth MF. Connectivity-based parcellation of human cingulate cortex and its relation to functional specialization. J Neurosci. 2009;29:1175–90.
Li W, Qin W, Liu H, Fan L, Wang J, Jiang T, et al. Subregions of the human superior frontal gyrus and their connections. Neuroimage. 2013;78:46–58.
Koski L, Paus T. Functional connectivity of the anterior cingulate cortex within the human frontal lobe: a brain-mapping meta-analysis. Exp Brain Res. 2000;133:55–65.
Scheidegger M, Walter M, Lehmann M, Metzger C, Grimm S, Boeker H, et al. Ketamine decreases resting state functional network connectivity in healthy subjects: implications for antidepressant drug action. PLoS ONE. 2012;7:e44799.
Duncan NW, Wiebking C, Tiret B, Marjanska M, Hayes DJ, Lyttleton O, et al. Glutamate concentration in the medial prefrontal cortex predicts resting-state cortical-subcortical functional connectivity in humans. PLoS ONE. 2013;8:e60312.
Mennecke A, Gossler A, Hammen T, Dorfler A, Stadlbauer A, Rosch J, et al. Physiological effects of cigarette smoking in the limbic system revealed by 3 tesla magnetic resonance spectroscopy. J Neural Transm. 2014;121:1211–9.
Hu S, Ide JS, Zhang S, Li CR. The right superior frontal gyrus and individual variation in proactive control of impulsive response. J Neurosci. 2016;36:12688–96.
du Boisgueheneuc F, Levy R, Volle E, Seassau M, Duffau H, Kinkingnehun S, et al. Functions of the left superior frontal gyrus in humans: a lesion study. Brain. 2006;129:3315–28.
Campbell DW, Wallace MG, Modirrousta M, Polimeni JO, McKeen NA, Reiss JP. The neural basis of humour comprehension and humour appreciation: the roles of the temporoparietal junction and superior frontal gyrus. Neuropsychologia. 2015;79:10–20.
Kong F, Hu S, Wang X, Song Y, Liu J. Neural correlates of the happy life: the amplitude of spontaneous low frequency fluctuations predicts subjective well-being. Neuroimage. 2015;107:136–45.
Japee S, Holiday K, Satyshur MD, Mukai I, Ungerleider LG. A role of right middle frontal gyrus in reorienting of attention: a case study. Front Syst Neurosci. 2015;9:23.
Leung HC, Gore JC, Goldman-Rakic PS. Sustained mnemonic response in the human middle frontal gyrus during on-line storage of spatial memoranda. J Cogn Neurosci. 2002;14:659–71.
Thiel CM, Zilles K, Fink GR. Nicotine modulates reorienting of visuospatial attention and neural activity in human parietal cortex. Neuropsychopharmacology. 2005;30:810–20.
Giessing C, Thiel CM, Rosler F, Fink GR. The modulatory effects of nicotine on parietal cortex activity in a cued target detection task depend on cue reliability. Neuroscience. 2006;137:853–64.
Hahn B, Ross TJ, Wolkenberg FA, Shakleya DM, Huestis MA, Stein EA. Performance effects of nicotine during selective attention, divided attention, and simple stimulus detection: an fMRI study. Cereb Cortex. 2009;19:1990–2000.
Hahn B, Ross TJ, Yang Y, Kim I, Huestis MA, Stein EA. Nicotine enhances visuospatial attention by deactivating areas of the resting brain default network. J Neurosci. 2007;27:3477–89.
Hahn B, Ross TJ, Stein EA. Neuroanatomical dissociation between bottom-up and top-down processes of visuospatial selective attention. Neuroimage. 2006;32:842–53.
Kenney JW, Adoff MD, Wilkinson DS, Gould TJ. The effects of acute, chronic, and withdrawal from chronic nicotine on novel and spatial object recognition in male C57BL/6J mice. Psychopharmacology. 2011;217:353–65.
Bell SL, Taylor RC, Singleton EG, Henningfield JE, Heishman SJ. Smoking after nicotine deprivation enhances cognitive performance and decreases tobacco craving in drug abusers. Nicotine Tob Res. 1999;1:45–52.
Lesage E, Sutherland MT, Ross TJ, Salmeron BJ, Stein EA. Nicotine dependence (trait) and acute nicotinic stimulation (state) modulate attention but not inhibitory control: converging fMRI evidence from Go-Nogo and Flanker tasks. Neuropsychopharmacology. 2020;45:857–65.
Flannery JS, Riedel MC, Poudel R, Laird AR, Ross TJ, Salmeron BJ, et al. Habenular and striatal activity during performance feedback are differentially linked with state-like and trait-like aspects of tobacco use disorder. Sci Adv. 2019;5:eaax2084.
Due DL, Huettel SA, Hall WG, Rubin DC. Activation in mesolimbic and visuospatial neural circuits elicited by smoking cues: evidence from functional magnetic resonance imaging. Am J Psychiatry. 2002;159:954–60.
Liechti ME, Lhuillier L, Kaupmann K, Markou A. Metabotropic glutamate 2/3 receptors in the ventral tegmental area and the nucleus accumbens shell are involved in behaviors relating to nicotine dependence. J Neurosci. 2007;27:9077–85.
Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, et al. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65:841–5.
McKenna MC. The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res. 2007;85:3347–58.
Li X, Semenova S, D’Souza MS, Stoker AK, Markou A. Involvement of glutamatergic and GABAergic systems in nicotine dependence: Implications for novel pharmacotherapies for smoking cessation. Neuropharmacology. 2014;76:554–65.
Wonnacott S, Barik J, Dickinson J, Jones IW. Nicotinic receptors modulate transmitter cross talk in the CNS: nicotinic modulation of transmitters. J Mol Neurosci. 2006;30:137–40.
McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–6.
Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–19.
Sebastiao AM, Ribeiro JA. Tuning and fine-tuning of synapses with adenosine. Curr Neuropharmacol. 2009;7:180–94.
Barrie AP, Nicholls DG. Adenosine A1 receptor inhibition of glutamate exocytosis and protein kinase C-mediated decoupling. J Neurochem. 1993;60:1081–6.
Wardas J. Neuroprotective role of adenosine in the CNS. Pol J Pharmacol. 2002;54:313–26.
Quarta D, Ferre S, Solinas M, You ZB, Hockemeyer J, Popoli P, et al. Opposite modulatory roles for adenosine A1 and A2A receptors on glutamate and dopamine release in the shell of the nucleus accumbens. Effects of chronic caffeine exposure. J Neurochem. 2004;88:1151–8.
Mosqueda-Garcia R, Tseng CJ, Appalsamy M, Beck C, Robertson D. Cardiovascular excitatory effects of adenosine in the nucleus of the solitary tract. Hypertension. 1991;18:494–502.
Corsi C, Pinna A, Gianfriddo M, Melani A, Morelli M, Pedata F. Adenosine A2A receptor antagonism increases striatal glutamate outflow in dopamine-denervated rats. Eur J Pharm. 2003;464:33–8.
Lin Z, Huang P, Huang S, Guo L, Xu X, Shen X, et al. Effect of adenosine and adenosine receptor antagonists on retinal muller cell inwardly rectifying potassium channels under exogenous glutamate stimulation. Biomed Res Int. 2018;2018:2749257.
Lopes LV, Sebastiao AM, Ribeiro JA. Adenosine and related drugs in brain diseases: present and future in clinical trials. Curr Top Med Chem. 2011;11:1087–101.
Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastiao AM. Adenosine: setting the stage for plasticity. Trends Neurosci. 2013;36:248–57.
Pardridge WM, Yoshikawa T, Kang YS, Miller LP. Blood-brain barrier transport and brain metabolism of adenosine and adenosine analogs. J Pharm Exp Ther. 1994;268:14–8.
Guzman DC, Garcia EH, Delgadillo VN, Mejia GB, Olguin HJ, Perez JH, et al. Effect of nicotine on ATPase, catalase and calcium levels in the presence of oligoelements in brain regions of young rats. Cardiovasc Hematol Disord Drug Targets. 2012;12:63–7.
Shallom JM, Katyare SS. Altered synaptosomal ATPase activity in rat brain following prolonged in vivo treatment with nicotine. Biochem Pharmacol. 1985;34:3445–9.
Bennett HJ, White TD, Semba K. Activation of cholinergic and adrenergic receptors increases the concentration of extracellular adenosine in the cerebral cortex of unanesthetized rat. Neuroscience. 2003;117:119–27.
Gossain VV, Sherma NK, Srivastava L, Michelakis AM, Rovner DR. Hormonal effects of smoking-II: Effects on plasma cortisol, growth hormone, and prolactin. Am J Med Sci. 1986;291:325–7.
Laghi Pasini F, Guideri F, Picano E, Parenti G, Petersen C, Varga A, et al. Increase in plasma adenosine during brain ischemia in man: a study during transient ischemic attacks, and stroke. Brain Res Bull. 2000;51:327–30.
Filip M, Zaniewska M, Frankowska M, Wydra K, Fuxe K. The importance of the adenosine A(2A) receptor-dopamine D(2) receptor interaction in drug addiction. Curr Med Chem. 2012;19:317–55.
Castane A, Soria G, Ledent C, Maldonado R, Valverde O. Attenuation of nicotine-induced rewarding effects in A2A knockout mice. Neuropharmacology. 2006;51:631–40.
Grieder TE, George O, Tan H, George SR, Le Foll B, Laviolette SR, et al. Phasic D1 and tonic D2 dopamine receptor signaling double dissociate the motivational effects of acute nicotine and chronic nicotine withdrawal. Proc Natl Acad Sci USA. 2012;109:3101–6.
Ballesteros-Yanez I, Castillo CA, Merighi S, Gessi S. The role of adenosine receptors in psychostimulant addiction. Front Pharmacol. 2017;8:985.
Brown RM, Short JL. Adenosine A(2A) receptors and their role in drug addiction. J Pharm Pharmacol. 2008;60:1409–30.
Fuxe K, Marcellino D, Borroto-Escuela DO, Guescini M, Fernandez-Duenas V, Tanganelli S, et al. Adenosine-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci Ther. 2010;16:e18–42.
Ferre S. Mechanisms of the psychostimulant effects of caffeine: implications for substance use disorders. Psychopharmacology. 2016;233:1963–79.
Nam HW, Bruner RC, Choi DS. Adenosine signaling in striatal circuits and alcohol use disorders. Mol Cells. 2013;36:195–202.
Ferre S, Ciruela F, Quiroz C, Lujan R, Popoli P, Cunha RA, et al. Adenosine receptor heteromers and their integrative role in striatal function. ScientificWorldJournal 2007;7:74–85.
Garcao P, Szabo EC, Wopereis S, Castro AA, Tome AR, Prediger RD, et al. Functional interaction between pre-synaptic alpha6beta2-containing nicotinic and adenosine A2A receptors in the control of dopamine release in the rat striatum. Br J Pharmacol. 2013;169:1600–11.
Jastrzebska J, Nowak E, Smaga I, Bystrowska B, Frankowska M, Bader M, et al. Adenosine (A)(2A)receptor modulation of nicotine-induced locomotor sensitization. A pharmacological and transgenic approach. Neuropharmacology. 2014;81:318–26.
Ribeiro JA, Sebastiao AM. Caffeine and adenosine. J Alzheimers Dis. 2010;20 Suppl 1:S3–15.
Sudakov SK, Bogdanova NG. Activation of peripheral kappa-opioid receptors normalizes caffeine effects modified in nicotine-dependent rats during nicotine withdrawal. Bull Exp Biol Med. 2016;161:743–45.
Metaxas A, Al-Hasani R, Farshim P, Tubby K, Berwick A, Ledent C, et al. Genetic deletion of the adenosine A(2A) receptor prevents nicotine-induced upregulation of alpha7, but not alpha4beta2* nicotinic acetylcholine receptor binding in the brain. Neuropharmacology. 2013;71:228–36.
Kaiser LG, Schuff N, Cashdollar N, Weiner MW. Age-related glutamate and glutamine concentration changes in normal human brain: 1H MR spectroscopy study at 4T. Neurobiol Aging. 2005;26:665–72.
Cleophas TJ, de Vogel EM. Crossover studies are a better format for comparing equivalent treatments than parallel-group studies. Pharm World Sci. 1998;20:113–7.
Maddock RJ, Buonocore MH. MR spectroscopic studies of the brain in psychiatric disorders. Curr Top Behav Neurosci. 2012;11:199–251.
Ottersen OP, Zhang N, Walberg F. Metabolic compartmentation of glutamate and glutamine: morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience. 1992;46:519–34.
Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF, et al. Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13 C NMR. Proc Natl Acad Sci USA. 1999;96:8235–40.
Li BS, Wang H, Gonen O. Metabolite ratios to assumed stable creatine level may confound the quantification of proton brain MR spectroscopy. Magn Reson Imaging. 2003;21:923–8.
Acknowledgements
The authors wish to thank all the participants in this study and the NIDA nursing staff. Dr. Sergi Ferre offered critical interpretation of the adenosine data. This research was supported by the Intramural Research Program of the NIDA, NIH.
Author information
Authors and Affiliations
Contributions
OAA, ES, TJR, and YY designed the study, OAA, BS, JC collected the data, ECC, MT acquired and analyzed functional imaging data, OAA, TJR, and HG analyzed spectroscopy data, HWN assayed and analyzed plasma data, OAA, MT, ECC, JC, BS performed data analysis, OAA, TJR, EAS, YY, JF, HG interpreted the results, OAA drafted the manuscript and ES, TJR, HG, ECC, JF, HWN, and YY revised the manuscript critically and all authors reviewed and contributed to the intellectual content of the manuscript and approved the final version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abulseoud, O.A., Ross, T.J., Nam, H.W. et al. Short-term nicotine deprivation alters dorsal anterior cingulate glutamate concentration and concomitant cingulate-cortical functional connectivity. Neuropsychopharmacol. 45, 1920–1930 (2020). https://doi.org/10.1038/s41386-020-0741-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-0741-9
This article is cited by
-
No difference in COVID-19 treatment outcomes among current methamphetamine, cannabis and alcohol users
Journal of Cannabis Research (2023)
-
Smoking, tobacco dependence, and neurometabolites in the dorsal anterior cingulate cortex
Molecular Psychiatry (2023)
