
Abstract
The etiologic pathways leading to neuropsychiatric diseases remain poorly defined. As genomic technologies have advanced over the past several decades, considerable progress has been made linking neuropsychiatric disorders to genetic underpinnings. Interest and consideration of nongenetic risk factors (e.g., lead exposure and schizophrenia) have, in contrast, lagged behind heritable frameworks of explanation. Thus, the association of neuropsychiatric illness to environmental chemical exposure, and their potential interactions with genetic susceptibility, are largely unexplored. In this review, we describe emerging approaches for considering the impact of chemical risk factors acting alone and in concert with genetic risk, and point to the potential role of epigenetics in mediating exposure effects on transcription of genes implicated in mental disorders. We highlight recent examples of research in nongenetic risk factors in psychiatric disorders that point to potential shared biological mechanisms—synaptic dysfunction, immune alterations, and gut–brain interactions. We outline new tools and resources that can be harnessed for the study of environmental factors in psychiatric disorders. These tools, combined with emerging experimental evidence, suggest that there is a need to broadly incorporate environmental exposures in psychiatric research, with the ultimate goal of identifying modifiable risk factors and informing new treatment strategies for neuropsychiatric disease.
Similar content being viewed by others
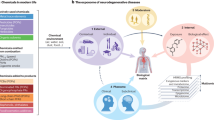

Introduction
Neuropsychiatric conditions such as anxiety, major depression, bipolar disorder, schizophrenia, obsessive-compulsive disorder, autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder (ADHD), and post-traumatic stress disorder affect hundreds of millions of people worldwide. The economic and social costs are substantial [1, 2]. While much progress has been made in identifying the complex genetic contributions to these disorders [3], genetics alone does not fully explain risk and there is significant variation that can be ascribed to environmental factors. In the broadest sense, environmental exposures can be viewed as nongenetic factors that include psychosocial factors, diet and nutrition, pharmaceuticals and medical interventions, health conditions, infectious agents, environmental chemicals, and physical features of the environment. The contribution of some of these broadly defined factors, such as drugs of abuse and developmental risk factors (e.g., preterm birth and early-life adversity), to the pathogenesis of neuropsychiatric abnormalities have been well-studied (and discussed elsewhere) [4,5,6,7] compared to non-pharmaceutical chemicals that individuals are exposed to such as pesticides, metals, and air pollutants. In some cases, work in model systems has facilitated exploration of how environmental perturbations impact brain processes relevant to mental health. Such studies have furthered our understanding of vulnerability to neuropsychiatric disorders and revealed factors contributing to resiliency. Detailed investigation of the mechanisms and comorbidities associated with the development of neuropsychiatric disorders after exposure to environmental contaminants offers the potential to afford similar gains.
Emerging evidence has suggested that exposures to environmental chemicals can contribute to the pathogenesis of neuropsychiatric abnormalities. One of the few historical examples comes from occupational exposure to mercurous nitrate in the process for curing felt for the production of hats [8]. Prolonged mercury exposure led to “Mad Hatter” syndrome with symptoms including depression, delirium, and tremor [9, 10]. Other evidence comes from studies of lead (Pb), a prevalent exposure risk that has been known for many decades to cause cognitive impairments, even at low levels [11]. Indeed, for children, the US Centers for Disease Control and Prevention (CDC) currently maintains that “no safe blood lead level…has been identified” (https://www.cdc.gov/nceh/lead/default.htm). Pb exposures in childhood have been associated with specific neuropsychiatric disturbances, including symptoms of ADHD, anxiety, and depression [12,13,14]. A 17-year prospective birth cohort study in Chicago reported that each 1 µg/dL increase in blood Pb was associated with a 0.09 point increase in anxiety/depression symptom scores [15]. Deficits in executive function, a hallmark of many neuropsychiatric disorders, have also been linked to developmental Pb exposure in experimental systems [16]. Other studies have shown that developmental exposure to pesticides is associated with neuropsychiatric behavioral phenotypes [17, 18].
A deeper understanding of the risk of psychiatric disorders related to environmental exposures and the underlying biological mechanisms could lead to more effective prevention and treatment. This review summarizes how genetic and environmental factors, along with epigenetic modifications, are being investigated to assess susceptibility and progression of neuropsychiatric disease. Next, three exemplar biological targets of environmental exposures that may contribute to etiology of mental health disorders—synaptic integrity and transmission, immune function, and signaling along the gut microbiome-brain axis are described. Finally, key challenges that remain to be addressed to enable a rigorous interrogation of the potential importance of environmental agents as determinants of risk for the development of this diverse and heterogeneous constellation of disorders are outlined.
The importance of including genetics in studies that assess environmental risks for neuropsychiatric disorders
Most psychiatric illnesses are considered to be complex diseases that involve genetic and environmental risk factors. Large genome-wide association studies (GWAS), made possible through the Psychiatric Genomics Consortium (https://www.med.unc.edu/pgc/about-us), have identified common genetic risk variants associated with specific psychiatric disorders [19,20,21,22,23], as well as across psychiatric disorders [24]. Common genetic risk variants for psychiatric outcomes are highly polygenic with many different genetic variants each contributing a small risk effect [25, 26]. Complementary whole-exome and genome-based studies have also identified a number of rare and/or de novo genetic variants that have larger risk effects on psychiatric outcomes. It is clear that genetic variation exerts a powerful influence on risk for major neuropsychiatric disorders; however, evidence as to the strength of these effects is conflicting. The wide variation in heritability estimates across and within neuropsychiatric disorders [27,28,29], and relatively small proportion of phenotypic variance explained by the genetic variants discovered to date, leaves substantial room for contributions from environmental factors in understanding neuropsychiatric risk.
The growing recognition that both genes and environmental factors contribute to neuropsychiatric disease risk underscores the need for studies that examine environmental exposures in the context of genetic risks, e.g., via gene–environment interactions (GxE). There are several conceptual models outlining how genes and environment can interact to influence disease susceptibility [30,31,32,33,34]; most neuropsychiatric studies have focused on the diathesis–stress interaction model [35], which postulates that inter-individual differences in genes result in different susceptibilities to environmental risk factors and candidate genes. Substance use disorders (SUDs) have been particularly proliferative in investigating GxE with numerous reports of interactions between social adversity or stress exposure and genetic variants that influence risk of drug or alcohol abuse [36,37,38,39,40,41]. Chemical exposures-gene interactions that influence risk of psychiatric and neurologic diseases have also been reported. For example, individuals with increased air pollutant or heavy metal exposure and a particular genetic variant have increased risk of neurodevelopmental disorders compared to individuals without the genetic variant, suggesting the genetic variant makes them more susceptible to the chemical exposure [42,43,44,45,46]. Interaction between cigarette smoking and genotype at the PON1 gene locus were observed to influence the risk of bipolar and major depression disorders [47]. Studies that assess chemical exposures and a wider range of gene-environment conceptual models are needed to advance our understanding of gene–environment interactions in neuropsychiatric disease and better inform prevention, intervention, and treatment efforts.
Integration of genetic and chemical exposure data offers new opportunities in psychiatric research but there are numerous challenges that need to be recognized and properly addressed in GxE studies, including the need for large sample sizes, consideration of exposure complexities and measurement error, potential correlation between genetic and environmental factors, and use of appropriate study designs and statistical methods [48]. Researchers are beginning to address and overcome several of these challenges. First, studies based on unified genome-wide genetic and exposure information in large samples are emerging to facilitate the study of GxE in neuropsychiatric disorder pathogenesis. One such study is the Initiative for Integrative Psychiatric Research (iPSYCH), a consortium integrating data from national registries and genomic analysis of neonatal blood spots [49]. Many other large-scale studies, including work related to efforts of the Nordic OCD & Related Disorders Consortium (NORDiC), have been launched that will link genomic data with nongenetic exposures as contributors to mental health [50, 51]. In addition to launching new studies that collect both genetic and environmental information, experimental designs that can leverage substantial extant genomics data and biorepository resources would enhance GxE efforts. This will likely involve development of new cost-effective methods to enable rigorous ascertainment of environmental exposure data from individuals with extant genetic data. Statistical challenges in GxE include low power for traditional statistical methods and inconsistent results that have often failed to replicate in independent samples for both candidate gene and genome-wide GxE studies. Over the decade new statistical methods have been developed to improve power, appropriately control for type I error rates, address exposure misclassification and gene–environment correlation, and to ensure efficient implementation of GxE analyses [52, 53]. In addition, the recent development of polygenic risk scores, representing an individual’s genome-wide genetic risk as a single summed variable, provides opportunities to evaluate environmental exposure risks in the context of aggregate genetic risk using a well-powered approach. Finally, there are several additional complexities related to potential confounding and correlations between genetic and environmental exposures in human observational studies. Exposure mixtures and multiple forms of genetic risks (e.g., inherited common single nucleotide polymorphisms, rare de novo variants, DNA structural variants), need to be considered and potentially at different developmental or life course stages. In addition, genetic variation can result in different exposure to environmental risks. For example, single nucleotide polymorphisms (SNPs) located within the nicotinic receptor genes on chromosome 15 influence smoking behaviors [54]. Importantly, correlations between polygenic risk liability and environmental risk factors for psychiatric health outcomes have been reported [55, 56]. Thus, assessing gene-exposure correlations using unified genetic and environmental exposure data could provide important insights into the contribution of each risk factor, and their potential interplay, on psychiatric health outcomes. Complementary GxE studies in animal and cellular models provide an opportunity to overcome issues of confounding, timing, gene-exposure correlation, and exposure mixtures that are present in human observational data because they use highly controlled experimental conditions. Additionally, they can provide important mechanistic insights not always possible in human observational research.
Epigenetics
Multiple lines of evidence support a role for epigenetics in the etiology of psychiatric disorders, particularly for chromatin remodeling and DNA methylation. Rare variant genetic studies of autism spectrum and bipolar disorders have shown chromatin remodeling genes are more frequently dysregulated among affected individuals [57,58,59,60]. Integration of genetic and epigenetic data has shown that genetic risk variants for ASD, schizophrenia, ADHD, and bipolar disorder control DNA methylation levels more often than non-psychiatric genetic variants [61,62,63,64,65,66,67,68]. Epigenetic changes have also been directly observed in postmortem brain [69,70,71], blood [72,73,74], semen [75, 76], saliva [77,78,79], placenta [80], and buccal [81,82,83] tissues in individuals with a psychiatric disorder. While these results support epigenetic involvement in psychiatric disease, there is some debate about the validity and reliability of these findings with respect to etiologic relevance. The concerns mainly stem from the inaccessibility of mechanistically relevant tissues or sorted cell types for brain disorders and the timing of sample collection to ascertain whether epigenetic changes are likely causal or consequential in the disease process. Because of the practical difficulty with addressing some of these concerns in human populations, complementary approaches using in vitro and animal-based model research will be necessary.
Epigenetic changes have also been associated with a diverse set of environmental exposures in human observational and animal model studies, including for chemical toxicants; these have been comprehensively reviewed elsewhere [84,85,86,87,88]. One of the best-known examples of a chemical exposure that directly alters epigenetic patterns comes from experiments in the Agouti mouse model. Exposure to bisphenol-A (BPA) in these mice leads to shifts in DNA methylation levels at the Agouti coat color locus and ultimately, an altered coat color [89]. BPA has also been associated with global and site-specific changes in DNA methylation and histone tail modifications in human observational studies [90,91,92]. A cross-organism study found mouse liver DNA methylation levels at the STAT3 locus were altered in response to perinatal exposure to BPA and were also relevant to adult health outcomes [93]. The authors also found similar DNA methylation changes in STAT3 related to BPA exposure levels measured in human fetal liver samples. Epigenetic changes in response to BPA, a type of endocrine disrupting chemical (EDC), are particularly relevant as an example given reports that EDC exposure may increase risk of psychiatric disease [94,95,96].
Studies of epigenetic changes related to chemical exposures and psychiatric disease have mainly been performed in parallel. Few studies have evaluated whether epigenetic changes related to environmental exposures are also associated with psychiatric disease or vice versa. Our ability to identify epigenetic changes related to both environmental chemicals and psychiatric disease and to formally evaluate potential epigenetic mechanisms in environmental effects on mental health is limited by small sample sizes, inaccessibility of brain tissue, potential exposure mixtures and confounding, and a lack of large prospective studies with unified epigenetic and environmental measures from relevant risk windows. This will require new epidemiology and animal and cellular model-system-based studies specifically designed to overcome these limitations. The National Institute of Environmental Health Sciences (NIEHS) is directly addressing several of these challenges through the Toxicant Exposures and Responses by Genomic and Epigenomic Regulators of Transcription (TaRGET) II Program [97]. Even if epigenetic changes are not in the causal pathway, they could provide a useful biomarker of current and past environmental exposures, including prenatal and early-life windows [98], or of psychiatric disease, which would also have great utility.
Beyond genetic risks, gene–environment interactions and epigenetic factors in psychiatric disorders, great progress has been made in understanding neurobiological mechanisms. Three examples of how environmental exposures may impact neurobiological substrates contributing to psychiatric disorders are discussed, including impacts on synaptic biology in schizophrenia, contributions of altered immune and microglial function to neurodevelopment, and emerging research on the microbiome and mood disorders. In each of these examples, environmental exposures can interact in multiple ways to influence function that contributes to risk for psychiatric disorders.
Modification of synaptic transmission by environmental exposure: evidence from studies on lead and schizophrenia
Perturbations in synaptic integrity, synaptic plasticity and neurotransmission are implicated in the pathophysiology of several psychiatric disorders. Mice that lack actin depolymerizing proteins (ADF) and n-cofilin, which are critical for synaptic function, showed increases in locomotion and impulsive behaviors, as well as impairments in working memory [99]. In addition, many neural circuits require a balance between excitatory and inhibitory neurotransmitters with new treatments for mood disorders targeting modulation of gamma-aminobutyric acid (GABA) and glutamate neurotransmission [100]. In this section, we will use schizophrenia as an example to show the relevance of studying synaptic mechanisms when considering potential targets of environmental toxicants.
Aside from the longstanding dopamine hypothesis of schizophrenia pathogenesis, altered glutamate and GABA neurotransmission have also been implicated [101]. Substantial evidence suggests that alterations in glutamatergic function can produce changes in dopaminergic and GABAergic systems that may underlie the positive and negative symptoms associated with the cognitive components of the disease [102,103,104]. A currently accepted pathophysiologic framework for schizophrenia relates to hypofunction of the N-methyl-d-aspartate subtype (NMDAR) of glutamatergic ionotropic receptor during critical periods of brain development. This relative deficit in NMDARs results in alterations of neurobiological processes essential for brain growth, wiring, synaptic plasticity, and cognitive function, all of which are affected in schizophrenia [105].
While a body of prior work has examined the association of cannabis and tobacco smoking with schizophrenia risk, emerging evidence points to a role for environmental Pb exposure. As reviewed by Guilarte et al. [101], early-life Pb exposure in animals recapitulates behavioral, neurobiological, and imaging changes relevant to schizophrenia, including loss of parvalbumin-positive GABAergic interneurons (PVGI) in the frontal cortex, hippocampus, and striatum [106,107,108], and hyperactivity in the subcortical dopaminergic system [109, 110]. A recent rodent study by Stansfield et al. [111] found that chronic exposure to environmentally relevant concentrations of Pb (average 22 µg/dL) during early brain development produced selective loss of the PVGI phenotypic markers parvalbumin and glutamic acid decarboxylase 67 (GAD67) in prefrontal cortex, hippocampus, and striatum. Pb exposure also enhanced locomotor activity in response to the psychostimulant cocaine and increased striatal dopamine metabolites and D2-dopamine receptor binding, findings indicative of a hyperactive striatal dopaminergic system, as seen in model systems and postmortem brain from individuals with schizophrenia.
Population-based studies providing the first association of early-life (prenatal) lead exposure and schizophrenia were described by Opler and colleagues [112, 113]. Using two different population-based cohorts, they found that prenatal lead exposure (maternal blood lead levels estimated to be >15 μg/dL) were associated with a significant increase in the risk of schizophrenia later in life. Following these landmark studies there have been more recent reports providing additional support to this association. For example, a recent proof-of-concept study using childhood-shed teeth and laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) showed higher levels of lead in teeth from diagnosed schizophrenic subjects relative to controls [114]. Furthermore, they found a positive correlation between early-life lead exposure and psychotic experiences in adulthood and a negative correlation with adult IQ. One of the advantages of the analysis of teeth using LA-ICP-MS is the ability to retrospectively reconstruct environmental exposures from the second trimester of pregnancy and through the first years of life. Using this approach, they found that the negative correlation of lead exposure with IQ was most pronounced if the exposure to lead occurred during the prenatal period. Arinola and colleagues also found increased plasma lead concentrations in newly diagnosed schizophrenic subjects [115]. Another study using a multidecade longitudinal design of lead-exposed children showed that higher levels of blood lead were associated with greater psychopathology and difficult personality traits as adults [116]. Childhood lead intoxication has also been associated with a high level of incarceration, delinquent behavior, and substance abuse, which are comorbid factors in schizophrenia [13, 117,118,119]. Lastly, recent assessments on the burden of lead exposure as a risk factor for mental illness in countries such as India have estimated that lead exposure accounts for a large portion of the prevalence of mental disorders including schizophrenia [120].
Neuroimmune responses and environmental chemicals
Immune perturbations have attracted special interest for their possible link to psychiatric disorders [121,122,123,124,125], consistent with emerging knowledge that cytokines, chemokines and other immune factors, as well as the cells that produce them and/or harbor cognate receptors for them within the brain, play important roles in fetal brain development, brain maturation and brain function in adulthood. Notably microglia, resident immune cells of the brain, are critical players in normal brain development. In turn, aberrant immune signaling early in life may contribute to the pathogenesis of schizophrenia, ASD, and other neuropsychiatric disorders. This is consistent with the idea that early-life environmental exposures might affect later risk for psychiatric disorders through effects on the immune system. Research using maternal immune activation model(s) provides some support for this general idea, as do studies of exposure to chronic stress [126, 127]. Many chemical exposures are also known to interact with immune pathways and molecules, or contribute to the development of abnormal immune responses such as brain-directed autoimmune reactions, yet the linkage to alterations in brain development and the implications for psychiatric conditions remain largely unexplored [128,129,130,131]. This section summarizes recent work that points to microglia as an attractive target for further studies to advance this line of inquiry.
Microglial development begins with colonization of the brain by fetal yolk sac-derived macrophage precursor cells in early embryonic stages, followed by migration and spread throughout the central nervous system (CNS) [132, 133]. Microglia are critical for normal brain development via the phagocytosis of extraneous synapses [134,135,136,137] and apoptotic cells [138,139,140], learning-dependent synapse formation [141, 142], cortical wiring [143], neuronal survival [144], and the induction of programmed cell death [139, 145]. Owing to the critical role that microglia play in normal brain development, the activation or “programming” of these dynamic, long-living cells in response to immune activation, especially early in life, can have persistent consequences for brain function throughout the lifespan [146]. Maternal immune activation (MIA) in mouse dams in response to a viral mimic, Polyinosinic:polycytidylic acid (Poly IC), at GD 15 decreased microglial phagocytic function and altered behavior consistent with schizophrenia-like traits in humans, changes which were reversed using a partial microglial inhibitor, minocycline [147]. These data point to a potential critical role for microglia in persistent changes in neural function, although other studies have failed to show an impact on microglial function in MIA models [148]. This said, MIA models have typically been limited to infectious stimuli, whereas MIA is likely elicited by a broader range of environmental chemicals or stimuli, and we point to this possibility briefly in the sections that follow.
Psychosocial stress is a well-studied nongenetic risk factor for psychiatric disorders. Multiple stress models are associated with features of microglial activation, highlighting the role of these cells as key sensors of stress-related signaling by neurons [149]. Exposure to stress induces a chronically activated phenotypic profile within microglia—referred to as “priming”—during which time these cells undergo morphological changes, demonstrate pro-inflammatory bias, and exhibit an exaggerated response to experimental immune stimulation by endotoxins [150]. This primed or “sensitized” profile of microglia in psychological stress is implicated in the long-term re-establishment of behavioral symptoms such as anxiety-like behaviors in mice initially exposed to repeated social defeat stress [151].
There is now a significant body of literature linking air pollutant exposures with microglial activation and other neuroimmune changes, e.g., increased oxidative stress and lipid peroxidation [152,153,154]. The association between ambient air pollution and neurodevelopmental disorders such as ASD points to the prenatal period as particularly important. Indeed, recent studies of air pollution exposure underscore the ability of environmental chemicals to impact brain development via effects on microglia [155]. For example, maternal diesel exhaust particle (DEP) exposure exerted sex-dependent effects on fetal brain microglial morphology and development. Results in males were consistent with activation and/or a delay in maturation in several brain regions, along with striking changes in neural–glial interactions suggestive of a defect in phagocytosis [156]. In addition, exposure of pregnant dams to ultrafine particles (UFP), another component of air pollution, leads to increases in corpus callosum size, hypermyelination and microglial activation in offspring [157].
Environmental exposures and the microbiome-gut-brain axis
The gut microbiome is increasingly understood to be a vital contributing component of the gut-brain axis. Mechanisms by which the microbiome may drive mental and brain health outcomes, and the need to understand how environmental chemicals can perturb these processes, are explored in this section.
Microbes and microbial metabolites influence neurobehavioral and neurotoxic outcomes through dysregulation of biological signaling pathways, or disruption of epithelial barrier integrity (“leaky gut”). Essential neurotransmitters for CNS functioning and mood regulation are interdependent with functions of the gut microbiome; for example, certain gut microbes are required to support peripheral production of serotonin and GABA, with serotonin serving as the key signaling molecule in both the enteric and central nervous systems [158, 159]. Critically, gut microbiota and metabolites, including short chain fatty acids, exert potent effects on peripheral immune processes via their effects on intestinal epithelial cells and modulation of G-protein coupled receptor-dependent signaling pathways [160]. In a leaky gut state, intestinal microbe membrane-derived lipopolysaccharide (LPS), also known as endotoxin, enters the peripheral circulation to promote an inflammatory state. Persistent low-grade immune-inflammatory processes have been implicated in the pathophysiology of major depressive disorder (MDD), schizophrenia, PTSD and mania [121, 122, 161,162,163], potentially linking the gut microbiome to psychiatric outcomes.
The role of environmental chemical exposure in gastrointestinal microbe–brain interactions is not well-characterized; however, inferential evidence in the literature supports microbial dysbiosis as a mediator of chemical-induced neurotoxicity. Toxicants may factor into the aforementioned mechanisms by targeting specific subpopulations of microbes to alter bacterial community diversity, creating or enhancing a leaky gut or neuroimmune state, or by undergoing biotransformation by bacteria to neurotoxic metabolites. The potential role of the gut-brain axis in chemical-induced neurotoxicity has been broadly reviewed previously [164, 165]. Despite the large body of literature pointing to interconnectivity of the microbiome, chemical exposure, and the brain, few studies have successfully linked all three factors in a manner that confers causality. This may be due to the many challenges faced by ascribing mechanism to something as variable as the microbiome.
Studies suggest that changes in intestinal microbiota diversity and community may be associated with psychiatric disorders such as ASD, schizophrenia, and depression [166,167,168]. While these studies demonstrate a clear gut–brain interaction, they do not consider the additional complexity of external environmental factors. A wide range of environmental factors and neurological outcomes have been linked to altered microbial composition. The anti-seizure benefits of a ketogenic diet were found to be dependent upon the microbiome in mice [169]. Light-stress in mice led to enhanced abundances of some bacterial species and metabolites, which correlated with reduced memory potential [170]. Some studies have focused on chemical stressors as a cause of microbial dysbiosis, although they tend to be more correlative in nature; for example, neurobehavioral changes due to developmental exposure to genistein, a phytoestrogen, were correlated with microbiome and metabolome changes in California mice [171].
The most significant challenge faced in understanding the role of the microbiome in psychiatric disorders, and human health more broadly, is moving beyond association and correlation to causation. This is not only true in rodents, but also in humans, where it is less evident, which factor is the mediator for disease and toxicity. Epidemiology studies have identified associations between microbial dysbiosis and environmental exposure, including traffic-related air pollution, persistent organic pollutants, mercury, lead and other chemicals [165, 172,173,174,175]. It is necessary to use a myriad of scientific approaches to arrive at a compelling display of evidence for gut microbiome–brain interaction. There have been several studies in rodents and humans examining this interplay in ASD. In an animal model of ASD, gut permeability was linked to serum levels of the microbial metabolite 4EPS; direct treatment of 4EPS was sufficient to induce autism-like behaviors in naïve mice [176]. Other studies have confirmed that autistic behaviors in mouse models of ASD can be attenuated by treatment with microbial metabolites or probiotics [177, 178]. A fecal microbial transfer (FMT) study in germ-free mice showed that mice harboring microbiota from human ASD donors, but not typical donors, displayed hallmark autistic behaviors [177]. In humans, there have been a few small studies assessing the utility of the gut microbiome to alter behavior. FMT in children with ASD who also presented with moderate gastrointestinal distress led to gastrointestinal and behavioral improvements [179]. Much more information is needed to understand which patient populations are most responsive, and whether probiotic treatment would be a valuable alternative. Importantly, there have also been many environmental exposures associated with the development of ASD [180,181,182,183], and more research into the role of the microbiome could help elucidate mechanisms.
ASD is just one example of a psychiatric outcome that may sit at the crossroads of microbial dysbiosis and environmental exposure. Continuing to clarify the relationship among environmental exposure, microbiome, and psychiatric outcomes is crucial because of the potential impact on human health and preventative medicine. Understanding these nuances can pave the way for developing novel interventions and biomarkers for exposure, gaining insight into inter-individual susceptibility, and ultimately allowing for focused, innovative, and holistic approaches to medical treatment.
Addressing key challenges
The previous sections highlighted three areas, synaptic biology, immunology, and gut microbiome, where provocative new data support the feasibility and significance of studies to identify the role of environmental chemicals in psychiatric conditions. The pace of continued progress will depend on the ability of researchers to harness new tools and concepts that are being developed to understand causation of complex disorders. Foremost is the development and implementation of the exposome concept. Another important advance is the changing conceptualization of psychiatric disorders; the field is moving from symptom-based diagnosis to identifying patterns of neurobiologic dysfunction and dimensional models of psychiatric traits. New population-based preclinical models of common genetic diversity and computational resources such as the Comparative Toxicogenomics Database provide additional opportunities to explore relationships among chemicals, genes, and psychiatric disorders. This section lays out the promise of these new tools and approaches for the study of environmental contributors to psychiatric disorders.
Environmental health science is moving away from a focus on single toxicants or single classes of exposure, recognizing that we are exposed to complex mixtures that vary over time in composition and intensity. The concept of the exposome, proposed in 2005 as a complement to the genome [184], has evolved to be defined as the “cumulative measure of environmental influences and biologic responses throughout the lifespan” [185]. Three broad and partially overlapping domains of the exposome are recognized [186]. The specific external exposome includes exposures such as environmental chemicals, pharmaceuticals, radiation, infectious agents, diet and physical activity. The general external exposome refers to larger social and economic factors such as urbanicity, education, and climate. The internal exposome comprises the chemical milieu within the body and includes biologically active compounds associated with endogenous processes (e.g., inflammation, metabolism, microbiome). Significant advances in use and integration of data obtained from geographic information systems, remote sensing, wearable personal sensors, and numerous mobile phone-based applications (e.g., activity tracking) have created new opportunities for measuring important aspects of the external exposome [187]. Metabolomic-based biomonitoring has seen increasing use in measuring the internal exposome, as it can provide an untargeted, agnostic approach to characterize the holistic cellular response to exposure(s) in the context of susceptibility factors (e.g., aging, genetic variation). In the same way that genomic approaches provide significant advantage over candidate gene approaches to understanding the complex genetic architecture of psychiatric disorders, exposomic approaches can deepen our knowledge of the multiple, interactive and time-dependent nongenetic factors that affect risk and resilience for these disorders. While many measurement and analytic challenges remain, meaningful preparatory steps can be taken to ensure that psychiatric cohorts are poised to capitalize on advances in exposomics [188,189,190]. Moreover, collaborative efforts between psychiatric and environmental health researchers would provide an excellent testbed for addressing the integration of different aspects of the external exposome (e.g., environmental chemicals that are the focus of environmental health science studies and larger social and economic environments that are often considered in psychiatric research).
The current phenomenological criteria used to clinically diagnose an individual with mental illness is subjective [191], leading to considerable variance and imprecision [192, 193]. For research purposes, this means that individuals with distinct neurobiological circuits and pathophysiology may be grouped together under a single diagnosis, obscuring important differences in risk factors among individuals. The corollary, that two individuals with different clinical diagnoses may have shared pathophysiology, also creates challenges for clinical research. To help address this issue, the National Institute of Mental Health (NIMH) has spearheaded an initiative to classify neurocircuit mediated symptoms, with relevance to psychiatric disorders, by creating the Research Domain Criteria (RDoC) initiative [194, 195]. Although studies of environmental influences are strongly emphasized and encouraged in RDoC research (https://www.nimh.nih.gov/research/research-funded-by-nimh/rdoc/constructs/rdoc-matrix.shtml), specific environmental effects are not listed explicitly. New partnerships between psychiatric and environmental health researchers are needed to implement RDoC for the study of nongenetic/environmental influences. Psychiatric researchers who are already collecting data in the framework of RDoC domains and constructs can expand their studies to include one or more exposures, thus contributing data to illuminate whether and how relationships among functional domains and constructs may depend on environmental context. Environmental health researchers can consider how the RDoC framework can be incorporated in their own lines of research; this may require additional collection of physiology, behavior, cognition, and symptom measures to enable systematic mapping to existing RDoC domains and constructs. Ultimately, the use of objective measures anchored to neurocircuits of neuropsychopathology as in RDoC will allow for more precise evaluation of environmental contributions to psychiatric symptoms and disease.
The increasing recognition of psychiatric disorders as dimensional rather that dichotomous [196,197,198,199] also has implications for the study of nongenetic influences. The low prevalence of most psychiatric disorders requires very large sample sizes for sufficiently powered population-based studies. Unfortunately, deep and rigorous exposure assessments on large numbers of study participants are often not feasible; however, if psychiatric traits are distributed continuously in the general population, it is possible to use an unselected sample to identify associations of exposures with variation in the trait because, in addition to individuals that meet diagnostic criteria, the much larger number of individuals in the population-based sampling frame with subclinical traits can contribute to identifying any association with exposure. This approach could be adapted for use in many existing smaller studies that have deep exposure characterization. The Epidemiology Resources web tool (https://tools.niehs.nih.gov/cohorts/) organizes and shares information about environmental epidemiology studies supported by the NIEHS, with the goal of facilitating new collaborations and ancillary studies such as those recommended in this section.
Model systems provide a critical tool in the investigation of environmental exposure effects on human health. Although rodents cannot fully recapitulate the complex neuropathology of psychiatric disorders, model systems can be used to test hypotheses about the impact of perturbations representing potential risk factors, including genomic or environmental variables, on brain processes relevant to mental health. Thus, these models can provide a platform to investigate the role of environmental mechanisms that shape neurodevelopment of circuits linked to etiology and symptomatology of human psychopathology. Although models that focus on a single exposure and/or genetic risk are prevalent, a promising trend is the use of models that better reflect multifactorial risk. “Two-hit” rodent models have been used to understand the interactions between multiple or repeated exposures at critical developmental timepoints. For example, a second juvenile exposure to polychlorinated biphenyls (PCBs) can have profound and differential sex-specific effects on gene expression, social or anxiety behavior in rats prenatally exposed to the same PCBs [200, 201]. Many other “two-hit” models, often focused on immune and/or stress exposures, support the important role of combined exposures in psychiatric phenotypes [202,203,204].
As was described in an earlier section, both rare and common forms of genetic risk contribute to psychiatric disorders. A recent GWAS study identified more than 100 genetic loci for schizophrenia [21]. The large number of common alleles confer modest effect; this suggests that reliance on models with disruption of single genes to test gene–environment interaction may not be informative. To confront the challenge of polygenic causation of complex diseases, population-based mouse models have been developed [205]. The Collaborative Cross (CC) is a large panel of recombinant inbred mouse strains intentionally designed to mimic the genetic diversity observed in human populations [206, 207]; this provides a powerful model to support systems genetic approaches for the study of quantitative biologic and behavioral traits. In the context of environmental exposures, studies using population-based mouse resources such as CC have the advantage of including both mice with increased sensitivity and those with reduced sensitivity (resilience). CC mice can be used to study gene–environment interaction, i.e., how common genetic variation modifies the effect of exposure(s) on psychiatric phenotypes. As these mice capture a very wide range of common genetic variation, researchers are freed from focusing on a preselected small number of candidate genes/loci thought to confer susceptibility, making it more likely that new, previously unrecognized GxE will be identified. Two recent examples in psychiatric research support the use of CC mice to study gene–environment interactions [208, 209].
As emphasized throughout this section, new approaches are needed to move beyond single gene and single exposure studies of mental illness. Data generation is increasing in areas of brain-based disorders, and spans genomics, transcriptomics, metagenomics, and exposomics. Given the diversity of information scope and scale, providing resources to mine, share and manage relevant information will require public resources and data standards. One resource that merits attention is the Comparative Toxicogenomics Database (CTD) [210]. CTD is a manually curated database that provides information describing chemical-gene interactions, chemical- and gene-disease relationships, chemical-phenotype relationships and detailed exposure data [211,212,213,214]. These data are integrated with pathway and functional information with tools that can help users inform hypotheses about the etiologies of phenotypes and diseases with a specific focus on environmental and exposure influences. In addition to curation and summation of data providing direct evidence for linkage of a gene or exposure with a disease, CTD provides statistically ranked inferred relationships among genes, exposures and diseases. This feature is particularly powerful for unearthing new clues about possible chemical-disease relationships. For example, if the curated literature indicates that an exposure influences the expression of a gene and, in separate publications, that gene is associated with a disease, then CTD provides an inference (via the shared gene) that the exposure is linked to the disease; this provides a new testable hypothesis that can be pursued further in laboratory or clinical settings. CTD has been employed in the analysis of a range of brain disorders [215,216,217,218] and can be helpful for guiding future hypotheses about relationships among exposures, genes and psychiatric disorders.
Concluding perspective
The landscape of psychiatric research has changed rapidly over the past decade. The growing evidence that environmental exposures disrupt normal brain and behavioral function to increase psychiatric risk suggests that greater collaboration between psychiatric and environmental health research communities is critical. Further, strategies to determine the most appropriate and productive directions in psychiatric research will undoubtedly require the inclusion of and appreciation for emerging areas discussed here, e.g., synaptic plasticity, immunology, gut microbiome-brain signaling, and others. The potential payoff for identifying environmental risk factors for psychiatric disorders is tremendous, as it affords a unique opportunity to not just modify symptom trajectory following diagnosis, but the possibility to prevent the development of the disease from the outset.
Funding and disclosure
This work was supported in part by the NIH, National Institute of Environmental Health Sciences (to JA Hollander, KA McAllister, AL Dzierlenga, AE Garton, CP Lawler) and National Institute of Mental Health (to JL Zehr), the NIH grants: R01ES014065 and P30ES025128 (to CJ Mattingly); R01ES06189 (to TR Guilarte); R01HD090051 (to M Hornig); U54HD079124 (to SS Moy); and the U.S. Department of Veterans Affairs grant IK2CX001397 (to ST Szabo). FN Jacka has received Grant/Research support from the Brain and Behavior Research Institute, the National Health and Medical Research Council (NHMRC), Australian Rotary Health, the Geelong Medical Research Foundation, the Ian Potter Foundation, Eli Lilly, Meat and Livestock Australia, Woolworths Limited, Fernwood Gyms, The Wilson Foundation, The A2 Milk Company, The University of Melbourne and has received speakers honoraria from Sanofi-Synthelabo, Janssen Cilag, Servier, Pfizer, Health Ed, Network Nutrition, Angelini Farmaceutica, Eli Lilly and Metagenics. FN Jacka has written two books for commercial publication that promotes the idea that good diet quality is important for mental and brain health. The views expressed in this review are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States Government. The remaining authors declare no competing interests.
References
Kessler RC. The costs of depression. Psychiatr Clin North Am. 2012;35:1–14.
Baldan F, Gnan C, Franzoni A, Ferino L, Allegri L, Passon N, et al. Genomic deletion involving the IMMP2L gene in two cases of autism spectrum disorder. Cytogenet Genome Res. 2018;154:196–200.
Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13:537–51.
Miller ML, Chadwick B, Dickstein DL, Purushothaman I, Egervari G, Rahman T, et al. Adolescent exposure to Delta(9)-tetrahydrocannabinol alters the transcriptional trajectory and dendritic architecture of prefrontal pyramidal neurons. Mol Psychiatry. 2019;24:588–600.
Ross S, Peselow E. Co-occurring psychotic and addictive disorders: neurobiology and diagnosis. Clin Neuropharmacol. 2012;35:235–43.
Nosarti C, Reichenberg A, Murray RM, Cnattingius S, Lambe MP, Yin L, et al. Preterm birth and psychiatric disorders in young adult life. Arch Gen Psychiatry. 2012;69:E1–8.
Copeland WE, Shanahan L, Hinesley J, Chan RF, Aberg KA, Fairbank JA, et al. Association of childhood trauma exposure with adult psychiatric disorders and functional outcomes. JAMA Netw Open. 2018;1:e184493.
United States. Public Health Service., Neal PA, Jones RR, Bloomfield JJ, DallaValle JM, Edwards TI. A study of chronic mercurialism in the hatters’ fur-cutting industry. Washington: U.S. Govt. Print. Off.; 1937. p. 70.
Feldman RG. Neurological manifestations of mercury intoxication. Acta Neurol Scand Suppl. 1982;92:201–9.
Fine EJ. The Alice in Wonderland syndrome. Prog Brain Res. 2013;206:143–56.
American Academy of Pediatrics Committee on Environmental Health. Lead exposure in children: prevention, detection, and management. Pediatrics. 2005;116:1036–46.
Needleman HL, Gunnoe C, Leviton A, Reed R, Peresie H, Maher C, et al. Deficits in psychologic and classroom performance of children with elevated dentine lead levels. N. Engl J Med. 1979;300:689–95.
Needleman HL, Riess JA, Tobin MJ, Biesecker GE, Greenhouse JB. Bone lead levels and delinquent behavior. JAMA. 1996;275:363–9.
Bouchard MF, Bellinger DC, Weuve J, Matthews-Bellinger J, Gilman SE, Wright RO, et al. Blood lead levels and major depressive disorder, panic disorder, and generalized anxiety disorder in US young adults. Arch Gen Psychiatry. 2009;66:1313–9.
Winter AS, Sampson RJ. From lead exposure in early childhood to adolescent health: a Chicago Birth Cohort. Am J Public Health. 2017;107:1496–501.
Driscoll LL, Strupp BJ. Assessment of attention and inhibitory control in rodent developmental neurotoxicity studies. Neurotoxicol Teratol. 2015;52:78–87.
Richardson JR, Taylor MM, Shalat SL, Guillot TS III, Caudle WM, Hossain MM, et al. Developmental pesticide exposure reproduces features of attention deficit hyperactivity disorder. FASEB J. 2015;29:1960–72.
Grandjean P, Landrigan PJ. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014;13:330–8.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81.
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Li Z, Chen J, Yu H, He L, Xu Y, Zhang D, et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat Genet. 2017;49:1576–83.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431–44.
Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–9.
Brainstorm C, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360:1–12.
Zhang Y, Qi G, Park JH, Chatterjee N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat Genet. 2018;50:1318–26.
Hilker R, Helenius D, Fagerlund B, Skytthe A, Christensen K, Werge TM, et al. Heritability of schizophrenia and schizophrenia spectrum based on the Nationwide Danish Twin Register. Biol Psychiatry. 2018;83:492–8.
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–62.
Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702–9.
Belsky J, Pluess M. Beyond diathesis stress: differential susceptibility to environmental influences. Psychol Bull. 2009;135:885–908.
Bronfenbrenner U, Ceci SJ. Nature-nurture reconceptualized in developmental perspective: a bioecological model. Psychol Rev. 1994;101:568–86.
Jolicoeur-Martineau A, Belsky J, Szekely E, Widaman KF, Pluess M, Greenwood C, et al. Distinguishing differential susceptibility, diathesis-stress, and vantage sensitivity: Beyond the single gene and environment model. Dev Psychopathol. 2020;32:73–83.
Zubin J, Spring B. Vulnerability–a new view of schizophrenia. J Abnorm Psychol. 1977;86:103–26.
Pluess M, Belsky J. Vantage sensitivity: individual differences in response to positive experiences. Psychol Bull. 2013;139:901–16.
Assary E, Vincent JP, Keers R, Pluess M. Gene-environment interaction and psychiatric disorders: Review and future directions. Semin Cell Dev Biol. 2018;77:133–43.
Kendler KS, Ohlsson H, Sundquist K, Sundquist J. Peer deviance, parental divorce, and genetic risk in the prediction of drug abuse in a nationwide Swedish sample: evidence of environment-environment and gene-environment interaction. JAMA Psychiatry. 2014;71:439–45.
Enoch MA. The influence of gene-environment interactions on the development of alcoholism and drug dependence. Curr Psychiatry Rep. 2012;14:150–8.
Kim J, Marciano MA, Ninham S, Zaso MJ, Park A. Interaction effects between the cumulative genetic score and psychosocial stressor on self-reported drinking urge and implicit attentional bias for alcohol: a human laboratory study. Alcohol Alcohol. 2019;54:30–7.
Rovis D, Cernelic Bizjak M, Vasiljev Marchesi V, Petelin A, Jenus T, Vidic S, et al. Increased risk-taking behaviour and brain-derived neurotrophic factor Val66Met polymorphism correlates to decreased serum brain-derived neurotrophic factor level in heroin users. Eur Addict Res. 2018;24:189–200.
Barr PB, Silberg J, Dick DM, Maes HH. Childhood socioeconomic status and longitudinal patterns of alcohol problems: variation across etiological pathways in genetic risk. Soc Sci Med. 2018;209:51–8.
Brody GH, Chen YF, Yu T, Beach SR, Kogan SM, Simons RL, et al. Life stress, the dopamine receptor gene, and emerging adult drug use trajectories: a longitudinal, multilevel, mediated moderation analysis. Dev Psychopathol. 2012;24:941–51.
Kim D, Volk H, Girirajan S, Pendergrass S, Hall MA, Verma SS, et al. The joint effect of air pollution exposure and copy number variation on risk for autism. Autism Res. 2017;10:1470–80.
Volk HE, Kerin T, Lurmann F, Hertz-Picciotto I, McConnell R, Campbell DB. Autism spectrum disorder: interaction of air pollution with the MET receptor tyrosine kinase gene. Epidemiology. 2014;25:44–7.
Alemany S, Vilor-Tejedor N, Garcia-Esteban R, Bustamante M, Dadvand P, Esnaola M, et al. Traffic-related air pollution, APOEepsilon4 status, and neurodevelopmental outcomes among school children enrolled in the BREATHE Project (Catalonia, Spain). Environ Health Perspect. 2018;126:087001.
Wang Z, Claus Henn B, Wang C, Wei Y, Su L, Sun R, et al. Genome-wide gene by lead exposure interaction analysis identifies UNC5D as a candidate gene for neurodevelopment. Environ Health. 2017;16:81.
Kim JI, Kim JW, Lee JM, Yun HJ, Sohn CH, Shin MS, et al. Interaction between DRD2 and lead exposure on the cortical thickness of the frontal lobe in youth with attention-deficit/hyperactivity disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2018;82:169–76.
Bortolasci CC, Vargas HO, Souza-Nogueira A, Barbosa DS, Moreira EG, Nunes SO, et al. Lowered plasma paraoxonase (PON)1 activity is a trait marker of major depression and PON1 Q192R gene polymorphism-smoking interactions differentially predict the odds of major depression and bipolar disorder. J Affect Disord. 2014;159:23–30.
McAllister K, Mechanic LE, Amos C, Aschard H, Blair IA, Chatterjee N, et al. Current challenges and new opportunities for gene-environment interaction studies of complex diseases. Am J Epidemiol. 2017;186:753–61.
Pedersen CB, Bybjerg-Grauholm J, Pedersen MG, Grove J, Agerbo E, Baekvad-Hansen M, et al. The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry. 2018;23:6–14.
Nievergelt CM, Maihofer AX, Mustapic M, Yurgil KA, Schork NJ, Miller MW, et al. Genomic predictors of combat stress vulnerability and resilience in U.S. Marines: A genome-wide association study across multiple ancestries implicates PRTFDC1 as a potential PTSD gene. Psychoneuroendocrinology. 2015;51:459–71.
Brander G, Rydell M, Kuja-Halkola R, Fernandez de la Cruz L, Lichtenstein P, Serlachius E, et al. Association of perinatal risk factors with obsessive-compulsive disorder: a population-based birth cohort, sibling control study. JAMA Psychiatry. 2016;73:1135–44.
Gauderman WJ, Mukherjee B, Aschard H, Hsu L, Lewinger JP, Patel CJ, et al. Update on the state of the science for analytical methods for gene-environment interactions. Am J Epidemiol. 2017;186:762–70.
Mechanic LE, Chen HS, Amos CI, Chatterjee N, Cox NJ, Divi RL, et al. Next generation analytic tools for large scale genetic epidemiology studies of complex diseases. Genet Epidemiol. 2012;36:22–35.
Wen L, Jiang K, Yuan W, Cui W, Li MD. Contribution of variants in CHRNA5/A3/B4 gene cluster on chromosome 15 to tobacco smoking: from genetic association to mechanism. Mol Neurobiol. 2016;53:472–84.
Leppert B, Havdahl A, Riglin L, Jones HJ, Zheng J, Davey Smith G, et al. Association of maternal neurodevelopmental risk alleles with early-life exposures. JAMA Psychiatry. 2019;76:834–42.
Krapohl E, Hannigan LJ, Pingault JB, Patel H, Kadeva N, Curtis C, et al. Widespread covariation of early environmental exposures and trait-associated polygenic variation. Proc Natl Acad Sci USA. 2017;114:11727–32.
Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215–33.
Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37:95–105.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94:677–94.
Han MR, Han KM, Kim A, Kang W, Kang Y, Kang J, et al. Whole-exome sequencing identifies variants associated with structural MRI markers in patients with bipolar disorders. J Affect Disord. 2019;249:159–68.
Zhao Y, Liang X, Zhu F, Wen Y, Xu J, Yang J, et al. A large-scale integrative analysis of GWAS and common meQTLs across whole life course identifies genes, pathways and tissue/cell types for three major psychiatric disorders. Neurosci Biobehav Rev. 2018;95:347–52.
Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 2016;19:48–54.
Gamazon ER, Badner JA, Cheng L, Zhang C, Zhang D, Cox NJ, et al. Enrichment of cis-regulatory gene expression SNPs and methylation quantitative trait loci among bipolar disorder susceptibility variants. Mol Psychiatry. 2013;18:340–6.
Ciuculete DM, Bostrom AE, Voisin S, Philipps H, Titova OE, Bandstein M, et al. A methylome-wide mQTL analysis reveals associations of methylation sites with GAD1 and HDAC3 SNPs and a general psychiatric risk score. Transl Psychiatry. 2017;7:e1002.
Hoffmann A, Ziller M, Spengler D. The Future is The Past: Methylation QTLs in Schizophrenia. Genes (Basel). 2016;7:1–18.
Hannon E, Dempster E, Viana J, Burrage J, Smith AR, Macdonald R, et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016;17:176.
Andrews SV, Ellis SE, Bakulski KM, Sheppard B, Croen LA, Hertz-Picciotto I, et al. Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder. Nat Commun. 2017;8:1011.
Hannon E, Schendel D, Ladd-Acosta C, Grove J, i P-BASDG, Hansen CS, et al. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med. 2018;10:19.
Birnbaum R, Weinberger DR. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat Rev Neurosci. 2017;18:727–40.
Nestler EJ, Pena CJ, Kundakovic M, Mitchell A, Akbarian S. Epigenetic basis of mental illness. Neuroscientist. 2016;22:447–63.
Ellis SE, Gupta S, Moes A, West AB, Arking DE. Exaggerated CpH methylation in the autism-affected brain. Mol Autism. 2017;8:6.
Andrews SV, Sheppard B, Windham GC, Schieve LA, Schendel DE, Croen LA, et al. Case-control meta-analysis of blood DNA methylation and autism spectrum disorder. Mol Autism. 2018;9:40.
Lin D, Chen J, Perrone-Bizzozero N, Bustillo JR, Du Y, Calhoun VD, et al. Characterization of cross-tissue genetic-epigenetic effects and their patterns in schizophrenia. Genome Med. 2018;10:13.
van Mil NH, Steegers-Theunissen RP, Bouwland-Both MI, Verbiest MM, Rijlaarsdam J, Hofman A, et al. DNA methylation profiles at birth and child ADHD symptoms. J Psychiatr Res. 2014;49:51–9.
Feinberg JI, Bakulski KM, Jaffe AE, Tryggvadottir R, Brown SC, Goldman LR, et al. Paternal sperm DNA methylation associated with early signs of autism risk in an autism-enriched cohort. Int J Epidemiol. 2015;44:1199–210.
Jenkins TG, Aston KI, Pflueger C, Cairns BR, Carrell DT. Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLoS Genet. 2014;10:e1004458.
Wilmot B, Fry R, Smeester L, Musser ED, Mill J, Nigg JT. Methylomic analysis of salivary DNA in childhood ADHD identifies altered DNA methylation in VIPR2. J Child Psychol Psychiatry. 2016;57:152–60.
Abdolmaleky HM, Nohesara S, Ghadirivasfi M, Lambert AW, Ahmadkhaniha H, Ozturk S, et al. DNA hypermethylation of serotonin transporter gene promoter in drug naive patients with schizophrenia. Schizophr Res. 2014;152:373–80.
Teroganova N, Girshkin L, Suter CM, Green MJ. DNA methylation in peripheral tissue of schizophrenia and bipolar disorder: a systematic review. BMC Genet. 2016;17:27.
Schroeder DI, Schmidt RJ, Crary-Dooley FK, Walker CK, Ozonoff S, Tancredi DJ, et al. Placental methylome analysis from a prospective autism study. Mol Autism. 2016;7:51.
Fisher HL, Murphy TM, Arseneault L, Caspi A, Moffitt TE, Viana J, et al. Methylomic analysis of monozygotic twins discordant for childhood psychotic symptoms. Epigenetics. 2015;10:1014–23.
Heinrich H, Grunitz J, Stonawski V, Frey S, Wahl S, Albrecht B, et al. Attention, cognitive control and motivation in ADHD: Linking event-related brain potentials and DNA methylation patterns in boys at early school age. Sci Rep. 2017;7:3823.
Januar V, Ancelin ML, Ritchie K, Saffery R, Ryan J. BDNF promoter methylation and genetic variation in late-life depression. Transl Psychiatry. 2015;5:e619.
Bakulski KM, Fallin MD. Epigenetic epidemiology: promises for public health research. Environ Mol Mutagen. 2014;55:171–83.
Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011;31:363–73.
Del Blanco B, Barco A. Impact of environmental conditions and chemicals on the neuronal epigenome. Curr Opin Chem Biol. 2018;45:157–65.
Estill MS, Krawetz SA. The epigenetic consequences of paternal exposure to environmental contaminants and reproductive toxicants. Curr Environ Health Rep. 2016;3:202–13.
Ryu HW, Lee DH, Won HR, Kim KH, Seong YJ, Kwon SH. Influence of toxicologically relevant metals on human epigenetic regulation. Toxicol Res. 2015;31:1–9.
Anderson OS, Nahar MS, Faulk C, Jones TR, Liao C, Kannan K, et al. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ Mol Mutagen. 2012;53:334–42.
Senyildiz M, Karaman EF, Bas SS, Pirincci PA, Ozden S. Effects of BPA on global DNA methylation and global histone 3 lysine modifications in SH-SY5Y cells: An epigenetic mechanism linking the regulation of chromatin modifiying genes. Toxicol Vitr. 2017;44:313–21.
Junge KM, Leppert B, Jahreis S, Wissenbach DK, Feltens R, Grutzmann K, et al. MEST mediates the impact of prenatal bisphenol A exposure on long-term body weight development. Clin Epigenetics. 2018;10:58.
Zheng H, Zhou X, Li DK, Yang F, Pan H, Li T, et al. Genome-wide alteration in DNA hydroxymethylation in the sperm from bisphenol A-exposed men. PLoS ONE. 2017;12:e0178535.
Weinhouse C, Bergin IL, Harris C, Dolinoy DC. Stat3 is a candidate epigenetic biomarker of perinatal Bisphenol A exposure associated with murine hepatic tumors with implications for human health. Epigenetics. 2015;10:1099–110.
Rivollier F, Krebs MO, Kebir O. Perinatal exposure to environmental endocrine disruptors in the emergence of neurodevelopmental psychiatric diseases: a systematic review. Int J Environ Res Public Health. 2019;16:1–19.
Brown JS Jr. Effects of bisphenol-A and other endocrine disruptors compared with abnormalities of schizophrenia: an endocrine-disruption theory of schizophrenia. Schizophr Bull. 2009;35:256–78.
Braun JM, Muckle G, Arbuckle T, Bouchard MF, Fraser WD, Ouellet E, et al. Associations of prenatal urinary bisphenol a concentrations with child behaviors and cognitive abilities. Environ Health Perspect. 2017;125:067008.
Wang T, Pehrsson EC, Purushotham D, Li D, Zhuo X, Zhang B, et al. The NIEHS TaRGET II Consortium and environmental epigenomics. Nat Biotechnol. 2018;36:225–7.
Ladd-Acosta C, Fallin MD. DNA methylation signatures as biomarkers of prior environmental exposures. Curr Epidemiol Rep. 2019;6:1–13.
Zimmermann AM, Jene T, Wolf M, Gorlich A, Gurniak CB, Sassoe-Pognetto M, et al. Attention-deficit/hyperactivity disorder-like phenotype in a mouse model with impaired actin dynamics. Biol Psychiatry. 2015;78:95–106.
Wilkinson ST, Sanacora G. A new generation of antidepressants: an update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems. Drug Disco Today. 2018;24:606–15.
Guilarte TR, Opler M, Pletnikov M. Is lead exposure in early life an environmental risk factor for Schizophrenia? Neurobiological connections and testable hypotheses. Neurotoxicology. 2012;33:560–74.
Nakao K, Jeevakumar V, Jiang SZ, Fujita Y, Diaz NB, Pretell Annan CA, et al. Schizophrenia-like dopamine release abnormalities in a mouse model of NMDA receptor hypofunction. Schizophr Bull. 2019;45:138–47.
Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2012;62:1574–83.
Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull. 2012;38:950–7.
Coyle JT. Schizophrenia: basic and clinical. Adv Neurobiol. 2017;15:255–80.
Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–24.
Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci. 2010;11:100–13.
Woo TU, Walsh JP, Benes FM. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2004;61:649–57.
Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharm Sci. 2011;32:507–13.
Del Arco A, Segovia G, Mora F. Blockade of NMDA receptors in the prefrontal cortex increases dopamine and acetylcholine release in the nucleus accumbens and motor activity. Psychopharmacol (Berl). 2008;201:325–38.
Stansfield KH, Ruby KN, Soares BD, McGlothan JL, Liu X, Guilarte TR. Early-life lead exposure recapitulates the selective loss of parvalbumin-positive GABAergic interneurons and subcortical dopamine system hyperactivity present in schizophrenia. Transl Psychiatry. 2015;5:e522.
Opler MG, Brown AS, Graziano J, Desai M, Zheng W, Schaefer C, et al. Prenatal lead exposure, delta-aminolevulinic acid, and schizophrenia. Environ Health Perspect. 2004;112:548–52.
Opler MG, Susser ES. Fetal environment and schizophrenia. Environ Health Perspect. 2005;113:1239–42.
Modabbernia A, Velthorst E, Gennings C, De Haan L, Austin C, Sutterland A, et al. Early-life metal exposure and schizophrenia: A proof-of-concept study using novel tooth-matrix biomarkers. Eur Psychiatry. 2016;36:1–6.
Arinola G, Idonije B, Akinlade K, Ihenyen O. Essential trace metals and heavy metals in newly diagnosed schizophrenic patients and those on anti-psychotic medication. J Res Med Sci. 2010;15:245–9.
Reuben A, Schaefer JD, Moffitt TE, Broadbent J, Harrington H, Houts RM, et al. Association of childhood lead exposure with adult personality traits and lifelong mental health. JAMA Psychiatry. 2019;76:418–25.
Nevin R. How lead exposure relates to temporal changes in IQ, violent crime, and unwed pregnancy. Environ Res. 2000;83:1–22.
Dietrich KN, Ris MD, Succop PA, Berger OG, Bornschein RL. Early exposure to lead and juvenile delinquency. Neurotoxicol Teratol. 2001;23:511–8.
Fishbein DH, Todd AC, Ricketts EP, Semba RD. Relationship between lead exposure, cognitive function, and drug addiction: pilot study and research agenda. Environ Res. 2008;108:315–9.
Anusa M, Rooban T. Burden of exposure to lead as a risk factor for mental illness in Indian children 1990-2015: A systematic analysis based on global burden of disease approach. Ann Indian Psychiatry. 2017;1:88–96.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57.
Berk M, Williams LJ, Jacka FN, O’Neil A, Pasco JA, Moylan S, et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013;11:200.
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56.
Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry. 2016;21:1696–709.
Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology. 2012;37:137–62.
Boulanger-Bertolus J, Pancaro C, Mashour GA. Increasing role of maternal immune activation in neurodevelopmental disorders. Front Behav Neurosci. 2018;12:230.
Leonard BE. The concept of depression as a dysfunction of the immune system. Curr Immunol Rev. 2010;6:205–12.
Corsini E, Sokooti M, Galli CL, Moretto A, Colosio C. Pesticide induced immunotoxicity in humans: a comprehensive review of the existing evidence. Toxicology. 2013;307:123–35.
Ferrario D, Gribaldo L, Hartung T. Arsenic exposure and immunotoxicity: a review including the possible influence of age and sex. Curr Environ Health Rep. 2016;3:1–12.
Kreitinger JM, Beamer CA, Shepherd DM. Environmental immunology: lessons learned from exposure to a select panel of immunotoxicants. J Immunol. 2016;196:3217–25.
Plusquin M, Guida F, Polidoro S, Vermeulen R, Raaschou-Nielsen O, Campanella G, et al. DNA methylation and exposure to ambient air pollution in two prospective cohorts. Environ Int. 2017;108:127–36.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5.
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–80.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–78.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–8.
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705.
Bialas AR, Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16:1773–82.
Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–57.
Wakselman S, Bechade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28:8138–43.
Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112:733–43.
Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. 2007;32:1106–15.
Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR III, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–609.
Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, et al. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8:1271–9.
Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16:543–51.
Frade JM, Barde YA. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron. 1998;20:35–41.
Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Front Neuroendocrinol. 2012;33:267–86.
Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry. 2017;7:e1120.
Smolders S, Notter T, Smolders SMT, Rigo JM, Brone B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav Immun. 2018;73:51–65.
Ramirez K, Shea DT, McKim DB, Reader BF, Sheridan JF. Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain Behav Immun. 2015;46:212–20.
Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology. 2017;42:318–33.
Wohleb ES, McKim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci. 2014;8:447.
Costa LG, Cole TB, Coburn J, Chang YC, Dao K, Roque PJ. Neurotoxicity of traffic-related air pollution. Neurotoxicology. 2017;59:133–9.
Mumaw CL, Levesque S, McGraw C, Robertson S, Lucas S, Stafflinger JE, et al. Microglial priming through the lung-brain axis: the role of air pollution-induced circulating factors. FASEB J. 2016;30:1880–91.
Jayaraj RL, Rodriguez EA, Wang Y, Block ML. Outdoor ambient air pollution and neurodegenerative diseases: the neuroinflammation hypothesis. Curr Environ Health Rep. 2017;4:166–79.
Allen JL, Oberdorster G, Morris-Schaffer K, Wong C, Klocke C, Sobolewski M, et al. Developmental neurotoxicity of inhaled ambient ultrafine particle air pollution: parallels with neuropathological and behavioral features of autism and other neurodevelopmental disorders. Neurotoxicology. 2017;59:140–54.
Bolton JL, Marinero S, Hassanzadeh T, Natesan D, Le D, Belliveau C, et al. Gestational exposure to air pollution alters cortical volume, microglial morphology, and microglia-neuron interactions in a sex-specific manner. Front Synaptic Neurosci. 2017;9:10.
Klocke C, Allen JL, Sobolewski M, Mayer-Proschel M, Blum JL, Lauterstein D, et al. Neuropathological consequences of gestational exposure to concentrated ambient fine and ultrafine particles in the mouse. Toxicol Sci. 2017;156:492–508.
O’Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res. 2015;277:32–48.
Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015;161:264–76.
Correa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MA. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunol. 2016;5:e73.
Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry. 2015;2:258–70.
Speer K, Upton D, Semple S, McKune A. Systemic low-grade inflammation in post-traumatic stress disorder: a systematic review. J Inflamm Res. 2018;11:111–21.
Becking K, Boschloo L, Vogelzangs N, Haarman BCM, Riemersma-van der Lek R, Penninx BWJH, et al. The association between immune activation and manic symptoms in patients with a depressive disorder. Transl Psychiatry. 2013;3:e314.
Dempsey JL, Little M, Cui JY. Gut microbiome: An intermediary to neurotoxicity. Neurotoxicology. 2019;75:41–69.
Claus SP, Guillou H, Ellero-Simatos S. The gut microbiota: a major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes. 2016;2:16003.
Wang M, Zhou J, He F, Cai C, Wang H, Wang Y, et al. Alteration of gut microbiota-associated epitopes in children with autism spectrum disorders. Brain Behav Immun. 2019;75:192–9.
Nguyen TT, Kosciolek T, Maldonado Y, Daly RE, Martin AS, McDonald D, et al. Differences in gut microbiome composition between persons with chronic schizophrenia and healthy comparison subjects. Schizophr Res. 2018;204:23–9.
Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol Psychiatry. 2016;21:786–96.
Olson CA, Vuong HE, Yano JM, Liang QY, Nusbaum DJ, Hsiao EY. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cell. 2018;174:497.
Kim YM, Snijders AM, Brislawn CJ, Stratton KG, Zink EM, Fansler SJ, et al. Light-stress influences the composition of the murine gut microbiome, memory function, and plasma metabolome. Front Mol Biosci. 2019;6:108.
Marshall BL, Liu Y, Farrington MJ, Mao J, Helferich WG, Schenk AK, et al. Early genistein exposure of California mice and effects on the gut microbiota-brain axis. J Endocrinol. 2019;242:139–57.
Alderete TL, Jones RB, Chen Z, Kim JS, Habre R, Lurmann F, et al. Exposure to traffic-related air pollution and the composition of the gut microbiota in overweight and obese adolescents. Environ Res. 2018;161:472–8.
Tang M, Xu C, Chen K, Yan Q, Mao W, Liu W, et al. Hexachlorocyclohexane exposure alters the microbiome of colostrum in Chinese breastfeeding mothers. Environ Pollut. 2019;254:112900.
Rothenberg SE, Keiser S, Ajami NJ, Wong MC, Gesell J, Petrosino JF, et al. The role of gut microbiota in fetal methylmercury exposure: insights from a pilot study. Toxicol Lett. 2016;242:60–7.
Eggers S, Safdar N, Sethi AK, Suen G, Peppard PE, Kates AE, et al. Urinary lead concentration and composition of the adult gut microbiota in a cross-sectional population-based sample. Environ Int. 2019;133:105122.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–63.
Sharon G, Cruz NJ, Kang DW, Gandal MJ, Wang B, Kim YM, et al. Human gut microbiota from Autism spectrum disorder promote behavioral symptoms in mice. Cell. 2019;177:1600–18 e1617.
Tabouy L, Getselter D, Ziv O, Karpuj M, Tabouy T, Lukic I, et al. Dysbiosis of microbiome and probiotic treatment in a genetic model of autism spectrum disorders. Brain Behav Immun. 2018;73:310–9.
Kang DW, Adams JB, Gregory AC, Borody T, Chittick L, Fasano A, et al. Microbiota transfer therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome. 2017;5:10.
Schmidt RJ, Kogan V, Shelton JF, Delwiche L, Hansen RL, Ozonoff S, et al. Combined prenatal pesticide exposure and folic acid intake in relation to Autism spectrum disorder. Environ Health Perspect. 2017;125:097007.
Shin HM, Schmidt RJ, Tancredi D, Barkoski J, Ozonoff S, Bennett DH, et al. Prenatal exposure to phthalates and autism spectrum disorder in the MARBLES study. Environ Health. 2018;17:85.
Barkoski JM, Busgang SA, Bixby M, Bennett D, Schmidt RJ, Barr DB, et al. Prenatal phenol and paraben exposures in relation to child neurodevelopment including autism spectrum disorders in the MARBLES study. Environ Res. 2019;179:108719.
Chun H, Leung C, Wen SW, McDonald J, Shin HH. Maternal exposure to air pollution and risk of autism in children: A systematic review and meta-analysis. Environ Pollut. 2020;256:113307.
Wild CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomark Prev. 2005;14:1847–50.
Miller GW, Jones DP. The nature of nurture: refining the definition of the exposome. Toxicol Sci. 2014;137:1–2.
Wild CP. The exposome: from concept to utility. Int J Epidemiol. 2012;41:24–32.
Turner MC, Nieuwenhuijsen M, Anderson K, Balshaw D, Cui Y, Dunton G, et al. Assessing the exposome with external measures: commentary on the state of the science and research recommendations. Annu Rev Public Health. 2017;38:215–39.
Guloksuz S, van Os J, Rutten BPF. The exposome paradigm and the complexities of environmental research in psychiatry. JAMA Psychiatry. 2018;75:985–6.
Guloksuz S, Rutten BPF, Pries LK, Ten Have M, de Graaf R, van Dorsselaer S, et al. The complexities of evaluating the exposome in psychiatry: a data-driven illustration of challenges and some propositions for amendments. Schizophr Bull. 2018;44:1175–9.
Pries LK, Lage-Castellanos A, Delespaul P, Kenis G, Luykx JJ, Lin BD, et al. Estimating exposome score for schizophrenia using predictive modeling approach in two independent samples: the results from the EUGEI study. Schizophr Bull. 2019;45:960–5.
American Psychiatric A, American Psychiatric A, Force DSMT. Diagnostic and statistical manual of mental disorders: DSM-5. Arlington, VA: American Psychiatric Association; 2017.
Casey BJ, Craddock N, Cuthbert BN, Hyman SE, Lee FS, Ressler KJ. DSM-5 and RDoC: progress in psychiatry research? Nat Rev Neurosci. 2013;14:810–4.
Insel TR, Cuthbert BN. Medicine. Brain disorders? Precisely. Science. 2015;348:499–500.
Cuthbert BN. Research domain criteria: toward future psychiatric nosologies. Dialogues Clin Neurosci. 2015;17:89–97.
Kozak MJ, Cuthbert BN. The NIMH research domain criteria initiative: background, issues, and pragmatics. Psychophysiology. 2016;53:286–97.
Guloksuz S, van Os J. The slow death of the concept of schizophrenia and the painful birth of the psychosis spectrum. Psychol Med. 2018;48:229–44.
Constantino JN. The quantitative nature of autistic social impairment. Pediatr Res. 2011;69:55R–62R.
Plomin R, Haworth CM, Davis OS. Common disorders are quantitative traits. Nat Rev Genet. 2009;10:872–8.
Coghill D, Sonuga-Barke EJ. Annual research review: categories versus dimensions in the classification and conceptualisation of child and adolescent mental disorders–implications of recent empirical study. J Child Psychol Psychiatry. 2012;53:469–89.
Bell MR, Thompson LM, Rodriguez K, Gore AC. Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 1. Sexually dimorphic effects on social and anxiety-like behaviors. Horm Behav. 2016;78:168–77.
Bell MR, Hart BG, Gore AC. Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 2. Sex-specific neuromolecular effects in the brain. Mol Cell Endocrinol. 2016;420:125–37.
Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. 2013;339:1095–9.
Monte AS, Mello BSF, Borella VCM, da Silva Araujo T, da Silva FER, Sousa FCF, et al. Two-hit model of schizophrenia induced by neonatal immune activation and peripubertal stress in rats: Study of sex differences and brain oxidative alterations. Behav Brain Res. 2017;331:30–7.
Gea LP, Colombo R, Rosa EDD, Antqueviezc B, Aguiar EZ, Hizo GH, et al. Anhedonic-like behavior correlates with IFNgamma serum levels in a two-hit model of depression. Behav Brain Res. 2019;373:112076.
Harrill AH, McAllister KA. New rodent population models may inform human health risk assessment and identification of genetic susceptibility to environmental exposures. Environ Health Perspect. 2017;125:086002.
Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, et al. The collaborative cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36:1133–7.
Threadgill DW, Miller DR, Churchill GA, de Villena FP. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 2011;52:24–31.
Schoenrock SA, Oreper D, Farrington J, McMullan RC, Ervin R, Miller DR, et al. Perinatal nutrition interacts with genetic background to alter behavior in a parent-of-origin-dependent manner in adult Collaborative Cross mice. Genes Brain Behav. 2018;17:e12438.
Molenhuis RT, Bruining H, Brandt MJV, van Soldt PE, Abu-Toamih Atamni HJ, Burbach JPH, et al. Modeling the quantitative nature of neurodevelopmental disorders using Collaborative Cross mice. Mol Autism. 2018;9:63.
Davis AP, Grondin CJ, Johnson RJ, Sciaky D, McMorran R, Wiegers J, et al. The Comparative Toxicogenomics Database: update 2019. Nucleic Acids Res. 2019;47:D948–54.
Davis AP, Grondin CJ, Johnson RJ, Sciaky D, King BL, McMorran R, et al. The Comparative Toxicogenomics Database: update 2017. Nucleic Acids Res. 2017;45:D972–8.
Davis AP, Wiegers TC, Wiegers J, Johnson RJ, Sciaky D, Grondin CJ, et al. Chemical-induced phenotypes at CTD help inform the predisease state and construct adverse outcome pathways. Toxicol Sci. 2018;165:145–56.
Grondin CJ, Davis AP, Wiegers TC, Wiegers JA, Mattingly CJ. Accessing an expanded exposure science module at the comparative toxicogenomics database. Environ Health Perspect. 2018;126:014501.
Grondin CJ, Davis AP, Wiegers TC, King BL, Wiegers JA, Reif DM, et al. Advancing exposure science through chemical data curation and integration in the comparative toxicogenomics database. Environ Health Perspect. 2016;124:1592–9.
Santos JX, Rasga C, Marques AR, Martiniano HFMC, Asif M, Vilela J, et al. A role for gene-environment interactions in Autism Spectrum Disorder is suggested by variants in genes regulating exposure to environmental factors. bioRxiv. 2019; 520544.
Carter CJ, Blizard RA. Autism genes are selectively targeted by environmental pollutants including pesticides, heavy metals, bisphenol A, phthalates and many others in food, cosmetics or household products. Neurochem Int. 2016;101:83–109.
Preciados M, Yoo C, Roy D. Estrogenic endocrine disrupting chemicals influencing NRF1 regulated gene networks in the development of complex human brain diseases. Int J Mol Sci. 2016;17:1–62.
Ma M, Liang X, Cheng S, Zhang L, Cheng B, Yang J, et al. Integrating genome-wide association study, chromosomal enhancer maps and element-gene interaction networks detected brain regions related associations between elements and ADHD/IQ. Behav Brain Res. 2018;353:137–42.
Acknowledgements
We would like to thank Dr. Bruce Cuthbert (NIMH) for his thoughtful feedback during the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hollander, J.A., Cory-Slechta, D.A., Jacka, F.N. et al. Beyond the looking glass: recent advances in understanding the impact of environmental exposures on neuropsychiatric disease. Neuropsychopharmacol. 45, 1086–1096 (2020). https://doi.org/10.1038/s41386-020-0648-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-0648-5
This article is cited by
-
Mental health of UK firefighters
Scientific Reports (2023)
-
Excessive Lysosomal Stress Response and Consequently Impaired Autophagy Contribute to Fluoride-Induced Developmental Neurotoxicity
Biological Trace Element Research (2023)
-
Pharmacological modulation of HIF-1 in the treatment of neuropsychiatric disorders
Journal of Neural Transmission (2023)
-
Controlling litter effects to enhance rigor and reproducibility with rodent models of neurodevelopmental disorders
Journal of Neurodevelopmental Disorders (2021)
-
Effects of air pollution exposure on social behavior: a synthesis and call for research
Environmental Health (2021)
