
Abstract
Enkephalins, endogenous ligands for delta opioid receptors (DORs), are highly enriched in the nucleus accumbens (NAc). They are implicated in depression but their role in the NAc, a critical brain region for motivated behavior, is not fully investigated. To provide insight into enkephalin function we used a chronic social defeat stress paradigm, where animals are either categorized as susceptible or resilient to stress based on their performance in a social interaction test. Compared to controls, susceptible animals showed reduced enkephalin levels in the NAc. Such decrease in enkephalin levels is not due to a change in mRNA of its precursor protein, proenkephalin, in susceptible mice but is consistent with increased mRNA levels of enkephalinases in the NAc of susceptible animals. Systemic administration of enkephalinase inhibitors or NAc infusion of the DOR agonist, SNC80, caused a resilient outcome to CSDS. Both treatments increased phosphorylation of ERK, which was downregulated by social defeat stress. To further validate these results, we also used Q175 knock-in mice, an animal model of Huntington’s disease in which enkephalin levels are reduced in striatum and comorbidity with mood disorders is common. Consistent with data in wild-type mice, Q175 animals showed reduced enkephalin levels in the NAc and enhanced susceptibility to a social defeat stress. Overall, our data implicate that depressionlike behavior induced by social defeat stress arises from disrupted DOR signaling resulting from lowered levels of enkephalins, which is partly mediated through elevated expression of enkephalinases.
Similar content being viewed by others


Introduction
Enkephalins are primary endogenous ligands for delta opioid receptors (DORs), and also bind to mu opioid receptors (MORs) but with considerably less affinity [1]. In addition to analgesic properties expected as ligands for opioid receptors [2], enkephalins are highly implicated in motivated behaviors and stress responses. Previous studies have found anxiety- and depression-like behaviors in animals that were treated with a DOR antagonist [3], or in animals devoid of precursor of enkephalins (proenkephalin) or DOR [4, 5]. Consistent with these studies, various DOR agonists and enkephalinase inhibitors have been tested in animal models and in clinical cases for treating symptoms of depression [6,7,8].
Enkephalin-containing neurons and DORs are present throughout the brain, including amygdala, striatum, and hypothalamus [9]. There are two different enkephalin pentapeptides that differ by one amino acid, methionine-(Met-) and leucine-(Leu-) enkephalins. A common precursor protein, proenkephalin, contains four copies of Met-enkephalin and one copy of Leu-enkephalin within its sequence, while another polypeptide prodynorphin contains three copies of Leu-enkephalin sequence as well [10, 11]. The role of enkephalin—DOR signaling in regulating depression-like behaviors has been studied in multiple brain areas. For example, footshock stress [12] and forced swimming decreased the level of Leu-enkephalin in the hypothalamus [13], and reduced levels of Met-enkephalin in the striatum and hypothalamus. NAc enkephalin levels were also decreased in animals that went through chronic mild stress, and increased by chronic treatment of antidepressants [14]. Additionally, prolonged predator odor and elevated platform stress combined with acute restraint stress downregulated Leu-enkephalin in the hippocampus [15].
Taken together, these previous studies suggest that enkephalin—DOR signaling play a key role in regulating depression-like behaviors and behavioral responses to stress. Despite enkephalins being highly expressed within the striatum [16, 17], their role has not been studied extensively in this region in stress, unlike the dynorphin system [18]. The NAc, ventral striatum, known for its role in motivated behaviors and stress responses [19,20,21], contains projection neurons called medium spiny neurons (MSNs) which represent more than 90% of the neurons in this region. The two major subtypes of MSNs, Dopamine receptor 1 (D1)- vs. Dopamine receptor 2 (D2)-MSNs, which display distinct roles in motivation and stress response [22,23,24,25], are each enriched with distinct precursor proteins that can be converted into endogenous opioids (D1-MSNs—prodynorphin; D2-MSNs—proenkephalin).
In this study, we utilized a chronic social defeat stress (CSDS) model, which produces robust and long-lasting reduced motivated behavior [19, 26]. Using this model, we previously demonstrated that animals that are susceptible to CSDS display distinct cellular and/or molecular properties in NAc MSN subtypes [27,28,29]. Previous work investigated the dynorphin system in CSDS, including demonstration of reduced prodynorphin, the precursor for both dynorphin and Leu-enkephalin, in NAc of CSDS susceptible and resilient mice [30]. However, enkephalins, which are mainly enriched in D2-MSN subtypes, have not been well studied in NAc in social stress. Thus, we investigated the enkephalin—DOR signaling pathway in NAc to determine if pharmacological interventions targeting enkephalins or DOR can alter behavioral responses to CSDS.
In addition, we examined enkephalins in the NAc of the Q175 mouse model of Huntington’s disease (HD), which have a spontaneous expansion of the CAG copy number in the huntingtin gene [31]. HD is an inherited neurodegenerative disorder that is characterized by motor dysfunction and cognitive impairment [32]. Importantly, HD associates pro-dromally with various psychiatric deficits such as depression, affective disorders, and mood disturbances that are usually observed prior to clinical symptoms [33]. Although these non-motor symptoms are suggested to be early indicators of HD and impact the life quality of HD patients greatly, the mechanism underlying such symptoms are still relatively unknown and not studied compared to motor symptoms [34,35,36]. Since it is well-known that enkephalin signaling is dysregulated in the striatum of HD patients and mouse models, we further probed Q175 mice for NAc enkephalin levels and examined their response to social stress.
Methods
Animals
All the procedures regarding animal use in this study were approved by the University of Maryland School of Medicine (UMSOM) Institutional Animal Care and Use Committee. Mice were given food and water ad libitum and housed in UMSOM animal facilities on a 12:12 h light:dark cycle. Male C57BL6/J mice (7–8-weeks-old, Charles River, Kingston, NY) were used for CSDS experiments, enkephalinase inhibitor injections, and SNC80 infusions. BAC transgenic D1-tdTomato [37] and D2-GFP [38] mice on a C57BL6 background were crossed together to create D1-tdTomato/D2-GFP reporter line, and these male mice (8–10 weeks old) were also used for CSDS experiments. Male CD-1 retired breeders (Charles River, >2 months) were used as aggressors for CSDS. zQ175 KI [31] heterozygous mice (Q175) and their C57BL6 age-matched littermates (4–8 months, Jackson Laboratory, Bar Harbor, ME) were used for HD experiments. All subject mice were randomly assigned to groups.
Social defeat stress
Social defeat stress was performed as previously by our lab [27,28,29] using a well-established protocol [19, 26]. Briefly, subject mice were placed in hamster cages with perforated clear dividers containing an aggressive resident mouse (CD1). For CSDS, mice were physically attacked by a resident for 10 min, then housed opposite the resident on the other side of the divider for 24 h sensory interaction. Defeat session was repeated with a novel resident mouse each day for 10 consecutive days. Twenty-four hours after the last session, mice were tested for social interaction (SI). Subject mice were placed in an open field containing a clear perforated box first without a social target for 2.5 min and immediately following this, for another 2.5 min with a novel CD1 mouse serving as social target. Time spent around the box (“interaction zone”) with or without an interactor was quantified utilizing a video tracking software (TopScan Lite 2.0, CleverSys, Reston, VA). The time spent with the social target in the interaction zone is reported as “interaction time”; the time spent with the empty box was not affected in any of our experiments. Animals were categorized as susceptible if the interaction time was two standard deviations below the mean of non-defeated controls, and as resilient if they do not fall in that category. CSDS animals were analyzed in separate groups of susceptible vs. resilient vs. non-defeated control animals when noted; otherwise, CSDS animals were grouped together for comparison with non-defeated controls.
Radioimmunoassay (RIA)
NAc tissue punches were collected 24 h after SI or 30 min after enkephlinase inhibitor injections, after which they were homogenized in 150 µl of 1N acetic acid. The homogenates were centrifuged, and the supernatant collected was lyophilized and resuspended in 500 µl of RIA buffer (0.1 M sodium phosphate, 50 mM sodium chloride, pH 7.4, 0.1% heat-treated BSA, and 0.1% sodium azide). For Met- and Leu-enkephalin assays, 50 µl of resuspended NAc homogenates were subjected to assay in triplicates using the protocols described previously [39]. Briefly, samples were incubated with 10,000 cpm of iodinated peptide and the appropriate dilution of rabbit antiserum in a final volume of 300 µl at 4 °C overnight. To separate the antibody-bound labeled peptide from the unbound labeled peptide, 1 ml of 25% polyethylene glycol and 100 µl of 7.5% carrier bovine γ-globulin were added. The samples were vortexed, kept on ice for 20 min, and then centrifuged for 20 min at 3,000 × g at 4 °C. The supernatant was aspirated, and the radioactivity in the pellets was determined using a gamma counter (Wallac 1470, PerkinElmer, Shelton, CT). Amounts of peptides were calculated using Prism 6 software (GraphPad, San Diego, CA) and then normalized to the total input protein amount measured prior to lyophilization. Polyclonal “Lucy” anti-Leu- [40] and “Betty” Met-Enkephalin antisera were generous gifts from Dr. Iris Lindberg at our department in UMSOM. 125I-labeled Met- and Leu-enkephalin were prepared by the chloramine-T method using Met- (M6638, Sigma, St. Louis, MO) and Leu-enkephalin acetate salt hydrates (L9133, Sigma).
RNA extraction and quantitative RT-PCR
NAc tissue punches from CSDS animals were collected 24 h after the SI test and stored at −80 °C. RNA extraction and RT-PCR were performed as previously [41]. Briefly, RNA was extracted using Trizol (Invitrogen, Carlsbad, CA) and the MicroElute Total RNA Kit (Omega Bio-tek, Norcross, GA) with a DNase step (Qiagen, Germantown, MD). All RNA quantity was measured on a Nanodrop (ThermoFisher Scientific, Waltham, MA). Complementary DNA was then synthesized using a reverse transcriptase iScript complementary DNA synthesis kit (Bio-Rad, Hercules, CA). mRNA expression changes were measured using quantitative PCR with PerfeCTa SYBR Green FastMix (Quantabio, Beverly, MA). Quantification of mRNA changes was performed using the 2–ΔΔCt method, using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a housekeeping gene. Primers used in this study included GAPDH forward (AGGTCGGTGTGAACGGATTTG) and reverse (TGTAGACCATGTAGTTGAGGTCA), proenkephalin (PENK) forward (CCCTTTTCCAGCAGTGACC) and reverse (GGGCAACTGTACAGCACAAA), membrane metallo-endopeptidase (MME) forward (AGGCTCGTGCAGTTTTGAAC) and reverse (GGGTGAAATAGGGGAACCGT), aminopeptidase puromycin sensitive (NPEPPS) forward (GGATGAGGTGGAGCTACCAAA) and reverse (CAACCCCCCACTGGTGTAAGA), angiotensin I converting enzyme (ACE) forward (AGCACGACCCTTTACTGGTT) and reverse (CTCACCTGGGCTGTTAGGAA), and alanyl aminopeptidase, membrane (ANPEP) forward (CCACTTGTCCCAGTATGACAC) and reverse (AGAGTAGGATGCTGGTGTCTG).
Stereotaxic surgery for intracranial cannula
Mice were anesthetized with isoflurane (3% induction, 1.5% maintenance) and affixed in a stereotaxic frame (Kopf Instruments, Tujunga, CA). Bilateral cannula (Plastics One, Roanoke, VA) were implanted over the NAc (from Bregma, AP: +1.6 mm, ML: ±1.5 mm: DV: −4.0 mm). Cannula were secured by a dental cement skull-cap.
Drugs
For enkephalinase inhibitor treatment, bestatin hydrochloride (B8385, Sigma) and thiorphan (BML-PI113, Enzo Life Sciences, Farmingdale, NY) was used. Bestatin, a protease inhibitor that inhibits multiple enzymes including ANPEP [42, 43], was dissolved in sterile water (12.5 mg/mL). Thiorphan, an active metabolite of acetorphan, an enkephalinase inhibitor which has strong affinity for MME [42, 43], was dissolved in 100% ethanol (50 mg/mL). The drugs were mixed and diluted in 0.9% saline immediately before each injection. The mixture at a final concentration of 5 mg/kg of bestatin and 2.5 mg/kg of thiorphan or vehicle (1.25% ethanol in saline) was delivered intraperitoneally 15 min before each defeat session for 10 days during CSDS. For the DOR agonist experiment, SNC80 (ALX-550-172, Enzo) was dissolved in sterile water (2.5 mg/mL) and diluted in 0.9% saline. The drug at a final concentration of 8 mg/kg or 0.9% saline was delivered intracranially over 10 min using a programmable syringe pump (Harvard Apparatus, Holliston, MA), immediately prior to the SI test.
Immunohistochemistry
D1-tdTomato/D2-GFP mice were deeply anesthetized with isoflurane and transcardially perfused with 0.1 M PBS followed by 4% paraformaldehyde (PFA). Animals were sacrificed 15 min following the SI test according to a previous literature [44], as pERK levels can change rapidly and are very sensitive to the time spent after the stimulus. Following perfusions, brains were extracted, post-fixed in 4% PFA overnight, and immersed in 30% sucrose overnight. Brains were sectioned coronally on a cryostat at 40 μm. Following rinses in PBS, the sections were incubated for 1 h in blocking buffer containing 3% normal donkey serum (NDS) and 0.3% Triton X-100 in PBS for 30 min. Sections were immunostained with a rabbit anti-phospho-p44/42 MAPK antibody (4370, Cell Signaling Technology, Danvers, MA) at 1:200 in blocking solution. Following overnight incubation at room temperature, the tissue was rinsed with PBS and followed by Cy5-conjugated goat anti-rabbit IgG (1:1,000; Jackson Immuno, West Grove, PA). Tissue sections were mounted onto slides, coverslipped, and imaged on a laser-scanning confocal microscope (CSU-W1, Nikon Instruments, Melville, NY).
Western blots
NAc tissue punches from CSDS animals were collected 15 min after SI test, due to the reason stated above, and stored at −80 °C. Tissue was homogenized in 30 µL of lysis buffer containing 320 mM sucrose, 5 nM HEPES buffer, 1% SDS, phosphatase inhibitor cocktails I and II (Sigma) and protease inhibitors (Roche, Mannheim, Germany) using an ultrasonic processor (Cole Parmer, Vernon Hills, IL). Protein concentrations were determined using DC protein assay (Bio-Rad) and then 15–20 mg of total protein were loaded onto Tris–HCl polyacrylamide gel (Bio-Rad). The samples were transferred to a nitrocellulose membrane and blocked for 1 h in blocking buffer, 5% nonfat dry milk in Tris-buffered saline (pH 7.6) with 0.1% Tween 20. Blocked membranes were incubated overnight at 4 °C in blocking buffer with primary antibodies at 1:1,000 concentration using either anti-phospho-p44/42 MAPK (Thr202/Tyr204) (#4370; Cell Signaling), anti-phospho-Akt (Ser473) (#4060, Cell Signaling), anti-p44/42 MAPK (#9102; Cell Signaling), anti-Akt (#4691, Cell Signaling) or 1:30,000 anti-GAPDH (2118; Cell Signaling). Membranes were then incubated with goat anti-rabbit peroxidase-labeled secondary antibodies (PI-1000, 1:20,000; Vector Laboratories, Burlingame, CA) in blocking buffer. The bands were visualized using an ECL substrate (34075; ThermoFisher) on a ChemiDoc Imaging System (Bio-Rad). Bands were quantified with ImageJ 1.52 and normalized to GAPDH levels to control for sample loading. Phosphorylation level was calculated using phosphorylated ERK or AKT/total ERK or AKT protein amount.
Statistical analysis
Data are presented as mean ± S.E.M, with the exception in Fig. 4, where S.D. is used. To estimate sample size, we utilized R version 3.3 with pwr power analysis package. Statistical data were analyzed using GraphPad Prism 6. Unpaired Student’s t-test and one-way or two-way analysis of variance (ANOVA) test followed by Sidak’s multiple comparison post hoc test were used to determine effects across all experiments.
Results
We first examined NAc enkephalin levels after CSDS. Using RIA we measured levels of enkephalins directly from NAc tissue punches of mice that underwent CSDS and the SI test. CSDS-susceptible mice display reduced SI compared to control or resilient mice (one-way ANOVA F(2,30) = 140.5, p < 0.0001; post-hoc, ***p < 0.001 for susceptible vs. control and susceptible vs. resilient ; Fig. 1a). CSDS-susceptible mice have reduced NAc Leu- (one-way ANOVA F(2,30) = 3.66, p = 0.03; post hoc, *p < 0.05 for susceptible vs. control) and Met-enkephalin (one-way ANOVA F(2,30) = 5.89, p = 0.006; post hoc, **p < 0.01 for susceptible vs. control; Fig. 1b). Resilient mice have similar enkephalin levels compared to control animals and showed trends toward higher levels compared to susceptible animals (post hoc, p = 0.10 for susceptible vs. control for both Leu- and Met-enkephalins; Fig. 1b). Further, enkephalin levels and social interaction scores were significantly correlated with each other (R2 = 0.22, p < 0.05 for Leu-enkephalin and R2 = 0.21, p < 0.05 for Met-enkephalin; Fig. 1c).

Enkephalin levels are decreased in the NAc of mice susceptible to CSDS. Transcription of enkephalin precursor proenkephalin is not altered, while enzymes that degrade enkephalins are reduced in CSDS-susceptible mice. a Social interaction scores from the animals used in enkephalin RIA experiment (***p < 0.001, post-hoc; one-way ANOVA: p < 0.001). b CSDS-susceptible mice display decreased Leu- (*p < 0.05, post-hoc; one-way ANOVA: p < 0.05) and Met-enkephalin levels (**p < 0.01, post-hoc; one-way ANOVA: p < 0.01) in the NAc compared to the non-defeated controls. c Leu- (p < 0.05, R2 = 0.21) and Met-enkephalin levels (p < 0.05, R2 = 0.21) within the NAc correlates with the social interaction scores. d Social interaction scores from the animals used in NAc qRT-PCR experiment (*p < 0.05, post-hoc; one-way ANOVA: p < 0.01). e NAc mRNA levels of proenkephalin (PENK) are not reduced in susceptible animals compared to the controls. f Increased mRNA levels of NPEPPS, ACE, ANPEP, and a decrease in MME were observed in susceptible animals (**p < 0.01, *p < 0.05, post hoc; one-way ANOVA: p < 0.05)
We next sought to investigate the mechanisms that could lead to decreased enkephalin levels in the NAc of CSDS-susceptible animals. Using the NAc of CSDS mice (one-way ANOVA F(2,15) = 6.81, p < 0.01; post hoc, *p < 0.05 for susceptible vs. control and susceptible vs. resilient; Fig. 1d), we analyzed the mRNA levels of genes that could underlie altered enkephalin levels. We observed no difference in mRNA for proenkephalin precursor protein (one-way ANOVA F(2,15) = 0.85, p = 0.45; Fig. 1e). We then analyzed the levels of enkephalinases, peptidases that can degrade enkephalins. Among the enkephalinases that we tested, levels of NPEPPS, ACE, and ANPEP mRNAs were increased in CSDS-susceptible animals (one-way ANOVA, NPEPPS F(2,14) = 5.27, p = 0.01; post hoc, *p < 0.05 for susceptible vs. control; ACE F(2,12) = 7.41, p < 0.01; post-hoc, **p < 0.01 for susceptible vs. control; ANPEP F(2,11) = 8.45, p < 0.01; post hoc, **p < 0.01 for susceptible vs. control; Fig. 1e) while MME was decreased (one-way ANOVA F(2,14) = 7.74, p < 0.01; post hoc, *p < 0.05 for susceptible vs. control; Fig. 1f).
Enhanced enkephalinases may modulate enkephalin levels in the NAc of CSDS-susceptible mice, thus we next tested if inhibiting enkephalinases could alter behavioral outcomes to CSDS. We first tested enkephalinase inhibitors to see if they can enhance the levels of enkephalins within the NAc. When injected intraperitoneally, the combination of two enkephalinase inhibitors bestatin (5 mg/kg) and thiorphan (2.5 mg/kg) was able to increase the level of both Met- and Leu-enkephalins (t-test, t(3) = 2.48, *p < 0.05 for Met-Enkephalin and t(3) = 3.97, *p < 0.05 for Leu-Enkephalin; Fig. 2a). We then injected the enkephalinase inhibitor cocktail or vehicle, prior to each defeat episode, to increase enkephalin levels during the defeat sessions (Fig. 2b). Following CSDS, mice treated with enkephalinase inhibitors spent significantly more time with a novel social target compared to the vehicle-treated controls (two-way ANOVA F(1,17) = 62.85, p < 0.0001 for defeat effect; F(1,17) = 9.85, p < 0.01 for interaction effect; post hoc, **p < 0.01 for vehicle vs. enkephalinase inhibitor in CSDS group; Fig. 2c). Enkephalinase inhibitors did not affect time in the interaction zone when the target was absent (data not shown).

Enkephalinase inhibitors can alleviate the effects of chronic social stress in mice. Enkephalinase inhibitor cocktail (5 mg/kg bestatin and 2.5 mg/kg thiorphan) was injected intraperitoneally into the subject mice. a A timeline of experiments. b An increase in the amount of enkephalins by enkephalinase cocktail in the NAc was confirmed using radioimmunoassay (*p < 0.05 for t-test). c Enkephalinase inhibitor treatment resulted in higher scores in SI test, rescuing the adverse effects of CSDS (**p < 0.01, post hoc; two-way ANOVA: p < 0.001 for effect of defeats and p < 0.01 for interaction)
While the enkephalinase inhibitors used in this experiment were able to increase the level of enkephalins, we wanted to confirm if increased enkephalins are potentially activating DOR signaling pathways in the NAc. To look at the effect of DOR activation, we focused on downstream signaling candidates of DOR, phospho(p)ERK1/2. We first used D1-tdTomato/D2-GFP mice to examine pERK1/2 in MSN subtypes after CSDS (t-test, t(10) = 2.73, *p < 0.05; Fig. 3a). The percentage of cells positive for pERK1/2 was decreased in both D1- and D2-MSNs within the NAc core and shell (NAc core, two-way ANOVA F(1,22) = 34.53, p < 0.0001 for defeat effect; F(1,22) = 7.40, p = 0.01 for cell-type effect; F(1,22) = 5.59, p = 0.02 for interaction effect ERK2; post hoc, **p < 0.01 for D1 vs. D2 in no-defeat control group, ***p < 0.001 for control vs. CSDS in D1 MSNs; *p < 0.05 for control vs. CSDS in D2-MSNs; NAc shell, two-way ANOVA F(1,22) = 26.14, p < 0.0001 for defeat effect; cell-type and interaction effect not significant; post-hoc, ***p < 0.001 for control vs. CSDS in D1 MSNs, *p < 0.05 for control vs. CSDS in D2 MSNs; Fig. 3c, d). In contrast, cells that did not express D1 or D2 in NAc displayed no difference in pERK colocalization between CSDS and control mice (Supplemental Fig. 1). After treatment of enkephalinase inhibitors, the phosphorylation level of pERK1/2 was increased (pERK1, two-way ANOVA F(1,24) = 12.97, p < 0.01 for drug treatment effect; post-hoc, **p < 0.01 for vehicle vs. inhibitor in CSDS group; pERK2, two-way ANOVA F(1,24) = 9.73, p < 0.01 for drug treatment effect; post-hoc, **p < 0.01 for vehicle vs. inhibitor in CSDS group; Fig. 3e,f), along with phospho(p)AKT (two-way ANOVA F(1,24) = 14.03, p < 0.01; post-hoc, **p < 0.01 for vehicle vs. inhibitor in no-defeat control group; Fig. 3e,f), another known downstream pathway of DOR that is independent of ERK1/2.

Molecular changes downstream of DOR activation following treatment of enkephalinase inhibitors. a Social interaction scores from the animals used in this experiment (*p < 0.05, t-test). b Anatomy of NAc subdivisions used for analysis as shown in our reporter mice (scale bar: 200 µm). c Following CSDS, the percentage of cells positive for phosphorylated ERK1/2 was markedly decreased (scale bar: 20 µm). d in both D1 and D2 neurons within NAc core and shell (***p < 0.001, **p < 0.01, and *p < 0.05, post hoc; two-way ANOVA: p < 0.001 for effect of CSDS, p < 0.05 for effect of cell type and interaction in NAc core; p < 0.001 for effect of CSDS, effect of defeats and interaction was not significant in NAc shell). Percentage of D1-MSNs positive for pERK was significantly higher than D2s in no-defeat controls within NAc core. e Phosphorylation of ERK1/2 and AKT, downstream molecular targets of DOR activation, was increased after treatment of enkephalinase inhibitors (**p < 0.01 and *p < 0.05, post-hoc; two-way ANOVA: p < 0.01 for drug treatment effect, effect of defeats and interaction was not significant). f Representative images of blots used for analysis
Since enkephalins are endogenous ligands for DORs and potential DOR signaling molecules are altered in CSDS mice, we then investigated whether directly modulating DORs within the NAc can induce behavioral changes to CSDS. In this experiment, we first categorized mice as susceptible or resilient to CSDS after the first SI test (pre-treatment). Following the cannulation surgeries in the NAc (Supplemental Fig. 2), mice were then infused with a DOR agonist (SNC80, 8 mg/kg) immediately before a second SI test (post-treatment). Following treatment with SNC80, the interaction scores were significantly increased in the susceptible mice (two-way repeated measures ANOVA, F(5,42) = 27.84, p < 0.0001 for treatment group effect; F(5,42) = 4.22, p < 0.01 for interaction effect; post hoc, *p < 0.05 for pre- vs. post-treatment in susceptible SNC80 group; Fig. 4). However, SNC80 infusion led to varying social interaction outcomes in the susceptible group during SI. When comparing animals with higher SI scores (18–24 s) vs. lower SI scores (0–16 s, which was the median) pre-SNC80 treatment, we observed a trend in improved SI scores after SNC80 although this was not statistically significant (Supplemental Fig. 3). SI scores of the resilient and control groups were unchanged after SNC80 treatment. SNC80 had no effect on time in the interaction zone with the target absent (data not shown). We next sought to examine if pERK induction may also be occurring following SNC80 treatment. We observed an increase in pERK colocalization within the NAc D1-MSNs but not D2-MSNs of CSDS mice exposed to SNC80 (Supplemental Fig. 4).

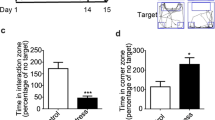
DOR activation in the NAc rescues social avoidance in mice susceptible to CSDS. a A timeline of experiments. After the animals were categorized as either resilient or susceptible to CSDS by SI (pre), they received bilateral infusion of DOR agonist SNC80 (8 mg/kg) prior to a second SI (post). b SNC80 infusion significantly increased the interaction time in the susceptible mice but it did not affect resilients or non-stressed controls (*p < 0.05, post hoc; two-way repeated measures ANOVA: p < 0.0001 for treatment effect and p < 0.01 for interaction effect)
We next sought to determine if an animal model of HD, Q175, displayed reduced enkephalin in the NAc. We chose an HD model because HD patients often report behavioral symptoms including major depression, and reduced striatal enkephalin levels is a molecular hallmark of HD. We first examined enkephalin levels within the NAc of Q175 animals. Their NAc tissue contained less Leu-enkephalin (t-test, t(16) = 3.51, p < 0.01; Fig. 5a). Reduction of Leu-enkephalin in Q175 animals is likely the result of reduced precursor protein (t-test, PENK, t(42) = 3.81, p = 0.0004; Fig. 5b) but not enkephalinases (t-test, MME, t(40) = 0.27, p = 0.78; ACE, t(36) = 0.11, p = 0.90; ANPEP, t(38) = 0.82, p = 0.41; NPEPPS, t(40) = 0.14, p = 0.88; Fig. 5c). Since Leu-Enkephalin is reduced in NAc of Q175 mice we next determined if they are vulnerable to social defeat stress. In this test, we utilized an abbreviated version of social defeat stress, a one-day subthreshold defeat stress (SSDS), which does not normally induce susceptibility in wildtype mice. The Q175 mice displayed a susceptible outcome to SSDS, as observed by significantly less SI time compared to non-stressed controls (two-way ANOVA, F(1,35) = 12.24, p < 0.01 for effect of stress; F(1,35) = 5.51, p < 0.05 for genotype effect; F(1,35) = 5.09, p < 0.05 for interaction effect; post-hoc, ***p < 0.001 for no-defeat control vs. SSDS in Q175 group; Fig. 5d). SSDS did not induce this behavioral effect in littermate controls (post hoc, p = 0.62 for no-defeat control vs. SSDS in WT group; Fig. 5d). Reduced amount of available enkephalins may be one of the factors that render Q175s more vulnerable to social stress.

Q175 mice are susceptible to social stress. An animal model of HD, zQ175 KI mice (Q175), were tested for stress susceptibility using a subthreshold social defeat stress (SSDS), an abbreviated version of CSDS that does not induce behavioral phenotype to wildtype mice. a Q175 heterozygous mice display a reduced Leu-enkephalin compared to the wildtype littermates (**p < 0.01, t-test). b In a qRT-PCR study, NAc mRNA levels of proenkephalin (PENK) was reduced in Q175 heterozygous mice (***p < 0.001, t-test). c However, mRNA levels of enkephalinases NPEPPS, ACE, ANPEP, and MME were all unaltered. d Heterozygous Q175 mice showed significantly enhanced susceptibility to SSDS (***p < 0.05, post hoc; two-way ANOVA: p < 0.01 for effect of stress and p < 0.05 for effect of genotype and interaction), while the behavior of wildtype littermates was not affected
Discussion
In the present study, we utilized the CSDS model to study the effects of stress on enkephalin dynamics in the NAc. First, we found reduced enkephalin levels in animals susceptible to CSDS. Stress-induced reduction of enkephalins using RIA has been reported in striatum and hypothalamus [13], although social stress was not used in this previous study. Our studies demonstrated reduced Leu- and Met-enkephalin in the NAc (ventral striatum) of CSDS susceptible mice. Interestingly, the levels of the precursor proenkephalin remained unchanged within the NAc. However, stress-induced changes in the levels of proenkephalin have been reported in other brain areas and also within the NAc of rats in response to repeated restraint stress [45, 46]. Further, previous work demonstrated reduced prodynorphin, which is also a precursor for Leu-Enkephalin, in the NAc of both CSDS susceptible and resilient mice [30]. While these changes are related to the dynorphin neuropeoptide and its kappa opioid receptor, it is plausible that reduced prodynorphin may partially underlie the reduction of Leu-Enekphalin in susceptible mice. mRNA analysis of enzymes that can degrade enkephalins demonstrated that many enkephalinases were increased in the NAc of CSDS-susceptible mice. These results indicate that these peptidases are a potential mechanism for reduced enkephalin levels in CSDS-susceptible mice. This was confirmed by the ability of enkephalinase inhibitors to cause resilient behavior to CSDS, which also enhanced enkephalin levels in the NAc. This is consistent with previous studies demonstrating enkephalinase inhibitors are effective in reducing depression-like behavior in rodents, including experimental drugs such as RB38 and RB101 [6, 7] and commercial drugs such as bestatin [47] and thiorphan [42]. The enkephalinase enzymes examined in this study; MME (also known as neprilysin or neutral endopeptidase), NPEPPS (also known as puromycin-sensitive aminopeptidase), ANPEP (also known as aminopeptidase N), and ACE are all proteases that are enriched in the brain [48,49,50]. All of these enzymes cleave enkephalin, each cleaving different bonds within the peptide [49]. Aside from enkephalins, these enzymes can also hydrolyze other neuropeptides, hormones, cytokines, and antigenic peptides [49]. Thus, further studies are warranted to examine the other properties of these proteases in stress susceptibility.
CSDS and treatment with enkephalin inhibitors affected DOR downstream signaling. DORs have been previously shown to activate ERK1/2 [51]. Although ERK1/2 can be activated via many other upstream signaling pathways (such as TrkB) [26, 52], our examination of pERK1/2 immediately after enkephalin inhibitor treatment suggests that ERK activity is a consequence of elevated enkephalins. We were surprised to find that the pERK1/2 levels in untreated CSDS animals were not decreased compared to the controls as we observed in the immunohistochemistry experiments examining pERK1/2 in MSN subtypes. This could reflect dilution of the signal when examining pERK1/2 from entire NAc punches, which includes non-MSN cells that displayed no difference in pERK1/2 levels when comparing control and CSDS mice; thus preventing detection of differences between control and CSDS mice. Previous work demonstrated pERK1/2 is enhanced in the NAc of CSDS susceptible mice [19, 53]. The differences in this study could reflect differences in temporal activation or depletion of pERK1/2 through differing mechanisms since pERK1/2 activation in CSDS occurs through BDNF-TrkB signaling [19, 53], as well as many other signaling pathways including other opioid receptors [54]. Thus, pERK and other signaling molecules examined in our study are highly promiscuous. Our studies examined pERK immediately after SI whereas these previous studies examined pERK as a consequence of enhanced BDNF signaling observed in susceptible mice 24 h after SI. Our studies using enkephalinases and SNC80, the latter showing enhanced pERK in D1-MSNs of CSDS mice implicate that enkephalins could be acting through DOR mainly through D1-MSNs to achieve stress resilient behavior.
Cell subtype examination of pERK1/2, 15 min after SI, demonstrated reduction in both MSN subtypes after CSDS. Enkephalin is enriched in D2-MSNs but enkephalins can act locally on DORs on all MSN subtypes and interneurons [55,56,57] thus it is not surprising that the pERK1/2 are reduced in both MSN subtypes. Our previous work demonstrated enhanced excitatory input in D2-MSNs of CSDS-susceptible mice and repeated high frequency activation of these neurons induced susceptible behavior to SSDS [27]. Whether enkephalins are released as a consequence of enhanced excitatory input and with the repeated activation is unknown. It is plausible that this heightened excitation in D2-MSNs triggers mechanisms that enhance enkephalinases leading to degradation of enkephalins thereby preventing enhanced enkephalin release from these neurons in stress-susceptible conditions. A previous study indicated that high frequency stimulation of D2-MSN inputs onto ventral pallidum (VP) neurons induces LTD at this synapse, and this is mediated through enkephalin release [58]. This enkephalin-mediated LTD effect was occluded by cocaine exposure and restoration at this synapse recovered cocaine-induced impaired motivation and anhedonia. It is plausible that reduced enkephalins produce similar plasticity effects to cocaine resulting in reduced motivational behavior, as observed by reduced SI after CSDS. This is consistent with social defeat stress causing enhanced cocaine intake [59, 60] and cocaine exposure promoting vulnerability to social defeat stress [61].
In our study, we infused DOR agonists directly into the NAc. Although we have not tested and thus cannot rule out the possible effects of downstream DOR activation in MSN projection target areas such as VP, our results show that local effects of enkephalin were capable of modulating the behavioral consequence of social defeat. One possible mechanism of such local modulation by DOR signaling is through cholinergic interneurons located throughout the striatum. Cholinergic interneurons are involved in dopamine transmission and striatal function [62] and enkephalin, as a DOR ligand, can modulate cholinergic interneurons, as D2-MSNs display heavy innervation onto them [63]. However, following DOR activation using SNC80, we see enhanced colocalization of pERK within the D1-MSNs. Whether this is a direct effect of SNC80 acting on DORs in D1-MSNs or an indirect effect through cholinergic or other mechanisms is unknown. Future studies are needed to parcel out the exact local MSN circuit effects of enkephalin—DOR signaling in stress.
Infusion of a DOR agonist into NAc significantly reverses susceptible behavior induced by CSDS. However, not all of the animals respond to this SNC80 treatment. While there was a trend for improved SI after SNC80 treatment in susceptible mice that displayed higher SI scores prior to treatment, this was not significant. It is plausible that such variability stems from the animals’ inherent difference in epigenetic, behavioral, and/or neuropharmacological properties just as some animals are resilient and some are susceptible to social defeat stress. Indeed, inbred animals respond differently to SSRIs with not all animals of one strain showing antidepressant-like outcomes [64, 65]. Thus, similar individual differences in SNC80 treatment response might be occurring. Agonists of DOR and MOR along with antagonists of kappa opioid receptor have been studied for their antidepressant-like properties in the past [66], but most of the preclinical experiments examining DOR have been conducted with rats in forced swim or learned helplessness test. Using the CSDS model, we confirm the antidepressant-like effect of DOR agonist. Our results agree with a recent study where SNC80 was administered subcutaneously in CSDS animals [46]. In the same study, changes in expression of proenkephalin mRNA were observed in multiple brain areas including BLA, suggesting that enkephalin levels are altered through the expression of proenkephalin in other brain areas. Our study is a first to specifically focus on enkephalins in the NAc, while previous studies have examined dynorphin, neuropeptide contained in D1-MSNs, in NAc in stress and motivation [18, 67].
In this present study, we further show that the Q175 mouse model of HD is susceptible to social stress. As expected from previous HD studies [68], Q175 mice have reduced proenkephalin mRNA, which likely underlie reduced enkephalin peptide in the NAc since these mice did not display alterations in enkephalinases. Our studies are consistent with previous work demonstrating reduced motivation through aberrant reward circuitry in Q175 mice [35, 36]. Our findings further support this model of pro-dromal psychiatric disorders in HD and validate a role for enkephalins as a therapeutic target. Proenkephalin mRNA is decreased in the striatum in the early stages of HD in both animal models and patients. Results from our present study demonstrate a similar outcome in NAc that may underlie reduced enkephalin peptides [69]. Previous studies suggest that the decrease in proenkephalin mRNA is due to an increased cellular response to endogenous glutamate release [69]. Striatal neurons containing proenkephalin seem to receive more glutamatergic cortical inputs and are more directly affected by cortical activity [70]. In HD, cellular pathology caused by mutant huntingtin protein in the presynaptic terminals can lead to an increased release of glutamate [70], and this overactivity of glutamate transmission may in turn result in decreased proenkephalin. Previous reports suggested that enkephalin in striatal neurons provides presynaptic inhibition of corticostriatal excitatory synaptic input [71] through DORs. Indeed, overexpressing proenkephalin in the striatum was able to delay or attenuate motor dysfunction of an animal model of HD [69], which could be attributed to the reduction of corticostriatal signaling via activation of presynaptic DORs through enkephalin released from striatum [72]. It’s plausible that similar mechanisms are occurring in NAc in Q175 mice and in stress-susceptible mice since our previous studies demonstrate enhanced excitatory input onto NAc D2-MSNs of susceptible mice [27], consistent with those observed in dorsal striatal D2-MSNs with mutant huntingtin. Future studies are needed to further explore these overlapping excitatory input and enkephalin mechanisms between stress and HD.
Finally, a limitation of our current study is the lack of NAc subregion manipulation. Recent studies demonstrate that dynorphin neurons in NAc ventral shell vs. dorsal shell show contrasting aversion vs. rewarding properties through kappa opioid receptor activation [67, 73]. Future work is needed to determine if subpopulations within the NAc shell respond differently to DOR activation as well. Overall, our data extend previous reports that enkephalin—DOR signaling is critical in regulating reduced motivational behavior. Our findings provide further insight into how this pathway acts within the NAc and implicates that enhancing enkephalins or DOR signaling in the NAc may be effective in preventing reduced motivational behavior after stress.
Funding and disclosure
This work was supported by NIMH R01MH106500 (MKL) and a NIGMS PRAT award 1FI2GM128622-01 (TCF). The authors declare no competing interests.
References
Mansour A, Hoversten MT, Taylor LP, Watson SJ, Akil H. The cloned μ, δ and κ receptors and their endogenous ligands: evidence for two opioid peptide recognition cores. Brain Res. 1995;700:89–98.
Belluzzi JD, Grant N, Garsky V, Sarantakis D, Wise CD, Stein L. Analgesia induced in vivo by central administration of enkephalin in rat. Nature. 1976;260:625–6.
Perrine SA, Hoshaw BA, Unterwald EM. Delta opioid receptor ligands modulate anxiety-like behaviors in the rat. Br J Pharmacol. 2006;147:864–72.
Konig M, Zimmer AM, Steiner H, Holmes P V., Crawley JN, Brownstein MJ, et al. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–8.
Filliol D, Ghozland S, Chluba J, Martin M, Matthes HWD, Simonin F, et al. Mice deficient for δ- and μ-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200.
Jutkiewicz EM, Torregrossa MM, Sobczyk-Kojiro K, Mosberg HI, Folk JE, Rice KC, et al. Behavioral and neurobiological effects of the enkephalinase inhibitor RB101 relative to its antidepressant effects. Eur J Pharmacol. 2006;531:151–9.
Tejedor-Real P, Micó JA, Maldonado R, Roques BP, Gibert-Rahola J. Effect of mixed (RB 38A) and selective (RB 38B) inhibitors of enkephalin degrading enzymes on a model of depression in the rat. Biol Psychiatry. 1993;34:100–7.
Germain L, Chouinard G. Treatment of recurrent unipolar major depression with captopril. Biol Psychiatry. 1988;23:637–41.
Le Merrer J, Becker JAJ, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev. 2009;89:1379–412.
Akil H, Watson SJ, Young E, Lewis ME, Khachaturian H, Walker JM. Endogenous opioids: biology and function. Annu Rev Neurosci. 1984;7:223–55.
Snyder SH, Pasternak GW. Historical review: opioid receptors. Trends Pharmacol Sci. 2003;24:198–205.
Rossier J, Guillemin R, Bloom F. Foot shock induced stress decreases Leu5-enkephalin immunoreactivity in rat hypothalamus. Eur J Pharmacol. 1978;48:465–6.
Nabeshima T, Katoh A, Wada M, Kameyama T. Stress-induced changes in brain Met-enkephalin, Leu-enkephalin and dynorphin concentrations. Life Sci. 1992;51:211–7.
de Felipe MDC, Jiménez I, Castro A, Fuentes JA. Antidepressant action of imipramine and iprindole in mice is enhanced by inhibitors of enkephalin-degrading peptidases. Eur J Pharmacol. 1989;159:175–80.
Li W, Papilloud A, Lozano-Montes L, Zhao N, Ye X, Zhang X, et al. Stress impacts the regulation neuropeptides in the rat hippocampus and prefrontal cortex. Proteomics. 2018;18:1–10.
Gerfen CR, Scott Young W. Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 1988;460:161–7.
Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci. 2006;9:443–52.
Knoll AT, Carlezon WA. Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73.
Krishnan V, Han M-H, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404.
Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;625:609–25.
Lobo MK, Zaman S, Damez-Werno DM, Koo JW, Bagot RC, DiNieri JA, et al. ΔFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. J Neurosci. 2013;33:18381–95.
Chandra R, Francis TC, Konkalmatt P, Amgalan A, Gancarz AM, Dietz DM, et al. Opposing role for Egr3 in nucleus accumbens cell subtypes in cocaine action. J Neurosci. 2015;35:7927–37.
Hikida T, Kimura K, Wada N, Funabiki K, Nakanishi S. Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron. 2010;66:896–907.
Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15:816–8.
Carlezon WA, Thomas MJ. Biological substrates of reward and aversion: A nucleus accumbens activity hypothesis. Neuropharmacology. 2009;56:122–32.
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–8.
Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, et al. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol Psychiatry. 2015;77:212–22.
Chandra R, Francis TC, Nam H, Riggs LM, Engeln M, Rudzinskas S, et al. Reduced Slc6a15 in nucleus accumbens D2-neurons underlies stress susceptibility. J Neurosci. 2017;37:6527–38.
Fox, ME, Chandra R, Menken MS, Larkin EJ, Nam H, Engeln M, et al. Dendritic remodeling of D1 neurons by RhoA/Rho-kinase mediates depression-like behavior. Mol. Psychiatry. 2018. https://doi.org/10.1038/s41380-018-0211-5
Donahue RJ, Landino SM, Golden SA, Carroll FI, Russo SJ, Carlezon WA. Effects of acute and chronic social defeat stress are differentially mediated by the dynorphin/kappa-opioid receptor system. Behav Pharmacol. 2015;26:654–63.
Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: ZQ175. PLoS ONE. 2012;7:e49838.
Paulsen JS, Ready RE, Hamilton JM, Mega MS, Cummings JL. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71:310–4.
Julien CL, Thompson JC, Wild S, Yardumian P, Snowden JS, Turner G, et al. Psychiatric disorders in preclinical Huntington’s disease. J Neurol Neurosurg Psychiatry. 2007;78:939–43.
Frank S. Treatment of Huntington’s disease. Neurotherapeutics. 2014;11:153–60.
Covey DP, Dantrassy HM, Zlebnik NE, Gildish I, Cheer JF. Compromised dopaminergic encoding of reward accompanying suppressed willingness to overcome high effort costs is a prominent prodromal characteristic of the Q175 mouse model of Huntington’s disease. J Neurosci. 2016;36:4993–5002.
Covey DP, Dantrassy HM, Yohn SE, Castro A, Conn PJ, Mateo Y, et al. Inhibition of endocannabinoid degradation rectifies motivational and dopaminergic deficits in the Q175 mouse model of Huntington’s disease. Neuropsychopharmacology. 2018;43:2056–63.
Ade KK, Wan Y, Chen M, Gloss B, Calakos N. An improved BAC transgenic fluorescent reporter line for sensitive and specific identification of striatonigral medium spiny neurons. Front Syst Neurosci. 2011;5:1–9.
Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–25.
Lindberg I, Smythe SJ, Dahl JL. Regional distribution of enkephalin in bovine brain. Brain Res. 1979;168:200–4.
Lindberg I, Dahl JL. Characterization of Enkephalin Release from Rat Striatum. J Neurochem. 1981;36:506–12.
Chandra R, Engeln M, Schiefer C, Patton MH, Martin JA, Werner CT, et al. Drp1 mitochondrial fission in D1 neurons mediates behavioral and cellular plasticity during early cocaine abstinence. Neuron. 2017;96:1327–41.e6.
Tejedor-Real P, Mico JA, Maldonado R, Roques BP, Gibert-Rahola J. Implication of endogenous opioid system in the learned helplessness model of depression. Pharmacol Biochem Behav. 1995;52:145–52.
Jia MR, Wei T, Xu WF. The analgesic activity of Bestatin as a potent APN inhibitor. Front Neurosci. 2010;4:1–10.
Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Herve D, Valjent E, et al. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–85.
Poulin JF, Laforest S, Drolet G. Enkephalin downregulation in the nucleus accumbens underlies chronic stress-induced anhedonia. Stress. 2014;17:88–96.
Henry MS, Bisht K, Vernoux N, Gendron L, Torres-Berrio A, Drolet G, et al. Delta opioid receptor signaling promotes resilience to stress under the repeated social defeat paradigm in mice. Front Mol Neurosci. 2018;11:1–15.
Ben Natan L, Chaillet P, Lecomte J-M, Marcais H, Uchida G, Costentin J. Involvement of endogenous enkephalins in the mouse ‘behavioral despair’ test. Eur J Pharmacol. 1984;97:301–4.
Turner AJ, Tanzawa K. Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J. 1997;11:355–64.
Pal Khaket T, Singh J, Attri P, Dhanda S. Enkephalin degrading enzymes: metalloproteases with high potential for drug development. Curr Pharm Des. 2012;18:220–30.
Karsten SL, Sang TK, Gehman LTT, Chatterjee S, Liu J, Lawless GM, et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of Tau-induced neurodegeneration. Neuron. 2006;51:549–60.
Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115:1363–81.
Shirayama Y, Chen AC-H, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–61.
Wook Koo J, Labonté B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. Essential Role of mesolimbic brain-derived neurotrophic factor in chronic social stress-induced depressive behaviors. Biol Psychiatry. 2016;80:469–78.
Kramer HK, Simon EJ. mu and delta-opioid receptor agonists induce mitogen-activated protein kinase (MAPK) activation in the absence of receptor internalization. Neuropharmacology. 2000;39:1707–19.
Blomeley CP, Bracci E. Opioidergic interactions between striatal projection neurons. J Neurosci. 2011;31:13346–56.
Svingos AL, Clarke CL, Pickel VM. Cellular sites for activation of δ-opioid receptors in the rat nucleus accumbens shell: relationship with Met5-enkephalin. J Neurosci. 1998;18:1923–33.
Laurent V, Leung B, Maidment N, Balleine BW. μ- and δ-opioid-related processes in the accumbens core and shell differentially mediate the influence of reward-guided and stimulus-guided decisions on choice. J Neurosci. 2012;32:1875–83.
Creed M, Ntamati NR, Chandra R, Lobo MK, Luscher C. Convergence of reinforcing and anhedonic cocaine effects in the ventral pallidum. Neuron. 2016;92:214–26.
Covington H, Miczek K. Repeated social-defeat stress, cocaine or morphine. Psychopharmacol (Berl). 2001;158:388–98.
Yap JJ, Chartoff EH, Holly EN, Potter DN, Carlezon WA, Miczek KA. Social defeat stress-induced sensitization and escalated cocaine self-administration: the role of ERK signaling in the rat ventral tegmental area. Psychopharmacol (Berl). 2015;232:1555–69.
Covington HE, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71:656–70.
Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294–307.
McGinty JF. Co-localization of GABA with other neuroactive substances in the basal ganglia. Prog Brain Res. 2007;160:273–84.
Armario A, Nadal R. Individual differences and the characterization of animal models of psychopathology: a strong challenge and a good opportunity. Front Pharmacol. 2013;4:1–13.
Vaugeois JM, Passera G, Zuccaro F, Costentin J. Individual differences in response to imipramine in the mouse tail suspension test. Psychopharmacol (Berl). 1997;134:387–91.
Lutz PE, Kieffer BL. Opioid receptors: distinct roles in mood disorders. Trends Neurosci. 2013;36:195–206.
Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, et al. Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron. 2015;87:1063–77.
Augood SJ, Faull RLM, Love DR, Emson PC. Reduction in enkephalin and substance P messenger RNA in the striatum of early grade Huntington’s disease: a detailed cellular in situ hybridization study. Neuroscience. 1996;72:1023–36.
Bissonnette S, Vaillancourt M, Hébert SS, Drolet G, Samadi P. Striatal pre-enkephalin overexpression improves Huntington’s disease symptoms in the R6/2 mouse model of Huntington’s disease. PLoS ONE. 2013;8:e75099.
Cepeda C, Wu N, André VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington’s disease. Prog Neurobiol. 2007;81:253–71.
Wang H, Pickel VM. Preferential cytoplasmic localization of delta-opioid receptors in rat striatal patches: comparison with plasmalemmal mu-opioid receptors. J Neurosci. 2001;21:3242–50.
Parent A, Sato F, Wu Y, Gauthier J, Lévesque M, Parent M. Organization of the basal ganglia: the importance of axonal collateralization. Trends Neurosci. 2000;23:20–7.
Massaly N, Copits BA, Wilson-Poe AR, Hipólito L, Markovic T, Yoon HJ, et al. Pain-induced negative affect is mediated via recruitment of the nucleus accumbens kappa opioid system. Neuron. 2019;102:1–10.
Acknowledgements
We thank Dr. Iris Lindberg (UMSOM) for her generous donation of anti-Leu- and Met-Enkephalin antisera. We thank Dr. Eric Nestler (ICAHN School of Medicine at Mount Sinai) for reagents used in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nam, H., Chandra, R., Francis, T.C. et al. Reduced nucleus accumbens enkephalins underlie vulnerability to social defeat stress. Neuropsychopharmacol. 44, 1876–1885 (2019). https://doi.org/10.1038/s41386-019-0422-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0422-8
This article is cited by
-
Maturation of nucleus accumbens synaptic transmission signals a critical period for the rescue of social deficits in a mouse model of autism spectrum disorder
Molecular Brain (2023)
-
Mediation of the behavioral effects of ketamine and (2R,6R)-hydroxynorketamine in mice by kappa opioid receptors
Psychopharmacology (2022)
-
Housing conditions during self-administration determine motivation for cocaine in mice following chronic social defeat stress
Psychopharmacology (2021)
-
Dendritic spine density is increased on nucleus accumbens D2 neurons after chronic social defeat
Scientific Reports (2020)
-
Accelerated development of cocaine-associated dopamine transients and cocaine use vulnerability following traumatic stress
Neuropsychopharmacology (2020)
