
Abstract
ILCs and T cells are closely related functionally but they significantly differ in their ability to circulate, expand, and renew. Cooperation and reciprocal functional regulation suggest that these cell types are more complementary than simply redundant during immune responses. How ILCs shape T-cell responses is strongly dependent on the tissue and inflammatory context. Likewise, indirect regulation of ILCs by adaptive immunity is induced by environmental cues such as the gut microbiota. Here, we review shared requirements for the development and function of both cell types and divergences in the orchestration of prototypic immune functions. We discuss the diversity of functional interactions between T cells and ILCs during homeostasis and immune responses. Identifying the location and the nature of the tissue microenvironment in which these interactions are taking place may uncover the remaining mysteries of their close encounters.
Similar content being viewed by others
Introduction
Innate lymphoid cells have been classified in multiple subsets based on functional and developmental similarities with T cells.1 Natural killer cells (NK) cells express eomesodermin, perforin, and granzymes, which are the hallmarks of cytotoxic CD8+ T cells. ILC1s are similar to Th1 cells, expressing T-bet and producing IFNγ in response to IL-12 and IL-18 at early stages of infections with intracellular microorganisms such as viruses2 or parasites.3 However, there is neither specific phenotype nor unique function that universally define ILC1s, as both transcription factor expression and functions vary depending on tissue location. For instance, in the liver, ILC1s might be more related to resident NK cell subsets than to helper ILCs, as they develop cytotoxicity.4 Similar to Th2 cells, ILC2s express high levels of GATA-3 and produce IL-5, IL-13, IL-9, and IL-4. ILC2s are activated in response to type-2 inducer cytokines such as IL-25, TSLP, and IL-33 in the context of parasitic infections and airway inflammation induced by respiratory viruses. ILC3s express RORγt and produce IL-22, IL-17, TNFα, and GM-CSF in response to IL-1β and IL-23. In humans and adult mice a significant proportion of ILC3s express the natural cytotoxic receptor 1 (NCR1). Similar to Th17 cells, both NCR1− and + ILC3s are enriched in the lamina propria (LP) of the small intestine where they participate in tissue homeostasis at the steady state and in protective immune responses against extracellular bacteria and fungi.5 ILC1, 2 and 3 are therefore considered the innate counterparts of Th1, Th2, and Th17, respectively, and designated as “helper” ILCs. Standing apart from the classical parallel between ILCs and T cells, lymphoid tissue inducer cells (LTis) are fetal ILC3s, they also develop in a RORγt dependent manner and produce IL-22 and IL-17. In contrast to Th17 cells, LTis are generated in the absence of microbiota as soon as the embryonic day 12.5 in mice and initiate the development of secondary lymphoid structures that are instrumental in the orchestration of adaptive immune responses.6
ILC subsets in mice and humans show striking similarities suggesting conservation throughout evolution.7 Despite their potent immune functions, ILCs are apparently redundant in humans when adaptive immunity is preserved.8 Accumulation of ILCs is however common in inflamed tissues of patients suffering from IBD (ILC1s and ILC3s),9 asthma (ILC2s)10 or psoriasis (ILC3s).11 ILCs are also deregulated in the context of tissue inflammation as evidenced in numerous mouse models mimicking these pathologies. Indeed, their active participation in such immunological disorders is considered as a potential therapeutic target in various inflammatory diseases.12
In this review, we will discuss T/ILC similarities and divergences to frame their relative contribution to immunological processes. We will underscore most recent advances focusing on their specificities and collaborative complementarities.
T cells and ILCs are closely related developmentally and functionally
Cell fate decision
Early in their ontogeny, T and ILC progenitors share similarities in their transcriptional programs. As lymphoid subsets, both T cells and ILCs are derived from the common lymphoid progenitor in mice and in humans. While T cells are strictly dependent on the successful recombination and expression of TCR genes, ILCs remain mostly unaffected in the absence of V(D)J recombination and do not undergo massive proliferation nor selection during differentiation.
During the past few years, the roles of transcription factors involved in ILC commitment have been identified and the sequence of events dictating their fate has been partially uncovered in mice.13 In mice, early Lin− CD127+ Flt3− lymphoid progenitors expressing the integrin α4β7 generate all ILC subsets and T cells but not B cells.14,15 The first stage deprived of T potential has been isolated as the early innate lymphoid progenitor (EILP) stage, where Lin− α4β7+ CD127− Flt3− progenitors have the capacity to generate all ILC subsets.16 At these early stages, the induction of the inhibitor of DNA binding Id2 titrates the activity of E-box proteins and inhibits the progression of B- and T-cell developmental programs.17,18 This EILP stage is surprisingly transiently down regulating the IL-7 receptor. This downregulation is also observed during early thymocyte development, just at the transition of double negative stages DN2 to DN3. The reason for the IL-7Rα decrease is unknown for both lineages. Human ILCs were generated from two distinct subsets of early lymphoid progenitors (ELPs) in a humanized mouse model. Both CD127+ and CD127− ELPs could generate NK cells and ILCs, while T-cell potential was lost in CD127+ ELPs.19 In human secondary lymphoid organs, a population of early tonsillar progenitors expressing the receptor for IL-1 and identified as Lin− CD34+ CD10− c-Kit+ IL-1R+ may be considered as the earliest committed common ILC progenitor.20,21 Nonetheless, human ILC progenitors have also been identified in the blood and tissues. The developmental relationships between human ILC progenitors have been reviewed elsewhere.22 We will further discuss their migratory behavior later in this review.
Notch signaling in the differentiation of ILCs and T cells
It has been largely documented that T-cell development requires signals emanating from the thymus microenvironment such as Notch ligands and MHCII expressed by thymic DCs and AIRE expressing epithelial cells. In contrast, most ILC subsets develop normally in athymic mice suggesting that ILCs do not require an intra-thymic stage in order to fully accomplish their developmental program. In addition, deletion of RBPJ (the main transcription factor downstream of the canonical Notch pathway) in IL-7Rα expressing cells showed that Notch signaling is not required for ILC lineage specification.23 Nonetheless, in vivo and in vitro studies showed that the strength and duration of Notch signals control cell fate decision at different stages of ILC differentiation.15,23,24,25 Intermediate strength of Notch signaling or transient activation by Notch ligands can favor ILC fate decision in common lymphoid progenitors with LTis and ILC3s being more frequently produced with stronger or longer activation of Notch than ILC2s and NK/ILC1s24 in vitro. Overall, Notch signaling can modulate ILC identity in many possible ways without being as critically and constantly required as it is for T-cell development.14,26,27,28,29
Shared requirements of ILCs and T cells for transcriptional regulators
Numerous transcription factors have been identified as instrumental for both ILC and T-cell development. Loss of all ILC subsets in adult tcf7 deficient mice indicates that TCF1 expression, instrumental in T-cell development, is also one of the earliest critical events in ILC fate decision.30,31 Moreover, both T cells and “helper” ILCs rely on GATA-3 expression at early stages of their development32,33 and PLZF expression marks the earliest ILCP stages restricted to all helper ILCs fate and is also absolutely needed for MAIT34 and NKT cell development.35 Of note, NK cells can be obtained from committed PLZF+ ILCP18 suggesting that ILC1s cannot be completely distinguished from NK cells based on their history of PLZF expression.36 Finally, Bcl11b first considered as the ultimate marker of T-cell committed progenitors was shown to be highly expressed in ILC2 precursors36 and required for ILC2 specification.37,38,39 Indeed, ILC2 potential is enriched in Bcl11b+ bone marrow (BM) progenitors, while the generation of alternate ILC lineages is significantly reduced compared to ILCPs.18
Despite the significant overlap in developmental requirements for ILCs and T cells, BM chimeras and genetically engineered mouse models helped to clarify the contribution of ILCs in immune responses of lympho-replete hosts. These findings are reviewed and discussed in the next sections of this review.
Identical functions by the use of redundant immune modules
Prototypic immune responses are orchestrated by transcription factors and effector molecules defined as “Immune modules”.40 Antiviral type 1 immune responses rely on T-bet, IL-12, and IFNγ, while type-2 antiparasitic responses rely on GATA-3, IL-25, IL-4, and IL-13. Type-17 antibacterial responses rely on RORγt, AhR, T-bet, and cytokines such as IL-1β, IL-23, IL-22, IL-17, and GM-CSF. Regulatory immune responses are characterized by TGFβ and IL-10 but the ontogeny of regulatory lymphocytes widely diverges from one lineage to the other. T cells and ILCs share these immune modules, overall raising the question of their complementarity and redundancy during immune responses.40
Essential for the differentiation of Th141 and ILC1s,3 T-bet promotes Th1 cytokine production through interactions with the promoters and regulatory elements of genes encoding IL-12RB2 and IFNγ.42,43 T-bet also induces the expression of Runx3 a transcription factor that is instrumental for the development of ILC1s and ILC3s.44 In T cells, T-bet supports the differentiation of Th1 cells through the induction of IFNγ and suppression of Th2 cell fate.40 Interestingly, a DNase I hypersensitivity (HS) site (HS IV) located in the Il4 gene was detected in both Th1 and Th2 cells45 and RUNX3 binding to HS IV was found to suppress IL-4 production in Th1 cells.46,42
IL-5 and IL-13 are type-2 immunity signature cytokines produced by ILC2s,47,48 Th2 and Th9.49 IL-4 is mainly produced by Th2, follicular helper T(Tfh) cells and basophils while ILC2s and Th9 are more significant producers of IL-9. During helminthes infection, Tfh produced IL-4 while Th2 cells secreted both IL-13 and IL-4. Likewise, on the innate side of antiparasitic immune responses, basophils produced IL-4 while type-2 ILCs secreted IL-13. Distinct expression patterns for IL-14 and IL-13 may explain their unique role in protective immunity and allergic immune responses.50 IL-4 and IL-9 sustain type-2 immunity programs with an impact on IgE class switching for IL-4 and the expansion of mastocytes for IL-9. TSLP, IL-25, and IL-33 participate in the maintenance and expansion of tissue-resident ILC2 subsets. GATA-3 was demonstrated to be instrumental for the development of Th2 and ILC2s and in the transcriptional activation of genes encoding IL-5, IL-13, and amphiregulin.32 In Th2 cells, Notch signaling and phosphorylated STAT6 reinforce polarization through the induction of GATA-3 expression.32,40
Immune responses directed against extracellular pathogens and fungi rely on effector cytokines such as IL-17A, IL-22, and GM-CSF. IL-17A promotes the recruitment of neutrophils51 and IL-22 induces the production of antimicrobial peptides by epithelial cells.52 GM-CSF sustains myeloid cell populations that secrete polarizing cytokines such as IL-1β and IL-23,53 which in turn favor the differentiation and survival of Th17 and Th22 cells as well as the production of IL-22 by ILC3s.54 In both ILC3s and T cells, RORγt induces the expression of IL-23R. The aryl hydrocarbon receptor (AhR) plays a role in Th22 differentiation, in postnatal maturation of gut associated tertiary lymphoid structures and in intestinal NCR1+ ILC3s differentiation.26 Such developmental and functional similarities raise the question of the complementarity and/or redundancy between ILC3s and Th22 cells. In line with the role played by Th22 in responses against enteropathogens, T-cell competent mice lacking NCR1+ ILC3s have been shown to be protected against Citrobacter rodentium55,56 suggesting that the latter are largely redundant in immunocompetent hosts. However, in early immune responses against C. rodentium, LTi-like ILC3s played a significant role before the onset of adaptive immunity.55,57 In addition, Id2 is required for colonization resistance against C. rodentium through IL-22 dependent regulation of the microbiota.58
Besides their protective role against enteropathogens, LTi-like cells retain the ability to interact with stromal cells after birth and the functional consequences of such interactions will be discussed later.
Although most T cells have an ILC counterpart, the existence of a separate regulatory ILC subset is controversial. Regulatory ILCs were described based on their ability to produce IL-10 in the context of tissue inflammation in the gut59 and in the lung.60 However, a recent study provided evidence that ILC2s are the main source of IL-10 in the mouse gastrointestinal tract.61 Despite functional similarities with regulatory T cells (Tregs), IL-10 secreting ILC subsets do not rely on Foxp3 for their generation and maintenance and do not seem to exert regulatory functions towards adaptive immune responses.
In summary, ILCs and T cells share immune modules that are associated with prototypic responses against viruses, intracellular and extracellular pathogens. The importance and the contribution of these populations are not equivalent and ILCs have been shown to be redundant in immunocompetent hosts. Several reasons might explain why ILCs have been conserved despite the onset of adaptive immunity. LTis are instrumental in the organogenesis of secondary and tertiary lymphoid tissues. LTis are not only required for proper adaptive immune responses but also to establish tolerance and maintain homeostasis in the gut. Second, the adaptive immune system remains largely immature during the first weeks after birth leaving neonates at risk of infection. ILCs populate lymphoid organs and nonlymphoid barrier tissues before and early after birth and may compensate the immaturity of adaptive immunity during the perinatal period (Table 1). In addition to these putative advantages during evolution, quiescent tissue-resident populations such as ILCs may partially escape chemically-induced cell death, compensate aplasia, and immunodeficiency resulting from aggressive cancer treatments and participate in tissue repair and remodeling in graft versus host disease.62,63,64
T cells and ILCs compete for space and signals
The expression of the common cytokine receptor γc, which is shared by IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 is instrumental for both T and ILC development. Both cell types rely on identical survival factors, notably IL-7, IL-15, and IL-2. As IL-7 is produced in limiting amounts by a small number of specialized stromal cells and epithelial cells,65 its availability regulates the abundance and distribution of each cell type in lymph nodes.66 All helper ILCs express the IL-7Rα chain but only ILC2s and ILC3s require IL-7 for their development and/or maintenance.67,68,69 In contrast, NK cells and ILC1s are only significantly reduced in IL-15 deficient mice.3,70
IL-7 reporter mice revealed il-7 gene expression by epithelial cells in the small intestine and in the lung suggesting that niches may sustain both T and ILC survival in these tissues.65 Indeed, IL-7 may participate in the maintenance of resident memory T cells in hair follicles71 as well as in the lung and airways.72 However, it has been recently shown that IL-15 can largely compensate IL-7Rα deficiency for ILC2 and ILC3 survival in the small intestine but not in other tissues.73
Activated T cells produce IL-2 as well as ILCs at the steady state. Tregs and ILC2s express the high affinity receptor for IL-2, CD25. Of note, IL-2 derived from ILC3s was recently shown to be instrumental in the generation and maintenance of induced Tregs in the small intestine (Fig. 1a).74 Reciprocally, IL-2 derived from activated CD4+ T cells was shown to promote the survival of type-2 ILCs in a mouse model of helminthes infection (Fig. 2c).75 Interestingly, Tregs were reported to restrain the availability of IL-2 derived from CD4+ T for the expansion and differentiation of immature CD25+ CD127+ NK cell precursors76 and this may also be the case for ILC2s.77 The role of autocrine IL-2 in ILC homeostasis remains unclear. Loss of IL-2 expression by ILC3s did not affect their numbers in the small intestine74 and the role played by autocrine IL-2 in the survival and activation of CD25+ ILC2s remains to be established.

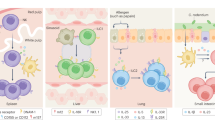
a In the small intestine, the commensal Segmented filamentous bacteria induce IL-22 production by ILC3s, which in turn promotes the local maturation of Th17 cells through the induction of SAA1/2 production by intestinal epithelial cells.132 GM-CSF derived from ILC3s sustains the function and survival of myeloid cells, which release IL-1β in response to microbiota.53 IL-1β increases the production of IL-2 by gut ILC3s that are in turn instrumental in the maintenance of induced regulatory T cells.74 b In mucosal draining lymph nodes, MHCII-expressing LTi-like cells suppress the activation or promote the cell death of microbiota-specific T cells through antigen presentation in the absence of co-stimulatory molecules. Highly reactive clones are depleted from the repertoire, thereby promoting peripheral tolerance toward gut microbiota-derived antigens.113 MHCII-expressing ILC3s also control IgA responses against commensal bacteria through interactions with follicular helper T cells (Tfh). Antigen presentation by ILC3s inhibits the production of IL-4 by Tfh and of IgA by B cells. PD1–PDL1 interactions may synergize with antigen presentation to suppress Tfh functions114.

a Allergen exposure induces the release of IL-33 by lung epithelial cells, which in turn stimulates IL-13 production by tissue-resident ILC2s. ILC2-derived IL-13 promotes the activation of dendritic cells (DC) as well as their migration towards the draining lymph nodes where naïve T cells are activated and converted into Th2 cells. Th2 cells eventually migrate back to the tissue in response to DC-derived CCL17128 (b). T-ILC2 interactions also take place in the lungs and promote adaptive immune responses: antigen presentation through MHCII and OX40-OX40L interactions favors Th2 differentiation and the maintenance of memory T cells.148 c Upon helminthes infection, a specialized subset of intestinal epithelial cells named Tuft cells release IL-25. IL-25 induces the activation of inflammatory ILC2s and their exit into the blood. Circulating inflammatory ILC2s reach the lung where they can promote parasite clearance through the release of IL-13 and the activation and mucus production by goblet cells.79 T-ILC interactions such as antigen presentation, OX40–OX40L125 binding and IL-2 production75,119 coordinate innate and adaptive immunity in the small intestine to promote parasite expulsion.
Divergence between ILCs and T cells as a source of complementarity
T cells and ILCs share master transcriptional regulators and effector cytokines. However, the timing of their maturation and activation as well as their ability to enter the bloodstream and tissues of residence are strikingly different and support complementarity and cooperation.
Tissue residency and trafficking
Given the large diversity of the naïve T-cell repertoire, the frequency of clones that are specific for one given antigen is extremely low. The ability to patrol and recirculate through the body is therefore necessary to increase the probability for antigen-specific naïve T cells to get activated by professional APCs in secondary lymphoid tissues.78 In contrast, and alike unconventional T cells lymphocytes with a restricted TCR repertoire, ILCs are mostly tissue-resident cells, and in both cell types, tissue residency appears to be associated with cytokine-induced activation (Table 1). Parabiosis experiments, in which the circulatory systems of two animals have been surgically joined, showed that very few ILCs are exchanged between partners even on the long term at the steady state, in stark contrast with T and B cells that equilibrated within a few weeks. Nonetheless, inflammation and infection promoted ILC2 migration but in lower numbers than their adaptive counterparts.77 Interestingly, a study from Huang et al. showed that, during the intestinal stage of helminthes infection, inflammatory ILC2s migrated from one parabiont to the other: these ILC2s originated from the small intestine and migrated to the lungs in a S1PR dependent manner (Fig. 2c).79 Moreover, in mice infected by Nippostrongylus brasiliensis, ILC2s circulating in the blood were shown to originate from diverse tissues (lung, intestine, and BM) depending on the stage of infection. Environmental constraints such as carrying capacity were proposed to explain extrusion after the local expansion of activated ILC2.80 Indeed, cells probably migrate from the tissue when their numbers have reached the maximum population size that can be sustained in this environment.
In naïve mice, helper ILCs are barely detected in the blood, except for ILC2s, although the possibility that circulating ILCs are phenotypically different from their tissue-resident counterparts (thereby preventing their detection) cannot be excluded. In humans, there is evidence that committed ILC precursors expressing Id2, Tox, Tcf7, and Runx3 but no transcription factors specific of mature helper ILCs are circulating.81 They may circulate through the blood and lymph nodes as CD62L expressing cells82 and seed the periphery to give rise to diverse tissue-resident ILC subsets. In mice, migratory mature ILCs have also been identified in peripheral lymph nodes. ILC1s were found to be the most abundant migratory subset in the blood and lymph nodes. Their entry was dependent on CD62L and CCR7 while their egress relied on S1PR as previously described for T cells.83
A few studies suggest that chemokine receptors such as CCR7 and CCR9 are involved in the trafficking of LTi-like cells and in the migration of ILC1s and ILC3s from the mesenteric lymph nodes to the small intestine.84,85 CCR9 and α4β7 expression by ILC2 precursors control homing to the small intestine while CXCR4 promotes retention in the BM. The downregulation of CXCR4 by IL-33 was shown to promote ILC2 egress from the BM.86 Migratory ILCs have been identified in cryptopatches at the steady state and shown to exit these structures in a mouse model of innate colitis.87 Yet, key chemokines involved in this process have not been clearly identified.
Overall, it appears that ILCs can adopt a migratory behavior using similar homing receptors and molecular interactions as T cells. Reciprocally, T cells can establish residency in lymphoid and nonlymphoid tissues. These resident T cells include unconventional T-cell subsets such as γδ T, CD8αα IEL, MAIT cells and iNKT cells or conventional T cells called tissue-resident memory (TRM). Resident memory T cells (TRM) cells and ILCs thus share the common feature of long-term residency associated with a slow turnover. By secreting cytokines and chemokines that recruit T cells independently of their antigen specificity TRM cells can also provide an innate type of immune response.88 Interactions between these different tissue-resident lymphocytes and their hematopoietic and non-hematopoietic partners likely participate in the maintenance of tissue homeostasis at the steady state and promote tissue repair during infection or injury. Conversely, memory T cells and ILCs can act in concert to foster immune pathologies such as allergy, inflammation, and autoimmunity.
Renewal and functional diversification
Whether ILCs colonize peripheral tissues as immature precursors or mature populations remains unclear. In mice, several studies indicated that, during embryogenesis, immature ILC precursors migrated from the fetal liver to the periphery where they promptly acquired specific lineage markers.24,89 To date, whether ILC development follows the same rules in newborn and adult mice is not known. What triggers the early priming of immune modules shared with T cells is unclear as well as the contribution of extrinsic cues in the adult or fetus. Genes defining each helper ILC lineage were shown to be transcriptionally active in fetal liver common ILC progenitors suggesting that the underlying epigenetic modifications are largely independent of microbial stimuli.90 Accordingly, all ILC subsets are detected in adult germ-free mice suggesting that they could be produced independently from the microbiota.91,92 However, analyses of their transcriptome at the single cell level suggested potential functional modifications, especially for intestinal ILC2 subsets.92 The differentiation of mouse ILCs thus differs from that of conventional helper T cells, for which a combination of antigen and cytokine-mediated activation is a prerequisite for polarization in secondary lymphoid organs.
Most studies on ILC ontogeny have been performed in mice. In humans, a population of multipotent ILC progenitors has been characterized in the blood, suggesting that immature ILCs may be recruited to tissues and differentiate locally in response to Notch ligands and/or the cytokine milieu.81 A recent study showed however that circulating human ILC progenitors contain a mix of specified ILC precursors with restricted potential as well as multipotent progenitors.93 Thus, circulating ILC precursors may not have the same potential for polarization as naïve T cells in humans. Progenitors with ILC potential have also been detected in the human fetal liver, small intestine, tonsils,94 BM, and cord blood19 while bi-potent NK-ILC precursors have been observed in the human fetal intestine.95 The respective contribution of these human ILC precursors to the generation and renewal of tissue-resident ILCs remains to be evaluated.
Successive waves of development lead to the colonization of peripheral tissues by γδ T cells before and shortly after birth. Likewise, ILCs are more abundant in lymphoid and nonlymphoid tissues before and during the first days after birth96 where they compete with T cells for stroma-derived IL-7.97 Accordingly, γδ T cells and ILCs are progressively outcompeted after birth by αβ T cells in peripheral lymph nodes as well as in tissues. In contrast to αβ T cells that are renewed and constantly replaced by new thymic emigrants, the maintenance of the peripheral pool of ILCs relies on local renewal of tissue-resident cells rather than distal input from the BM.77,98 These processes are however heterogeneous among ILCs. ILC2s are renewed at a higher rate than ILC1s and ILC3s at the steady state.79 Genetic fate mapping approaches were used in order to describe the dynamics of ILC2 renewal at the steady state and during infection.80,98 At the steady state, de novo generation of ILC2s was shown to take place mostly during the perinatal period and the contribution of BM derived-precursors was found to be moderate in the adult.98 In the context of N. brasiliensis infection, the expansion of peripheral ILC2s relies on the migration of activated ILC2s from infected tissues rather than newly generated cells.80,98
Type 2 ILCs and alveolar macrophages are derived from fetal/neonatal progenitors and were both shown to be long-lived tissue-resident subsets. Such features may promote the establishment of lung perinatal type-2 immunity, in combination with environmental cytokines such as IL-33. Indeed, during postnatal alveologenesis, IL-33 promotes Th2 cell mediated immunity through the activation of pulmonary DCs and ILC2s.99 In turn, ILC2s support the maintenance of alveolar macrophage type-2 polarization through IL-13 secretion.100
Memory and trained immunity
In T cells, adaptive immunological memory relies on the selection of highly specific clones among naïve lymphocytes expressing a vast diversity of antigen-specific receptors. T-cell activation in the context of antigen presentation and polarizing cytokines induces chromatin remodeling at loci encoding immune effectors.101 Epigenetic marks such as DNA methylation patterns and posttranslational modifications of histone proteins remain partially imprinted in memory T cells allowing higher accessibility for transcriptional regulators and faster induction of gene expression upon rechallenge.101
Innate lymphoid cells might also remember immunological challenges and thereby acquire adaptive features. The development of memory responses by conventional NK cells was first described during murine cytomegalovirus (MCMV) infection.102 NK memory generation was dependent on Ly49H interaction with its virally-induced ligand m157 and a memory pool of Ly6Chi DNAM1lo CD27− CD11bhi KLRG1hi MCMV-induced NK cells was shown to arise from KLRG1lo Ly49H+ effector NK cells.103,104 Development of memory NK cell was shown to be positively regulated by IL-12, DNAX accessory molecule 1 (DNAM1), Eomes, T-bet and STAT4 and antagonized by Bim105 and was linked to epigenetics alterations allowing enhanced chromatin accessibility at the Prf1 (encoding perforin-1) locus.106 In humans, memory-like NK cells have also been observed after cytomegalovirus (HCMV) infection. Their memory phenotype and their increased IFNγ secretion was also linked to epigenetic modifications.105 Memory acquisition has also been observed in liver-resident NK cells and ILC1s which can protect upon secondary challenge by viruses.107,108 Likewise, lung ILC2s stimulated by IL-33 were suggested to develop into “memory-like cells” that produced high amounts of IL-5 and IL-13, showed long-term residency in the lungs and high responsiveness to IL-25.109 Whether these IL-33-experienced ILC2 cells are true memory cells remain however uncertain. Future studies will be necessary to determine the molecular and epigenetic features that characterize these various memory-like populations of ILCs.
Overall, it appears that both T cells and ILCs can participate in protective immune responses and in the maintenance of tissue homeostasis with the capacity to colonize and establish long-term residency within mucosal tissues and barrier sites. However, T cells are more numerous and subdivided into resident and circulating subsets with a much higher capacity to renew, expand and mount specific memory responses. Therefore, the cooperation between these two cell types is expected to be essential to coordinate appropriate immune responses while maintaining tissue integrity, function, and homeostasis.
Reciprocal regulation of effector functions between ILCs and T cells
Functional interactions between T cells and ILCs include direct cell–cell contacts or rely on secreted molecules. Indirect interactions involving a third partner such as dendritic cells, macrophages, epithelial cells, or stromal cells have also been reported.110 They are shaped by environmental cues such as alarmins and commensal bacteria, which play a critical role in the regulation of ILCs and T cells.111
Regulation of T-cell responses by ILCs
Antigen presentation by ILCs to T cells
In the mesenteric lymph nodes and large intestine, CCR6+ ILC3s express high levels of MHCII, while they lack the expression of co-stimulatory molecules. Ablation of MHCII molecules in ILC3s was shown to result in microbiota-dependent spontaneous colitis.112 Indeed, antigen presentation by ILC3s to their cognate T cells induced the cell death of highly reactive T-cell clones and resulted in peripheral tolerance towards microbiota antigens.113 In colon-draining lymph nodes, MHCII+ ILC3s could also limit Tfh-mediated Ig class switch and mucosal IgA responses.114 These observations indicate that MHCII+ ILC3s participate in the regulation of adaptive immune responses against commensal bacteria antigens (Fig. 1a, b). Whether these mechanisms are redundant with the action of induced Tregs remains unclear. Of note, ablation of MHCII expression by ILC3s did not induce spontaneous colitis in other studies,115,116 perhaps due to differences in the microbiota between mouse colonies. Interestingly, expression of co-stimulatory molecules by ILC3s and antigen presentation to T cells were differentially regulated in the spleen and small intestine. Thus, IL-1β induced the expression of CD40 and CD86 in splenic but not intestinal ILC3s116 and activated splenic NCR− ILC3s efficiently induced T-cell activation and proliferation in vitro. Reciprocally, adaptive immune responses were significantly impaired in mice lacking MHCII expression in ILC3s.116 Collectively, these observations indicate that the tissue microenvironment profoundly affects the outcome of antigen presentation by type 3 ILCs through the regulation of the expression of co-stimulatory molecules. This conclusion is also supported by a recent study showing that IFNγ promotes MHCII expression in spleen ILC3s and stimulates their APC function, while microbiota-induced IL-23 suppresses MHCII expression in small intestinal ILC3s through pSTAT3.117
Splenic ILC3s were also shown to express CD1d and to internalize and present lipid antigens to iNKT cells. As a result, NKT cells produced IL-4 and INFγ while activation by αGal-Cer was able to increase IL-22 expression by ILC3s through CD1d signaling both in vitro and in vivo.118 ILC3s are rare in the spleen compared with dendritic cells and B cells that express CD1d, therefore the frequency at which NKT-ILC interactions occurs in this compartment and their physiological relevance remain to be fully evaluated.
Antigen presentation by ILC2s to CD4+ T cells has also been described. ILC2s express MHCII at lower levels than ILC3s or B cells but a fraction also expresses co-stimulatory molecules. MHCII+ ILC2s promoted worm expulsion and instructed adaptive immune response in a mouse model of helminthes infection (Fig. 2c). Although murine ILC2s were shown to process and present antigens to T cells, they were unable to induce T-cell proliferation in vitro. It has been suggested that low MHCII expression by ILC2s may skew T-cell differentiation towards type-2 immunity but investigations are needed to clarify how MHCII expression by ILC2s potentiates adaptive immune responses in vivo.75,119
In human, ILC2s express CD1a and group 4 phospholipase A2 (PLA2G4) in the epidermis at the steady state. Both CD1a and PLA2G4 expression were potentiated by TSLP in vitro thereby enhancing lipid presentation by ILC2s since PLA2G4 activity participates in the processing of lipid ligands presented by CD1a. Accordingly, CD1a+ ILC2s were shown to efficiently present lipids derived from house dust mite and Staphylococcus aureus and to induce the production of cytokines such as IFNγ, IL-22, and IL-13 by T cells. Thus, lipid presentation by ILC2s may participate in skin inflammation in patients suffering from atopic dermatitis.120
Role of ILCs in the survival of memory CD4+ T cells and in T-cell activation
In addition to MHCII and co-stimulatory molecules, ILCs express surface molecules that can impact adaptive immune responses depending on the context in which cellular interactions take place. LTi-like cells were shown to express OX40L and CD30L in adult mice121 and RORγt deficiency was associated with reduced survival of CD4+ memory T cells. It was thus proposed that LTi-like cells provide OX40L and CD30L signals that are instrumental for memory CD4+ T cells survival.122,123 However, models in which OX40L is selectively inactivated in ILCs still remain to be generated in order to clarify whether ILCs are a relevant source of this ligand to support CD4+ memory T-cell survival in vivo. Inducible expression of OX40L by MHCII+ ILC3s might also support the activation of pathogenic T cells during colitis. Thus, it was shown that microbiota-induced TNF-like Ligand 1A (TL1A) promoted OX40 ligand (OX40L) expression on MHCII+ ILC3s which, in turn, supported antigen-specific T-cell proliferation in vitro and in vivo expansion of pathogenic Th1 in a mouse model of chronic colitis.124 Finally, OX40L may also play a role in the interaction between ILC2s and Th2 cells in mice. Intranasal administration of IL-33 induced OX40L expression in lung ILC2s, while ILC2s targeted deletion of OX40L, alike ILC2 depletion, impaired Th2 and Treg responses to IL-33. These observations were reproduced in mouse models of allergy and helminthes infection, demonstrating the physiological relevance of IL-33 dependent OX40L expression by ILC2s125 (Fig. 2a).
Role of ILC-derived cytokines in the recruitment and activation of T cells
ILCs and helper T cells activate immune and non-hematopoietic cells through the production of effector cytokines. Cytokines produced by each ILC subset can also regulate adaptive immune responses.
ILC1s and T cells: studies on the cross talk between ILC1s and T cells are rare and the specific contribution of ILC1s was not investigated in depth. ILC1s have been associated with increased intestinal inflammation through a positive feedback loop leading to the secretion of IFNγ by ILCs and T cells in Crohn’s disease patients and in a colitis mouse model.9,126 It was also proposed that IFNγ released by ILC1s and NCR1+ ILC3s promotes the migration of T cells across the parenchyma via chemokines and matrix metalloproteases in a Th17-induced neuro-inflammation model.127
ILC2s and T cells: during IL-33-mediated lung inflammation induced by repeated intranasal administration of papain, ILC2s-derived IL-13 was shown to be essential for initiating lung Th2 responses. Thus, the early production of IL-13 induced the migration of dendritic cells (DCs) from the lungs to the draining lymph nodes, an instrumental step for Th2 cell priming.128 Of note however, ILC2s-derived IL-13 was sufficient to initiate allergic airway hyper reactivity in the absence of T cells.129 ILC2-derived IL-13 could also promote memory Th2 responses through the induction of CCL17 expression by pulmonary DCs. Thus, transient depletion of ILC2s before rechallenge with allergens (papain or Alternaria alternata) resulted in drastic reduction of lung memory Th2 cells 130 days after priming (Fig. 2b). Altogether these observations point at a critical role for ILC2-derived IL-13 in the priming and recall of adaptive Th2 response during allergic inflammation through its action on lung DCs.130 In a model of chronic arthritis, ILC2s dampened inflammation via their production of IL-9. Autocrine production of IL-9 induced the expression of ICOSL and of glucocorticoid-induced TNFR-related protein ligand (GITRL) on ILC2s allowing their interactions with adjacent Tregs. IL-9 also activated Tregs and promoted their proliferation. Conversely, in a model of allergic lung inflammation, upregulation of GITRL in ILC2s played a pro-inflammatory role. GITRL signals through GITR, also expressed by lung ILC2s, induced ILC2 expansion and Th2 cytokine production in a T-cell-independent manner.131
ILC3s and T cells: cooperation between commensal bacteria, phagocytes and ILC3s might also promote the differentiation and the maintenance of intestinal Tregs. Thus, microbiota-induced production of IL-1β by macrophages was shown to boost IL-2 production by ILC3s, while disruption of the il2 gene in NCR1-expressing ILCs reduced the numbers of induced Tregs, overall suggesting that ILC3-derived IL-2 is instrumental for Tregs maintenance in the small intestine.74 A role of ILC3-derived GM-CSF in the maintenance of intestinal Tregs was also suggested.53 Production of cytokines by ILC3s might also promote Th17 cells differentiation. Thus, colonization of the small intestine by commensal segmented filamentous bacteria (SFB) was shown to induce strong innate and adaptive immune responses with ILC3s being an early source of IL-22, which promoted containment of the gut microbiota by stimulating the production of antimicrobial peptides by intestinal epithelial cells. IL-22-dependent activation of epithelial cells upon colonization by SFB induced the release of serum amyloid A (SAA1 and 2) which, in turn, favored the local differentiation of Th17 cells, thus revealing an indirect positive regulation of microbiota-specific adaptive immune responses by ILCs132 (Fig. 1a).
Postnatal regulation of lymph node architecture and function by adult LTi-like cells
LTi cells are instrumental in the orchestration of adaptive immune responses through their role in the development of secondary and tertiary lymphoid structures. Interactions between LTα1β2 expressed by LTi cells and LTβR expressed by stromal and lymphatic endothelial cells initiate the organogenesis of lymph nodes133 but also the maturation of isolated lymphoid follicles subsequently to colonization by commensal bacteria.134 Whether LTi cells retain a lymphoid tissue organizing function throughout life remains unclear. A few observations suggest that functional interactions between LTi cells and lymphoid tissue stromal cells take place long after birth. Adoptive transfer of fetal or adult LTi-like cells into LTα deficient hosts partially restored the B/T-cell zone segregation in the spleen of adult recipients while adoptively transferred splenocytes or DCs failed to do so. Restoration of the B/T-cell zone segregation correlated with the production of CCL21 and VCAM1 by stromal cells, both indicative of lymphoid tissue re-organization.135 Similarly, LTαβ+ LTi cells may contribute to restore the architecture of lymph nodes following their destruction by cytotoxic T cells during acute infection by LCMV. Thus, restoration of fibroblastic reticular cells in the T-cell zone was significantly delayed in BM chimeras lacking RORγt+ cells and upon LTβR blockade.136,137
Regulation of ILCs by T cells
At the steady state and in adult immunocompetent hosts, ILCs are rare tissue-resident populations with slow renewal. During the neonatal and perinatal periods when the adaptive immune system is still immature, ILCs are more abundant: they proliferate and display a high renewal capacity and an activated phenotype. Similar characteristics were observed in immunodeficient hosts, especially those lacking T cells.91,138,139 It is therefore reasonable to assume that once adaptive immune responses have reached maturity and achieved containment of potential infectious threats and tolerance towards innocuous antigens, ILC populations exert redundant functions, shrink and rest.139 However, the mechanisms through which adaptive immune responses complete this transition are still poorly understood.
Regulation of gut resident ILC3s by T cells
Korn et al. were the first to report the role of conventional CD4+ T cells in the control of IL-22 production by intestinal ILC3s.138 They observed increased expression of antimicrobial peptides and IL-22-producing ILC3 in the intestine of RAG-deficient mice compared to control mice and observed that these changes were corrected by adoptive transfer of CD4+ T cells.138 A few years later, Mao et al. confirmed these observations and reported that STAT3 phosphorylation was transiently induced in intestinal epithelial cells upon weaning in immune-competent hosts.139 While STAT3 phosphorylation remained high in adult RAG-deficient mice, it was lost upon treatment with broad-spectrum antibiotics or in a germ-free context. The authors further showed that STAT3 phosphorylation was the consequence of IL-22 production by ILC3s in response to microbiota-induced production of IL-23 by CCR2+ myeloid cells and that adoptive transfers of Th17 or Tregs could significantly reduce ILC3 activation. Th17 differentiation decreased microbial colonization in reconstituted immunodeficient hosts, while Tregs reduced the production of IL-23 by myeloid populations.139
Overall, these observations show that adaptive immune responses are crucial in regulating the activity of intestinal ILC3s through the containment of commensal bacteria and the suppression of pro-inflammatory signals emanating from myeloid cells.
Regulation of ILC2s by T cells
ILC2s express both ICOS and ICOSL in mice and humans.140,141 Interestingly, Tregs have been reported to suppress the activity of mouse ILC2s in response to intranasal administration of IL-33 in a TGFβ- and ICOS-dependent manner.137 Conversely, ICOS expression was shown to be necessary for lung ILC2 survival and function at steady state and upon induction of airway hyper reactivity by IL-33 intranasal administration.140 Therefore, ICOS–ICOSL interactions might either support ILC2 survival and function or suppress inflammatory cytokine production depending on the context. IL-2 derived from activated CD4+ T cells was also shown to support ILC2 survival and activation in vivo during parasitic infection75 and lung inflammation.119 In contrast, IFNγ could inhibit the survival of hepatic ILC2s activated by IL-33.142 These observations indicate that T-cell-derived cytokines participate in the regulation of ILC2 function and survival during immune challenges or tissue inflammation. Whether conventional T cells also participate in the control of ILC2 homeostasis at steady state has not been reported yet.
Concluding remarks
Many functional interactions between T cells and ILCs are taking place, but how and when remains difficult to determine. Major advances on ILC–T interactions will result from the determination of the highly coordinated sequence of cellular processes. Time-lapse movies achieved by intravital imaging should clarify where T cells and ILCs interact. ILCs are rare populations lacking lineage markers and their unambiguous identification using classical immunostaining and histology remains challenging. Few studies managed to track them in their local environment using Kaede mice143 but these approaches cannot be generalized to all tissues or experimental conditions as photo-conversion is transient and only a limited number of cells can be followed. Migratory subsets of ILCs were identified for each ILC subgroup with ILC1s being the most abundant subset to traffic continuously within the blood and lymph. Hence, circulating ILC subsets may support the priming of CD4+ T cells during immune responses.83 Interactions within the tissues in which ILCs reside remain poorly understood. Only rare studies explored the ILC microenvironment by imaging. Lung intravital microscopy studies were pioneers in patterning ILC2 migration and showing their highly dynamic properties during lung inflammation.144 3D microscopy was also used to identify advential stromal cells as a tissue niche for lung ILC2s.145 Development of long-term tracking Cre induced models as well as “rainbow” reporter mouse models should soon be available and may help uncovering the encounters between ILCs and adaptive immune cells. High-throughput RNA sequencing on human ILCs across different tissues revealed that contrary to mice, tissue environment does not determine transcriptional heterogeneity in human ILCs146 as strongly as it does in mice. Human ILC biology remains to be linked to its transcriptional signature. Spatial transcriptomics already used in diagnosis, especially in the cancer area, should be a precious technology for the analysis of human ILCs and their role in disease.147
References
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Weizman, O.-E. et al. ILC1 confer early host protection at initial sites of viral infection. Cell 171, 795–808.e12 (2017).
Klose, C. S. N. et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356 (2014).
Tang, L. et al. Differential phenotypic and functional properties of liver-resident NK cells and mucosal ILC1s. J. Autoimmun. 67, 29–35 (2016).
Artis, D. & Spits, H. The biology of innate lymphoid cells. Nature 517, 293 (2015).
Lane, P. J. L. et al. Lymphoid tissue inducer cells: bridges between the ancient innate and the modern adaptive immune systems. Mucosal Immunol. 2, 472–477 (2009).
Mjösberg, J. & Spits, H. Human innate lymphoid cells. J. Allergy Clin. Immunol. 138, 1265–1276 (2016).
Vély, F. et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 17, 1291 (2016).
Bernink, J. H. et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 14, 221–229 (2013).
Mjösberg, J. M. et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 12, 1055–1062 (2011).
Teunissen, M. B. M. et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR+ ILC3 in lesional skin and blood of psoriasis patients. J. Investig. Dermatol. 134, 2351–2360 (2014).
Ebbo, M., Crinier, A., Vély, F. & Vivier, E. Innate lymphoid cells: major players in inflammatory diseases. Nat. Rev. Immunol. 17, 665 (2017).
Ishizuka, I. E., Constantinides, M. G., Gudjonson, H. & Bendelac, A. The innate lymphoid cell precursor. Annu. Rev. Immunol. 34, 299–316 (2016).
Possot, C. et al. Notch signaling is necessary for adult, but not fetal, development of RORγt+ innate lymphoid cells. Nat. Immunol. 12, 949 (2011).
Cherrier, M., Sawa, S. & Eberl, G. Notch, Id2, and RORγt sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J. Exp. Med. 209, 729 (2012).
Harly, C., Cam, M., Kaye, J. & Bhandoola, A. Development and differentiation of early innate lymphoid progenitors. J. Exp. Med. 215, 249 (2018).
Rothenberg, E. V. Transcriptional control of early T and B cell developmental choices. Annu. Rev. Immunol. 32, 283–321 (2014).
Xu, W. et al. An Id2(RFP)-reporter mouse redefines innate lymphoid cell precursor potentials. Immunity 50, 1054–1068.e3 (2019).
Alhaj Hussen, K. et al. Molecular and functional characterization of lymphoid progenitor subsets reveals a bipartite architecture of human lymphopoiesis. Immunity 47, 680–696.e8 (2017).
Freud, A. G. et al. Evidence for discrete stages of human natural killer cell differentiation in vivo. J. Exp. Med. 203, 1033–1043 (2006).
Scoville, S. D. et al. A progenitor cell expressing transcription factor RORγt generates all human innate lymphoid cell subsets. Immunity 44, 1140–1150 (2016).
Scoville, S. D., Freud, A. G. & Caligiuri, M. A. Cellular pathways in the development of human and murine innate lymphoid cells. Curr. Opin. Immunol. 56, 100–106 (2019).
Chea, S. et al. Single-cell gene expression analyses reveal heterogeneous responsiveness of fetal innate lymphoid progenitors to notch signaling. Cell Rep. 14, 1500–1516 (2016).
Koga, S. et al. Peripheral PDGFRα+gp38+ mesenchymal cells support the differentiation of fetal liver–derived ILC2. J. Exp. Med. 215, 1609 (2018).
Gentek, R. et al. Modulation of signal strength switches notch from an inducer of T cells to an inducer of ILC2. Front. Immunol. 4, 334 (2013).
Lee, J. S. et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 13, 144 (2011).
Rankin, L. C. et al. The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat. Immunol. 14, 389 (2013).
Viant, C. et al. Transforming growth factor–β and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci. Signal. 9, ra46 (2016).
Chea, S. et al. Notch signaling in group 3 innate lymphoid cells modulates their plasticity. Sci. Signal. 9, ra45 (2016).
Yang, Q. et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat. Immunol. 16, 1044 (2015).
Harly, C. et al. The transcription factor TCF-1 enforces commitment to the innate lymphoid cell lineage. Nat. Immunol. https://doi.org/10.1038/s41590-019-0445-7 (2019).
Tindemans, I., Serafini, N., Di Santo, J. P. & Hendriks, R. W. GATA-3 function in innate and adaptive immunity. Immunity 41, 191–206 (2014).
Ting, C., Olson, M., Barton, K. & Leiden, J. Transcription factor GATA-3 is required for development of the T-cell lineage. Nature 384, 474–478 (1996).
Rahimpour, A. et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J. Exp. Med. 212, 1095–1108 (2015).
Constantinides, M. G., McDonald, B. D., Verhoef, P. A. & Bendelac, A. A committed precursor to innate lymphoid cells. Nature 508, 397 (2014).
Walker, J. A. et al. Polychromic reporter mice reveal unappreciated innate lymphoid cell progenitor heterogeneity and elusive ILC3 progenitors in bone marrow. Immunity 51, 104–118.e7 (2019).
Califano, D. et al. Transcription factor Bcl11b controls identity and function of mature type 2 innate lymphoid cells. Immunity 43, 354–368 (2015).
Walker, J. A. et al. Bcl11b is essential for group 2 innate lymphoid cell development. J. Exp. Med. 212, 875 (2015).
Yu, Y. et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J. Exp. Med. 212, 865 (2015).
Robinette, M. L. & Colonna, M. Immune modules shared by innate lymphoid cells and T cells. J. Allergy Clin. Immunol. 138, 1243–1251 (2016).
Szabo, S. J. et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000).
Djuretic, I. M. et al. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat. Immunol. 8, 145–153 (2007).
Zhu, J., Yamane, H., Cote-Sierra, J., Guo, L. & Paul, W. E. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 16, 3–10 (2006).
Ebihara, T. et al. Runx3 specifies lineage commitment of innate lymphoid cells. Nat. Immunol. 16, 1124–1133 (2015).
Agarwal, S. & Rao, A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 9, 765–775 (1998).
Ansel, K. M. et al. Deletion of a conserved Il4 silencer impairs T helper type 1–mediated immunity. Nat. Immunol. 5, 1251–1259 (2004).
Neill, D. R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Moro, K. et al. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature 463, 540–544 (2010).
Zhu, J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine 75, 14–24 (2015).
Liang, H.-E. et al. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat. Immunol. 13, 58–66 (2012).
Ye, P. et al. Requirement of interleukin 17 receptor signaling for lung Cxc chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194, 519–528 (2001).
Sonnenberg, G. F., Fouser, L. A. & Artis, D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 12, 383–390 (2011).
Mortha, A. et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343, 1249288–1249288 (2014).
Takatori, H. et al. Lymphoid tissue inducer–like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41 (2008).
Song, C. et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J. Exp. Med. 212, 1869 (2015).
Rankin, L. C. et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat. Immunol. 17, 179 (2015).
Sonnenberg, G. F., Monticelli, L. A., Elloso, M. M., Fouser, L. A. & Artis, D. CD4+ lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122–134 (2011).
Guo, X. et al. Innate lymphoid cells control early colonization resistance against intestinal pathogens through ID2-dependent regulation of the microbiota. Immunity 42, 731–743 (2015).
Wang, S. et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell 171, 201–216.e18 (2017).
Seehus, C. R. et al. Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat. Commun. 8, 1900–1900 (2017).
Bando, J. K. et al. ILC2s are the predominant source of intestinal ILC-derived IL-10. J. Exp. Med. 217, e20191520 (2019).
Hanash, A. M. et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 37, 339–350 (2012).
Lindemans, C. A. et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564 (2015).
Bruce, D. W. et al. Type 2 innate lymphoid cells treat and prevent acute gastrointestinal graft-versus-host disease. J. Clin. Investig. 127, 1813–1825 (2017).
Kim, G. Y., Hong, C. & Park, J.-H. Seeing is believing: illuminating the source of in vivo interleukin-7. Immune Netw. 11, 1–10 (2011).
Martin, C. E. et al. Interleukin-7 availability is maintained by a hematopoietic cytokine sink comprising innate lymphoid cells and T cells. Immunity 47, 171–182.e4 (2017).
Vonarbourg, C. et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to nk cell receptor-expressing RORγt+ innate lymphocytes. Immunity 33, 736–751 (2010).
Moro, K. et al. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature 463, 540 (2009).
Satoh-Takayama, N. et al. IL-7 and IL-15 independently program the differentiation of intestinal CD3−NKp46+ cell subsets from Id2-dependent precursors. J. Exp. Med. 207, 273 (2010).
Kennedy, M. K. et al. Reversible defects in natural killer and memory Cd8 T cell lineages in interleukin 15–deficient mice. J. Exp. Med. 191, 771 (2000).
Adachi, T. et al. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med. 21, 1272–1279 (2015).
Yeon, S.-M. et al. IL-7 plays a critical role for the homeostasis of allergen-specific memory CD4 T cells in the lung and airways. Sci. Rep. 7, 11155–11155 (2017).
Robinette, M. L. et al. IL-15 sustains IL-7R-independent ILC2 and ILC3 development. Nat. Commun. 8, 14601 (2017).
Zhou, L. et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409 (2019).
Oliphant, C. J. et al. MHCII-mediated dialog between Group 2 innate lymphoid cells and CD4+ T cells potentiates Type 2 immunity and promotes parasitic helminth expulsion. Immunity 41, 283–295 (2014).
Gasteiger, G., Hemmers, S., Bos, P. D., Sun, J. C. & Rudensky, A. Y. IL-2–dependent adaptive control of NK cell homeostasis. J. Exp. Med. 210, 1179 (2013).
Gasteiger, G., Fan, X., Dikiy, S., Lee, S. Y. & Rudensky, A. Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985 (2015).
Jenkins, M. K., Chu, H. H., McLachlan, J. B. & Moon, J. J. On the composition of the preimmune repertoire of T cells specific for peptide–major histocompatibility complex ligands. Annu. Rev. Immunol. 28, 275–294 (2010).
Huang, Y. et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 359, 114 (2018).
Ricardo-Gonzalez, R. R. et al. Tissue-specific pathways extrude activated ILC2s to disseminate type 2 immunity. J. Exp. Med. 217, e20191172 (2020).
Lim, A. I. et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell 168, 1086–1100.e10 (2017).
Bar-Ephraim, Y. E. et al. CD62L Is a functional and phenotypic marker for circulating innate lymphoid cell precursors. J. Immunol. https://doi.org/10.4049/jimmunol.1701153 (2018).
Dutton, E. E. et al. Peripheral lymph nodes contain migratory and resident innate lymphoid cell populations. Sci. Immunol. 4, eaau8082 (2019).
Mackley, E. C. et al. CCR7-dependent trafficking of RORγ+ ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat. Commun. 6, 5862–5862 (2015).
Kim, M. H., Taparowsky, E. J. & Kim, C. H. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity 43, 107–119 (2015).
Stier, M. T. et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J. Exp. Med. 215, 263–281 (2018).
Pearson, C. et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. eLife 5, e10066 (2016).
Schenkel, J. M. et al. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346, 98 (2014).
Bando, J. K., Liang, H.-E. & Locksley, R. M. Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat. Immunol. 16, 153 (2014).
Ishizuka, I. E. et al. Single-cell analysis defines the divergence between the innate lymphoid cell lineage and lymphoid tissue–inducer cell lineage. Nat. Immunol. 17, 269 (2016).
Sawa, S. et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 12, 320–326 (2011).
Gury-BenAri, M. et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell 166, 1231–1246.e13 (2016).
Nagasawa, M. et al. KLRG1 and NKp46 discriminate subpopulations of human CD117+CRTH2− ILCs biased toward ILC2 or ILC3. J. Exp. Med. 216, 1762 (2019).
Montaldo, E. et al. Human RORγt+CD34+ cells are lineage-specified progenitors of Group 3 RORγt+ innate lymphoid cells. Immunity 41, 988–1000 (2014).
Li, N. et al. Mass cytometry reveals innate lymphoid cell differentiation pathways in the human fetal intestine. J. Exp. Med. 215, 1383 (2018).
Sawa, S. et al. Lineage relationship analysis of RORγt+ innate lymphoid cells. Science 330, 665 (2010).
Bank, U. et al. Cutting edge: innate lymphoid cells suppress homeostatic T cell expansion in neonatal mice. J. Immunol. 196, 3532 (2016).
Schneider, C. et al. Tissue-resident Group 2 innate lymphoid cells differentiate by layered ontogeny and in situ perinatal priming. Immunity 50, 1425–1438.e5 (2019).
de Kleer, I. M. et al. Perinatal activation of the interleukin-33 pathway promotes Type 2 immunity in the developing lung. Immunity 45, 1285–1298 (2016).
Saluzzo, S. et al. First-breath-induced Type 2 pathways shape the lung immune environment. Cell Rep. 18, 1893–1905 (2017).
Youngblood, B., Hale, J. S. & Ahmed, R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology 139, 277–284 (2013).
Sun, J. C., Beilke, J. N. & Lanier, L. L. Adaptive immune features of natural killer cells. Nature 457, 557–561 (2009).
Nabekura, T. & Lanier, L. L. Antigen-specific expansion and differentiation of natural killer cells by alloantigen stimulation. J. Exp. Med. 211, 2455–2465 (2014).
Lee, J. et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 42, 431–442 (2015).
Rapp, M., Wiedemann, G. M. & Sun, J. C. Memory responses of innate lymphocytes and parallels with T cells. Semin. Immunopathol. 40, 343–355 (2018).
Lau, C. M. et al. Epigenetic control of innate and adaptive immune memory. Nat. Immunol. 19, 963–972 (2018).
Wang, X., Tian, Z. & Peng, H. Tissue-resident memory-like ILCs: innate counterparts of TRM cells. Protein Cell https://doi.org/10.1007/s13238-019-0647-7 (2019).
Weizman, O.-E. et al. Mouse cytomegalovirus-experienced ILC1s acquire a memory response dependent on the viral glycoprotein m12. Nat. Immunol. 20, 1004–1011 (2019).
Martinez-Gonzalez, I. et al. Allergen-experienced Group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity 45, 198–208 (2016).
Withers, D. R. Innate lymphoid cell regulation of adaptive immunity. Immunology 149, 123–130 (2016).
Sonnenberg, G. F. & Hepworth, M. R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 19, 599–613 (2019).
Hepworth, M. R. et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498, 113–117 (2013).
Hepworth, M. R. et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 348, 1031–1035 (2015).
Melo-Gonzalez, F. et al. Antigen-presenting ILC3 regulate T cell–dependent IgA responses to colonic mucosal bacteria. J. Exp. Med. 216, 728 (2019).
Goto, Y. et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 40, 594–607 (2014).
von Burg, N. et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc. Natl Acad. Sci. USA 111, 12835–12840 (2014).
Lehmann, F. M. et al. Microbiota-induced tissue signals regulate ILC3-mediated antigen presentation. Nat. Commun. 11, 1794 (2020).
Saez de Guinoa, J. et al. CD1d-mediated activation of group 3 innate lymphoid cells drives IL-22 production. EMBO Rep. 18, 39–47 (2017).
Mirchandani, A. S. et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J. Immunol. 192, 2442 (2014).
Hardman, C. S. et al. CD1a presentation of endogenous antigens by group 2 innate lymphoid cells. Sci. Immunol. 2, eaan5918 (2017).
Kim, M.-Y. et al. OX40 ligand and CD30 ligand are expressed on adult but not neonatal CD4+CD3− inducer cells: evidence that IL-7 signals regulate CD30 ligand but Not OX40 ligand expression. J. Immunol. 174, 6686 (2005).
Kim, M.-Y. et al. CD4+CD3− accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where T cells collaborate with B cells. Immunity 18, 643–654 (2003).
Withers, D. R. et al. Cutting edge: lymphoid tissue inducer cells maintain memory CD4 T cells within secondary lymphoid tissue. J. Immunol. 189, 2094 (2012).
Castellanos, J. G. et al. Microbiota-induced TNF-like ligand 1A drives Group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during colitis. Immunity 49, 1077–1089.e5 (2018).
Halim, T. Y. F. et al. Tissue-restricted adaptive Type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on Group 2 innate lymphoid cells. Immunity 48, 1195–1207.e6 (2018).
Fuchs, A. et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 38, 769–781 (2013).
Kwong, B. et al. T-bet-dependent NKp46+ innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat. Immunol. 18, 1117 (2017).
Halim, T. Y. F. et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014).
Barlow, J. L. et al. Innate IL-13–producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J. Allergy Clin. Immunol. 129, 191–198.e4 (2012).
Halim, T. Y. F. et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat. Immunol. 17, 57–64 (2016).
Nagashima, H. et al. GITR cosignal in ILC2s controls allergic lung inflammation. J. Allergy Clin. Immunol. 141, 1939–1943.e8 (2018).
Sano, T. et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163, 381–393 (2015).
van de Pavert, S. A. & Mebius, R. E. New insights into the development of lymphoid tissues. Nat. Rev. Immunol. 10, 664–674 (2010).
Tsuji, M. et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29, 261–271 (2008).
Kim, M.-Y. et al. Function of CD4+CD3− cells in relation to B- and T-zone stroma in spleen. Blood 109, 1602–1610 (2006).
Scandella, E. et al. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue–inducer cells with stroma of the T cell zone. Nat. Immunol. 9, 667 (2008).
Rigas, D. et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J. Allergy Clin. Immunol. 139, 1468–1477.e2 (2017).
Korn, L. L. et al. Conventional CD4+ T cells regulate IL-22-producing intestinal innate lymphoid cells. Mucosal Immunol. 7, 1045–1057 (2014).
Mao, K. et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 554, 255 (2018).
Maazi, H. et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity 42, 538–551 (2015).
Paclik, D., Stehle, C., Lahmann, A., Hutloff, A. & Romagnani, C. ICOS regulates the pool of group 2 innate lymphoid cells under homeostatic and inflammatory conditions in mice. Eur. J. Immunol. 45, 2766–2772 (2015).
Steinmann, S. et al. Hepatic ILC2 activity is regulated by liver inflammation-induced cytokines and effector CD4+ T cells. Sci. Rep. 10, 1071 (2020).
Tomura, M. et al. Monitoring cellular movement in vivo with photoconvertible fluorescence protein ‘Kaede’ transgenic mice. Proc. Natl Acad. Sci. 105, 10871 (2008).
Puttur, F. et al. Pulmonary environmental cues drive group 2 innate lymphoid cell dynamics in mice and humans. Sci. Immunol. 4, eaav7638 (2019).
Dahlgren, M. W. et al. Adventitial stromal cells define Group 2 innate lymphoid cell tissue niches. Immunity 50, 707–722.e6 (2019).
Yudanin, N. A. et al. Spatial and temporal mapping of human innate lymphoid cells reveals elements of tissue specificity. Immunity 50, 505–519.e4 (2019).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78 (2016).
Drake, L. Y., Iijima, K. & Kita, H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy 69, 1300–1307 (2014).
Acknowledgements
We thank Serge van de Pavert for critical discussions and for review of the paper. M.C. is supported by INSERM and Association François Aupetit dedicated to patients suffering from IBD. R.G. work is supported by Institut Pasteur, Institut National de la Santé et de la Recherche Médicale, Université de Paris, and ANR project NASHILCCD8 (#18-CE15-0024-01).
Author information
Authors and Affiliations
Contributions
M.C. and R.G. both wrote the paper. M.C. and G.R. designed the figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cherrier, M., Ramachandran, G. & Golub, R. The interplay between innate lymphoid cells and T cells. Mucosal Immunol 13, 732–742 (2020). https://doi.org/10.1038/s41385-020-0320-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-020-0320-8
