
Abstract
Protein kinases (PKs) have emerged as one of the most intensively investigated drug targets in current pharmacological research, with indications ranging from oncology to neurodegeneration. Tau protein hyperphosphorylation was the first pathological post-translational modification of tau protein described in Alzheimer’s disease (AD), highlighting the role of PKs in neurodegeneration. The therapeutic potential of protein kinase inhibitors (PKIs)) and protein phosphatase 2 A (PP2A) activators in AD has recently been explored in several preclinical and clinical studies with variable outcomes. Where a number of preclinical studies demonstrate a visible reduction in the levels of phospho-tau in transgenic tauopathy models, no reduction in neurofibrillary lesions is observed. Amongst the few PKIs and PP2A activators that progressed to clinical trials, most failed on the efficacy front, with only a few still unconfirmed and potential positive trends. This suggests that robust preclinical and clinical data is needed to unequivocally evaluate their efficacy. To this end, we take a systematic look at the results of preclinical and clinical studies of PKIs and PP2A activators, and the evidence they provide regarding the utility of this approach to evaluate the potential of targeting tau hyperphosphorylation as a disease modifying therapy.
Similar content being viewed by others

Introduction
Neurofibrillary pathology constitutes one of the two major histopathological hallmarks of Alzheimer’s disease (AD). The pathology is primarily composed of truncated and aberrantly hyperphosphorylated protein tau in the form of paired helical filaments (PHFs) or straight filaments (SFs) [1,2,3,4,5,6,7]. Remarkably, the density and stereotyped spatiotemporal distribution of this neurofibrillary pathology consistently correlate with the degree of cognitive decline, memory impairment, and brain atrophy [8,9,10,11,12,13]. Tau positron emission tomography (Tau-PET), and cerebrospinal fluid (CSF), and plasma biomarkers of tau further complement these findings [14,15,16,17,18]. On that account, tau pathology and subsequent neurofibrillary degeneration appear to play a leading role in the pathophysiology of AD.
Tau, recognized as an intrinsically disordered protein (IDP), undergoes various order-to-disorder or disorder-to-order transitions while retaining a flexible conformation. This flexibility is essential for its role in various cellular processes, such as regulation of microtubule (MT) dynamics, MT-mediated axonal transport, mRNA translation, cellular signalling, chromatin remodelling, neuroprotection, and neuronal development [19,20,21,22,23,24,25,26,27,28]. The monomeric conformational tau ensemble is modulated by a variety of factors, such as degradation processes, chaperone-mediated refolding, and several post-translational modifications (PTMs) [29,30,31,32]. In physiology, these modulations aid dynamic stability; in pathology, however, genetic mutations or dysregulation in these modulation results in weaker interactions between tau and its natural binding-partners, resulting in its accumulation. This creates conditions favourable for its unfolding, refolding, and misfolding into a tremendously large conformational ensemble that could potentially be capable of template-directed misfolding and aggregation [33, 34].
Phosphorylation of tau is one of the most actively investigated PTMs, with significant impact on solubility, localization, function, interaction with other proteins and susceptibility to additional PTMs [35, 36]. The longest of the ‘classic’ six human tau isoforms (tau40, 2N4R) encompasses ~85 potential serine (Ser), threonine (Thr), and tyrosine (Tyr) phosphosites [37], mainly localized in the proline-rich region (residues 172–251) and the C-terminal tail region (residues 368–441) [37]. Relatively few phosphosites, but important in the context of pathology, are also present in the microtubule-binding region (MTBR; residues 244–369) (Fig. 1) [38,39,40]. In healthy individuals, only two to three phosphate molecules were detected per molecule of tau; in AD, this stoichiometry is increased manifold. Further data also suggest phosphorylation to be sufficient for the induction of tau filament formation [41,42,43]. The characterization of tau filaments via electron cryo-microscopy (cryo-EM) has revealed the presence of unique conformational folds that are conserved among individuals with the same tauopathy [44, 45]. These conformational folds are reported to encompass unique site-specific phosphorylation signatures [46,47,48], further suggesting that there may be a causative link between dysregulation of tau phosphorylation/dephosphorylation and different tauopathies.

The phosphosites identified for AD pathology (in red), physiology (in green) and overlapping sites for both physiology and pathology (in purple) are shown here. Therapeutically targeted protein kinases (PKs) and protein phosphatase (PPs) are colour coded to their respective inhibitors or modulators explored clinically or preclinically.
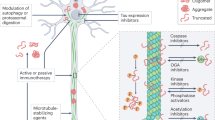
A well-grounded rationale for support modulating tau phosphorylation by targeting protein kinases (PK) or protein phosphatase 2A (PP2A) to counteract AD. Although preclinical efficacy studies provide promising data, most if not all the clinical trials testing PKIs in AD failed to demonstrate any efficacy in humans, with only a few still unconfirmed positive trends. Other tau-targeted therapies in AD include active/passive immunization to target pathological tau species, and a range of small molecules intended to either reduce tau expression, aggregation, and propagation, or promote MT stabilization and tau degradation [49,50,51]; whether they are efficacious has not been conclusively assessed yet.Therefore, the aim of this study is to identify whether inhibition of tau hyperphosphorylation is a promising strategy for the development of disease-modifying therapies (DMTs) against AD.
Protein kinases selected as a target for Alzheimer’s therapy
In AD, a concerted activity of several PKs is known to phosphorylate tau at nearly 40 AD-relevant epitopes [38, 39] (Table 1). PKs are stratified based on their proline-, non-proline -, or tyrosine-directed kinase activities.
Proline-Directed Protein Kinases (PDPKs) target phosphorylation at serine and threonine that precedes a proline residue (Ser/Thr-Pro motif). Glycogen synthase kinase-3 (GSK3), cyclin-dependent protein kinase-5 (CDK5), p38 mitogen-activated protein kinases (p38 MAPK) and Dual specificity tyrosine-phosphorylation regulated kinase 1A (DYRK1A) belong to this PDPK subclass.
GSK3, with over 100 substrates, is a ubiquitously expressed multifunctional enzyme [52]. It regulates a wide array of molecular pathways, including glycogen synthesis, WNT signalling, transcriptional network maintenance, apoptosis, and axonal MT remodelling [53,54,55,56,57,58,59]. Expressed as two isoforms, α and β [60], it demonstrates an unusual preference for target substrates that are pre-phosphorylated or “primed” at Ser/Thr-XXX-Ser/Thr motifs. Tau fulfils this requirement, having 24 such motifs suitable for priming, facilitated by the activity of PKA, CK1, CK2, MAPKs, or CDK5 [61,62,63]. Differing in kinetics and epitope preferences, both isoforms phosphorylate tau in vitro [64,65,66,67,68].
Cdk5 has been demonstrated to be one of the most functionally diverse kinases within neurons. Cdk5 is crucial for a number of cellular and developmental processes, including neuronal migration, synaptic plasticity, microtubule regulation, pain signalling, and apoptotic cell death in neuronal diseases [69,70,71,72,73]. The expression is abundant in all tissue types, but its highest expression with associated kinase activities are primarily detected in the CNS [74]. Cdk5 complexing with neuron-specific regulatory subunits p35 and p39 plays a role in physiological tau phosphorylation. Under pathological conditions, cellular stress induces calcium-dependent protease calpain cleavage of p35 and p39 into p25 and p29, and a p10 fragment respectively. This results in pathological tau hyperphosphorylation [75, 76]. Cdk5 is involved in priming for GSK3β; their crosstalk is reported to be dependent on ageing [77]. Together with GSK3β, CDK5 activity is central to AD pathophysiology [77].
P38 MAPKs, regulate various cellular functions such as metabolism, secretion, migration, differentiation, apoptosis, and senescence in response to various extracellular stimuli [78]. In mammals, p38 MAPK is expressed as four isoforms: α, β, γ and δ [79], which differ in their expression patterns in neurons, astrocytes, microglia, and endothelial cells. However, they serve non-redundant functions (based on substrate specificities and sensitivities) depending on cell type and context [80]. P38 plays a physiological role in tau phosphorylation; however, in AD pronounced alteration in its levels and distribution is observed. It is capable of phosphorylating tau protein in vitro in a manner similar to the hyperphosphorylation of PHF-tau [81]. The finding that PHF-tau co-immunoprecipitates with p38, and that p38 co-purifies with PHF-tau, strongly suggests that they are physically associated [82].
DYRK1A is a dual-specificity proline/arginine-directed kinase that possesses both Tyr and Ser/Thr kinase activities. It plays a key role in neurogenesis, neuronal trafficking, aging, and outgrowth of axons and dendrites. DYRK1A is expressed ubiquitously, with the highest expression being observed in the brain and heart [83]. DYRK1A regulates numerous cellular pathways, among them tau phosphorylation [84, 85].
Non-Proline-Directed Protein Kinases (Non-PDPK) group comprises kinases that phosphorylate tau at Ser/Thr-X motifs. MAP/microtubule affinity-regulating kinase 4 (MARK4) belongs to this subclass.
MARK4, belongs to AMP-activated protein kinases (AMPKs) subfamily of calcium/calmodulin dependent protein kinases (CAMK). It plays a central role in regulation of cell shape and polarity during differentiation, chromosome partition in mitosis, and intracellular transport by acting on microtubule-associated proteins (MAPs), including MAP2, MAP4 and tau [86, 87]. It is predominantly expressed in the brain. MARK4-mediated phosphorylation subsequently catalyses the detachment of tau from MTs to regulate the transition between stable and dynamic MTs [88, 89]. De novo mutation in MARK4 is associated with early onset of AD, indicating the role of MARK4 in the development of AD [90]. Synergistic activity of Cdk5 with MARK4 is reported to augment tau phosphorylation at Cdk5 specific sites, in addition to those for MARK4, suggesting that Cdk5 could directly or indirectly phosphorylate and activate MARK4, creating a feedback loop [91, 92].
Tyrosine protein kinase (TPK) phosphorylates tau at Tyr18, Tyr29, Tyr197, Tyr310 and Tyr394. Src family kinase (SFK) members such as Src, c-Abl, and Fyn belong to this subclass of PKs.
Src is a proto-oncoprotein [93] that is ubiquitously expressed with low tissue specificity. Src is largely localized to the plasma membrane and cell junctions, with minor presence in nucleoplasm and cytoplasm where it interacts with different classes of cell receptors involved in signalling pathways that control immune response, cell adhesion, cell cycle progression, apoptosis, migration, and transformation [94]. Src interacts with the proline rich sequence in the N-terminus of tau [95] where it can bind and phosphorylate Tyr18 [96, 97].
c-Abl is a proto-oncoprotein [98], alternative splicing of which results in two transcript variants, c-Abl-1a and c-Abl-1b [99, 100]. Its subcellular localization determines its function; cytoplasmic localization appears necessary for the transforming and cell survival functions whereas nuclear localization typically occurs in response to stress or overexpression and results in growth inhibitory functions, including cell cycle arrest and apoptosis [101, 102]. c-Abl is shown to co-immunoprecipitate with and directly phosphorylate tau [103] and modulate tau through activating Cdk5 by phosphorylating at Tyr15, as seen in AD models [104, 105].
Fyn is a non‐receptor or cytoplasmic TPK that mediates multiple transduction pathways in the central nervous system (CNS) including synaptic transmission, myelination, axon guidance, and oligodendrocyte formation. It is ubiquitously expressed with its highest expression seen in the CNS and lymphoid tissue. Levels of Fyn are reported to alter in AD [106]. Besides its commonly reported indirection associated with tau hyperphosphorylation, Fyn is shown to physically interact with tau and phosphorylate it at Tyr18 [107].
Rho-associated kinase (ROCK) belongs to the AGC (PKA/PKG/PKC) family of Ser-Thr specific PKs. It is regulated by the small GTPase RhoA and integrates activating signals from surface receptors, leading to both cell proliferation and migration [108]. RhoA/ROCK signalling pathway activation appears to affect a range of processes involved in the pathogenesis of AD, including tau hyperphosphorylation, Aβ aggregation, neuroinflammation, and synaptic damage, promoting neurodegeneration [109,110,111,112]. ROCK phosphorylates tau at Thr245, Thr377, Ser409 and to some extent Ser262 [113].
Protein kinase inhibitors evaluated in AD
A large majority of PKIs investigated in AD with the intent of disease modification are designed/repurposed to target PKs to impede their enzymatic activity and thus reduce tau hyperphosphorylation. Here, we focus the discussion on those drugs that underwent clinical trials and briefly mention the ones in preclinical studies with the prime intention of targeting tau hyperphosphorylation.
Saracatinib (AZD0530) is a member of a large class of biologically active compounds called quinazolines, where quinazoline is substituted by (5-chloro-2H-1,3-benzodioxol-4-yl)amino, (oxan-4-yl)oxy and 2-(4-methylpiperazin-1-yl)ethoxy groups at positions 4, 5 and 7, respectively. It is a highly potent, orally available, small molecule that selectively inhibits the Src (Src, Fyn, Yes, and Lyn) and Abl families of tyrosine kinases [114, 115]. The compound was originally developed by Astra Zeneca as a treatment for various oncological disorders. Despite showing promising results in preclinical studies and a favourable safety profile, it was withdrawn in Phase 2 due to lack of efficacy. Clinical exploration of saracatinib in AD up to Phase 2a was conducted with the rationale to target Fyn kinase mediated Aβ toxicity. A separate line of evidence linked Fyn to tau; suggesting that the phosphorylation of tau at Tyr-18 was associated with the interaction of Fyn [107].
Lithium (Li+) salts are used for treating and preventing psychiatric disorders, primarily relapses in both type I and type II bipolar disorders (BDs), suicidal behaviours during major depressive disorders, and schizophrenic disorders. Li+ inhibits Mg2+-dependent enzymes by displacing Mg2+ from their binding sites within specific catalytic protein domains [116], thereby reducing the stability and activity of enzymes including GSK3 relevant both for neuropsychiatric and neurodegenerative disorders [117, 118]. Thus Li+ salts, such as lithium sulphate, lithium carbonate, and lithium chloride sparked substantial attention as a treatment possibility in AD. Chronic lithium treatment was shown to affect numerous tau kinases, with different biological effects depending on the concentration range and regional specificity [119]. However, the putative mechanism of action of lithium remains unknown.
Nilotinib (AMN107), an aminopyrimidine-derivative, is FDA approved drug for the treatment of Philadelphia chromosome positive chronic myeloid leukemia (CML). It is an orally available, selective, ATP competitive, reversible inhibitor of BCR/Abl tyrosine kinase with antineoplastic activity [120]. It was initially developed by Novartis Pharmaceuticals against imatinib-resistant mutants of BCR/Abl protein [121]. Low doses of nilotinib were further shown to penetrate the BBB, and reduce CSF tau, independent of Abl inhibition [122, 123]. The rationale to proceed for clinical use of nilotinib against AD was apparently based on the neuroprotective effects [124] and reduction of inflammation in non-tauopathic cellular and animal models [125, 126]. However, the availability of preclinical data that would show nilotinib counteracting tau hyperphosphorylation is limited, with such studies either not done or not reported.
Tideglusib (NP031112/NP12) belongs to the thiadiazolidine class of compounds; it’s composed of 1,2,4-thiadiazolidine-3,5-dione with a naphthalen-1-yl group at position 2 and a benzyl group at position 4. It is an orally active, potent, selective, irreversible, and non-ATP competitive small-molecule inhibitor of GSK3β developed against AD by the Zeltia group [127]. It’s considered a tau-protein kinase inhibitor with neuroprotective and anti-inflammatory effects [128, 129]. To the best of our knowledge there is no available in vitro data and merely a single in vivo study [130] showed clear reduction in tau hyperphosphorylation; several studies provide evidence of neuroprotection, and mitigating inflammation though [131].
PKIs tested in preclinical studies. Several potentially interesting compounds were tested for efficacy in both in vitro and in vivo studies. The compounds including SAR502250 [132], AR-A014418 [133, 134], MMBO [135], (R)-rescovitine [136], MW181 [137], SB239063 [137], bosutinib [138], SM07883 [139], Dyrk1-inh [140], DYR219 [141], BAY61-3606 [142] and Fasudil [143] are yet to be tested at clinical front for AD, whereas, lithium, tideglusib, nilotinib, and dl-3-n-butylphthalide have already made a headway to clinical trials.
Involvement of protein phosphatase 2A (PP2A) in tau hyperphosphorylation
Protein Ser/Thr phosphatases comprise three major families: phosphoprotein phosphatases (PPPs), metal-dependent protein phosphatases (PPMs), and the aspartate-based phosphatases. Protein phosphatase (PP)1, PP2A, PP2B, and PP5 are PPPs that were found to dephosphorylate tau in vitro at various phosphorylation sites [144]. Among them, PP2A was shown to be the major phosphatase in the brain [145, 146] and accounts for >70% of total tau phosphatase activity [145]. Activity and/or expression of PP1, PP2A, and PP5 are decreased [145, 147,148,149,150], whereas PP2B truncation and activity are increased in AD brains [151].
PP2A regulates numerous signalling pathways, playing an important role in development, cell proliferation and death, cell mobility, cytoskeleton dynamics, and the control of the cell cycle [152]. PP2A is one of the most abundant enzymes, accounting for up to 1% of total cellular protein mass [153]. Its function is regulated by several endogenous proteins. PP2A dephosphorylates tau at almost all phosphorylation sites [145]. Among them, pThr205, pThr212, pSer262, and pSer409 are most conductive to PP2A activity [145]. The best characterized inhibitors of PP2A are PP2A-inhibitor 1 (I1PP2A, also known as Acidic Nuclear Phosphoprotein 32 A), PP2A-inhibitor 2 (I2PP2A, also known as SET), and CIP2A (cancerous inhibitor of PP2A).
PP2A activity is significantly decreased in the AD cortex and hippocampus [145, 147, 150]. The level of tau hyperphosphorylation at many sites is negatively correlated with PP2A activity [151]. Up-regulation of I1PP2A and I2PP2A, and mislocalization and cleavage of I2PP2A could underlie the inactivation of PP2A in neocortical neurons in AD [154]. Increased expression of CIP2A in the AD brain leads to tau mislocalization to dendrites and spines, and to synaptic degeneration [155]. Collectively, these studies directly point to a central role of PP2A dysfunction in AD pathogenesis, identifying PP2A as a druggable target of therapeutic interest [154, 156, 157]. Due to the pivotal cellular role of PP2A and its broad range of substrates, indiscriminately activating PP2A with potent compounds risks toxicity, though.
A comprehensive literature search was performed to identify pertinent in vivo efficacy studies utilizing PP2A activators such as antineoplastic cancer drug sodium selenate [158,159,160,161]; the anti-diabetic drug, glimepiride [162] and metformin [163,164,165]; a zinc ionophore and an experimental drug candidate, copper/zinc chaperone (PBT2) [166]; anti-depressant, S-adenosylmethionine [167,168,169]; anti-microbial clioquinol (an intracellular zinc chelator) [170] an immunomodulating medication, fingolimod [171]; and caffeine [172], demonstrated substantial reduction of tau phosphorylation, insoluble tau, and tangle load, along with the alleviation of cognitive/behavioural phenotypes (Supplementary Table S1). Even though the pharmacological modulation of tau pathology is evident from these efficacy studies, the precise mechanism of action (MOA) of most of these multi-targeted drugs remains elusive. For instance, the PP2A stimulator metformin can target an upstream regulatory kinase, AMPK, that can inhibit GSK3β, and thus the observed effects cannot be evenly segregated [173, 174]. Among the aforementioned PP2A activators, only sodium selenate stated PP2A activation as its MOA in clinical trials.
Efficacy studies on selected kinase inhibitors
To evaluate in vivo efficacy of putative PKIs a wide range of preclinical studies has utilized aged, amyloid-beta (Aβ), Down syndrome, TMT chloride, colchicine, scopolamine, streptozotocin, anaesthesia, hypothermia, or hypoxia/stroke rodent models of AD [173,174,175,176,177,178]. Whereas on one hand, some of these models develop hyperphosphorylation, amyloid plaques (some transgenic models), and inflammation-induced synaptic dysfunction leading to visible behavioural deficits, they lack the targeted pathological hallmarks, i.e., NFTs. As a result, their relevance and translational value remain unknown.
We performed a comprehensive literature search to identify studies investigating the effects of PKIs exclusively in rodent tauopathy models (Tables 2). We collected pertinent publications from three independent databases: PubMed, Web of Science, and Google Scholar. We used “Alzheimer’s disease” AND “protein kinase inhibitors” as key words for the literature search. The search was restricted to English language and not by date. The inclusion criteria included: transgenic rodent models expressing mutated, truncated, or wild-type tau; original experimental studies; studies demonstrating target engagement, i.e., behavioural and biochemical/histological studies to demonstrate reduction in tau hyperphosphorylation. Exclusion criteria comprised duplicated references; review articles; literature with incorrect or incomplete data; absence of behavioural and biochemical/histological evaluation of tau phosphorylation.
The 17 shortlisted studies targeted GSK3β (via SAR502250 [132]; tideglusib [130]; lithium chloride [133, 179, 180]; AR-A014418 [133]; MMBO [135]); CDK-5 (via ®-roscovitine [136]), p38MAPK (via MW181; SB239063 [137]); c-Abl and Src (via nilotinib [138]; bosutinib [138]), Fyn (AZD0530 [181]), DYRK1A (via Dryk1-inh [140]; DYR219 [141]; SM07883 [139]), MARK4 (dl-NBP [182]), SYK (BAY61 [142]) and Rho-kinase (Fasudil) [143] inhibition, which resulted in cumulative reduction of tau hyperphosphorylation and alleviation of the AD cognitive/behavioural phenotype.
The studies were performed utilizing transgenic mouse models: 3xTg-AD, JNPL3 (P301L), PS19 (P301S), APPsw-Tauvlw, PrP T44 Tau, and hTau and rTG4510. The commonly used triple mutant 3xTg-AD mouse model contains two mutations associated with familial AD, APP (KM670/671NL; Swedish), and PSEN1 (M146V), and one with fronto-temporal dementia (FTD), MAPT (P301L) [183]. The double-transgenic APPsw-Tauvlw mice over-express human mutant APP (KM670/671NL; Swedish) and triple mutant human MAPT (G272V, P301L, R406W) mimicking several features of the AD phenotype to a remarkable extent [184]. However, these combinations of mutations do not actually exist in AD. The JNPL3(P301L) mouse model expresses mutant human MAPT (4 R/0 N; P301L), where the hemizygotes express human tau at levels comparable to endogenous murine tau and homozygotes at approximately twice the endogenous levels [185]. The PS19 mouse has P301S tau (4 R/1 N) mutant overexpressed fivefold higher than that of endogenous tau. It develops tangles in the neocortex, amygdala, hippocampus, brain stem and spinal cord at 6 months, with progressive accumulation thereafter [186]. The rTg4510 mice express mutant human MAPT (4 R/0 N; P301L) downstream of a tetracycline operon–responsive element (TRE), to an activator line expressing a tetracycline-controlled transactivator under control of the CaMKIIα promoter [187]. The expression is largely restricted to the forebrain and can be inactivated by administration of the tetracycline analogue doxycycline [188]. These mutations are not present in AD, though.
To address these limitations, models such as PrP T44 Tau, and hTau mice were used; PrP T44 Tau mice express the shortest human tau isoform (T44, also known as fetal tau) [189] and hTau mice express all six classic human tau isoforms with endogenous murine tau knocked out [190]. Among these, the 3xTg AD model shows tau pathology in the hippocampus, particularly pyramidal neurons, where JNPL3(P301L) homozygous and heterozygous mice and P301S mice develop neuronal inclusions reminiscent of tangles and Pick-bodies by 4.5 and 6, and 6 months respectively [185, 186]. APPsw-Tauvlw, PrP T44 Tau, hTau, and rTG4510 mice line develop pathology in various brain areas: APPsw-Tauvlw model manifests neuritic plaques [184], PrPT44 model develops tau-rich filamentous inclusions in cortical and brainstem neurons at 6 months [189], and hTau model develop aggregates and PHF detectable at 9 months [190] and rTG4510 accumulate an early burden of tau pathology in the form of argyrophilic inclusions, with tangles being observed in the cortex by 4 months and in the hippocampus by 5.5 months [187, 188]. Tau inclusions are characterized by Gallyas silver staining, Bielschowsky silver staining, Thioflavin S, or Congo red and presence of sarkosyl-insoluble tau. Neurofibrillary lesions correspond to motor and/or cognitive decline, associated neurodegeneration and neuroinflammation [191].
The efficacy of PKI-treatment was examined in the above studies at a behavioural, biochemical, and histological/immunochemical level, with methods tailored to the impairment observed in individual models. With respect to the behavioural aspect, the cognitive functions such as learning and memory were assessed using Morris water maze (MWM), radial arm water maze (RAWM), novel object recognition test (NORT), open field test (OFT), fear conditioning test (FCT), T-/Y-maze, or Barnes maze (BM). Motor functions and deficits were assessed using wire-hang test, whereas to assess anxiety and depression-like behaviour, forced-swim or tail suspension tests were used. Improvements in cognitive functions of learning and memory were assessed by improved escape latency in the MWM for tideglusib [130], Dyrk1-inh [140], and partially dl-NBP with increased freezing time in FCT [182], improved performance on NORT for SAR502250 [132] and MMBO [135], improvement in hippocampal dependent spatial working memory deficits in Y-maze test for MW181 and SB239063 [137]. Motor functions demonstrated improvement in clasping phenotype in the tail suspension test for LiCl [133, 179, 180]. Anxiety and depression-like behaviour showed effective improvement in the wire suspension test for SM07883 [139]. In the remaining studies, the animals were not subjected to behavioural testing.
In biochemical, histological and immunochemical analysis, almost all inhibitors demonstrated dose-dependent or independent decrease in hyperphosphorylation of tau at sites that were specific for the inhibited kinases, which is indicative of the pharmacodynamic action of these inhibitors directly on tau. For LiCl [180] and SB239063 [137] biochemical analysis were not performed, thus their pharmacodynamic effect on tau hyperphosphorylation remains unknown. Altogether, up to 13 phospho-sites were analysed in these preclinical efficacy studies (Ser199, Thr181, Ser262, Ser396, Ser202, Thr205, Thr212, Ser214, Thr231, Ser235, Ser396, Ser404 and Ser422). It is worth noticing that although tau has ~85 putative phospho-sites and a large portion of them (>40 sites) are found to be hyperphosphorylated in AD, of which each kinase targets a handful of sites that it can phosphorylate, the choice of analysed phosphor-sites (as low as one or two in some studies [132, 141]) limits the interpretability of these results. In addition, the lack of consideration of the PK in question and their respective p-sites was also apparent in some studies.
As tau hyperphosphorylation is hypothesised to lead to aggregation, confirming the reduction of detergent-insoluble tau is of utmost importance; it is imperative that studies show a clear impact of kinase inhibition on the reduction of oligomeric and filamentous tau, as mere tau hyperphosphorylation could be a physiologically protective phenomenon. In this context, LiCl [180], MW181 and SB239063 [137], SM07883 [139], and Fasudil [143] demonstrate significant reduction in levels of sarcosyl-insoluble tau. Meanwhile, with Tideglusib [130], the levels in the selected age groups were pointed out to be too low to be reliably quantified [192, 193]. BAY-61 [140, 142] and AZD0530 [181] were also demonstrated to reduce detergent and tris-buffered saline (TBS) insoluble fractions. In the remaining studies, although the data from immunoblots was consistent with immunohistochemical staining, demonstrating efficient reduction in tau hyperphosphorylation [137, 180], yet the quantification of insoluble fractions was not performed. In the process of evaluating the efficacy of the compounds in selected preclinical studies, a critical limitation remained their inability to assess the effect of the studied compounds on the tangle load (i.e., mature tau pathology) which represents a likely prerequisite for successful therapy of AD and related tauopathies. It may be overly ambitious to require a treatment to remove existing neurofibrillary pathology, but compounds should be able to reduce the accumulation of new neurofibrillary lesions.
From a methodological point of view, many studies are either flawed, or do not report on key aspects such as methods to counteract bias. Sample size calculations are missing across all studies. Randomisation is reported for a few studies [137, 139, 180,181,182], however, the method of randomisation is not reported. Blinding of the experimenters to treatment groups, a key step in preventing bias, was limited to a few studies [137, 138, 140, 143, 180]. Sex proportionality is maintained at large [130, 133, 136,137,138,139, 142, 179, 180, 182].
To conclude, future studies should most importantly assess the effect of putative therapies based on their effect on neurofibrillary pathology load as a primary outcome measure, and incorporate important design elements [194] such as effect size estimates and sample size calculation, the method of randomisation and allocation concealment, blinding, sex proportionality, exclusion and inclusion criteria (weight, health of animals…), in order to increase their rigour, transparency, and probability of succeeding in clinical development.
Clinical development of protein kinase inhibitors and phosphatase modulators for Alzheimer’s disease
We have evaluated the available data from clinical trials for kinase inhibitors, with special emphasis on efficacy and safety of PK-targeted therapeutics for AD, and the druggability of targeted PKs. To this end, we conducted a comprehensive search through ClinicalTrials.gov, the Cochrane Central Register of Controlled Trials (CENTRAL), and the EU clinical trial register; and queried to identify Phase I/II/III/IV clinical trials testing PKIs against conditions mentioned as mild to moderate Alzheimer’s disease and/or Alzheimer’s disease in addition to mild cognitive impairment (MCI). Within our selected time frame, as of 1st July 2022, 26 clinical trials were completed, suspended, terminated, or withdrawn; these trials targeted only a handful of kinases. Only 10 trials tested direct effects on tau hyperphosphorylation and had changes in Tau-PET or plasma/CSF biomarker assessment of tau as their primary or secondary measures. These trials tested lithium, tideglusib, saracatinib and nilotinib. Notably, other potentially interesting kinase inhibitors, such as neflamapimod and valproate, were never evaluated in a study suitable to assess their impact on tau hyperphosphorylation in AD. All identified trials were interventional, randomised, placebo controlled, with parallel arm design, and were double-, triple, or quadruple-blind Table 3.
GSK3β was targeted using tideglusib, salts of lithium and valproate. Tideglusib (NP-031112), an orally available drug with a thiadiazolidinone scaffold underwent two clinical trials. The pilot phase I/IIa trial (NCT00948259, N = 30, 4 & 6 weeks), was primarily designed to assess safety and tolerability. The drug was administered once daily (q.d.) at an escalating dose of 400, 600, 800, 1000 mg for 4, 4, 6, and 6 weeks respectively, in mild to moderate AD subjects. The overall safety findings in the study indicated that tideglusib can be administered for weeks/months if serum transaminase levels are closely monitored; due to the pronounced hepatotoxicity only 50% of the subjects continued the treatment though. Although there was no statistically significant evidence for efficacy, a difference of 4.7 points in the ADAS-cog+ and 1.7 points in the MMSE in favour of tideglusib was observed in subjects who could be escalated to 1000 mg [195]. A subsequent trial (NCT01350362, N = 306, 26w), called ARGO, tested daily doses of 500 mg and two different regimes of once a day (q.d.) and once every other day (q.o.d.) of 1000 mg; both doses demonstrated a reasonable safe profile, except 14–18% of subjects experienced diarrhoea, and 8.9–16% exhibited elevated transaminase levels. However, neither any significant long-term differences nor definite trends in any efficacy variable (ADAS-cog11, MMSE, Word Fluency test, ADCS-ADL, NPI, EQ-5D, and QUI) were observed, suggesting a lack of efficacy on the clinical front. In addition, CSF levels of Aβ1-42, total tau, pT181-tau and pS396-tau also remained unchanged, putting the expected pharmacological actions of tideglusib as a GSK-3β inhibitor in question. A significant reduction in BACE1 in the CSF of the treatment group was observed, but whether this had an effect on BACE1-mediated cleavage of amyloid precursor protein (APP) is uncertain. Overall, a non-significant trend was observed in the active treatment arm compared to placebo in the levels pT181-tau but the study was underpowered both to address the question of target-engagement of tideglusib, and to assess clinical efficacy [196]. Subsequently, tideglusib showed negative results in a trial in PSP [197].
Lithium is currently used as a mood stabiliser. A total of five completed phase II clinical trials in AD and one ongoing trial are on the record with lithium salts. Of these, only three have published their rather inconclusive results. Two short-term Phase II pilot clinical trials conducted to examine the acute effect of lithium on neuroprotective activity (ISRCTN2046462, N = 71, 10w) & (NCT00088387, N = 35, 6w) demonstrated no significant improvement in cognitive or neuropsychiatric measures (MMSE, ADAS-Cog score, NPI). The effect on GSK3β activity or concentration of blood or CSF derived biomarkers such as Aβ1-42, pT181-tau, pT231-tau and total tau were also not significant [198]. A subsequent open label trial (GO300913, N = 22, 1 y) with lithium carbonate (serum levels 0.3–0.8 mmol/l) produced no significant improvement on the MMSE in elderly individuals with mild to moderate AD. Lithium was deemed safe despite a high discontinuation rate, as the side effects were mild and reversible [199]. All these studies failed to find a positive effect of lithium on cognition, CSF p-tau and other AD-related blood or CSF biomarkers, conceivably because of small samples, short observation periods that were insufficient to detect any impact of neuroprotective agents, and the inclusion of subjects in more advanced stages of cognitive deterioration, i.e., mild, and moderate AD. In contrast, a later study (NCT01055392, N = 80, 2 y) sought to address the methodological aspects of trials on disease-modification in AD. It recruited patients with prodromal AD, i.e., amnestic mild cognitive impairment (aMCI) and administered a low dose (150 mg) of lithium. The statistical analysis performed by the authors doesn’t allow to assess whether treatment slowed progression over time; however, authors report a non-significant trend towards decrease of CSF p-tau (Fisher’s, P = 0.2) [200]. Another trial (NCT02129348, N = 77, 12 w) aiming to evaluate the efficacy and side-effects of low dose (150–600 mg) of lithium to treat agitation in AD subjects reported an excellent safety profile. GCI behaviour change on lithium (31.6%; n = 12) was observed to be similar to placebo (20.5% [n = 8]; X2(1) = 0.72; P = 0.40); however, there was moderate or marked improvement (CGI) in the lithium group (10/38 = 36.8%) compared to placebo (0/39 = 0%, Fisher’s exact test p < 0.001) [201, 202]. An actively recruiting Phase IV clinical trial LATTICE (NCT03185208, N = 80, 2 y) involving lithium carbonate as a possible treatment to prevent cognitive impairment in the elderly may further elucidate the effect of long-term lithium administration on CSF p-tau.
Saracatinib and nilotinib are small molecule inhibitors of Fyn and Bcr-Abl, respectively. In phase Ib and II clinical trials (NCT01864655, N = 24, 4 w & NCT02167256, N = 159, 52 w) saracatinib was found to be reasonably safe and well tolerated in participants with mild AD, but neither showed any statistically significant treatment differences for change in either CSF total tau or p-tau, nor impact on any other primary or secondary outcomes (cognition, brain glucose metabolism) over the course of treatment [203, 204]. Nilotinib’s phase II trial (NCT02947893, N = 42, 26 w) showed only an effect on CSF amyloid, CNS amyloid in the frontal cortex (but not temporal), and the level of the dopamine metabolite homovanillic acid (HVA) compared to the placebo group; no effects on tau were observed in the comparison between treatment arms. The major limitation of this study remains the lack of multiplicity adjustment, and other flaws in statistical analysis [205]. An active large-scale Phase III trial of nilotinib (NCT05143528, N = 1275, 5 y) is currently recruiting participants with early AD, who will be randomly assigned to receive 1 of 2 different doses (84 and 112 mg) of nilotinib or placebo for 72 weeks. A biomarker sub-study will investigate the effects of drug on the amyloid brain burden as well as tau and other markers of AD pathology.
Sodium selenate is the first drug targeting PP2A in clinical trials in AD and related disorders. A Phase IIa clinical trial compared sodium selenate (VEL015) to placebo or low-dose selenate in 40 subjects with mild–moderate AD over 24 weeks [206]. Sodium selenate was found to be generally safe, with minor side effects of headache, fatigue and nausea. No significant differences in cognitive measures between groups over the treatment period were observed, though. Additional exploratory diffusion-weighted MRI endpoints found less degeneration in the white matter of patients treated with sodium selenate than placebo [206]. Furthermore, a pooled analysis of all subjects showed that the patients who had higher selenium levels in their blood and cerebrospinal fluid (CSF) showed less cognitive decline than those with lower selenium levels [207]. The open-label extension study was not conclusive. A new phase II trial on sodium selenate in behavioural variant of FTD will use higher doses (15 mg taken three times daily) in a cohort of 120 patients [208, 209]. This trial is currently in recruitment and is expected to be completed by 2024. In summary, reduced phosphatase activity is likely responsible for a part of tau hyperphosphorylation. Due to the paucity of clinical evidence, it is too early to say whether enhancing phosphatase activity can yields an efficacious treatment.
It is important to note that published preclinical studies did not bring conclusive results on therapeutic effect on prevention of tangle formation or diminishing of tangle load by inhibiting tau kinases in transgenic animal models. Therefore, it is important to address this point in the clinical development by using suitable biomarker assessment as prescribed by the amyloid/tau/neurodegeneration (ATN) classification [210]. None of the above studies have a complete A/T/N classification for the whole patient cohort, which is particularly peculiar for e.g., the most recent study of nilotinib, which screens all patients for amyloid, yet does not appear to confirm tau pathology for inclusion. Recent results show that even mild AD dementia cohorts that are amyloid-positive can be tau-negative in ~30% of cases, hampering the efficacy readouts of tau-targeted compounds [211]. Currently, whether kinase inhibitors can efficiently halt the clinical progression of the disease via preventing the tangle formation cannot be conclusively stated. Most of the studies were not designed to answer questions of efficacy—the sample sizes were not sufficient to evaluate clinical endpoints with any degree of rigor. Studies on the various immunotherapeutic approaches, such as monoclonal antibodies, universally arrive at the conclusion that sample sizes of several hundred subjects are necessary to evaluate clinical efficacy of disease-modifying therapies for AD [212, 213]. Sample sizes required for biomarker assessments are usually smaller, yet many of the studies conducted on PKI were likely underpowered even in this aspect. The duration of observation in most of the discussed trials was also insufficient. A disease-modifying therapy, much unlike symptomatic treatments (e.g., acetylcholinesterase inhibitors), will not manifest its effects in a few months’ time. Even if a therapy stopped all neurodegeneration caused by tau pathology on day one of treatment, this would not become readily apparent until a concurrently randomised placebo control group declines. The reasons are manifold—a part of decline is driven by various common comorbidities such as vascular, TDP43 [214], or alpha-synuclein pathology [215]; the cognitive performance of patients varies on a day-to-day basis [216]; the clinical assessment tools are not perfect [217]; the margin of error of biomarker assessment methods is frequently close to the mean annual change on various disease markers such as hippocampal volume [218]; placebo groups often don’t decline right away. In fact, of the above, solely NCT01055392 with its 24-months had a duration usual for studies of potentially disease-modifying compounds [219]; the 36-moth duration chosen for the most recent nilotinib trial is also appropriate for an early AD population. As for populations selected for evaluation of these compounds, only a few confirmed the presence of tau pathology via CSF biomarkers or tau PET at enrolment, thus the question remains: what proportion of these subjects actually had tau pathology as their dominant neurodegenerative component? Finally, it must be mentioned that several of the studies had critical flaws in methodology, including incorrect statistical evaluation of the results, or claims of blinding where the study in essence was unblinded.
Future drug development of tau hyperphosphorylation modulators
Further clinical studies are warranted to show whether this therapeutic approach is able to impact the clinical course of AD and non-AD tauopathies. To achieve this, numerous improvements in the design and methodology of these studies are quintessential. As for study populations, it should be obvious that to evaluate treatments that aim to impact tau hyperphosphorylation, one should select subjects whose neurodegeneration is primarily driven by tau pathology. Thus, inclusion criteria for trials should include a biomarker assessment that confirms this—CSF and/ tau and phospho-tau markers, tau PET, or blood tau markers. Markers of tau pathology and neurodegeneration should be evaluated longitudinally over a sufficient time frame. Markers that reflect current neurodegeneration intensity (e.g., CSF tau) can be expected to respond sooner than markers that reflect disease progression (e.g., tau PET); with the latter, expecting a small molecule to clear tau deposits present at baseline may be too ambitious, and expecting the compound to slow tangle accumulation may be more realistic.
When calculating sample sizes, especially for clinical endpoints one should consider a) that even if a compound halts the disease in its tracks, an improvement above baseline is not to be expected with disease-modifying compounds, b) even excellent drugs should not be expected to prevent all of the clinical decline in subjects. A slowing of the disease by 25% should be considered a success. Meanwhile, the design of many of the reviewed trials (sample size, observation period) indicates that investigators were expecting the subjects to not just slow or cease deterioration, but to improve above baseline.
Accordingly, trials should choose endpoints that are appropriate for the stage of development, i.e., at early development stages, a primary target engagement endpoint such as CSF phospho-tau is more appropriate than clinical endpoints. A robust effect on a tau marker in an early study should be valued higher than a seemingly massive (most likely coincidental) effect on a clinical endpoint in a tiny group. Consequently, the success criteria for phase 1 and smaller phase 2 studies should be primarily based on showing proof-of-mechanism, i.e., biomarker impact. Meanwhile, the efficacy in later-stage studies need to be established based on clinical endpoints, as no surrogate efficacy biomarkers are established yet.
As for lead selection for clinical development, compounds should be selected based on multiple criteria. Primarily, compounds should show a robust effect in proper models that actually recapitulate the development of neurofibrillary pathology. An aspect of pharmacokinetics that has often been neglected is that PKI intended for AD treatment need to have decent blood-brain-barrier penetration (see ref. [220])—which was not always the case for PKI repurposed from oncology, as seen with nilotinib.
As AD tau is hyperphosphorylated at more than 40 sites, it may be beneficial to study combinations of PKIs, or multi-target inhibitors, e.g., a combination of PDPKs and non-PDPKs [39]. In the ideal case, the combinations would include brain-specific kinases to limit systemic side effects. What should also be on the forefront of our thinking is the possibility of combining PKIs with modulators of phosphatase activity, constituting a two-pronged approach aimed at reducing hyperphosphorylation.
Finally, the studies should be adequately powered, and statistical rigor should be maintained; this especially applies to the assessment of cognition and function.
To conclude, the intimate connection tau hyperphosphorylation has to tau pathology genesis and progression is apparent in numerous preclinical studies. The translation to clinical practice has so far eluded grasp, with the entirety of the available evidence being insufficient to conclude unequivocally whether PKIs are efficacious in AD and tauopathies. It is definitely warranted to pursue this therapeutic concept further, but with adequate rigor, and employing the lessons gathered from the many failures in AD drug development.
References
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–9.
Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K. Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimer’s Dis. 2013;33:S123–139.
Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet. 1986;2:421–6.
Iqbal K, Grundke-Iqbal I, Smith AJ, George L, Tung YC, Zaidi T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:5646–50.
Lee VM, Balin BJ, Otvos L Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–8.
Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993;12:365–70.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–7.
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59.
Alafuzoff I, Iqbal K, Friden H, Adolfsson R, Winblad B. Histopathological criteria for progressive dementia disorders: clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol. 1987;74:209–25.
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–9.
Elahi FM, Miller BL. A clinicopathological approach to the diagnosis of dementia. Nat Rev Neurol. 2017;13:457–76.
Therriault J, Zimmer ER, Benedet AL, Pascoal TA, Gauthier S, Rosa-Neto P. Staging of Alzheimer’s disease: past, present, and future perspectives. Trends Mol Med. 2022;28:726–741.
Ossenkoppele R, Hansson O. Towards clinical application of tau PET tracers for diagnosing dementia due to Alzheimer’s disease. Alzheimers Dement. 2021;17:1998–2008.
Wolters EE, Ossenkoppele R, Verfaillie SCJ, Coomans EM, Timmers T, Visser D, et al. Regional [(18)F]flortaucipir PET is more closely associated with disease severity than CSF p-tau in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2020;47:2866–78.
La Joie R, Bejanin A, Fagan AM, Ayakta N, Baker SL, Bourakova V, et al. Associations between [(18)F]AV1451 tau PET and CSF measures of tau pathology in a clinical sample. Neurology. 2018;90:e282–e290.
Cho H, Choi JY, Hwang MS, Kim YJ, Lee HM, Lee HS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol. 2016;80:247–58.
Scholl M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron. 2016;89:971–82.
Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, et al. Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr Biol. 2018;28:2181–2189.e2184.
Dehmelt L, Halpain S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005;6:204.
Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, et al. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J Neurosci. 2016;36:1001–7.
Hamdane M, Bretteville A, Sambo AV, Schindowski K, Begard S, Delacourte A, et al. p25/Cdk5-mediated retinoblastoma phosphorylation is an early event in neuronal cell death. J Cell Sci. 2005;118:1291–8.
Qu MH, Li H, Tian R, Nie CL, Liu Y, Han BS, et al. Neuronal tau induces DNA conformational changes observed by atomic force microscopy. Neuroreport. 2004;15:2723–7.
Qi H, Cantrelle FX, Benhelli-Mokrani H, Smet-Nocca C, Buee L, Lippens G, et al. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry. 2015;54:1525–33.
Frost B, Hemberg M, Lewis J, Feany MB. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. 2014;17:357–66.
Camero S, Benitez MJ, Barrantes A, Ayuso JM, Cuadros R, Avila J, et al. Tau protein provides DNA with thermodynamic and structural features which are similar to those found in histone-DNA complex. J Alzheimer’s Dis. 2014;39:649–60.
Brandt R. The tau proteins in neuronal growth and development. Front Biosci. 1996;1:d118–130.
DeVos SL, Hyman BT. Tau at the Crossroads between Neurotoxicity and Neuroprotection. Neuron. 2017;94:703–4.
Pevalova M, Filipcik P, Novak M, Avila J, Iqbal K. Post-translational modifications of tau protein. Bratisl Lek Listy. 2006;107:346–53.
Gorantla NV, Chinnathambi S. Tau Protein Squired by Molecular Chaperones During Alzheimer’s Disease. J Mol Neurosci. 2018;66:356–68.
Jiang S, Bhaskar K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front Mol Neurosci. 2020;13:586731.
Ahmadi S, Zhu S, Sharma R, Wilson DJ, Kraatz HB. Interaction of metal ions with tau protein. The case for a metal-mediated tau aggregation. J Inorg Biochem. 2019;194:44–51.
Chiti F, Dobson CM. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem. 2017;86:27–68.
Mamun AA, Uddin MS, Mathew B, Ashraf GM. Toxic tau: structural origins of tau aggregation in Alzheimer’s disease. Neural Regen Res. 2020;15:1417–20.
Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol. 2016;12:15–27.
Iqbal K, Liu F, Gong CX. Recent developments with tau-based drug discovery. Expert Opin Drug Disco. 2018;13:399–410.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–26.
Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–9.
Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, Seereeram A, et al. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. 2007;282:23645–54.
Hasegawa M, Morishima-Kawashima M, Takio K, Suzuki M, Titani K, Ihara Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J Biol Chem. 1992;267:17047–54.
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–6.
Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–84.
Despres C, Di J, Cantrelle FX, Li Z, Huvent I, Chambraud B, et al. Major Differences between the Self-Assembly and Seeding Behavior of Heparin-Induced and in Vitro Phosphorylated Tau and Their Modulation by Potential Inhibitors. ACS Chem Biol. 2019;14:1363–79.
Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A, et al. Structure-based classification of tauopathies. Nature. 2021;598:359–63.
Scheres SH, Zhang W, Falcon B, Goedert M. Cryo-EM structures of tau filaments. Curr Opin Struct Biol. 2020;64:17–25.
Samimi N, Sharma G, Kimura T, Matsubara T, Huo A, Chiba K, et al. Distinct phosphorylation profiles of tau in brains of patients with different tauopathies. Neurobiol Aging. 2021;108:72–79.
Rawat P, Sehar U, Bisht J, Selman A, Culberson J, Reddy PH. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int J Mol Sci. 2022;23:12841.
Kitoka K, Skrabana R, Gasparik N, Hritz J, Jaudzems K. NMR Studies of Tau Protein in Tauopathies. Front Mol Biosci. 2021;8:761227.
Jadhav S, Avila J, Scholl M, Kovacs GG, Kovari E, Skrabana R, et al. A walk through tau therapeutic strategies. Acta Neuropathol Commun. 2019;7:22.
Chang CW, Shao E, Mucke L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science. 2021;371:eabb8255.
Guo Y, Li S, Zeng L-H, Tan J. Tau-targeting therapy in Alzheimer’s disease: critical advances and future opportunities. Ageing Neurodegener Dis. 2022;2:11.
Sutherland C. What Are the bona fide GSK3 Substrates? Int J Alzheimers Dis. 2011;2011:505607.
Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, et al. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991;64:573–84.
Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–4.
Cho JH, Johnson GV. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J Neurochem. 2004;88:349–58.
Zaoui K, Benseddik K, Daou P, Salaun D, Badache A. ErbB2 receptor controls microtubule capture by recruiting ACF7 to the plasma membrane of migrating cells. Proc Natl Acad Sci USA. 2010;107:18517–22.
Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V, et al. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 2003;22:494–501.
Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–7.
Yin L, Wang J, Klein PS, Lazar MA. Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science. 2006;311:1002–5.
Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–8.
Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–77.
Singh TJ, Haque N, Grundke-Iqbal I, Iqbal K. Rapid Alzheimer-like phosphorylation of tau by the synergistic actions of non-proline-dependent protein kinases and GSK-3. FEBS Lett. 1995;358:267–72.
Polakis P. Casein kinase 1: a Wnt’er of disconnect. Curr Biol. 2002;12:R499–R501.
Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, et al. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol. 1994;4:1077–86.
Wagner U, Utton M, Gallo JM, Miller CC. Cellular phosphorylation of tau by GSK-3 beta influences tau binding to microtubules and microtubule organisation. J Cell Sci. 1996;109:1537–43.
Sang H, Lu Z, Li Y, Ru B, Wang W, Chen J. Phosphorylation of tau by glycogen synthase kinase 3beta in intact mammalian cells influences the stability of microtubules. Neurosci Lett. 2001;312:141–4.
Sperbera BR, Leight S, Goedert M, Lee V-Y. Glycogen synthase kinase-3β phosphorylates tau protein at multiple sites in intact cells. Neurosci Lett. 1995;197:149–53.
Lovestone S, Hartley CL, Pearce J, Anderton BH. Phosphorylation of tau by glycogen synthase kinase-3 beta in intact mammalian cells: the effects on the organization and stability of microtubules. Neuroscience. 1996;73:1145–57.
Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–83.
Lopes JP, Agostinho P. Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol. 2011;94:49–63.
Zhang J, Li H, Yabut O, Fitzpatrick H, D’Arcangelo G, Herrup K. Cdk5 suppresses the neuronal cell cycle by disrupting the E2F1-DP1 complex. J Neurosci. 2010;30:5219–28.
Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–17.
Chang KH, Vincent F, Shah K. Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J Cell Sci. 2012;125:5124–37.
Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–59.
Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–4.
Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–72.
Engmann O, Giese KP. Crosstalk between Cdk5 and GSK3beta: Implications for Alzheimer’s Disease. Front Mol Neurosci. 2009;2:2.
Origlia N, Arancio O, Domenici L, Yan SS. MAPK, beta-amyloid and synaptic dysfunction: the role of RAGE. Expert Rev Neurother. 2009;9:1635–45.
Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–17.
Falcicchia C, Tozzi F, Arancio O, Watterson DM, Origlia N. Involvement of p38 MAPK in Synaptic Function and Dysfunction. Int J Mol Sci. 2020;21:5624.
Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62.
Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–8.
Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J Biol Chem. 1998;273:25893–902.
Liu F, Liang Z, Wegiel J, Hwang YW, Iqbal K, Grundke-Iqbal I, et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008;22:3224–33.
Ryoo SR, Jeong HK, Radnaabazar C, Yoo JJ, Cho HJ, Lee HW, et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J Biol Chem. 2007;282:34850–7.
Trinczek B, Brajenovic M, Ebneth A, Drewes G. MARK4 is a novel microtubule-associated proteins/microtubule affinity-regulating kinase that binds to the cellular microtubule network and to centrosomes. J Biol Chem. 2004;279:5915–23.
Gu GJ, Lund H, Wu D, Blokzijl A, Classon C, von Euler G, et al. Role of individual MARK isoforms in phosphorylation of tau at Ser(2)(6)(2) in Alzheimer’s disease. Neuromol Med. 2013;15:458–69.
Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–44.
Tournebize R, Heald R, Hyman A. Role of chromosomes in assembly of meiotic and mitotic spindles. Prog Cell Cycle Res. 1997;3:271–84.
Oba T, Saito T, Asada A, Shimizu S, Iijima KM, Ando K. Microtubule affinity-regulating kinase 4 with an Alzheimer’s disease-related mutation promotes tau accumulation and exacerbates neurodegeneration. J Biol Chem. 2020;295:17138–47.
Naz F, Islam A, Ahmad F, Hassan MI. Atypical PKC phosphorylates microtubule affinity-regulating kinase 4 in vitro. Mol Cell Biochem. 2015;410:223–8.
Saito T, Oba T, Shimizu S, Asada A, Iijima KM, Ando K. Cdk5 increases MARK4 activity and augments pathological tau accumulation and toxicity through tau phosphorylation at Ser262. Hum Mol Genet. 2019;28:3062–71.
Le Beau MM, Westbrook CA, Diaz MO, Rowley JD. Evidence for two distinct c-src loci on human chromosomes 1 and 20. Nature. 1984;312:70–71.
Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–9.
Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111:3167–77.
Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J Neurosci. 2002;22:10–20.
Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, et al. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci. 2004;24:2304–12.
Wang JY, Ledley F, Goff S, Lee R, Groner Y, Baltimore D. The mouse c-abl locus: molecular cloning and characterization. Cell. 1984;36:349–56.
Heisterkamp N, Groffen J, Stephenson JR, Spurr NK, Goodfellow PN, Solomon E, et al. Chromosomal localization of human cellular homologues of two viral oncogenes. Nature. 1982;299:747–9.
Jhanwar SC, Neel BG, Hayward WS, Chaganti RS. Localization of the cellular oncogenes ABL, SIS, and FES on human germ-line chromosomes. Cytogenet Cell Genet. 1984;38:73–5.
Van Etten RA, Jackson P, Baltimore D. The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell. 1989;58:669–78.
Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44.
Derkinderen P, Scales TM, Hanger DP, Leung K-Y, Byers HL, Ward MA, et al. Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci. 2005;25:6584–93.
Cancino GI, Perez de Arce K, Castro PU, Toledo EM, von Bernhardi R, Alvarez AR. c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol Aging. 2011;32:1249–61.
Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, et al. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–46.
Ho GJ, Hashimoto M, Adame A, Izu M, Alford MF, Thal LJ, et al. Altered p59Fyn kinase expression accompanies disease progression in Alzheimer’s disease: implications for its functional role. Neurobiol Aging. 2005;26:625–35.
Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–25.
Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho-associated kinase. Exp Cell Res. 2000;261:44–51.
Chen J, Sun Z, Jin M, Tu Y, Wang S, Yang X, et al. Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-kappaB pathway. J Neuroimmunol. 2017;305:108–14.
Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Disco. 2005;4:387–98.
Koch JC, Tatenhorst L, Roser AE, Saal KA, Tonges L, Lingor P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharm Ther. 2018;189:1–21.
Gao Y, Yan Y, Fang Q, Zhang N, Kumar G, Zhang J, et al. The Rho kinase inhibitor fasudil attenuates Abeta(1-42)-induced apoptosis via the ASK1/JNK signal pathway in primary cultures of hippocampal neurons. Metab Brain Dis. 2019;34:1787–801.
Amano M, Kaneko T, Maeda A, Nakayama M, Ito M, Yamauchi T, et al. Identification of Tau and MAP2 as novel substrates of Rho-kinase and myosin phosphatase. J Neurochem. 2003;87:780–90.
Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–88.
Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, et al. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009;3:248–61.
Jakobsson E, Arguello-Miranda O, Chiu SW, Fazal Z, Kruczek J, Nunez-Corrales S, et al. Towards a Unified Understanding of Lithium Action in Basic Biology and its Significance for Applied Biology. J Membr Biol. 2017;250:587–604.
Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–5.
Freland L, Beaulieu JM. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front Mol Neurosci. 2012;5:14.
De-Paula VJ, Forlenza OV. Lithium modulates multiple tau kinases with distinct effects in cortical and hippocampal neurons according to concentration ranges. Naunyn-Schmiedeberg’s Arch Pharmacol. 2022;395:105–13.
Salomoni P, Calabretta B. Targeted therapies and autophagy: new insights from chronic myeloid leukemia. Autophagy. 2009;5:1050–1.
Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD. AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer. 2006;94:1765–9.
Pagan FL, Hebron ML, Wilmarth B, Torres-Yaghi Y, Lawler A, Mundel EE, et al. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharm Res Perspect. 2019;7:e00470.
Pagan FL, Hebron ML, Wilmarth B, Torres-Yaghi Y, Lawler A, Mundel EE, et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020;77:309–17.
Nishioka H, Tooi N, Isobe T, Nakatsuji N, Aiba K. BMS-708163 and Nilotinib restore synaptic dysfunction in human embryonic stem cell-derived Alzheimer’s disease models. Sci Rep. 2016;6:33427.
Wu J, Xu X, Zheng L, Mo J, Jin X, Bao Y. Nilotinib inhibits microglia-mediated neuroinflammation to protect against dopaminergic neuronal death in Parkinson’s disease models. Int Immunopharmacol. 2021;99:108025.
Fowler AJ, Hebron M, Balaraman K, Shi W, Missner AA, Greenzaid JD, et al. Discoidin Domain Receptor 1 is a therapeutic target for neurodegenerative diseases. Hum Mol Genet. 2020;29:2882–98.
Martinez A, Alonso M, Castro A, Pérez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J Med Chem. 2002;45:1292–9.
Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M. Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib. J Biol Chem. 2012;287:893–904.
Noori MS, Bhatt PM, Courreges MC, Ghazanfari D, Cuckler C, Orac CM, et al. Identification of a novel selective and potent inhibitor of glycogen synthase kinase-3. Am J Physiol Cell Physiol. 2019;317:C1289–C1303.
Sereno L, Coma M, Rodriguez M, Sanchez-Ferrer P, Sanchez MB, Gich I, et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–67.
Luna-Medina R, Cortes-Canteli M, Sanchez-Galiano S, Morales-Garcia JA, Martinez A, Santos A, et al. NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: potential therapeutic role in brain disorders. J Neurosci. 2007;27:5766–76.
Griebel G, Stemmelin J, Lopez-Grancha M, Boulay D, Boquet G, Slowinski F, et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci Rep. 2019;9:18045.
Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–5.
Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278:45937–45.
Onishi T, Iwashita H, Uno Y, Kunitomo J, Saitoh M, Kimura E, et al. A novel glycogen synthase kinase-3 inhibitor 2-methyl-5-(3-{4-[(S)-methylsulfinyl]phenyl}-1-benzofuran-5-yl)-1,3,4-oxadiazole decreases tau phosphorylation and ameliorates cognitive deficits in a transgenic model of Alzheimer’s disease. J Neurochem. 2011;119:1330–40.
Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–53.
Maphis N, Jiang S, Xu G, Kokiko-Cochran ON, Roy SM, Van Eldik LJ, et al. Selective suppression of the alpha isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res Ther. 2016;8:54.
Hebron ML, Javidnia M, Moussa CE-H. Tau clearance improves astrocytic function and brain glutamate-glutamine cycle. J Neurol Sci. 2018;391:90–99.
Melchior B, Mittapalli GK, Lai C, Duong-Polk K, Stewart J, Guner B, et al. Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: A potential therapeutic for Alzheimer’s disease. Aging Cell. 2019;18:e13000.
Branca C, Shaw DM, Belfiore R, Gokhale V, Shaw AY, Foley C, et al. Dyrk1 inhibition improves Alzheimer’s disease-like pathology. Aging Cell. 2017;16:1146–54.
Velazquez R, Meechoovet B, Ow A, Foley C, Shaw A, Smith B, et al. Chronic Dyrk1 Inhibition Delays the Onset of AD-Like Pathology in 3xTg-AD Mice. Mol Neurobiol. 2019;56:8364–75.
Schweig JE, Yao H, Coppola K, Jin C, Crawford F, Mullan M, et al. Spleen tyrosine kinase (SYK) blocks autophagic Tau degradation in vitro and in vivo. J Biol Chem. 2019;294:13378–95.
Hamano T, Shirafuji N, Yen SH, Yoshida H, Kanaan NM, Hayashi K, et al. Rho-kinase ROCK inhibitors reduce oligomeric tau protein. Neurobiol Aging. 2020;89:41–54.
Liu F, Liang Z, Gong CX. Hyperphosphorylation of tau and protein phosphatases in Alzheimer disease. Panminerva Med. 2006;48:97–108.
Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–50.
Goedert M, Jakes R, Qi Z, Wang JH, Cohen P. Protein phosphatase 2A is the major enzyme in brain that dephosphorylates tau protein phosphorylated by proline-directed protein kinases or cyclic AMP-dependent protein kinase. J Neurochem. 1995;65:2804–7.
Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–7.
Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–8.
Liu F, Iqbal K, Grundke-Iqbal I, Rossie S, Gong CX. Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer’s disease. J Biol Chem. 2005;280:1790–6.
Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, et al. Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol. 2004;63:287–301.
Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–62.
Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–39.
Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–84.
Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am J Pathol. 2005;166:1761–71.
Shentu YP, Huo Y, Feng XL, Gilbert J, Zhang Q, Liuyang ZY, et al. CIP2A Causes Tau/APP Phosphorylation, Synaptopathy, and Memory Deficits in Alzheimer’s Disease. Cell Rep. 2018;24:713–23.
Qian W, Shi J, Yin X, Iqbal K, Grundke-Iqbal I, Gong CX, et al. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J Alzheimer’s Dis: JAD. 2010;19:1221–9.
Wang Y, Yang R, Gu J, Yin X, Jin N, Xie S, et al. Cross talk between PI3K-AKT-GSK-3beta and PP2A pathways determines tau hyperphosphorylation. Neurobiol Aging. 2015;36:188–200.
Corcoran NM, Martin D, Hutter-Paier B, Windisch M, Nguyen T, Nheu L, et al. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J Clin Neurosci. 2010;17:1025–33.
Ahmed T, Van der Jeugd A, Caillierez R, Buee L, Blum D, D’Hooge R, et al. Chronic Sodium Selenate Treatment Restores Deficits in Cognition and Synaptic Plasticity in a Murine Model of Tauopathy. Front Mol Neurosci. 2020;13:570223.
Jin N, Zhu H, Liang X, Huang W, Xie Q, Xiao P, et al. Sodium selenate activated Wnt/beta-catenin signaling and repressed amyloid-beta formation in a triple transgenic mouse model of Alzheimer’s disease. Exp Neurol. 2017;297:36–49.
van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Gotz J, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci USA. 2010;107:13888–93.
Zaki MO, El-Desouky S, Elsherbiny DA, Salama M, Azab SS. Glimepiride mitigates tauopathy and neuroinflammation in P301S transgenic mice: role of AKT/GSK3beta signaling. Inflammopharmacology. 2022;30:1871–90.
Zhao S, Fan Z, Zhang X, Li Z, Shen T, Li K, et al. Metformin Attenuates Tau Pathology in Tau-Seeded PS19 Mice. Neurotherapeutics. 2023;20:452–63.
Barini E, Antico O, Zhao Y, Asta F, Tucci V, Catelani T, et al. Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy. Mol Neurodegener. 2016;11:16.
Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci USA. 2010;107:21830–5.
Sedjahtera A, Gunawan L, Bray L, Hung LW, Parsons J, Okamura N, et al. Targeting metals rescues the phenotype in an animal model of tauopathy. Metallomics. 2018;10:1339–47.
Beauchamp LC, Liu XM, Sedjahtera A, Bogeski M, Vella LJ, Bush AI, et al. S-Adenosylmethionine Rescues Cognitive Deficits in the rTg4510 Animal Model by Stabilizing Protein Phosphatase 2A and Reducing Phosphorylated Tau. J Alzheimer’s Dis: JAD. 2020;77:1705–15.
Sontag E, Nunbhakdi-Craig V, Sontag JM, Diaz-Arrastia R, Ogris E, Dayal S, et al. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci. 2007;27:2751–9.
Wei W, Liu YH, Zhang CE, Wang Q, Wei Z, Mousseau DD, et al. Folate/vitamin-B12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J Alzheimer’s Dis: JAD. 2011;27:639–50.
Xiong Y, Jing XP, Zhou XW, Wang XL, Yang Y, Sun XY, et al. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol Aging. 2013;34:745–56.
Fagan SG, Bechet S, Dev KK. Fingolimod Rescues Memory and Improves Pathological Hallmarks in the 3xTg-AD Model of Alzheimer’s Disease. Mol Neurobiol. 2022;59:1882–95.
Laurent C, Eddarkaoui S, Derisbourg M, Leboucher A, Demeyer D, Carrier S, et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol Aging. 2014;35:2079–90.
Tan X, Liang Z, Li Y, Zhi Y, Yi L, Bai S, et al. Isoorientin, a GSK-3beta inhibitor, rescues synaptic dysfunction, spatial memory deficits and attenuates pathological progression in APP/PS1 model mice. Behav Brain Res. 2021;398:112968.
Chen Q, Tu Y, Mak S, Chen J, Lu J, Chen C, et al. Discovery of a novel small molecule PT109 with multi-targeted effects against Alzheimer’s disease in vitro and in vivo. Eur J Pharm. 2020;883:173361.
Halkina T, Henderson JL, Lin EY, Himmelbauer MK, Jones JH, Nevalainen M, et al. Discovery of Potent and Brain-Penetrant Tau Tubulin Kinase 1 (TTBK1) Inhibitors that Lower Tau Phosphorylation In Vivo. J Med Chem. 2021;64:6358–80.
Dillon GM, Henderson JL, Bao C, Joyce JA, Calhoun M, Amaral B, et al. Acute inhibition of the CNS-specific kinase TTBK1 significantly lowers tau phosphorylation at several disease relevant sites. PLoS One. 2020;15:e0228771.
Ashour NH, El-Tanbouly DM, El Sayed NS, Khattab MM. Roflumilast ameliorates cognitive deficits in a mouse model of amyloidogenesis and tauopathy: Involvement of nitric oxide status, Abeta extrusion transporter ABCB1, and reversal by PKA inhibitor H89. Prog Neuropsychopharmacol Biol Psychiatry. 2021;111:110366.
Yoneyama M, Shiba T, Hasebe S, Umeda K, Yamaguchi T, Ogita K. Lithium promotes neuronal repair and ameliorates depression-like behavior following trimethyltin-induced neuronal loss in the dentate gyrus. PLoS One. 2014;9:e87953.
Caccamo A, Oddo S, Tran LX, LaFerla FM. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol. 2007;170:1669–75.
Nakashima H, Ishihara T, Suguimoto P, Yokota O, Oshima E, Kugo A, et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 2005;110:547–56.
Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015;77:953–71.
Chang Y, Yao Y, Ma R, Wang Z, Hu J, Wu Y, et al. Dl-3-n-Butylphthalide Reduces Cognitive Deficits and Alleviates Neuropathology in P301S Tau Transgenic Mice. Front Neurosci. 2021;15:620176.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21.
Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–91.
Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, et al. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem. 2005;280:3963–73.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51.
Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci. 2005;25:10637–47.
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81.
Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–62.
Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–90.
Zilka N, Korenova M, Novak M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol. 2009;118:71–86.
Perez DI, Gil C, Martinez A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med Res Rev. 2011;31:924–54.
Ribe EM, Perez M, Puig B, Gich I, Lim F, Cuadrado M, et al. Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol Dis. 2005;20:814–22.
Huang W, Percie du Sert N, Vollert J, Rice ASC. General Principles of Preclinical Study Design. Handb Exp Pharm. 2020;257:55–69.
del Ser T, Steinwachs KC, Gertz HJ, Andres MV, Gomez-Carrillo B, Medina M, et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimer’s Dis. 2013;33:205–15.
Lovestone S, Boada M, Dubois B, Hull M, Rinne JO, Huppertz HJ, et al. A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimer’s Dis. 2015;45:75–88.
Tolosa E, Litvan I, Hoglinger GU, Burn D, Lees A, Andres MV, et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord. 2014;29:470–8.
Hampel H, Ewers M, Burger K, Annas P, Mortberg A, Bogstedt A, et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922–31.
Macdonald A, Briggs K, Poppe M, Higgins A, Velayudhan L, Lovestone S. A feasibility and tolerability study of lithium in Alzheimer’s disease. Int J Geriatr Psychiatry. 2008;23:704–11.
Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198:351–6.
Devanand DP, Strickler JG, Huey ED, Crocco E, Forester BP, Husain MM, et al. Lithium Treatment for Agitation in Alzheimer’s disease (Lit-AD): Clinical rationale and study design. Contemp Clin Trials. 2018;71:33–39.
Devanand DP, Crocco E, Forester BP, Husain MM, Lee S, Vahia IV, et al. Low Dose Lithium Treatment of Behavioral Complications in Alzheimer’s Disease: Lit-AD Randomized Clinical Trial. Am J Geriatr Psychiatry. 2022;30:32–42.
van Dyck CH, Nygaard HB, Chen K, Donohue MC, Raman R, Rissman RA, et al. Effect of AZD0530 on Cerebral Metabolic Decline in Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2019;76:1219–29.
Nygaard HB, Wagner AF, Bowen GS, Good SP, MacAvoy MG, Strittmatter KA, et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res Ther. 2015;7:35.
Turner RS, Hebron ML, Lawler A, Mundel EE, Yusuf N, Starr JN, et al. Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer’s Disease. Ann Neurol. 2020;88:183–94.
Malpas CB, Vivash L, Genc S, Saling MM, Desmond P, Steward C, et al. A Phase IIa Randomized Control Trial of VEL015 (Sodium Selenate) in Mild-Moderate Alzheimer’s Disease. J Alzheimer’s Dis. 2016;54:223–32.
Cardoso BR, Roberts BR, Malpas CB, Vivash L, Genc S, Saling MM, et al. Supranutritional Sodium Selenate Supplementation Delivers Selenium to the Central Nervous System: Results from a Randomized Controlled Pilot Trial in Alzheimer’s Disease. Neurotherapeutics. 2019;16:192–202.
Vivash L, Malpas CB, Churilov L, Walterfang M, Brodtmann A, Piguet O, et al. A study protocol for a phase II randomised, double-blind, placebo-controlled trial of sodium selenate as a disease-modifying treatment for behavioural variant frontotemporal dementia. BMJ Open. 2020;10:e040100.
Vivash L, Bertram KL, Malpas CB, Marotta C, Harding IH, Kolbe S, et al. Sodium selenate as a disease-modifying treatment for progressive supranuclear palsy: protocol for a phase 2, randomised, double-blind, placebo-controlled trial. BMJ Open. 2021;11:e055019.
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
Novak P, Kovacech B, Katina S, Schmidt R, Scheltens P, Kontsekova E, et al. ADAMANT: a placebo-controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer’s disease. Nat Aging. 2021;1:521–34.
Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. 2021;7:e12179.
Holland D, McEvoy LK, Desikan RS, Dale AM. Alzheimer’s Disease Neuroimaging I. Enrichment and stratification for predementia Alzheimer disease clinical trials. PLoS One. 2012;7:e47739.
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503–27.
Jellinger KA, Attems J. Challenges of multimorbidity of the aging brain: a critical update. J Neural Transm (Vienna). 2015;122:505–21.
Lim YY, Jaeger J, Harrington K, Ashwood T, Ellis KA, Stoffler A, et al. Three-month stability of the CogState brief battery in healthy older adults, mild cognitive impairment, and Alzheimer’s disease: results from the Australian Imaging, Biomarkers, and Lifestyle-rate of change substudy (AIBL-ROCS). Arch Clin Neuropsychol. 2013;28:320–30.
Hobart J, Cano S, Posner H, Selnes O, Stern Y, Thomas R, et al. Putting the Alzheimer’s cognitive test to the test I: traditional psychometric methods. Alzheimers Dement. 2013;9:S4–9.
Holland D, Desikan RS, Dale AM, McEvoy LK. Rates of decline in Alzheimer disease decrease with age. PLoS One. 2012;7:e42325.
Cummings J, Lee G, Nahed P, Kambar M, Zhong K, Fonseca J, et al. Alzheimer’s disease drug development pipeline: 2022. Alzheimers Dement. 2022;8:e12295.
He Q, Liu J, Liang J, Liu X, Li W, Liu Z, et al. Towards Improvements for Penetrating the Blood-Brain Barrier-Recent Progress from a Material and Pharmaceutical Perspective. Cells. 2018;7:24.
Serenó L, Coma M, Rodriguez M, Sanchez-Ferrer P, Sánchez MB, Gich I, et al. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–67.
Acknowledgements
This work was supported by grant number APVV-20-0447. We would like to express our sincere gratitude to Professor Michal Novak, the founder of tau research in Slovakia, for his leadership and support.
Funding
Open access funding provided by The Ministry of Education, Science, Research and Sport of the Slovak Republic in cooperation with Centre for Scientific and Technical Information of the Slovak Republic.
Author information
Authors and Affiliations
Contributions
NZ: concept and main idea, chapters “Introduction” and “Efficacy Studies On Selected Kinase Inhibitors”. NB: chapters “Introduction”, “Protein Kinases Selected As A Target For Alzheimer’s Therapy”, “Protein Kinase Inhibitors Evaluated In AD”, “Clinical Development Of Protein Kinase Inhibitors and Phosphatase Modulators For Alzheimer’s Disease” and “Efficacy Studies On Selected Kinase Inhibitors”. TS: chapter “Efficacy Studies On Selected Kinase Inhibitors”. KI: chapter “Protein Phosphatase 2a (PP2A) In Tau Phosphorylation”. FL: chapter “Protein Phosphatase 2a (PP2A) In Tau Phosphorylation”. IH: chapters “Protein Kinases Selected As A Target For Alzheimer’s Therapy” and “Protein Kinase Inhibitors Evaluated In AD”. PN: critical comments, chapters “Clinical Development Of Protein Kinase Inhibitors And Phosphatase Modulators For Alzheimer’s Disease” and “Future Drug Development Of Tau Phosphorylation Modulators”.
Corresponding authors
Ethics declarations
Competing interests
NB, PN, TS, and NZ have received salary from AXON Neuroscience SE or one of its subsidiaries. PN has received payments from F. Hoffmann La Roche. Other authors do not declare any conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’ks Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Basheer, N., Smolek, T., Hassan, I. et al. Does modulation of tau hyperphosphorylation represent a reasonable therapeutic strategy for Alzheimer’s disease? From preclinical studies to the clinical trials. Mol Psychiatry 28, 2197–2214 (2023). https://doi.org/10.1038/s41380-023-02113-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-023-02113-z
This article is cited by
-
Neuronal double-stranded DNA accumulation induced by DNase II deficiency drives tau phosphorylation and neurodegeneration
Translational Neurodegeneration (2024)
-
Recent advances in Alzheimer’s disease: mechanisms, clinical trials and new drug development strategies
Signal Transduction and Targeted Therapy (2024)
-
CDK5 Deficiency Does not Impair Neuronal Differentiation of Human Induced Pluripotent Stem Cells but Affects Neurite Outgrowth
Molecular Neurobiology (2024)
-
Identification of high-performing antibodies for the reliable detection of Tau proteoforms by Western blotting and immunohistochemistry
Acta Neuropathologica (2024)
-
Recent Insights into the Neurobiology of Alzheimer’s Disease and Advanced Treatment Strategies
Molecular Neurobiology (2024)
