
Abstract
Childhood apraxia of speech (CAS), the prototypic severe childhood speech disorder, is characterized by motor programming and planning deficits. Genetic factors make substantive contributions to CAS aetiology, with a monogenic pathogenic variant identified in a third of cases, implicating around 20 single genes to date. Here we aimed to identify molecular causation in 70 unrelated probands ascertained with CAS. We performed trio genome sequencing. Our bioinformatic analysis examined single nucleotide, indel, copy number, structural and short tandem repeat variants. We prioritised appropriate variants arising de novo or inherited that were expected to be damaging based on in silico predictions. We identified high confidence variants in 18/70 (26%) probands, almost doubling the current number of candidate genes for CAS. Three of the 18 variants affected SETBP1, SETD1A and DDX3X, thus confirming their roles in CAS, while the remaining 15 occurred in genes not previously associated with this disorder. Fifteen variants arose de novo and three were inherited. We provide further novel insights into the biology of child speech disorder, highlighting the roles of chromatin organization and gene regulation in CAS, and confirm that genes involved in CAS are co-expressed during brain development. Our findings confirm a diagnostic yield comparable to, or even higher, than other neurodevelopmental disorders with substantial de novo variant burden. Data also support the increasingly recognised overlaps between genes conferring risk for a range of neurodevelopmental disorders. Understanding the aetiological basis of CAS is critical to end the diagnostic odyssey and ensure affected individuals are poised for precision medicine trials.
Similar content being viewed by others


Introduction
Childhood apraxia of speech (CAS) is a rare neurodevelopmental disorder, occurring in 0.1% of the population [1]. CAS stems from deficits in speech planning and programming, affecting a child’s ability to sequence sounds and syllables accurately and with correct prosody, resulting in highly unintelligible speech [1, 2]. The first evidence implicating a specific gene in aetiology of CAS was provided in 2001, via a family study revealing that pathogenic variants in FOXP2 were responsible for the speech disorder [3]. For almost two decades, FOXP2 was the only gene associated with CAS, in the absence of intellectual disability. Technological advances and reduced costs for DNA analyses have recently enabled efficient genome sequencing and bioinformatic follow-up, paving the way for high throughput discovery of genes involved in CAS. In particular, two independent cohort studies have performed genome-wide sequencing on 52 individuals with CAS [4, 5].
In the first cohort, aetiologic variants were identified in eight of 19 probands ascertained for CAS, yielding a genetic diagnostic rate of 42% with pathogenic variants revealed in: CHD3, SETD1A, WDR5, KAT6A, SETBP1, ZFHX4, TNRC6B and MKL2 [4]. In the second cohort, comprising 33 probands with CAS, nine additional genes were implicated: CDK13, EBF3, GNAO1, GNB1, DDX3X, MEIS2, POGZ, UPF2 and ZNF142. One individual also had a contiguous gene deletion, yielding a genetic diagnostic rate of 33% (11/33) across this second cohort [5]. In these studies, there was no evidence of recurrent point mutations and no genes which appeared to carry a higher burden of mutations, except for SETBP1; for which two individuals were found to carry variants across the two cohorts [6].
Taken together, the two cohort studies provided novel insights into the neurobiology of childhood speech disorders. First, the discovery of 17 new genes involved in CAS aetiology, with a combined diagnostic yield of 37%, revealing for the first time, that many children do have a single gene diagnosis explaining their speech condition. Second, many of the highly penetrant variants implicated shared pathways broadly involved in transcriptional regulation (e.g. POGZ, SETBP1, SETD1A, KAT6A), suggesting a key role for transcriptional dysregulation in aberrant speech development [4, 5]. Other molecular pathways of significance were also revealed with high confidence and likely novel pathogenic variants, such as in GNAO1 and GNB1, both part of G-protein signalling pathways [5]. Third, the studies demonstrated that pathogenic variants more commonly arise de novo rather than being inherited, and that speech disorders are genetically heterogeneous [5], as is the case for other neurodevelopmental disorders [7,8,9]. Fourth, the candidate genes newly implicated in CAS were frequently associated with other neurodevelopmental disorders, such as epilepsy (e.g. GNAO1, GNB1, SETD1A) and/or intellectual disability (e.g. CDK13, CHD3, DDX3X, POGZ, SETBP1) [4, 5]. These novel insights into CAS aetiology, including genetic heterogeneity, demonstrate the need to study additional, larger cohorts to reveal further causative genes, increase the genetic diagnostic yield, and further unravel molecular pathways underlying severe childhood speech disorder. A much deeper knowledge of the molecular basis of severe speech conditions such as CAS is essential to move the field toward precision therapies.
Here, we aimed to identify the molecular basis of severe speech disorder, in a large cohort of probands ascertained for a primary diagnosis of CAS. Each proband underwent comprehensive phenotypic analysis and genome sequencing to identify pathogenic variants of major effect. We also analysed the molecular co-expression of all genes associated with CAS, and the overlap of genes associated with CAS and other neurodevelopmental disorders.
Methods
Ethical consent
The study was approved by the Human Research Ethics Committee of The Royal Children’s Hospital, Melbourne, Australia (#37353). Written informed consent was obtained from parents or legal guardians.
Participants and phenotyping
Probands under age 18 years were ascertained with a clinical diagnosis of CAS and where parents and clinicians reported the current primary clinical concern as poor speech development due to childhood apraxia of speech [5]. Probands with moderate to severe intellectual disability as determined via psychometric testing, were excluded. Participants were recruited via medical and speech pathology clinicians or direct parent referral. Medical and developmental history and secondary neurodevelopmental outcomes were recorded with validation via relevant professional reports (e.g. paediatrician, multi-disciplinary assessment team for ASD diagnosis, physiotherapist, occupational therapist, academic outcomes) (Tables 1, 2; Supplementary Table 1).
A diagnosis of CAS was then confirmed based on meeting the three American Speech-Language-Hearing Association consensus criteria for CAS: (1) inconsistent errors on consonants and vowels in repeated productions of syllables or words; (2) lengthened and disrupted coarticulatory transitions between sounds and syllables, and (3) inappropriate prosody [1]. Criteria were operationally defined and rated [10] from phonetic transcriptions of standardised single word speech sub-tests (phonology and inconsistency) [11] and a 5-min conversational speech sample [5]. Dysarthria was diagnosed in the presence of oral tone or coordination disturbance and dysarthric features identified during conversation using the Mayo Clinic Dysarthria rating scale [12, 13]. Language and cognition were also assessed with standardised tools [14,15,16,17].
Genetic testing
Genomic DNA was extracted from whole blood or saliva using a Qiagen (Valencia, CA) QIAamp DNA Maxi kit or a prepIT L2P kit (DNA Genotek Inc., Ontario, Canada), respectively. Probands underwent chromosomal microarray testing on Illumina (SanDiego, CA) platforms, with the reportable effective resolution of arrays being 200 Kb. Results were analyzed with Karyostudio software version 1.3 or 1.4 (Illumina), using genome reference sequence NCBI36/hg18 (v1.3, pre-2013) or GRCh37/hg19 (v1.4, 2013 onwards).
Genome sequencing was conducted on 204 individuals from 70 families comprising 71 probands (two probands were monozygotic twins hence for the genetic analysis and results we report on 70 probands), 127 parents and 6 other relatives. Illumina TruSeq DNA Nano or NovaSeq PE150 PCR free library preparation was completed prior to sequencing on the Illumina NovaSeq 6000 to average 30-fold depth with ~100 Gb data generated per sample at the Australian Genome Research Facility or Novogene (HK) Company Limited. Sanger sequencing or droplet digital PCR (ddPCR) were used to segregate variants in additional family members who had not undergone microarray or genome sequencing.
Variant analysis
Variant discovery was performed using trio or parent–child pair (where one parent was unavailable for testing) designs (Fig. 1). Exceptions to this were two singletons, and four larger families. 150 bp sequence pair-end reads were mapped to the hg19 reference genome using the Burrow-Wheeler Aligner (BWA-MEM, bwa v0.7.17) [18]. Read sorting and indexing was undertaken using SAMtools (v1.9) and Genome Analysis Toolkit (GATK, v4.1.4.1) was used to mark duplicates. Base quality score recalibration was performed, and variants were called using HaplotypeCaller, with individuals called separately, as implemented by GATK. Sequencing quality control was performed using fastQC.

Workflow covers recruitment of patients (CAS in red, affected relative in blue, unaffected in black, not sequenced in white), DNA sequencing, analysis and filtering of genomic data, identification of potential causative variants, geneticist review, molecular validation, segregation and integration of all findings. Please note that affectedness status refers to a parent having speech therapy as a child but not necessarily for a diagnosis of CAS which is not historically well reported for that generation. *Only the damaging effects of small intragenic variants are predicted bioinformatically.
Genotype calling and quality filtering were performed separately in multiple genome sequencing batches. Joint calling was performed by merging per-sample gvcf files and applying GATK’s GenotypeGVCFs tool. Variants with excess heterozygosity (Z score >4.5) were removed, then variant quality score recalibration was carried out for single nucleotide variants (SNVs) and indels separately, with a truth sensitivity filter of 99.7 to flag variants for exclusion. Filtering of low quality SNV calls excluded those flagged by low threshold or any of the following filters: low quality by depth (QD < 2); evidence of strand bias (Fisher strand [FS] > 60 or strand odds ratio [SOR] > 3); and evidence of significant differences between alternate and reference alleles for read mapping qualities (rank sum < −12.6) or position bias (ReadPosRankSum < −8). Indels filtering was performed in a similar manner to missense variant filtering, with exceptions being to exclude variants with FS > 200; SOR > 10; or ReadPosRankSum < −20. Finally, familial relationships were confirmed using peddy [19]. Filtering and other scripted analysis was conducted using R version 3.5.2.
Analysis was restricted to variants: (1) not present in gnomAD or with gnomAD allele count ≤2 (in all populations), (2) not present in unaffected family members from our cohort, and (3) potentially de novo, or consistent with an appropriate inheritance model matching the phenotypic pedigree (e.g. dominant, recessive). Compound heterozygous models were considered for variants present in gnomAD with a mean allele frequency <0.05%. Only variants with read depth >10 and genotype quality >20 in the proband and their sequenced family members were considered. Identified variants were annotated with the variant effect predictor (VEP v93.3) algorithm, using the assembly version GRCh37.p13 and categorized based on the following series of annotations.
Genome-wide analysis of LoF and predicted damaging missense variants
We analyzed the genome sequencing data for loss of function (LoF) and predicted damaging missense variants genome-wide. Predicted LoF candidates were defined by using VEP annotations that were required to meet three criteria: (1) annotated as frameshift, stop or start lost, stop gained, splice acceptor or donor variant, (2) in a gene predicted intolerant to LoF variation (ExACpLI ≥ 0.9 or LoFtool <0.1), and (3) at least one of the following: (a) CADD Phred score ≥20 predicted damaging, or (b) predicted to affect splicing (AdaBoost score ≥0.6 or random forest score ≥0.6 using the dbscSNV VEP plugin). For frameshift variants, the variant was only required to be in a LoF intolerant gene.
Predicted damaging missense variants had to meet at least three criteria: (1) PolyPhen-2 prediction as “probably” or “possibly damaging”, (2) SIFT prediction as “deleterious” or “deleterious low confidence”, (3) a CADD Phred score ≥20 predicted damaging, or (4) a missense tolerance ratio significantly different from 1 (false discovery rate <0.05).
Criteria for identification and reporting of candidate variants
We applied a two-stage approach for shortlisting candidate variants, from our identified LoF and damaging missense variants: (1) we selected variants located within genes of interest, a gene list collated based on several relevant criteria, informed by previous CAS studies [4, 5], and described below. Pathogenicity of these variants was assessed using the American College of Medical Genetics (ACMG) guidelines [20], and via review by a clinical geneticist; (2) where no candidate variant was identified, we then applied a genome-wide, wholly agnostic to gene, search for candidate variants, to be followed up with ACMG and clinical geneticist review. The size of the cohort and the inability to perform statistical analyses to implicate novel genes, such as via burden analysis, necessitated the usage of these constraints.
We report candidate variants as follows:
-
1.
High-confidence variants: LoF and predicted damaging missense variants, that were classified with the American College of Medical Genetics (ACMG) guidelines as pathogenic (class 5) or likely pathogenic (class 4) [20], and where the phenotype associated with the gene was consistent with that of the proband.
-
2.
Low confidence variants: LoF and predicted damaging missing variants, that were either classified as of uncertain significance (3) according to ACMG guidelines, or classified likely pathogenic (class 4), but where the gene was not consistent with the proband’s phenotype, or otherwise lacked evidence for pathogenicity.
All reported variants were inspected with the Integrative Genome Viewer (v 2.7).
Collated list of genes of interest
The list of genes of interest, used in shortlisting candidate variants (n = 2145 genes, Supplementary Table 2), was collated from the following sources: genes from recent CAS cohort studies [4, 5] as well as previously confirmed single genes implicated in CAS such as FOXP2 or GRIN2A [3, 21] and previously confirmed single genes associated with speech disorder or delay (n = 81). Additionally, high-confidence genes known to harbour pathogenic variants in intellectual disability (n = 1399), epilepsy (n = 611), autism spectrum disorder (ASD, n = 131) and cleft palate (n = 156), recognised by Victorian Clinical Genetics Services, were extracted from PanelApp using an application programming interface (https://panelapp.agha.umccr.org/) [22]. There were 233 overlapping genes across these groups making 2145 genes in total. High-confidence ASD-related genes from the Simons Foundation Autism Research Initiative database were also included [23] (n = 419). Finally, brain-expressed genes associated with primate-human accelerated evolution were included; this set comprised of 415 genes overlapping human accelerated regions (HARs) that are also significantly over-expressed in brain, compared to other tissues [24], and 45 genes overlapping with HARs, that were found to be exclusively expressed in human brain cells, and not in other primates [25]. This final set of genes were included, as HARs have previously been implicated in ASD and cognitive development, and thus may be involved in the evolutionary development of speech.
Copy number and structural variants
Manta (regions up to 5 Mb) [26] (v 1.6.0) and qDNAseq (bin size 10 kb with CNVs up to 5 Mb) [27] (v 1.18.0) were used to detect CNVs and other structural variants. Manta detects structural variants based on abnormal alignment of read pairs. qDNAseq detects structural variants based on read depth. Variants occurring in more than two families were filtered out to avoid false positives due to technical artefact. SVAnnot (v 2.5) was used to annotate the variants, filtering by gnomAD SV abundance with SVs with frequency >0.05% excluded. Candidate structural variants were identified using the same approach as for SNVs, with pathogenicity assessed via ACMG guidelines and clinical review.
Variant validation
High-confidence variants were validated using Sanger sequencing or ddPCR. For Sanger sequencing, gene variants were amplified using gene specific primers (oligonucleotide sequences available on request) designed to the reference human gene transcripts (NCBI Gene). Amplification reactions were cycled using a standard protocol on a Veriti Thermal Cycler (Applied Biosystems, Carlsbad, CA) at 60 °C annealing temperature for 1 min. Bidirectional sequencing of all exons and flanking regions was completed with a BigDye v3.1 Terminator Cycle Sequencing Kit (Applied Biosystems). Sequencing products were resolved using a 3730xl DNA Analyzer (Applied Biosystems). All sequencing chromatograms were compared to the published cDNA sequence; nucleotide changes were detected using Codon Code Aligner (CodonCode Corporation, Dedham, MA). For ddPCR, probes and primers were designed in-house and synthesized by Integrated DNA Technologies (Coraville, IA) and assays were performed [28, 29] using a Bio-Rad QX200 Droplet Digital PCR System (Hercules, CA) and QuantaSoft software v1.7.4.0917.
Analysis of novel sources of genetic contributions to CAS
Three forms of genetic analysis for CAS that have not been previously applied were undertaken: (1) short tandem repeat (STR) analysis of both known and novel pathogenic repeats (Supplementary Table 3); (2) examination of common variants implicated in ASD and non-syndromic cleft palate, and their relevance to CAS, via associations with polygenic risk scores (PRS) and (3) estimation of mitochondrial gene abundance (see Supplementary methods).
Brain gene co-expression and gene set enrichment analysis
Gene co-expression analyses were undertaken for our novel high-confidence genes identified in the present manuscript (n = 15) and then extended to include a further 19 genes previously implicated in cohorts ascertained for CAS across the Eising et al. [4]. (CHD3, SETD1A, WDR5, KAT6A, SETBP1, ZFHX4, TNRC6B, MKL2, ARID1A, TRIO) and Hildebrand et al. [5]. cohorts (CDK13, EBF3, GNAO1, GNB1, DDX3X, MEIS2, POGZ, UPF2 and ZNF142), totalling 34 genes. ARID1A and TRIO are not yet confirmed candidate genes for CAS as parental data were not available, so de novo status and hence pathogenicity could not be confirmed in the original study [4, 20]. Yet both genes had previously been implicated in neurodevelopmental conditions where speech was a core phenotype and hence were included in expression analyses in Eising et al. [4] and also included in our meta-analyis here. Analyses were conducted using a Monte Carlo sampling approach [4, 5] with data from the BrainSpan Atlas of the Developing Human Brain [30] (Supplementary Table 4). Co-expression analyses were also performed to prioritize genes of uncertain significance for CAS (see Supplementary methods).
Gene set enrichment analyses were undertaken using gene sets in Gene Ontology molecular function, cellular component, and biological processes databases as well as Reactome pathway databases [31]. g:Profiler was used to test for gene set enrichment [32] with a Bonferroni-corrected p-value threshold of 0.05 to determine pathways enriched for genes implicated in CAS.
Results
Phenotypic data
One hundred and seventeen probands were recruited and 46 participants were excluded based on having an inappropriate phenotype (i.e. not having CAS; having another neurodevelopmental condition that affected the child’s development more than the speech presentation). The 71 included probands (53 males, 18 females, 1 monozygotic twin pair) from the 70 families had an average age of 5 years 7 months (range 2 years 2 months to 16 years 8 months). Phenotypic information for the cohort is shown in Tables 1, 2; Supplementary Tables 1a, b. Pathogenic variants were confirmed in 18 probands (12 males, 6 females; average age of 5 years 7 months) (Tables 1, 2) as described further in the following section. The phenotypes of the probands with (n = 18) versus without (n = 52) variants are presented in Fig. 2.

Phenotypic features of CAS cohort with (blue, n = 18) and without (orange, n = 52) pathogenic variants. Data based on children with psychometric assessments by health professionals (i.e., cognition, language, motor, formal ASD diagnoses). Data from Tables 1, 2; Supplementary Tables 1a, b. Dots indicate percent of children with (blue) and without (orange) pathogenic variants who had psychometric test results confirming diagnoses.
All probands were ascertained based on a clinical diagnosis of CAS. Following our speech assessment protocol, 15 of the 18 probands with pathogenic variants had CAS in isolation (n = 7) or co-occurring with other speech disorders (n = 8) (Table 1). Three probands (IDs 2, 5, and 6; Table 1) had other severe speech disorder presentations (dysarthria, n = 1; phonological and articulation disorder, n = 1; inconsistent phonological disorder, n = 1). Expressive language disorder was implicated in 15/17 probands (mild, n = 1; moderate, n = 4; severe or unable to be scored due to severity, n = 10; Fig. 2). One proband was minimally verbal and unable to complete a valid expressive language assessment. Receptive language disorder was noted in 13/18 individuals (mild, n = 1; moderate, n = 3; severe, n = 9). Of those old enough to read and write (>5 years, n = 8), all had reading and spelling impairment. Two probands with pathogenic variants (2/18, 11%; IDs 10, 18) had CAS accompanied by fine motor and related linguistic deficits, but without other neurodevelopmental disorder diagnoses.
Of the 18 children identified to carry high confidence variants, 14 had formal cognitive assessment with profiles ranging from an average full-scale IQ (FSIQ) (n = 3), to borderline FSIQ results (n = 6), to mild intellectual disability (n = 5). For 3 children, FSIQ could not be calculated due to significant performance variation across verbal and nonverbal subscales, which is a common experience for children with severe speech production deficits. The remainder (n = 4) did not have IQ testing because concerns with learning or cognition had not been raised or pursued by the family or treating physician and children were attending mainstream childcare or school settings (IDs 13–16; IDs 13, 14, 16 were <4 years of age when few children receive formal cognitive testing). Other features included neurodevelopmental diagnoses or features secondary to CAS including mild ASD (n = 2), difficulties with attention (n = 4), and anxiety and mood-related symptoms (n = 1). Dysmorphic features such as epicanthic folds and pointed chin (Table 1), rated by a clinical geneticist with 24 years of clinical experience, were present in just over half of the probands with high confidence variants (11/18). Gross motor (n = 16) and fine motor (n = 14) delays were common and associated with a slower trajectory in learning to ride a bicycle, balance appropriately, draw, write, and cut compared to typical peers. Two of the 18 children with pathogenic variants (IDs 1, 12) had a history of seizures.
Single nucleotide and indel variants
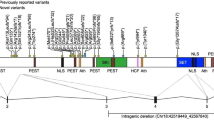
A high confidence variant was identified in 18/70 (26%) of probands (Table 3a, Fig. 3). These included three frameshift, two splice acceptor, six nonsense, and six missense variants, as well as one multiple exon duplication, and they were found in 18 different genes (ARHGEF9, BRPF1, DDX3X, DIP2C, ERF, HRNPNK, KDM5C, PHF21A, PURA, RBFOX3, SETBP1, SETD1A, SETD1B, SHANK3, SPAST, TAOK2, TRIP12, ZBTB18). All high-confidence variants were de novo except in PURA, ERF and RBFOX3, which were inherited (Table 3a, Fig. 3). Many of these genes that we newly implicate in CAS, as well as genes previously described in earlier sequenced CAS cohorts [4, 5] are also associated with other neurodevelopmental disorders (Fig. 4a and Fig. 4b, Supplementary Table 5).



Families analysed by Genome Sequencing. Pedigrees (A–F, M–R, Y-D1) from 18 families with 18 different high confidence variants. Sequence chromatograms (G, I, J, L, S, V, W, X, E1) showing de novo or inherited variants. Sanger sequencing was not performed for the variants in eight of the families (H, K, T, U, F1, G1, H1, I1) because they had variants in known genes with sufficient coverage in the genome sequencing to be confident they were real, heterozygous variants. The large duplication in Family 17 (J1) could not be validated by Sanger sequencing.

A Candidate Genes for CAS identified in this study and Hildebrand et. al. (*) also have been shown to cause other neurodevelopmental disorder traits. B Venn diagram showing the overlap of these genes and multiple neurodevelopmental disorder traits.
The 13 nonsense, splice-site, frameshift or exon duplication variants were all in genes intolerant to loss-of-function variation (ARHGEF9, DDX3X, DIP2C, ERF, HNRNPK, KDM5C, PHF21A, RBFOX3, SETD1A, SETBP1, SHANK3, TRIP12, ZBTB18), according to gnomADpLI or LoFtool scores (Table 3a). The five missense variants were all predicted to be damaging by at least three in silico tools (SIFT, PolyPhen, CADD, MTR). Four of the 18 high confidence variants (in ERF, SETD1A, SHANK3 and ZBTB18) were recurrent, with these variants having been reported previously in individuals with neurodevelopmental disorders [33,34,35,36,37]. The remaining 14 high-confidence variants have not been previously reported: of these, ten were pathogenic and four were likely pathogenic [20].
In five probands, we identified low confidence, LoF variants in genes predicted to be intolerant to haploinsufficiency (Table 3b, ABCC11, ATP7B, CTDSPL2, ERCC6L, ZNF512B). These variants are all predicted to cause loss of function of the gene; however at present, none of these genes are established to cause CAS or other neurodevelopmental disorders and therefore are variants of unknown significance. Nor did any of the cases have dysmorphology associated with any of these conditions. Of note, a frameshift variant in ATP7B of uncertain significance was also identified in our previous CAS cohort [5]; in the present cohort, the identified variant (proband 49) is shared with their father; however the father has a history of self-reported but undiagnosed dyslexia without CAS, and the mother has a brief history of speech therapy as a child but also without a CAS diagnosis, so the variant does not fully segregate with CAS or speech affected status. Thus, the relevance of ATP7B in CAS remains unclear.
In eight probands (8/70; 11.4%), we report nine rare (gnomAD allele count < =2) low confidence, predicted damaging missense variants (Table 3c). These are a selected subset of predicted damaging variants, located in genes that were of relevance due to known disease association, or biological relevance, but were of uncertain pathogenicity, or the gene was not consistent with the proband’s phenotype. Two of the nine variants were in genes previously associated with speech and/or language disorders - ROBO2, and ZFHX4; however, one of these was inherited from an unaffected parent (ROBO2, Proband 52), and for the other, it was not possible to determine whether the variant was de novo, as parental DNA was unavailable (ZFHX4, proband 43). The remaining seven variants were located in CHD5, FGFR1, JARID2, MEF2C, RAPGEF2, TET3, UBQLN2 and ZEB2. For each of these, the variant was deemed to be of uncertain significance (ACMG), and/or the phenotype associated with the gene was not consistent with the proband’s phenotype. For all probands without a high confidence variant, the remaining predicted damaging missense variants, identified though our genome-wide search, are listed in Supplementary Table 6.
Copy number and structural variation
There were no diagnostic findings on clinical microarray analysis. In one individual (ID17) a de novo pathogenic tandem 59,799 bp duplication was identified in TRIP12, spanning exons 7 to 37 out of 42 (NM_001348323.3). If tandem, this duplication would be predicted to disrupt the reading frame causing loss of function (Supplementary Fig. 1), however we could not confirm this using the microarray probe data and independent confirmation by sequencing would be required.
Analysis of novel sources of genetic contributions to CAS
No expansion of either known or novel repeats were identified in the CAS probands. The polygenic risk score analysis did not identify any statistically significant findings, with the strongest trend being observed for ASD where probands were enriched for ASD risk, nearly achieving nominal significance (Two sample t test, p = 0.054). The non-syndromic cleft palate PRS also showed increased risk for CAS probands, but this was not significant (two sample t test, p = 0.226).
Mitochondrial abundance analyses identified two CAS probands with high confidence, likely pathogenic variants in genes known to have mitochondrial function as outliers (DDX3X and HNRNPK) but mitochondrial abundance did not appear to be a biomarker for CAS overall (see Supplementary results, Supplementary Fig. 2A–C).
Brain gene co-expression and gene set enrichment analyses
The median absolute correlation between our 18 high-confidence genes was |ρ | = 0.4194 (Fig. 5A). Thirty-two of the 153 pairwise correlations were among the top 5% most highly correlated gene pairs genome-wide |ρ | > 0.647, (Fig. 5B), and there was evidence that this set of genes was more highly co-expressed than expected by chance (p = 0.0038). Gene set enrichment analyses of a subset of seven highly co-expressed genes (BRPF1, DIP2C, KDM5C, PHF21A, SETBP1, SETD1A, SETD1B) indicate they are involved in chromatin organization (GO:0006325; p = 1.238 × 10–3). (Supplementary Table 7)

A Gene co-expression matrix for the 18 high-confidence candidate genes with pairwise Spearman’s rank correlation coefficients between genes shown, based on 280 samples from 24 individuals (8 weeks post conception to 10 months after birth) from the BrainSpan resource. Genes ordered by hierarchical clustering, using the median linkage method. B Network of gene co-expression. Nodes represent genes; edges represent gene–pair correlations that exceed the threshold for the top 5% most highly correlated gene pairs genome-wide (|ρ | > 0.64) (blue—positive correlation, red—negative correlation). C Gene co-expression matrix for the 18 high-confidence candidate genes (black) as well as the genes from [4] (green) and [5] (blue). D Network of gene co-expression. Nodes represent genes; edges represent gene–pair correlations that exceed the threshold for the top 5% most highly correlated gene pairs genome-wide (|ρ | > 0.64) (blue—positive correlation, red – negative correlation). Black nodes—novel genes from this work, green nodes genes from [4] and blue nodes from [5]. E Network of gene co-expression. Nodes represent genes; edges represent gene–pair correlations that exceed the threshold for the top 5% most highly correlated gene pairs genome-wide (|ρ | > 0.64) (blue—positive correlation, red—negative correlation). Black nodes are a set of co-expressed genes including genes from the present study and from previous studies [4, 5]. Blue nodes—the top prioritized genes from cytogenic variants described in Table 4.
The median pairwise correlation of gene expression for the 34 genes, was significantly higher than expected by chance (median | ρ | = 0.4095, p = <2 × 10–4, Fig. 5D). Gene set enrichment analyses of the highly co-expressed cluster of 15 genes from the present study and past cohorts [4, 5] (Fig. 5C) further highlighted the significant over-representation of genes involved in chromatin organization (GO:0006325; Bonferroni-corrected p = 2.304 × 10–6) as well as transcriptional regulation (GO:0003676; Bonferroni-corrected p = 1.103 × 10–4, 25,396 sets tested, Supplementary Table 7).
Finally, our co-expression model was used to prioritize candidate genes, beyond our high confidence set. Firstly we re-examined the set of low-confidence variants identified in the present study (Table 3b, c), and variants of uncertain significance from our previous cohort [5]. Amongst the low confidence findings, TET3 was the only gene identified for proritization (FDR < 0.1), while three genes identified in our previous cohort were prioritized (BRWD3, MCMBP and ZKSCAN1). All four prioritzed genes are associated with chromatin organization and/or DNA binding. Second, we sought to prioritize genes contained in each of 21 large copy number variant regions, identified through a literature search. All regions span multiple genes, and the associated phenotypes include speech disorder as a clinical feature (Supplementary Tables 8 and 9). Prioritization analysis identified at least one gene in each region (FDR < 0.1), with more than one candidate for 18/21 regions (Table 4) (Fig. 5E). In several instances, the prioritized gene from our co-expression network had already been proposed as the likely causal gene (see Supplementary results).
Discussion
Our findings almost double the current number of genes implicated in causation of CAS and provide further novel insights into the biology of childhood speech disorder. We identified high confidence variants, thereby providing a clinical genetic diagnosis, for 18 children ascertained for CAS, revealing 15 genes that have not previously been associated with this severe speech disorder (ARHGEF9, BRPF1, DDX3X, DIP2C, ERF, HRNPNK, KDM5C, PHF21A, PURA, RBFOX3, SETBP1, SETD1A, SETD1B, SHANK3, SPAST, TAOK2, TRIP12, ZBTB18). We identified a clinical genetic diagnosis in one-quarter of individuals tested; a diagnostic yield comparable to or even higher than other neurodevelopmental disorders with a substantial burden of de novo variants [38]. We provide independent confirmation with unrelated cases for three genes previously implicated in CAS; SETD1A [4], DDX3X [5] and SETBP1 [5, 6]. We highlight chromatin organization and transcriptional regulation as critical biological mechanisms underpinning speech development.
The high confidence variants in this study were all located in genes previously associated with other common neurodevelopmental phenotypes including epilepsy, intellectual disability and ASD [4, 5]. These complex speech and neurodevelopmental presentations match our current understanding of genes that have been associated with ASD, epilepsy and/or intellectual disability, where pleiotropy, or overlapping comorbid phenotypes, are common [39]. However, for 15 of the 18 genes, this is the first time they have been specifically associated with CAS. Our work highlights the current bias in the literature to gene discovery cohorts across intellectual disability, autism and epilepsies relative to speech disorder. Although there is arguably some circularity here because our variant curation pipeline did prioritise variants previously associated with neurodevelopmental disorders. Still, we have expanded the phenotypic spectrum for a number of genes previously implicated in neurodevelopmental disorders, linking them with specific speech diagnoses, as well as markedly increasing the list of genes that should be prioritized for clinical testing in individuals with CAS.
Probands for whom we could provide a genetic diagnosis had a higher proportion of motor, language and cognitive impairments, secondary to the primary concern of CAS, compared to those probands without genetic diagnoses at a group level. We provide preliminary evidence for a threshold effect where monogenic conditions may be more likely when individuals with CAS have additional neurodevelopmental conditions, although further work on larger cohorts is needed to confirm this hypothesis. Only two probands (11%) with genetic diagnoses (SETD1B (ID10), ERF (ID18)) had CAS without co-occurring neurodevelopmental disorder diagnoses. One was aged 10;8 years, had average IQ and was attending a school for children with specific speech and language impairment. The other child was only 4;7 years and had not yet had IQ testing because no concerns had been raised by his treating physician, family or preschool teacher regarding his general learning ability; however, it is possible that other neurodevelopmental diagnoses could still be made into the future.
These findings expand the spectrum of phenotypes associated with these conditions. SETD1B has been previously associated with epilepsy, intellectual disability and language delay, and ERF-related craniosynostosis syndrome often includes speech and language delay, learning difficulties or behavioural problems; however variable expressivity and incomplete penetrance have previously been observed [40]. Our data suggest that monogenic causes can underpin the less commonly occurring more “specific” CAS phenotypes. We also reinforce the observation that, just as recent reports have suggested there are no ‘autism-(specific) genes,’ [39] it appears “speech-specific” monogenic conditions are also rare. This has recently been acknowledged for individuals with FOXP2 variants, where the phenotypic spectrum has been expanded from a relatively specific speech condition to include learning difficulties in at least some of the affected individuals [41, 42]. These observations of neurodevelopmental phenotypic comorbidity across genetic conditions and in our own data imply that it may be short-sighted to exclude children with autism, epilepsy or moderate to severe intellectual disability from studies examining the genetic basis of CAS.
In terms of the biological pathways associated with speech disorder, we found a significant over-representation of perturbed chromatin and transcriptional regulation pathways, consistent with prior studies [4, 5]. The five chromatin-related genes habouring high confidence variants in the current cohort were significantly co-expressed during brain development and are co-expressed with similar genes previously implicated in CAS[4, 5]. KDM5C encodes a histone demethylase involved in regulation of gene expression [43] and DNA methylation [44], and LoF mutations in this gene have been shown to cause intellectual disability in females [45]. BRPF1, encoding a histone acetyl transferase, has also been associated with intellectual disability and dysmorphic features [46]. SETD1A encodes a histone methyltranferase and has previously been associated with schizophrenia, intellectual disability, and speech and/or language delays [33]. SETD1B is also a histone methyltransferase associated with neurodevelopmental disorder [47]. PHF21A is a member of the BRAF35/histone deacetylase complex that mediates repression of neuron-specific genes [48] and has previously been associated with ASD and intellectual disability [49]. HNRNPK encodes an RNA-binding protein known to interact with many molecular partners in multiple processes that regulate gene expression: chromatin remodelling, transcription, and mRNA splicing, translation, and stability [50]. De novo truncating variants in HNRNPK have been shown to cause Au-Kline neurodevelopmental syndrome including intellectual disability, ADHD, speech impairment, cardiac anomalies and a variety of dysmorphic features [37]. These results support earlier findings [4, 5] that chromatin modifiers and transcriptional regulators are critical for speech development. More generally, chromatin modifiers play important roles in neurodevelopmental disorders as they are key regulators of progenitor expansion, differentiation, cell-type specification, migration and maturation, with early errors in chromatin remodelling known to impact development of brain networks [51].
Other genes confirmed to harbour pathogenic variants in CAS and associated with chromatin organization and transcriptional regulation were not as highly co-expressed during brain development. Among these, DDX3X, regulates gene splicing and is associated with neurodevelopmental disorders characterised by intellectual disability, ASD [52] and more recently, CAS [5]. RBFOX3, a gene showing neuron-specific expression, is also involved in splicing and has been associated with epilepsy aphasia syndrome and impaired language [53]. PURA is implicated in the control of both DNA replication and transcription [54], and PURA syndrome is noted to include ‘absent speech’ as a feature [55]. ZBTB18 encodes a transcriptional repressor shown to play a critical role in orchestrating brain development, and has been associated with non-syndromic intellectual disability [56].
The remaining candidate genes that we newly implicate in CAS (TAOK2, SPAST and ARHGEF9) encode proteins with distinct functions. TAOK2 is located in the 16p11.2 deletion region, a well-recognised CNV associated with CAS, among other neurodevelopmental phenotypes [10, 57]. The protein encoded by TAOK2 has established roles in dendrite growth and synapse development [58]. ARHGEF9 encodes collybistin, a brain-specific guanine nucleotide exchange factor. The gene has also been implicated in X-linked epileptic encephalopathy and neurodevelopmental disorder, where there is skewed X-inactivation in favour of the abnormal X-chromosome [59]. Finally, SPAST promotes microtubule growth in the cytoskeleton, playing an important role in neuronal development, and has been implicated in spastic paraplegia with dysarthric speech [60]. Although previous reports describing individuals with SPAST variants have not thoroughly characterised speech in the early years of life, it is possible that CAS was part of the early profile of such cases. A pattern of CAS alongside dysarthria is not uncommon in other genetic forms of persistent speech disorder, for example in Koolen de Vries Syndrome [61], SETBP1-haploinsufficiency disorder [6], or EBF3-related core motor disturbance and ataxia [5].
Whilst we were able to attain a diagnostic rate of 26%, other highly penetrant risk variants may have remained hidden due to our strict definition of high confidence variants, which was largely based on ACMG guidelines. By definition, for an identified variant to be deemed high confidence, a causative link to a relevant disorder must be already established. Hence it is likely that a proportion of our low confidence candidate variants are truly causal but currently lack sufficient evidence. Identification of additional variants in these low confidence genes in future cohorts of individuals with CAS would elevate thes findings to declare these genes as truly implicated in CAS. Currently, they provide candidate genes for future studies. Our filtering strategy may also have been overly conservative for genes harbouring variants with a recessive mode of inheritance. For example, we utilised pLI scores for prioritising LoF variants; these scores are relevant identifying genes that are intolerant of heterozygous protein truncating variants, but may be less appropriate for identifying genes that are intolerant to homozygous variants. We also performed an expanded variant identification analysis including short tandem repeats which yielded no hits, suggesting at this time, that they do not play a major role in CAS [62, 63].
Our data confirm that a substantial proportion of children with CAS or equally marked and persistent speech disorders may have a monogenic condition. As such CAS can be viewed as a critical clinical indicator for single gene disorders, due to its sensitivity as a rare phenotype (1 per 1000) [4], relative to more common speech diagnoses such as articulation or phonological disorder (1 in 20) [64]. While some individuals may have relatively ‘specific’ CAS in the absence of other neurodevelopmental disorders, our findings support the increasing overlap between genes conferring risk for a range of neurodevelopmental disorders including CAS, epilepsy, ASD and intellectual disability. This observation is important because well defined speech diagnoses are not typically reported in published clinical studies where the focus lies on other diagnoses like intellectual disability, epilepsy or ASD. If there is mention of speech or language impairment, the phrase ‘speech delay’ is typically used, which is a highly non-specific term that could imply general language understanding or expression difficulties (e.g. in semantic or syntactic domains), and hence may not even be referring to ‘speech’ impairment itself (e.g. difficulty with producing speech sounds) [12]. Thus, our work highlights the importance of specifically describing speech and language phenotypes, that is, at the very least being specific with the presence of clinical speech diagnoses of phonological disorder, stuttering, CAS and dysarthria, and specifying whether language is also impaired, and if so, in what domains, and by conducting genotype-phenotype correlation studies. We also propose considering a core diagnosis of CAS as a red flag for a monogenic condition. Understanding the aetiological basis of CAS is critical to end the diagnostic odyssey, identify comorbidities and ensure patients are poised for precision medicine trials.
Data availability
Data can be made available by contacting the corresponding author.
Change history
30 September 2022
Supplementary Information file was updated.
19 January 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41380-022-01879-y
References
American Speech-Language-Hearing A. Childhood apraxia of speech [Technical report]. 2007.
Morgan AT, Murray E, Liégeois FJ. Interventions for childhood apraxia of speech. Cochrane Database Syst Rev. 2018;5:CD006278.
Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 2001;413:519–23.
Eising E, Carrion-Castillo A, Vino A, Strand EA, Jakielski KJ, Scerri TS, et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol Psychiatry. 2019;24:1065–78.
Hildebrand MS, Jackson VE, Scerri TS, Van Reyk O, Coleman M, Braden RO, et al. Severe childhood speech disorder: Gene discovery highlights transcriptional dysregulation. Neurology 2020;94:e2148–e2167.
Morgan A, Braden R, Wong MMK, Colin E, Amor D, Liégeois F, et al. Speech and language deficits are central to SETBP1 haploinsufficiency disorder. Eur J Hum Genet. 2021;29:1216–25.
Lelieveld SH, Reijnders MRF, Pfundt R, Yntema HG, Kamsteeg E-J, de Vries P, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–6.
Stessman HAF, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. 2017;49:515–26.
Epi KC, Epilepsy Phenome/Genome P, Allen AS, Berkovic SF, Cossette P, Delanty N, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–21.
Mei C, Fedorenko E, Amor DJ, Boys A, Hoeflin C, Carew P, et al. Deep phenotyping of speech and language skills in individuals with 16p11. 2 deletion. Eur J Hum Genet. 2018;26:676–86.
Dodd B, Zhu H, Crosbie S, Holm A, Ozanne A. Diagnostic evaluation of articulation and phonology (DEAP). Psychology Corporation: London, 2002.
Braden RO, Amor DJ, Fisher SE, Mei C, Myers CT, Mefford H, et al. Severe speech impairment is a distinguishing feature of FOXP1-related disorder. Dev Med Child Neurol. 2021;63:1417–26.
Braden RO, Boyce JO, Stutterd CA, Pope K, Goel H, Leventer RJ, et al. Speech, Language, and Oromotor Skills in Patients With Polymicrogyria. Neurology 2021;96:e1898–e1912.
Wiig EH, Secord WA, Semel E. Clinical evaluation of language fundamentals: CELF-5. Pearson, 2013.
Dumont R, Willis JO. Kaufman Assessment Battery for Children–Second Edition. Encyclopedia of Special Education 2008:1196–7.
Wechsler D. Wechsler Intelligence Scale for Children Fifth Edition: WISC-V. Pearson; 2013.
Wechsler D. Wechsler preschool and primary scale of intelligence—fourth edition. The Psychological Corporation San Antonio, TX 2012.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bioGN] 2013.
Pedersen BS, Quinlan AR. Who’s Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy. Am J Hum Genet. 2017;100:406–13.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Turner SJ, Mayes AK, Verhoeven A, Mandelstam SA, Morgan AT, Scheffer IE. GRIN2A: an aptly named gene for speech dysfunction. Neurology 2015;84:586–93.
Martin AR, Williams E, Foulger RE, Leigh S, Daugherty LC, Niblock O, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. 2019;51:1560–5.
Banerjee-Basu S, Packer A. SFARI Gene: an evolving database for the autism research community. Dis Model Mech. 2010;3:133–5.
Wei Y, de Lange SC, Scholtens LH, Watanabe K, Ardesch DJ, Jansen PR, et al. Genetic mapping and evolutionary analysis of human-expanded cognitive networks. Nat Commun. 2019;10:1–11.
Khrameeva E, Kurochkin I, Han D, Guijarro P, Kanton S, Santel M et al. Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Res. 2020;30:776–89.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016;32:1220–2.
Scheinin I, Sie D, Bengtsson H, van de Wiel MA, Olshen AB, van Thuijl HF, et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 2014;24:2022–32.
Hildebrand MS, Harvey AS, Malone S, Damiano JA, Do H, Ye Z, et al. Somatic GNAQ mutation in the forme fruste of Sturge-Weber syndrome. Neurol Genet. 2018;4:e236.
Damiano JA, Do H, Ozturk E, Burgess R, Kalnins R, Jones NC, et al. Sensitive quantitative detection of somatic mosaic mutation in “double cortex” syndrome. Epileptic Disord. 2017;19:450–5.
Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature 2014;508:199–206.
Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–D503.
Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc. 2019;14:482–517.
Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19:571–7.
Popp B, Ekici AB, Thiel CT, Hoyer J, Wiesener A, Kraus C, et al. Exome Pool-Seq in neurodevelopmental disorders. Eur J Hum Genet. 2017;25:1364–76.
De Rubeis S, Siper PM, Durkin A, Weissman J, Muratet F, Halpern D, et al. Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol Autism. 2018;9:31.
Twigg SRF, Vorgia E, McGowan SJ, Peraki I, Fenwick AL, Sharma VP, et al. Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat Genet. 2013;45:308–13.
Au P, You J, Caluseriu O, Schwartzentruber J, Majewski J, Bernier F, et al. Care for Rare Canada, C, Valle D., Parboosingh JS et al. GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in HNRNPK. Hum Mutat. 2015;36:1009–14.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014;515:216–21.
Myers SM, Challman TD, Bernier R, Bourgeron T, Chung WK, Constantino JN, et al. Insufficient Evidence for “Autism-Specific” Genes. Am J Hum Genet. 2020;106:587–95.
Glass GE, O’Hara J, Canham N, Cilliers D, Dunaway D, Fenwick AL, et al. ERF-related craniosynostosis: The phenotypic and developmental profile of a new craniosynostosis syndrome. Am J Med Genet Part A. https://doi.org/10.1002/ajmg.a.61073.
Reuter MS, Riess A, Moog U, Briggs TA, Chandler KE, Rauch A, et al. FOXP2 variants in 14 individuals with developmental speech and language disorders broaden the mutational and clinical spectrum. J Med Genet. 2017;54:64–72.
Morgan A, Fisher SE, Scheffer I, Hildebrand M. FOXP2-related speech and language disorders. In: Pagon RAA, M. P; Ardinger H. H; Wallace, S. E; Amemiya, A; Bean, L. J. H; Bird, T. D; Fong, C, T; Mefford, H. C; Smith R. J. H; Stephens, K. (ed). GeneReviews: Seattle, Washington, 2017.
Brookes E, Laurent B, Õunap K, Carroll R, Moeschler JB, Field M, et al. Mutations in the intellectual disability gene KDM5C reduce protein stability and demethylase activity. Hum Mol Genet. 2015;24:2861–72.
Aref-Eshghi E, Kerkhof J, Pedro VP, Groupe DIF, Barat-Houari M, Ruiz-Pallares N, et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am J Hum Genet. 2020;106:356–70.
Ounap K, Puusepp-Benazzouz H, Peters M, Vaher U, Rein R, Proos A, et al. A novel c.2T > C mutation of the KDM5C/JARID1C gene in one large family with X-linked intellectual disability. Eur J Med Genet. 2012;55:178–84.
Mattioli F, Schaefer E, Magee A, Mark P, Mancini GM, Dieterich K, et al. Mutations in Histone Acetylase Modifier BRPF1 Cause an Autosomal-Dominant Form of Intellectual Disability with Associated Ptosis. Am J Hum Genet. 2017;100:105–16.
Roston A, Evans D, Gill H, McKinnon M, Isidor B, Cogné B, et al. SETD1B-associated neurodevelopmental disorder. J Med Genet. 2021;58:196–204.
Hakimi M-A, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci USA. 2002;99:7420–5.
Kim H-G, Rosenfeld JA, Scott DA, Bénédicte G, Labonne JD, Brown J, et al. Disruption of PHF21A causes syndromic intellectual disability with craniofacial anomalies, epilepsy, hypotonia, and neurobehavioral problems including autism. Mol Autism. 2019;10:35.
Fukuda T, Naiki T, Saito M, Irie K. hnRNP K interacts with RNA binding motif protein 42 and functions in the maintenance of cellular ATP level during stress conditions. Genes Cells. 2009;14:113–28.
Mossink B, Negwer M, Schubert D, Nadif, Kasri N. The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective. Cell Mol Life Sci. 2021;78:2517–63.
Beal B, Hayes I, McGaughran J, Amor DJ, Miteff C, Jackson V, et al. Expansion of phenotype of DDX3X syndrome: six new cases. Clin Dysmorphol. 2019;28:169–74.
Turner SJ, Morgan AT, Perez ER, Scheffer IE. New genes for focal epilepsies with speech and language disorders. Curr Neurol Neurosci Rep. 2015;15:35.
Hunt D, Leventer RJ, Simons C, Taft R, Swoboda KJ, Gawne-Cain M, et al. Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J Med Genet. 2014;51:806–13.
Reijnders MRF, Leventer RJ, Lee BH, Baralle D, Selber P, Paciorkowski AR et al. PURA-Related Neurodevelopmental Disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G et al. (eds). GeneReviews®. University of Washington, Seattle: Seattle (WA), 2017.
van der Schoot V, de Munnik S, Venselaar H, Elting M, Mancini GMS, Ravenswaaij-Arts CMA, et al. Toward clinical and molecular understanding of pathogenic variants in the ZBTB18 gene. Mol Genet Genom Med. 2018;6:393–400.
Fedorenko E, Morgan A, Murray E, Cardinaux A, Mei C, Tager-Flusberg H, et al. A highly penetrant form of childhood apraxia of speech due to deletion of 16p11.2. Eur J Hum Genet. 2016;24:302–6.
de Anda FC, Rosario AL, Durak O, Tran T, Gräff J, Meletis K, et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat Neurosci. 2012;15:1022–31.
Alber M, Kalscheuer VM, Marco E, Sherr E, Lesca G, Till M et al. ARHGEF9 disease. Neurology Genetics. 2017;3:e148.
Ogasawara M, Saito T, Koshimizu E, Akasaka N, Sasaki M. A p.Arg499His mutation in SPAST is associated with infantile onset ascending spastic paralysis complicated with dysarthria and anarthria. Neuropediatrics 2019;50:391–4.
Morgan AT, van Haaften L, van Hulst K, Edley C, Mei C, Tan TY, et al. Early speech development in Koolen de Vries syndrome limited by oral praxis and hypotonia. Eur J Hum Genet. 2018;26:75–84.
Folker JE, Murdoch BE, Rosen KM, Cahill LM, Delatycki MB, Corben LA, et al. Differentiating profiles of speech impairments in Friedreich’s ataxia: a perceptual and instrumental approach. Int J Lang Commun Disord. 2012;47:65–76.
Chan JCS, Stout JC, Vogel AP. Speech in prodromal and symptomatic Huntington’s disease as a model of measuring onset and progression in dominantly inherited neurodegenerative diseases. Neurosci Biobehav Rev. 2019;107:450–60.
Reilly S, McKean C, Morgan A, Wake M. Identifying and managing common childhood language and speech impairments. BMJ. 2015;350:h2318.
Kohlenberg TM, Trelles MP, McLarney B, Betancur C, Thurm A, Kolevzon A. Psychiatric illness and regression in individuals with Phelan-McDermid syndrome. J Neurodev Disord. 2020;12:7.
Acknowledgements
We thank the families for participating in this study.
Funding
This work was funded by a National Health and Medical Research Council (NHMRC) Centre of Research Excellence Grant (CRE-SLANG; 1116976, AM, MH, IS, MB, DA, SEF) and an NHMRC project grant (1160893, AM, MB, DA, MH, SEF). AM was supported by an NHMRC Practitioner Fellowship (1105008) and Investigator grant (1195955). KB was supported by Australian Research Council Future Fellowship (120100355). MB was supported by a NHMRC Senior Research Fellowship (1102971) and a NHMRC Investigator grant (1195236). IS was supported by an NHMRC Investigator grant (1172897). SEF is supported by the Max Planck Society. Additional funding was provided by the Independent Research Institute Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Program. Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
AM, MBa, IS, MH, SF designed the study, funded by their joint collaborative NHMRC grant. AM led the phenotyping work with IS, DA and supervised RB, OvR, TH, SD, ML, LM, EB. MBa led the bioinformatics work and supervised AK, VJ, MBe, ER, LW. MH led the molecular analysis and supervised MJC. AB conducted microarray analysis. AM, MB, AK created the original draft of the manuscript. Other authors contributed clinical patients and phenotyping information (RW, DC, MW, GD, LD, BPF, KB, EH, SZ). All authors contributed to and refined the draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Only two probands (11%) with genetic diagnoses (SETD1B (ID10), ERF (ID18)) had CAS without co-occurring neurodevelopmental disorder diagnoses. One was aged 10;8 years, had average IQ and was attending a school for children with specific speech and language impairment. The other child was only 4;7 years and had not yet had IQ testing because no concerns had been raised by his treating physician, family or preschool teacher regarding his general learning ability; however, it is possible that other neurodevelopmental diagnoses could still be made into the future (pg 13).
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaspi, A., Hildebrand, M.S., Jackson, V.E. et al. Genetic aetiologies for childhood speech disorder: novel pathways co-expressed during brain development. Mol Psychiatry 28, 1647–1663 (2023). https://doi.org/10.1038/s41380-022-01764-8
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01764-8
This article is cited by
-
CDK13-related disorder: a deep characterization of speech and language abilities and addition of 33 novel cases
European Journal of Human Genetics (2023)
