
Abstract
Alzheimer’s disease (AD) is a genetically complex disease for which nearly 40 loci have now been identified via genome-wide association studies (GWAS). We attempted to identify groups of rare variants (alternate allele frequency <0.01) associated with AD in a region-based, whole-genome sequencing (WGS) association study (rvGWAS) of two independent AD family datasets (NIMH/NIA; 2247 individuals; 605 families). Employing a sliding window approach across the genome, we identified several regions that achieved association p values <10−6, using the burden test or the SKAT statistic. The genomic region around the dystobrevin beta (DTNB) gene was identified with the burden and SKAT test and replicated in case/control samples from the ADSP study reaching genome-wide significance after meta-analysis (pmeta = 4.74 × 10−8). SKAT analysis also revealed region-based association around the Discs large homolog 2 (DLG2) gene and replicated in case/control samples from the ADSP study (pmeta = 1 × 10−6). In conclusion, in a region-based rvGWAS of AD we identified two novel AD genes, DLG2 and DTNB, based on association with rare variants.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is a heterogeneous, genetically complex neurodegenerative disorder [1]. Over the past 15 years, around 120 genome-wide association studies (GWAS) have been performed to elucidate the genetic architecture underlying AD according to the GWAS catalog [2]. The latest GWAS which has utilized over 1 million individuals ascertained from clinical and proxy-based AD cases and controls has identified 38 independent loci to be associated with AD [3]. GWAS heritability which is tagged by common variants is estimated to be 24-33% [4, 5] - less than a half of the heritability calculated from twin studies [1]. Identification of rare variants associated with AD may help explain the missing heritability, and lead to new biological insights [6]. Several rare variant loci previously associated with AD [7], including TREM2 [8, 9], have been identified with whole-exome sequencing (WES) studies [10].
Identification of association signals that are driven by rare variants remains cumbersome due to low power and relatively small sample sizes. Hence, aggregation methods, such as burden tests [11, 12] and variance component tests (SKAT) [13, 14], have been developed to jointly test regions of rare variants for association. Combining variant data increases the association signal and reduces the number of statistical tests. While burden tests are most powerful for signals with consistent effect directions, SKAT is more powerful for signals with different effect directions or when the fraction of causal variants within a region is small. Previously, aggregated gene-based association analyses have been successful in identifying exome-wide significant associations with sporadic AD [15,16,17,18]. This includes burden rare variant signals in genes with variants previously associated with AD, such as ABCA7, PILRA, SORL1, TREM2, as well as novel genes, such as ZNF655. Recently, we have performed a rare variant region-based analysis in whole-genome sequencing (WGS) data [19] using a family-based design and a burden family-based association test (FBAT), which was based on estimating the correlation between rare variants based on the observed empirical distribution [20]. Furthermore, an applicable SKAT approach in FBAT was not available at the time of the first manuscript [19]. In the current study, we utilize two novel and complementary region tests (burden and variance component (SKAT)) within a recently developed general framework for exact region-based association testing in family-based designs [21]. The former relates to an improved haplotype algorithm for rare variants recently developed by Hecker et al [22, 23]. This approach alleviates the need to use approximations of the correlation between rare variants: The joint conditional distribution of the rare variants can now be simply obtained by the haplotype approach. Using the proposed region-based testing framework and a systematic region definition, we performed a rvGWAS combining two AD family-based cohorts (605 families; 1509 affecteds; 738 unaffecteds) focusing on rare variants. For replication, we used case/control subjects from NIA ADSP, which included WGS data from a Non-Hispanic White (NHW) subcohort (983 cases; 686 controls), an African-American (AA) subcohort (450 cases; 501 controls), and a Hispanic (HISP) subcohort (486 cases; 613 controls).
Using a p value cutoff of 5 × 10−6, the burden test and SKAT identified several genomic regions showing association with AD risk. A region identified by the burden test in the DTNB gene (p = 7 × 10−8) was replicated in the NHW samples. SKAT analysis revealed an association with variants encompassing a region around DLG2 (p = 4 × 10−6), which replicated in the NHW and the AA samples.
Methods
Study populations
Discovery family-based dataset
Our discovery dataset consisted of two WGS family-based cohorts: the National Institute of Mental Health (NIMH) family AD cohort [24] and families from the National Institute of Aging Alzheimer’s Disease Sequencing Project [25] (NIA ADSP). Whole-genome sequencing and variant calling in NIMH are described elsewhere [26]. Variant calls for the families from the NIA ADSP cohort were obtained from the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS; URLs) under accession number: NG00067. Both cohorts consisted of multiplex AD families with affected and unaffected siblings (Supplementary table 1). A subject was considered to be affected if he/she was included in one of the following categories: “Definite AD”,”Probable AD” or”Possible AD”. Subjects were counted as unaffected by AD if they either had no diagnosis of dementia, or suspected dementia (46 subjects), or non-AD dementia (10 subjects). Given FBAT’s robustness to model misspecifications [27] (including case vs. control status) this strategy will generally result in an increase of statistical power (owing to the larger sample size). It is important to note that even in families where the non-AD or suspected dementia is actually the result of an overlooked diagnosis of AD, this will not lead to spurious findings but merely reduce statistical power. It is important to note that NIA ADSP families by design did not include individuals with two APOE-ε4 alleles. After standard quality control, both cohorts were merged together.
NIA ADSP case-control dataset
WGS variant calls for the NIA ADSP replication case-control dataset were obtained from the NIAGADS under accession number: NG00067 and consisted of the ADSP Discovery-Extension Case-Control WGS dataset [25] and the ADNI Case-Control WGS dataset. Samples were remapped to GRCh38 and jointly called with the families from the NIA ADSP cohort. Full details can be found on NIAGADS (https://dss.niagads.org/datasets/ng00067/) and elsewhere [28]. Briefly, a subject was considered affected, if he/she met the NINCDS-ADRDA criteria for possible, probable, or definite AD, had documented age at onset or age at death (for pathologically verified cases), and APOE genotyping. All controls were 60 or more years old and were free of dementia.
Quality control
Briefly, we have excluded individuals based on genotyping rate, inbreeding coefficient, and family mismatches using identity by descent (IBD) sharing coefficients. After sample-based quality control, we have combined two WGS family-based cohorts: NIMH (1393 individuals in 446 families) and families from NIA ADSP (854 individuals in 159 families). In the merged dataset we excluded multiallelic variants, monomorphic variants, singletons (i.e., variants with only one alternate allele across the dataset and variants seen only in one family), indels, and variants which had one missing allele among 2 alleles in an individual. The remaining variants were filtered based on Mendel errors, genotyping rate (95%), Hardy–Weinberg equilibrium (p < 1e−08), calling quality in TOPMed (URLs), which is a large WGS database with >100,000 individuals sequenced jointly, and alternate allele frequency as defined in gnomAD (AF ≤ 1% in either whole gnomAD or nonFinnish European sample) (URLs).
WGS regional-based analysis
We have performed a whole-genome scan for our combined family-based AD dataset using a newly developed exact framework in FBAT for region-based association testing [21]. We grouped rare variants in nonoverlapping consecutive sets of ten based on our discovery family-based dataset. For each set of rare variants, we considered the burden test and the SKAT test using Affection Status (coded as 0/1 for unaffected/affected) minus offset as phenotype. We selected an offset of 0.15 which approximately corresponds to the population prevalence of AD. We have used FBAT [29] (URLs), R [30], snakemake [31] and bash commands to implement and run the described analyses.
Replication
Replication significance level was set to 0.05. In addition, we compute the combined meta p values and highlight regions that reached overall Bonferroni-corrected genome-wide significance (p < 6.24 × 10−8). We have used the SKAT package to perform Burden and SKAT-O tests on the same sets of rare variants in the case-control replication cohorts. We chose SKAT-O because it is the optimal test in an extended family of SKAT tests and combines the power of a burden and SKAT test and it implements a small-sample adjustment procedure (n < 2000) [14]. As covariates, we used sequencing center, age, sex, and principal components (to account for population structure). To recover more recent admixture and better correct for population stratification in WGS data we calculated principal components based on 100,000 rare variants using the Jaccard index [32]. Those rare variants were randomly selected from a pruned subset of rare variants (R2 < = 0.01). We have also performed meta-analysis among datasets with similar ethnical background using the Fisher’s combined probability test.
RNA-Seq and microarray analysis
We explored DLG2 and DTNB genes’ expression based on the Human Protein Atlas (HPA) RNA-seq data (URLs) and tested for differential expression of synaptic and immune-related genes including DLG2 and DTNB genes between normal controls (N = 173, aged 20–99 years) and AD cases (N = 80) in the brain regions including hippocampus, entorhinal cortex, superior frontal cortex, and post-central gyrus using microarray dataset GSE48350, which is available from the Gene Expression Omnibus Web site (URLs). Differential expression was tested using the “GEO2R” tool.
Network construction
We used Cytoscape 3.8.0 and the StringDB protein-protein interaction resource [33] (URLs) using only identified protein-protein interactions. Using a background that agglomerates protein-protein interaction datasets, we seeded the network with DLG2 and DTNB and identified direct associations between proteins and DLG2 and DTNB in a global network. Results were combined using the Genemania server (Utilizing significantly co-expressed genes across several experimental datasets) [34] to further capture functional relationships and to build a combined protein-protein/gene co-expression network.
Functional enrichment
Functional enrichment within the network was performed using the remote StringDB server linked to Cytoscape “String App Enrichment function” [35], producing enrichments using the hypergeometric test, with P values corrected for multiple testing using the method of Benjamini and Hochberg in known molecular pathways and GO terms as described in Franceschini et al. [36].
Results
In a region-based whole-genome sequencing rvGWAS focusing on rare genomic variants, we combined two AD family-based cohorts, the NIMH Alzheimer’s disease genetics initiative study (NIMH) and the family component of the NIA ADSP sample. The combined sample consisted of 1509 affected and 738 unaffected siblings in families of predominantly European ancestry (Supplementary Table 1, Methods). 8,011,126 variants passed strict quality control and alternate allele frequency (AF) filter of ≤1% (based on gnomAD [37]). We grouped rare variants into consecutive non-overlapping regions/windows of ten variants and performed a rare variant WGS scan over the whole genome (801,124 windows). We employed a recently developed framework for exact regional-based analysis within FBAT [21] to analyze these sets of rare variants using both the burden test and SKAT. These tests are able to detect different configurations of disease regions - dense regions with the same effect directions (burden test) or less dense signals with varying effect directions (SKAT).
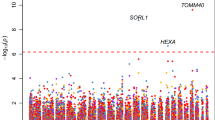
Since we restricted our analysis to rare variants (i.e., AF < 0.01) and given our modest sample size in the family-discovery cohort, we have used a relatively liberal p value threshold p < 5 × 10−6 to identify “suggestive associations” by burden test or SKAT. A stricter Bonferroni-corrected significance threshold would be p = 6.24 × 10−8. Seven loci exhibited suggestive evidence for association with AD risk (Fig. 1, Supplementary Fig. 1, Table 1). For replication analysis, we selected the unrelated, multiethnic WGS AD subset from the NIA ADSP dataset (Methods). This dataset consists of three subpopulations: NHW (n = 1669), AA (n = 951), HISP (n = 1099) (Sample sizes after quality control; Supplementary Table 1). A region located downstream to DTNB, with a burden p value of 7 × 10−8 and a SKAT p value of 1.4 × 10−6 in the discovery dataset, showed a burden p value of 0.0324 and a SKAT-O p value of 0.054 in the replication ADSP NHW dataset (Table 1 and Supplementary Table 2). Another region, located in an intron of DLG2 with a SKAT p value of 4 × 10−6 in the discovery family-based dataset, showed replication with a significant p value of 0.0143 in the ADSP NHW dataset and a p value of 0.053 in the ADSP AA dataset (Table 1 and Supplementary Table 3). Two other regions showed nominally significant replication p values in AA (SEMA3C, p = 0.046) and HISP (ISX, p = 0.014), but not in NHW.

Dashed line corresponds to suggestive threshold of 5 × 10−6.
During the peer review of this paper NIA ADSP released a larger AD WGS case-control dataset with a 4.5-fold sample size increase for the NHW subpopulation, which we used for additional replication analyses. To this end, we recalculated the burden and the SKAT-O test statistics for our two top hits in n = 7413 NHW individuals (previously n = 1669). The analyses revealed only minor changes with respect to our original replication results, i.e., the burden p value for DTNB increased to 0.0698 while the SKAT-O p value for DLG2 decreased to 0.0038.
Both DLG2 and DTNB are highly expressed in the brain based on RNA-data from three different sources: Internally generated Human Protein Atlas (HPA) RNA-seq data, RNA-seq data from the Genotype-Tissue Expression (GTEx) project, and CAGE data from the FANTOM5 project, as well as the consensus dataset for each gene derived from the Human Protein Atlas [38] (Supplementary Figs. 2, 3). It is important to mention that besides being expressed in different brain tissues, DTNB is also highly expressed in salivary gland tissue, which might be due to the fact that the resulting protein, β-dystrobrevin, is only found in non-muscle tissues [39]. In the Alzheimer’s Disease Dataset analysis [40] (GSE48350) from the GEO database [41] expression of DLG2 and DTNB is significantly decreased in AD compared to control subjects in at least one of two microarray ids corresponding to the genes (Supplementary Table 4).
Network analysis revealed a network of 33 proteins interacting with DLG2 and DTNB that were enriched for neuronal synaptic functions (Supplementary Fig. 4). Functional enrichment of the subnetwork of proteins directly interacting with DLG2 and DTNB revealed 694 enriched GO process/ pathway terms (Supplementary table 5). The most enriched part of the network was for proteins interacting with DLG2 that are connected to neurexins and neuroligins, as well as trafficking of AMPA receptors. DLG2 also interacted with 4 proteins (NOS1, ERBB4, DLGAP2, NRXN3) which were among the top 1000 leading AD-associated single rare variants and regions [19], and 4 proteins (GRIN1, GRIN2A, GRIN2B, GAPDH) associated with AD in the KEGG Alzheimer’s pathway. DLG2 and DTNB also share protein-protein or co-expression interactions through KIF1B, MLC1, and SH3D19.
Discussion
Here, we describe a comprehensive region-based analysis of Alzheimer’s disease using WGS datasets. We specifically searched for novel AD association signals driven by regions of rare variants in a large family-based cohort. To account for different disease region specifications, we employed both the burden test and SKAT. This yielded seven regions of suggestive evidence (p < 5 × 10−6) for association with AD risk in the family datasets. These results were followed up with replication analysis in independent case-control samples of different ethnicities. Two loci, DTNB and DLG2, showed consistent evidence of replication in the NHW subpopulation. The DLG2 region was also confirmed in the African-American sample.
DLG2 encodes a member of the membrane-associated guanylate kinase family, also known as post-synaptic density protein, PSD-93. Down-regulation of synaptic scaffolding proteins, including DLG2, has been described as an early event in AD [42]. DLG2 has been proposed as a potential target for AD based on an integrated metabolomics-genetics-imaging systems approach in Agora (URLs); agonism of DLG2 is predicted to reduce disease progression. An expression dataset of AD in the GEO database revealed reduced expression of DLG2 in AD versus controls. A common variant in DLG2, rs683250, was previously associated with increases of shape asymmetry in controls as compared to individuals with dementia [43]. This same variant is in linkage disequilibrium (LD, D’ = 1) with all rare variants of the DLG2 region found to be associated with AD here. DLG2 variant, rs286043 (AF = 0.03), which exhibited suggestive evidence for association with AD risk in IGAP (p = 5e−06), is in LD with 4 out of 10 variants from our DLG2 AD-associated region, suggesting possible allelic heterogeneity. DLG2 has previously been associated with schizophrenia [44] and autism [45, 46]. Along these lines, DLG2 deficiency in mice has been reported to lead to reduced sociability and increased repetitive behavior along with aberrant synaptic transmission in the dorsal striatum [47].
β-Dystrobrevin (DTNB) is associated with neurons in the cortex, hippocampus, and cerebellum, as well as other brain regions, implying that it might be an important protein involved in some neuronal pathways in the brain [39], and has also been reported to be enriched in the post-synaptic density (PSD), a protein complex associated with postsynaptic membranes of excitatory synapses [48, 49]. Kinesin superfamily motor proteins (KIF) are responsible for anterograde protein transport within the axon of various cellular cargoes, including synaptic and structural proteins [50]. Dysregulated KIF expression has also been associated with early AD pathology [51], and β-Dystrobrevin interacts directly with kinesin heavy chain in the brain [52]. Expression of α-Dystrobrevin (DTNA), which a paralog of DTNB, has been associated with dementia status and P-tau levels in temporal cortex [53]. Dystrobrevin-binding protein 1, also known as dysbindin, has been reported to be associated with schizophrenia [54, 55]. Thus, both novel AD gene candidates identified in this study have been associated with post-synaptic function. They have also shown association with risk for schizophrenia. While schizophrenia and Alzheimer’s disease have, generally, different etiologies (including genetics), other studies have shown that there are some important intersections between both diseases, especially related to post-synaptic density proteins [56,57,58]. The two novel AD genes identified here might be located at one of such functional intersections.
Family-based designs are completely robust to potential misspecification of disease model and population stratification. This led us to define the family-based portion of our study as our “discovery” dataset. In contrast, the “replication” portion of our study utilized datasets from unrelated cases and controls. Two regions (DLG2 and DTNB) were validated in the replication cohort. Utilizing the increased sample size of the latest NIAGADS release, the replication evidence became stronger for DLG2 and slightly decreased for DTNB.
Concurrent to our analyses, we became aware of an independent WES study of AD cerebrospinal fluid (CSF) biomarker levels by Neumann et al. [59]. Intriguingly, that study also identified rare variants in DTNB showing experiment-wide rare-variant association signals with the CSF biomarkers analyzed. Thus, there are now two studies using independent datasets, sequencing techniques and different AD-related outcome phenotypes converging on highly significant rare-variant association signals in DTNB, emphasizing the likely crucial – and hitherto unrecognized – role of this gene in AD pathogenesis.
Our approach utilized two region-based tests (burden and SKAT) in a family-based design, in which the joint distribution of rare variants is not estimated, but rather obtained by the haplotype algorithm for FBAT, which is robust against population structure and admixture, and allows for construction of exact or simulation-based p values. Previously, we performed region-based rare variant testing, but with different region definitions, and using only burden tests with empirical estimation of the variant correlations and asymptotic p values [19]. We also note that by utilizing a window size of 10 consecutive variants, we could have missed sparsely distributed signals. Since the number of possible haplotypes increases exponentially with the number of variants tested, larger window sizes were computationally infeasible.
In summary, we identified two novel loci associated with AD, based on association with rare variants in DLG2 and DTNB in a family-based AD WGS sample using methods that are robust to population structure. Both novel AD genes identified here encode post-synaptic density proteins and have been implicated for roles in schizophrenia. These loci showed replication in an independent AD WGS dataset with unrelated cases and controls and, additionally, DTNB was recently highlighted in independent work [59] on the effect of rare-variants on AD CSF biomarker levels. In this separate work Neumann et al. using WES reported rare-variant association signals between DTNB and AD CSF biomarker levels in two independent datasets, which makes further studies on the role of DTNB in AD pathogenesis warranted.
URLs
FBAT, https://sites.google.com/view/fbatwebpage; gnomAD, https://gnomad.broadinstitute.org/; Agora AMP-AD, https://agora.ampadportal.org/genes; TOPMED, https://www.nhlbiwgs.org/; Human Protein Atlas, https://www.proteinatlas.org/; GEO database, https://www.ncbi.nlm.nih.gov/geo/; NIAGADS, https://www.niagads.org/; StringDB, https://string-db.org/.
Data availability
The NIMH dataset analyzed during the current study is available from the corresponding author on reasonable request. The family component and the case-control component of the NIA ADSP WGS dataset is available from DSS NIAGADS under accession number: NG00067. Data used in preparation of this article were in part obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report.
Code availability
All scripts used to generate the region-based analyses are available from the authors upon request.
References
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–74.
Buniello A, Macarthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012.
Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53:1276–82.
Lee SH, Harold D, Nyholt DR, Goddard ME, Zondervan KT, Williams J, et al. Estimation and partitioning of polygenic variation captured by common snps for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum Mol Genet. 2013;22:832–41.
Ridge PG, Mukherjee S, Crane PK, Kauwe JSK. Alzheimer’s disease: Analyzing the missing heritability. PLoS One. 2013;8:1–10.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature 2009;461:747–53.
Grozeva D, Saad S, Menzies GE, Sims R. Benefits and challenges of rare genetic variation in Alzheimer’s disease. Curr Genet Med Rep. 2019;7:53–62.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–16.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–27.
Lill CM, Rengmark A, Pihlstrøm L, Fogh I, Shatunov A, Sleiman PM, et al. The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement. 2015;11:1407–16.
Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384.
Li B, Leal S. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–21.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93.
Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–37.
Bis JC, Jian X, Kunkle BW, Chen Y, Hamilton-Nelson KL, Bush WS, et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Mol Psychiatry. 2018. 2018. https://doi.org/10.1038/s41380-018-0112-7.
Patel T, Brookes KJ, Turton J, Chaudhury S, Guetta-Baranes T, Guerreiro R, et al. Whole-exome sequencing of the BDR cohort: evidence to support the role of the PILRA gene in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2018;44:506–21.
Raghavan NS, Brickman AM, Andrews H, Manly JJ, Schupf N, Lantigua R, et al. Whole-exome sequencing in 20,197 persons for rare variants in Alzheimer’s disease. Ann Clin Transl Neurol. 2018;5:832–42.
Ma Y, Jun GR, Zhang X, Chung J, Naj AC, Chen Y, et al. Analysis of whole-exome sequencing data for Alzheimer disease stratified by APOE genotype. JAMA Neurol. 2019;76:1099–108.
Prokopenko D, Morgan SL, Mullin K, Hofmann O, Chapman B, Kirchner R, et al. Whole‐genome sequencing reveals new Alzheimer’s disease–associated rare variants in loci related to synaptic function and neuronal development. Alzheimer’s Dement. 2021;17:1509–27.
De G, Yip WK, Ionita-Laza I, Laird N. Rare variant analysis for family-based design. PLoS One. 2013;8:e48495.
Hecker J, Townes FW, Kachroo P, Laurie C, Lasky-Su J, Ziniti J, et al. A unifying framework for rare variant association testing in family-based designs, including higher criticism approaches, SKATs, and burden tests. Bioinformatics. 2020;36:5432–8.
Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM. Family-based tests for associating haplotypes with general phenotype data: application to asthma genetics. Genet Epidemiol. 2004;26:61–69.
Hecker J, Xu X, Townes FW, Loehlein Fier H, Corcoran C, Laird N, et al. Family-based tests for associating haplotypes with general phenotype data: Improving the FBAT-haplotype algorithm. Genet Epidemiol. 2018;42:123–6.
Blacker D, Albert MS, Haines JL, Rodes L, Terwedow H, Go RC, et al. ApoE-4 and age at onset of Alzheimer’s disease: the NIMH genetics initiative. Neurology. 1997;48:139–47.
Beecham GW, Bis JC, Martin ER, Choi S-H, DeStefano AL, van Duijn CM, et al. The Alzheimer’s disease sequencing project: study design and sample selection. Neurol Genet. 2017;3:e194.
Prokopenko D, Hecker J, Kirchner R, Chapman BA, Hoffman O, Mullin K, et al. Identification of novel Alzheimer’s disease loci using sex-specific family-based association analysis of whole-genome sequence data. Sci Rep. 2020;10:1–9.
Laird NM, Lange C. Family-based designs in the age of large-scale gene-association studies. Nat Rev Genet. 2006;7:385–94.
Leung YY, Valladares O, Chou YF, Lin HJ, Kuzma AB, Cantwell L, et al. VCPA: genomic variant calling pipeline and data management tool for Alzheimer’s disease sequencing project. Bioinformatics. 2019;35:1768–70.
Laird N, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19 Suppl 1:S36-42.
Team RC. R: a language and environment for statistical computing. https://www.r-project.org.
Köster J, Rahmann S. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2.
Prokopenko D, Hecker J, Silverman E, Pagano M, Nöthen MM, Dina C, et al. Utilizing the Jaccard index to reveal population stratification in sequencing data: a simulation study and an application to the 1000 Genomes Project. Bioinformatics. 2016;32:1366–72.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:214–20.
Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. Cytoscape StringApp: network analysis and visualization of proteomics data. J Proteome Res. 2019;18:623–32.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:808–15.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419.
Blake DJ, Nawrotzki R, Loh NY, Górecki DC, Davies KE. Β-dystrobrevin, a member of the dystrophin-related protein family. Proc Natl Acad Sci USA. 1998;95:241–6.
Berchtold NC, Coleman PD, Cribbs DH, Rogers J, Gillen DL, Cotman CW. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol Aging. 2013;34:1653–61.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets - update. Nucleic Acids Res. 2013;41:991–5.
Xu J, Patassini S, Rustogi N, Riba-garcia I, Hale BD, Phillips AM, et al. Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun Biol. https://doi.org/10.1038/s42003-018-0254-9.
Wachinger C, Nho K, Saykin AJ, Reuter M, Rieckmann A. A longitudinal imaging genetics study of neuroanatomical asymmetry in Alzheimer’s disease. Biol Psychiatry. 2018;84:522–30.
Ingason A, Giegling I, Hartmann AM, Genius J, Konte B, Friedl M, et al. Expression analysis in a rat psychosis model identifies novel candidate genes validated in a large case-control sample of schizophrenia. Transl Psychiatry. 2015;5:e656.
Egger G, Roetzer KM, Noor A, Lionel AC, Mahmood H, Schwarzbraun T, et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics. 2014;15:117–27.
Ruzzo EK, Pérez-Cano L, Jung JY, Wang LK, Kashef-Haghighi D, Hartl C. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178:850–.e26.
Yoo T, Kim SG, Yang SH, Kim H, Kim E, Kim SY. A DLG2 deficiency in mice leads to reduced sociability and increased repetitive behavior accompanied by aberrant synaptic transmission in the dorsal striatum. Mol Autism. 2020;11:1–14.
Blake DJ, Hawkes R, Benson MA, Beesley PW. Different dystrophin-like complexes are expressed in neurons and glia. J Cell Biol. 1999;147:645–57.
Kaizuka T, Takumi T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J Biochem. 2018;163:447–55.
Hirokawa N, Noda Y. Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol Rev. 2008;88:1089–118.
Andersson ME, Sjölander A, Andreasen N, Minthon L, Hansson O, Bogdanovic N, et al. Kinesin gene variability may affect tau phosphorylation in early Alzheimer’s disease. Int J Mol Med. 2007;20:233–9.
Macioce P, Gambara G, Bernassola M, Gaddini L, Torreri P, Macchia G, et al. β-Dystrobrevin interacts directly with kinesin heavy chain in brain. J Cell Sci. 2003;116:4847–56.
Simon MJ, Wang MX, Murchison CF, Roese NE, Boespflug EL, Woltjer RL, et al. Transcriptional network analysis of human astrocytic endfoot genes reveals region-specific associations with dementia status and tau pathology. Sci Rep. 2018;8:1–16.
Baek JH, Kim JS, Ryu S, Oh S, Noh J, Lee WK, et al. Association of genetic variations in DTNBP1 with cognitive function in schizophrenia patients and healthy subjects. Am J Med Genet Part B Neuropsychiatr Genet. 2012;159 B:841–9.
Yang Y, Zhang L, Guo D, Zhang L, Yu H, Liu Q, et al. Association of DTNBP1 with schizophrenia: findings from two independent samples of Han Chinese population. Front Psychiatry. 2020;11:1–9.
Krivinko JM, Erickson SL, Ding Y, Sun Z, Penzes P, MacDonald ML, et al. Synaptic proteome compensation and resilience to psychosis in Alzheimer’s disease. Am J Psychiatry. 2018;175:999–1009.
Inestrosa NC, Montecinos-Oliva C, Fuenzalida M. Wnt signaling: role in Alzheimer disease and schizophrenia. J Neuroimmune Pharm. 2012;7:788–807.
Cong Q, Soteros BM, Wollet M, Kim JH, Sia GM. The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat Neurosci. 2020;23:1067–78.
Neumann A, Küçükali F, Bos I, Vos SJB, Engelborghs S, De Pooter T, et al. Rare variants in IFFO1, DTNB and NLRC3 associate with Alzheimer’s disease CSF profile of neuronal injury and inflammation. MedRxiv. 2021. https://www.medrxiv.org/content/10.1101/2021.07.10.21260177v1.
Acknowledgements
This work was supported by Cure Alzheimer’s Fund, JPB Foundation and NIH R56AG057191 (DWF and YK). The computations in this paper were run in part on the FASRC Cannon cluster supported by the FAS Division of Science Research Computing Group at Harvard University with support from John Morrissey and in part on computers provided by Dell HPC Research Computing Solutions with support by Glen Otero. The funding body has no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. Please refer to the Supplementary Note for full acknowledgements.
Author information
Authors and Affiliations
Consortia
Contributions
DP, CL, RET contributed to the study concept and design. DP, SL, KM, SM, YK, DWF, LB, WH, CL, RET contributed to data analysis and/or interpretation. JH, NL, CL contributed to statistical support. DP, SL, WH, CL, LB and RET wrote the original draft of the paper, and all authors critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This research project is approved by the Institutional Review Board (IRB) (2015P000111) at Massachusetts General Hospital. Informed consent was obtained from all subjects. All methods were carried out in accordance with relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prokopenko, D., Lee, S., Hecker, J. et al. Region-based analysis of rare genomic variants in whole-genome sequencing datasets reveal two novel Alzheimer’s disease-associated genes: DTNB and DLG2. Mol Psychiatry 27, 1963–1969 (2022). https://doi.org/10.1038/s41380-022-01475-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01475-0
This article is cited by
-
Omics-based biomarkers discovery for Alzheimer's disease
Cellular and Molecular Life Sciences (2022)
