
Abstract
Alzheimer’s disease (AD) is the most common multifactorial neurodegenerative disease among elderly people. Genome-wide association studies (GWAS) have been highly successful in identifying genetic risk factors. However, GWAS investigate common variants, which tend to have small effect sizes, and rare variants with potentially larger phenotypic effects have not been sufficiently investigated. Whole-genome sequencing (WGS) enables us to detect those rare variants. Here, we performed rare-variant association studies by using WGS data from 140 individuals with probable AD and 798 cognitively normal elder controls (CN), as well as single-nucleotide polymorphism genotyping data from an independent large Japanese AD cohort of 1604 AD and 1235 CN subjects. We identified two rare variants as candidates for AD association: a missense variant in OR51G1 (rs146006146, c.815 G > A, p.R272H) and a stop-gain variant in MLKL (rs763812068, c.142 C > T, p.Q48X). Subsequent in vitro functional analysis revealed that the MLKL stop-gain variant can contribute to increases not only in abnormal cells that should die by programmed cell death but do not, but also in the ratio of Aβ42 to Aβ40. We further detected AD candidate genes through gene-based association tests of rare variants; a network-based meta-analysis using these candidates identified four functionally important hub genes (NCOR2, PLEC, DMD, and NEDD4). Our findings will contribute to the understanding of AD and provide novel insights into its pathogenic mechanisms that can be used in future studies.
Similar content being viewed by others


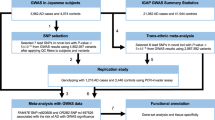
Introduction
Alzheimer’s disease (AD) is the most common multifactorial neurodegenerative disease among elderly people [1, 2]. To date, there are no curative treatments for patients who already have AD, and available treatments are only able to delay the progression of the disease [3]. AD is classified into two types according to the age of onset: early-onset AD (EOAD), diagnosed in people <65 years, and late-onset AD (LOAD), diagnosed in people ≥65 years [4]. EOAD accounts for up to 5% of all AD cases, most of which are caused by rare autosomal dominant mutations in one of three genes: amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) [5]. The majority of AD cases are sporadic LOAD, a heterogeneous disorder with complex interactions between genetic and environmental risk factors, which are influenced by multiple common variants with low effect sizes [6, 7]. Estimates of the genetic heritability of LOAD range between 60% and 80% [8]. The ε4 polymorphism in the protein encoded by the apolipoprotein E (APOE) gene, located on chromosome 19, is considered to be the strongest genetic risk factor for LOAD [9]. However, the APOE ε4 effect accounts for only 27.3% of the overall heritability [10], and a large proportion of the heritability remains unexplained.
Genome-wide association studies (GWAS) have been highly successful in identifying additional genetic risk factors associated with LOAD [7, 11]. Kunkle et al. identified 25 AD risk loci with genome-wide significance by using genomes from over 90000 individuals, including more than 30000 with LOAD [7]. Jansen et al. identified 29 AD risk loci through genome-wide meta-analysis in a case–control study of clinically diagnosed AD (>600,000 individuals) [11]. These findings can explain a fraction of the missing heritability of LOAD. However, GWAS mostly investigate common variants with a minor allele frequency (MAF) ≥ 0.01, which tend to have small effect sizes. Rare variants (MAF < 0.01), which potentially have larger phenotypic effects, have not been sufficiently investigated in LOAD.
The development of next-generation sequencing technologies [12] enables us to detect rare variants. Whole-exome sequencing (WES) and whole-genome sequencing (WGS) have been used to identify rare causal mutations of Mendelian diseases [13, 14] and driver mutations in tumors [15,16,17]. Although WGS is still less cost-effective than WES, WGS is able to detect more coding variants than WES owing to the limitations of WES capture methods and the advantages of the broader coverage of WGS [18]. Here, we applied WGS to samples from clinically characterized individuals with probable AD and cognitively normal elder controls (CN). We comprehensively investigated pathogenic rare variants associated with AD with single-nucleotide polymorphism (SNP) genotyping data from a large independent Japanese AD case–control cohort. We identified two candidates for rare AD variants: a missense variant in OR51G1 and a stop-gain variant in MLKL. Subsequent in vitro functional analyses demonstrated that the MLKL loss-of-function variant plays a crucial role in the pathogenesis of AD. Furthermore, gene-based association tests of rare variants detected AD candidate genes. A subsequent network-based meta-analysis using the candidates revealed functionally important modules (i.e., hub genes). Our findings contribute to the understanding of AD and provide novel insights into its mechanisms of pathogenesis for further investigation in future studies.
Results
WGS of Japanese subjects
A total of 3777 samples from 1744 Japanese individuals with AD and 2033 Japanese CN, all at least 60 years old, were included in this study. We performed WGS on 938 of the samples by using the Illumina HiSeq X Ten and NovaSeq 6000 platforms. On average, 365 million read pairs were obtained from the WGS. Of these, 98.3% were mapped to the human reference genome (GRCh37) and 7.6% were removed as duplicate PCR read pairs (Supplementary Table S1). Variant calling was performed by using Genome Analysis Toolkit (GATK) [19]. A total of 15,361,434 genetic variants (single-nucleotide variants [SNVs] and short insertions and deletions [Indels]) from 933 samples passed stringent quality control (QC) criteria for both genotypes and samples (see Materials and Methods). The QC-passed samples consisted of 139 AD and 794 CN samples. Of the 15,361,434 genetic variants, 130,533 were found in protein-coding regions. The average ages of the individuals from whom the AD and CN samples were obtained was 70.5 years (SD = 8.1 years) and 76.5 years (SD = 4.5 years), respectively, and the female-to-male ratios were 1.44:1 and 1.24:1, respectively.
Variant-based association study for AD pathogenic genes
We classified the 130,533 coding variants into three variant types: rare (n = 79,842, 61.2%), infrequent (n = 17,100, 13.1%), and common (n = 33,591, 25.7%) (see Materials and Methods). We functionally annotated the coding variants. Of the 130,533 variants, 988 were assigned to frameshift deletion; 521 to frameshift insertion: 974 to nonframeshift deletion; 447 to nonframeshift insertion; 856 to splicing variant; 1074 to stop-gain; 76 to stop-loss; 58,611 to synonymous SNV; and 66,986 to nonsynonymous SNV. Rare variants had more potentially deleterious mutations (frameshift deletion/insertion; splicing variants; stop-gain/stop-loss; nonsynonymous SNV) than infrequent and common variants (Fig. 1).

Coding variants were assigned the indicated functional annotations by using ANNOVAR. Of the annotations, frameshift deletion/insertion, splicing variants, stop-gain/stop-loss, and nonsynonymous SNVs were defined as potentially deleterious mutations. On the basis of the minor allele frequency (MAF), available from ToMMo 8.3KJPN, the variants were classified into three groups: rare (MAF < 0.01), infrequent (0.01 ≤ MAF ≤ 0.05), and common (0.05 < MAF).
We used functional rare variants for the detection of association signals (discovery data set). The associations were assessed with Fisher’s exact tests. A total of nine genetic markers, located within FAM126A, ZFHX4, LGR5, ZFC3H1, OR51G1, OR4X2, ARHGEF4, PRUNE2, and MLKL, were identified as possible associations (Pdiscovery < 5.0 × 10−4, Table 1). Also, when we examined association signals in all coding variants, one common variant in APOE, which has been reported as an AD susceptibility locus in several populations [7, 11], reached the GWAS significance level (rs429358, odds ratio [OR] = 4.23, 95% confidence interval [CI] 2.90–6.15, Pdiscovery < 5.0 × 10−8, Fig. S1).
Replication study and meta-analysis
For validation assessments, the nine possible association signals identified above were genotyped by using an independent Japanese case–control cohort of 1604 AD and 1235 CN participants (replication data set, Table 1). Of the nine, an OR51G1 variant (rs146006146, c.815 G > A, p.R272H) showed modest evidence of association in the replication data set (Preplication < 0.05, Table 1). Subsequent meta-analysis combining results from the discovery and replication datasets showed a significant association with the same direction of effect in both datasets (n = 3731, Pmeta = 1.36 × 10−3, Table 1). We further assessed the association by using logistic regression analysis with adjustment for sex and age (Pmeta = 6.82 × 10−3, OR = 2.20, 95% CI = 1.24 to 3.91). All of the OR51G1 variants in WGS were validated by using multiplex PCR-based Invader assay or Sanger sequencing.
The OR51G1 variant has never been observed in European, American, or African populations except for the population of Asian origin in the Genome Aggregation Database (gnomAD) [20], where the MAF is 0.016 in East Asians. This variant has been observed in other Asian databases, where the MAFs are 0.0049 in the Korean Reference Genome Database (KRGDB) [21] and 0.0089 in the Tohoku Medical Megabank Organization (ToMMo 8.3KJPN) [22], a Japanese database. We have also reported anther olfactory receptor gene (OR2B2) from a GWAS analysis of a large number of Japanese AD patients [23]. These results imply that the OR51G1 variant is an Asian-specific rare AD pathogenic variant.
Gene-based association study for AD pathogenic genes
In order to identify significant genes with multiple causal variants, we conducted genome-wide gene-based burden testing on rare coding variants. The MLKL gene (mixed lineage kinase domain-like) reached a Bonferroni-corrected level of significance (Pbon = 0.010, Table S2). Six rare variants were involved in the association. Three of them (rs763812068, rs778326056, and rs55867815) showed AD associations with P < 0.05 (Table 2). Because rs55867815 had been examined in the variant-based association study and meta-analysis combining results from the discovery and replication datasets showed no significant association (n = 3759, Pmeta = 0.39, Table 1), the remaining two (rs763812068 and rs778326056) were further genotyped by using the replication data set. The rs763812068 variant showed a significant association with the same direction of effect in the discovery and replication datasets in a subsequent meta-analysis combining results from both datasets (Pmeta = 0.046, n = 3761, Fig. 2a). This is a stop-gain variant (c.142 C > T, p.Q48X) with a high Combined Annotation Dependent Depletion (CADD) Phred-scaled score [24] of 33, defined as a loss-of-function variant, and it was observed only in AD cases (Fig. 2a). All of the stop-gain variants in WGS were validated by using Sanger sequencing (Fig. 2b). All four MLKL stop-gain variants were observed in women with AD, of whom three were APOE ε4–positive (Fig. 2c). This rare variant appears in Asian populations (0.0013 in gnomAD [20], 0.0005 in KRGDB [21], 0.0007 in ToMMo 8.3KJPN [22]), although it appears rarely in the European population (5.0 × 10−5 in gnomAD [20], Fig. 2d).

Two variants (rs763812068 and rs778326056) were genotyped by using a replication data set. a For rs763812068, a subsequent meta-analysis combining results from the discovery and replication datasets showed a significant association, with the same direction of effect in both datasets (Pmeta = 0.046). Abbreviations: A1 allele 1, A2 allele 2. b MLKL stop-gain variants (rs763812068) detected with WGS were validated by using Sanger sequencing. c All four of the stop-gain variants were observed in females with AD, most of whom were APOE ε4-positive. d The stop-gain variant appears especially in Asian populations. The source databases are noted: KRGDB, Korean Reference Genome Database; ToMMo, Tohoku Medical Megabank Organization; gnomAD, Genome Aggregation Database.
Functional analysis of the MLKL stop-gain variant
We conducted in vitro functional analyses for the MLKL stop-gain variant (p.Q48X). The cellular localization of variant proteins was first examined by using immunocytochemistry of human HEK293 cells. Wild-type proteins and missense variant proteins (p.P326A) were uniformly distributed around the cell nucleus, whereas we did not observe stop-gain variant proteins in the cells (Fig. 3a). Stop-gain variant proteins were likely degraded to fragments by a nonsense fragment degradation system [25] that acts on abnormal proteins.

a Localization of Myc-MLKL was visualized by immunocytochemistry in human HEK293 cells. Myc: Myc vector. b Proportions of cell death using SYTOX Green nuclear stain in human HEK293 cells transfected with the MLKL variant proteins. c The ratio of Aβ42 to Aβ40 was calculated from the ELISA results. Statistical analysis of the ratio of Aβ42 to Aβ40 between two groups was performed by using Welch’s t-test. Statistical significance was set at P < 0.05. *: Welch’s t-test P < 0.05; n.s: not significant.
MLKL is a pseudokinase that functions as the key effector of necroptosis, a form of inflammatory programmed cell death [26]. Therefore, we next assessed the proportion of cell death by using HEK293 cells transfected with the MLKL variant proteins. The p.Q48X carrier had a significantly lower proportion of cell death than the wild type (Fig. 3b, Welch’s t-test P = 0.00030) and p.P326A carrier (Fig. 3b, Welch’s t-test P = 0.00078). This experiment was independently performed three times with five replicates of each variant carrier (Fig. S2). These results suggest that p.Q48X variant proteins contribute to an accumulation of abnormal cells that should die by programmed cell death but do not because MLKL has been inactivated.
Because amyloid-β (Aβ) is implicated in AD pathogenesis [27], we examined whether the MLKL variant proteins promote Aβ generation. We transfected the MLKL variant proteins into an AD model HEK293 cell line (APPswe-293 cells, see Materials and Methods) and measured the ratio of Aβ42 to Aβ40 by using an enzyme-linked immunosorbent assay (ELISA). The ratio of Aβ42 to Aβ40 was significantly decreased in the wild type compared with the mock-infected cells (Welch’s t-test P = 0.031), and was increased slightly in the p.Q48X and p.P326A carriers compared with the wild type, whereas it showed no statistically significant difference in p.Q48X and p.P326A carriers compared with the mock-infected cells (Welch’s t-test P = 0.45, p.Q48X; Welch’s t-test P = 0.93, p.P326A, Fig. 3c). These results indicated that MLKL can play a key role in the decrease of the ratio of Aβ42 to Aβ40. In other words, these findings suggest that the MLKL p.Q48X variant can increase the abundance of abnormal cells that would otherwise die by programmed cell death and also contribute to increases in the ratio of Aβ42 to Aβ40.
Network-based meta-analysis for AD pathogenic genes
Protein-protein interaction (PPI) network analysis is effective for identifying functionally important modules (i.e., hub genes) involved in the pathogenesis of AD [28]. Through genome-wide gene-based burden testing on rare coding variants, we detected 67 candidate pathogenic genes with Benjamini-Hochberg (BH) corrected PBH < 0.1 (Table S2). We performed a PPI network analysis of the candidate variants by using NetworkAnalyst [29] (http://www.networkanalyst.ca) with the STRING Interactome database [30]. A PPI network with 245 nodes and 254 edges was obtained. The most highly ranked hub genes were recognized in terms of network topology measures of degree of centrality (DC) and betweenness of centrality (BC). Eight top-ranked genes with DC ≥15 and BC ≥3000 were identified as hub genes (NCOR2, DC = 56, BC = 17,272; PLEC, DC = 43, BC = 9545; DMD, DC = 32, BC = 9809.5; NEDD4, DC = 29, BC = 14,454; PIK3C2G, DC = 28, BC = 6210; ESR2, DC = 26, BC = 8177; EXO1, DC = 17, BC = 3768; TTN, DC = 17, BC = 3311.5, Fig. 4). Four of them have not been verified as expressed in brain tissues through the Human Protein Atlas database [31], which provides quantitative transcriptomics at the tissue and organ level and is publicly accessible at http://www.proteinatlas.org (Fig. S3), but we considered the remaining four (i.e., NCOR2, DMD, NEDD4, and PLEC) to be strong candidate hub genes associated with AD pathogenesis.

Through genome-wide gene-based burden testing on rare coding variants, candidate pathogenic genes with PBH < 0.1 were detected. A PPI network analysis was performed on the candidates by using NetworkAnalyst with the STRING Interactome database. Eight genes with degree of centrality (DC) ≥15 and betweenness of centrality (BC) ≥3,000 were identified as hub genes. Hub genes size corresponds to the DC. Hub genes are labeled with the gene names.
Quantitative RT-PCR assay of hub genes by using blood samples
For the four hub genes described above (NCOR2, DMD, NEDD4, and PLEC), we examined the differential gene expression between AD and CN blood samples, because these genes could have the potential to act as blood-based biomarkers for AD. This would have significant advantages of being more time-and cost-efficient and less invasive than current diagnostic methods for AD. We measured the expression of these genes by using our 20 blood samples (10 ADs and 10 CNs) through quantitative RT-PCR (qRT-PCR). NCOR2 and DMD showed low levels of expression in blood, as shown in the Human Protein Atlas (HPA) database [31] (Fig. S3), so that we could not compare the difference in expression between AD and CN. The remaining two (PLEC and NEDD4) were expressed in blood, but they showed no statistically significant difference in expression between AD and CN (Welch’s t-test P = 0.39, PLEC; Welch’s t-test P = 0.77, NEDD4, Fig. 5). Unfortunately, these genes cannot be used as blood-based biomarkers for AD prediction, although they might be associated with AD in brain tissues.

The expression of all four hub genes in brain tissues was validated in the Human Protein Atlas database. Differential gene expression between AD and CN subjects (n = 20; 10 AD and 10 CN) was investigated. No differences were significant.
Discussion
By using WGS of 938 subjects and an independent case–control cohort of > 2800 subjects, we identified two novel rare heterozygous variants and four candidate genes in the pathogenesis of AD via variant-based and gene-based association studies and a network-based meta-analysis. A similar analysis was recently conducted on a large number of samples in populations primarily of European descent, and several AD candidate loci have been reported [32]. However, our findings were not included in their results because they seem to be Asian specific.
From our variant-based association study, we first discovered an Asian-specific rare heterozygous missense variant in OR51G1 showing a nominally significant association with the same direction of effect in the both the discovery and replication sets. OR51G1 is an olfactory receptor gene, and previous studies have reported a relationship between olfactory dysfunction and risk of cognitive decline [33], including AD [34, 35]. We have also reported another olfactory receptor gene (OR2B2) from a GWAS analysis using a large number of Japanese AD patients [23]. These data support a link between our finding and AD pathogenesis.
From our gene-based association study, we discovered a rare heterozygous stop-gain variant, defined as a loss-of-function variant in the MLKL gene, which was observed only in samples from individuals with AD among more than 3700 Japanese samples. MLKL encodes a pseudokinase that functions as the key effector of necroptosis, a form of inflammatory programmed cell death [26]. We found that the MLKL stop-gain variant can decrease the proportion of cells undergoing cell death, presumably leading to the persistence of abnormal cells, and also contribute to increases in the ratio of Aβ42 to Aβ40. In a systematic review, Wang et al. reported that the MLKL variant might contribute to late-onset, APOE ε4-negative AD in the Chinese population [36]. Faergeman et al. reported from a WGS-based pedigree analysis that a frameshift variant in MLKL caused neurodegenerative spectrum disorder; their patients gradually developed mild cognitive impairment [37]. These results strongly support the likelihood that the MLKL stop-gain variant contributes to AD pathogenesis, but in contrast to the findings of Wang et al., three of the four carriers identified in this study were APOE ε4-positive patients, suggesting that this variant contributes to AD pathogenesis irrespective of APOE ε4 allele status in the Japanese population.
From genome-wide gene-based burden testing on rare coding variants, we further detected candidate pathogenic genes. Subsequently, a PPI network analysis based on the candidates elucidated four functionally important hub genes (NCOR2, DMD, NEDD4, and PLEC) verified to be expressed in brain tissues through the HPA database [31]. NCOR2 (nuclear receptor co-repressor 2) binds to several nuclear receptors to suppress the expression of their regulatory targets [38]. The regulatory targets of NCOR2, such as NR4A, are involved in learning and memory circuits in the adult brain [39]. Zhou et al. reported that loss of function of NCOR1 and NCOR2 accompanied memory deficits through a novel GABAergic hypothalamus-CA3 projection [40]. PLEC encodes plectin, a universally expressed multifunctional cytolinker protein that is crucial for the intermediate filament network including crosstalk between actomyosin and microtubules [41]. The major isoform, P1c, is expressed in neural and epidermal cells [42]. Valencia et al. reported that lack of P1c in neurons induced long-term memory impairment [41]. The DMD (dystrophin) gene is expressed in brain tissues. Anand et al. reported that dystrophin deficiency triggers alterations in the function of central cholinergic synapses and their regulation of neuronal metabolism, and induces cognitive deficits [43]. NEDD4 encodes the NEDD4 E3 ubiquitin protein ligase, which ubiquitinates the primary mediator of synaptic transmission, AMPA receptors (AMPARs), which are crucial for synaptic plasticity and brain functions [44]. Zhang et al. reported that AMPAR ubiquitination acts as the key molecular event leading to the loss of AMPARs and suppresses synaptic transmission in AD [45]. These data show that all four hub genes can at least be linked to memory impairment.
The two hub genes we were able to evaluate in blood samples (NEDD4 and PLEC) showed no statistical difference in expression in the blood between AD and CN. Therefore, these genes cannot be used as blood-based biomarkers for AD prediction. However, as the expression of all four hub genes may show a statistical difference between AD and CN in brain tissues, cell lines, and animal models, we will continue to explore their potential involvement in the pathogenesis of AD. Although to our knowledge this study is the largest WGS-based rare-variant analysis in Japanese AD cases, further investigation using larger sample sizes will likely reveal additional pathogenic mutations or genes in AD.
In summary, we highlighted two novel ethnicity-specific rare variants (rs146006146, c.815 G > A, p.R272H in OR51G1; rs763812068, c.142 C > T, p.Q48X in MLKL) and four hub genes (NCOR2, DMD, NEDD4, and PLEC) associated with AD pathogenesis. Our findings in this study will not only contribute to the understanding of AD but will also provide insights for future studies into its pathogenic mechanism. In addition, understanding the differences in genetic variant profiles among ethnicities will be important for individualizing treatment and for understanding the pathogenic mechanisms of AD. Association studies between AD and CN using WGS data with a larger sample size would likely lead to the identification of additional novel AD mutations in the future.
Materials and methods
Clinical samples
All 3777 blood samples used in this study and the associated clinical data were obtained from the NCGG Biobank. Of the samples, 938 were used for WGS analyses: 140 samples were from patients with AD and 798 samples were from CN. Donors with AD were diagnosed with probable or possible AD by using the criteria of the National Institute on Aging Alzheimer’s Association workgroups [46, 47]. The CN subjects had subjective cognitive complaints but normal cognition on a neuropsychological assessment with a comprehensive neuropsychological test and a Mini-Mental State Examination score >23. Diagnosis of all subjects was based on medical history, physical examination and diagnostic tests, neurological examination, neuropsychological tests, and brain imaging with magnetic resonance imaging or computerized tomography by experts, including neurologists, psychiatrists, geriatricians, or neurosurgeons, all experts in dementia who were familiar with its diagnostic criteria. The remaining 1604 AD subjects and 1235 CN subjects were used as an independent replication cohort for validation assessments. All of the subjects were ≥60 years old.
WGS data analysis
All WGS data were downloaded from the NCGG Biobank database. DNA concentration was measured by using PicoGreen DNA assay, and fragmentation of DNA was assessed with agarose gel electrophoresis. High-quality DNA was used for DNA libraries. A WGS library was constructed by using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina, Inc., San Diego, CA, USA) in accordance with the manufacturer’s instructions. WGS was conducted at Macrogen Japan Corporation, Takara Bio Inc., and GENEWIZ Inc. DNA was sequenced by using the Illumina HiSeq X Ten or NovaSeq 6000 platform with paired-end reads of 151 bp in accordance with the manufacturer’s instructions.
Read sequences were mapped with BWA-MEM (version 0.7.15) [48] to the human reference genome (GRCh37). Duplicate PCR reads were identified and removed by using picard (version 2.21.4) [49]. Variant calling was conducted by using GATK (version 4.1.0.0) [19]. Individual variant calling was performed by using GATK HaplotypeCaller. Multisample individual variants were joint-called together with in-house data (WGS = 1572) by using GATK GenotypeGVCFs. Variant quality score recalibration was applied according to the GATK Best Practice recommendations [50]. We filtered out SNVs that satisfied the following criteria: (1) Depth (DP) < 10, (2) GenotypeQuality (GQ) < 20, (3) Quality by Depth (QD) < 2, QUAL < 30, StrandOddsRatio (SOR) > 4, FisherStrand (FS) > 60, RMSMappingQuality (MQ) < 40, MappingQualityRankSumTest (MQRankSum) < −12.5, ReadPosRankSumTest (ReadPosRankSum) <−8, and (4) ExcessHet >20, and short Indels with (1) DP < 10, (2) GQ < 20, (3) QD < 2, QUAL < 30, FS > 200, SOR > 10, ReadPosRankSum < −20, and (4) ExcessHet > 20.
QC of the all variants was performed by using PLINK software [51]. We first applied QC filters to the subjects: (1) sex inconsistencies (--check-sex), (2) PI_HAT > 0.25, where PI_HAT is a statistic for the proportion of identity by descent (--genome), (3) genotype missingness (--mind 0.05), and (4) exclusion of outliers from the clusters of East Asian populations in a principal component analysis that was conducted together with population data from 1000 Genomes Phase 3. We next applied QC filters to the variants: (1) genotyping efficiency or call rate (--geno 0.05), (2) minor count (--mac 2), and (3) Hardy-Weinberg equilibrium (--hwe 1 × 10−5). Furthermore, we excluded all variants located in low-complexity regions, available from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/low_-complexity_regions/hs37d5-LCRs.20140224.bed.gz.
Annotation of variants and variant-based or gene-based association tests
Functional annotations of the variants were made with ANNOVAR (version 20191024). We annotated protein-coding variants with frameshift Indel, nonframeshift Indel, stop-gain/-loss, nonsynonymous SNV, synonymous SNV, and splicing variants. Variant frequency data were found in a public database from 8380 healthy Japanese individuals, called ToMMo 8.3KJPN [22]. We classified the variants into three groups on the basis of MAF: rare (MAF < 0.01), infrequent (0.01 ≤ MAF ≤ 0.05), and common (0.05 < MAF). GWAS of those variants were conducted with Fisher’s exact test by using PLINK software (--model fisher) [51].
Candidate pathogenic variants that satisfied P < 5.0 × 10−4 were genotyped by using an independent replication cohort. SNP genotyping was performed with the multiplex PCR-based Invader assay (Third Wave Technologies, Madison, WI). Association analysis in the replication cohort as well as the combined analysis of the WGS and replication study were conducted with Fisher’s exact test by using PLINK software (--model fisher) [51]. Variant frequency data were found in public databases: gnomAD [20], KRGDB [21], and ToMMo 8.3KJPN [22].
Gene-based association tests of rare variants-burden tests that assume all variants in the target region have effects on the phenotype in the same direction [52], were implemented in the R programming language (R Development Core Team, http://www.r-project.org/). Genes with Bonferroni-corrected Pbon < 0.05 were considered to be significantly enriched.
Construction of plasmids and stable cell lines
Complementary DNA (cDNA) was synthesized from mRNA extracted from the buffy coat of patients. Wild-type MLKL was amplified by PCR from the cDNA cloned into a pCMV-Myc vector. The sequence of MLKL was confirmed by Sanger sequencing. Variant-type Myc-MLKL plasmids, p.Q48X and p.P326A, were constructed from the wild-type Myc-MLKL plasmid by using site-directed mutagenesis with PrimeSTAR Max DNA Polymerase (Takara Bio Inc, Shiga, Japan). Primers with each mutation (underlined) were designed in accordance with the manufacturer’s instructions: 5′-CCAGGACTAAGGAAAGAGGAGCGTGC-3′ and 5′-TTTCCTTAGTCCTGGAGCATCTCCAG-3′ for p.Q48X; and 5′-AGAAGCAGCTGAACTCCACGGAAAAA-3′ and 5′-AGTTCAGCTGCTTCTGAATGGTGTAG-3′ for p.P326A. The mutagenesis reaction mix contained 1× PrimeSTAR Max Premix, 0.2 μM of each primer set, and wild-type Myc-MLKL plasmid (70 pg) in a total reaction volume of 50 μL. The cycling conditions were 30 cycles of 98 °C for 10 s, 55 °C for 15 s, and 72 °C for 25 s. Escherichia coli strain DH5α was transformed with the resulting solutions, and the variant sequences were confirmed by using Sanger sequencing.
Human APP695 was cloned from human brain cDNA (Clontech, Palo Alto, CA) into a pcDNA™3.1/Hygro (+) vector (Thermo Fisher Scientific, Waltham, MA). Then, the Swedish mutation (APPswe) was induced by using site-directed mutagenesis as described above with the primer set 5′-AAGTGAATCTGGATGCAGAATTCCGAC-3′ and 5′-CATCCAGATTCACTTCAGAGATCTCC-3′. HEK293 cells were transfected with the APPswe plasmid, and cells stably expressing the Swedish mutant of APP695 (HEK-APPswe) were selected in medium containing hygromycin B (Nacalai Tesque, Kyoto, Japan) for 1 week.
Immunocytochemistry
HEK293 cells were seeded at a density of 2.0 × 104 cells/well on BioCoat Poly-D-Lysine 4-well Culture Slides (Corning, NY, USA), cultured for 24 h, and transfected with wild-type, p.Q48X, or p.P326A Myc-MLKL plasmids and FuGENE HD Transfection Reagent (Promega, Madison, WI). Twenty-four hours after transfection, the cells were fixed and incubated with Anti-Myc-tag mAb-Alexa Fluor 488 (MBL, Nagoya, Japan) in accordance with the manufacturer’s protocol. The slides were mounted with SlowFade Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific), and fluorescence images were obtained with a BIOREVO BZ-9000 fluorescence microscope (Keyence, Osaka, Japan).
Cell death
HEK293 cells were plated in 96-well plates (1.5 × 104 cells/well) and cultured for 24 h before transfection with Myc-MLKL plasmids and FuGENE HD (Promega). After 24 h of culture, SYTOX Green (Thermo Fisher Scientific) was added at a final concentration of 5 μM, and the fluorescence intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 520 nm by using an Infinite M200 PRO plate reader (Tecan, Männedorf, Switzerland). Then the maximum fluorescence intensity for the calculation of cell death rate was measured after all cells were lysed by adding Triton X-100 (final 0.1%). This experiment was performed independently three times with five replicates of each variant carrier.
Aβ ELISA
HEK293-APPswe cells were plated into 6-well culture plates (4.0 × 105 cells/well) and were cultured for 24 h before transfection with Myc-MLKL plasmids and FuGENE HD (Promega). The supernatant was collected 24 h after transfection, and the concentrations of Aβ40 and Aβ42 were analyzed by using a human β-amyloid ELISA kit (Wako, Osaka, Japan). This experiment was performed independently three times with 12 replicates of each variant carrier. The ratio of Aβ42 to Aβ40 was calculated on the basis of the average of the replicates. Statistical analysis of the ratio of Aβ42 to Aβ40 between two groups was performed with Welch’s t-test. Statistical significance was set at a P value < 0.05.
Network-based meta-analysis
Significant genes with multiple causal variants were detected by using genome-wide gene-based burden testing on rare coding variants. Network-based analysis was performed on the genes with a Benjamini-Hochberg (BH) corrected PBH < 0.1 with NetworkAnalyst 3.0 [29] and the STRING Interactome database [30], which provides comprehensive information about interactions between proteins, including prediction and experimental interaction data. The confidence cutoff score was set to 900. The large-scale PPI network was visualized with Cytoscape v3.8.2 (http://www.cytoscape.org/) [53].
qRT-PCR validation of genes
cDNA was synthesized by using a PrimeScript II 1st Strand cDNA Synthesis Kit (Takara Bio, Shiga, Japan). qRT-PCR analysis was performed by using TB Green Premix Ex Taq II (Takara Bio, Shiga, Japan) and the Quantstudio7 Flex Real-Time PCR System (Thermo Fisher, Waltham, MA). The following commercially available PCR primers (forward and reverse, 5ʹ to 3ʹ) were used for gene expression analysis: NCOR2 (GACAAGGAGGCAGAGAAGCCT, GTGACCAGGTGGAGCGTAG), DMD (GTTGGAAGAACTCATTACCGCTGC, CTGTCCTAAGACCTGCTCAGC), NEDD4 (CTGAGGAATTAGAGCCTGGCTG, GCACGTTGTGCTTGCAGTTG), and PLEC (GAGTTTGAGAGGCTGGAGTGTC, GGTCTGCACGTCGTTGAAGAG). The qRT-PCR conditions were as follows: one cycle of 50 °C for 2 min, 95 °C for 20 s followed by 40 cycles of 95 °C for 1 s, 60 °C for 20 s, and 72 °C for 30 s. The human beta-2-microglobulin (hB2M) was preselected as a reference gene for normalization of target gene expression levels. Gene expression levels from qRT-PCR were calculated relative to that of the reference gene hB2M by using the semiquantitative method [54]. Gene expression levels were obtained for 10 AD and 10 CN randomly selected samples.
Data availability
All datasets used or analyzed are available from the corresponding author on reasonable request.
Code availability
We used open source program languages R (version 3.4.1) and Ruby (version 2.4.0) to analyze data and create plots. Code is available upon request from the corresponding authors.
References
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6.
Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9:63–75 e62.
Lee G, Nho K, Kang B, Sohn KA, Kim D.for Alzheimer’s Disease Neuroimaging I Predicting Alzheimer’s disease progression using multi-modal deep learning approach. Sci Rep.2019;9:1952.
Ayodele T, Rogaeva E, Kurup JT, Beecham G, Reitz C. Early-onset Alzheimer’s disease: what is missing in research? Curr Neurol Neurosci Rep. 2021;21:4.
Giau VV, Bagyinszky E, Yang YS, Youn YC, An SSA, Kim SY. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci Rep. 2019;9:8368.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414–30.
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–74.
Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Ridge PG, Hoyt KB, Boehme K, Mukherjee S, Crane PK, Haines JL, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. 2016;41:200 e213–200 e220.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13.
Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45.
Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:19096–101.
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35.
Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43:442–6.
Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–42.
Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7.
Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci USA. 2015;112:5473–8.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Jung KS, Hong KW, Jo HY, Choi J, Ban HJ, Cho SB, et al. KRGDB: the large-scale variant database of 1722 Koreans based on whole genome sequencing. Database (Oxford) 2020;2020:baaa030.
Tadaka S, Saigusa D, Motoike IN, Inoue J, Aoki Y, Shirota M, et al. jMorp: Japanese multi omics reference panel. Nucleic Acids Res. 2018;46:D551–D557.
Shigemizu D, Mitsumori R, Akiyama S, Miyashita A, Morizono T, Higaki S, et al. Ethnic and trans-ethnic genome-wide association studies identify new loci influencing Japanese Alzheimer’s disease risk. Transl Psychiatry. 2021;11:151.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Karousis ED, Nasif S, Muhlemann O. Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley Interdiscip Rev RNA. 2016;7:661–82.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Liu W, Wu A, Pellegrini M, Wang X. Integrative analysis of human protein, function and disease networks. Sci Rep. 2015;5:14344.
Santiago JA, Potashkin JA. Network-based metaanalysis identifies HNF4A and PTBP1 as longitudinally dynamic biomarkers for Parkinson’s disease. Proc Natl Acad Sci USA. 2015;112:2257–62.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map Hum proteome Sci. 2015;347:1260419.
Prokopenko D, Morgan SL, Mullin K, Hofmann O, Chapman B, Kirchner R, et al. Whole-genome sequencing reveals new Alzheimer’s disease-associated rare variants in loci related to synaptic function and neuronal development. Alzheimers Dement. 2021;17:1509–27.
Wilson RS, Schneider JA, Arnold SE, Tang Y, Boyle PA, Bennett DA. Olfactory identification and incidence of mild cognitive impairment in older age. Arch Gen Psychiatry. 2007;64:802–8.
Schubert CR, Carmichael LL, Murphy C, Klein BE, Klein R, Cruickshanks KJ. Olfaction and the 5-year incidence of cognitive impairment in an epidemiological study of older adults. J Am Geriatr Soc. 2008;56:1517–21.
Roberts RO, Christianson TJ, Kremers WK, Mielke MM, Machulda MM, Vassilaki M, et al. Association between olfactory dysfunction and amnestic mild cognitive impairment and Alzheimer disease dementia. JAMA Neurol. 2016;73:93–101.
Wang B, Bao S, Zhang Z, Zhou X, Wang J, Fan Y, et al. A rare variant in MLKL confers susceptibility to ApoE varepsilon4-negative Alzheimer’s disease in Hong Kong Chinese population. Neurobiol Aging. 2018;68:160 e161–160 e167.
Faergeman SL, Evans H, Attfield KE, Desel C, Kuttikkatte SB, Sommerlund M, et al. A novel neurodegenerative spectrum disorder in patients with MLKL deficiency. Cell Death Dis. 2020;11:303.
Schoch H, Abel T. Transcriptional co-repressors and memory storage. Neuropharmacology. 2014;80:53–60.
Hawk JD, Bookout AL, Poplawski SG, Bridi M, Rao AJ, Sulewski ME, et al. NR4A nuclear receptors support memory enhancement by histone deacetylase inhibitors. J Clin Invest. 2012;122:3593–602.
Zhou W, He Y, Rehman AU, Kong Y, Hong S, Ding G, et al. Loss of function of NCOR1 and NCOR2 impairs memory through a novel GABAergic hypothalamus-CA3 projection. Nat Neurosci. 2019;22:205–17.
Valencia RG, Mihailovska E, Winter L, Bauer K, Fischer I, Walko G, et al. Plectin dysfunction in neurons leads to tau accumulation on microtubules affecting neuritogenesis, organelle trafficking, pain sensitivity and memory. Neuropathol Appl Neurobiol. 2021;47:73–95.
Fuchs P, Zorer M, Rezniczek GA, Spazierer D, Oehler S, Castanon MJ, et al. Unusual 5’ transcript complexity of plectin isoforms: novel tissue-specific exons modulate actin binding activity. Hum Mol Genet. 1999;8:2461–72.
Anand A, Tyagi R, Mohanty M, Goyal M, Silva KR, Wijekoon N. Dystrophin induced cognitive impairment: mechanisms, models and therapeutic strategies. Ann Neurosci. 2015;22:108–18.
Lin A, Hou Q, Jarzylo L, Amato S, Gilbert J, Shang F, et al. Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J Neurochem. 2011;119:27–39.
Zhang Y, Guo O, Huo Y, Wang G, Man HY. Amyloid-beta induces AMPA receptor ubiquitination and degradation in primary neurons and human brains of Alzheimer’s disease. J Alzheimers Dis. 2018;62:1789–801.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–9.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinforma. 2013;43:11 10 11–11 10 33.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–75.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinforma. 2003;4:2.
Marone M, Mozzetti S, De Ritis D, Pierelli L, Scambia G. Semiquantitative RT-PCR analysis to assess the expression levels of multiple transcripts from the same sample. Biol Proced Online. 2001;3:19–25.
Acknowledgements
We thank NCGG Biobank for providing the study materials, clinical information, and technical support. This study was supported by grants from the Japan Agency for Medical Research and Development (AMED) (Grant Number JP18kk0205009 to S.N., Grant Number JP18kk0205012 to SN, Grant Number JP19dk0207045 to SN, and KO, Grant Number JP20354793 to DS and SN, and Grant Number JP20km040550 to KO); JSPS KAKENHI (Grant Numbers JP17H06307 and JP21H02470 to DS); The DAIKO Foundation (to DS); Research Funding for Longevity Sciences from the NCGG (29–45 to KO, and 30–29 to DS); and a grant for Research on Dementia from the Japanese Ministry of Health, Labour, and Welfare (to KO).
Author information
Authors and Affiliations
Contributions
DS developed the method and performed the analyses; YA performed the WGS, SNP genotyping and functional analysis using cell lines; RM performed the qRT-PCR validation of gene expression; SA provided technical assistance; DS wrote the manuscript; and DS, SN, and KO organized this work. All authors contributed to and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the ethics committee of the National Center for Geriatrics and Gerontology (NCGG). The design and performance of the current study involving human subjects were clearly described in a research protocol. Participation in the NCGG Biobank was voluntary, and all participants completed informed consent in writing before registering.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shigemizu, D., Asanomi, Y., Akiyama, S. et al. Whole-genome sequencing reveals novel ethnicity-specific rare variants associated with Alzheimer’s disease. Mol Psychiatry 27, 2554–2562 (2022). https://doi.org/10.1038/s41380-022-01483-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01483-0
This article is cited by
-
CRISPR/Cas9-mediated knock-in cells of the late-onset Alzheimer’s disease-risk variant, SHARPIN G186R, reveal reduced NF-κB pathway and accelerated Aβ secretion
Journal of Human Genetics (2024)
-
The HLA-DRB1*09:01-DQB1*03:03 haplotype is associated with the risk for late-onset Alzheimer’s disease in APOE \({{\varepsilon }}\)4–negative Japanese adults
npj Aging (2024)
-
Identification of potential blood biomarkers for early diagnosis of Alzheimer’s disease through immune landscape analysis
npj Aging (2022)
-
Omics-based biomarkers discovery for Alzheimer's disease
Cellular and Molecular Life Sciences (2022)
