
Abstract
Familial Alzheimer’s disease (fAD) mutations alter amyloid precursor protein (APP) cleavage by γ-secretase, increasing the proportion of longer amyloidogenic amyloid-β (Aβ) peptides. Using five control induced pluripotent stem cell (iPSC) lines and seven iPSC lines generated from fAD patients, we investigated the effects of mutations on the Aβ secretome in human neurons generated in 2D and 3D. We also analysed matched CSF, post-mortem brain tissue, and iPSCs from the same participant with the APP V717I mutation. All fAD mutation lines demonstrated an increased Aβ42:40 ratio relative to controls, yet displayed varied signatures for Aβ43, Aβ38, and short Aβ fragments. We propose four qualitatively distinct mechanisms behind raised Aβ42:40. (1) APP V717I mutations alter γ-secretase cleavage site preference. Whereas, distinct presenilin 1 (PSEN1) mutations lead to either (2) reduced γ-secretase activity, (3) altered protein stability or (4) reduced PSEN1 maturation, all culminating in reduced γ-secretase carboxypeptidase-like activity. These data support Aβ mechanistic tenets in a human physiological model and substantiate iPSC-neurons for modelling fAD.
Similar content being viewed by others


Introduction
Familial Alzheimer’s disease (fAD) describes a hereditary (high penetrance, autosomal dominant) subgroup of AD that represents less than 1% of all AD cases. Nevertheless, the pathological hallmarks of extracellular amyloid beta (Aβ) enriched plaques and intracellular neurofibrillary tangles of hyper-phosphorylated tau are shared between fAD and sporadic AD (sAD). fAD-causing mutations in the genes coding for amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) affect the production of Aβ in the central nervous system (CNS), implicating altered APP cleavage and processing in the AD disease mechanism [1]. Study of these mutations offers a powerful way of interrogating key underlying disease mechanisms for all forms of AD [2].
Aβ peptides are produced via sequential proteolytic cleavage of APP by β-secretase and then γ-secretase activity [3] (Fig. 1a). Neither of the enzymes that perform this activity (BACE1 and γ-secretase respectively) are limited to proteolysis at a set residue of the APP sequence, but have multiple potential cleavage sites that in turn produce Aβ peptides of different lengths. BACE1 may cleave APP at Asp1 (of the Aβ domain), or Glu11, producing the classic or N-terminally truncated form of Aβ, respectively. γ-Secretase performs an initial endopeptidase-like ε-cleavage at Leu49 or Thr48, and then continues to make carboxypeptidase-like cleavages which produce shorter fragments [4]. As Aβ peptides can be truncated both N-terminally and C-terminally, we will hereafter use the convention of referring to peptides cleaved at the canonical N-terminal Asp1 amino acid by the C-terminal truncation (e.g. Aβ1–42 as Aβ42), while peptides with N-terminal truncations will be directly specified.

Ratios act as internal normalisers for relative Aβ peptide abundance in conditioned media from stem cell models of AD. a The Aβ domain of APP, highlighting the canonical cleavage sites of α-, β-, and γ-secretase within the Aβ peptide sequence itself, as well as the proposed pathways of carboxypeptidase-like activity following alternative ε-cleavage. Letters coloured blue, pink, and green indicate amino acids with hydrophilic, hydrophobic, and amphipathic properties, respectively. b Western blotting of full-length APP in iPSC-derived neuronal lysates and c quantification of APP western blot band intensities. d qPCR expression analysis of APP in iPSC-derived neurons. Replicates are shown within histogram bars and error bars represent SEM. e–h Normalisation of Aβ peptides measured in cell media using ratios e Aβ42:40, f Aβ42:38, g Aβ38:40 and h Aβ43:40. e–h Display an average percentage coefficient of variance (%CV) < 4% over 6 days and < 7% over 100 days for ratios. For h Aβ43:40 average %CV was 7.1% over 6 days and 20.7% over 100 days. Data is based on multiple independent inductions per line, specifically APP V717I-1 clone 1 (n = 3), APP V717I-1 clone 3 (n = 2), PSEN1 int4del clone 4 (n = 1), PSEN1 int4del clone 6 (n = 2), Ctrl1 (n = 1), Ctrl 2 (n = 1) and Shef6 (n = 2)
The tripeptide hypothesis proposes that γ-secretase cleavage activity occurs in a stepwise manner, every three residues, producing two alternative peptide production pathways: Aβ49 > 46 > 43 > 40 and Aβ48 > 45 > 42 > 38 [4,5,6] (Fig. 1a). Many questions still surround this hypothesis, including where Aβ39/38/37 and other detectable peptides fit within this model, how rigid these pathways are [7], and whether there may be others [6].
Different fAD-associated mutations are thought to have qualitatively different effects on Aβ cleavage and processing [8]. APP mutations are proposed to favour processing through the Aβ48 pathway, whereas PSEN1/2 mutations are thought to reduce carboxypeptidase-like activity and lead to an accumulation of longer Aβ fragments [8]. The Aβ42:38 ratio is a potential readout of γ-secretase carboxypeptidase-like cleavage efficiency [9] and the Aβ38:40 has been used to compare the theorised Aβ49 versus Aβ48 dependent pathways [5]. The Aβ42:40 ratio has been shown to effectively distinguish fAD and sAD from healthy controls in cerebrospinal fluid (CSF) samples, improving diagnostic accuracy and prediction of AD [10,11,12,13,14,15]. However, few studies have explored these dynamics in human neurons and at endogenous expression levels.
Induced pluripotent stem cell (iPSC) models are a powerful tool for exploring APP processing in tissue specific cells from individuals with fAD-causing mutations [16]. Yagi et al. found increased levels of secreted Aβ42 in neurons with the PSEN1 A246E and PSEN2 N141I mutations [17]. Further work on neurons bearing pathogenic PSEN1 mutations has shown an increased ratio of Aβ42:40 [18,19,20,21,22,23]. Similarly, iPSC-derived neurons with an APP V717I genotype show a raised Aβ42:40 ratio [21] as well as increases in Aβ42 and Aβ38 [24]. APP duplication iPSCs also exhibit high levels of Aβ40 [25]. Finally, 3D cerebral organoids provide a potentially valuable additional model, for example in modelling CSF due to the presence of choroid plexus tissue [26], and early reports suggest that increased amyloid aggregation in 3D cultures [27], as well as an ability to identify candidate proteins involved in AD pathogenesis [28]. However, a comprehensive analysis of the full spectrum of Aβ peptide production across multiple different fAD mutations to explore the mechanisms proposed by Chávez-Gutiérrez et al. [8] has yet to be performed.
The present study examined the full spectrum of secreted Aβ peptide ratios in both 2D cortical neurons and 3D cerebral organoids generated from seven fAD patient-derived pluripotent stem cell lines. We present the first analysis of Aβ43 in human neurons, and our findings highlight multiple qualitatively distinct fAD mutation-specific effects on APP processing. This study validates the use of in vitro iPSC-derived neuronal models, further develops understanding of APP processing, and provides a basis for stratifying mutation effects with regards to disease modelling and drug screening readouts in heterogeneous patient cohorts.
Materials and methods
Cell culture
Fibroblasts were cultured as previously described [29]. Ethical permission was obtained from the National Hospital for Neurology and Neurosurgery and the Institute of Neurology Joint Research Ethics Committee (09/H0716/64) and informed consent was obtained for all samples. Episomal reprogramming was performed as described by Okita et al. [30], using plasmids #27077, #27078, and #27080 which were obtained from Addgene. Fibroblasts were nucleofected using the Lonza P2 Nucleofection kit (Amaxa) as per manufacturer’s instructions. Re-programmed cells were grown on MEFs (ATCC) and nascent iPSC colonies were picked and transferred into feeder-free conditions (essential eight growth media and geltrex matrix (Thermo Fisher)) for further expansion. iPSCs were routinely passaged using ethylenediaminetetraacetic acid (EDTA). Karyotype screens and G-band analysis were performed on newly generated lines (The Doctors Laboratory, London, UK). Lines were routinely screened for the absence of mycoplasma contamination using the MycoAlert assay (Lonza).
Differentiation to cortical neurons was performed as described in Shi et al. [31], and all reagents were purchased from Thermo Fisher Scientific unless specified. Briefly, iPSCs were grown to 100% confluence and media were changed to neural induction media (N2B27 containing 10 μM SB431542 (Tocris) and 1 μM dorsomorphin (Tocris)). N2B27 media consist of a 1:1 mixture of Dulbecco's modified eagle medium F12 (DMEM-F12) and Neurobasal supplemented with 0.5× N2, 0.5× B27, 0.5× non-essential amino acids, 1 mM l-glutamine, 25U pen/strep, 10 μM β-mercaptoethanol and 25U insulin. At days 10 and 18, precursors were passaged using dispase and plated in laminin-coated wells (Sigma) in N2B27 media. The final passage was performed at day 35 using accutase, plated at a final density of 50,000 cells per cm2 and maintained in N2B27 media until the required timepoint.
Cerebral organoids were produced following the protocol described by Lancaster et al. [26]. Briefly, iPSCs were dissociated to single cells using EDTA followed by accutase, and 9000 cells were seeded in ultra-low attachment U-bottomed 96 well plates to generate embryoid bodies (EBs). EBs were grown in DMEM-F12 supplemented with 20% knockout serum replacement, 3% embryonic stem cell (ESC) quality foetal bovine serum, 1x non-essential amino acids and 3.5 μM β-mercaptoethanol supplemented from day 0 to 4 with rho-associated protein kinase (ROCK) inhibitor at 50 μM (Y27632 Millipore) and 4 ng/ml basic fibroblast growth factor (FGF)2 (Peprotech). On day 6, the media were changed to neural induction media containing DMEM-F12 1x N2 supplement, 1x non-essential amino acids, and 1 μg/ml of heparin. At day 12, EBs were embedded in matrigel (BD), media were changed to N2B27 (as above but with B27 supplement minus vitamin A) and moved to a 5 cm dish. At day 18, EBs were moved to the orbital shaker and fed with N2B27 (containing vitamin A) for the remainder of the experiment.
100 days post neural induction was taken as the time point for mature neurons in both 2D and 3D paradigms.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 15 min and stored in phosphate buffered saline (PBS) at 4 °C until immunostaining was performed. Organoids were fixed for 1 h in 4% paraformaldehyde and infused with 30% sucrose solution overnight at 4 °C before being embedded in optimal cutting tissue compound (OCT) (Bright) and stored at −80 °C. 10 µm sections were taken on the cryostat and added onto Superfrost slides (VWR). Before staining, OCT was removed via washing in PBS for 20 min. Frozen frontal cortex was sectioned on the cryostat to 10 µm sections and fixed on the slide for 15 min in 4% paraformaldehyde before immunostaining. After three washes in 0.3% triton-X-100 in PBS (PBST), cells and sections were blocked in 3% bovine serum albumin in PBST. Cells were incubated in primary antibodies in blocking solution overnight (Table 1). After three washes in PBST, secondary antibodies (Alexafluor 488, 568, 594, and 648; Thermo Scientific) were added in blocking solution for one hour in the dark. 4',6-diamidino-2-phenylindole (DAPI) was added to the cells as a nuclear counterstain at 1 μM and the cells were washed three times in PBST. Cells were mounted on slides using DAKO mounting media or imaged in PBS. For Aβ immunofluorescence, after fixation, all samples were treated with formic acid for 5 min for antigen retrieval. Images were captured at room temperature on the Opera Phenix (Perkin-Elmer) or a Zeiss LSM microscope at ×20, ×40, and ×63 magnification using Harmony and Leica LAS software, respectively. No post-hoc manipulation was performed.
Quantitative PCR
RNA was harvested using Trizol reagent (Sigma) as per the manufacturer’s instructions. Two micrograms of total RNA was reverse transcribed using Superscript IV (Thermo Fisher) and random hexamers. qPCR was performed using Power Sybr Green (Thermo Fisher) and an MX300P real time PCR cycler (Agilent). Primers used for qPCR are shown in Table 2.
Western blot
Cells were lysed in RIPA lysis buffer containing protease inhibitors (Roche). Lysates were loaded with Protein Orange G (Li-Cor) and NuPAGE reducing agent and denatured at 95 °C for 5 min. Following centrifugation at 9300 g for 3 min at 4 °C, electrophoresis was conducted with a NuPage 10% Bis-Tris gel in NuPage MES SDS running buffer at 150 V for 1 h. Transfer to nitrocellulose membrane was conducted at 30 volts for 1 h at 4 °C. The membrane was blocked in 3% BSA and incubated with primary antibody overnight (Table 1). The membrane was washed three times with 0.1% PBS-Tween solution and incubated in secondary antibody for 1 h, washed three times in 1× PBS and imaged using an Odyssey Fc (Licor Biosciences).
Collection of cell culture media
Cortical neurons and 3D organoids were incubated in N2B27 for 48 h prior to media collection. Media were centrifuged at 2000 relative centrifugal force (RCF) for 5 min at 21 °C, aliquoted at 1 mL into Sarstedt 2 mL PP tubes (cat. 72.694.406), and stored at −80 °C.
Collection of CSF
Anonymised human CSF was provided by the University College London Dementia Research Centre. The CSF used was approved for biomarker research by the National Hospital for Neurology and Neurosurgery/UCLH joint REC (12 LO 1504). CSF samples were collected by lumbar puncture prior to noon according to a standard operating procedure. A volume of up to 20 mL of CSF was collected at ambient room temperature into two 10 mL polypropylene tubes (Sarstedt, Nümbrecht, Germany, cat. 62.9924.284) directly from a 22G spinal needle, without use of a manometer. Samples were centrifuged at 1750 RCF for 5 min at 21 °C, aliquoted to 2 mL tubes (Elkay Laboratory Products, Basingstoke, UK, cat. 021-4204-500) and stored at −80 °C within 1–4 h of collection.
Immunoassays
Aβ38/40/42 were measured by electrochemiluminescence (ECL) using a Meso Scale Discovery (MSD) V-PLEX Aβ peptide panel 1(6E10) kit, according to manufacturer’s instructions. Briefly, samples were diluted 1:2 with diluent 35 and added in duplicate to microplate wells coated with mouse monoclonal peptide specific capture antibodies for human Aβx-38/x-40/x-42. Samples were incubated with anti-Aβ antibody (6E10 clone) as the detection antibody conjugated with an electrically excitable SULFO-TAG. Measurements were made using an MSD SECTOR 6000. Concentrations were calculated from ECL signal using a four-parameter logistic curve-fitting method with the MSD Workbench software package.
Lactate dehydrogenase assay
CM samples were thawed at 21 °C for one hour and assayed for lactate dehydrogenase reaction using a Randox LDH P-L 401 kit (Randox, Crumlin, UK). The assay was performed on a Randox Monza according to manufacturer protocol. Briefly, the reaction mix was created by reconstituting lyophilised NADH (0.18 mmol/L) in 3 mL of R1a buffer/substrate (phosphate buffer (50 mmol/L, pH 7.5 and pyruvate (0.6 mmol/L)). After running a blank (de-ionised H2O), 10 μL sample/quality control was diluted in 500 μL reaction mix, vortexed and analysed.
Immunoprecipitation mass spectrometry
Aβ peptides were immunoprecipitated using Aβ-specific antibodies coupled to magnetic beads [32]. Briefly, 4 mg of the anti-Aβ antibodies 6E10 and 4G8 (Signet Laboratories, Dedham, MA, USA) were separately added to 50 mL each of magnetic Dynabeads M-280 Sheep Anti-Mouse IgG (Invitrogen, Carlsbad, CA, USA). The 6E10 and 4G8 antibody-coated beads were mixed and added to the samples to which 0.025% Tween20 in phosphate-buffered saline (pH 7.4) had been added. After washing, using the KingFisher magnetic particle processor, the Aβ peptides were eluted using 100 μL 0.5% formic acid. Mass spectrometry measurements were performed using a Bruker Daltonics UltraFleXtreme matrix-assisted-laser-desorption/ionisation time-of-flight/time-of-flight (MALDI TOF/TOF) instrument (Bruker Daltonics, Bremen, Germany). All samples were analysed in duplicate. It should be noted that although relative quantification of Aβ peptides is valid, this does not represent a reflection on absolute abundance due to different ionisation efficiencies and peptide hydrophobicity.
Statistical analysis
Statistical analysis was performed in R (version 1.1.456), graphs were created using the ggplot2 package. Mean concentrations for each biomarker were calculated, within group variation was calculated as standard error of the mean (SEM). For time course experiment data, variation between different inductions of the same line was calculated by coefficient of variance. Data normality was assessed by histogram, qq-plot, and Shapiro–Wilk test. Given non-normal data distribution, pairwise comparisons between Non-AD controls and each fAD genotype were conducted by Mann–Whitney U test (alpha 0.05). Spearman’s rank correlation coefficient was conducted to assess the relationship between cell media concentrations of Aβ peptides or total tau and LDH, Aβ ratios and age of onset, and Non-AD and fAD line Aβ peptide spectra.
The use of 7 fAD lines, together with 5 controls, represents sufficient statistical power for a false discovery rate of 0.2 [33]. For comparison of biomarkers in different tissues/fluids of the same patient the sample size was limited to one, therefore it was not possible to conduct tests of statistical significance between sample types.
Assay variation
Percent coefficient of variance (CV) of the intra- and inter-assay variability respectively were Aβ38 (4.7%, 13.3%), Aβ40 (5.9%, 9.4%), Aβ42 (4.7%, 12.6%), Aβ43 (7.4%, 13.2%), T-tau (7.9%, 12.6%), calculated from concentrations of an internal control CSF sample. The intra- and inter-assay CV was calculated according to ISO 5725-2 standards [34].
Results
To explore Aβ processing in vitro, we employed a 2D cortical differentiation protocol to a panel of five control lines (four control iPSCs and one human embryonic stem cell line), hereafter referred to as non-AD, and seven fAD patient-derived lines consisting of two independent APP V717I patient lines and five PSEN1 mutation lines (int4del, Y115H, M139V, M146I, and R278I, see Table 3 and Fig. S1). All lines generated cortical neurons with a similar efficiency, quantified by similar population expression of cortical layer markers TBR1 and CTIP2 after 100 days of differentiation (Fig. S1). APP expression was not altered at the protein or RNA level by the presence of fAD mutations (Fig. 1b–d).
Consistent Aβ ratios over time and between independent inductions
Absolute concentrations of secreted Aβ42, Aβ40, Aβ38 (using ECL), and Aβ43 (using ELISA) were quantified in 2D iPSC-derived neuronal conditioned media (CM) from three non-AD and two fAD iPSC lines. Measurements were taken at three successive 48-h intervals at 100 days of differentiation and again at day 200. We have previously characterised Aβ profiles through neuronal differentiation and day 100 represents a timepoint when neurogenesis is largely complete and a neuronal Aβ secretome is detectable [35].
Secreted Aβ43, Aβ42, Aβ40, and Aβ38 did not correlate to levels of cell death. This was in contrast to secreted tau that displayed a strong correlation to cell death (Fig. S2). Although acceptable measurement consistency (<20% coefficient of variance (CV)) was observed within each induction over the course of 6 days, a large degree of variability (>20% CV) was observed over longer time periods (day 100 versus day 200) (Fig. S2). Furthermore, different iPSC clones of the same reprogrammed fibroblast line and independent inductions of the same iPSC line were highly variable, in some cases even over the course of 6 days.
However, when iPSC-neuron-derived Aβ peptides were analysed as a ratio to the most abundant product, Aβ40, results were highly consistent when comparing distinct clones from the same patient, independent neural inductions, and different time points from the same induction (Fig. 1e–h). Thus, Aβ production ratios appear tightly regulated within a narrow physiological range, which remains stable over substantial periods of time once cells have reached maturity 100 days post-induction. Employing Aβ ratios overcomes experimental variability through internal normalisation, allowing meaningful comparisons between fAD lines.
Aβ ratios are consistent in different biosamples of the same individual patient donor
To further investigate the consistency of Aβ ratios in patient-derived material, we compared relative Aβ levels in iPSC conditioned media, iPSC-neuronal lysates, CSF, and post-mortem brain tissue homogenate from the same patient donor (APP V717I-1) (Fig. 2a–c and Fig. S1).

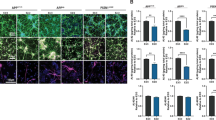
Aβ ratios are consistent between matched in vitro and in vivo samples from the same patient donor. a Aβ42:40 and b Aβ38:40 measured in conditioned media (n = 12), cell lysates (n = 8), lumbar CSF (n = 1) and brain tissue homogenate (n = 1) from the same individual. c Post-mortem tissue, 3D cerebral organoids and 2D iPSC-neurons from the same individual were immunostained for Aβ and MAP2
Results showed that Aβ38:40 levels were highly consistent between samples (Fig. 2b). Notably, these data were increased compared with Non-AD CM (Fig. 1g). Unfortunately, Aβ38 levels were below the detection threshold in brain homogenate. Aβ42:40 ratios were less consistent between the different cellular compartments and cellular states (Fig. 2a). Interestingly, the Aβ42:40 ratio secreted by cultured neurons closely matched that observed in the soluble fraction of matched cortical tissue. A comparative reduction in CSF may be consistent with preferential deposition of Aβ42 into insoluble amyloid plaques, thus affecting downstream CSF ratios. Deposition of amyloid into dense-core and diffuse plaques was observed in post-mortem brain of this individual, but not in our in vitro models (Fig. 2c). Results from cultured neuron lysates, representing soluble intracellular and membrane bound Aβ, show depletion of Aβ42:40 equivalent to that of CSF compared to that secreted into culture media.
These data promote the use of Aβ ratios to compare different cell states and show the value of iPSC-based disease modelling.
Mutation-specific effects on APP cleavage, highlighting relative increments in Aβ42 and Aβ43
We expanded our investigation to include additional iPSC lines and samples derived from 3D cerebral organoids. The expression of APP was largely consistent between 2D and 3D differentiation paradigms, with 3D organoids showing increased variability likely due to the increased cellular diversity and heterogeneity of this system (Fig. S1). Estimates of total levels of Aβ peptides in the CM were made relative to cell pellet protein content (Fig. S2). The data displayed non-significant trends, such as increased Aβ42 and decreased Aβ38 released from fAD neurons, however a large degree of variability was shown. Because of this variability, investigations were focused on the ratios of Aβ peptides.
In both 2D and 3D cultures, Aβ42:40 was increased in all fAD mutations, to approximately twice that of controls (Fig. 3a). The PSEN1 R278I mutation displayed the smallest increase in Aβ42:40.

fAD neurons display mutation-specific Aβ profile differences. Conditioned media was collected at 100 days post-neuronal induction for analysis. Results from the ratios a Aβ42:40, b Aβ42:38, c Aβ38:40, d Aβ43:40, e Aβ42:43, f Aβ38:43 are displayed. 2D data were generated from multiple inductions per line, specifically APP V717I-1 clone 1 (n = 7), APP V717I-1 clone 3 (n = 3), APP V717I-2 (n = 2), PSEN1 Int4del clone 4 (n = 5), PSEN1 Int4del clone 6 (n = 5), PSEN1 Y115H (n = 6), PSEN1 M139V (n = 6), PSEN1 M146I (n = 3), PSEN1 R278I (n = 6). Control data were generated from the following inductions: Ctrl 1 (n = 5), Ctrl2 (n = 6), Ctrl3 (n = 7), Ctrl4 (n = 6), and SHEF6 (n = 4). 3D data consisted of two inductions of each line, except APP V717I-1 clone 3, SHEF6, and M139V for which no data is available. Significance levels: * = < 0.05, ** = < 0.01, *** = < 0.001
Aβ42:38, a putative biomarker for γ-secretase cleavage efficiency, was significantly increased in all PSEN1 mutation lines versus non-AD (Fig. 3b). Specifically, PSEN1 int4del and PSEN1 Y115H demonstrated similar Aβ42:38, whereas PSEN1 M139V, PSEN1 M146I, and PSEN1 R278I exhibited decreasing changes versus non-AD. APP V717I mutant neurons also showed a small, yet significant, increase in Aβ42:38. 3D cultures closely followed the results of their 2D counterparts in the fAD lines, although greater variability between 3D and 2D control lines was observed.
Aβ38:40 is a proposed marker of γ-secretase cleavage pathway. Interestingly, this ratio was able to distinguish neuronal lines based on mutation status (Fig. 3c). Compared to non-AD cells Aβ38:40 was significantly increased in APP V717I, whilst in PSEN1 mutations the ratio was unchanged versus non-AD in PSEN1 Y115H, PSEN1 M146I and PSEN1 R278I, and decreased in PSEN1 int4del, PSEN1 M139V. Once more results from 3D cultures closely aligned with those of 2D.
Ratios of Aβ43 to other Aβ peptides have rarely been described in the literature. Results showed that in comparison to non-AD, all PSEN1 mutations, except PSEN1 M146I, demonstrated increased Aβ43:40 (Fig. 3d). The magnitude of increase in PSEN1 R278I is particularly noteworthy given the comparatively small degree of change in the Aβ42:40 ratio we observed in this line, and the previous reports of elevated Aβ43 in this mutation [36, 37]. APP V717I was not observed to differ from non-AD. Results for Aβ42:43 and Aβ38:43 generally mirror results for Aβ38:40. These ratios compare products on the two cleavage product pathways (Fig. 3c, e, f, see also Fig. 1a) where ratios were raised in APP V717I versus non-AD. This was not the case for PSEN1 mutations, where changes in the Aβ production pathway shift are not supported (Fig. 3c, e, f).
When Aβ peptide ratios were compared with age of onset for each genotype, the Aβ42:40 ratio showed negative trend (Fig. S3), depicting younger age of onset with higher Aβ42:40 ratios. Aβ43:40 showed a weak positive trend (Fig. S3), suggesting that Aβ43 is less well correlated to age of onset. Combining Aβ42 and Aβ43 relative to Aβ40 and Aβ38 did not enhance correlation, suggesting that Aβ42 shows the strongest link with age of onset. It should be noted that none of these correlations reached significance, likely due to the low-sample size.
In summary, our panel of fAD iPSC-derived neurons display a series of mutation-specific alterations in the relative production of different Aβ peptides, highlighting varied size and scale of changes in γ-secretase endopeptidase and carboxypeptidase-like cleavage of APP. The APP V717I mutation specifically alters the endopeptidase cleavage pathway, with Aβ38:40 and Aβ42:43 results reinforcing the proposed shift in product lineages [8]. PSEN1 mutations lead to higher relative proportions of either Aβ42 or Aβ43 via reduced carboxypeptidase-like activity, and may implicate a role for Aβ43 in AD pathogenesis.
Mass spectrometry highlights varied reduction in γ-secretase activity relative to other secretases as a result of fAD mutations
To investigate the secretome of Aβ peptides and to infer different activities of the α-, β-, and γ-secretases, MALDI TOF/TOF mass spectrometry was performed on Aβ peptides immunoprecipitated from 2D iPSC-neuronal supernatants. Given that Aβ profiles were consistent in 2D and 3D cultures, only media from 2D cultures were analysed. As demonstrated in Fig. 1, using ratios of Aβ peptides to Aβ40 acts as an internal normalisation to represent the data, and therefore was utilised to probe the mass spectrometry data.
Mass spectrometry confirmed the broad findings for relative amounts of Aβ38/40/42 generated using the MSD immunoassay previously described, although Aβ43 was not detectable with this method of analysis (Supplementary Table 1). Aβ42:40 was significantly raised in all fAD mutations except PSEN1 R278I (Fig. 4a), which had been the mutation where this ratio was least altered in the immunoassay. Aβ42:38 was significantly increased versus non-AD in all PSEN1 mutations except PSEN1 Y115H (although a clear tendency for increase, proportional to the immunoassay results, was observed) and R278I where the level of significance in the immunoassay had once again been weak (Fig. 4b). Aβ38:40 was significantly increased in the APP V717I line versus non-AD, unchanged versus non-AD in PSEN1 Y115H, PSEN1 M146I and PSEN1 R278I, and decreased in PSEN1 int4del and M139V (Fig. 4c), mirroring the immunoassay results.

Mutation specific differences of Aβ secretomes from multiple proteolytic pathways. Results from mass spectrometric analysis of cell media for Aβ peptides generated by a–f BACE1 and γ-secretase activity g–i, BACE1 and BACE2 activity, and j–l BACE1 and α-secretase activity normalised as ratios. Mean data were generated at day 100 from multiple independent inductions per line, specifically non-AD (n = 10) consisting of pooled data of Ctrl1 (n = 2), Ctrl2 (n = 2), Ctrl3 (n = 2), Ctrl4 (n = 2), and Shef6 (n = 2). fAD data were generated from the following, APP V717I-1 clone 1 (n = 5), APP V717I-1 clone 3 (n = 2), PSEN1 int4del clone 4 (n = 2), PSEN1 int4del clone 6 (n = 2), PSEN1 Y115H (n = 2), PSEN1 M139V (n = 2), PSEN1 M146I (n = 2), and PSEN1 R278I (n = 2). Significance levels: * = < 0.05, ** = < 0.01, *** = < 0.001
Moving beyond Aβ38/40/42, mass spectrometry highlighted several interesting patterns among less well studied peptides. Aβ39 ratios displayed a similar, though not identical, pattern to Aβ38 whereby specifically APP V717I showed increased Aβ39:40 relative to non-AD and PSEN1 mutant neurons (Fig. 4d). Aβ38 is often used as the final fragment in the Aβ48 > 45 > 42 > 38 pathway, although Aβ39 is reported as an alternative fragment generated from Aβ42 [6]. Aβ42:39 was found to be raised significantly in a subset of PSEN1 mutant neurons as well as APP V717I versus non-AD (Fig. 4e), supporting findings of Aβ42:38 by MSD.
No differences in Aβ37:40 were observed between non-AD and any fAD mutation (Fig. 4f), indicating that the disease mechanism that results in impaired γ-secretase carboxypeptidase-like efficiency mainly affects the processing of longer Aβ peptides, which is in line with the proposed mechanistic model for fAD [38].
Relative to the γ-secretase-dependent peptide Aβ40, PSEN1 int4del neurons exhibited significantly raised levels of BACE1–BACE2 products (Aβ19/20) and BACE1–BACE1/BACE2 products (Aβ34) (Fig. 4g–i) [35, 39, 40], as well as a non-significant skew toward increased α-secretase products (Aβ15/16) (Fig. 4k, l), and Aβ17, attributed to either γ-secretase cleavage [41] or endothelin-converting enzyme cleavage [42] (Fig. 4j). Similarly, PSEN1 Y115H significantly increased BACE1-α-secretase products and displayed a tendency for increased BACE1–BACE2 products (Fig. 4g–l). These effects were not evident in other fAD neurons, which displayed grossly similar secretomes to non-AD samples (Fig. S3).
Finally, N-terminally truncated Aβ peptides (in ratio to Aβ40) in fAD cell lines did not significantly differ from non-AD. The exceptions to this were the APP V717I and PSEN1 M139V lines, which displayed increased Aβ2-40:40 (p = 0.01 and p = 0.04, respectively) as well as PSEN1 M139V that also exhibited a decrease in Aβ11-40:40 (p = 0.03), (Fig. S3). Generally, similar mutation-specific effects were seen for Aβ11-x as for Aβ1-x peptides described above that would be worth pursuing with greater sample sizes; e.g. carboxypeptidase-like defects caused by PSEN1 mutations and endopeptidase effects in APP mutant cells. For example, the fAD lines tended to increase Aβ11-42 in ratio to Aβ11-40 (Supplementary Table 1 and Fig. S3), an effect previously observed by Siegel et al. [43].
Together these data report on fAD mutation dependent effects on APP. Mass spectrometry data reinforce the findings that APP mutations alter endopeptidase cleavage and that PSEN1 mutations reduce γ-secretase carboxypeptidase-like activity. Additionally PSEN1 int4del and Y115H mutations appear to display a greater deficiency in γ-secretase activity than other PSEN1-mutation bearing neurons, shown by an increase in α- and β-secretase-dependent products relative to γ-secretase-dependent peptides.
γ-Secretase protein levels are altered in a subset of PSEN1 mutant lines
PSEN1 mutations have been shown to alter γ-secretase stability [38, 44], and so we investigated total PSEN1 protein levels in our human neuronal model using western blotting (Fig. 5a). Neurons harbouring the PSEN1 mutation R278I displayed a band at 46 kDa; relating to full length PSEN1 that has not undergone autocatalysis and maturation [9, 36, 37]. As a result, this line exhibited reduced mature PSEN1 levels (Fig. 5b). Despite proper maturation, the PSEN1 mutations M139V and M146I showed a high degree of variability in PSEN1 protein levels. In a subset of neural inductions, PSEN1 levels were considerably lower than control neurons, however, this was inconsistent and did not reach significance (Fig. 5a, b). PSEN1 int4del and Y115H lines showed consistent PSEN1 protein levels that were similar to APP V717I mutant neurons and non-AD lines (Fig. 5a).

PSEN1 protein levels are variably altered in a subset of PSEN1 mutant neurons. a Representative western blot of 3 control neuron lysates and 6 fAD lysates. The asterisk depicts immature, full length PSEN1 protein at 46 kDa. b Quantification of independent neuronal lysates, replicates are depicted within histogram
These data suggest that mature PSEN1 protein levels are variably altered when harbouring M139V, M146I or R278I amino acid substitutions. These lines showed Aβ profiles that were most similar to control secretomes (Fig. 4 and S3). The PSEN1 mutant lines that showed the greatest reduction in γ-secretase-dependent Aβ peptides, int4del and Y115H, displayed PSEN1 protein levels similar to controls. These data suggest that three alternative mechanisms behind PSEN1 partial loss of function, lack of PSEN1 protein maturation, lack of PSEN1 stability and reduced catalytic activity of γ-secretase.
Discussion
In this study, we systematically investigated the profile of Aβ species using in vitro, patient-derived stem cell models of fAD. The main finding was that different fAD mutations have qualitatively distinct effects on APP processing and Aβ production by γ-secretase; affecting the APP ε-cleavage pathway and carboxypeptidase-like activity in different ways (summarised in Table 4). Other key findings were that employing ratios of each Aβ peptide relative to Aβ40 provides a robust internal normalisation to overcome inherent variability between cultured cell lines and between inductions of the same line. Additionally, Aβ ratio profiles in cultured cortical neurons, CSF and brain tissue from the same patient with an fAD genotype and diagnosis were compatible with the current understanding of AD-associated Aβ brain deposition and clearance. Our work shows that fAD mutation effects are consistent between 2D cortical neurons and 3D cerebral organoids and we provide the first investigations into Aβ43 as well as smaller Aβ peptides in iPSC neuronal models without overexpression.
The data presented offer a human neuronal validation of two hypotheses advocated by Chavez-Gutierrez et al. [8]. Firstly, that pathogenic APP mutations favour increased γ-secretase Aβ48 > 45 > 42 > 38 ε-cleavage. The concomitant increase in Aβ38:40, Aβ42:40, and Aβ42:43 observed in APP V717I cells is consistent with this idea. Interestingly, this pattern extended to Aβ39:40, Aβ39 being the true tripeptide postcedent of Aβ42 [6]. Study of Aβ39 has been largely limited to models of oligomerisation [45, 46] and further investigation is called for.
The second tenet substantiated by our data is that mutations in PSEN1 lead to inefficient γ-secretase carboxypeptidase activity. This predisposes neurons to the accumulation of longer Aβ fragments, demonstrated by consistently increased Aβ42:40 alongside increased Aβ42:38 in PSEN1 mutation cell lines. The observed increase in Aβ43:40 reinforces the idea of reduced carboxypeptidase efficiency on the Aβ49 > 46 > 43 > 40 pathway, and it is interesting that different mutations specifically effect one pathway or both. Reduced carboxypeptidase activity can be further explained by three distinct mechanisms. Firstly, reduced γ-secretase activity is suggested for PSEN1 int4del and Y115H lines, potentially due to their location near the substrate docking domain [47, 48]. Secondly, incomplete maturation of PSEN1 protein with R278I mutations leads to reduced levels of mature protein [36, 37]. Finally, PSEN1 M139V and M146I mutations lead to variably altered PSEN1 levels, consistent with altered protein stability [38, 44] and supported by studies of PSEN1 abundance in brain tissue of early onset AD patients [49, 50]. This variability in PSEN1 protein levels is likely to reduce the pool of functional γ-secretase enzyme. This finding demonstrates the advantage of using a more physiological model, such as iPSC-derived neurons, as protein instability could explain why the pathogenic M139V protein shows close to wild-type biochemical enzyme kinetics [8]. Reduced carboxypeptidase activity is especially relevant, given the recent suggestion that shorter Aβ peptides may be protective, meaning reductions in Aβ38 and Aβ40 could lie behind certain PSEN1-associated pathology [51]. It remains unclear why PSEN1 mutations affect the carboxypeptidase-like activity of γ-secretase without an apparent change to endopeptidase activity, but can be explained due to enzyme-substrate interaction destabilisation as proposed by Szaruga et al. [38].
Three findings described by the data add to the complexities of the tripeptide hypothesis. Firstly, PSEN1 int4del and M139V mutations reduce Aβ38:40, potentially suggesting that alterations to the ε-cleavage pathway. It is important to note that altered γ-secretase efficiency between the two tripeptide pathways can also explain these findings. Secondly, APP V717I mutations may lead to small yet significant increases in Aβ42:38 and Aβ42:39, suggesting that reduced carboxypeptidase activity in these neurons. Thirdly, Aβ37:40 ratios were not significantly altered where this might have been expected in the PSEN1 lines, suggesting either that the effect of reduced γ-secretase carboxypeptidase-like efficiency may diminish beyond a focal point in the APP C-terminal sequence or that Aβ37 is γ-secretase independent.
Given the divergence of different phenotypes in different lines, the use of additional lines in future work will provide added validation; for example, investigating extra C-terminal PSEN1 mutations and alternative APP mutation bearing lines. Larger studies may make it possible to correlate the functional outcome of the different APP processing defects to clinical symptoms. For example, whether mutations pre- and post-codon 200 of PSEN1 could predict age of onset [52]. Despite these limitations, our study represents one of the largest series of fAD lines, thereby allowing comparisons of the effects of different mutations on the Aβ secretome.
Conclusion
This study employed multiple patient-derived iPSC-neurons to model APP processing and Aβ production in the context of fAD APP and PSEN1 mutations. Ratios of secreted Aβ peptide fragments revealed consistent fundamental differences between mutations, leading to quantitatively and qualitatively divergent Aβ secretomes, explained by three partial loss-of-function mechanisms and one ε-cleavage pathway shift. Common cellular functional outcomes of these changes are needed to further our understanding of the disease mechanism and these patient differences are a fundamental consideration for clinical trials involving γ- and β-secretase inhibitors. This work demonstrates that iPSCs offer a valuable model to investigate underlying cellular dysfunction that results from inherited fAD mutations; representing early changes that may progress with age to neurodegeneration.
References
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421–30.
O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204.
Fernandez MA, Biette KM, Dolios G, Seth D, Wang R, Wolfe MS. Transmembrane substrate determinants for γ-secretase processing of APP CTFβ. Biochemistry. 2016;55:5675–88.
Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, et al. Gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of -carboxyl terminal fragment. J Neurosci. 2009;29:13042–52.
Matsumura N, Takami M, Okochi M, Wada-Kakuda S, Fujiwara H, Tagami S, et al. γ-Secretase associated with lipid rafts: multiple interactive pathways in the stepwise processing of β-carboxylterminal fragment. J Biol Chem. 2014;289:5109–21.
Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014;289:31043–52.
Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, et al. The mechanism of gamma-secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–74.
Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda-Falla D, Lleo A, et al. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J Exp Med. 2015;212:2003–13.
Slemmon JR, Shapiro A, Mercken M, Streffer J, Romano G, Andreasen N, et al. Impact of cerebrospinal fluid matrix on the detection of Alzheimer’s disease with Abeta42 and influence of disease on the total-Abeta42/Abeta40 ratio. J Neurochem. 2015;135:1049–58.
Vanderstichele H, Bibl M, Engelborghs S, Le Bastard N, Lewczuk P, Molinuevo JL, et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: a consensus paper from the Alzheimer’s Biomarkers Standardization Initiative. Alzheimers Dement. 2012;8:65–73.
Toombs J, Paterson RW, Schott JM, Zetterberg H. Amyloid-beta 42 adsorption following serial tube transfer. Alzheimers Res Ther. 2014;6:5.
Janelidze S, Zetterberg H, Mattsson N, Palmqvist S, Vanderstichele H, Lindberg O, et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: better diagnostic markers of Alzheimer disease. Ann Clin Transl Neurol. 2016;3:154–65.
Dorey A, Perret-Liaudet A, Tholance Y, Fourier A, Quadrio I. Cerebrospinal fluid Aβ40 improves the interpretation of Aβ42 concentration for diagnosing Alzheimer’s disease. Front Neurol. 2015;6:247.
Blennow K, Zetterberg H, Fagan AM. Fluid biomarkers in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006221.
Arber C, Lovejoy C, Wray S. Stem cell models of Alzheimer’s disease: progress and challenges. Alzheimers Res Ther. 2017;9:42.
Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–9.
Mahairaki V, Ryu J, Peters A, Chang Q, Li T, Park TS, et al. Induced pluripotent stem cells from familial Alzheimer’s disease patients differentiate into mature neurons with amyloidogenic properties. Stem Cells Dev. 2014;23:2996–3010.
Sproul AA, Jacob S, Pre D, Kim SH, Nestor MW, Navarro-Sobrino M, et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-Derived neural progenitors. PLoS ONE 2014;9:e84547.
Woodruff G, Young JE, Martinez FJ, Buen F, Gore A, Kinaga J, et al. The presenilin-1 ΔE9 mutation results in reduced γ-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013;5:974–85.
Moore S, Evans LDB, Andersson T, Portelius E, Smith J, Dias TB, et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015;11:689–96.
Ochalek A, Mihalik B, Avci HX, Chandrasekaran A, Téglási A, Bock I, et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res Ther. 2017;9:90.
Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc Natl Acad Sci USA. 2017;114:E476–85.
Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LNP, et al. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet. 2014;23:3523–36.
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–20.
Lancaster MA, Knoblich J. Generation of cerebral organoids from human pluripotent stem. Nat Protoc. 2014;10:2329–40.
Raja WK, Mungenast AE, Lin Y-T, Ko T, Abdurrob F, Seo J, et al. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS ONE 2016;11:e0161969.
Chen M, Lee HK, Moo L, Hanlon E, Stein T, Xia W. Common proteomic profiles of induced pluripotent stem cell-derived three-dimensional neurons and brain tissue from Alzheimer patients. J Proteom. 2018;182:21–33.
Wray S, Self M, Lewis PA, Taanman J-W, Ryan NS, Mahoney CJ, et al. Creation of an open-access, mutation-defined fibroblast resource for neurological disease research. PLoS ONE 2012;7:e43099.
Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–12.
Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7:1836–46.
Portelius E, Tran AJ, Andreasson U, Persson R, Brinkmann G, Zetterberg H, et al. Characterization of amyloid β peptides in cerebrospinal fluid by an automated immunoprecipitation procedure followed by mass spectrometry. J Proteome Res. 2007;6:4433–9.
Germain PL, Testa G. Taming human genetic variability: transcriptomic meta-analysis guides the experimental design and interpretation of iPSC-based disease modeling. Stem Cell Rep. 2017;8:1784–96.
British Standards Institution. Accuracy (trueness and precision) of measurement methods and results. Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method [Internet]. Mol. Ecol. 1994 Available from: http://www.iso.org/iso/iso_catalogue/catalogue_tc/catalogue_detail.htm?csnumber=11833. Accessed 11 Feb. 2017.
Bergstrom P, Agholme L, Nazir FH, Satir TM, Toombs J, Wellington H, et al. Amyloid precursor protein expression and processing are differentially regulated during cortical neuron differentiation. Sci Rep. 2016;6:29200.
Veugelen S, Saito T, Saido TC, Chávez-Gutiérrez L, De Strooper B. Familial Alzheimer’s disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron. 2016;90:410–6.
Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci. 2011;14:1023–32.
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, et al. Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell. 2017;170:443–.e14.
Yan R, Han P, Miao H, Greengard P, Xu H. The transmembrane domain of the Alzheimer’s β-secretase (BACE1) determines its late Golgi localization and access to β-amyloid precursor protein (APP) substrate. J Biol Chem. 2001;276:36788–96.
Shi X-P, Tugusheva K, Bruce JE, Lucka A, Wu G-X, Chen-Dodson E, et al. Beta-secretase cleavage at amino acid residue 34 in the amyloid beta peptide is dependent upon gamma-secretase activity. J Biol Chem. 2003;278:21286–94.
Portelius E, Price E, Brinkmalm G, Stiteler M, Olsson M, Persson R, et al. A novel pathway for amyloid precursor protein processing. Neurobiol Aging. 2011;32:1090–8.
Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer’s amyloid beta peptide by endothelin- converting enzyme. J Biol Chem. 2001;276:24540–8.
Siegel G, Gerber H, Koch P, Bruestle O, Fraering PC, Rajendran L. The Alzheimer’s disease γ-secretase generates higher 42:40 ratios for β-amyloid than for p3 peptides. Cell Rep. 2017;19:1967–76.
Wanngren J, Lara P, Öjemalm K, Maioli S, Moradi N, Chen L, et al. Changed membrane integration and catalytic site conformation are two mechanisms behind the increased Aβ42/Aβ40 ratio by presenilin 1 familial Alzheimer-linked mutations. FEBS Open Bio. 2014;4:393–406.
Anand P, Nandel FS, Hansmann UHE. The Alzheimer beta-amyloid (Abeta(1-39)) dimer in an implicit solvent. J Chem Phys. 2008;129:195102.
Cloe AL, Orgel JPRO, Sachleben JR, et al. The Japanese mutant Aβ (ΔE22-Aβ 1-39) forms fibrils instantaneously, with low-thioflavin T fluorescence: seeding of wild-type Aβ 1-40 into atypical fibrils by Δe22- Aβ 1-39. Biochemistry. 2011;50:2026–39.
Takagi-Niidome S, Sasaki T, Osawa S, Sato T, Morishima K, Cai T, et al. Cooperative roles of hydrophilic loop 1 and the C-terminus of presenilin 1 in the substrate-gating mechanism of gamma-secretase. J Neurosci. 2015;35:2646–56.
Somavarapu AK, Kepp KP. The dynamic mechanism of presenilin-function: sensitive gate dynamics and loop unplugging control protein access. Neurobiol Dis. 2016;89:147–56.
Verdile G, Gnjec A, Miklossy J, Fonte J, Veurink G, Bates K, et al. Protein markers for Alzheimer in the frontal cortex and cerebellum. Neurology. 2004;63:1385–92.
Mathews PM, Cataldo AM, Kao BH, Rudnicki AG, Qin X, Yang JL, et al. Brain expression of presenilins in sporadic and early-onset, familial Alzheimer’s disease. Mol Med. 2000;6:878–91.
Moore BD, Martin J, de Mena L, Sanchez J, Cruz PE, Ceballos-Diaz C, et al. Short Aβ peptides attenuate Aβ42 toxicity in vivo. J Exp Med. 2018;215:283–301.
Ryan NS, Nicholas JM, Weston PSJ, Liang Y, Lashley T, Guerreiro R, et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol. 2016;15:1326–35.
Acknowledgements
We gratefully acknowledge the support of the Leonard Wolfson Experimental Neurology Centre, the NIHR UCL Hospitals Biomedical Research Centre. The Dementia Research Centre is an Alzheimer’s Research UK Coordinating Centre. SW is supported by an Alzheimer’s Research UK Senior Research Fellowship (ARUK-SRF2016B-2). NF acknowledges the support of the UK Dementia Research Institute at UCL. NSR is supported by a University of London Chadburn Academic Clinical Lectureship in Medicine. The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115439, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007–2013) and EFPIA companies’ in kind contribution. This publication reflects only the author’s views and neither the IMI JU nor EFPIA nor the European Commission are liable for any use that may be made of the information contained therein. This work was supported by the UK Medical Research Council funding to the MRC Dementia Platform UK (MR/M02492X/1) and Medical Research Council core funding to the High-Content Biology Platform at the MRC-UCL LMCB university unit (MC_U12266B). We would like to thank Dr. Rita Louro Guerreiro, Lee Darwent, and Celia Kun Rodrigues for help with sequencing of iPSC clones.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Arber, C., Toombs, J., Lovejoy, C. et al. Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol Psychiatry 25, 2919–2931 (2020). https://doi.org/10.1038/s41380-019-0410-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-019-0410-8
This article is cited by
-
Mutual interaction of neurons and astrocytes derived from iPSCs with APP V717L mutation developed the astrocytic phenotypes of Alzheimer’s disease
Inflammation and Regeneration (2024)
-
Humanized brain organoids-on-chip integrated with sensors for screening neuronal activity and neurotoxicity
Microchimica Acta (2024)
-
Simple model systems reveal conserved mechanisms of Alzheimer’s disease and related tauopathies
Molecular Neurodegeneration (2023)
-
3D bioprinting patient-derived induced pluripotent stem cell models of Alzheimer’s disease using a smart bioink
Bioelectronic Medicine (2023)
-
Altered ubiquitin signaling induces Alzheimer’s disease-like hallmarks in a three-dimensional human neural cell culture model
Nature Communications (2023)
