
Abstract
Cutaneous deep penetrating melanocytic neoplasms frequently simulate melanoma and might occasionally progress to metastatic melanoma. Distinguishing deep penetrating nevi (DPN) and deep penetrating melanocytomas (DPM) from malignant deep penetrating tumors (MDPT) is difficult based on histopathology alone, and diagnostic criteria for MDPT are currently lacking. Using a molecular workup, we aimed to provide readily available diagnostic tools for classification of deep penetrating tumors. We used clinical follow-up and Single Nucleotide Polymorphism (SNP) array for tumor classification of 20 deep penetrating neoplasms to identify associations with histopathological, immunohistochemistry, and NGS findings. Ten neoplasms were classified as MDPT, four as DPM, and six as DPN. Two MDPT showed metastases. The following parameters were statistically significantly associated with MDPT: severe nuclear atypia (risk ratio [RR] 2.9, p < 0.05), absence of a nevus component (RR 10.0, p = 0.04), positive PRAME expression (RR 9.0, p = 0.02), complete loss of p16 expression (RR 3.5, p = 0.003), TERT-p and APC mutations (RR 11.0, p = 0.01 and RR 2.7, p = 0.002, respectively), and ≥1 additional pathogenic mutation (RR 9.0, p = 0.02). Ki-67 expression ≥ 5% was not significantly associated with MDPTs, although it was <5% in all DPNs. Three MDPT did not show nuclear β-catenin expression despite having a CTNNB1 (n = 2) or an APC mutation (n = 1). Our findings suggest that complete loss of p16 and positive PRAME expression, a driver mutation in APC, ≥ 1 additional pathogenic mutation, especially in TERT-p, support an MDPT diagnosis in deep penetrating neoplasms. Besides severe nuclear atypia and possibly severe inflammation, we did not identify specific histopathological criteria for malignancy. Non-aberrant nuclear β-catenin expression might not exclude a deep penetrating signature in MDPT.
Similar content being viewed by others


Introduction
Deep penetrating nevi (DPN) are acquired cutaneous melanocytic neoplasms with a wedge-shaped configuration penetrating the reticular dermis or subcutis1,2. DPN often pose a diagnostic challenge since their ambiguous presentation can simulate melanoma and distinct entities such as cellular blue and Spitz tumors3. Also, malignant and metastatic progression of deep penetrating tumors has been reported in some cases4,5,6,7. DPN typically have driver mutations in the MAPK pathway (e.g., in BRAF or MAP2K1), with additional mutations in the WNT pathway (in APC or CTNNB1) resulting in deep penetrating histomorphology7,8. Based on these additional mutations, DPN are now classified as intermediate melanocytic tumors or low-grade melanocytomas in 2018 WHO Classification of Skin Tumors, despite their primarily benign behavior and name. Atypical DPN are now referred to as high-grade deep penetrating melanocytoma (DPM) based on the presence of atypical histopathological features, including large size, asymmetry, sheet-like arrangements of melanocytes, >2 dermal mitoses per mm2, and nuclear atypia9. In contrast, there are no histopathological or molecular criteria defined for their rare malignant counterpart (“melanoma in DPN”). Distinguishing malignant deep penetrating tumors (MDPT) from melanoma-mimicking DPN and DPM is difficult based on histopathological assessment alone but has substantial treatment implications10. Molecular analyses, including next-generation sequencing (NGS) and single nucleotide polymorphism (SNP) array, allow more accurate tumor classification by detecting driver and pathogenic mutations and copy number variations11. However, these techniques are often only available in specialized centers. Therefore, we present the extensive diagnostic workup of 20 deep penetrating neoplasms to provide readily available diagnostic tools for MDPT.
Materials and methods
Study population
The Department of Pathology of the University Medical Center Utrecht is a tertiary referral center for dermatopathology. Ambiguous melanocytic tumors sent in for consultation by pathologists from July 1, 2018 to July 31, 2021 were reviewed to identify specimens with deep penetrating histomorphology for which a diagnostic workup was performed including high-resolution SNP array, NGS, IHC, and expert histopathological assessment. Specimens of cutaneous, primary, melanocytic neoplasms with a molecularly confirmed deep penetrating signature were potentially eligible for this study. Non-cutaneous, recurrent, or metastatic melanocytic lesions were excluded.
Study design
The main objective of this study was to identify readily available diagnostic criteria for MDPT by using clinical outcome and the number of genome-wide CNVs, based on a recent meta-analysis of our group11, as gold standard. First, NGS was used to identify a pathogenic APC or CTNNB1 mutation in all tumors for molecular confirmation of their assignment to the deep penetrating subset of WHO-pathway 1. Second, the biological behavior of the tumor was assessed. Tumors with locoregional or distant metastases or ≥3 genome-wide CNVs were classified as malignant (MDPT)11. Tumors without eventful follow-up were classified as DPN in the absence of CNVs, as DPM if they had two CNVs, and, if they had a single CNV, as DPN or DPM based on histopathological evaluation using the current WHO-criteria for DPM (>2 dermal mitoses per mm2, large size, asymmetry, sheet-like growth, and nuclear atypia)9. Then, NGS, IHC, and additional histopathological findings were compared between MDPT, DPM, and DPN. At least two experts reviewed immunohistopathological and molecular data. This study was performed in compliance with the UMC Utrecht’s medical research ethics committee guidelines for case studies.
Follow-up
Follow-up data regarding recurrence and locoregional and distant metastasis were gathered from the nationwide network and registry of histo- and cytopathology in the Netherlands (PALGA), including all the country’s pathological examinations.
DNA isolation
For DNA isolation, formalin-fixed paraffin-embedded (FFPE) tissue blocks from all cases were cut at 4 μm. After HE staining, viable tumor tissue areas with the highest tumor cell percentage (TCP) were selected and marked by a pathologist and macro-dissected. Non-lesional areas (e.g., adjacent nevi) were not included in the sampling. DNA from the samples was subsequently extracted and purified using the Cobas® DNA Sample Preparation Kit (Roche, Basel, Switzerland), according to the manufacturer’s protocols. TCPs were estimated by a pathologist.
SNP array analysis
SNP array was performed using the Infinium CytoSNP-850K v1.2 BeadChip (Illumina, San Diego, California), according to the manufacturer’s protocol. This exon-centric oligo-array employs 850,000 single nucleotide polymorphism probes with enriched coverage for 3,262 cancer genes enabling copy number calling with a resolution of 10 kb in selected genomic regions. Briefly, 8–200 ng of isolated DNA was treated with the Infinium HD FFPE DNA Restore Kit (Illumina, San Diego, CA). After FFPE restoration, DNA was amplified, fragmented, and hybridized to the Infinium CytoSNP-850K v1.2 BeadChip. After extension and staining, the Beadchip was scanned using the iScan (Illumina, San Diego, California) to generate probe intensity output files. Probe fluorescence was compared with a reference human genome. The results were visualized, analyzed, and interpreted using NxClinical Software (BioDiscovery, El Segundo, California). NxClinical provides information on B-allele frequency and intensity, allowing accurate calling of CNVs (chromosomal gains and losses) and copy-neutral loss of heterozygosity using a pre-defined ruleset11, and chromothripsis (defined as at least ten gains or losses on a chromosomal segment or arm). A TCP of ≥ 30% was considered sufficient for reliable interpretation of the SNP array results.
NGS
As previously described, amplicon-based targeted Next-Generation Sequencing was performed with Ion Ampliseq™ custom-designed panels12. All panels included amplicons covering (hotspot regions of) APC, BRAF, CDKN2A, CTNNB1, HRAS, IDH1, KIT, NRAS, the TERT promoter region (TERT-p), and TP53 (see supplementary Tables S1–S3 for the complete list of genes). Library preparation was performed using the Ion Ampliseq™ Library kit 2.0 (Thermo Fisher Scientific, Waltham, USA), according to the manufacturer’s protocol, on the Janus Express (PerkinElmer, Waltham, USA). Template preparation and chip loading was performed by the Ion Chef System using the Ion 510™ & Ion 520™ & Ion 530™ Chef kit and protocol. Sequencing was performed on the Ion Torrent S5, followed by variant calling by the Torrent Variant Caller. Variant annotation was done by an in-house bioinformatics pipeline using Ensembl API12. Variants were visually inspected using the Integrative Genomics Viewer (IGV) and assigned to one of the following classes: benign (class 1), likely benign (class 2), variant of uncertain significance (VUS, class 3), likely pathogenic (class 4), or pathogenic (class 5). In this study, all class 4 and 5 variants are reported.
Histopathological evaluation and IHC
All WHO-criteria9 mentioned above and an additional 14 pre-defined histopathological parameters were reviewed in the samples. The following parameters were rated as absent or present: nevus component, asymmetry, well-defined lateral margins, ulceration (defined as the complete absence of the epidermis including the stratum corneum with a fibrinopurulent stromal reaction), ascent of melanocytes (reaching the stratum granulosum), severe atypia (defined as nuclei ≥ 1.5 times the size of resting basal keratinocytes, hyperchromasia, presence of nucleoli, and severe nuclear variation), sheet-like growth, severe inflammation (defined as significant patchy or diffuse lymphocytic or lymphoplasmacytic infiltrate), presence of plasma cells, regression, satellites, perineural growth pattern, lymphangioinvasive growth, and necrosis. Also, invasion depth (mm), diameter (mm), number of dermal mitoses per mm2, epidermal change (absent, hyperplasia, effacement), and cytology (epithelioid, spindle-cell, nevoid, other) were assessed. We performed IHC using the following antibodies, if indicated: HMB-45, Ki-67, MART-A, SOX-10, S-100, p16, TP53, PRAME, β-catenin, BAP1, ALK, ROS1, NTRK, PRKAR1A, and BRAF V600E. Positive β-catenin staining was defined as aberrant nuclear staining (solely cytoplasmic or membranous staining was considered negative)8. Positive PRAME expression was defined as ≥ 75% nuclear staining13. For statistical analysis, p16 and Ki-67 outcomes were categorized as complete loss (absent/present) or partial or complete loss (absent/present) and as <5% and ≥5%, respectively.
Statistical analyses
To identify associations between histopathological, IHC, and NGS parameters and classification of deep penetrating tumors, risk ratios (RR), 95% confidence intervals (CI), and P values were calculated. Primary analyses investigated associations with tumors classified as MDPT versus non-MDPT (DPN/DPM). These analyses were performed for parameters for which at least two tumors were positive. Results were considered to indicate an association if P values were < 0.05. Secondary analyses investigated whether identified associations also existed with tumors classified as MDPT versus DPM. Tumors with missing values for a specific parameter were excluded from that analysis.
Results
Patients and clinical features
During the 3-year study period, 35 primary melanocytic neoplasms sent in for consultation were diagnosed as part of the deep penetrating spectrum, representing ~4% of the consulting practice. NGS was performed on 28 (80%) tumors, of which 21 (75%) had additional SNP array testing. One tumor (case 21) was excluded from statistical analysis because the NGS variant allele frequencies and sample quality were suggestive of a low TCP (<25%), rendering the SNP results unreliable. Therefore, a total of 20 tumors were included in this study. Table 1 shows the clinical characteristics and the initial differential and final diagnosis after full diagnostic workup. Eleven patients (55%) were female, and the median age was 43 (range 15–75) years. Most lesions were located on the trunk or shoulder (n = 9; 45%) or extremities (n = 8; 40%). Follow-up ranged from three to 36 months (median 10 months). The option of a deep penetrating tumor was not initially considered in 12 (60%) patients, and the differential diagnosis included melanoma for two DPN and one DPM. A re-excision was performed in 18 cases (90%) and a sentinel lymph node biopsy (SNB) in seven MDPT (70%).
Classification of deep penetrating tumors using follow-up and SNP array
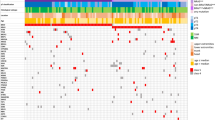
Follow-up was available for all cases, of which 18 (90%) did not show evidence of disease and two (10%) had metastases (Table 1). Case 14 (Fig. 1) had a satellite and axillary lymph node metastasis (5/13 lymph nodes positive, largest metastatic deposit 8.6 mm) and was clinically classified as MDPT stage IIIc (pT3aN3c). The patient was treated with NKTR-214 and adjuvant nivolumab. Case 18 also had a satellite and subsequent sentinel lymph node metastasis (largest deposit 0.2 mm) and was clinically classified as stage IIIc melanoma (pT2aN2c). The patient was treated with adjuvant pembrolizumab. Ten (50%) tumors had ≥ 3 CNVs and were classified as MDPT, including the two cases with metastases. Another 10 cases had < 3 CNVs, of which five (50%) lacked CNVs, and one (10%) had two CNVs, resulting in classification as DPN and DPM, respectively. The remaining four (40%) tumors had a single CNV. They were classified as DPN (n = 1, case 6, Fig. 2, harboring a small focal gain of 7p13.3 with the CHN2 gene segment) or DPM (n = 3, including case 9, Fig. 3) based on the absence or presence of atypical histopathological features, respectively. In total, ten tumors were classified as MDPT, four as DPM, and six as DPN. The most frequent CNVs in MDPT were heterozygous loss of 9p21 (CDKN2A; in 45%) and 9q (in 45%) and gain of 7q including 7q34 (BRAF; in 45%) and 6p including 6p24 (RREB1; in 36%); see Supplementary Table S4 for all CNV findings and Fig. 4. Two out of four MDPT with an APC mutation had concurrent loss of 5q22.2 (APC), yielding functional bi-allelic loss. The tumor excluded from statistical analysis (case 21) did not have CNVs, likely due to a low TCP, but demonstrated positive PRAME and complete loss of p16 expression, and high mitotic activity (6 per mm2), and was, therefore, classified as malignant irrespective of the CNV analysis.

A Skin excision of the back of a 74-year-old man with a strongly pigmented deep penetrating, asymmetrical lesion with epidermal effacement. B Detail showing the epitheloid, enlarged, polymorphic melanocytes with prominent nucleoli and nuclear variation. A mitosis is easily identified (white arrow). C The lesion shows strong nuclear PRAME expression. D Lymph node metastatic deposit of 8.6 mm. E In a different section, a satellite is clearly visibly (white arrow). F p16 expression is completely lost in the lesional cells. G β-catenin staining shows membranous and partly cytoplasmic and nuclear staining. H Detail of metastatic deposit with tumor cells. I Detail showing the satellite of the lesion. In addition, this MDPT had severe inflammation with the presence of plasma cells, NGS showed a CTNNB1S33P and KITL576P mutation (the TERT-p region was not interpretable), and SNP array identified 3 CNVs and one copy-neutral loss-of-heterozygosity. BAP1 and PRKAR1A IHC were not aberrant (not shown).

A Skin excision from the lower back of a 33-year-old man with an asymmetrical, compound melanocytic lesion, measuring 10 mm, with a DPN on the left and an unrelated nevus on the right. B The DPN component is wedge-shaped and shows a nested growth of slightly pigmented melanocytes with heavily pigmented melanophages in between. C The common nevus shows a nested growth of small ‘nevoid’ melanocytes. D BRAF V600E staining is positive in the common nevus on the right and is negative in the DPN on the left. E β-catenin staining shows strong cytoplasmatic and nuclear staining in the DPN. F SNP array showed a single gain of 7p13.3 with the CHN2 gene segment. This case did not show any signs of malignancy. NGS of the DPN showed CTNNB1D32N and MAP2K1P105_I107delinsL mutations.

A Skin excision from the shoulder of a 19-year-old man with a wedge-shaped, compound melanocytic lesion with deep expanding nodular growth to a depth of 3,3 mm. The lesional cells are spindle-shaped and arranged in fusiform nests. The melanocytes have gray-bluish pigmentation and the nuclei are slightly but relatively uniformly enlarged with the presence of small nucleoli. There are some heavily pigmented melanophages in between. No necrosis or ulceration is present. B There were 10 mitoses per mm2 in the dermal component; the mitotic activity was most pronounced superficially in the lesion (arrows). C p16 shows partial loss of expression. D β-Catenin is strongly positive in the cytoplasm and in some nuclei. E Ki-67 demonstrates a proliferation fraction of 26%, with a decreasing gradient towards the base. PRAME was negative (not shown). F SNP array demonstrated a single CNV (monosomy of chromosome 9). NGS showed CTNNB1P37S and MAP2K1Q56_G61delinsL mutations.

A Skin excision from the back of a 39-year-old woman with a wedge-shaped, compound melanocytic lesion penetrating the deep reticular dermis along adnexa. B Detail which shows the epitheloid, enlarged melanocytes and a mitosis. C SNP array showed 14 CNVs including those frequently found in melanoma, such as loss of 6q (MYB) and loss of 9p21(CDKN2A). In addition (not shown), this MDPT had a moderately high proliferation fraction (10%), 6 mitoses per mm2, complete loss of p16 expression, and TERT-pC250T, CTNNB1S37F and BRAFV600E mutations.
Comparison of tumor classification with NGS results
Selected IHC, NGS, and SNP array findings are shown in Table 2. MAPK pathway mutations, mainly in BRAF and MAP2K1, were found in 16 (80%) tumors (Table 2). One MDPT (case 16) had an NRASQ61R mutation. Additional pathogenic mutations in IDH1, KIT, PIK3CA, TERT-p, or TP53, were found in nine MDPT (90%) and one DPM (25%, case 7) from a patient with a known p16-Leiden CDKN2A and low-frequent TP53G245D and KITT553N mutations. The following parameters were statistically significantly associated with MDPT (Table 3 and Supplementary Table S5): TERT-p and APC mutations, respectively 11.0 times (p = 0.01) and 2.7 times (p = 0.002) more likely than wild-type, and presence of ≥ 1 additional pathogenic mutation (9.0 times more likely than absence of mutations, p = 0.02). Secondary analyses also suggested that TERT-p mutations and ≥ 1 additional pathogenic mutation were associated with MDPTs, although not statistically significant (Table 3). APC mutations remained statistically significantly associated with MDPTs (1.7 times more likely, p < 0.05).
Comparison of tumor classification with IHC and histopathological parameters
Histopathological assessment for all tumors is shown in Table 4. Only severe nuclear atypia and absence of a nevus component were statistically significantly associated with MDPTs, 2.9 times more likely than lack of severe atypia (p < 0.05) and 10.0 times more likely than presence of a nevus component (p = 0.04), respectively. Secondary analyses supported that severe inflammation was associated with MDPTs (2.0 times more likely, p < 0.05). β-catenin, performed on five DPN, four DPM, and eight MDPT, did not show nuclear expression in one DPN (20%) and three (38%) MDPT. Of the IHC parameters, positive PRAME (9.0 times more likely than negative PRAME, p = 0.02), complete (3.5 times more likely, p = 0.003), and complete or focal loss of p16 expression (7.4 times more likely, p = 0.04) were statistically significantly associated with MDPT (Table 3). Secondary analyses also indicated that complete loss of p16 expression was statistically significantly associated with MDPT (2.0 times more likely, p < 0.05) and suggested an association for positive PRAME expression (4.0 times more likely, p = 0.1). Ki-67 expression ≥ 5% was not significantly associated with MDPTs, although it was < 5% in all DPNs.
Discussion
Given the paucity of data on MDPT, criteria for this rare melanoma type currently lack in the WHO classification of skin tumors9. Yet, we commonly encounter MDPT and suspect them of being underrecognized because their diagnosis usually requires molecular workup. This study is the first to report and use genome-wide SNP array data to classify deep penetrating tumors based on the number of CNVs. A recent meta-analysis from our group demonstrated its excellent diagnostic ability to differentiate malignant from intermediate melanocytic tumors: a cut-off of ≥3 CNVs had a sensitivity of 85% and a specificity of 84%, which increased to 94% and ≥ 97% for cut-offs of ≥ 5 and ≥ 7 CNVs, respectively11. Since microarray techniques are usually only available in specialized referral centers, we aimed to identify more readily available diagnostic criteria for classification of deep penetrating tumors, such as histopathological assessment, IHC, and mutational analysis.
Our study found positive PRAME and complete loss of p16 expression and APC and TERT-p mutations exclusively in MDPT, reflected in the statistically significant associations we identified. Also, severe nuclear atypia, absence of a nevus component, ≥1 additional pathogenic mutation (mostly in TERT-p), and complete or focal loss of p16 were significantly associated with MDPT but also found in DPM. Nuclear expression of β-catenin was negative in one DPM and two MDPT. Several of these parameters were also associated with MDPT when excluding DPNs from the analyses. Based on these results and previous research, we propose criteria for classifying deep penetrating neoplasms (Table 5) and a diagnostic workup as discussed below.
The first diagnostic step when encountering a deep penetrating neoplasm should be WHO-pathway confirmation, given the significant clinical implications10, for which nuclear β-catenin expression can be used. Previous research reported nuclear β-catenin expression in 98% of genetically unconfirmed DPN8 and 100% of atypical DPN with CTNNB1 or APC mutations14. Our results add that β-catenin IHC might be less reliable in MDPT since three (38%) tumors did not show nuclear β-catenin expression despite having a CTNNB1 (n = 2) or an APC mutation (n = 1). In such cases, NGS is needed to confirm or exclude a deep penetrating signature. However, CTNNB1 and APC mutations have been found in other melanoma types15 and are insufficient for an MDPT diagnosis in the absence of deep penetrating histomorphology.
Regarding MAPK driver mutations, Yeh et al. showed most deep penetrating lesions have BRAF or MAP2K1 mutations, but this study only included three genetically confirmed MDPT7. Our NGS results show this also holds for MDPT. One MDPT had an NRASQ61R mutation, an uncommon driver in deep penetrating lesions but reported before in some MDPT6,7 and an atypical DPN with locoregional metastases14. We did not identify a primary driver mutation in the MAPK pathway in four lesions (20%), which has been previously reported in 24% of atypical DPN14 and 6% of DPN7, most likely because most gene panels do not cover all MAPK pathway genes.
After pathway assignment, the tumor should be assessed for malignant features. We identified severe nuclear atypia to be significantly associated with MDPT but also found this in one DPN. This is in line with a recent review that did not identify any histopathological features objectively supporting an MDPT diagnosis, with the possible exception of mitotic activity1. Yet, we found the absence of mitoses does not equal a DPN diagnosis since two DPM and five MDPT had ≤ 2 mitoses per mm2. Most MDPT did not have an adjacent nevus, possibly not initially present or lost in malignant progression. Consequently, its presence is not required for this diagnosis and we prefer the term MDPT over ‘melanoma in DPN’ in analogy to the terminology in Spitz tumors.
In contrast to the histopathological findings, our results show that IHC can be an important tool for tumor classification. Positive PRAME and complete loss of p16 expression strongly support malignancy, whereas focal loss of p16 might exclude a DPN diagnosis but not differentiate MDPT from DPM. Still, negative PRAME and positive p16 expression do not exclude malignancy, and we advocate that cases with suspect histopathology but non-aberrant IHC should be referred for additional molecular analysis. In concordance with previous research in melanoma16,17 and some MDPT cases6,7, our results show that TERT-p mutations strongly support an MDPT diagnosis.
The main limitation of our study is the lack of definite proof for malignancy in most MDPT and the relatively short follow-up duration. Clinical follow-up with unfavorable outcomes (distant metastasis or death) remains the gold diagnostic standard for malignancy. This did not occur in any of our cases; only one patient had a locoregional spread to lymph nodes and one had a positive SNB. The latter cannot be regarded as definitive proof of malignancy since some melanocytic neoplasms might be associated with nodal involvement without distant metastases or death, as has been described in Spitz tumors18 and pigmented epithelioid melanocytoma9. Yet, follow-up is an imperfect gold standard since not all overt melanomas metastasize or only show metastasis after decades of follow-up. Therefore, we consider SNP array for genome-wide CNV detection a reasonable alternative in a diagnostic setting. Since CNV detection highly depends on DNA quality and TCP, SNP array also has limitations, and we had to exclude one tumor with an insufficient TCP from statistical analyses. Another limitation of our study is its modest size. Consequently, due to limited statistical power, we may not have been able to identify all relevant associations. However, our study still constitutes the most extensive series of MDPT to date.
To conclude, our findings suggest that complete loss of p16 expression, positive PRAME expression, a driver mutation in APC, and ≥ 1 additional pathogenic mutation, especially in TERT-p, support an MDPT diagnosis in deep penetrating tumors. Besides severe nuclear atypia and possibly severe inflammation, we did not identify specific histopathological criteria for malignancy, but overall histopathological assessment remains the cornerstone for interpreting these findings. Non-aberrant nuclear β-catenin expression might not exclude a deep penetrating signature in MDPT. Future research should further unravel the entire genomic landscape of this rare melanoma type and identify molecular alterations most predictive for malignancy and clinical outcomes.
Data availability
Raw data are available upon reasonable request.
References
Cosgarea, I., Griewank, K. G., Ungureanu, L., Tamayo, A. & Siepmann, T. Deep penetrating nevus and borderline-deep penetrating nevus: a literature review. Front. Oncol. 10, 837 (2020).
Robson, A., Morley-Quante, M., Hempel, H., McKee, P. H. & Calonje, E. Deep penetrating naevus: clinicopathological study of 31 cases with further delineation of histological features allowing distinction from other pigmented benign melanocytic lesions and melanoma. Histopathology 43, 529–537 (2003).
Cerroni, L. et al. Melanocytic tumors of uncertain malignant potential: results of a tutorial held at the XXIX symposium of the international society of dermatopathology in Graz, October 2008. Am. J. Surg. 34, 314–326 (2010).
Magro, C. M. et al. Deep penetrating nevus-like borderline tumors: a unique subset of ambiguous melanocytic tumors with malignant potential and normal cytogenetics. Eur. J. Dermatol. 24, 594–602 (2014).
Abraham, R. M., Ming, M. E., Elder, D. E. & Xu, X. An atypical melanocytic lesion without genomic abnormalities shows locoregional metastasis. J. Cutan. Pathol. 39, 21–24 (2012).
Isales, M. C. et al. Molecular analysis of atypical deep penetrating nevus progressing to melanoma. J. Cutan. Pathol. 47, 1150–1154 (2020).
Yeh, I. et al. Combined activation of MAP kinase pathway and β-catenin signaling cause deep penetrating nevi. Nat. Commun. 8, 644 (2017).
de la Fouchardière, A. et al. β-Catenin nuclear expression discriminates deep penetrating nevi from other cutaneous melanocytic tumors. Virchows Arch. 474, 539–550 (2019).
World Health Organization. Elder D. E., Massi D., Scolyer R. A., Willemze R., eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC (2018).
de la Fouchardiere, A. et al. ESP Dermatopathology Working Group; EORTC Melanoma Group; EURACAN. ESP, EORTC, and EURACAN Expert Opinion: practical recommendations for the pathological diagnosis and clinical management of intermediate melanocytic tumors and rare related melanoma variants. Virchows Arch. 479, 3–11 (2021).
Ebbelaar, C. F., Jansen, A. M. L., Bloem, L. T. & Blokx, W. A. M. Genome-wide copy number variations as molecular diagnostic tool for cutaneous intermediate melanocytic lesions: a systematic review and individual patient data meta-analysis. Virchows Arch. 479, 773–783 (2021).
Strengman, E. et al. Amplicon-based targeted next-generation sequencing of formalin-fixed, paraffin-embedded tissue. Methods Mol. Biol. 1908, 1–17 (2019).
Lezcano, C., Jungbluth, A. A. & Busam, K. J. Comparison of immunohistochemistry for PRAME with cytogenetic test results in the evaluation of challenging melanocytic tumors. Am. J. Surg. 44, 893–900 (2020).
Manca, A. et al. NGS-based analysis of atypical Deep Penetrating Nevi. Cancers 13, 3066 (2021).
Oulès B., et al. Clinicopathologic and molecular characterization of melanomas mutated for CTNNB1 and MAPK. Virchows Arch. (2021), Epub ahead of print.
Thomas, N. E. et al. Utility of TERT promoter mutations for cutaneous primary melanoma diagnosis. Am. J. Dermatopathol. 41, 264–272 (2019).
Motaparthi, K. et al. TERT and TERT promoter in melanocytic neoplasms: current concepts in pathogenesis, diagnosis, and prognosis. J. Cutan. Pathol. 47, 710–719 (2020).
Lallas, A. et al. Atypical Spitz tumours and sentinel lymph node biopsy: a systematic review. Lancet Oncol 15, 178–183 (2014).
Lezcano, C., Jungbluth, A. A., Nehal, K. S., Hollmann, T.J., Busam, K. J. PRAME expression in melanocytic tumors. Am. J. Surg. 42, 1456–1465 (2018).
Lopez, D. R., Forcucci, J. A., O’Connor, H., Maize, J. C. PReferentially expressed antigen in MElanoma (PRAME) expression in BRCA1-associated protein (BAP1)-inactivated melanocytic tumors and deep penetrating nevi: A pilot study. J. Cutan. Pathol. 48, 597–600 (2021).
Teunissen, B. T., Knuiman, G. J., Eijkelenboom, A., Wauters, C. A. P., Wouda, S., Blokx, W. A. M. CTNNB1-mutated melanocytic lesions with DPN like features: a distinct subtype of melanocytic tumors? A report of two cases. Virchows Arch. 472, 683–687 (2018).
Garrido, M. C. et al. Combination of congenital and deep penetrating nevus by acquisition of β-catenin activation. Am. J. Dermatopathol. 42, 948–952 (2020).
Alomari, A. K. et al. DNA copy number changes correlate with clinical behavior in melanocytic neoplasms: proposal of an algorithmic approach. Mod. Pathol. 33, 1307–1317 (2020).
Author information
Authors and Affiliations
Consortia
Contributions
C.E., A.S., L.B., A.J., and W.B. designed the study. All authors contributed to data collection, data analysis, and data interpretation. C.E. and L.B. performed statistical analyses. C.E. wrote the first draft of the manuscript. All authors contributed to the writing of the manuscript, approved the final version, and agree to be accountable for all aspects of the report.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval/consent to participate
This study was performed in compliance with the UMC Utrecht’s medical research ethics committee guidelines for case studies.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ebbelaar, C.F., Schrader, A.M.R., van Dijk, M. et al. Towards diagnostic criteria for malignant deep penetrating melanocytic tumors using single nucleotide polymorphism array and next-generation sequencing. Mod Pathol 35, 1110–1120 (2022). https://doi.org/10.1038/s41379-022-01026-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01026-6
This article is cited by
-
PRAME Staining in Sinonasal Mucosal Melanoma: A Single-Center Experience
Head and Neck Pathology (2022)
