
Abstract
Dedifferentiation and transdifferentiation are rare and only poorly understood phenomena in cutaneous melanoma. To study this disease more comprehensively we have retrieved 11 primary cutaneous melanomas from our pathology archives showing biphasic features characterized by a conventional melanoma and additional areas of de-/trans-differentiation as defined by a lack of immunohistochemical expression of all conventional melanocytic markers (S-100 protein, SOX10, Melan-A, and HMB-45). The clinical, histologic, and immunohistochemical findings were recorded and follow-up was obtained. The patients were mostly elderly (median: 81 years; range: 42–86 years) without significant gender predilection, and the sun-exposed skin of the head and neck area was most commonly affected. The tumors were deeply invasive with a mean depth of 7 mm (range: 4–80 mm). The dedifferentiated component showed atypical fibroxanthoma-like features in the majority of cases (7), while additional rhabdomyosarcomatous and epithelial transdifferentiation was noted histologically and/or immunohistochemically in two tumors each. The background conventional melanoma component was of desmoplastic (4), superficial spreading (3), nodular (2), lentigo maligna (1), or spindle cell (1) types. For the seven patients with available follow-up data (median follow-up period of 25 months; range: 8–36 months), two died from their disease, and three developed metastases. Next-generation sequencing of the cohort revealed somatic mutations of established melanoma drivers including mainly NF1 mutations (5) in the conventional component, which was also detected in the corresponding de-/trans-differentiated component. In summary, the diagnosis of primary cutaneous de-/trans-differentiated melanoma is challenging and depends on the morphologic identification of conventional melanoma. Molecular analysis is diagnostically helpful as the mutated gene profile is shared between the conventional and de-/trans-differentiated components. Importantly, de-/trans-differentiation does not appear to confer a more aggressive behavior.
Similar content being viewed by others


Introduction
Investigating the molecular mechanisms that drive melanoma development facilitates the discovery of new therapies, improved diagnosis, and furthers our understanding of the contribution that genetic changes play in tumor morphology and behavior. It is now well established that the proto-oncogene BRAF is mutated in around 40–45% of melanomas, with p.V600E mutations more commonly found in melanomas from low chronic sun-damaged (CSD) skin and non-p.V600E mutations more frequently reported in melanomas from high-CSD sites. In the same way, activating KIT mutations are more common in cases from sun-protected sites such as acral and mucosal melanomas, NF1 mutations are more frequent in desmoplastic melanomas, and GNAQ or GNA11 mutations are primarily found in uveal melanomas and melanomas that develop in blue nevi. It is also well established that the landscape of DNA copy-number changes and chromosomal aberrations vary depending on the melanoma subtype [1, 2].
Dedifferentiation and transdifferentiation (implying dedifferentiation with heterologous elements) in melanoma are unusual phenomena occurring mostly in metastatic deposits [3, 4]. It is exceptionally rare in primary cutaneous melanoma and only poorly documented in the literature, mainly in case reports (11 cases; Supplementary Table 1) [3,4,5,6,7,8,9,10,11]. By definition, primary cutaneous de-/trans-differentiated melanomas are biphasic tumors composed of an element of conventional melanoma with a retained expression of melanocytic markers, such as S-100 protein, SOX10, and variably Melan-A and HMB-45. The invasive de-/trans-differentiated components are characterized by the lack of expression of all conventional melanocytic markers (S-100 protein, SOX10, Melan-A, and HMB-45). The dedifferentiated component resembles atypical fibroxanthoma (AFX) histopathologically, while the transdifferentiated component shows histological and immunohistochemical evidence of transdifferentiation towards other lineages, including rhabdomyosarcomatous elements [12]. Melanomas with osteocartilaginous and schwannian differentiation may be better regarded as related differentiation rather than as transdifferentiation, as S-100 protein and SOX10 expression are retained in these cases. Schwannian differentiation is frequently encountered in melanocytic tumors such as neurotization in benign nevi [13] and osteocartilaginous differentiation has been reported in acral lentiginous melanoma [14].
The biological mechanisms underlying de-/trans-differentiation in melanoma and its prognostic significance are unknown. In this study, we detail the clinicopathologic spectrum and behavior of primary cutaneous de-/trans-differentiated melanomas. In addition, we profile the mutational landscape of these tumors and explore somatic point mutations, DNA copy-number changes, and also the transcriptome of the largest cohort of cases described to date.
Materials and methods
Case selection
Eleven de-/trans-differentiated melanoma cases were identified from nine clinical centers. All cases underwent a central pathological review of H&E stains and immunohistochemistry. To meet inclusion criteria, all tumors were primary cutaneous melanomas. Histopathologically, tumors were biphasic and contained a background of a conventional melanoma, either in situ or invasive, with immunohistochemical expression of S-100 protein and SOX10, and variable expression of Melan-A and HMB-45. The de-/trans-differentiated component was characterized by loss of expression of all melanocytic markers by immunohistochemistry. Immunohistochemistry was performed according to the manufacturer’s protocols. The antibodies used, their source and dilution, are shown in Supplementary Table 2. Histopathological features including the histopathologic subtype of the conventional melanoma, in situ component, heterologous elements, Breslow thickness, Clark level, ulceration, necrosis, mitoses (per mm2), lymphovascular and perineural invasions, regression, and tumor-infiltrating lymphocytes (TILs) were collected from each case by TB. Clinical data and follow-up were obtained from patient records.
Next-generation sequencing analyses
DNA and RNA extraction
We used a tissue microarray (TMA) coring needle to collect specifically-identified tumor and normal material from formalin-fixed paraffin-embedded (FFPE) tissue blocks following a detailed assessment of the H&E slide of each case. DNA and RNA were extracted using the All Prep DNA/RNA FFPE Kit (Qiagen), according to the manufacturer’s instructions.
DNA was extracted from seven cases/patients (21 samples in total), as detailed in Table 1. For five of these patients, tumor material from both the conventional and the de-/trans-differentiated components was obtained, as well as normal tissue; one patient had two samples of normal tissue that were sequenced (16/21 samples). For the remaining two patients, only material from the dedifferentiated component was collected, along with normal tissue, as material from the conventional component was insufficient. Additionally, for one of these cases, we analyzed material from a metastasis (5/21 samples). RNA of sufficient quality for sequencing was extracted from FFPE samples from six cases/patients (18 samples in total) (Table 1).
Whole exome sequencing, data processing, and variant analysis
Paired-end whole-exome sequencing was performed using the Agilent whole exome capture kit (SureSelect All Exon V5) on the HiSeq4000 platform (Illumina) at the Wellcome Sanger Institute. The DNA samples had average coverage of 6.2 Gb per sample, with an average of 86% of the exome being covered by >30×. The RNA samples were more variable, ranging from 3 to 200× coverage per sample. Sequencing reads were mapped with BWA-mem (via Canapps, v0.7.17) against the GRCh37 human reference genome [15]. Data processing and analysis were performed using GATK (v4.1.1.) and Mutect2 was used to call single-nucleotide variants (SNVs) [16]. Variant calls were subsequently filtered using the strategy outlined in Supplementary Table 3 as well as for a median-base quality (MBQ) > 30, and a total read depth >10 reads. Read processing in R (v3.6.0) was performed using the VariantAnnotation package (v1.30.1) [17]. The effects of variants on genes and proteins were annotated using the variant effect predictor (VEP) from Ensembl (processed on https://grch37.ensembl.org/Homo_sapiens/Tools/VEP on 09/2020, Ensembl database version 102, genome assembly GRCH37.p13, dbSNP version 153, ClinVar version 12/2019 and gnomAD version r2.1 exomes only dataset). Somatic variants were further filtered using data from gnomAD [18] to an allele frequency <0.01 and for variants in the canonical transcript of each gene, as defined by Ensembl. All SNVs shown in the figures were visually inspected by looking at the raw sequence alignments (Supplementary Table 4). Where a variant was called in one sample from a patient, we inspected that position in all additional samples from the same patient and considered it “true” if it was supported by two or more reads (even in the absence of the call being made by Mutect2). In this way manual inspection found a BRAF p.V600E mutation in PD42798c that was missed by Mutect2 because only 2/6 reads covering this position were mutant. Of note, there was a lower concordance of mutations in samples PD42798a and PD42798c when compared to other cases. We used Sequenza (v3.0.0) for somatic DNA copy-number analysis [19, 20]. Samples PD42798 and PD42799 were excluded due to their predicted low cellularity (≤0.2). Mutational Signatures were analyzed using the MutationalPatterns package (v3.0.1) in R (v4.0.3) [21].
Transcriptome sequencing and data processing
RNA-seq data were mapped to the GRCh37 reference genome using STAR (via Canapps, v2.5.0c) [22]. Read counts were tabulated using FeatureCounts (1.6.4). Differential gene analysis was performed using the DESeq2 package (v1.24.0) in R (v3.6.1) [23]. STAR-Fusion (v1.8.1) was used to identify putative fusion events. Of note, PD42798c failed RNA sequencing.
Results
Clinical data and follow-up
All eleven patients were adults, with a median age of 81 years (range: 42–86 years) and a mean age of 76 years (Table 1). The male:female ratio was 1.2:1. The majority of tumors presented on the head and neck followed by the extremities, but the anatomical distribution was wide and included the scalp (3), arm (3), ear, nose, chin, neck, and back (1 each).
Follow-up was available for seven patients, three cases were recent, and no follow-up could be obtained for one patient. The follow-up interval ranged from 8 to 36 months (median: 25 months, mean: 24 months). Two patients (33%) died from disease 8 and 24 months after presentation. Metastatic disease was documented in three patients (43%) and observed as a widespread metastatic disease in one patient; for the second as a satellite metastasis, nodal metastases, and lung metastasis; and for the third patient as metastases to lymph nodes, lung, and brain. No local recurrences were reported.
Histological and immunohistochemical features
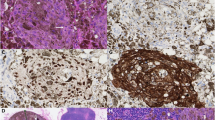
All eleven tumors were deeply invasive melanomas with two cases invading into the reticular dermis (Clark level IV), while the majority of tumors (9) showed Clark level V invasion. The Breslow thickness ranged from at least 4.0–80 mm with a median tumor thickness of 7 mm and a mean depth of 15.4 mm. A background of conventional melanoma was identified in all eleven melanomas and included “pure” desmoplastic melanoma (DM) (4), superficial spreading melanoma (SSM) (3), nodular melanoma (NM) (2), lentigo maligna melanoma (LMM) (1) and spindle cell melanoma (1). An in situ component was seen in six tumors. By immunohistochemistry, the conventional components expressed S-100 protein and SOX10. In addition, there was an expression of Melan-A in the in situ and the epithelioid invasive components. All desmoplastic melanomas lacked Melan-A and HMB-45 expression. The transition to the invasive de-/trans-differentiated component was abrupt rather than gradual and the de-/trans-differentiated component typically was the dominant aspect of the tumors, representing more than 50% of the tumor in most biopsies. Seven tumors showed areas of dedifferentiation (Fig. 1) characterized by a sheet-like growth of large pleomorphic epithelioid and spindle cells with variably abundant eosinophilic cytoplasm containing vesicular nuclei with prominent eosinophilic nucleoli reminiscent of AFX (Fig. 1a–b). Multinucleated pleomorphic tumor cells were also admixed. The tumor cells of the dedifferentiated component were consistently negative for S-100 protein (Fig. 1e) and SOX10, and no expression was seen for Melan-A (Fig. 1f) or HMB-45. Heterologous elements were observed in four tumors and included rhabdomyosarcomatous (2) and epithelial (2) transdifferentiation. The heterologous areas lacked S-100 protein, SOX10, Melan-A, and HMB-45 expression. Rhabdomyosarcomatous transdifferentiation (Fig. 2) was characterized by the presence of sheets of round to spindled cells with abundant brightly eosinophilic cytoplasm containing pleomorphic vesicular nuclei with prominent nucleoli (Fig. 2c). By immunohistochemistry, the tumor cells expressed desmin (Fig. 2e), myogenin, and MyoD1 (Fig. 2f). The tumor cells in the areas of epithelial transdifferentiation (Fig. 3) were pleomorphic and spindled and arranged in intersecting fascicles (Fig. 3d). They contained a moderate amount of cytoplasm with hyperchromatic to vesicular nuclei and eosinophilic nucleoli. By immunohistochemistry, the tumor cells expressed cytokeratins AE1/AE3 (Fig. 3f) and MNF116, but they were negative for p63 and p40 expression. Ulceration was present in 8 tumors (Fig. 1a), tumor necrosis in 1, lymphovascular invasion in 4, and perineural invasion in 3 of the eleven tumors. None of the tumors showed evidence of regression. The mitotic count varied from 3 to 40 per mm2 (median, 12 mm2; mean, 14.5 mm2) (Fig. 1b). TILs were present in 7 tumors; they were brisk in one and non-brisk in six tumors. The histopathologic and immunohistochemical features are summarized in Table 1 and Supplementary Table 5, and Supplementary Table 6, respectively.

This ulcerated tumor shows a nodular growth pattern within the dermis (a). It is composed of sheets of pleomorphic epithelioid cells with brisk mitotic activity (b). The adjacent epidermis shows melanoma in situ (c). The invasive tumor expresses CD10 (d) but it is negative for S-100 protein (e) and Melan-A (f). Melan-A staining also highlights the background of melanoma in situ (f).

This tumor invades adjacent skeletal muscle and is composed of two morphologically distinct components (a). The left-sided component is composed of intersecting fascicles of spindle cells (b), while the right-sided component shows a sheet-like proliferation of large round cells showing marked nuclear pleomorphism and containing ample cytoplasm with brightly eosinophilic inclusions, suggestive of rhabdomyosarcomatous transdifferentiation (c). S-100 protein expression is confined to the spindle cell component (d). Desmin (e) and MyoD1 (f) are only expressed in the pleomorphic component confirming the rhabdomyosarcomatous transdifferentiation.

This large tumor invades subcutis and into the underlying fascia (a). It is composed of two morphologically distinct components (b) characterized by a spindle cell tumor in a desmoplastic stroma to the right (c) and a pleomorphic spindle cell tumor to the left (d). The desmoplastic spindle cell tumor expresses S-100 protein (e) and is negative for cytokeratin AE1/AE3 (f). In contrast, the pleomorphic component lacks S-100 protein expression (e) but is strongly and diffusely positive for cytokeratin AE1/AE3 (f).
Molecular analyses
Somatic mutational landscape
DNA sequencing was performed on samples from 7/11 patients (Table 1) and used for somatic variant calling. A BRAF p.V600E mutation was found in the dedifferentiated part of a dedifferentiated NM, while non-p.V600E BRAF mutations were present in three of the seven cases (in the conventional melanoma of a dedifferentiated SSM, in both conventional and transdifferentiated components of a transdifferentiated DM, and in the dedifferentiated component of a dedifferentiated LMM). An NRAS p.Q61K mutation was found in both components of an NM with epithelial transdifferentiation. NF1 was mutated in five cases: in both components of a dedifferentiated SSM, a DM with epithelial transdifferentiation and two heterologous rhabdomyosarcomatous melanomas (a DM and an SSM), as well as in the dedifferentiated component of a dedifferentiated LMM (for which insufficient material from the conventional melanoma was available for analysis). No tumor was wild type for BRAF, NRAS, and NF1. BRAF and NRAS hotspot mutations as well as mutation of NRAS and NF1 were mutually exclusive, whereas BRAF and NF1 mutations were not (except for the BRAF p.V600E mutation) as shown in Fig. 4. Other known melanoma driver genes [1] that were mutated in our cohort are shown in Fig. 4, while known genetic alterations related to desmoplastic melanoma [24] are in shown Supplementary Figure 1, and genes mutated in at least half of patients are shown in Supplementary Fig. 2. Importantly, most somatic mutations were shared in all components of the tumor (primary [conventional and de-/trans-differentiated components of the melanoma] and/or metastasis [dedifferentiated component]) suggesting minimal genetic mutational divergence between these elements of the tumor. The mutational burden varied significantly between individual tumor samples with a range from 31 to 10,202 mutations (mean: 3885.4 mutations, median: 4113 mutations) or from 0.62 to 202.5 mutations/megabase (Mb) (mean: 77.1 mutations/Mb, median: 81.62 mutations/Mb). The highest (>50 mutations/Mb) mutational burden level was associated with COSMIC (catalog of somatic mutations in cancer) mutational signature seven which is caused by ultraviolet (UV) light, although this mutational signature also dominated the mutation catalog of tumors with a low mutational burden as well (Supplementary Fig. 3).

The top panel shows the somatic mutation rate for each of the tumor samples that were sequenced in this study. This figure also provides information on the melanoma subtype and presentation and the mutation status of key melanoma driver genes (bottom panel).
Somatic DNA copy-number alterations (CNAs)
As above for somatic point mutations, the conventional and the de-/trans-differentiated components of the tumor samples from the same individual were almost identical in their somatic DNA CNA profiles (Fig. 5). However, some additional structural variants were observed in the de-/trans-differentiated component of the tumor or in the metastasis, of which several were recurrent (variable alterations in chromosomes 6, 9, and 17).

DNA copy-number variants were called with Sequenza [19, 20]. Recurrent DNA copy-number alterations are highlighted with a red rectangle and selected significant DNA copy-number alterations private to the conventional or de-/trans-differentiated melanoma relative to each other are indicated with a blue asterisk.
Transcriptomic landscape
RNA sequencing was performed on samples from 6/11 patients (Table 1). For four cases, we generated data allowing a comparison between the conventional and the de-/trans-differentiated components. The expression of MITF, SOX10, MLANA, TYR, DCT, EDNRB, and EDN3, amongst others, were lower in the de-/trans-differentiated component (except in those cases that were desmoplastic melanomas [PR42796 and PR42800]). In contrast, AXL was higher in the de-/trans-differentiated component (PR42795 and PR42797). Interestingly, DES, MYOG, and MYOD1 (myogenic genes) were expressed in the heterologous rhabdomyosarcomatous melanoma (PR42800) (Fig. 6). No significant gene fusions between protein-coding genes were identified in samples from six patient’s tumors which were analyzed.

H&E (a) and desmin immunohistochemistry (b) of melanoma with rhabdomyosarcomatous transdifferentiation. The conventional melanoma is marked by a red asterisk, rhabdomyosarcomatous transdifferentiation by a green asterisk, and normal adjacent skeletal muscle by a blue asterisk. Expression of key skeletal muscle marker genes with transcripts per kilobase million (TPM) determined using DESeq2 (c). Note, the normal tissue was adjacent to normal skeletal muscle as indicated in (a). Importantly, MYOD1 and MYOG RNA are expressed in normal skeletal muscle (i.e., the control tissue) but an expression of these markers at the protein level is absent. Conversely, the presence of MYOD1 and MYOG RNA in the conventional melanoma with no associated protein expression may be explained by contamination from the adjacent (or underlying) normal skeletal muscle. CASZ1 somatic mutation status (d). CASZ1 gene expression has previously been linked to a rhabdomyosarcomatous transdifferentiation program.
Discussion
Dedifferentiation and transdifferentiation are rarely seen in primary cutaneous invasive melanoma. Clinically, these tumors usually arise on chronically sun-damaged skin of elderly patients, with an overall male predilection [3,4,5,6,7,8,9,10,11]. Although de-/trans-differentiation can be observed in any melanoma subtype, they present most commonly in the context of a desmoplastic melanoma, and dedifferentiation is more frequently observed than transdifferentiation [3,4,5,6,7,8,9,10,11]. The tumors are thick, often showing invasion of subcutaneous tissues. Histologically, there is a sharp demarcation between the de-/trans-differentiated areas and the conventional melanoma, which is generally only present as an in situ component or a minor component of the invasive tumor. The dedifferentiated areas are morphologically and immunohistochemically indistinguishable from AFX or pleomorphic dermal sarcoma (PDS). The transdifferentiated components in this study showed either rhabdomyosarcomatous transdifferentiation both morphologically and immunohistochemically as previously reported [5, 8, 9], or epithelial transdifferentiation. Epithelial transdifferentiation is characterized by a pleomorphic cellular component with immunohistochemical expression of cytokeratins, but no epithelial phenotype on morphology. Cytokeratin expression has been documented in melanoma [25], usually seen in conventional melanomas with retained S-100 protein expression, however, true epithelial transdifferentiation has not been reported in primary cutaneous melanoma, previously only described in metastatic deposits showing morphological and immunohistochemical evidence of epithelial transdifferentiation [26].
Driver mutation analysis showed that NF1 was the most frequently mutated gene, observed in tumors on chronically sun-damaged skin of the elderly and in all melanomas with a desmoplastic conventional melanoma component. The NF1 mutated tumors had a high mutation burden, were enriched for TP53 mutations, and lacked BRAF p.V600E or NRAS p.Q61 mutations, similar to other NF1 mutated melanomas [24, 27]. Mutations in other genes previously reported in desmoplastic melanomas and known to activate the MAPK and PI3K signaling cascades [24] were also identified (Supplementary Fig. 1), such as BRAF non-p.V600E and ERRB2, which is rarely altered in melanoma [28]. BRAF p.V600E and NRAS p.Q61K mutations were associated with nodular de-/trans-differentiated melanoma located on intermittently sun-exposed skin. Importantly, all somatic mutations were typically identified in both the conventional and the de-/trans-differentiated components of the tumor, as were DNA CNAs, demonstrating shared mutational and CNA landscapes despite the divergent morphological and immunohistochemical phenotype.
The loss of melanocytic differentiation within the de-/trans-differentiated component, as highlighted morphologically and immunohistochemically, was further demonstrated by MITFlow, SOX10low, and more variably AXLhigh expression at the RNA level compared to the conventional component (Supplementary Fig. 4) [29]. The same expression pattern as MITF was also observed for other genes playing a role in melanocyte differentiation such as MLANA, TYR, DCT, EDNRB, and EDN3 [29,30,31,32]. Interestingly, but not surprisingly, these genes displayed a similar “de-/trans-differentiated” gene expression profile within the two desmoplastic conventional melanoma components (PR42796a and PR42800a), which are characterized by a lack of some morphological and immunohistochemical melanocytic features. In addition, the rhabdomyosarcomatous component demonstrated strong expression of rhabdomyogenic RNA transcripts (DES, MYOD1, and MYOG), correlating with the morphologic features and the immunohistochemical profile (Fig. 6). As an internal control, adjacent normal skeletal muscle also expressed these rhabdomyogenic transcripts. Of note, expression of the protein products of these genes is not usually observed in normal skeletal muscle by immunohistochemistry (however, focal immunostaining of myogenin has been observed in normal or regenerative skeletal muscle) [33], although transcript expression has been reported (https://www.proteinatlas.org). We noted that CASZ1, a gene previously reported to be associated with upregulation of the myogenic regulatory factors MYOD1 and MYOG, and thought to contribute to rhabdomyosarcomatous tumorogenesis [34], was mutated within exon 12 of two tumors with rhabdomyosarcomatous transdifferentiation, potentially suggesting a role for this zinc finger transcription factor in rhabdomyosarcomatous transdifferentiation in these cases (Fig. 6, Supplementary Fig. 5 and Supplementary Table 4).
Notably, these observations were based on a small number of cases and further analyses of larger cohorts are required.
Given the small sample size, it is difficult to draw firm conclusions on the behavior of these tumors. Metastatic disease and disease-related mortality were only noted in tumors arising in a background of nodular and superficial spreading melanoma, while those associated with a desmoplastic melanoma pursued an indolent disease course despite a tumor thickness of up to 80 mm [35]. At the very least, the presence of de-/trans-differentiation does not result in a profoundly worse outcome for patients with these tumors. In contrast to conventional melanoma, rhabdomyosarcomas are more aggressive tumors (survival range of localized rhabdomyosarcoma in adults: 40–46% [36]). Of note, rhadomyosarcomatous transdifferentiation has been reported as a marker of poor prognosis in MPNST [37,38,39]. These associations will need to be explored with larger tumor collections.
The diagnosis of primary cutaneous de-/trans-differentiated melanoma is challenging and should be included in all differential diagnoses of tumors arising on sun-damaged skin of elderly patients. The key to the correct diagnosis is adequate tissue sampling and careful examination of the dermo-epidermal junction, especially at the edges of the lesion, where the conventional component is usually located. This may be particularly challenging in ulcerated tumors. The conventional component can be easily highlighted by S-100 protein and SOX10 immunohistochemical stains. The shared mutational landscape between the conventional and de-/trans-differentiated components may be an additional useful diagnostic tool and used to reliably exclude the possibility of a collision tumor.
The differential diagnosis of these tumors depends on the de-/trans-differentiated component. Dedifferentiated melanoma can be misdiagnosed as an AFX or a PDS. AFX is often polypoid, surrounded by an epidermal collarette with no evident epidermal connection, and located in the dermis with pushing rather than infiltrative deep margin. PDS shares many of the morphologic features with AFX but shows additional invasion of subcutis, necrosis, and/or, lymphovascular or perineural invasion [40]. Both tumors can express various non-specific markers such as CD10 and SMA, but do not express specific markers such as cytokeratins, S-100 protein, SOX10, desmin, or CD34 [40]. AFX and PDS share many genetic features, such as TP53, TERT promoter, FAT1, NOTCH1/2, and PIK3CA mutations, as well as a UV mutation signature and CNAs such as losses of 9p (AFX: 65% and PDS: 66%) and 13q (AFX: 65% and PDS: 93%), and gains of 8q (AFX: 18% and PDS: 33%). However, there are subtle differences, specifically, CDKN2A mutation is more related to AFX, and RAS mutations and CDKN2A deletion to PDX [41,42,43,44,45]. In addition, upregulation of genes in biological pathways such as tumor-associated macrophages response, GPCR (G-protein coupled receptors) signaling, and epithelial to mesenchymal transition (EMT) has been reported by gene expression profiling in AFX [46]. More importantly, they are both diagnoses of exclusion. Melanoma with rhabdomyosarcomatous transdifferentiation can be misinterpreted for rhabdomyosarcoma or MPNST with rhabdomyoblastic differentiation (malignant Triton tumor). By immunohistochemistry, the tumor cells express rhabdomyogenic markers such as desmin, myogenin, and MyoD1 in all three tumors. However, MPNST usually occurs at a younger age, is deeply located, expresses S-100 protein more focally than melanoma, and can be associated with a large nerve and/or a pre-existing neurofibroma. Also, MPNST with heterologous differentiation occurs more commonly in patients with neurofibromatosis type-1 (NF1) [39]. Rhabdomyosarcoma is extremely rare in the skin [47], as either a primary or a metastasis. In contrast to either melanoma with rhabdomyosarcomatous transdifferentiation or most malignant Triton tumors, the rhabdomyosarcomatous component of a primary or metastatic rhabdomyosarcoma is present throughout the entire tumor. Depending on its variants, rhabdomyosarcoma can mainly exhibit (i) a complex karyotype (including loss of heterozygosity of 11p15.5) [48], as well as RAS pathway (NRAS, KRAS, HRAS, NF1, FGFR4), PIK3CA, BCOR, FBXW7, or TP53 mutations in embryonal rhabdomyosarcoma [49]; (ii) in most cases, fusions involving PAX3/7-FOXO1 in alveolar rhabdomyosarcoma [50]; (iii) MYOD1 mutation [51,52,53] which can be associated with PIK3CA mutation [52, 54] or VGLL2 [54]/NCOA2 [55] rearrangements in spindle cell/sclerosing rhabdomyosarcoma; (iv) ALK upregulation and fusions involving TFCP2 in epithelioid and spindle cell rhabdomyosarcoma [56]; (v) a complex karyotype in pleomorphic rhabdomyosarcoma [57]. MPNST, including malignant Triton tumor, is mostly characterized by a complex karyotype [58]. In addition, NF1, CDKN2A, SUZ12, and/or EED mutation have also been reported [59,60,61], the latter two alterations lead to an expression loss of H3K27me3 immunohistochemistry [62,63,64]. Deletion of these genes, as well as CNAs, have also been described [60, 65, 66]. Finally, poorly differentiated/spindle cell squamous cell carcinoma (SCC) or metaplastic SCC may be misinterpreted as melanoma with divergent epithelial transdifferentiation, however, evidence of a better-differentiated area of SCC, as well as cytokeratins and p63 expression, are useful in distinguishing these tumors. Like AFX and PDX, SCCs may also carry TP53 mutations and a UV signature similar to what we saw in the primary cutaneous de-/trans-differentiated melanoma cases described here. CDKN2A mutation and CNAs such as losses of 3p (42%), 13q (42%) and 9p (63%), and gains of 3q (21%) and 8q (26%) are other events reported in SCCs [44, 45]. Squamomelanomcytic tumor enters the differential diagnosis with melanoma with epithelial transdifferentiation. It is characterized by a well-circumscribed dermal tumor with overt squamous differentiation colonized by morphologically and immunohistochemically distinct atypical melanocytes [67,68,69]. In contrast, melanoma with cytokeratin expression contains a subclone of cells that might be morphologically different and express both melanocytic and epithelial markers; the same phenomenon can be encountered with rhabdomyogeninc markers in which case a diagnosis of melanoma with rhabdomyogenic differentiation should be made.
In conclusion, primary cutaneous de-/trans-differentiated melanoma is a rare histopathologic variant of melanoma, which mostly occurs on sun-damaged skin of elderly men. This biphasic tumor preferentially contains a desmoplastic melanoma as the conventional component and a dedifferentiated, rather than transdifferentiated, component losing the expression of all conventional melanocytic markers. This melanoma subtype is often NF1 mutated. Although phenotypically distinct, both components demonstrate almost the same mutational and DNA CNA landscapes, showing the high plasticity potential of melanocytes and refuting the previous hypothesis of a collision tumor. This feature may be a useful diagnostic tool in complex cases. Despite its invasiveness and its phenotypic similarities with other aggressive tumors, its prognosis does not seem to be worse than other melanoma subtypes.
Data availability statement
The whole-exome and transcriptome sequencing data have been deposited at the European Genome-Phenome Archive (EGA) (https://www.ega-archive.org at the European Bioinformatics Institute): EGAD00001007033 (DNA) and EGAD00001007034 (RNA). The scripts used in this paper are available at https://github.com/team113sanger/dediff-melanoma. Associated data are available at https://doi.org/10.6084/m9.figshare.c.5432205.
References
Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545:175–80.
Rabbie R, Ferguson P, Molina-Aguilar C, Adams DJ, Robles-Espinoza CD. Melanoma subtypes: genomic profiles, prognostic molecular markers and therapeutic possibilities. J Pathol. 2019;247:539–51.
Agaimy A, Specht K, Stoehr R, Lorey T, Markl B, Niedobitek G, et al. Metastatic malignant melanoma with complete loss of differentiation markers (undifferentiated/dedifferentiated melanoma): analysis of 14 patients emphasizing phenotypic plasticity and the value of molecular testing as surrogate diagnostic marker. Am J Surg Pathol. 2016;40:181–91.
Agaimy A, Stoehr R, Hornung A, Popp J, Erdmann M, Heinzerling L, et al. Dedifferentiated and undifferentiated melanomas: report of 35 new cases with literature review and proposal of diagnostic criteria. Am J Surg Pathol. 2021;45:240–54.
Gharpuray-Pandit D, Coyne J, Eyden B, Banerjee SS. Rhabdomyoblastic differentiation in malignant melanoma in adults: report of 2 cases. Int J Surg Pathol. 2007;15:20–25.
Wilsher MJ. Collision tumour: atypical fibroxanthoma and invasive melanoma. Pathology. 2009;41:699–701.
Kiuru M, McDermott G, Berger M, Halpern AC, Busam KJ. Desmoplastic melanoma with sarcomatoid dedifferentiation. Am J Surg Pathol. 2014;38:864–70.
Shenjere P, Fisher C, Rajab R, Patnaik L, Hazell S, Thway K. Melanoma with rhabdomyosarcomatous differentiation: two further cases of a rare pathologic pitfall. Int J Surg Pathol. 2014;22:512–9.
Antonov NK, Niedt GW. Malignant melanoma with rhabdomyosarcomatous differentiation: a case report. Am J Dermatopathol. 2016;38:456–60.
Erstine EM, Tetzlaff MT, Ko JS, Prieto VG, Cheah AL, Billings SD. Living on the edge: diagnosing sarcomatoid melanoma using histopathologic cues at the edge of a dedifferentiated tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2017;39:593–8.
Lefferts JA, Loehrer AP, Yan S, Green DC, Deharvengt SJ, LeBlanc RE. CD10 and p63 expression in a sarcomatoid undifferentiated melanoma: a cautionary (and molecularly annotated) tale. J Cutan Pathol. 2020;47:541–7.
Banerjee SS, Eyden B. Divergent differentiation in malignant melanomas: a review. Histopathology. 2008;52:119–29.
Chen YM, Klonowski PW, Lind AC, Lu DS. Differentiating neurotized melanocytic nevi from neurofibromas using melan-A (MART-1) immunohistochemical stain. Arch Pathol Lab Med. 2012;136:810–15.
Murali R, McCarthy SW, Bothman J, Cachia A, Janarthanan P, Sharma R, et al. Melanoma exhibiting cartilaginous differentiation. Histopathology. 2010;56:815–21.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C & Lichtenstein L. Calling somatic SNVs and indels with Mutect2. BioRxiv. https://doi.org/10.1101/861054861054. 2019.
Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, Morgan M. Variant annotation: a bioconductor package for exploration and annotation of genetic variants. Bioinformatics. 2014;30:2076–8.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Rashid M, van der Horst M, Mentzel T, Butera F, Ferreira I, Pance A, et al. ALPK1 hotspot mutation as a driver of human spiradenoma and spiradenocarcinoma. Nat Commun. 2019;10:2213.
Wong K, van der Weyden L, Schott CR, Foote A, Constantino-Casas F, Smith S, et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat Commun. 2019;10:353.
Blokzijl F, Janssen R, van Boxtel R, Cuppen E. Mutational patterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Shain AH, Garrido M, Botton T, Talevich E, Yeh I, Sanborn JZ, et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat Genet. 2015;47:1194–9.
Banerjee SS, Harris M. Morphological and immunophenotypic variations in malignant melanoma. Histopathology. 2000;36:387–402.
Jalas JR, Vemula S, Bezrookove V, Leboit PE, Simko JP, Bastian BC. Metastatic melanoma with striking adenocarcinomatous differentiation illustrating phenotypic plasticity in melanoma. Am J Surg Pathol. 2011;35:1413–8.
Cirenajwis H, Lauss M, Ekedahl H, Torngren T, Kvist A, Saal LH, et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol Oncol. 2017;11:438–51.
Gottesdiener LS, O’Connor S, Busam KJ, Won H, Solit DB, Hyman DM, et al. Rates of ERBB2 alterations across melanoma subtypes and a complete response to trastuzumab emtansine in an ERBB2-amplified acral melanoma. Clin Cancer Res. 2018;24:5815–9.
Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018;33:890–904 e895.
Osawa M, Egawa G, Mak SS, Moriyama M, Freter R, Yonetani S, et al. Molecular characterization of melanocyte stem cells in their niche. Development. 2005;132:5589–99.
Loftus SK, Baxter LL, Buac K, Watkins-Chow DE, Larson DM, Pavan WJ. Comparison of melanoblast expression patterns identifies distinct classes of genes. Pigment Cell Melanoma Res. 2009;22:611–22.
Zaidi MR, Fisher DE & Rizos H. Biology of melanocytes and primary melanoma. In: Balch CM et al., editors. Cutaneous melanoma. 6th ed. Switzerland, Springer Nature Switzerland AG, 2020. p. 3–40.
Cessna MH, Zhou H, Perkins SL, Tripp SR, Layfield L, Daines C, et al. Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics. Am J Surg Pathol. 2001;25:1150–7.
Liu Z, Zhang X, Lei H, Lam N, Carter S, Yockey O, et al. CASZ1 induces skeletal muscle and rhabdomyosarcoma differentiation through a feed-forward loop with MYOD and MYOG. Nat Commun. 2020;11:911.
Lens MB, Newton-Bishop JA, Boon AP. Desmoplastic malignant melanoma: a systematic review. Br J Dermatol. 2005;152:673–8.
Nakagawa N, Tsuda T, Yamamoto M, Ito T, Futani H, Yamanishi K. Adult cutaneous alveolar rhabdomyosarcoma on the face diagnosed by the expression of PAX3-FKHR gene fusion transcripts. J Dermatol. 2008;35:462–7.
Woodruff JM, Chernik NL, Smith MC, Millett WB, Foote FW Jr. Peripheral nerve tumors with rhabdomyosarcomatous differentiation (malignant “Triton” tumors). Cancer. 1973;32:426–39.
Brooks JS, Freeman M, Enterline HT. Malignant “Triton” tumors. Natural history and immunohistochemistry of nine new cases with literature review. Cancer. 1985;55:2543–9.
Ramanathan RC, Thomas JM. Malignant peripheral nerve sheath tumours associated with von Recklinghausen’s neurofibromatosis. Eur J Surg Oncol. 1999;25:190–3.
Miller K, Goodlad JR, Brenn T. Pleomorphic dermal sarcoma: adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am J Surg Pathol. 2012;36:1317–26.
Griewank KG, Schilling B, Murali R, Bielefeld N, Schwamborn M, Sucker A, et al. TERT promoter mutations are frequent in atypical fibroxanthomas and pleomorphic dermal sarcomas. Mod Pathol. 2014;27:502–8.
Griewank KG, Wiesner T, Murali R, Pischler C, Muller H, Koelsche C, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma harbor frequent NOTCH1/2 and FAT1 mutations and similar DNA copy number alteration profiles. Mod Pathol. 2018;31:418–8.
Helbig D, Quaas A, Mauch C, Merkelbach-Bruse S, Buttner R, Emberger M, et al. Copy number variations in atypical fibroxanthomas and pleomorphic dermal sarcomas. Oncotarget. 2017;8:109457–67.
Koelsche C, Stichel D, Griewank KG, Schrimpf D, Reuss DE, Bewerunge-Hudler M, et al. Genome-wide methylation profiling and copy number analysis in atypical fibroxanthomas and pleomorphic dermal sarcomas indicate a similar molecular phenotype. Clin Sarcoma Res. 2019;9:2.
Miller TI, Zoumberos NA, Johnson B, Rhodes DR, Tomlins SA, Chan MP, et al. A genomic survey of sarcomas on sun-exposed skin reveals distinctive candidate drivers and potentially targetable mutations. Hum Pathol. 2020;102:60–9.
Lai K, Harwood CA, Purdie KJ, Proby CM, Leigh IM, Ravi N, et al. Genomic analysis of atypical fibroxanthoma. PLoS ONE. 2017;12:e0188272.
Marburger TB, Gardner JM, Prieto VG, Billings SD. Primary cutaneous rhabdomyosarcoma: a clinicopathologic review of 11 cases. J Cutan Pathol. 2012;39:987–95.
Davicioni E, Anderson MJ, Finckenstein FG, Lynch JC, Qualman SJ, Shimada H, et al. Molecular classification of rhabdomyosarcoma–genotypic and phenotypic determinants of diagnosis: a report from the Children’s Oncology Group. Am J Pathol. 2009;174:550–64.
Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–31.
Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2002;20:2672–9.
Szuhai K, de Jong D, Leung WY, Fletcher CD, Hogendoorn PC. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol. 2014;232:300–7.
Agaram NP, Chen CL, Zhang L, LaQuaglia MP, Wexler L, Antonescu CR. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer. 2014;53:779–87.
Agaram NP, LaQuaglia MP, Alaggio R, Zhang L, Fujisawa Y, Ladanyi M, et al. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol. 2019;32:27–36.
Alaggio R, Zhang L, Sung YS, Huang SC, Chen CL, Bisogno G, et al. A molecular study of pediatric spindle and sclerosing rhabdomyosarcoma: identification of novel and recurrent VGLL2-related fusions in infantile cases. Am J Surg Pathol. 2016;40:224–35.
Mosquera JM, Sboner A, Zhang L, Kitabayashi N, Chen CL, Sung YS, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer. 2013;52:538–50.
Le Loarer F, Cleven AHG, Bouvier C, Castex MP, Romagosa C, Moreau A, et al. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol. 2020;33:404–19.
Li G, Ogose A, Kawashima H, Umezu H, Hotta T, Tohyama T, et al. Cytogenetic and real-time quantitative reverse-transcriptase polymerase chain reaction analyses in pleomorphic rhabdomyosarcoma. Cancer Genet Cytogenet. 2009;192:1–9.
Fletcher CD, Dal Cin P, de Wever I, Mandahl N, Mertens F, Mitelman F, et al. Correlation between clinicopathological features and karyotype in spindle cell sarcomas. A report of 130 cases from the CHAMP study group. Am J Pathol. 1999;154:1841–7.
De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–51.
Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227–32.
Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1170–2.
Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 2016;29:4–13.
Prieto-Granada CN, Wiesner T, Messina JL, Jungbluth AA, Chi P, Antonescu CR. Loss of H3K27me3 expression is a highly sensitive marker for sporadic and radiation-induced MPNST. Am J Surg Pathol. 2016;40:479–89.
Cleven AH, Sannaa GA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, Rubin BP. et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol. 2016;29:582–90.
Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Schoenaers J, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–32.
Pemov A, Hansen NF, Sindiri S, Patidar R, Higham CS, Dombi E et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define pre-malignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol. 2019. https://doi.org/10.1093/neuonc/noz028.
Pool SE, Manieei F, Clark WH Jr., Harrist TJ. Dermal squamo-melanocytic tumor: a unique biphenotypic neoplasm of uncertain biological potential. Hum Pathol. 1999;30:525–9.
Amin SM, Cooper C, Yelamos O, Lee CY, Sholl LM, de la Fouchardiere A, et al. Combined cutaneous tumors with a melanoma component: a clinical, histologic, and molecular study. J Am Acad Dermatol. 2015;73:451–60.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Acknowledgements
DJA is supported by Cancer Research UK, The Wellcome Trust, and the Medical Research Council, UK. IF is supported by Wallonia-Brussels International, Belgium. MJA is supported by The Helmsley Charitable Trust, and the Medical Research Council, UK.
Funding
This project was supported by the Medical Research Council [MR/V000292/1 - The Genomic Atlas of Dermatological Tumors (DERMATLAS)].
Author information
Authors and Affiliations
Contributions
IF, MJA, TB, and DJA designed the study and oversaw the research. TB, OM, DG-P, JH, K Wiedemeyer, TM, SDB, JSK, LF, KM, IKP, BLA, NDSA, and LvdW were responsible for tissue collection. IF and TB reviewed all HE sections and immunohistochemistry and collated the clinical data. H.C. obtained tumor and normal material from the blocks. IF, AD, OE, K Wong, and LR performed data analysis. VH performed the PCR validation and sequencing. IF, MJA, TB, and DJA wrote and edited the manuscript which was approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Ethical approval was obtained from the Lothian NRS Bioresource/Edinburgh Experimental Cancer Medicine Center Biobank (reference SR946). Genetic analysis was further approved by the Sanger Human Materials and Data Management Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ferreira, I., Droop, A., Edwards, O. et al. The clinicopathologic spectrum and genomic landscape of de-/trans-differentiated melanoma. Mod Pathol 34, 2009–2019 (2021). https://doi.org/10.1038/s41379-021-00857-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00857-z
