
Abstract
Patients with endometrial cancer differ in terms of the extent of T-cell infiltration; however, the association between T-cell subpopulations and patient outcomes remains unexplored. We characterized 285 early-stage endometrial carcinoma samples for T-cell infiltrates in a tissue microarray format using multiplex fluorescent immunohistochemistry. The proportion of T cells and their subpopulations were associated with clinicopathological features and relapse-free survival outcomes. CD3+ CD4+ infiltrates were more abundant in the patients with higher grade or non-endometrioid histology. Cytotoxic T cells (CD25+, PD-1+, and PD-L1+) were strongly associated with longer relapse-free survival. Moreover, CD3+ PD-1+ stromal cells were independent of other immune T-cell populations and clinicopathological factors in predicting relapses. Patients with high stromal T-cell fraction of CD3+ PD-1+ cells were associated with a 5-year relapse-free survival rate of 93.7% compared to 79.0% in patients with low CD3+ PD-1+ fraction. Moreover, in patients classically linked to a favorable outcome (such as endometrioid subtype and low-grade tumors), the stromal CD3+ PD-1+ T-cell fraction remained prognostically significant. This study supports that T-cell infiltrates play a significant prognostic role in early-stage endometrial carcinoma. Specifically, CD3+ PD-1+ stromal cells emerge as a promising novel prognostic biomarker.
Similar content being viewed by others


Introduction
Endometrial cancer is one of the most common malignancies in women in the Western world, with endometrioid the most prevalent histological subtype1. Most cases are diagnosed in the early stages (I and II), when a large majority of patients will be cured. However, between 10% and 20% of these patients will experience a relapse, which is conditioned by clinicopathological factors, including age, histology, differentiation, myometrial invasion, lymphovascular space infiltration (LVSI), and disease stage. Despite these prognostic factors and the various risk stratifications proposed by the European Society of Medical Oncology (ESMO), European Society of Gynecological Oncology (ESGO) and European Society for Radiotherapy and Oncology (ESTRO), the outcome predictions remain imperfect2. The introduction of molecular biomarkers could significantly improve the diagnostic and prognostic precision. A number of these markers have been recently recommended for routine diagnosis in the current endometrial cancer World Health Organization classification and the ESGO-ESTRO-European Society of Pathology (ESP) guidelines3,4.
Four distinct molecular subgroups of endometrial cancer have been discovered in The Cancer Genome Atlas (TCGA)5. These have been confirmed by the ProMisE study using surrogate markers6,7. The POLE mutated subgroup is associated with the most favorable prognosis and is composed of tumors with an alteration in the exonuclease domain (EDM) of the POLE gene. The p53 abnormal (p53abn) subgroup is composed of tumors with alterations in the TP53 gene and confers the worst prognosis. Of the two intermediate risk subgroups, the Mismatch Repair deficient (MMRd) subtype is characterized by alterations in the MMR genes, and the p53 wild type or nonspecific molecular profile (p53wt/NSMP) comprises the remaining patients. Other additional biomarkers have been explored, such as CTNNB1 mutation or L1CAM expression, but have not yet been integrated into the international guidelines3,8,9.
The relevance of the microenvironment in cancer development and progression is widely accepted, but the role of T-cell infiltrates in endometrial carcinoma is unclear10. CD8+ cytotoxic T cells and CD4+ helper T cells are the most frequent tumor-infiltrating lymphocytes (TILs). A high density of cytotoxic T cells in the tumor microenvironment has previously been demonstrated to be associated with a superior prognosis11,12,13. The POLE and MMRd subtypes are characterized by more TILs infiltrates and a higher mutational rate and have therefore been suggested as being more sensitive for immunotherapy14,15,16. Regulatory T cells (Foxp3+) and PD-(L)1 checkpoint inhibitor-expressing cells are known to inhibit the antitumor activity of T cells, but their relation to the prognosis in the tumor microenvironment is inconsistent, with controversies regarding tumor types17.
A more precise definition of T-cell subpopulations would better establish the impact of immune infiltrates on the prognosis of endometrial cancer. In situ analysis of T-cell subpopulations using novel fluorescent technologies can overcome many of the limitations of conventional immunohistochemistry (IHC). Multiplexed fluorescence immunohistochemistry (mfIHC) enables the quantification of cells with marker combination phenotypes, such as T cells (CD3+) and their subpopulations (e.g., CD3+ CD8+ for cytotoxic T cells) in a spatial manner18,19.
In this study, we profiled T cells and double-marker defined T-cell subpopulations in surgically resected early-stage endometrial cancer samples and analyzed their association with the prognosis.
Material and methods
Patients and clinicopathological characteristics
A retrospective cohort of patients diagnosed with early-stage (International Federation of Gynecology and Obstetrics [FIGO] stage I or II) endometrial carcinoma at Hospital Universitario La Paz between 2003 and 2015 were identified as eligible for the study.
The patients were included consecutively if they had a minimum follow-up of 5 years and a paraffin-embedded tumor sample.
The study analyzed the following clinicopathological characteristics: age, histological subtype, FIGO stage, differentiation grade, LVSI, and myometrial infiltration. A risk classification was also performed applying 2016 ESMO-ESGO-ESTRO criteria2.
The study endpoint was relapse-free survival (RFS), defined as the time between the surgery and the first evidence of relapse or death from disease.
The study was approved by the local ethics committee (HULP#PI3778) and was conducted in accordance with the ethical standards of the Declaration of Helsinki of the World Medical Association.
Tissue microarray construction
Hematoxylin-eosin-stained slides were reviewed by an experienced pathologist, in order to select the most suitable area for tissue microarrays (TMAs). Selection was focused in the central tumor areas and based on the presence of abundant tumor cells. 1.2-mm tissue punches were arranged using a TMA workstation (Beecher Instruments, Silver Spring, MD, USA), as described previously20.
Molecular classification
We classified the cases based on the surrogate markers described in the ProMisE study: POLE mutated, MMRd, p53wt or NSMP, and p53abn [18]. DNA extracted from formalin-fixed paraffin-embedded (FFPE) samples was analyzed by Sanger sequencing to identify mutations within POLE-EDM, particularly in exons 9, 13, and 14. The expression of p53 and MMR proteins by IHC was also evaluated. The primary antibodies employed in this study (p53, Agilent Cat# GA616; MLH1, Agilent Cat# IR079; PMS2, Agilent Cat# IR087; MSH2, Agilent Cat# IR085; and MSH6, Agilent Cat# IR086) and the Envision kit for visualization were purchased from Agilent Technologies.
Multiplex fluorescent immunohistochemistry
MfIHC is based on a methodology originally described by Blom et al. in 2017 [16]. Prior to staining, we processed 4-µm paraffin sections. We employed 2 different antibody panels with 2 staining and imaging cycles; Supplementary Table S1 lists the antibodies, their dilutions, and the secondary fluorescent detection reagents. In the first-round of staining, we amplified the antibodies with 488 and 555 Alexa fluorophore detection using horseradish peroxidase-conjugated secondary antibodies and tyramide signal amplification following the manufacturer’s instructions (Life Technologies). We then denatured the antibodies through a 20-min heat inactivation and applied a second set of secondary labeled anti-rabbit, Alexa Fluor 647 (Thermo Fisher Scientific Cat# A-21236 and Cat# A-21245) and anti-mouse Alexa Fluor 750 antibodies (Thermo Fisher Scientific Cat# A-21037 and Cat# A-21039). Slides were co-stained with 4′,6-diamidino-2-phenylindole (DAPI, Roche Life Science) and mounted with ProLong Gold antifade mountant (Thermo Fisher Scientific). Coverslips were removed by soaking the slides in wash buffer at 4 °C after whole-slide imaging. We then bleached the Alexa Fluor staining by soaking the slides in TBS buffer containing 25 mM sodium hydroxide and 4.5% hydrogen peroxide. The antibodies from the first staining round were denatured by heating the slides in 10 mM Tris/1 mM EDTA pH9 solution for 20 min at 99 °C. The second-round staining consisted of one antibody with anti-rabbit Alexa Fluor 647 detection (none in panel 2) and a cocktail of antibodies for the detection of epithelial cells using anti-mouse Alexa Fluor 750 labeling. The second-round staining was co-stained with DAPI and mounted as mentioned before prior to imaging.
Fluorescence tissue imaging
TMAs were whole-slide imaged using a Zeiss Axio Scan.Z1 digital slide scanner with a 20X (0.8NA) Plan Apochromat objective lens, a Zeiss™ Colibri.7 LED Light Source, and a Hamamatsu ORCA-Flash 4.0 V2 Digital CMOS Camera (16 bit) with the following filter specifications: DAPI cube (Zeiss Filter Set 02), FITC cube (Zeiss Filter Set 38 HE), Cy3 cube (Chroma Technology Corp. 49004 ET CY3/R), Cy5 cube (Chroma Technology Corp. 49006 ET CY5), and Cy7 cube (Chroma Technology Corp. 49007 ET CY7).
Image export and registration
Resized (25%) 16-bit TIFF-images (BigTIFF) (whole slide images) were exported using original raw channel data. TMA spots were annotated using FIJI Roi1 1-Click Tool Macro, and the spot co-ordinates were saved in CSV format using the region of interest (ROI) Manager List tool. TMA spots were cropped using the ROI extraction script R2019b in MatLab (MATLAB). The images from the second imaging round were registered and overlayed with the corresponding images from the first imaging round using the DAPI channel information and a MatLab (MATLAB) registration script. These scripts are available upon request.
Image analysis
For the image preprocessing, highly autofluorescent red blood cells were detected and removed using the following procedure: We generated 3-channel RGB TIFFs using the 3.1.9 GrayToColor module (Panel 1: red_CD8 (647), green_Foxp3 (488), blue_CD3 (555); Panel 2: red_PD-L1 (555), green_CD3 (750), blue_CD8 (647)) from CellProfiler CellProfiler (Image Analysis Software). We employed Ilastik-1.3.3.post2 machine learning software for teaching the following tissue segments from the RGB images: empty, red blood cells, and good tissue. The “good tissue” TIFF mask was included in the final image analysis pipeline in 3.1.9 CellProfiler (Image Analysis Software), thereby removing all red blood cells and empty areas in the analysis.
For the final image analysis, we classified each tissue-confined pixel in 3.1.9 CellProfiler (Image Analysis Software) as negative or positive for each marker using a semi-automated procedure by applying a spot-wise adaptive Otsu threshold with a visually determined lower bound limit. The segmentation of epithelium and stroma was done by first detecting the epithelium using adaptive Otsu thresholding of the epithelial antibody cocktail channel image. Then the remaining tissue segment was classified as the stroma. We exported the classified pixel numbers for each spot in CSV format and normalized the pixel marker positivity to total tissue pixels and to CD3-positive pixels to obtain positive tissue fractions and positive T-cell fractions. In other words, in this image analysis procedure, we measured markers in each pixel and classified them as positive or negative for double marker combinations. The classification was semi-automated using the adaptive Otsu thresholding (CellProfiler) plus a careful visual inspection to apply a lower threshold limit for each marker. The analysis does not produce exact cell numbers, but double marker positive fractions from total tissue or from marker-defined positive areas, as done in Pollari et al.19 and Autio et al.21. This image analysis approach was validated in Blom et al. to produce highly similar results with a cell segmentation/counting approach22. As a final quality check, we removed spots with fewer than 200,000 good tissue pixels (mean, 485,000) or 10,000 epithelium pixels (mean, 310,000) from the survival analyses. Thus, the final number of patients in the analysis was 285. Replicate spots from the same patient were averaged.
T-cell immune infiltrate quantification
T-cell frequencies were recorded as fractions from all cells (Figs. 2–4A) or as fractions from T-cells (CD3+ cells only) (Figs. 4B–5). In addition, these cell fractions were measured either from the epithelial tissue compartment (Fig. 4C left bar and 4D left graph) on from the stromal tissue compartment (Fig. 4C right bar and 4D right graph, Fig. 5A, B). Due to the low signal-to-noise ratio in the PD-L1 staining, the automated adaptive thresholding for this marker was unsuccessful. Thus, PD-L1-positive T cells (CD3+ PD-L1+) were visually counted from all the TMA cores.
Statistical analysis
We employed R (R Core Team 2017), a language and environment for statistical computing (R Foundation for Statistical Computing), and SPSS Statistics (SPSS, version 25.0, IBM). Continuous data were compared between two groups using a nonparametric Mann–Whitney U test or among >2 groups using a Kruskal–Wallis test (asymptotic, 2-tailed). Categorical values were compared between groups using Fisher’s exact test. We employed Spearman’s rank correlation coefficient to assess the correlation between continuous nonparametric variables. We employed a univariate Cox regression analysis to identify the cell fraction association with RFS. Prior to the Cox regression, we multiplied the variables by 100 to make the hazard ratios (HRs) more informative. We performed a Bonferroni correction to adjust for multiple comparisons and compared the Kaplan–Meier survival curves between groups using a log-rank test. In the Cox regression analyses, we checked the proportional hazard assumption using Schoenfeld residuals.
Results
Patient characteristics
The final analysis included 285 patients diagnosed with early-stage endometrial carcinoma who underwent a long-term follow-up and had good-quality FFPE tissues. During a median follow-up of 6.2 years, 44 of the 285 patients (15.4%) experienced a relapse. Table 1 presents the main clinicopathological characteristics and their association with RFS.
T-cell infiltrate characterization
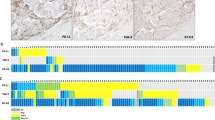
We employed mfIHC for the in-situ characterization of T-cell infiltrates (Fig. 1) and measured the T-cell fractions from all cells in each patient by averaging the replicate cores except for CD3+ PD-L1+, which were scored by visual cell counting (Supplementary Table S2). T-helper cells (CD3+ CD4+) and cytotoxic T cells (CD3+ CD8+) were the most abundant T-cell subtypes, followed by CD3+ CD25+, CD3+ PD-1+, and CD3+ FOXP3+ cells.

Two antibody panels (A and C) with indicated markers were used for the in-situ T-cell profiling of early-stage endometrial cancer using tissue microarrays (no. of patients, 285). B and D correspond to the amplified merged images. Grayscale (16-bit) images visualize individual markers, and color images represent their multi-channel composites (Merge). Scale bar, 20 µm.
T-cell associations with clinicopathological characteristics
We explored the distribution of the T-cell infiltrates in pathologically and molecularly defined patient subgroups. T-helper cells were significantly more frequent in the non-endometrioid carcinomas than in the endometrioid carcinomas, as well as in higher grade (grade 3 vs. grade 1–2) tumors (Fig. 2A–C).

A–C Distribution of T cells in tumors with different histological subtype (endometroid vs. other) and differentiation grade (low vs. high). D–F Distribution of T cells in tumors with different ProMisE molecular subtypes. Cell fractions in (A), (B), and (D) and cell counts in (C), (E), and (F) were calculated as normalized to the total cell area in each tumor (replicate spots were averaged). Non-parametric Mann–Whitney U test, independent samples, *p < 0.05, **p < 0.01.
There were no differences in T-cell populations between the molecular ProMisE subgroups, except for CD3+ PD-1+ and CD3+ PD-L1+, which showed higher frequencies in MMRd tumors than in p53wt/NSMP or p53abn (Fig. 2D–F). Moreover, there was a reciprocal correlation between these T-cell subtypes (Spearman, 0.781), and both also correlated with tumor T-cell density (Spearman 0.721 and 0.832), and all were statistically significant (p < 0.001) (Supplementary Fig. S1A).
Additional correlations are summarized in Supplementary Fig. S1 (B–F). None of the explored T-cell populations differed by myometrial invasion or LVSI status, despite elevated total cell fractions of CD3+ CD8+ cells in patients with no myometrial invasion. In addition, CD3+ CD25+ T cells varied between the ESMO risk groups but not consistently.
Prognostic value of T cells
Regarding the association between T-cell infiltrates and disease progression, total T cells and their specific subtypes (CD3+ CD4+, CD3+ CD8+, CD3+ CD25+, CD3+ PD-1+, CD3+ PD-L1+) were all more frequently present in non-relapsed tumors (Fig. 3A, B). Among them, higher total cell fractions of CD3+ CD8+, CD3+ CD25+, CD3+ PD-1+, and CD3+ PD-L1+ cells also predicted longer RFS when patients were dichotomized by the median cut-off values (Fig. 3C, D).

A–B Distribution of T-cell subtypes in the tumors of patients with (relapse) and without progression. C Hazard ratios (HR) and 95% confidence intervals (95% CI) (univariate Cox regression, RFS) for patients with high total cell fraction of T-cell subtypes (median cut-offs). The cut-off percentage value describes the total cell fraction at the median cut-off, except for CD3+ PD-1+, which describes the number of cells at the median cut-off. *p < 0.05, **p < 0.01, ***p < 0.001. D Kaplan–Meier survival curves for patients with low and high T-cell subtypes (total cell fractions) with RFS probability as the clinical endpoint (median cut-off). P, log rank.
CD3+ PD-1+ cells independently predict relapse-free survival
A multivariate Cox regression analysis demonstrated that the total cell fraction of CD3+ PD-1+ subpopulation was independent not only of the other T-cell subtypes but also of the clinicopathological factors (Fig. 4A). To further rule out the possibility that the prognostic effect of CD3+ PD-1+ cells is dependent on the number of T cells, we normalized the CD3+ PD-1+ cells with total T cell numbers. Accordingly, a higher fraction of CD3+ PD-1+/CD3+ cells (median cut-off, 7.72%) remained a strong predictor of RFS (Fig. 4B).

A Multivariate Cox regression analysis (RFS) combining clinicopathological variables and T-cell subtypes as total cell fractions. HR Hazard ratio, CI confidence interval. *ProMisE groups: POLE, MMRd, p53wt/NSMP, p53abn. Cell fractions were calculated from all the cells and represent low and high fractions using median cut-offs. B Relapse-free survival Kaplan–Meier plot for CD3+ PD-1+ T-cell fraction (median cut-off). C Distribution of CD3+ PD-1+ T-cell fraction in epithelial and stromal compartments (normalized to total CD3+ cells). D Kaplan–Meier plots for epithelial and stromal CD3+ PD-1+ cells as normalized to total CD3+ cells (median cut-off).
For further characterization, we separately evaluated CD3+ PD-1+/CD3+ cells in the epithelial and stromal compartments, which were enabled by the mfIHC-based spatial quantification. CD3+ PD-1+/CD3+ cells showed no significant difference in epithelial vs. stromal distribution (Fig. 4C), whereas only the stromal (and not the epithelial) CD3+ PD-1+/CD3+ cell fraction predicted patient outcomes (Fig. 4D). The median proportion of stromal CD3+ PD-1+/CD3+ cells was 3.36% in the patient cohort, and this cut-off value also separated the patients with higher expression into a much more favorable RFS group. Overall, the patient group with high stromal CD3+ PD-1+/CD3+ expression was associated with a 5-year RFS rate of 93.7% compared to 79.0% in the patient group with low CD3+ -PD-1+/CD3+ expression (Table 2).
Additional analyses demonstrated that stromal CD3 + PD-1+/CD3+ cells were positive for CD8 in 48% of cases, with the remaining 52% being putative CD4+. However, we observed that the prognostic effect of the stromal CD3 + PD-1+/CD3+ cell fraction was independent of its CD8 status (Supplementary Fig. S2).
In a further analysis that included patients classically linked to a favorable outcome (such as endometrioid subtype and low-grade tumors), the stromal CD3+ PD-1+/CD3+ cell fraction still stratified patients into two groups with statistically significant and clinically relevant RFS differences (Fig. 5A). In the cohort of endometrioid tumors, the 5-year RFS rate for the patients with a high and low presence of stromal CD3+ PD-1+/CD3+ infiltrates was 97% and 82%, respectively. Table 2 shows the 5-year RFS rates for the various patient cohorts based on this biomarker’s expression. In all of the cohorts, there were clinically relevant differences between the patients with high and low stromal CD3+ PD-1+/CD3+ infiltrates. However, these T cells were not prognostic in the patients with non-endometrioid or high-grade tumors (Supplementary Fig. S3).

A Kaplan–Meier survival analysis of patients with low and high stromal CD3+ PD-1+/CD3+ cell fraction in patient subgroups as indicated. The cut-off (3.36%) was derived from the median value in the total patient population. B Multivariate Cox regression analysis for patients with median dichotomized stromal CD3+ PD-1+/CD3+ cell fraction adjusted with relevant clinicopathological variables. HR hazard ratio, CI confidence interval.
In addition, the stromal CD3+ PD-1+/CD3 cell fraction remained a favorable outcome biomarker in ESMO-ESGO-ESTRO-defined patient subgroups with either low, intermediate, or high-intermediate risk classes. Furthermore, this T-cell fraction maintained its prognostic significance when analyzed in patients with intermediate risk molecular ProMisE categories (MMRd and p53wt/NSMP) (Fig. 5A). Supplementary Fig. S4 summarizes the additional subgroup analyses regarding the risk classifiers. CD3+ PD-1+/CD3+ infiltrates remained significant even when adjusted for the clinicopathological factors in a multivariate model (Fig. 5B).
Discussion
This study profiles different T-cell populations in their spatial context in the tumor microenvironment of early-stage endometrial carcinoma by using mfIHC and quantitative computerized image analysis. The data from this study suggest that a lower abundance of T-cell infiltrates is associated with a higher risk of relapse and that higher levels of stromal CD3+ PD-1+ cells could represent a biomarker for an excellent prognosis.
T-cell expression in endometrial carcinoma has been previously investigated in several studies10,11,13,23, with high variability in cell positivity scores. Most of these studies employed single-color IHC, visual scoring, and estimation of the positive cell fraction out of all the cells or immune cells. The differing methods and cut-offs employed for defining positivity make it cumbersome to compare T-cell expression among the studies. In this study, we analyzed T cells and their subtypes separately as a fraction of all cells but also normalized them by total T cells only.
Previous studies have reported contradictory results related to the correlation between T cells and histological grade24,25. Overall, our results suggest that T-cell subtypes have a similar distribution across distinct pathological groups, except for the CD3+ CD4+ subtype, which shows higher frequencies in higher grade and non-endometrioid tumors.
Previous studies on the PD-(L)1+ axis have focused on PD-L1+ T cells26,27,28. When encompassing advanced stages, the PD-L1+ immune population has been associated with a non-endometrioid histology, higher grade, and stage, with most PD-L1-negative cases in stage I of the disease29. These findings agree with the small number of PD-L1 cells observed in our early-stage patient series. There was no significant correlation between the PD-L1-positive T cells and the pathological features.
In a cohort of 200 patients with stages I-III endometrial cancer, higher frequencies of CD4+ Foxp3+ cells were shown to be enriched in higher stage tumors and conferred worse RFS30. In our early-stage cohort, CD3+ Foxp3+ cells constituted only ~1% of all the cells and was not associated with survival. Taken together, these findings suggest that T cells would be more frequent in more advanced endometrial cancer, where they could play a negative prognostic role.
Importantly, the only T-cell subpopulations that were significantly different across the four ProMisE categories were CD3+ PD-1+ and CD3+ PD-L1+, with higher frequencies in tumors with MMRd than in either p53wt/NSMP or p53abn cases. These findings are highly consistent with previous studies in more advanced stage series27,29,31.
Accumulating evidence suggests that the abundance of TILs represents a biomarker of superior prognosis for several solid cancers32,33. A recent meta-analysis of 17 prognostic studies indicated that a higher number of CD8+ cells translates to a more favorable outcome in endometrial cancer17.
Most of the aforementioned studies were based on conventional, single antibody IHC, which yields inaccurate T-cell phenotyping. CD8, Foxp3, PD-1, PD-L1, and CD25 markers are also expressed in other types of immune cells, such as in natural killer cells and myeloid cells. Co-staining approaches should therefore be required for a reliable scoring and measurement of these T-cell subtypes. Consistent with this finding, we analyzed double-marker combinations in pair with CD3 by mfIHC. All measured combinations (CD3 with CD4, CD8, CD25, PD-1, and PD-L1), except for CD3+ Foxp3+, were less abundant in the tumor microenvironment of patients who experienced a relapse. In addition, higher numbers of CD8+, CD25+, PD-1+, and PD-L1+ T cells predicted longer time for relapses. In line with these findings, two recent studies indicated that higher CD8+ cell numbers in endometrial cancer are linked with favorable prognoses10,34. Talhouk et al. found higher fractions of CD3+ CD8+ and CD3+ CD8− (putative CD3+ CD4+) cells associated with more favorable survival but without statistical significance in a multivariate analysis that included ProMisE subtypes and other clinical characteristics10. In addition to CD3+ CD8+, we found that CD3+ CD25+, CD3+ PD-1+, and CD3+ PD-L1+ are prognostic markers for RFS. However, CD3+ PD-1+ cells were the only T-cell subpopulation independently correlated with the prognosis when the clinicopathological characteristics and other T-cell subtypes were included in the model. Higher CD3+ PD-1+ cell numbers translated into more favorable outcomes, even when they were normalized to the total number of T cells (CD3+ PD-1+/CD3+ cell fraction) to achieve a more robust result. Interestingly, only the stromal (but not the epithelial T-cell) fraction of CD3+ PD-1+ cells was prognostic.
Despite the incorporation of the ProMisE subgroups into the risk stratification in the latest ESGO-ESTRO-ESP guidelines, the improvement in prognostic prediction is still challenging, and research on new biomarkers is a priority in endometrial cancer. An accurate prognosis could help identify patients who will need chemotherapy, radiotherapy, or even other alternative therapies in addition to surgery. Along these lines, the novel biomarker described in our study, defined by the high expression of the stromal CD3+ PD-1+/CD3+ cell fraction, is associated with a low risk of relapse in early-stage endometrial cancer. This association is even more evident in patients with an endometrioid histology (with a 5-year RFS rate of 97%). In our opinion, this is a highly relevant clinical finding because it can guide to select a patient group with excellent prognosis, with almost no relapses, and taking into account that the endometrioid subtype represents ~80% of early-stage endometrial cancer. In this study cohort of early-stage carcinomas, patients with endometrioid histology and with high stromal CD3+ PD-1+ T-cell fraction (>3.36%) represent 46.7% of all the patients in the cohort. Therefore, CD3+ PD-1+ could have a similar clinical implication to that of the POLE mutation, thereby avoiding unnecessary adjuvant therapies but with the advantage of selecting ~50% of patients with the endometrioid subtype, while the POLE mutation represents <10% of cases in the published series5,35.
In recent years, risk stratification for endometrial cancer has been performed based on criteria published by ESMO-ESGO-ESTRO 2016 consensus, which established four groups according to clinicopathological characteristics to create recommendations for adjuvant therapy. However, this classification has limitations in that there are patients with low to intermediate risk who relapse and patients with intermediate-high to high risk who do not. To improve the risk stratification, the ESGO-ESTRO-ESP guidelines suggest adding the molecular characteristics3. In this study, we identified stromal CD3+ PD-1+ cells to accurately stratify patients included in the low, intermediate, and intermediate-high groups combined, with differences in RFS. Therefore, this biomarker could ultimately facilitate the allocation of adjuvant therapy.
The molecular classification based on ProMisE established 4 other groups with prognostic implications6,7. Among them, the POLE-mutated group encompasses patients with excellent prognoses and an extremely rare possibility of relapse, while the p53abn group had those with the highest risk of relapse. High expression of stromal CD3+ PD-1+ was also confirmed as a marker of a good prognosis in intermediate risk molecular groups, MMRd and NSMP/p53wt, either separately or together.
In terms of the study limitations, we used TMAs to facilitate the analysis of large patient numbers. However, whole tissue sections are better suited for the comparison of tumor regions, such as the infiltrative border and the tumor core. Second, the use of digital pathology techniques is precise but not yet implemented in routine diagnostics. Thus, it would be desirable to implement CD3+ PD-1+ detection also using more classical methods, such as the chromogenic IHC.
Our findings suggest that the density of stromal PD-1-positive T cells (CD3+ PD-1+) in the tumor microenvironment predicts a much longer RFS in early-stage endometrial cancer. These results are clinically relevant because this biomarker could identify a high proportion of patients who could potentially be cured with surgery alone, at least in those with an endometrioid subtype. Moreover, high CD3+ PD-1+ levels define an extremely good prognosis group in the insufficiently defined low and intermediate risk categories of the ESMO-ESGO-ESTRO classifier. Although these results are relevant and encouraging, future studies are needed to validate the findings.
Data availability
The datasets are available from the corresponding authors upon request.
References
Braun, M. M., Overbeek-Wager, E. A. & Grumbo, R. J. Diagnosis and management of endometrial cancer. Am. Fam. Physician 93, 468–474 (2016).
Colombo, N. et al. ESMO-ESGO-ESTRO consensus conference on endometrial cancer: diagnosis, treatment and follow-up. Ann. Oncol. 27, 16–41 (2016).
Concin, N. et al. ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int. J. Gynecol. Cancer 31, 12–39 (2021).
Cree, I. A., White, V. A., Indave, B. I. & Lokuhetty, D. Revising the WHO classification: female genital tract tumours. Histopathology 76, 151–156 (2020).
Cancer Genome Atlas Research, N. et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013).
Kommoss, S. et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 29, 1180–1188 (2018).
Talhouk, A. et al. Confirmation of ProMisE: a simple, genomics-based clinical classifier for endometrial cancer. Cancer 123, 802–813 (2017).
Liu, Y. et al. Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. J. Natl Cancer Inst. 106, dju245 (2014).
Kommoss, F. K. et al. L1CAM further stratifies endometrial carcinoma patients with no specific molecular risk profile. Br. J. Cancer 119, 480–486 (2018).
Talhouk, A. et al. Molecular subtype not immune response drives outcomes in endometrial carcinoma. Clin. Cancer Res. 25, 2537–2548 (2019).
Kondratiev, S., Sabo, E., Yakirevich, E., Lavie, O. & Resnick, M. B. Intratumoral CD8+ T lymphocytes as a prognostic factor of survival in endometrial carcinoma. Clin. Cancer Res. 10, 4450–4456 (2004).
Versluis, M. A. et al. Prediction model for regional or distant recurrence in endometrial cancer based on classical pathological and immunological parameters. Br. J. Cancer 113, 786–793 (2015).
de Jong, R. A. et al. Presence of tumor-infiltrating lymphocytes is an independent prognostic factor in type I and II endometrial cancer. Gynecol. Oncol. 114, 105–110 (2009).
Piulats, J. M. et al. Molecular approaches for classifying endometrial carcinoma. Gynecol. Oncol. 145, 200–207 (2017).
Eggink, F. A. et al. Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE-mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. Oncoimmunology 6, e1264565 (2017).
Samstein, R. M. et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206 (2019).
Guo, F., Dong, Y., Tan, Q., Kong, J. & Yu, B. Tissue infiltrating immune cells as prognostic biomarkers in endometrial cancer: a meta-analysis. Dis. Markers 1805764 (2020).
Blom, S. et al. Systems pathology by multiplexed immunohistochemistry and whole-slide digital image analysis. Sci. Rep. 7, 15580 (2017).
Pollari, M. et al. Adverse prognostic impact of regulatory T-cells in testicular diffuse large B-cell lymphoma. Eur. J. Haematol. 105, 712–721 (2020).
Ruz-Caracuel, I. et al. Myoinvasive pattern as a prognostic marker in low-grade, early-stage endometrioid endometrial carcinoma. Cancers (Basel) 11, 1845 (2019).
Autio, M. et al. Immune cell constitution in the tumor microenvironment predicts the outcome in diffuse large B-cell lymphoma. Haematologica 106, 718–729 (2021).
Blom, S. et al. Fibroblast as a critical stromal cell type determining prognosis in prostate cancer. Prostate 79, 1505–1513 (2019).
Workel, H. H. et al. CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur. J. Cancer 60, 1–11 (2016).
Jung, I. K. et al. Tumor-infiltration of T-lymphocytes is inversely correlated with clinicopathologic factors in endometrial adenocarcinoma. Obstet. Gynecol. Sci. 57, 266–273 (2014).
Yamagami, W. et al. Immunofluorescence-detected infiltration of CD4+FOXP3+ regulatory T cells is relevant to the prognosis of patients with endometrial cancer. Int. J. Gynecol. Cancer 21, 1628–1634 (2011).
Sungu, N. et al. Expression of immunomodulatory molecules PD-1, PD-L1, and PD-L2, and their relationship with clinicopathologic characteristics in endometrial cancer. Int. J. Gynecol. Pathol. 38, 404–413 (2019).
Pasanen, A. et al. PD-L1 expression in endometrial carcinoma cells and intratumoral immune cells: differences across histologic and TCGA-based molecular subgroups. Am. J. Surg. Pathol. 44, 174–181 (2020).
Mo, Z. et al. Expression of PD-1, PD-L1 and PD-L2 is associated with differentiation status and histological type of endometrial cancer. Oncol. Lett. 12, 944–950 (2016).
Li, Z. et al. Programmed death ligand 1 expression among 700 consecutive endometrial cancers: strong association with mismatch repair protein deficiency. Int. J. Gynecol. Cancer 28, 59–68 (2018).
Xi, Z. et al. Evaluation of PTEN and CD4+FOXP3+ T cell expressions as diagnostic and predictive factors in endometrial cancer: A case control study. Med. (Baltim) 98, e16345 (2019).
Engerud, H. et al. High degree of heterogeneity of PD-L1 and PD-1 from primary to metastatic endometrial cancer. Gynecol. Oncol. 157, 260–267 (2020).
Pages, F. et al. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29, 1093–1102 (2010).
Barnes, T. A. & Amir, E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 118, e5 (2018).
Horeweg, N. et al. Prognostic integrated image-based immune and molecular profiling in early-stage endometrial cancer. Cancer Immunol. Res. 8, 1508–19 (2020).
Stelloo, E. et al. Improved risk assessment by integrating molecular and clinicopathological factors in early-stage endometrial cancer-combined analysis of the PORTEC cohorts. Clin. Cancer Res. 22, 4215–4224 (2016).
Acknowledgements
The authors would like to thank the IdiPAZ biobank core facility for essential services support, as well as the FIMM Digital microscopy and Molecular Pathology Unit supported by HiLIFE and Biocenter Finland for the scanning.
Author information
Authors and Affiliations
Contributions
These are the main contributions by authors: Conceptualization and resources—M.M., T.P., A.R. Data curation—M.M., T.P., J.R.P., A.B., O.B., V.H.S., R.T., J.E., A.H., L.G.C., R.C., A.G., A.H. Software and visualization—T.P., O.B., R.T., A.H. Methodology and Formal analysis—M.M., T.P., J.R.P., A.B., O.B., V.H.-S., R.T., J.E. Supervision and funding acquisition—M.M., J.F., A.R. Funding acquisition—M.M., J.F., A.R. Writing and editing original draft—M.M., T.P., A.R. All authors read, reviewed and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
M.M. reports having received honoraria (MSD, AstraZeneca and GSK), research grant/funding to her institution (Eisai and PharmaMar), and travel/accommodation/expenses (AstraZeneca, GSK, PharmaMar, Roche and Pfizer) outside the submitted work. O.B. reports having received honoraria (Novartis, Sanofi) outside the submitted work. A.G. reports having received honoraria (Clovis, MSD, AstraZeneca, GSK, PharmaMar and Roche) and travel/accommodation/expenses (Merck Sharp & Dohme, PharmaMar, Roche, Eisai, Pfizer, Pierre-Fabre and Tesaro-A GSK Company) outside the submitted work. A.R. reports having received honoraria and providing advisory/consultancy services (MSD, AstraZeneca, Roche, GSK, Clovis, PharmaMar, Lilly, Amgen), as well as having received research grant/funding to his institution (Eisai, PharmaMar, Roche), travel/accommodation/expenses (AstraZeneca, Tesaro: A GSK Company, PharmaMar, Roche), and participating in a speakers bureau (MSD, AstraZeneca, Roche, GSK, Clovis, PharmaMar) outside the submitted work. The remaining authors declare no conflicts of interest.
Ethics approval
The study was approved by the local ethics committee (HULP#PI3778) and was conducted in accordance with the ethical standards of the Declaration of Helsinki of the World Medical Association.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Mendiola, M., Pellinen, T., Ramon-Patino, J.L. et al. Prognostic implications of tumor-infiltrating T cells in early-stage endometrial cancer. Mod Pathol 35, 256–265 (2022). https://doi.org/10.1038/s41379-021-00930-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00930-7
