
Abstract
Biphasic squamoid alveolar papillary renal cell carcinoma (BSA-PRCC) is a recently studied lesion considered a morphologic variant of papillary renal cell carcinoma (RCC), more closely related to type 1. Considering the role of proto-oncogene MET in both sporadic type 1 papillary RCC and hereditary papillary RCC, we aimed to explore the role of MET activation in the oncogenesis of BSA-PRCC. We identified 17 patients with either unique (n = 14) or multiple (n = 3) BSA-PRCC, all localized, and performed an integrative analysis of MET status in 18 formalin-fixed paraffin-embedded tumors combining next-generation sequencing analysis, fluorescent in situ hybridization and immunohistochemistry. Trisomy 7 was found in 86% of tumors (14/16) without MET amplification at 7q31 (15/15). A pathogenic MET genetic variant was identified in 60% (9/15) of cases, at the germline level in 57% (4/7) of tested patients or at the somatic level (5/11). MET expression was observed in all tumors with a higher value of combined score in large cells (mean 97%, range 80–100%) than in small cells (mean 74%, range 10–100%) and was lower in two cases without MET copy number gain. In conclusion, our study provides additional evidence to consider biphasic squamoid alveolar papillary RCC as a morphological variant of type 1 papillary renal RCC. Our data strongly suggest that MET represents a major oncogenic driver gene in BSA-PRCC, harboring a higher frequency of MET mutation that encourages to further explore the benefice of anti-MET targeted therapies for aggressive BSA-PRCC.
Similar content being viewed by others

Introduction
Biphasic squamoid alveolar papillary renal cell carcinoma (BSA-PRCC), also named biphasic papillary renal cell carcinoma (RCC), has been mainly described as a morphological variant of RCC after the 2016 WHO classification [1]. Based on the first morphological description published in 2012 [2], a total of 57 patients harboring BSA-PRCC have been reported, including two large cohorts of 21 and 28 patients, respectively [3,4,5,6,7,8]. Many features are shared with papillary RCC, suggesting that BSA-PRCC is a variant of papillary RCC, more closely related to type 1 papillary RCC [5,6,7]. Its incidence has been estimated at less than 1% of papillary RCC [3, 5, 6]. As highlighted by Trpkov et al. [6], the peculiar dual morphology characterizing this variant has been previously mentioned in some solid papillary RCC with a “micronodular” or “glomeruloid” architecture or illustrated in some textbook [9,10,11]. BSA-PRCC is composed of an admixture of two cell populations: a population of unistratified low-grade small cells with scant cytoplasm arranged in solid tubulo-papillary and alveolar-like structures reminiscent of type 1 papillary RCC, and a remarkable population of large eosinophilic squamoid-like cells with large nuclei and prominent nucleoli. The latter cell population forms nests in the center of alveolar-like structures and exhibits frequent emperipolesis.
While keratin 7 and alpha-methyl-CoA-racemase (AMACR) are expressed in both populations, expression of cyclin D1 and CD57 are strikingly restricted to the larger central cells [3,4,5,6,7,8].
Chromosomal imbalance analyses have identified gains of chromosomes 7 and 17 in all (16/16) tested cases [3, 5, 6], and the loss of Y in 80% (4/5) of tumors developed in male patients [5]. Those gains and losses are commonly found in type 1 papillary RCC rather than in type 2 [12, 13], which represents an additional argument to consider BSA-PRCC as a variant of type 1 papillary RCC. Multifocal BSA-PRCC or association with other tumors occurred in 7% (4/57) and 18% (10/57) of cases, respectively [6,7,8]. Metastatic potential, reported in 3.5% (1/28) [6] to 24% (5/21) [5] of patients, is the most clinically relevant feature. Hereditary BSA-PRCC has been described, including one patient with Birt–Hogg–Dubé syndrome [6] and one family with hereditary papillary RCC [7]. In contrast to Birt–Hogg–Dubé syndrome, hereditary papillary RCC is not associated with extra-renal manifestations and presents an increased risk to develop type 1 papillary RCCs only, with frequently multiple and bilateral tumors [14,15,16]. This rare autosomal dominant disorder is due to germline activating MET mutation. MET is a proto-oncogene located at 7q31, encoding the transmembrane tyrosine-kinase receptor MET [17]. Interestingly, alterations in MET have been identified in 81% of cases of type 1 papillary RCC in The Cancer Genome Atlas (TCGA) study [18]. Copy number gain of MET due to gain of chromosome 7 is the main genetic alteration encountered in type 1 papillary RCC, while MET mutations have been identified in 17–21.6% of cases, mostly located in the tyrosine-kinase domain (82%) [17,18,19,20]. Few cases with MET amplification have been identified in advanced stage with metastasis [21]. Moreover, in inherited papillary RCC, allelic duplication on 7q occurred nonrandomly in alleles harboring MET mutation [22, 23].
In order to evaluate the role of MET alterations in BSA-PRCC, we performed a comprehensive study of MET status in BSA-PRCC combining analyses of MET copy number variation, mutational status, and protein expression in a cohort of 17 patients.
Materials and methods
Patient cohort
A total of 17 patients were retrieved from 3 institutional pathological departments of Assistance Publique-Hôpitaux Paris-Centre/Université de Paris, including Necker-Enfants Malades Hospital (n = 4), Georges Pompidou European Hospital (n = 11) and Cochin Hospital (n = 2). These patients suffered from at least one BSA-PRCC diagnosed according to the criteria previously described [5, 6], including positive AMACR and CK7 in both cell populations and a restricted expression of BCL1 in large central cells (Supplementary Fig. 1). A total of 18 tumors were collected, including 13 cases diagnosed after the first description of the entity, and 5 identified retrospectively. Clinical and pathological data were collected from local electronic medical patient records including age, sex, type of surgery, tumor size, number of tumors, stage, familial history of renal carcinoma, presence of chronic kidney disease, other renal tumors, time of follow-up, and clinical outcome. When multiple tumors were present, the stage of the more aggressive tumor was documented for each surgery.
Seven patients, who benefited from genetic counseling and signed an informed consent, were tested for germline mutations. Among them were two related patients (mother and son, cases #5 and #6, respectively) with hereditary papillary RCC, who have been previously reported [7].
This study was approved by the French national ethic committee (Comité de Protection des Personnes Number 19.05.27.61541; national identification 2018-A03147-48).
Immunohistochemical and pathological features
All slides routinely stained by hematoxylin, eosin, and saffron have been reviewed by three pathologists (TD, MS, and VV). Five to 29 slides have been reviewed per case. The following microscopic features were determined: surface occupied by the squamoid large cell component, percentage of necrosis, WHO/ISUP nucleolar grade of both cell populations, presence of foamy macrophages and/or calcifications. Routine immunohistochemistries (IHCs) performed at the time of diagnosis on full slide were also reviewed, including staining with BCL1 CCND1/cyclin D1 (SP4; 1/100; Thermo Fisher Scientific, Waltham, MA, USA), CK7 (OV-TL 12/30 [1] Isotype IgG1 kappa; 1/50; Dako, Carpinteria, CA, USA), AMACR (BC4A4; 1/200; Dako, Carpinteria, CA, USA), and FH (1/2000; Santa Cruz Biotechnologies Inc., Dallas, TX, USA). Standard immunohistochemical staining was completed when necessary.
Tumor sample selection for in situ analysis of MET
Representative areas of tumor containing both cellular populations were selected from FFPE blocks to perform MET IHC and FISH analysis. A total of 18 tumors were studied. Four manual tissue microarray (TMA) blocks (one spot of 5 mm width/patient) were prepared, including 17 FFPE tumors from 16 patients. For patients #5 and #6 with multiple BSA-PRCC, the most representative tumor was selected. For one patient (#12) with two tumors, one spot from each tumor was selected for the TMA. Tumor from one patient (#17) was analyzed on whole slide.
MET and chromosome 7 copy number analysis by fluorescent in situ hybridization (FISH)
Copy number analysis of centromere 7 was performed by FISH analyses on 4 μm FFPE tissue sections from the four TMAs. Hybridization was performed using the ZytoLight FISH-Tissue Implementation Kit (Zytovision GmbH, Bremerhaven, Germany) and either the ZytoLight SPEC MET/CEN 7 Dual Color Probe (Zytovision GmbH) or the ZytoLight SPEC CDKN2A/CEN 3/7/17 Quadruple Color Probe (Zytovision GmbH), according to the manufacturer’s instructions.
Slides were read by one expert pathologist (PAJ) on an epifluorescence microscope (BX51, Olympus, Tokyo, Japan) armed with appropriate fluorescent cubes. Images were analyzed using the Soft Imaging System (Cell, Olympus) software. One hundred nonoverlapping nuclei were counted for each case. Gains were defined by the presence of three or more copies of the studied locus in more than 20% of nuclei and losses by the presence of only one copy of the studied locus in more than 30% of nuclei. MET amplification was defined by the presence of more than six copies of MET on average [24].
MET genotyping using next-generation sequencing (NGS) and data analysis
For five patients, germline DNA was extracted from leukocytes using standard protocols by Chemagic DNA Blood kit in combination with a Chemagic 360 equipment (Perkin Elmer). Germline genetic testing was then performed using an in-house targeted NGS panel. The NGS panel was used to genotype the main kidney cancer-predisposing genes including MET, VHL, FH, TSC1, TSC2, FLCN, BAP1, PBRM1, MITF, PTEN, HNF1B, SMARCB1, SDHA, SDHB, SDHC, and SDHB (TruSeq Custom Amplicon Low Input, Illumina® or SeqCap EZ HyperCap, Roche®). DNA libraries were prepared according to the manufacturer’s recommendations and 2 × 150 bp paired-end sequenced on MiSeq platform (Illumina, CA, USA) using v2 chemistry according to the standard protocol. Demultiplexing was performed using MiSeq Reporter (Illumina, CA, USA). Alignment to the human genome hg19 (GRch37) and variant calling was performed using SeqNext (JSI Medical Systems) and PolyDiag (Paris Descartes University, France) software.
Seventeen tumors from 16 patients were tested for somatic mutations in MET tyrosine-kinase domain. Areas of pure tumor containing both cell populations were selected in order to realize five, 20 μm sections for DNA extraction from each FFPE sample. FFPE DNA was extracted on a Maxwell® 16 Forensic Instrument (Promega, France) using Maxwell® 16 FFPE Plus LEV DNA Purification Kit (Promega, France). DNA quantification was done by Qubit™ Fluorometric Quantitation using the Qubit dsDNA BR Assay Kit (Life Technologies–Thermo Fisher Scientific, Saint Aubin, France). Libraries were prepared from 30 ng of tumor DNA using the ROSMETALK Designed.bed panel covering MET tyrosine-kinase site (exons 16–20) and Ion AmpliSeq™ library preparation kit v2 (Ion AmpliSeq library kit v2, Ion library equalizer kit, Life Technologies™). Libraries were normalized using the Ion Library Equalizer™ Kit, pooled, processed on an Ion Chef™ System for template preparation and chip loading (Ion PI™ Hi-Q™ Chef Kit, Ion PI™ Chip Kit v3, Thermo Fisher Scientific), and sequenced using an Ion Proton™ System. The FASTQs sequencing data were aligned to the human genome (hg19) and processed using IonTorrent Suite V5.0.4.0. This package included the Torrent Variant Caller V5.0.4.0 provided by Life Technology, which was used along with the built-in “Somatic—low stringency” parameters to automatically call variants with AF > 2%. Annotation pipeline used a pipeline developed internally on a galaxy platform using SAFIR.
The NM_0011207500.2 transcript was used for annotations. The variant analysis was mainly performed using Alamut Visual 2.7 (Interactive Biosoftware) as an interface. To assess the frequency and the impact of MET variants identified by NGS, the following databases were consulted: MET Leiden Open Variation Database (LOVD) (https://databases.lovd.nl/shared/genes/MET), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), COSMIC (https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=MET), and genome aggregation database gnomAD (https://gnomad.broadinstitute.org/gene/ENSG00000105976?dataset=gnomad_r2_1). MET variants were considered pathogenic if they had been reported like this in the MET-LOVD and ClinVar, or variant of uncertain significance (VUS) if they had never been reported in the LOVD-MET, ClinVar and gnomAD databases.
MET expression by immunohistochemistry
Evaluation of MET expression was performed by IHC using prediluted anti-Total MET (SP44, Rabbit Monoclonal Primary Antibody ready-diluted, Ventana Medical System, Tucson, AZ, USA) on FFPE samples. Staining was assessed on autostainers (Benchmark Ultra Ventana; Ventana Medical System, Tucson, AZ, USA) according to manufacturer’s instructions. The reactivity of the antibodies was revealed with UltraView Universal Dab (Ventana Medical System, Tucson, AZ, USA) proprietary detection kit. Staining was performed on 4 TMAs from FFPE samples for 17 tumors (16 patients) and 1 whole slide for 1 tumor (1 patient). MET expression was evaluated separately on small and central large cells using the previously published combined score CS (CS, 0–300) as follows: staining intensity (range 0–3; 0: absent, 1: weak, 2: medium, 3: strong) multiplied by the percentage of positive cells (0–100) [25]. Analysis was assessed blinded of genetic status by one junior pathologist (TD) and two experts in uropathology (VV, MS).
Results
Clinicopathological data
Clinicopathological data are summarized in Table 1. Male to female ratio was 1.4:1. Mean age at diagnosis of the first renal tumor was 51.6 years (26–84 years). Eight patients out of 17 (47%) were diagnosed before the age of 50. Two patients had chronic renal failure. For one patient with a renal transplant (#3), the tumor arose on his native kidney. The specimens were composed of 5 radical nephrectomies (1 right, 2 left, 1 bilateral) and of 17 partial nephrectomies (8 right, 9 left). BSA-PRCC tumor size ranged from 1.5 to 15 centimeters. Six patients (35%) had multiple tumors. Among them, three patients (17%) had synchronous multiple and bilateral BSA-PRCC. Three patients had one BSA-PRCC and other tumors: one patient had multiple and bilateral type 1 papillary RCCs, one patient had an oncocytoma, and a papillary RCC (1.6 cm each), and another one had a contralateral tumor without histological documentation. In addition, six patients (33%) also presented either single or multiple papillary adenomas. In addition to patients #5 and #6, two patients had a familial history of renal carcinoma. Patient #17’s father died of advanced unclassified RCC. Reviewing of his lymph node biopsy demonstrated the same immunohistochemical phenotype as BSA-PRCC including positive PAX8, AMACR, CK7, and BCL1, without evocative morphology. Patient #7 had a cousin diagnosed with a history of clear cell RCC. No patient had metastasis at diagnosis. Follow-up data were available for 14 out of 17 patients with a mean follow-up of 32.6 months (from 1 to 151 months). One of the patients was lost to follow-up after developing a contralateral tumor. Only one patient developed metastases, occurring 13 months after surgery, with mediastinal lymph node involvement and pulmonary carcinomatous lymphangitis. After a first line of treatment with sunitinib for 5 months, followed by four of nivolumab, the patient died of disease.
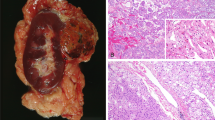
Main microscopic features and immunohistochemical studies are detailed in Table 1 and Supplementary Table 1 and illustrated in Fig. 1 and Supplementary Figs. 1 and 2. On gross examination, tumors were predominantly whitish or pale yellow and well-delimited (Fig. 1a). Eleven cases were pT1a, 5 pT1b, 2 pT2b, and two were pT3a due either to renal sinus fat infiltration (Fig. 1b) or perirenal fat invasion. No lymph node was detected on five radical nephrectomies. On microscopy, the dual component was either focal or diffuse, representing between 5 and 100% of the tumor surface (mean 20%). Generally, large squamoid-like cells were arranged in cohesive clusters of variable sizes localized in small spaces lined by small cells, giving them an alveolar or glomerular-like structure (Fig. 1c–h). The large cells had a vast eosinophilic cytoplasm and a big, sometimes angular nucleus with anisokaryosis. Emperipolesis was visible in all cases in various proportions, restricted to some clusters of large squamoid-like cells in some tumors (Fig. 1f–h). Rare mitotic figures were observed, restricted to the large cells. The nucleolar WHO/ISUP grade evaluated in large cells at 3 (in 17/18) or 2 (in 1/18), was higher than in small cells (grade 1 (in 2/18), 2 (in 14/18), or 3 (in 2/18)). Interestingly, a small component of grade 4 with sarcomatoid features was found in the tumor of patient #1, who subsequently developed metastasis. Neither sarcomatoid nor biphasic components were identified in his nodal mediastinal biopsy (Supplementary Fig. 2). Foamy macrophage clusters were found in 12 cases. Necrosis was observed in seven cases on 5–20% of the tumor surface, mostly at the center of the eosinophilic cell clusters. Upon routine IHC, the large eosinophilic cells of the biphasic component were always stained by the BCL1 (cyclin D1) antibody, unlike the conventional papillary component (Supplementary Fig. 1). FH expression was retained in all (8/8) tested cases (Supplementary Table 1).

a Gross picture of one of the two tumors (LPN) of patient #12 showing a well circumscribed white tumor with mastic and solid aspect. On hematoxylin, eosin saffron we illustrate: at low magnification ((b) ×2.5 magnification) pT3a tumor with sinus infiltration from patient #7 (yellow arrow surrounding large squamoid cells); at higher magnification the characteristic biphasic alveolo squamoid morphology showing: example of abrupt transition between areas with biphasic morphology and large cluster of squamoid-like cells (above the blue line) and classical papillary RCC type 1 (below the blue line) ((c) ×10 magnification) and different sizes of clusters of large eosinophilic cells nested arranged in alveolar-like spaces surrounded by small cells with either small clusters ((d) ×5 magnification; (f) ×2 magnification; (g) ×40 magnification), or larger clusters ((c) and (e) ×20 magnification; (h) ×40 magnification); (g–h) illustrate emperipolesis on large squamoid-like cells (green arrow in (f–h)).
Chromosome 7 and 17 gains
Centromere 7 enumeration using FISH was performed for 17/18 tumors and interpretable for 16/17 cases. Chromosome 7 trisomy was found in 14/16 cases (87.5%) (Table 2) in both small cells and large cells at the center of alveolar-like structure. The two remaining cases were disomic for chromosome 7. No MET amplification was found in 15 evaluated tumors (Fig. 2). For the 6 cases evaluated for centromeres 17 and 3, a trisomy 17, co-occurring with trisomy 7, was observed in all cases, while centromere 3 was found to be diploid.

FISH with a MET probe (green) showing three copies of MET in large squamoid cells (arrows) and small cells (arrow heads). Insert: FISH with a MET probe (green) and a chromosome 7 centromeric probe (red) showing chromosome 7 trisomy without specific MET gain (original objective: ×40 magnification).
MET mutational status
The results of germline and somatic analyses are summarized in Table 2. Patients #5 and #6, who are relatives affected by hereditary papillary RCC, carried a previously reported c.3335A>G, p.(His1112Arg) pathogenic MET variant [7]. The same MET germline mutation was also identified in an unrelated 46-year-old woman (#17) presenting multiple papillary adenomas, synchronous bilateral, and multiple type I papillary RCC as well as one BSA-PRCC. No genetic analysis could be performed on her father. Patient #12, a 57-year-old woman with two bilateral synchronous BSA-PRCC and without familial history of renal carcinoma, was found to carry the c.3712G>A, p.(Val1238Ile) pathogenic variant at the germline level. In the three other patients who benefited from germline genetic testing, no variant of interest was identified either in MET or other predisposing genes included in the NGS panel. Somatic genotyping of the MET tyrosine-kinase domain was interpretable for 14 tumor DNAs (Table 2). Among them, three tumors originated from patients carrying a germline MET mutation. In all cases, the germline MET mutation was found in the corresponding tumor DNA with an allelic ratio > 0.5. In the 11 remaining analyzed tumors, a MET variant was identified in 54% of cases (6/11). All but one was classified as pathogenic according to reports in ClinVar and LOVD-MET databases. They included c.3736G>A p.(Asp1246Asn), c.3334C>T p.(His1112Tyr), c.3803C>T p.(Met1268Thr), c.3389T>C p.(Leu1130Ser), and c.3328G>A p.(Val1110Ile) variants. The last variant was found in two different cases. All identified variants, except c.3389T>C p.(Leu1130Ser), were listed as “oncogenic validated mutations” in the Cancer Genome Interpreter (https://www.cancergenomeinterpreter.org/mutations) and have been previously reported in COSMIC database, mainly in RCC and interestingly at least once in papillary RCC.
Immunohistochemical evaluation of MET expression
MET was expressed in all tumors (18/18), as shown in Table 3 and illustrated in Fig. 3. Detailed combined score for each tumor on both small and large cells are available in Supplementary Table 2. In all cases, staining was observed on large cells, involving 80–100% of cells (mean 97%) with variable intensity: weak in two cases (11%), moderate in six (33%), and strong in ten (56%). In small cells, the staining was positive in all tumors (from 10–100%, mean 74%) and was weak in seven (37%), moderate in seven (37%), and strong in four (21%). The mean percentage of stained large cells was higher than that of small cells (97% versus 74%, respectively). Larger cells exhibited a higher CS score (mean ratio small/large = 0.59). Staining on large cells often exhibited combined cytoplasmic and membranous staining, whereas the staining was mostly membranous and cup shaped on small cells. In one patient with two tumors (case #12), a higher MET expression level in large cells compared with small cells (CS 160 and 300 versus 100 and 180, respectively) was observed on both tumors.

a example of the three intensity scoring system (score 1—score 2—score 3) (×20 magnification); b representative spots of three cases with MET copy number gain without MET mutation (patient #1, #3, #10), three cases with both MET copy number gain and MET mutation (patient #2, #4, #11), and two cases with unknown mutation status: one with MET copy number gain (patient #9) and one without MET copy number gain (patient #8) (×2.5 magnification).
Integrative analysis
A correlation between MET expression and MET genetic alterations was performed (Table 3). The two cases without trisomy 7 (#8 and #13) had lower CS scores on both small cells (CS < 50) (respectively 30 and 10) and large cells (CS ≤ 100) (respectively 80 and 100) compared with CS of tumors with trisomy 7. In cases without trisomy 7, the lower CS observed in large cells was mostly due to a lower intensity of staining (intensity 1) compared with those with trisomy 7 (intensity 2 or 3) (Supplementary Table 2). However, no difference was observed in MET expression on both large and small cells when compared to the mutational status of MET.
Discussion
This study reports a large cohort of BSA-PRCC allowing to enrich the knowledge about this rare and recently studied lesion considered a morphological variant of RCC, providing more clinicopathological data from 17 patients and additional evidence to consider BSA-PRCC as a variant of type 1 papillary RCC. Main features of BSA-PRCC and type 1 papillary RCC are summarized in Table 4. In our cohort, we confirmed the presence of trisomy 7 leading to MET copy number gain and highlighted for the first time the high incidence of MET mutation suggesting that MET plays a major role in the oncogenesis of this peculiar morphological variant. Pathological features were comparable to previous descriptions [3,4,5,6,7,8] with the same immunophenotype CK7 and AMACR positivity on both cellular type and BCL1 restricted to larger cells. The surface represented by the biphasic component was variable, ranging from 5 to 100% with a lower mean compared to previous cohorts (20% rather than 58%) [6]. The repartition of biphasic appearance was heterogeneous among cases as well as the size of clusters of large eosinophilic cells that could vary from small to larger ones. Emperipolesis was constantly observed in each case but not in each cluster of large eosinophilic cells. Emperipolesis is a peculiar biological event morphologically described as penetration of intact hematopoietic cells into another intact cell [26]. In contrast to BSA-PRCC, the occurrence of emperipolesis is rare in other RCC and has been recently described in 46% of clear cell RCC with a syncytial-type multinucleated giant tumor cell component [27] and in rare high-grade RCC [28]. In a recent case report of BSA-PRCC, this particular image has been interpreted as cytophagocytosis of apoptotic neutrophils rather than emperipolesis [29].
Clinical features were characterized by a slight male predominance and a mean age inferior to that reported in prior series (51.6 versus 58.6 years), together with a higher proportion of patients diagnosed before 50 years old (47% versus 10.9%) [3, 4, 6]. As previously underlined, most cases were localized tumors. However, we also have identified three patients (17.6%) presented with aggressive tumors, including two locally advanced tumors and one with grade 4 due to sarcomatoid areas. This patient subsequently developed metastases and failed to respond to anti-VEGFR and anti-PD1 therapy. Combined with cases previously described in the literature [5, 6], metastases occurred in 9.4% (7/74) of all BSA-PRCC, which is comparable to the incidence observed in type 1 papillary RCC (7%) but lower than in type 2 papillary RCC (30%) [30]. This metastatic potential underlines the need for finding biomarkers predicting response to targeted therapy. Our results also confirmed the high incidence of multifocal tumors (28%), including either patients with multiple BSA-PRCC (3/18) or patients with BSA-PRCC associated with papillary RCC (2/18) or other tumors (1/18).
While the role of MET alterations in type 1 papillary RCC is well-known, our study is the first one to decipher MET status in a large cohort of BSA-PRCC. Trisomy 7, including MET locus, leads to an increase of MET copy number [5]. In our series, no MET amplification was identified, but chromosome 7 trisomy was found by FISH analysis in 87.5% (14/16) of cases, showing that this is a frequent, but not constant, hallmark in BSA-PRCC. As reported in papillary RCC, MET gains in BSA-PRCC are most likely due to chromosome 7 copy number gain, rather than a gene amplification mechanism [31].
The association with trisomy 7 and 17 supports the view that BSA-PRCC is a distinct morphological variant of papillary RCC [5, 6]. Even if the frequency of trisomy 7 is variable among series of papillary RCC (ranged from 31 to 81%) [13, 20], the incidence of trisomy 7 in BSA-PRCC is closer to the one reported in type 1 papillary RCC (81%) than in type 2 papillary RCC (46%) [20, 32].
Germline MET pathogenic variants were identified in 57% (4/7) of tested patients. This high germline mutation rate could be overestimated due to the limited number of cases with a sporadic presentation benefiting from a germline genotyping in our cohort. These results should be confirmed in a larger cohort, especially in patients with sporadic presentation of BSA-PRCC. Interestingly, we reviewed the figure available in a recent case report of hereditary type 1 papillary RCC and identified morphological features consistent with the diagnosis of BSA-PRCC [33]. The incidence of MET germline mutation justifies that a germline MET analysis should be offered to patients with BSA-PRCC, especially in the context of associated papillary RCC, multiple tumors and/or family history of RCC. Indeed, the four patients had either multiple BSA-PRCC (3/4) or multiple papillary RCC type 1 (1/4). Two of them had a familial history of RCC and occurred before the age of 50. The germline mutational status is essential for guiding genetic counseling, which indicates or exempts specific management and follow-up as well as predictive genetic testing in relatives. Unfortunately, in the patient who developed BSA-PRCC, papillary RCC, and oncocytomas, suggesting a possible Birt–Hogg–Dubé syndrome as previously identified [6], the germline analysis could not be performed.
The tumor DNA analysis allowed finding a somatic MET variant in 6 out of 11 cases (54%), including 5 pathogenic variants (45%) and 1 variant of unknown significance. The somatic origin of the mutation was confirmed in three cases (absent in the corresponding germline DNA) and supposed in two cases, due to an allelic ratio <0.5. The c.3389T>C p.(Leu1130Ser) variant was not previously reported, explaining why it was classified as variation of unknown significance. Its allelic ratio in tumor DNA was >0.5, suggesting its presence in the germline DNA, but the analysis was not performed in the leukocyte DNA.
It is noteworthy that finding the same MET somatic mutation in multiple tumors of one patient, together with an allelic ratio of >0.5, suggests that the mutation’s origin is germline and that patient should seek genetic counseling.
Altogether, the mutation rate in the MET tyrosine-kinase catalytic domain was 60% (9/15) in our cohort of BSA-PRCC. This frequency is higher than in the TCGA cohort of papillary RCC, in which a MET mutation was found in 17% of type 1 papillary RCC, 2% of type 2, and 12% of unclassified papillary RCC [18]. Similarly, smaller cohorts of type 1 papillary RCC have identified MET mutations from 17% (4/23) [34] to 21.6% (11/51) [20]. Interestingly, we reviewed the available digital slides for all patients from the KIRP cohort with MET mutation, and three had morphologic features compatible with BSA-PRCC (TCGA-A4-7734/AL-3472/ MH-A561) [35]. In contrast to BSA-PRCC, no MET mutation has been identified in oncocytic papillary RCC, another rare morphological variant of papillary RCC with inconstant trisomy 7 but high expression of MET on IHC [36].
The high incidence of chromosome 7 gains and MET mutations in BSA-PRCC cases strongly support the idea that MET activation plays an essential role in the oncogenesis of BSA-PRCC.
MET expression has been poorly investigated in papillary RCC. It has been shown that increased expression of MET can be due to different mechanisms involving mutation, translocation, or copy number gain of chromosome 7 [18]. RNAseq analysis showed that MET expression was higher in type 1 papillary RCC than type 2, and was overexpressed in papillary RCC compared to normal kidneys or clear cell RCC [20]. Using IHC, a large study of MET expression performed in 145 sporadic RCC including 20 papillary RCC showed that MET expression was strong and diffuse (2+) in 90% of papillary RCC and expressed in 92 and 86% in type 1 and type 2 papillary RCC, respectively [37]. In our study, MET was expressed in all cases of BSA-PRCC, in both large and small cells with a stronger expression on large cells. The expression of MET failed to predict MET mutation in BSA-PRCC. However, we interestingly noted that in the two cases displaying the lower MET expression on both small cells (CS < 50) and large cells (CS ≤ 100), trisomy 7 was absent.
Finally, the main interest of the analysis of MET status in tumors is its implication for therapy. Indeed, the MET pathway is now being targeted by multiple novel agents in clinical development in various cancers [38]. In nonclear cell RCC patients, savolitinib (a highly selective MET TKI), crizotinib (TKI targeting c-MET, ROS1, and ALK), and cabozantinib (TKI targeting Vascular Endothelial Growth Factor Receptor-2, AXL, and MET) showed a clinically meaningful benefit in phase II/III trials or retrospective studies [34, 39,40,41]. It has been shown that MET alterations influence sensitivity to MET inhibition; thus, detection of MET alterations could be used as a predictive biomarker and improve the accurate selection of eligible patients for anti-MET drugs. For example, in the phase II trial evaluating savolitinib in papillary RCC patients, MET-driven tumors were predictive of partial response compared to MET-independent tumors (18% versus 0%), and were associated with a significantly better median progression-free survival (PFS) (6.2 months versus 1.4 months) [39]. In the CREATE trial (EORTC 90101), the 4 MET-driven type 1 papillary RCC patients reported better overall response rate (ORR) and 1-year PFS rate than the 16 MET-independent type 1 papillary RCC patients (ORR of 50% versus 6.3% and 1-year PFS rate of 75% versus 27%, respectively) [34]. Considering the high incidence of MET alterations in BSA-PRCC found in this study, it suggests that patients with aggressive BSA-PRCCs should be tested for MET mutation in order to evaluate their predictive value in response to anti-MET therapies.
In conclusion, our study provides more evidence to classify BSA-PRCC as a variant of type 1 papillary renal RCC and expands the subtype of RCC that could be encountered in hereditary papillary RCC disease. Our data strongly suggest that MET represents a major oncogenic driver gene in BSA-PRCC. This finding paves the way to explore the benefice of anti-MET targeted therapies for aggressive BSA-PRCC.
References
Moch H, Humphrey P, Ulbright T, Reuter V. WHO classification of tumours of the urinary system and male genital organs. 4th ed. Lyon: IARC Press; 2016.
Petersson F, Bulimbasic S, Hes O, Slavik P, Martínek P, Michal M, et al. Biphasic alveolosquamoid renal carcinoma: a histomorphological, immunohistochemical, molecular genetic, and ultrastructural study of a distinctive morphologic variant of renal cell carcinoma. Ann Diagn Pathol. 2012;16:459–69.
Zhou L, Xu H, Zhou Y, Zhou J, Zhang P, Yang X, et al. Biphasic squamoid alveolar renal carcinoma with positive CD57 expression: a clinicopathologic study of three cases. Pathol Int. 2019;69:519–25.
Troxell ML, Higgins JP. Renal cell carcinoma in kidney allografts: histologic types, including biphasic papillary carcinoma. Hum Pathol. 2016;57:28–36.
Hes O, Condom Mundo E, Peckova K, Lopez JI, Martinek P, Vanecek T, et al. Biphasic squamoid alveolar renal cell carcinoma. Am J Surg Pathol. 2016;40:664–75.
Trpkov K, Athanazio D, Magi-Galluzzi C, Yilmaz H, Clouston D, Agaimy A, et al. Biphasic papillary renal cell carcinoma is a rare morphological variant with frequent multifocality: a study of 28 cases. Histopathology. 2018;72:777–85.
Chartier S, Méjean A, Richard S, Thiounn N, Vasiliu V, Verkarre V. Biphasic squamoid alveolar renal cell carcinoma: 2 cases in a family supporting a continuous spectrum with papillary type I renal cell carcinoma. Am J Surg Pathol. 2017;41:1011–2.
López JI. Case report: multifocal biphasic squamoid alveolar renal cell carcinoma. F1000Research. 2016;5:607.
Renshaw AA, Zhang H, Corless CL, Fletcher JA, Pins MR. Solid variants of papillary (chromophil) renal cell carcinoma: clinicopathologic and genetic features. Am J Surg Pathol. 1997;21:1203–9.
Mantoan Padilha M, Billis A, Allende D, Zhou M, Magi-Galluzzi C. Metanephric adenoma and solid variant of papillary renal cell carcinoma: common and distinctive features. Histopathology. 2013;62:941–53.
Ulamec M, Skenderi F, Trpkov K, Kruslin B, Vranic S, Bulimbasic S, et al. Solid papillary renal cell carcinoma: clinicopathologic, morphologic, and immunohistochemical analysis of 10 cases and review of the literature. Ann Diagn Pathol. 2016;23:51–7.
Klatte T, Pantuck AJ, Said JW, Seligson DB, Rao NP, LaRochelle JC, et al. Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma. Clin Cancer Res. 2009;15:1162–9.
Pitra T, Pivovarcikova K, Alaghehbandan R, Hes O. Chromosomal numerical aberration pattern in papillary renal cell carcinoma: review article. Ann Diagn Pathol. 2019;40:189–99.
Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med. 2005;353:2477–90.
Carlo MI, Hakimi AA, Stewart GD, Bratslavsky G, Brugarolas J, Chen YB, et al. Familial kidney cancer: implications of new syndromes and molecular insights. Eur Urol. 2019;76:754–64.
Verkarre V, Morini A, Denize T, Ferlicot S, Richard S. Hereditary kidney cancers: the pathologist’s view in 2020. Ann Pathol. 2020;40:148–67.
Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73.
The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–45.
Durinck S, Stawiski EW, Pavía-Jiménez A, Modrusan Z, Kapur P, Jaiswal BS, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2015;47:13–21.
Albiges L, Guegan J, Le Formal A, Verkarre V, Rioux-Leclercq N, Sibony M, et al. MET Is a potential target across all papillary renal cell carcinomas: result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin Cancer Res. 2014;20:3411–21.
Pal SK, Ali SM, Yakirevich E, Geynisman DM, Karam JA, Elvin JA, et al. Characterization of clinical cases of advanced papillary renal cell carcinoma via comprehensive genomic profiling. Eur Urol. 2018;73:71–8.
Fischer J, Palmedo G, Von Knobloch R, Bugert P, Prayer-Galetti T, Pagano F, et al. Duplication and overexpression of the mutant allele of the MET proto-oncogene in multiple hereditary papillary renal cell tumours. Oncogene. 1998;17:733–9.
Zhuang Z, Park WS, Pack S, Schmidt L, Vortmeyer AO, Pak E, et al. Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat Genet. 1998;20:66–9.
Rochigneux P, Thomassin-Piana J, Laibe S, Brunelle S, Salem N, Escudier B, et al. Long-term efficacy of crizotinib in a metastatic papillary renal carcinoma with MET amplification: a case report and literature review. BMC Cancer. 2018;18:1159.
Lalani A-KA, Gray KP, Albiges L, Callea M, Pignon J-C, Pal S, et al. Differential expression of c-Met between primary and metastatic sites in clear-cell renal cell carcinoma and its association with PD-L1 expression. Oncotarget. 2017;8:103428–36.
Gupta N, Jadhav K, Shah V. Emperipolesis, entosis and cell cannibalism: demystifying the cloud. J Oral Maxillofac Pathol. 2017;21:92–8.
Williamson SR, Kum JB, Goheen MP, Cheng L, Grignon DJ, Idrees MT. Clear cell renal cell carcinoma with a syncytial-type multinucleated giant tumor cell component: Implications for differential diagnosis. Hum Pathol. 2014;45:735–44.
Rotterova P, Martinek P, Alaghehbandan R, Prochazkova K, Damjanov I, Rogala J, et al. High-grade renal cell carcinoma with emperipolesis: clinicopathological, immunohistochemical and molecular-genetic analysis of 14 cases. Histol Histopathol. 2018;33:277–87.
Suárez-Vilela D, Izquierdo FM, Méndez-Álvarez JR, Velasco-Alonso J. The squamoid cells in biphasic squamoid alveolar renal carcinoma present cytophagocytosis (not emperipolesis) of apoptotic neutrophilic granulocytes. Am J Surg Pathol. 2018;42:420–2.
Pignot G, Elie C, Conquy S, Vieillefond A, Flam T, Zerbib M, et al. Survival analysis of 130 patients with papillary renal cell carcinoma: prognostic utility of type 1 and type 2 subclassification. Urology. 2007;69:230–5.
Yin X, Zhang T, Su X, Ji Y, Ye P, Fu H, et al. Relationships between chromosome 7 gain, MET gene copy number increase and MET protein overexpression in Chinese papillary renal cell carcinoma patients. PLoS ONE. 2015;10:1–10.
Brunelli M, Eble JN, Zhang S, Martignoni G, Cheng L. Gains of chromosomes 7, 17, 12, 16, and 20 and loss of Y occur early in the evolution of papillary renal cell neoplasia: a fluorescent in situ hybridization study. Mod Pathol. 2003;16:1053–9.
Mikhaylenko DS, Klimov AV, Matveev VB, Samoylova SI, Strelnikov VV, Zaletaev DV, et al. Case of hereditary papillary renal cell carcinoma type I in a patient with a germline MET mutation in Russia. Front Oncol. 2020;9:1–6.
Schöffski P, Wozniak A, Escudier B, Rutkowski P, Anthoney A, Bauer S, et al. Crizotinib achieves long-lasting disease control in advanced papillary renal-cell carcinoma type 1 patients with MET mutations or amplification. EORTC 90101 CREATE trial. Eur J Cancer. 2017;87:147–63.
cBioPortal for Cancer Genomics. https://www.cbioportal.org/study/summary?id=kirp_tcga.
Xia Q, Rao Q, Shen Q, Shi S-S, Li L, Liu B, et al. Oncocytic papillary renal cell carcinoma: a clinicopathological study emphasizing distinct morphology, extended immunohistochemical profile and cytogenetic features. Int J Clin Exp Pathol. 2013;6:1392–9.
Choi JS, Kim M-K, Seo JW, Choi Y-L, Kim DH, Chun YK, et al. MET expression in sporadic renal cell carcinomas. J Korean Med Sci. 2006;21:672.
Peltola KJ, Penttilä P, Rautiola J, Joensuu H, Hänninen E, Ristimäki A, et al. Correlation of c-Met expression and outcome in patients with renal cell carcinoma treated with sunitinib. Clin Genitourin Cancer. 2017;15:487–94.
Choueiri TK, Plimack E, Arkenau H-T, Jonasch E, Heng DYC, Powles T, et al. Biomarker-based phase II trial of savolitinib in patients with advanced papillary renal cell cancer. J Clin Oncol. 2017;35:2993–3001.
Campbell MT, Bilen MA, Shah AY, Lemke E, Jonasch E, Venkatesan AM, et al. Cabozantinib for the treatment of patients with metastatic non-clear cell renal cell carcinoma: a retrospective analysis. Eur J Cancer. 2018;104:188–94.
Martínez Chanzá N, Xie W, Asim Bilen M, Dzimitrowicz H, Burkart J, Geynisman DM, et al. Cabozantinib in advanced non-clear-cell renal cell carcinoma: a multicentre, retrospective, cohort study. Lancet Oncol. 2019;20:581–90.
Acknowledgements
We wanted to thank Ivana Stankovic and Sophie Hajosi from the Department of Pathology, Georges Pompidou European Hospital for technical support, Marine Largeau, and Cécile Badoual, MD, PhD, from the Biological Resources Center and Tumor Bank Platform (BB-0033-00063) of Georges Pompidou European Hospital for providing biological samples.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Denize, T., Just, P.A., Sibony, M. et al. MET alterations in biphasic squamoid alveolar papillary renal cell carcinomas and clinicopathological features. Mod Pathol 34, 647–659 (2021). https://doi.org/10.1038/s41379-020-0645-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0645-6
This article is cited by
-
Genetically driven predisposition leads to an unusually genomic unstable renal cell carcinoma
Discover Oncology (2024)
-
Biphasic papillary (biphasic squamoid alveolar) renal cell carcinoma: a clinicopathologic and molecular study of 17 renal cell carcinomas including 10 papillary adenomas
Virchows Archiv (2024)
-
New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia
Modern Pathology (2021)
