
Abstract
While the genomics of BRAF, NRAS, and other key genes influencing MAP kinase (MAPK) activity have been thoroughly characterized in melanoma, mutations in MAP2K1 (MEK1) have received significantly less attention and have consisted almost entirely of missense mutations considered secondary oncogenic drivers of melanoma. Here, we investigated melanomas with in-frame deletions of MAP2K1, alterations characterized as MAPK-activating in recent experimental models. Our case archive of clinical melanoma samples with comprehensive genomic profiling by a hybrid capture-based DNA sequencing platform was searched for MAP2K1 genetic alterations. Clinical data, pathology reports, and histopathology were reviewed for each case. From a cohort of 7119 advanced melanomas, 37 unique cases (0.5%) featured small in-frame deletions in MAP2K1. These included E102_I103del (n = 11 cases), P105_A106del (n = 8), Q58_E62del (n = 6), I103_K104del (n = 5), I99_K104del (n = 3), L98_I103del (n = 3), and E41_F53del (n = 1). All 37 were wild type for BRAF, NRAS, and NF1 genomic alterations (“triple wild-type”), representing 2.0% of triple wild-type melanomas overall (37/1882). Median age was 66 years and 49% were male. The majority arose from primary cutaneous sites (35/37; 95%) and demonstrated a UV signature when available (21/25; 84%). Tumor mutational burden was typical for cutaneous melanoma (median = 9.6 mut/Mb, range 0–35.7), and frequently mutated genes included TERTp (63%), CDKN2A (46%), TP53 (11%), PTEN (8%), APC (8%), and CTNNB1 (5%). Histopathology revealed a spectrum of appearances typical of melanoma. For comparison, we evaluated 221 cases with pathogenic missense single nucleotide variants in MAP2K1. The vast majority of melanomas with missense SNVs in MAP2K1 showed co-mutations in BRAF (58%), NF1 (23%), or NRAS (18%). In-frame deletions in MAP2K1, previously shown in experimental models to be strongly MAPK-activating, characterized a significant subset of triple wild-type melanoma (2.0%), suggesting a primary oncogenic role for these mutations. Comprehensive genomic profiling of melanomas enables detection of this alteration, which may have implications for potential therapeutic options.
Similar content being viewed by others

Introduction
Most melanomas harbor driver mutations which promote activation of the MAP Kinase pathway, most frequently through mutations in BRAF, NRAS, or NF1 [1,2,3,4]. Increasingly, gene fusions involving various kinases, including ALK, ROS1, BRAF, RAF1, NTRK1, and NTRK3 have been identified within melanomas, and these fusion-positive melanomas often display spitzoid morphology [5,6,7,8,9]. In addition to these primary driver mutations, various secondary oncogenic driver mutations have been identified, including gain of function alterations, such as TERT promoter mutation and CDK4 amplification, and loss of function alterations, such as CDKN2A, TP53, and PTEN mutations [2].
Melanomas also contain MAP2K1 (MEK1) mutations, with prior reports consisting almost entirely of missense mutations characterized as secondary oncogenic driver mutations [2, 10, 11]. Most reported MAP2K1 mutations have accompanied other driver mutations of melanoma including BRAF, NRAS, and NF1 [4, 10,11,12]. Prompted by recent studies of mutual exclusivity among BRAF mutation and MAP2K1 mutation in Langerhans cell histiocytosis (LCH) [13,14,15], as well as work showing distinct functional classes of MAP2K1 mutations [16], we probed a database of clinical specimens with comprehensive genomic profiles for the relationship between MAP2K1 mutations and other driver mutations in melanoma. Herein, we report a series of melanomas with MAP2K1 in-frame deletions distinct from other driver mutations of melanoma.
Materials and methods
Cohort and genomic analyses
Cases evaluated for this study had undergone comprehensive genomic profiling (CGP) performed in a Clinical Laboratory Improvement Amendments-certified, College of American Pathologists-accredited laboratory (Foundation Medicine, Inc., Cambridge, MA, USA). The Western Institutional Review Board granted approval for this study (Protocol No. 20152817), including a waiver of informed consent and a HIPAA waiver of authorization. Briefly, hematoxylin and eosin (H&E)-stained histologic slides were reviewed to verify the presence of diagnostic lesional tissue. From formalin-fixed, paraffin-embedded tissue blocks, ≥60 ng of DNA was extracted from 40 μm sections. The samples were assayed by CGP using adapter ligation, and hybrid capture was performed for exons from 287 (version 1) to 315 (version 2) cancer-related genes plus select introns from 19 (version 1) to 28 (version 2) genes frequently rearranged in cancer (Supplemental Table 1). Sequences were analyzed for all classes of genomic alterations including short variant alterations (base substitutions, insertions, and deletions), copy number alterations (focal amplifications and homozygous deletions), and select gene fusions or rearrangements, by methods previously described [17,18,19]. A somatic-germline-zygosity computational method was applied for somatic, germline, and artifact determination; the method fit an optimal copy number model to the log-ratio and minor allele frequency data and compared the observed variant allele frequency against that of the model’s expected variant allele frequency [18]. Tumor mutational burden (TMB, mutations/Mb) was determined on 0.8–1.1 Mbp of sequenced DNA [19]. Microsatellite instability (MSI) was determined on up to 114 loci [20].
Cell-free circulating tumor DNA (ctDNA) was evaluated on 315 blood specimens (“liquid biopsy”) collected from 307 patients with clinical history of melanoma using the hybrid capture-based Illumina Hi-Seq technology. Maximum somatic allele frequency was used to estimate the fraction of ctDNA, per methods previously described [21, 22].
Mutational signatures
For all samples containing at least 20 nondriver somatic missense alterations, mutational signatures were assessed through analysis of the trinucleotide context and profiling by Sanger COSMIC signatures of cancer mutational processes [23]. A positive signature was designated for samples with at least a 40% fit to a mutational process [23]. The COSMIC UV signature is characterized by C>T and CC>TT base substitutions at dypirimidine sites [24].
Clinical-pathological analysis of melanoma cohort harboring in-frame deletions in MAP2K1
From a total of 7119 consecutive routine clinical melanoma specimens, each from a different patient, the cohort of melanomas harboring in-frame deletions in MAP2K1 comprised 37 cases. Samples which underwent assays with CGP (Foundation Medicine, Cambridge, MA, USA) were collected from patients receiving clinical care at other institutions. Clinicopathological data including patient age, gender, tumor site, tumor diameter, and stage were extracted from accompanying pathology reports.
H&E stained sections from each of the 37 cases were assessed retrospectively by two board-certified dermatopathologists (JYT, MCM) and an additional board-certified anatomic pathologist (EAW). Histologic parameters assessed on primary tumors included configuration at low magnification, symmetry, presence and patterns of epidermal involvement, ulceration, pattern of dermal growth, Breslow depth, mitotic rate, grade of solar elastosis [25], and tumor infiltrating lymphocytes. Primary and metastatic cases were assessed for cytologic features, including predominant cytomorphology (epithelioid vs. spindled), cytoplasmic color and abundance, chromatin quality, nucleolar prominence, and degree of pleomorphism. Accompanying pathology reports were utilized for diagnostically corroborating details, including immunohistochemical findings and melanoma history.
Quantitative data were analyzed using the Fisher exact test owing to the categorical quality of the data and the size of the cohort. For TMB comparison between two groups, the nonparametric Mann–Whitney U test was used. A two‐tailed P value of <0.05 was considered to be statistically significant; the Bonferroni correction was applied for multiple simultaneous comparisons.
Results
Clinical-pathologic features
Of 7119 melanomas with prior hybrid capture-based DNA sequencing, 37 distinct cases (0.5%) featured small in-frame deletions that resulted in known or likely activation of MAP2K1. Among patients with MAP2K1 in-frame deleted melanomas, the ages ranged from 28 to 89 years, with a median of 66 years. There were 18 males and 19 females. Age and gender did not show significant differences compared with the melanoma cohort overall. Nearly all cases were clinically advanced: most cases were documented at stage IV (n = 25 of 37; 68%), with the majority of the remaining cases documented at stage IIIA-C (n = 10 of 37; 27%). The other two cases were stage IIA and IIC.
Sequencing was performed on four primary cutaneous melanomas (Table 1), three primary site recurrences, and 30 metastatic disease samples. Of the metastatic samples, sites included regional lymph nodes (n = 5), in-transit metastasis (n = 1), and distant lymph nodes (n = 6). Additional distant metastatic sites included skin (subcutaneous [n = 6], dermal [n = 2]), lung (n = 5), brain (n = 2), and one each involving liver, omentum, and pericardium. Thirty-five cases were consistent with either primary cutaneous melanoma or metastatic melanoma from a cutaneous primary, while two cases were of unknown primary site.
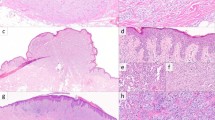
Histopathologic evaluation of melanomas revealed variable morphologies. Of the four primary cutaneous melanomas (excluding the primary site recurrences), two were superficial spreading type, and two were nodular type (Fig. 1). All primary tumors showed exophytic nodular components with irregular nested growth at the base and marked asymmetry (Fig. 1a, d). All showed epidermal involvement, with the two superficial spreading melanomas showing extension beyond the dermal component (Fig. 1b, e). Two lesions were ulcerated, three showed confluent growth along the basal epidermis with rete effacement, three showed pagetoid growth, and two had expansile intraepidermal nests (Fig. 1b). Dermal components consisted of variably sized nodules and nests in all cases (Fig. 1f), and one case contained elongated nests in some foci (Fig. 1c). Tumor infiltrating lymphocytes were present and non-brisk in all cases. As defined by the World Health Organization [25], solar elastosis scores were 0 in one case, 2 in two cases, and 3 in one case. Cytomorphologically, three were epithelioid and one was an admixture of epithelioid and spindled melanocytes. Melanocytes had variable amounts of cytoplasm which was amphophilic in three cases and densely eosinophilic in one case. Only one case showed cytoplasmic pigmentation, which was focal. Nuclear size was medium to large, chromatin was heterogenous (of varying density), nucleoli were variably prominent, and nuclear pleomorphism was moderate to severe.

a Histopathologic examination of case 1 reveals a polypoid proliferation of melanocytes with a slight epidermal collarette (H&E, ×20). b, c Case 1 shows expansile nested growth along the epidermis with associated epidermal effacement, and elongated nests in the deep aspect. d Case 3 is a melanocytic tumor with plaque-like and tumorigenic components (H&E, ×200). e The radial growth phase of Case 3 shows a confluent proliferation of atypical melanocytes along the basal epidermis (H&E, ×200). f The deep aspect of the tumorigenic component consists of nodules of atypical epithelioid cells (H&E, ×200). Metastatic lesions showed predominantly epithelioid cytomorphology (g), with occasional cases displaying spindled (h), and rhabdoid morphology (i). (H&E, ×400).
Among the 33 recurrent and metastatic tumors, 28 cases (85%) showed epithelioid cytomorphology (Fig. 1g), while five (15%) had mixed epithelioid and spindled cells (Fig. 1h). Among the epithelioid cases, three had rhabdoid cytology. Of note, each case with rhabdoid cytology was MAP2K1 Q58_E62del mutant (Fig. 1i). Cytoplasm tended to be moderate to abundant in amount, and only seven cases (21%) contained cytoplasmic melanin which was focal in five cases and diffuse in two. Nuclear size was medium to large, and 13 cases (39%) had prominent nucleoli, with the remaining cases showing small to indistinct nucleoli.
Comprehensive genomic profiling
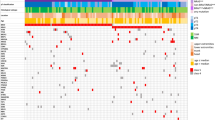
MAP2K1 in-frame deletions included E102_I103del (n = 11 cases), P105_A106del (n = 8), Q58_E62del (n = 6), I103_K104del (n = 5), I99_K104del (n = 3), L98_I103del (n = 3), and E41_F53del (n = 1) (Fig. 2a). Most deletions involved the portion of the gene corresponding to the kinase domain [16]. All 37 were wild type for BRAF, NRAS, and NF1 genomic alterations (“triple wild-type”) (Fig. 2b), representing 2.0% of triple wild-type melanomas overall (37/1882) and showing significant enrichment for the triple wild-type subcategory compared with the melanoma cohort overall (p < 0.0001, Fisher’s exact test). In melanomas with in-frame deletions, median age was 66 years, 51% were female, and the large majority were confirmed by history to be cutaneous primary (35/37; 95%) and demonstrated a UV signature (21/25; 84%) when available. TMB was typical for cutaneous melanoma (median = 9.6 mut/Mb, range 0–35.7). The most frequent other genes with known or likely pathogenic alterations identified included TERTp (63%), CDKN2A (46%), TP53 (11%), PTEN (8%), APC (8%), ERBB4 (8%), MITF (8%), CTNNB1 (5%), and MYC (5%) (Fig. 2b, Supplemental Table 4).

a Schematic of functional domains of MAP2K1 (transcript NM_002755), to include number of cases with each in-frame deletion (black bars; length~number of cases) and sites of pathogenic missense mutations (black arrows). The majority of in-frame deletions are present in the catalytic core. b Summary of clinical features and molecular alterations in MAP2K1 in-frame deletion melanomas. NES nuclear export signal, NRR negative regulatory region, ATP ATP binding site, Pro rich proline-rich domain, TERTp TERT promoter.
Comparison of tumors sequenced from the primary sites, regional metastases, and distant metastases was performed. Tumors sequenced from primary and regional sites (n = 13) versus distant metastases (n = 24) showed similar percentages of genomic alterations, including in TERTp, CDKN2A, TP53, and PTEN (54% vs. 68%, 39% vs. 50%, 15% vs. 8%, and 15% vs. 4%, respectively).
We identified 327 melanomas (4.6% of the entire melanoma cohort) harboring a missense single nucleotide variant (SNV) mutation in MAP2K1. Of these cases, 221 (3.1% of the melanoma cohort) contained a known or likely pathogenic missense SNV, including E203K (n = 58), P124L (n = 56), P124S (n = 45), D67N (n = 12), C121S (n = 10), F53L (n = 8), Q56P (n = 6), F53Y (n = 6), and K57E (n = 4) (Fig. 2a). In contrast to cases with in-frame deletions (Table 2), melanomas with pathogenic missense SNVs in MAP2K1 contained frequent co-mutation in BRAF (58%), NF1 (23%), and NRAS (18%), as well as TERTp (80%), CDKN2A (60%), and TP53 (23%) (Supplemental Fig. 1). Median age was 62 years, 67% were male, and most demonstrated a UV signature (165/198; 83%) when available. TMB was elevated (median = 25.2 mut/Mb). Notably, only 16% were triple wild-type (35/221) (Supplemental Fig. 1). These cases featured occasional additional pathogenic genomic alterations in genes in RAS signaling activation, including ARAF missense mutations (n = 2), activating RAF1 fusion (n = 2), MAPK1 amplification (n = 1), and KIT amplification (n = 1). Three of the four cases with two pathogenic MAP2K1 alterations were triple wild-type; the remaining case harbored a BRAF L597R mutation.
Other genomic alterations of MAP2K1 were rare in the internal series of melanomas. Seven cases with various in-frame MAP2K1 indels were identified: Q45_F53>L, Q46_T55>H, Q46_K59>L, Q46_E62>L, F53_Q58>L, Q56_G61>R, and L98_K104>M. All cases were of known cutaneous primary. One case had a concurrent BRAF L597R mutation, and one case had two truncating NF1 mutations (NF1 R440* and NF1 W1662*).
Three cases had MAP2K1 amplification, each with copy number ranging from 8 to 10. One case was sequenced from primary cutaneous site, one from colon metastasis of cutaneous primary, and another from liver metastasis of unknown primary site. One case had a concurrent BRAF V600E mutation, another had NRAS amplification, and the last had two truncating NF1 mutations (NF1 L1480fs*2 and NF1 Y1783*). No pathogenic large structural rearrangements of MAP2K1 were identified in any case.
Four cases harbored a truncating mutation in MAP2K1 (Q10*, Q153*, V173fs*2, and Y300*) and three cases had splice site variants in MAP2K1 (Splice Site 1023–1G>A, Splice Site 1068+1G>A, and Splice Site 961–1G>A). Each of these variants of uncertain significance occurred in BRAF, NRAS, or NF1 mutant melanomas.
ctDNA was evaluated from blood specimens (“liquid biopsies”) in a cohort of 307 melanoma patients. Genomic alterations in MAP2K1 were identified in five patients (Supplemental Table 2). A single case of stage IV melanoma of unknown primary was identified to have an in-frame deletion in MAP2K1 (E102_103del) at allele frequency (AF) of 3.85%.
Discussion
While previously reported SNVs of MAP2K1 in melanoma have been characterized as secondary oncogenic drivers typically paired with common driver mutations of the MAPK pathway, MAP2K1 deletions, as described here, delineate a unique subset of triple wild-type melanoma (2.0% of all triple wild-type melanoma in this cohort). We propose that the lack of co-mutations in MAPK-activating oncogenic drivers, including those involving RAF, RAS, and NF1, and prior studies characterizing particular functional subtypes of MAP2K1 mutation, support MAP2K1 in-frame deletions as defining for a distinct subgroup of melanomas, with potential therapeutic implications.
Several in vitro and in vivo studies have demonstrated that MAP2K1 in-frame deletions, identical to those in this study, promote activation of the MAP kinase pathway [15, 16, 26,27,28] (Supplemental Table 3). In particular, recent mechanistic studies by Gao et al. have characterized three distinct classes of mutations in MAP2K1 (MEK1): Class 1 mutants depend on phosphorylation by RAF enzyme for activity, Class 2 mutants show continuous activity independent from RAF stimulation but may be further activated by RAF, and Class 3 mutants demonstrate activity in an entirely RAF-independent manner [16]. Class 1 (RAF dependent) MAP2K1 mutations consist of various point mutations, Class 2 (RAF regulated) mutations consist mostly of point mutations in various positions as well as deletions spanning the 51–58 positions encoding the negative regulatory region, and Class 3 (RAF independent) mutations consist of deletions involving the 98–104 positions encoding the kinase domain [16]. While the majority of Class 1 (RAF dependent) MAP2K1 mutations are associated with co-mutation in BRAF, NRAS, or NF1, only a small proportion of class 2 (RAF regulated) cases and no class 3 (RAF regulated) cases share these co-mutations in a limited pan-cancer analysis [16].
Most of the melanomas with MAP2K1 in-frame deletion in our study, with the exception of 6 cases with Q58_E62del, involve the kinase domain locus corresponding to Class 3 (RAF independent) MAP2K1 mutations. Analogous to the reports from Gao et al., none of our cases with deletions had co-mutation involving RAF, RAS, or NF1 proteins [16]. This close correlation with prior functional studies and the independence from other MAPK driver mutations support the concept that MAP2K1 in-frame deletions characterize a distinct subtype of triple wild-type melanoma.
Previously reported genomic alterations in MAP2K1 in melanoma have consisted almost entirely of point mutations, with only a single report of in-frame deletion [12], and generally have been characterized as secondary driver mutations. Hodis et al. reported that, out of 121 melanomas, seven cases had MAP2K1 mutations, composed of point mutations and a single in-frame deletion; while four co-occurred with BRAF mutation, one co-occurred with NRAS mutation, and two occurred independently of BRAF, NRAS, and NF1 mutation, co-mutations were not specified by MAP2K1 mutation type [12]. In a genome-wide sequencing study of 183 melanomas, Hayward et al. reported five cases with MAP2K1 mutations, all defined as missense in supplemental materials, and all co-occurring with either BRAF, NRAS, or NF1 mutations [4]. In another study of 127 melanomas, eight had MAP2K1 point mutations and two had MAP2K2 point mutations [10]; these alterations were associated with constitutive ERK phosphorylation, and most co-occurred with either BRAF or NRAS mutation. Other series revealed MAP2K1 point mutations in two of 253 melanomas [29] and MAP2K1 mutations of unspecified type in 21 of 356 melanomas [30]. A case report of cutaneous melanoma metastases sampled over time revealed a MAP2K1 (F53Y) missense mutation paired with a BRAF fusion [11]. The authors of this study reviewed the TCGA database and identified MAP2K1 alterations in 7% of melanomas, consisting mostly of point mutations with some amplifications and no in-frame deletions, with nearly all co-occurring with BRAF or NRAS mutations [11]. These various point mutations in MAP2K1 have largely corresponded to alterations described above as Class 1 (RAF dependent) or Class 2 (RAF regulated) [16].
With regard to BRAF inhibitor resistances, Gao et al. argue, based on their findings and prior study [31], that Class 1 MAP2K1 mutations are unlikely to contribute to RAF inhibitor resistance, while Class 2 MAP2K1 mutations, which have been associated with acquired BRAF inhibitor resistance [32, 33], promote RAF inhibitor resistance through RAF-independent enzymatic activity [16]. Class 3 mutations, independent of BRAF, RAS, and NF1 mutations, promote the permanently activated confirmation of the MEK1 enzyme. To our knowledge, there is limited clinical data regarding RAF inhibition of Class 3 mutants given the lack of associated BRAF mutation, the essential indication for therapeutic RAF inhibition.
Some experimental models have investigated MEK inhibitors for MAP2K1 in-frame deletions [15, 16, 26, 27]. Gao et al. reported in vitro experiments where Classes 1 and 2 MAP2K1 mutants were effectively inhibited by allosteric MEK1 inhibitors, while Class 3 mutants, represented by in-frame deletions, were resistant [16]. They further showed that a new experimental MEK inhibitor, which competes for ATP binding to MEK protein, was effective against all three classes. Others models have demonstrated sensitivity of cancer cells with MAP2K1 in-frame deletion to MEK inhibition [15, 26,27,28]. Perhaps analogous to in-frame deleted cases, other investigations have shown MAP2K1 point mutations associated with RAF-independent MEK activity (involving helix-A) promote resistance to both BRAF and MEK inhibition, indicating a need for alternative therapies, such as ERK inhibitors [34].
While prior in vitro and clinical studies have correlated MAP2K1 missense mutations with resistance to targeted inhibitors in the context of BRAF-mutated melanomas [10, 32, 35,36,37,38,39,40,41], targeted therapy for melanoma with MAP2K1 in-frame deletion has not been reported to our knowledge. Case reports of MEK inhibitory therapy for patient with LCH with MAP2K1 p.E102_103del alteration showed lasting clinical improvement of lesions [42, 43], while a case of colonic adenocarcinoma with the same deletion showed progression on MEK and ERK inhibitory therapy [44] (Supplemental Table 3). In one report, a patient with triple wild-type (BRAF/NRAS/NF1 WT) metastatic melanoma with two missense mutations in MAP2K1 showed a partial response to MEK inhibitor trametinib, followed by progression of disease after two months [45].
Insight into MAP2K1-mutated tumors may inform the therapeutic approach to emerging small molecule inhibitors, such as ERK inhibitors [46, 47]. Future clinical studies are needed to identify effective therapies optimized for the various mutational patterns of MAP2K1.
Beyond melanoma, MAP2K1 deletions have been reported rarely among pigmented epithelioid melanocytoma (PEM), deep penetrating nevi (DPN), and Spitz tumors, while various MAP2K1 alterations have been identified in a range of non-melanocytic neoplasms. In a study of 13 cases of PEM, Cohen et al. characterized two cases with MAP2K1 in-frame deletions [48]. Interestingly, these MAP2K1 altered cases did not show co-occurring PRKAR1A mutations or PRKCA fusions, despite loss of PRKAR1a expression by immunohistochemistry [48]. Isales et al. also noted a case of PEM with MAP2K1 in-frame deletion, exclusive from PRKCA fusion, NTRK1/NTRK3 fusion, and BRAF/NRAS/NF1 mutations seen in the other cases [49]. In a thorough characterization of DPN, six cases showed in-frame deletion of MAP2K1, all co-occurring with CTNNB1 mutation, and all mutually exclusive from BRAF and HRAS mutations [28].
Interestingly, a recent study characterized a subset of Spitzoid melanocytic tumors with activating structural alterations in MAPK genes [50]. In addition to in-frame fusions of MAP3K8-DIPC2, MAP3K8-PCDH7, MAP3K8-UBL3, MAP3K8-SVIL, and ATP2A2-MAP3K3, one case showed an in-frame deletion of MAP2K1. This MAP2K1-mutated case consisted of an atypical Spitz tumor on the leg of a 30-year-old man with dermal growth, expansile nests, associated epidermal hyperplasia, epithelioid cytomorphology with extensive pigmentation, moderate nuclear atypia, and low mitotic rate. Follow-up showed no recurrence at 9 months [50].
With respect to histomorphology, the melanomas in this series were diverse. The qualities shared by the primary tumors, including exophytic nodules, irregular nested bases, and nuclear pleomorphism, are common to many cutaneous melanomas with tumorigenic vertical growth phases. In addition, the cytomorphology varied, with most displaying epithelioid morphology and a small minority showing spindled and rhabdoid cytomorphology. We note that a small proportion of cases were pigmented, although our analysis did not reveal tendency towards pigmented epithelioid cytomorphology as noted in previous study of BRAF-mutated melanomas [51]. Furthermore, we did not observe characteristic PEM-like, DPN-like, or spitzoid cytomorphology. Additional study and accumulation of increased numbers of primary cases may allow for improved recognition of common morphologic features.
MAP2K1 mutations occur in ~20% of cases of LCH and are mutually exclusive from BRAF mutation, the most common pathogenic mutation in LCH [13,14,15, 52]. These MAP2K1 mutations in LCH are predominantly in-frame deletions (corresponding to either class 2 or class 3 MAP2K1 mutations), with a minor component of point mutations [13,14,15, 53]. McGinnis et al. reported that, of their five cases of LCH with MAP2K1 mutation, two also had BRAF V600E mutation [54]. Interestingly, these authors describe that these two BRAF/MAP2K1 co-mutated cases had point mutations in MAP2K1, whereas the MAP2K1 in-frame deletions were apparently exclusive from BRAF mutants [54].
A similar mutual exclusivity of BRAF and MAP2K1 mutations has also been reported in other neoplasms, including hairy cell leukemia (HCL) and various epithelial malignancies. While classical HCL harbors BRAF V600E in nearly all cases, ~30% of variant HCL has MAP2K1 mutation [55,56,57]. These MAP2K1 mutations in HCL consist mostly of point mutations, with rarely reported in-frame deletion [56, 57]. KRAS and MAP2K1 point mutations have been shown to occur in a mutually exclusive manner in Rosai–Dorfman disease [58]. MAP2K1 mutation has been identified among subsets of smoking-associated lung carcinomas (mutually exclusive from EGFR, BRAF, KRAS, and NRAS mutations) [59, 60], lung adenocarcinoma in situ and early invasive disease [61], and KRAS/NRAS/BRAF/PIK3CA wild-type (“quadruple-wild-type”) colorectal carcinomas [62]. Study of extracranial arteriovenous malformations revealed point mutations and, rarely, in-frame deletions of MAP2K1 [63].
Our study also provides a proof of concept that liquid biopsy can detect ctDNA of in-frame deletion of MAP2K1 in melanoma, with a single case showing this alteration. Four additional melanoma cases demonstrated missense mutations in MAP2K1. Liquid biopsy may be a valuable method in these tumors, and further investigation may be warranted.
With respect to limitations, the cases described here were collected from patients with advanced malignancies, submitted for detection of therapeutically targetable mutations. Thus, these cases likely exemplify the aggressive end of the biologic spectrum without representing early, thin, and/or indolent melanomas. We speculate that we did not identify PEM-like, DPN-like, or atypical Spitz tumors as previously reported [49, 50], because these tumors are typically not aggressive and are unlikely to be submitted for therapy-oriented genomic sequencing. Another limitation of this study is that, owing to the nature of our sample bank, we were unable to directly assess MAPK activation in our particular cases. Nevertheless, three lines of evidence support our inference that the in-frame deletions of MAP2K1 in our melanoma cases were activating. First is extensive mechanistic literature that MAP2K1 in-frame deletions identical to those in our study activate MAPK signaling [15, 16, 26,27,28] (Supplemental Table 3). Second is evidence of MAPK pathway activation in prior study of non-melanoma tumors with these same MAP2K1 in-frame deletions [15] (Supplemental Table 3). The third line of evidence is the mutual exclusivity that we found in our cohort from well-characterized mutations of the MAPK pathway (Fig. 2b, Table 2). Follow-up data, which was not available in this study, will be important to obtain for this cohort of MAP2K1-mutated melanoma to correlate with prognosis and therapeutic outcomes. In particular, therapeutic responses of this subgroup to inhibitors of the MAP Kinase pathway, including MAP2K1 and ERK inhibitors, require further study. With respect to MAP2K1 missense mutations, it is possible that the BRAF-mutated cases, through the effects of combined BRAF and MEK inhibitor therapy, are enriched for these point mutations of MAP2K1, selected through acquired resistance. Finally, while H&E slides and accompanying pathology reports were reviewed from all cases, we were not able to review immunohistochemical slides, limiting our ability to correlate genomic findings with immunophenotype.
Melanomas with MAP2K1 in-frame deletions show distinct mutational profiles and correlate closely with prior functional studies of MAP2K1 deletion. Additional studies on therapeutic approaches to MAP2K1-mutated melanomas are needed.
References
Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N. Engl J Med. 2005;353:2135–47.
Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol. 2014;9:239–71.
Broekaert SMC, Roy R, Okamoto I, Van Den Oord J, Bauer J, Garbe C, et al. Genetic and morphologic features for melanoma classification. Pigment Cell Melanoma Res. 2010;23:763–70.
Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017;545:175–80.
Turner J, Couts K, Sheren J, Saichaemchan S, Ariyawutyakorn W, Avolio I, et al. Kinase gene fusions in defined subsets of melanoma. Pigment Cell Melanoma Res. 2017;30:53–62.
Wiesner T, Kutzner H, Cerroni L, Mihm MC, Busam KJ, Murali R. Genomic aberrations in spitzoid melanocytic tumours and their implications for diagnosis, prognosis and therapy. Pathology 2016;48:113–31.
Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 2014;5:3116.
Williams EA, Shah N, Montesion M, Sharaf R, Pavlick DC, Sokol ES, et al. Melanomas with activating RAF1 fusions: clinical, histopathologic, and molecular profiles. Mod Pathol. 2020. https://doi.org/10.1038/s41379-020-0510-7. [Epub ahead of print].
Botton T, Talevich E, Mishra VK, Zhang T, Shain AH, Berquet C, et al. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell Rep. 2019;29:573–88.e7.
Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet 2012;44:133–9.
Davidson G, Coassolo S, Kieny A, Ennen M, Pencreach E, Malouf GG, et al. Dynamic evolution of clonal composition and neoantigen landscape in recurrent metastatic melanoma with a rare combination of driver mutations. J Investig Dermatol. 2019;139:1769–78.e2.
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63.
Nelson DS, van Halteren A, Quispel WT, van den Bos C, Bovée JVMG, Patel B, et al. MAP2K1 and MAP3K1 mutations in Langerhans cell histiocytosis. Genes Chromosomes Cancer. 2015;54:361–8.
Brown NA, Furtado LV, Betz BL, Kiel MJ, Weigelin HC, Lim MS, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood. 2014;124:1655–8.
Chakraborty R, Hampton OA, Shen X, Simko SJ, Shih A, Abhyankar H, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;124:3007–15.
Gao Y, Chang MT, McKay D, Na N, Zhou B, Yaeger R, et al. Allele-specific mechanisms of activation of mek1 mutants determine their properties. Cancer Discov. 2018;8:648–61.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1021–31.
Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Pathog. 2018;14:e1005965.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
Trabucco SE, Gowen K, Maund SL, Sanford E, Fabrizio DA, Hall MJ, et al. A novel next-generation sequencing approach to detecting microsatellite instability and pan-tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J Mol Diagn. 2019;21:1053–66.
Clark TA, Chung JH, Kennedy M, Hughes JD, Chennagiri N, Lieber DS, et al. Analytical validation of a hybrid capture–based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J Mol Diagnostics. 2018;20:686–702.
Gregg JP, Li G, Pavlick D, Chung J, Cooke M, He J, et al. Comprehensive genomic profiling of ctDNA in patients with colon cancer and its fidelity to the genomics of the tumor biopsy. J Clin Oncol. 2018;36:569.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–13.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature . 2013;500:415–21.
Elder DE, Barnhill RL, Bastian BC, Cook MG, de la Fouchardiere A, Gerami P, et al. Melanocytic tumours. In: Elder DE, Massi D, Scolyer RA, and Willemze RE, editor. World Health Organization 2018 Classification of Skin Tumours. Lyon, France: International Agency for Research on Cancer, 2018. p. 65–77.
Yuan J, Ng WH, Tian Z, Yap J, Baccarini M, Chen Z, et al. Activating mutations in MEK1 enhance homodimerization and promote tumorigenesis. Sci Signal. 2018;11:eaar6795.
Kohsaka S, Hayashi T, Nagano M, Ueno T, Kojima S, Kawazu M, et al. Identification of novel CD74-NRG2α fusion from comprehensive profiling of lung adenocarcinoma in Japanese never or light smokers. J Thorac Oncol. 2020;S1556-0864:30098–8.
Yeh I, Lang UE, Durieux E, Tee MK, Jorapur A, Shain AH, et al. Combined activation of MAP kinase pathway and β-catenin signaling cause deep penetrating nevi. Nat Commun. 2017;8:644.
Emelyanova M, Ghukasyan L, Abramov I, Ryabaya O, Stepanova E, Kudryavtseva A, et al. Detection of BRAF, NRAS, KIT, GNAQ, GNA11 and MAP2K1/2 mutations in Russian melanoma patients using LNA PCR clamp and biochip analysis. Oncotarget. 2017;8:52304–20.
Zhao X, Little P, Hoyle AP, Pegna GJ, Hayward MC, Ivanova A, et al. The prognostic significance of low-frequency somatic mutations in metastatic cutaneous melanoma. Front Oncol. 2019;8:584.
Shi H, Moriceau G, Kong X, Koya RC, Nazarian R, Pupo GM, et al. Preexisting MEK1 Exon 3 mutations in V600E/KBRAF melanomas Do Not confer resistance to BRAF inhibitors. Cancer Discov. 2012;2:414–24.
Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96.
Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–74.
Emery CM, Monaco KA, Wang P, Balak M, Freeman A, Meltzer J, et al. BRAF-inhibitor associated MEK mutations increase RAF-dependent and -independent enzymatic activity. Mol Cancer Res. 2017;15:1431–44.
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009;106:20411–6.
Carlino MS, Fung C, Shahheydari H, Todd JR, Boyd SC, Irvine M, et al. Preexisting MEK1P124 mutations diminish response to BRAF inhibitors in metastatic melanoma patients. Clin Cancer Res. 2015;21:98–105.
Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51:2792–9.
Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109.
Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, et al. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of us food and drug administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 2018;11:7095–107.
Luebker SA, Koepsell SA. Diverse mechanisms of BRAF inhibitor resistance in melanoma identified in clinical and preclinical studies. Front Oncol 2019;9:1–8.
Paraiso KHT, Smalley KSM. Making sense of MEK1 mutations in intrinsic and acquired BRAF inhibitor resistance. Cancer Discov. 2012;2:390–2.
Papapanagiotou M, Griewank KG, Hillen U, Schimming TT, Moeller LC, Führer D. et al.Trametinib-induced remission of an MEK1-mutated langerhans cell histiocytosis. JCO Precis Oncol. 2017;1:1–5.
Lorillon G, Jouenne F, Baroudjian B, de Margerie-Mellon C, Vercellino L, Meignin V, et al. Response to trametinib of a pulmonary Langerhans cell histiocytosis harboring a MAP2K1 deletion. Am J Respir Crit Care Med. 2018;198:675–8.
Wang C, Sandhu J, Fakih M. A case of class 3 MEK1 mutated metastatic colorectal cancer with a non-durable tumor marker response to MEK and ERK inhibitors. J Gastrointest Oncol. 2019;10:1140–3.
Homicsko KG, Aedo V, Missiaglia E, Bisig B, Michielin O. Targeting MAP2K1 mutation with trametinib in a triple wild-type melanoma patient. J Clin Oncol. 2019;37:e21027–e21027.
Smalley I, Smalley KSM. ERK inhibition: a new front in the war against mapk pathway-driven cancers? Cancer Disco. 2018;8:140–2.
Maust JD, Whitehead CE, Sebolt-Leopold JS. Oncogenic mutants of MEK1: a trilogy unfolds. Cancer Discov. 2018;8:534–6.
Cohen JN, Joseph NM, North JP, Onodera C, Zembowicz A, LeBoit PE. Genomic analysis of pigmented epithelioid melanocytomas reveals recurrent alterations in PRKAR1A, and PRKCA Genes. Am J Surg Pathol. 2017;41:1333–46.
Isales MC, Mohan LS, Quan VL, Garfield EM, Zhang B, Shi K, et al. Distinct genomic patterns in pigmented epithelioid melanocytoma: a molecular and histologic analysis of 16 Cases. Am J Surg Pathol. 2018;43:480–8.
Quan VL, Zhang B, Mohan LS, Shi K, Isales MC, Panah E, et al. Activating structural alterations in MAPK genes are distinct genetic drivers in a unique subgroup of spitzoid neoplasms. Am J Surg Pathol. 2019;43:538–48.
Viros A, Fridlyand J, Bauer J, Lasithiotakis K, Garbe C, Pinkel D, et al. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med 2008;5:941–52.
Tran G, Huynh TN, Paller AS. Langerhans cell histiocytosis: a neoplastic disorder driven by Ras-ERK pathway mutations. J Am Acad Dermatol. 2018;78:579–90.e4.
Alayed K, Medeiros LJ, Patel KP, Zuo Z, Li S, Verma S, et al. BRAF and MAP2K1 mutations in Langerhans cell histiocytosis: A study of 50 cases. Hum Pathol. 2016;52:61–7.
McGinnis LM, Nybakken G, Ma L, Arber DA. Frequency of MAP2K1, TP53, and U2AF1 Mutations in BRAF-mutated Langerhans cell histiocytosis. Am J Surg Pathol. 2018;42:885–90.
Durham BH, Getta B, Dietrich S, Taylor J, Won H, Bogenberger JM, et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood. 2017;130:1644–8.
Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46:8–10.
Mason EF, Brown RD, Szeto DP, Gibson CJ, Jia Y, Garcia EP, et al. Detection of activating MAP2K1 mutations in atypical hairy cell leukemia and hairy cell leukemia variant. Leuk Lymphoma. 2017;58:233–6.
Garces S, Medeiros LJ, Patel KP, Li S, Pina-Oviedo S, Li J, et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod Pathol .2017;30:1367–77.
Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68:5524–8.
Arcila ME, Drilon A, Sylvester BE, Lovly CM, Borsu L, Reva B, et al. MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking. Clin Cancer Res. 2015;21:1935–43.
Chen H, Carrot-Zhang J, Zhao Y, Hu H, Freeman SS, Yu S, et al. Genomic and immune profiling of pre-invasive lung adenocarcinoma. Nat Commun. 2019;10:5472.
Rachiglio AM, Lambiase M, Fenizia F, Roma C, Cardone C, Iannaccone A, et al. Genomic profiling of KRAS/NRAS/BRAF/PIK3CA wild-type metastatic colorectal cancer patients reveals novel mutations in genes potentially associated with resistance to anti-EGFR agents. Cancers. 2019;11:1–12.
Couto JA, Huang AY, Konczyk DJ, Goss JA, Fishman SJ, Mulliken JB, et al. Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am J Hum Genet. 2017;100:546–54.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
EAW, NS, MM, RS, DCP, ESS, BA, JV, JAE, JSR, and JYT are employees of Foundation Medicine, Inc., a wholly owned subsidiary of Roche Holdings, Inc. and Roche Finance Ltd, and these employees have equity interest in an affiliate of these Roche entities. KJW receives research funding from Novo Nordisk for studies unrelated to cancer. MCM declares no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Williams, E.A., Montesion, M., Shah, N. et al. Melanoma with in-frame deletion of MAP2K1: a distinct molecular subtype of cutaneous melanoma mutually exclusive from BRAF, NRAS, and NF1 mutations. Mod Pathol 33, 2397–2406 (2020). https://doi.org/10.1038/s41379-020-0581-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0581-5
This article is cited by
-
Molecular Mutations in Histiocytosis: A Comprehensive Survey of Genetic Alterations
Molecular Biotechnology (2024)
-
Clinical study of MAP2K1-mutated Langerhans cell histiocytosis in children
Journal of Cancer Research and Clinical Oncology (2022)
