
Abstract
We performed harmonized molecular and clinical analysis on 1,048 melanomas and discovered markedly different global genomic properties among subtypes (BRAF, (N)RAS, NF1, triple wild-type (TWT)), subtype-specific preferences for secondary driver genes and active mutational processes previously unreported in melanoma. Secondary driver genes significantly enriched in specific subtypes reflected preferential dysregulation of additional pathways, such as induction of transforming growth factor-β signaling in BRAF melanomas and inactivation of the SWI/SNF complex in (N)RAS melanomas, and select co-mutation patterns coordinated selective response to immune checkpoint blockade. We also defined the mutational landscape of TWT melanomas and revealed enrichment of DNA-repair-defect signatures in this subtype, which were associated with transcriptional downregulation of key DNA-repair genes, and may revive previously discarded or currently unconsidered therapeutic modalities for genomically stratified melanoma patient subsets. Broadly, harmonized meta-analysis of melanoma whole exomes revealed distinct molecular drivers that may point to multiple opportunities for biological and therapeutic investigation.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
All of the datasets used in the present study are publicly available. The raw sequence data can be obtained through dbGaP (https://www.ncbi.nlm.nih.gov/gap) and the ICGC Data Access Compliance Office (https://icgc.org/daco), or as described in the original papers (Supplementary Table 1). The accession codes can also be found in Supplementary Table 1. Publicly available databases used in the present study include MSigDB v.6.2 (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), ExAC (http://exac.broadinstitute.org), gnomAD (https://gnomad.broadinstitute.org), the Broad Institute Single Cell portal (https://singlecell.broadinstitute.org/single_cell) and ConsensusPathDB v.34 (http://cpdb.molgen.mpg.de).
Code availability
All software and bioinformatic tools used in the present study are publicly available.
References
Hodis, E. et al. A landscape of driver mutations in melanoma. Cell 150, 251–263 (2012).
Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 161, 1681–1696 (2015).
Flaherty, K. T. et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819 (2010).
Flaherty, K. T. et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 367, 107–114 (2012).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Van Allen, E. M. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015).
Wolchok, J. D. et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 377, 1345–1356 (2017).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Krauthammer, M. et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 44, 1006–1014 (2012).
Cirenajwis, H. et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol. Oncol. 11, 438–451 (2017).
Nsengimana, J. et al. β-Catenin-mediated immune evasion pathway frequently operates in primary cutaneous melanomas. J. Clin. Invest. 128, 2048–2063 (2018).
Krauthammer, M. et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 47, 996–1002 (2015).
Van Allen, E. M. et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 4, 94–109 (2014).
Wagle, N. et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 4, 61–68 (2014).
Miao, D. et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 50, 1271–1281 (2018).
Hayward, N. K. et al. Whole-genome landscapes of major melanoma subtypes. Nature 545, 175–180 (2017).
Liu, D. et al. Integrative molecular modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 25, 1916–1927 (2019).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Dietlein, F. et al. Identification of cancer driver genes based on nucleotide context. Nat. Genet. 52, 208–218 (2020).
Mularoni, L., Sabarinathan, R., Deu-Pons, J., Gonzalez-Perez, A. & López-Bigas, N. OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 17, 128 (2016).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 1, 1–16 (2017).
Johannessen, C. M. et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 504, 138–142 (2013).
Pan, D. et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018).
Yu, Q. et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 9, 23–32 (2006).
Boland, C. R., Richard Boland, C. & Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 138, 2073–2087.e3 (2010).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 174, 1034–1035 (2018).
Alliston, T. et al. Repression of bone morphogenetic protein and activin-inducible transcription by Evi-1. J. Biol. Chem. 280, 24227–24237 (2005).
Buonamici, S. et al. EVI1 abrogates interferon-alpha response by selectively blocking PML induction. J. Biol. Chem. 280, 428–436 (2005).
Lee, J. J. et al. Targeted next-generation sequencing reveals high frequency of mutations in epigenetic regulators across treatment-naïve patient melanomas. Clin. Epigenet. 7, 59 (2015).
Menzies, A. M. et al. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin. Cancer Res. 18, 3242–3249 (2012).
Flaherty, K. et al. Genomic analysis and 3-y efficacy and safety update of COMBI-d: a phase 3 study of dabrafenib (D) + trametinib (T) vs D monotherapy in patients (pts) with unresectable or metastatic BRAF V600E/K-mutant cutaneous melanoma. J. Clin. Orthod. 34, 9502–9502 (2016).
Long, G. V. et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 28, 1631–1639 (2017).
Kadoch, C. & Crabtree, G. R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci. Adv. 1, e1500447 (2015).
Miao, D. et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806 (2018).
Arafeh, R. et al. Recurrent inactivating RASA2 mutations in melanoma. Nat. Genet. 47, 1408–1410 (2015).
van der Weyden, L. & Adams, D. J. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 1776, 58–85 (2007).
Akino, K. et al. The Ras effector RASSF2 is a novel tumor-suppressor gene in human colorectal cancer. Gastroenterology 129, 156–169 (2005).
Endoh, M. et al. RASSF2, a potential tumour suppressor, is silenced by CpG island hypermethylation in gastric cancer. Br. J. Cancer 93, 1395–1399 (2005).
Curtin, J. A., Busam, K., Pinkel, D. & Bastian, B. C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 24, 4340–4346 (2006).
Robertson, A. G. et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 32, 204–220.e15 (2017).
Newell, F. et al. Whole-genome landscape of mucosal melanomas reveals diverse drivers and therapeutic targets. Nat. Commun. 10, 3163 (2019).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Gao, Q. et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 23, 227–238.e3 (2018).
Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S. & Swanton, C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016).
Gehring, J. S., Fischer, B., Lawrence, M. & Huber, W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics 31, 3673–3675 (2015).
Greenman, C. et al. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Abkevich, V. et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 107, 1776–1782 (2012).
Timms, K. M. et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 16, 475 (2014).
Birkbak, N. J. et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2, 366–375 (2012).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462 (2012).
Marquard, A. M. et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res 3, 9 (2015).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019).
Alexandrov, A. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Polak, P. et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 49, 1476–1486 (2017).
Jaffe, A. E. et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 41, 200–209 (2012).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Tsukuda, T. et al. INO80-dependent chromatin remodeling regulates early and late stages of mitotic homologous recombination. DNA Repair 8, 360–369 (2009).
Lademann, C. A., Renkawitz, J., Pfander, B. & Jentsch, S. The INO80 complex removes H2A.Z to promote presynaptic filament formation during homologous recombination. Cell Rep. 19, 1294–1303 (2017).
Bakr, A. et al. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nucleic Acids Res. 43, 3154–3166 (2015).
Fenton, A. L., Shirodkar, P., Macrae, C. J., Meng, L. & Koch, C. A. The PARP3- and ATM-dependent phosphorylation of APLF facilitates DNA double-strand break repair. Nucleic Acids Res. 41, 4080–4092 (2013).
Ceccaldi, R., Rondinelli, B. & D’Andrea, A. D. Repair pathway choices and consequences at the double-stranded break. Trends Cell Biol. 26, 52–64 (2016).
Balmus, G. et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 10, 87 (2019).
Zhang, J., Ma, Z., Treszezamsky, A. & Powell, S. N. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat. Struct. Mol. Biol. 12, 902–909 (2005).
Scully, R. & Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 750, 5–14 (2013).
Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016).
Iles, N., Rulten, S., El-Khamisy, S. F. & Caldecott, K. W. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell. Biol. 27, 3793–3803 (2007).
Macrae, C. J., McCulloch, R. D., Ylanko, J., Durocher, D. & Koch, C. A. APLF (C2orf13) facilitates nonhomologous end-joining and undergoes ATM-dependent hyperphosphorylation following ionizing radiation. DNA Repair 7, 292–302 (2008).
Flaherty, K. T. et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J. Clin. Oncol. 31, 373–379 (2013).
Wilson, M. A. et al. Correlation of somatic mutations and clinical outcome in melanoma patients treated with carboplatin, paclitaxel, and sorafenib. Clin. Cancer Res. 20, 3328–3337 (2014).
Rafiei, S. et al. ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Clin. Cancer Res. 80, 2094–2100 (2020).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.1–33 (2013).
Cibulskis, K. et al. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 27, 2601–2602 (2011).
Taylor-Weiner, A. et al. DeTiN: overcoming tumor-in-normal contamination. Nat. Methods 15, 531–534 (2018).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Costello, M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013).
Saunders, C. T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
McGranahan, N. et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 7, 283ra54 (2015).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Conway, J. R., Lex, A. & Gehlenborg, N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940 (2017).
Riaz, N. et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949.e16 (2017).
Roh, W. et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 9, eaah3560 (2017).
Hugo, W. et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016).
Rodig, S. J. et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 10, eaar3342 (2018).
Mills, R. E. et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 21, 830–839 (2011).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Ignatiadis, N., Klaus, B., Zaugg, J. B. & Huber, W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 13, 577–580 (2016).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Rauen, K. A. The RASopathies. Annu. Rev. Genomics Hum. Genet. 14, 355–369 (2013).
Kamburov, A. et al. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 39, 712–717 (2011).
Acknowledgements
This work was supported by the National Cancer Institute (grant no. F31CA239347 to J.R.C.), National Institutes of Health (grant nos. 5T32HG002295-15 to J.R.C., and R01CA227388-02 and R21CA242861 to E.M.V.A.), and the Damon Runyon Clinical Investigator Award (to E.M.V.A.). F.D. was supported by the Claudia Adams Barr Program for Innovative Cancer Research and the AWS Cloud Credits for Research Program. The results presented in the present study are in part based on data generated by the TCGA Research Network (https://www.cancer.gov/tcga). This publication and the underlying research are partly facilitated by Hartwig Medical Foundation and the Center for Personalized Cancer Treatment which have generated, analyzed and made available data for this research. We thank Petra Ross-Macdonald for providing clinical outcomes data used for the immunotherapy response validation analyses.
Author information
Authors and Affiliations
Contributions
J.R.C., A.T.-W., S.A., F.D., B.R. and M.X.H. contributed to the analysis of genomic data. J.R.C., C.A.M., B.S., D.S., D.L. and E.M.V.A. contributed to the aggregation of raw sequence data. N.V., T.K., D.L. and E.M.V.A. contributed to analysis and data interpretation of the immunotherapy response. J.R.C., A.T.-W., S.A., N.V., F.D., B.R., J.L.W., R.H., F.S.H., B.S., D.S., D.L. and E.M.V.A. contributed to the interpretation of results and preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
E.M.V.A. is a consultant for Tango Therapeutics, Genome Medical, Invitae, Enara Bio, Monte Rosa Therapeutics, Manifold Bio and Janssen, provides research support to Novartis and Bristol-Myers Squibb, has equity in Tango Therapeutics, Genome Medical, Syapse, Enara Bio, Monte Rosa Therapeutics and Microsoft, receives travel reimbursement from Roche/Genentech, and has institutional patents filed on chromatin mutations and immunotherapy response, and methods for clinical interpretation.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Overlap between SMGs from the entire cohort and subtype analyses.
a, Overlap between the subtype-specific SMGs and the SMGs that were identified via the entire cohort (M1000). Most of the SMGs identified in the entire cohort analysis were not identified through the subtype specific analysis (115 of 178, 64.6%).
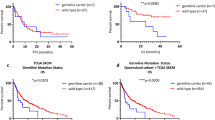
Extended Data Fig. 2 MECOM/BMP5 immunotherapy validation (overall survival and RECIST response).
External validation analysis of overall survival for MECOM/BMP5 mutations using the Roh, Riaz, Hugo, and Rodig whole-exome cohorts (n = 194 total) for a, all melanomas, b, BRAF melanomas, and c, non-BRAF melanomas, excluding post treatment biopsies. These cohorts were chosen because they were immunotherapy treated, whole-exome sequenced, cohorts not included in our discovery cohort. Due to the diverse treatment regimens in each of these trials and cohorts, we were unable to correct for drug. Further, since we did not have access to raw sequencing data from all these studies, we could not calculate and correct for tumor purity and utilized published variant calls. The hazard rate ratios of MECOM/BMP5 mutations when correcting for only mutational load was (a) 0.59 (multivariate Cox proportional-hazards, p = 0.09) for all melanomas, (b) 0.46 (multivariate Cox proportional-hazards, p = 0.16) for BRAF melanomas, and (c) 0.68 (multivariate Cox proportional-hazards, p = 0.31) for non-BRAF melanomas. These results are similar to what was observed in the discovery cohort (Supplementary Table 8), although this validation cohort size was not powered to achieve statistical significance. d, The association between the BRAF subtype and MECOM/BMP5 mutations for clinical benefit to immunotherapy (via RECIST) in our limited validation cohort was similar to our discovery cohort findings, but not statistically significant. The p-values shown in a-c) are derived from the log-rank test.
Extended Data Fig. 3 PBAF complex immunotherapy validation (overall survival and RECIST response).
External validation analysis of overall survival for PBAF mutations using the Roh, Riaz, Hugo, and Rodig cohorts (n = 194), which are immunotherapy treated, whole-exome sequenced, cohorts not included in our discovery cohort. a, Survival curves between PBAF-mutants and non-PBAF mutants. b, Survival curves between PBAF-mutants and non-PBAF mutants where PBAF mutants are classified by having mutations in ARID2, PBRM1, SMARCA4, and SMARCB1, which are the 4 PBAF complex genes commonly used in clinical sequencing panels. This limited validation cohort lacked sufficient samples with co-mutation of (N)RAS and PBAF complex genes (n = 9), and thus validation analysis was only performed on all tumors. Due to the unique treatment regimens in each of these cohorts, we were unable to correct for drug. Further, because we did not have access to raw sequencing data from these studies, we could not calculate and correct for tumor purity. When correcting only for mutational load the hazard ratio of PBAF mutations in the whole-exome cohorts, (a) when considering all genes in the PBAF complex, was 1.07 (multivariate Cox proportional-hazards, p = 0.80). The differences in these findings relative to the primary larger cohort may indicate differences in patient population and study size relative to our discovery cohort. (b) When considering only mutations in ARID2, PBRM1, SMARCA4, and SMARCB1 as PBAF-mutant, the HRR was 0.86 (multivariate Cox proportional-hazards, p = 0.61). The p-values for a-b) are derived from the log-rank test.
Extended Data Fig. 4 NMF validation of deconstructSigs results on genomic subtypes via SomaticSignatures.
a, NMF statistics for BRAF melanomas. b, Cosine similarity between COSMIC signatures and signatures decomposed via NMF for BRAF melanomas. c, NMF statistics for (N)RAS melanomas. d, Cosine similarity between COSMIC signatures and signatures decomposed via NMF for (N)RAS melanomas. e, NMF statistics for NF1 melanomas. f, Cosine similarity between COSMIC signatures and signatures decomposed via NMF for NF1 melanomas. g, NMF statistics for TWT melanomas. h, Cosine similarity between COSMIC signatures and signatures decomposed via NMF for TWT melanomas. The cophenetic correlation coefficient and residual sum of squares (RSS) suggests 3 is the optimal number of signatures for each genomic subtype.
Extended Data Fig. 5 NMF simulations via SomaticSignatures on TWT melanomas removing 35 random non-signature 3 samples each simulation.
A total of 35 signature 3 samples were identified via deconstructSigs in our signature analysis. To ensure that our NMF validation in TWT melanomas (Supplementary Fig. 17) is actually identifying signature 3 because it is indeed present, and not because it’s a flat signature, we performed 1000 simulations removing 35 random non-signature 3 samples each time. Signature 3 was identified 927 times (92.7%), which corroborates the deconstructSigs results and suggests signature 3 is the third most dominant signature in TWT melanomas. Performing 1000 simulations when removing the 35 signature 3 samples each time never yielded the identification of signature 3 via NMF.
Extended Data Fig. 6 DSB repair deficiency - unweighted sum of HRD associated CNA events.
a, Distribution of the unweighted sum of HRD associated CNA events (loss of heterozygosity, telomeric allelic imbalance, large scale transitions) in signature 3 (yellow) and non-signature 3 (purple) melanomas in the entire cohort. Signature 3 tumors were significantly enriched in HRD associated copy number events via a Mann-Whitney U test (p = 6.21 × 10−5, two-sided). b, Density plot of HRD associated copy number events in the entire cohort. c, Distribution of HRD associated copy number events in signature 3 and non-signature 3 melanomas in TWT melanomas (Mann-Whitney U, p = 5.49 × 10−3, two-sided). d, Density plot of HRD associated copy number events in the TWT melanomas. In (a) and (c) the data is represented as a boxplot where the middle line is the median, the lower and upper edges of the box are the first and third quartiles, the whiskers represent the interquartile range (IQR) multiplied by 1.5, and beyond the whiskers are outlier points.
Extended Data Fig. 7 Indel mutational signatures on the 390 WGS tumors.
Cosine similarity between COSMIC indel mutational signatures and the suggested solution NMF results from SigProfileExtractor. Indel mutational signatures revealed that a, BRAF, b, (N)RAS, and c, NF1 melanomas were associated with indel signatures ID1, ID2 and ID13 (associated with UV), while d, TWT melanomas were associated with indel signatures ID1, ID8 (associated with NHEJ), and ID13. e, Mutational signature 3 was associated with indel signatures ID1 and ID8, and was the sole mutational signature associated with ID8. f, Interestingly, when removing signature 3 tumors from the TWT melanoma cohort, TWT melanomas were still associated with indel signature ID8. Thus, the increased genomic instability of TWT melanomas in general is enough to result in ID8.
Extended Data Fig. 8 Comparison of transcriptional profiles between DSB repair deficient and DSB repair intact TWT melanomas.
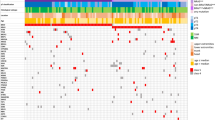
a, The workflow used to identify transcriptional differences between putative DSB repair deficient (presence of signature 3) and non-DSB repair deficient (no contribution of signature 3) TWT tumors. b, Pearson correlation between signature 3 contribution and normalized gene expression in TWT melanomas (Methods) identified 9 positive and 10 negative signifi- cant correlations for DNA-repair genes (Pearson’s, p-value cutoff < 0.05; Methods). Genes highlighted in purple function in DSB repair pathways, including HR. Opacity was used to show the density of non-significant points along both axes.
Extended Data Fig. 9 Differential expression analysis between signature 3 and non-signature 3 TWT melanomas.
a, DESeq2 log2 fold-change vs edgeR log2 fold-change for cumulative set of DNA-repair genes. b, Significance vs log2 fold-change of significantly differentially expressed DNA repair genes as determined by DESeq2. Yellow points indicate genes whose expression was significantly correlated with signature 3 contribution and significantly differentially expressed. Green points indicate genes that were only significantly differentially expressed. Genes highlighted in purple function in DSB repair. Opacity was used to show the density of non-significant points along both axes.
Extended Data Fig. 10 Methylation and signature 3 contribution.
a, Pearson correlation between signature 3 contribution and methylation β-values plotted on the x-axis vs. difference in median methylation between signature 3 and non-signature 3 TWT samples on the y-axis. Six probe sites were significantly correlated with signature 3 contribution, had a significant difference in median β-values (via Mann-Whitney U), and had methylation β-values significantly associated with gene expression. Of the six probe sites, INO80 was the only gene involved in HR repair. Opacity was used to show the density of non-significant points along both axes. b, Expression of INO80 was significantly correlated with methylation β-values at INO80-ch.15.415873F (Pearson’s, r = −0.51, p = 8.516 × 10−5). Points in yellow are from signature 3 TWT samples and points in purple are from non-signature 3 TWT samples.
Supplementary information
Supplementary Information
Supplementary Figs. 1–31 and Note
Supplementary Table 1
Supplementary Tables 1–13
Supplementary Data 1
Complete list of all somatic mutations in this cohort (MAF file).
Supplementary Data 2
MutSigCV2, OncodriveFML and MutPanning results for entire cohort and genomic subtypes (including V600E and V600K); q values are Benjamini–Hochberg-corrected P values.
Supplementary Data 3
GISTIC2.0 amplifications and deletions thresholded by gene results.
Supplementary Data 4
DeconstructSigs signature contributions for whole cohort and validation set.
Supplementary Data 5
Differential expression results between signature 3 and nonsignature 3 TWT melanomas; padj is the Benjamini–Hochberg-corrected P value.
Rights and permissions
About this article

Cite this article
Conway, J.R., Dietlein, F., Taylor-Weiner, A. et al. Integrated molecular drivers coordinate biological and clinical states in melanoma. Nat Genet 52, 1373–1383 (2020). https://doi.org/10.1038/s41588-020-00739-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-020-00739-1
This article is cited by
-
A Fucose-Containing Sulfated Polysaccharide from Spatoglossum schröederi Potentially Targets Tumor Growth Rather Than Cytotoxicity: Distinguishing Action on Human Melanoma Cell Lines
Marine Biotechnology (2024)
-
Integrated genomic analyses of acral and mucosal melanomas nominate novel driver genes
Genome Medicine (2022)
-
Targeting TGF-β signal transduction for fibrosis and cancer therapy
Molecular Cancer (2022)
-
Cutaneous and acral melanoma cross-OMICs reveals prognostic cancer drivers associated with pathobiology and ultraviolet exposure
Nature Communications (2022)
