
Abstract
The near-infrared luminescence of Ca6Ba(PO4)4O:Mn5+ is demonstrated and explained. When excited into the broad and strong absorption band that spans the 500–1000 nm spectral range, this phosphor provides an ultranarrow (FWHM = 5 nm) emission centered at 1140 nm that originates from a spin-forbidden 1E → 3A2 transition with a 37.5% internal quantum efficiency and an excited-state lifetime of about 350 μs. We derived the crystal field and Racah parameters and calculated the appropriate Tanabe–Sugano diagram for this phosphor. We found that 1E emission quenches due to the thermally-assisted cross-over with the 3T2 state and that the relatively high Debye temperature of 783 K of Ca6Ba(PO4)4O facilitates efficient emission. Since Ca6Ba(PO4)4O also provides efficient yellow emission of the Eu2+ dopant, we calculated and explained its electronic band structure, the partial and total density of states, effective Mulliken charges of all ions, elastic constants, Debye temperature, and vibrational spectra. Finally, we demonstrated the application of phosphor in a luminescence intensity ratio thermometry and obtained a relative sensitivity of 1.92%K−1 and a temperature resolution of 0.2 K in the range of physiological temperatures.
Similar content being viewed by others
Introduction
Mn5+ optical centers have the [Ar]3d2 electron configuration and always encounter a strong crystal field when tetrahedrally coordinated in crystals. Their lower electronic states have the 3A2 < 1E < 1A1 < 3T2 < 3T1 progression in energy. The ground state (3A2) is orbitally non-degenerate and the first excited state 1E has almost no nuclear displacement with respect to the 3A2 state and can be split by the low-symmetry ligand field1. The 1E energy of approximately 8000–9000 cm−1 is strongly affected by a nephelauxetic effect. At low temperatures, emission occurs solely from the spin-forbidden 1E → 3A2 electronic transition of a genuine electric dipole origin. At these temperatures, the emission from the spin-allowed 3T2 → 3A2 transition is almost negligible since the low energy orbital of the 3T2 state is localized more than 1000 cm−1 above the 1E state, which results in its low population. Therefore, Mn5+ emissions appearing in the near infrared (NIR) spectral range at wavelengths longer than 1100 nm and have a narrow spectral band (FWHM < 10 nm) that can be split into two bands with an energy difference of up to 300 cm−1. These emission bands are usually accompanied by vibronic sidebands and have decay times of a few tens to a few hundred microseconds.
The ultranarrow-band NIR luminescence of Mn5+ is especially favorable for NIR lasers2,3,4 and the development of narrow-band NIR light sources for the selective identification of chemical analytes5. Recent research suggests that Mn5+ activated nanoparticles are excellent probes for deep-tissue luminescence imaging and luminescence thermometry in the second biological transparency window (1000–1350 nm) and that they show high photo- and chemical stabilities6,7. One of their features, especially favorable from the biomedical application perspective, is that they exhibit broad and strong absorption bands from the spin-allowed electronic transitions that span the 500–1000 nm range8,9,10,11,12,13, which facilitates their excitation by the wavelength of the first optical biological window. They have a higher quantum efficiency (QE) than Ag2S or Ag2Se quantum dots, and do not contain toxic heavy metals like InAs or PbS quantum dots, nor suffer from photobleaching and photoblinking. In comparison to the lanthanide activated NIR bioimaging nanoprobes, Mn5+ nanoparticles are brighter due to higher values of the d-d absorption cross-section compared to the spin-forbidden f-f absorptions in the NIR-excited lanthanides. All these advantages of Mn5+ indicate that it is an ion with a very important and, so far, unexplored application potential worth large-scale intensive research.
Emissions from Mn5+ centers have been demonstrated in a considerably smaller number of host materials compared to Mn2+ and Mn4+ optical centers14. To facilitate Mn5+ emission, the material needs to provide both the tetrahedral environment for Mn5+ and a sufficiently large energy bandgap compared to the Mn5+ transitions’ energies. More importantly, the host material must provide the stabilization of the Mn 5+ valence state, which imposes more constraints on the materials’ structure and composition than the 2+ and 4+ valance states. For these reasons, the majority of Mn5+ emitting host materials contain electropositive ions such as alkaline earth metals and PO43- and VO43- groups (or SiO44− group with a charge compensation). The typical examples of such materials are Mn5+ activated Li3PO415, Sr5(VO4)3F4,16, Ba5(PO4)3Cl17, Sr5(PO4)3Cl1,17,18, Ca2PO4Cl1,17, Ca2VO4Cl1,17, Sr2VO4Cl1,17, Y2SiO519, and M2SiO4 (M = Ba, Sr, Ca)20.
Recently, Kim et al.21 have introduced the Ca6Ba(PO4)4O:Mn5+ as a new blue pigment that shows coloration due to intense Mn5+ absorption, but its luminescent properties have not been analyzed. Also recently, the efficient yellow emission from Eu2+ activated Ca6Ba(PO4)4O has been demonstrated22,23,24, implying that Ca6Ba(PO4)4O is an interesting host material for luminescent ions. Considering Ca6Ba(PO4)4O structure and composition, one can observe seven electropositive ions (six Ca and one Ba) surrounding the rigidly connected PO4 tetrahedra. Thus, one may assume that the Mn5+ emission, in this new host-activator combination, would be efficient and of high energy due to the nephelauxetic effect. For this reason, we prepared the Ca6Ba(PO4)4O:Mn5+ powder for this research and observed the intense NIR emission under the 650 nm excitation. The emission energy of 8772 cm−1 is among the highest energies detected for Mn5+ activated phosphors. Considering the potential use of Ca6Ba(PO4)4O for both yellow and NIR phosphors and the absence of data on its electronic and vibrational properties, we calculated and explained its electronic band structure, the partial and total density of states (DOS), effective Mulliken charges of all constituent ions, elastic constants, Debye temperature, and vibrational spectra of undoped and Mn5+ doped material. Then, we proceeded with the detailed characterization and analysis of Ca6Ba(PO4)4O:Mn5+ luminescence that includes the measurements of material absorption, excitation and emission spectra, emission decays, concentration quenching, quantum yield, the temperature dependence of emission band shift, bandwidth and decay, and the calculation of the crystal field parameters. This versatile characterization and analysis of Ca6Ba(PO4)4O:Mn5+ undoubtedly confirm the high potential of this material in NIR applications.
Experimental
The conventional solid-state reaction was employed for the preparation of Ca6BaP4-4xMn4xO17 (x = 0.005, 0.0075, 0.0125, 0.01, 0.015, 0.02) powder samples. Stoichiometric amounts of CaCO3 (Alfa Aesar, 98%), BaCO3 (Alfa Aesar, 99.8%), (NH4)H2PO4 (Alfa Aesar, 98%), and MnO (Aldrich, 99.99%) were thoroughly mixed in an agate mortar for 1 h with an appropriate amount of ethanol. Mixtures of the raw materials were placed in alumina crucibles and heated in an air atmosphere at 600 °C for 6 h, ground in an agate mortar, and further calcinated at 1280 °C for 10 h.
The crystal structure of powders was examined by powder X-ray diffraction (PXRD) using the Rigaku SmartLab instrument (Cu-Kα1,2 radiation; λ = 0.1540 nm) at room temperature. Data were recorded over the 6°−130° 2θ range, with a 0.01° step size and 1 min/° counting time. All PXRD data were analyzed by the Rietveld method implemented in TOPAS Academic software25,26. Raman scattering measurements were performed using micro – Raman system TriVista 557 equipped with a triple monochromator and CCD detector (monochromator configuration 900/900/1800 points per millimeter) with 1.5 cm−1 resolution. For excitation, Ar laser line at 514.5 nm has an incident power of less than 60 mW to minimize the heating effect. Laser beam was focused on the samples by means of microscopic lenses with 100× magnification. Spectra were recorded in the range of 100–1200 cm−1. Measurements of diffuse reflectance were performed on the Thermo Evolution 600 spectrometer equipped with an integrated sphere, using BaSO4 as a reference over the 220–1350 nm wavelength range. The photoluminescence emission and emission decays were measured using the FLS1000 Fluorescence spectrometer (Edinburgh Instruments; 0.1 nm spectral resolution) supplied with R5509-72 photomultiplier tube from Hamamatsu in nitrogen-flow cooled housing for near-infrared range detection. For measurements of the emission spectra and decays, the 668 nm laser diode excitation is used in the continuous and pulsed mode, respectively. The temperature of the sample was controlled using a THMS 600 heating-cooling stage from Linkam (0.1 K temperature stability and 0.1 K set point resolution). Emission decays at low temperatures were recorded at 1136 nm for all samples. The luminescence quantum efficiencies were measured using FLS980 Fluorescence Spectrometer from Edinburgh Instruments equipped with 450 W Xenon lamp, R5509-72 photomultiplier tube from Hamamatsu in nitrogen-flow cooled housing for near-infrared range detection, and calibrated integrating sphere for the direct absolute efficiency reading. The Al2O3 powder was used as a scattering reference. Thermometry was performed using a custom-made Peltier-based heating stage in the 20–100 °C range (0.02 °C precision). An Ocean Insight LSM-635A LED was used as the excitation source and is controlled by the Ocean Insight LDC-1 single channel driver and controller. The bifurcation optical Y cable was used for measuring PL emission spectra by Ocean Insight NIRQuest+ Spectrometers.
Results and discussion
The structure of Ca6Ba(PO4)4O and Ca6Ba(PO4)4O:Mn5+
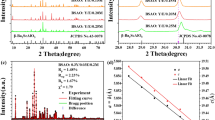
The starting model used for Rietveld refinement and detailed structural analysis of the two key materials – the Ca6Ba(PO4)4O host and the sample containing 0.5%Mn – was the previously published crystal structure of Ca6Ba(PO4)4O determined from synchrotron powder diffraction data22. Refined parameters included the zero-point error, background polynomial terms, peak shape function terms, unit cell parameters, an isotropic atomic displacement parameter per atom type and atomic fractional coordinates, using bond valence sum restraints on the two P atoms. The key crystallographic parameters are summarized in Table 1. Ca6Ba(PO4)4O and Ca6Ba(PO4)4O:0.5%Mn adopt monoclinic space group C2/m, with one Ba, two Ca, two P and seven O atoms in the asymmetric unit. Ba atoms are 12-coordinate, the two crystallographically unique Ca atoms are 7- and 8-coordinate, while both unique P atoms adopt tetrahedral coordination environments. Dopant Mn5+ ions replace P5+ on these two sites, which lie on a mirror plane (Wyckoff site 4 m in space group C2/m). In Ca6Ba(PO4)4O, the average cation-oxygen bond lengths in the two tetrahedra are 1.534(13) and 1.529(15) Å, while bond angles range from 105.1(8) to 112.9(7)° and 105.3(7) to 112.8(9)°, with bond valence sums of 5.0(1) for both P atoms. In Ca6Ba(PO4)4O:0.5%Mn, the coordination environments remain similar, as expected given a low doping level. Average bond lengths are 1.534(22) and 1.504(27) Å, while bond angles range from 103.4(16) to 112.8(12)° and from 105.5(13) to 112.5(11)°, with bond valence sums of 5.0(2) and 5.4(2) for P1 and P2 sites, (see Tables 2 and 3), respectively. The final Rietveld fits obtained are shown in Fig. 1a, b, while the unit cell and the P atom environments are given in Fig. 1c, d, respectively.

a Rietveld fits for Ca6Ba(PO4)4O, Rwp = 5.31%. b Rietveld fit for Ca6Ba(PO4)4O:Mn5+, Rwp = 8.59%. In each case the blue curve represents the observed pattern, the red curve is the calculated pattern and the difference curves are shown in grey, while blue tick marks represent the positions of the Bragg peaks. c The crystal structure of Ca6Ba(PO4)4O, with the unit cell viewed along the b-axis; d Geometries of two PO4 tetrahedra with average P–O bond lengths in Ca6Ba(PO4)4O:Mn5+
Electronic properties
A detailed calculation of the electronic properties of the Ca6Ba(PO4)4O was performed to verify if its electronic structure is suitable to facilitate the Mn5+ emission. The electronic configurations for all chemical elements in Ca6Ba(PO4)4O were as follows: O – 2s22p4, P - 3s23p3, Ca - 3s23p64s2, Ba - 5s25p66s2. The following parameters were used for the calculations: energy 10−5 eV per atom; maximal force 0.03 eVÅ−1; maximal stress 0.05 GPa; maximal displacement 0.001 Å. K-points set was 2 × 2 × 2 for geometry optimization and 3 × 3 × 3 for the DOS calculations; the energy cut-off was 340 eV.
The structures obtained by geometry optimizations using the generalized gradient approximation (GGA) and the local density approximation (LDA) calculations are shown in Table 1 and are in excellent agreement, LDA especially, with the data obtained by Rietveld analysis of PXRD.
The band gaps in both GGA and LDA calculations, Fig. 2a, are direct, equal to 4.365 eV (GGA) and 4.475 eV (LDA) and have similar values to the reported bandgaps of 4.75 eV for the Ca4(PO4)2O and 5.0 eV for the Ba2Ca(PO4)227,28. Since the density-functional theory-based calculations (DFT) always tend to underestimate the true band gaps, the calculated values should be considered as lower band gap estimates.

a The electronic band structure of Ca6Ba(PO4)4O. b Partial and total density of states of Ca6Ba(PO4)4O
The conduction band is due to the Ca 4s, 3d and Ba 6s states, Fig. 2b. The top of the valence band is remarkably flat, thus indicating extremely high effective masses of holes. The valence band consists of two sub-bands, which are separated by a narrow gap of about 0.5 eV. The upper one – from about −4.2 eV to 0 eV is made predominantly by the oxygen 2p states. The lower one (from −4.7 eV to about −7 eV) consists of the O 2p states and P 3p states, highly hybridized with each other. The P 3s states make another narrow band between −7.5 eV and −8.5 eV. The O 2s and P 3s states are spread between −22 eV and −17.5 eV, making several clearly seen maxima. The Ba 5s and 5p states are peaked at about −26 eV and −11 eV, respectively, whereas the Ca 3s and 3p states are localized deep in energy at about −38 eV and −20 eV, respectively.
The considered crystal has a high degree of covalency of chemical bonds, which can be assessed by calculating effective Mulliken charges. Since there are two crystallographically inequivalent P5+ ions, two Ca2+ ions and seven O2− ions and since two types of calculations (GGA/LDA) were run, we give the ranges, in which these charges fall for all ions. They are −1.07-1.09 (in units of proton charge) for the oxygen ions, +2.25 + 2.34 for the phosphorus ions, +1.25 + 1.31 for the calcium ions, and +1.39 + 1.47 for the barium ions. The deviation from the formal charges is especially large for the P and O ions, whereas the Ca and Ba effective charges are closer to their formal ones. This is consistent with the P–O bonds being more covalent than the Ca–O and Ba–O ones.
With the calculated elastic constants, it is possible to estimate the Debye temperature using the following equations:
where h = 6.626 × 10−34 J·s is the Planck’s constant; kB = 1.381 × 10−23 JK−1 is the Boltzmann constant; NA = 6.022 × 1023 mol−1 is the Avogadro’s number; ρ is the crystal’s density; n is the number of atoms per one formula unit (twenty eight in the case of Ca6Ba(PO4)4O), and M is the formula weight. The average, transverse and longitudinal sound velocities are denoted by vm, vt, vl, correspondingly. The B (bulk modulus) and G (shear modulus) values are calculated as the average values of the corresponding Voigt and Reuss (denoted with the V and R subscripts, respectively) values from Table 4. With these equations and calculated elastic parameters, the Debye temperature for Ca6Ba(PO4)4O was estimated to be 496 K (GGA) and 551 K (LDA).
Vibrational spectra
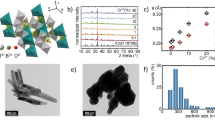
For an undistorted tetrahedron (Td symmetry), the four fundamental vibrational modes (all Raman active) are a1 + e + 2t21,29,30. The ν1(a1) is the totally symmetric stretching mode, ν2(e) is bending deformation, and ν3(t2) and ν4(t2) vibrations are the two stretching t2 modes. With symmetry lowered from Td to Cs, several bands appear for each vibrational mode. Raman scattering spectra of the Ca6Ba(PO4)4O and Ca6Ba(PO4)4O:Mn5+ powders are shown in Fig. 3a, b, respectively. The following assignment is made for the Ca6Ba(PO4)4O: νL (cm−1) = 123, 153, 183, 215.4, 262.5, and 294.7 are the lattice modes; ν1(a1) (cm−1) = 944 and 985.3; ν2(e) = 434.5 (cm−1); ν3(t2) (cm−1) = 1040.1, and 1074.2; ν4(t2) (cm−1) = 584.3 and 608.3. In Raman scattering spectra of the Ca6Ba(PO4)4O:Mn5+ powder, Fig. 3b, additional vibrations from the MnO43− ion are clearly visible: ν1(a1) (cm−1) = 807; ν2(e) (cm−1) = 314.4; ν3(t2) (cm−1) = 851.4; ν4(t2) (cm−1) = 343.9, 356.2, and 368.5; ν1+ νL/ ν3+ νL (cm−1) = 1128.5 and >1128.5. They agree with the data reported by Gonzalez-Vilchez and Griffith29 for vibrational modes of the MnO43− molecular ion.

a Raman scattering spectra of Ca6Ba(PO4)4O powder. b Raman scattering spectra of Ca6Ba(PO4)4O:Mn5+ powder
Photoluminescence properties
Figure 4a, b displays the absorption, color (inset in Fig. 4a), and emission spectra of Ca6Ba(PO4)4O:0.5%Mn5+ powder. The observed absorption and emission, the blue coloration of the Mn5+ doped powder, and the characteristic vibrational modes of MnO43− in the Raman scattering spectrum of Ca6Ba(PO4)4O:Mn5+ (Fig. 3b) all unambiguously demonstrate the presence of Mn5+ in the sample.

a The Kubelka−Munk transformation of the Ca6Ba(PO4)4O:Mn5+ diffuse reflectance The inset shows photographs of the Ca6Ba(PO4)4O (white) and Ca6Ba(PO4)4O:Mn5+ (blue) powders. b Emission spectra measured at −190 °C and 10 °C. c Tanabe–Sugano diagram for 3d2 electron configuration in tetrahedral coordination. d Emission spectrum of the Ca6Ba(PO4)4O:Mn5+ measured at room temperature (black line) and the fit to the Gaussian of the 3T2 →3A2 emission peak (red line) showing its maximum 1062 nm/9416 cm−1. Spectra shown in logarithmic scale. e Temperature dependence of the excited state lifetime (symbols – experimental data, solid line – the fit to Eq. (4)). The inset shows emission decay measured at 208 K. f Temperature dependence of the 1E emission peak spectral position (symbols – experimental data, solid line – the fit to Eq. (5)). g The estimate of configurational diagram based on the spectroscopic data with calculated Stokes shift (EStokes) and Huang–Rhys parameter (S). h Photoluminescence internal quantum efficiency (QE) of Ca6Ba(PO4)4O:Mn5+ powders for different concentrations of Mn. The inset shows linear dependence of the log10(QE/concentration) vs log10(concentration) for data equal and above critical concentration (0.75%) with a slope of −1.97 indicating that a multipolar dipole-dipole mechanism is responsible for the concentration quenching of emission. i The linear dependence of Ca6Ba(PO4)4O:Mn5+ emission intensity on excitation power
Figure 4a depicts the Kubelka–Munk transformation of the Ca6Ba(PO4)4O:Mn5+ powder diffuse reflection measured between 220 and 1350 nm. The O2− → Mn5+ charge-transfer band appears at around 301 nm (33,222 cm−1) as expected for the tetraoxo-coordinated Mn5+ 31 and the peak at the lower wavelength (225 nm) is associated with the intrinsic host absorption. The strong absorption around 639 nm (15,649.5 cm−1) is associated with the 3A2 → 3T1(3F) electronic transition, which is electric dipole-allowed in an undistorted tetrahedral symmetry and is composed of three overlapping components due to the removal of the orbital degeneracy of the 3T1(3F) state with the site symmetry lowering from Td to Cs. The weak shoulder at about 943 nm (10,604.5 cm−1) corresponds to the symmetry forbidden 3A2 → 3T2(3F) transition (in Td site symmetry) and becomes partially allowed with a symmetry lowering. The electric dipole-allowed 3A2 → 3T1(3P) transition that corresponds to a two-electron jump is located at approximately 369 nm (27,100 cm−1) and is barely visible due to the much more intense charge transfer band. The spin-forbidden transitions to the singlet states 1A1(1G) at 740 nm (13,513.5 cm−1) and 1E(1D) at 1140 nm (8772 cm−1) are weak, sharp, and only weakly depend on the host materials properties. The transitions to 1T1,2 singlet states are difficult to observe in the spectrum since they are very weak and superimposed on the main and stronger bands.
Emission spectra of Ca6Ba(PO4)4O:Mn5+ powder measured at −190 °C and 10 °C are shown in Fig. 4b with blue and red lines, respectively, and are typical for emissions from transitions of 3d2 electronic configuration in a tetrahedral environment as described by the Tanabe–Sugano diagram, Fig. 4c. The spectra show ultranarrow emission bands (FWHM = 3 nm (20 cm−1) at −190 °C; FWHM = 5 nm (35 cm−1) at 10 °C) from the 1E → 3A2 intraconfigurational transition (1140 nm), followed by vibrational sidebands with progressions of ≈320 cm−1 ((ν2(e)) and ≈800 cm−1 ((ν1(a1)). This indicates the coupling of the 1E excited state and the non-totally symmetric ν2(e) mode of MnO43−, i.e., a dynamic Jahn–Teller effect.
The very small splitting of 1E emission band is due to only weakly distorted MnO4 tetrahedra (see Table 3) and it is barely visible with our instrument resolution at the emission spectrum measured at low temperatures (−190 °C). The low-intensity broad emission band from the 3T2(3F) → 3A2 transition is centered at 1062 nm (9416 cm−1) and can be resolved only spectral deconvolution, Fig. 4d.
Temperature dependence of the 1E lifetime and emission peak spectral position are shown in Fig. 4e, f, respectively. The 1E level emission decays show lifetime values of about 350 μs at room temperature and 560 μs at low temperatures. The temperature dependence of lifetime, Fig. 4e, shows that a low-temperature lifetime value is approximately the value of a radiative lifetime. Considering that the excited state is separated in energy from the ground state by 8772 cm−1, almost eight quanta of the highest vibrational frequencies of the phosphor (≈1100 cm−1) are needed to bridge the gap. Thus, a multiphonon non-radiative relaxation is not probable as the emission quenching mechanism. The 1E emission deactivation through the crossing with a charge transfer band is also not probable due to very high energy difference. Therefore, we assume that the thermal quenching of the 1E state population takes place by a thermally activated cross-over via 3T2 state, see Fig. 4g, similarly to Mn4+ activated phosphors. The temperature dependence of the emission lifetime, shown in Fig. 4e, can be described by the following equation32,33,34,35:
where \(\tau _{R0}\) = 560 ± 19 μs is the radiative lifetime at T = 0 K, kB = 0.69503476 cm−1K−1 is the Boltzmann constant, \(h\nu\) = 448 ± 90 cm−1 is the average energy of phonon coupled to the 1E → 3A2 transition, 1/\(\tau _{NR}\) = 1527 ± 120 ms−1 is the nonradiative decay rate, \(\Delta E\) = 1631 ± 200 cm−1 is the activation energy of the process (the cross-over via the 3T2 state), and T represents the temperature. The smaller the configuration coordinate parabola offset between the ground state (3A2) and the 3T2 state, the larger the cross-over energy \(\Delta E\) (activation energy of the process) needed to activate the non-radiative de-excitation process. Thus, the 1E → 3A2 emission of Mn5+ activated phosphors, which have large 3T2 energies and smaller Stokes shifts, will start to quench at higher temperatures.
The shift of 1E emission band with energy is shown in Fig. 4f. It can be described by the following equation:36,37
where \(\theta _D\) = 783 ± 12 K is the Debye temperature of the host material, \(x = \hbar \omega _D/k_BT = \theta _D/T\), \(\omega _D\) is Debye cut-off frequency, and α = −650 ± 17 cm−1 represents the electron-phonon coupling coefficient. The relatively high Debye temperature indicates a rigid structure which favors efficient emissions from optical centers38.
The concentration dependence of an internal quantum efficiency (QE) is given in Fig. 4h. The largest value of 37.5 ± 2.0% is recorded for the 0.5% Mn5+ doped sample, after which the concentration quenching of emission occurs. This is a relatively high value for an NIR-emitting phosphor, and comparable to one obtained in Ba3(PO4)27. The log10(QE/concentration) vs log10(concentration) plot has a −1.97 slope, which is close to −2, which undoubtedly indicates that a multipolar electric dipole-dipole mechanism is responsible for the concentration quenching of emission. The linear dependence of Ca6Ba(PO4)4O:Mn5+ emission intensity on excitation power, Fig. 4i, is expected for the typical downshifting photoluminescence emission process.
The 3d2 electronic configuration of Mn5+ in a tetrahedral environment is described by the Tanabe–Sugano model for 3d8 electronic configuration in octahedral symmetry39, see Fig. 4c. The crystal field and Racah parameters are calculated from the following equations using data from diffuse reflection and emission spectra40,41:
By comparing the obtained Dq, B and C parameters with literature data, Table 5, one can observe that Ca6Ba(PO4)4O provides the smallest Dq and the largest B parameters amongst all phosphate hosts, and that Li3VO4 is the only host with a smaller Dq (considering available data).
By considering the obtained parameters and the configuration coordinate diagram, Fig. 4g, the relatively small value of Huang–Rhys parameter S = 0.53 is found for the assumed coupling to the ν1+ νL/ ν3+ νL vibrational mode with energy \(\hbar \omega = 1128.5\;{{{\mathrm{cm}}}}^{ - 1}\).
The Slater parameters are calculated from Racah parameters by the simple relations42,43:
Both values are considerably reduced from the free-ion values of \(F^{\left( 2 \right)} = 91427{{{\mathrm{cm}}}}^{ - 1}\) and \(F^{(4)} = 56625\;{{{\mathrm{cm}}}}^{ - 1}\)18.
As it follows from the Tanabe–Sugano diagram for the 3d2 configuration in the tetrahedral crystal field (Fig. 4c), the energy separation between the ground state 3A2 and the first excited state 1E (in the strong crystal field) is practically independent on the crystal field strength (both states are parallel to each other). At the same time, this energy interval is very close to the energy interval between the 3F and 1D states of the free ion, which is determined by the Racah parameters B and C, which vary from host to host because of the covalent effects. As a result, the nephelauxetic effect is dominating in this case.
Application in luminescence thermometry
We have tested the performance of Mn5+ activated Ca6Ba(PO4)4O (the sample containing 0. 5% Mn since it showed the best quantum efficiency) as a NIR luminescent thermometer operating in the second biological window and in the physiological temperature range. As can be seen from Fig. 5a, when temperature increases, the broad emission peak from the 3T2 level in the 950 nm to 1030 nm range also increases in intensity, while the intensity of the narrow emission peak from the 1E level around 1140 nm decreases with temperature. This occurs due to thermalization between 1E and 3T2 levels where the energy difference between these two levels (\(\Delta E_T\)) is bridged by thermally exited electrons. Consequently, a simple Boltzmann-type relation for the luminescence intensity ratio (LIR) between the two abovementioned emission intensities applies44,45:
where B is a temperature-invariant constant and T represents temperature. The fit of Eq. (13) (full line, Fig. 5b) to experimental LIR data (diamond markers, Fig. 5b) is almost perfect (R2 = 0.997) and provides an energy difference \(\Delta E_T\;{{{\mathrm{of}}}}\;1216\;{{{\mathrm{cm}}}}^{ - 1}\) that agrees with the energy difference obtained from spectroscopy (Fig. 4g). To experimentally determine the uncertainty in the LIR (error budget), 50 emission spectra were acquired at each temperature. Then, the measurement distribution mean was used as the LIR value while a standard deviation (\(\sigma _{LIR}\)) was used as an uncertainty in LIR as shown in the insert of Fig. 5b on the LIR value distribution measured at 30 °C).

a Photoluminescence emission spectra of Ca6Ba(PO4)4O:Mn5+ powder measured at different temperatures. b Luminescence intensity ratio (LIR) as a function of temperature (experimental data - diamond markers; fit to Eq. (13) - full line). The insert shows the LIR distribution histogram measured at 303.15 K (30 °C) – filled diamond marker. c Calculated absolute and relative sensitivities (marked values at 303.15 K (30 °C)). d Experimentally obtained values for temperature resolution – filled diamond marker represents the value at 303.15 K (30 °C)
The absolute (Sa) and relative (Sr) sensitivities of the thermometer were then calculated from the following equations:
and presented in Fig. 5c (blue dots represent values obtained at a temperature of 30 °C). The relative sensitivity value varies from 2.35%K−1 to 1.26%K−1 over the measurement range, being 1.92%K−1 at 30 °C. These are relatively high values46, especially for luminescence thermometers operating in the second biological transparency window (>1000 nm). For example, Gschwend et al.47 achieved a relative sensitivity of 0.43%K−1 for an LIR thermometer based on Mn5+-activated Ba3(PO4)2, while Shen et al.48 achieved a relative sensitivity of 1.3%K−1 for an LIR thermometer based on Ag2S quantum dots.
The temperature resolution (uncertainty in measured temperature, \(\delta T\)) is determined as a ratio between experimentally obtained LIR uncertainty (\(\sigma _{LIR}\)) and absolute sensitivity (\(S_a\)) for a given temperature, Fig. 5d:
and it has an average value of 0.21 K. Finally, repeatability of measurement (RM) is quantified as46:
where \(\overline {LIR}\) is the average LIR measured at a certain temperature over all \(LIR_i\) acquired. Based on experimental data, an RM value of 0.97 (97%) is obtained.
Conclusion
Because of its rigid structure, appropriate crystal sites for doping, and sufficiently large energy band gap to accommodate the energy levels of dopant ions, Ca6Ba(PO4)4O is an excellent host for Eu2+, Sm2+ and Mn5+ luminescence centers. In this host, Mn5+ provides ultranarrow emission in the near-infrared spectral range at 1140 nm that can be easily excited over the broad spectral range that spans 500–1000 nm covering the entirety of the first biological transparency window and making this material an excellent near-infrared phosphor and non-toxic blue/turquoise pigment. The phosphor has an internal quantum efficiency of 37.5%, which is a high value considering the quantum efficiencies of inorganic NIR phosphors. The thermal quenching of the 1E emission takes place by a thermally activated cross-over via the 3T2 state with an activation energy of 1631 cm−1. The optimal Mn5+ doping concentration is 0.5%. For higher doping concentrations, quantum efficiency decreases due to non-radiative deexcitation caused by a dipole-dipole electric interaction. Based on the available literature data, the Ca6Ba(PO4)4O:Mn5+ phosphor provides the smallest Dq and the largest B parameters amongst all phosphate hosts. This material is one of the best single-doped ratiometric luminescence thermometry sensors for use in a second biological transparency window due to the opposite temperature dependence of the 1E and 3T2 emission intensities. It provides a relative sensitivity of 1.92%K−1 and a temperature resolution of 0.2 K in the range of physiological temperatures, with a measurement repeatability of 97%. These findings, particularly the high value of quantum efficiency and strong absorption, tuneability of emission wavelength by changing the Mn5+ environment, long emission decay times that allow for time-gated measurements, and strong temperature susceptibility of emission, demonstrate the great potential of Mn5+-activated phosphors for NIR applications. When reduced to nano dimension, Ca6Ba(PO4)4O:Mn5+ has great potential for bioimaging and biothermal imaging applications in the second biological transparency window. Future research will concentrate on the synthesis and applications of Ca6Ba(PO4)4O:Mn5+ nanoparticles.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Capobianco, J. A. et al. Near-infrared intraconfigurational luminescence spectroscopy of the Mn5+ (3d2) ion in Ca2PO4Cl, Sr5(PO4)3Cl, Ca2VO4Cl and Sr2VO4Cl. J. Lumin. 54, 1–11, https://doi.org/10.1016/0022-2313(92)90043-9 (1992).
Merkle, L. D. et al. Laser action from Mn5+ in Ba3(VO4)2. Appl. Phys. Lett. 61, 2386–2388, https://doi.org/10.1063/1.108172 (1992).
Moncorgé, R., Manaa, H. & Boulon, G. Cr4+ and Mn5+ active centers for new solid state laser materials. Optical Mater. 4, 139–151, https://doi.org/10.1016/0925-3467(94)90071-X (1994).
Merkle, L. D., Guyot, Y. & Chai, B. H. T. Spectroscopic and laser investigations of Mn5+:Sr5(VO4)3F. J. Appl. Phys. 77, 474–480, https://doi.org/10.1063/1.359585 (1995).
Ma, L. J. et al. Systematic discovery about NIR spectral assignment from chemical structural property to natural chemical compounds. Sci. Rep. 9, 9503, https://doi.org/10.1038/s41598-019-45945-y (2019).
Gschwend, P. M. et al. Bi2O3 boosts brightness, biocompatibility and stability of Mn-doped Ba3(VO4)2 as NIR-II contrast agent. J. Mater. Chem. B 9, 3038–3046, https://doi.org/10.1039/d0tb02792h (2021).
Cao, R. P. et al. Near-infrared emission Ba3(PO4)2:Mn5+ phosphor and potential application in vivo fluorescence imaging. Spectrochimica Acta Part A 128, 671–673, https://doi.org/10.1016/j.saa.2014.02.081 (2014).
Medina, E. A. et al. Intense turquoise colors of apatite-type compounds with Mn5+ in tetrahedral coordination. Solid State Sci. 52, 97–105, https://doi.org/10.1016/j.solidstatesciences.2015.12.001 (2016).
Laha, S. et al. Stabilization of a tetrahedral (Mn5+O4) chromophore in ternary barium oxides as a strategy toward development of new turquoise/green-colored pigments. Inorg. Chem. 55, 3508–3514, https://doi.org/10.1021/acs.inorgchem.5b02957 (2016).
Wang, Y. Q. et al. Synthesis and optical properties of intense blue colors oxides based on Mn5+ in tetrahedral sites in Ba7Al2-xMnxO10+y. Ceram. Int. 47, 686–691, https://doi.org/10.1016/j.ceramint.2020.08.177 (2021).
Cui, K. Y. et al. Synthesis and properties of novel inorganic green chromophore based on Bi24Al2-xMnxO39. Ceram. Int. 47, 8944–8948, https://doi.org/10.1016/j.ceramint.2020.12.016 (2021).
Zhou, Y. C. et al. Synthesis and properties of novel turquoise-green pigments based on BaAl2-xMnxO4+y. Dyes Pigments 155, 212–217, https://doi.org/10.1016/j.dyepig.2018.03.042 (2018).
Dardenne, K., Vivien, D. & Huguenin, D. Color of Mn(V)-substituted apatites A10((B, Mn)O4)6F2, A = Ba, Sr, Ca; B = P, V. J. Solid State Chem. 146, 464–472, https://doi.org/10.1006/jssc.1999.8394 (1999).
Zhou, Q. et al. Mn2+ and Mn4+ red phosphors: synthesis, luminescence and applications in WLEDs. A review. J. Mater. Chem. C 6, 2652–2671, https://doi.org/10.1039/C8TC00251G (2018).
Hazenkamp, M. F. et al. Excited state absorption and laser potential of Mn5+-doped Li3PO4. Chem. Phys. Lett. 265, 264–270, https://doi.org/10.1016/S0009-2614(96)01434-0 (1997).
Kück, S., Schepler, K. L. & Chai, B. H. T. Evaluation of Mn5+-doped Sr5(VO4)3F as a laser material based on excited-state absorption and stimulated-emission measurements. J. Optical Soc. Am. B 14, 957–963, https://doi.org/10.1364/JOSAB.14.000957 (1997).
Herren, M. et al. Near-infrared luminescence of manganate(V)-doped phosphates and vanadates. J. Luminescence 53, 452–456, https://doi.org/10.1016/0022-2313(92)90196-G (1992).
Capobianco, J. A. et al. Crystal-field analyis of Mn5+ (3d2) in Sr5(PO4)3Cl. Optical Mater. 1, 209–216, https://doi.org/10.1016/0925-3467(92)90029-M (1992).
Hömmerich, U. et al. The optical center MnO43- in Y2SiO5:Mn, X (X=Al, Ca). Chem. Phys. Lett. 213, 163–167, https://doi.org/10.1016/0009-2614(93)85435-Q (1993).
Zhang, X. W. et al. Structural variation and near infrared luminescence in Mn5+-doped M2SiO4 (M = Ba, Sr, Ca) phosphors by cation substitution. J. Mater. Sci. Mater. Electron. 29, 6419–6427, https://doi.org/10.1007/s10854-018-8622-2 (2018).
Kim, S. W. et al. Discovery of novel inorganic Mn5+-doped sky-blue pigments based on Ca6BaP4O17: crystal structure, optical and color properties, and color durability. Dyes Pigments 139, 344–348, https://doi.org/10.1016/j.dyepig.2016.12.038 (2017).
Komuro, N. et al. Synthesis, structure and optical properties of europium doped calcium barium phosphate – a novel phosphor for solid-state lighting. J. Mater. Chem. C 2, 6084–6089, https://doi.org/10.1039/c4tc00732h (2014).
Ji, H. P. et al. Facile solution-precipitation assisted synthesis and luminescence property of greenish-yellow emitting Ca6Ba(PO4)4O:Eu2+ phosphor. Mater. Res. Bull. 75, 233–238, https://doi.org/10.1016/j.materresbull.2015.11.055 (2016).
Meng, Y. J. et al. Thermal stability of the luminescence of Ca6Ba(PO4)4O:Eu2+ yellow phosphor for white light-emitting diodes. Appl. Phys. A 122, 636, https://doi.org/10.1007/s00339-016-0141-5 (2016).
Rietveld, H. M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 2, 65–71, https://doi.org/10.1107/S0021889869006558 (1969).
Coelho, A. A. et al. The TOPAS symbolic computation system. Powder Diffr. 26, S22–S25, https://doi.org/10.1154/1.3661087 (2011).
Deng, D. G. et al. Ca4(PO4)2O:Eu2+ red-emitting phosphor for solid-state lighting: structure, luminescent properties and white light emitting diode application. J. Mater. Chem. C 1, 3194–3199, https://doi.org/10.1039/C3TC30148F (2013).
Yu, H. et al. Ba2Ca(PO4)2:Eu2+ emission-tunable phosphor for solid-state lighting: luminescent properties and application as white light emitting diodes. J. Mater. Chem. C 1, 5577–5582, https://doi.org/10.1039/C3TC30998C (2013).
Gonzalez-Vilchez, F. & Griffith, W. P. Transition-metal tetra-oxo-complexes and their vibrational spectra. J. Chem. Soc. Dalton Trans. 1416–1421, https://doi.org/10.1039/DT9720001416 (1972).
Griffith, W. P. Raman studies on rock-forming minerals. Part II. Minerals containing MO3, MO4, and MO6 groups. J. Chem. Soc. A 286-291, https://doi.org/10.1039/J19700000286 (1970).
Lachwa, H. & Reinen, D. Color and electronic structure of manganese(V) and manganese(VI) in tetrahedral oxo coordination. A spectroscopic investigation. Inorg. Chem. 28, 1044–1053, https://doi.org/10.1021/ic00305a011 (1989).
Ristić, Z. et al. Near-infrared luminescent lifetime-based thermometry with Mn5+-activated Sr3(PO4)2 and Ba3(PO4)2 phosphors. ACS Appl. Electron. Mater. 4, 1057–1062, https://doi.org/10.1021/acsaelm.1c01207 (2022).
Senden, T., van Dijk-Moes, R. J. A. & Meijerink, A. Quenching of the red Mn4+ luminescence in Mn4+-doped fluoride LED phosphors. Light Sci. Appl. 7, 8, https://doi.org/10.1038/s41377-018-0013-1 (2018).
Dramićanin, M. D. et al. Li2TiO3:Mn4+ deep-red phosphor for the lifetime-based luminescence thermometry. Chem. Select 4, 7067–7075, https://doi.org/10.1002/slct.201901590 (2019).
Srivastava, A. M. et al. Unusual luminescence of octahedrally coordinated divalent europium ion in Cs2M2+P2O7 (M2+=Ca, Sr). J. Luminescence 129, 919–925, https://doi.org/10.1016/j.jlumin.2009.03.018 (2009).
McCumber, D. E. & Sturge, M. D. Linewidth and temperature shift of the R lines in ruby. J. Appl. Phys. 34, 1682–1684, https://doi.org/10.1063/1.1702657 (1963).
Yen, W. M., Scott, W. C. & Schawlow, A. L. Phonon-induced relaxation in excited optical states of trivalent praseodymium in LaF3. Phys. Rev. J. Archive 136, A271–A283, https://doi.org/10.1103/PhysRev.136.A271 (1964).
Pust, P. et al. Narrow-band red-emitting Sr[LiAl3N4]:Eu2+ as a next-generation LED-phosphor material. Nat. Mater. 13, 891–896, https://doi.org/10.1038/nmat4012 (2014).
Tanabe, Y. & Sugano, S. On the absorption spectra of complex ions. I. J. Phys. Soc. Jpn 9, 753–766, https://doi.org/10.1143/JPSJ.9.753 (1954).
Sugano, S., Tanabe, Y. & Kamimura, H. Multiplets of transition-metal ions in crystals (New York: Academic Press, 1970).
Brik, M. G. et al. Spin-forbidden transitions in the spectra of transition metal ions and nephelauxetic effect. ECS J. Solid State Sci. Technol. 5, R3067–R3077, https://doi.org/10.1149/2.0091601jss (2016).
Schmidtke, H. H. The variation of slater-condon parameters Fk and racah parameters B and C with chemical bonding in transition group complexes. In: Optical spectra and chemical bonding in inorganic compounds (eds Mingos, D. M. P. & Schönherr, T.) 19–35 (Springer, Berlin, Heidelberg, 2004) https://doi.org/10.1007/b11303.
Racah, G. Theory of complex spectra. III. Phys. Rev. J. Arch. 63, 367–382, https://doi.org/10.1103/PhysRev.63.367 (1943).
Dramićanin, M. Lanthanide and transition metal ion doped materials for luminescence temperature sensing. In: Luminescence thermometry: methods, materials, and applications (ed. Dramićanin, M.) (Elsevier, Amsterdam, 2018), https://doi.org/10.1016/B978-0-08-102029-6.00006-3.
Marciniak, L. et al. Luminescence thermometry with transition metal ions. A review. Coordination Chem. Rev. 469, 214671, https://doi.org/10.1016/j.ccr.2022.214671 (2022).
Dramićanin, M. D. Trends in luminescence thermometry. J. Appl. Phys. 128, 040902, https://doi.org/10.1063/5.0014825 (2020).
Gschwend, P. M. et al. Simultaneous nanothermometry and deep‐tissue imaging. Adv. Sci. 7, 2000370, https://doi.org/10.1002/advs.202000370 (2020).
Shen, Y. L. et al. Ag2S nanoheaters with multiparameter sensing for reliable thermal feedback during in vivo tumor therapy. Adv. Funct. Mater. 30, 2002730, https://doi.org/10.1002/adfm.202002730 (2020).
Brik, M. G. et al. Crystal field parameters and energy level structure of the MnO43- tetroxo anion in Li3PO4, Ca2PO4Cl and Sr5(PO4)3Cl crystals. J. Luminescence 129, 801–806, https://doi.org/10.1016/j.jlumin.2009.02.018 (2009).
Shen, Y. R., Riedener, T. & Bray, K. L. Effect of pressure on site-symmetry distortions of Mn5+ and Cr4+ in Y2SiO5. Phys. Rev. B 61, 9277–9286, https://doi.org/10.1103/PhysRevB.61.9277 (2000).
Brik, M. G. et al. Spectroscopic and crystal field studies of YAlO3 single crystals doped with Mn ions. J. Phys. Condensed Matter 21, 025404, https://doi.org/10.1088/0953-8984/21/2/025404 (2009).
Scott, M. A. et al. Optical spectroscopy of (MnO4)3- and (VO4)5- in Sr10(VO4)6F2. J. Phys. Condensed Matter 9, 9893–9908, https://doi.org/10.1088/0953-8984/9/45/017 (1997).
Andreici, E. L. Modeling of crystal field and spin-hamiltonian parameters for tetrahedral coordinated Mn5+ doped In Li3VO4. AIP Conf. Proc. 1472, 101–107, https://doi.org/10.1063/1.4748075 (2012).
Acknowledgements
The authors from Serbia acknowledge funding from the Ministry of Education, Science, and Technological Development of the Republic of Serbia. M.D.D. recognizes the support from the Chinese National Foreign Experts Program for “Belt and Road Initiative” Innovative Talent Exchange (Grant No. DL2021035001L). M.G.B. thanks the support from the Program for Foreign Experts (Grant No. W2017011) offered by Chongqing University of Posts and Telecommunications, Estonian Research Council grant PUT PRG111, European Regional Development Fund (TK141), NCN project 2018/31/B/ST4/00924 and 2021/40/Q/ST5/00336. C.-G.M. acknowledges the support of the National Natural Science Foundation of China (Grant No. 52161135110) and China-Poland Intergovernmental Science and Technology Cooperation Program (Grant No. 2020[15]/10).
Author information
Authors and Affiliations
Contributions
M.D.D.: Conceptualization, Methodology, Formal analysis, Writing- Original draft preparation; Writing- Revised draft preparation. Ł.M., Z.R., V.Đ.: Investigation, Formal analysis. Z.R., I.E., M.G.B.: Investigation, Formal analysis, Writing- Revised draft preparation. S.K., W.P., J.P., J.M., N.R., C.-G.M.: Investigation.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dramićanin, M.D., Marciniak, Ł., Kuzman, S. et al. Mn5+-activated Ca6Ba(PO4)4O near-infrared phosphor and its application in luminescence thermometry. Light Sci Appl 11, 279 (2022). https://doi.org/10.1038/s41377-022-00958-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41377-022-00958-7
