
Abstract
Near-infrared (NIR)-emitting phosphor-converted light-emitting diodes have attracted widespread attention in various applications based on NIR spectroscopy. Except for typical Cr3+-activated NIR-emitting phosphors, next-generation Cr3+-free NIR-emitting phosphors with high efficiency and tunable optical properties are highly desired to enrich the types of NIR luminescent materials for different application fields. Here, we report the Fe3+-activated Sr2−yCay(InSb)1−zSn2zO6 phosphors that exhibit unprecedented long-wavelength NIR emission. The overall emission tuning from 885 to 1005 nm with broadened full-width at half maximum from 108 to 146 nm was realized through a crystallographic site engineering strategy. The NIR emission was significantly enhanced after complete Ca2+ incorporation owing to the substitution-induced lower symmetry of the Fe3+ sites. The Ca2InSbO6:Fe3+ phosphor peaking at 935 nm showed an ultra-high internal quantum efficiency of 87%. The as-synthesized emission-tunable phosphors demonstrated great potential for NIR spectroscopy detection. This work initiates the development of efficient Fe3+-activated broadband NIR-emitting phosphors and opens up a new avenue for designing NIR-emitting phosphor materials.
Similar content being viewed by others

Introduction
Near-infrared (NIR) light has been widely used in plant cultivation, night vision, food analysis, photovoltaics, and biomedicine1,2,3. With the increasing popularity of smart devices, it is necessary to develop portable NIR light sources. Traditional tungsten–halogen lamps are not compact, and NIR light-emitting diodes (LEDs) suffer from narrow spectral bandwidths4,5. Therefore, NIR-emitting phosphor-converted LEDs (pc-LEDs) have attracted great attention because of their small size, high efficiency, and tunable broadband emission6,7. However, exploitation of efficient and broadband NIR-emitting phosphors is one of the key obstacles encountered in the development of NIR-emitting pc-LEDs. The currently reported broadband NIR-emitting phosphors are mainly based on Cr3+ because it can usually produce broadband emission in the range 650–1200 nm when it is located in a weak octahedral coordination crystal field8,9,10,11. In addition, it can efficiently absorb at 460 nm due to a spin-allowed 4A2g → 4T1g transition, which matches well with the commercial blue LED chips. Moreover, substantial progress has been achieved in the tunable NIR luminescence of Cr3+-doped phosphor materials12,13,14. However, there is a potential risk of oxidation of Cr3+ to Cr6+ 15,16. On the one hand, mixed Cr6+ seriously affects the NIR luminescence efficiency17. On the other hand, this might increase the chromium toxicity of the phosphors, thereby limiting their practical applications in certain fields, particularly in long-term in vivo applications18. Therefore, there is an urgent requirement to find alternatives to the Cr3+ activator to achieve NIR emission. Recently, several studies have focused on Bi3+-, Eu2+-, and Mn2+-activated NIR-emitting phosphors15,16,19. Their emission wavelengths are near the deep-red light region, which has inferior penetration ability in biological tissues. Hence, efforts should be made to further tune the emission to a longer wavelength.
Another activator, Fe3+, is an essential element ion of the human body; moreover, it is non-toxic and can be regarded as a friendly dopant20,21. Thus, its optical properties are worth exploring. Although the luminescence of Fe3+ with intraconfigurational d–d transitions has been reported, the emission commonly occurs in the red and far-red light regions depending on the local environment of Fe3+ in the host materials22,23,24,25,26. Almost no Fe3+-doped phosphors exhibit NIR emission exceeding 800 nm, except for the recently reported CaAl12O19:Fe3+ (808 nm), SrAl12O19:Fe3+ (811 nm), and CaGa2O4:Fe3+ (809 nm)27,28,29. Typically, tetrahedrally coordinated Fe3+ ions emit in the 650−750 nm range, whereas octahedrally coordinated ones are expected to exhibit longer NIR emission wavelengths30. However, the d–d transitions of Fe3+ in the octahedral sites are more strictly restricted by the Laporte selection rule because of the higher symmetry of the octahedra23,31. Therefore, the achievement of long-wavelength NIR emission of Fe3+ with high luminescence efficiency is a challenging task.
The double-perovskite compounds generally show excellent optical properties, physical stability, and chemical stability32. NIR emission of Cr3+ has been obtained in the double-perovskite structures6,33. The double-perovskite hosts with typically octahedral sites have never been used for Fe3+ doping, the crystal structures of which can be flexibly modulated by cation substitution. Thus the tunable luminescence of Fe3+ can be expected. Furthermore, the octahedral In3+ sites are common for Cr3+ doping7,34,35. Given that Fe3+ and Cr3+ have the same valence state and similar ionic radii, an In-based double-perovskite Sr2InSbO6 was chosen as the initial host for Fe3+ doping in this work. Here, a series of Fe3+-activated Sr2-yCay(InSb)1−zSn2zO6 NIR-emitting phosphors with uncommon long-wavelength NIR emission of Fe3+ were synthesized. Tunable emission from 885 to 935 and then up to 1005 nm were achieved by the premeditated cation substitution of Ca2+ for Sr2+ and further cosubstitution of [Sn4+–Sn4+] for [In3+–Sb5+]. The full-width at half maximum (FWHM) was broadened from 108 to 146 nm during this process. The complete introduction of Ca2+ significantly improved the luminescence efficiency, thereby reaching an ultra-high internal quantum efficiency (IQE) of 87% for Ca2InSbO6:Fe3+. The structure-related emission-tunable properties and the corresponding luminescence mechanism were analyzed. The feasibility of the as-synthesized phosphors in applications such as night vision, nondestructive biological monitoring, and NIR spectroscopy detection were also investigated.
Results
Crystal structure and phase identification
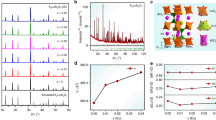
As shown in Fig. 1a, Sr2InSbO6 (SISO) adopts a double-perovskite structure with the general formula A2BB’O6, which consists of alternately arranged [InO6] and [SbO6] octahedra with larger Sr cations occupying the voids between the octahedra. The cation substitution of Ca2+ for Sr2+ at A sites leads to composition transformation from SISO to Ca2InSbO6 (CISO), followed by the cosubstitution of [Sn4+–Sn4+] for [In3+–Sb5+] at B and B’ sites to obtain the CaSnO3 (CSO) phase. The detailed substitution process was revealed by the X-ray diffraction (XRD) patterns of Sr2-yCayInSbO6:Fe3+ (y = 0–2) and Ca2(InSb)1-zSn2zO6:Fe3+ (z = 0–1) (Fig. S1). The Bragg reflections shifting toward higher angles indicates the lattice shrinkage, which is attributed to the smaller Ca2+ and [Sn4+–Sn4+] substituting Sr2+ and [In3+–Sb5+], respectively (Table S1). To verify the phase purity of the as-synthesized phosphors, the Rietveld refinement was performed, as shown in Figs. 1b, S2, and S3. The refined crystallographic parameters and main bond lengths are listed in Tables S2 and S3, respectively. As expected, the cell parameters (a, b, c) and volume (V) decreased with the introduction of Ca2+ and Sn4+ (Figs. 1b and S4), demonstrating the lattice shrinkage. These results also indicate the successful realization of the designed substitution. The elemental mappings of scanning electron microscopy (SEM) in Fig. S5 show the evenly distributed composition elements of SISO:Fe3+, CISO:Fe3+, and CSO:Fe3+. Figure 1c shows the high-resolution transmission electron microscopy (HRTEM) images of SISO:Fe3+ and CISO:Fe3+. The (101) and (121) crystal planes were differentiated in the selected area electron diffraction (SAED) patterns. The interplanar spacing value of the (101) plane was decreased from 4.07 Å to 3.93 Å, which further confirmed the lattice shrinkage after Ca2+ completely substituted Sr2+. Structural changes in Sr2-yCayInSbO6:Fe3+ were also reflected in the Raman spectra (Fig. 1d). The strong bands in the 200–350 cm−1 region are assigned to the stretching vibrations of Sr/Ca–O36,37. Peak broadening and splitting appeared after Ca2+ incorporation, indicating a reduced structural symmetry. Furthermore, the peak shifting to higher wavenumbers is associated with the shortened Sr/Ca–O bond lengths, as derived from the Rietveld refinements (Table S3). Another intense band (700–800 cm−1) can be attributed to the symmetric stretching of the [BO6] and [B’O6] octahedra38, which shifts to lower wavenumbers with Ca2+ substituting Sr2+. A similar phenomenon was found for A2NdSbO6 (A = Ba, Sr, Ca), which is attributed to the increase in the Sb–O bond length39. These structural changes lay the foundation for the luminescence tuning of Fe3+.

a Schematic illustration of the composition transformation from SISO to CISO and to CSO via cation substitution. b XRD Rietveld refinement of SISO:Fe3+. The inset shows the cell parameters and volume variations of Sr2-yCayInSbO6:Fe3+ (y = 0–2). c HRTEM images with the SAED patterns of SISO:Fe3+ and CISO:Fe3+. d Raman spectra of Sr2-yCayInSbO6:Fe3+ (y = 0–2)
Photoluminescence properties
X-ray absorption fine structure (XAFS) was investigated to analyze the local coordination environment of Fe3+. As these hosts were isomorphic, only the coordination environment of Fe3+ in CISO was analyzed. In order to obtain the clear signal of Fe, Ca2In0.88SbO6:0.12Fe3+ with a larger Fe doping concentration was chosen to obtain the XAFS data, as shown in Fig. 2a. The Fe K-edge X-ray absorption near-edge structure (XANES) spectrum (the inset of Fig. 2a) of the sample is close to that of the standard Fe2O3, indicating that the valence state of iron in the sample is mainly trivalent (Fe3+). Divalent Fe2+ also coexists in the sample, the amount of which is determined to be 17.6% of total doped Fe by quantitative analysis of the XANES spectra. These Fe2+ ions could be transformed to Fe3+ ions by changing the reaction conditions such as sintering under O2 atmosphere. The fitting results of the Fourier-transformed extended X-ray absorption fine structure(EXAFS) spectrum are shown in Table S4. The first, second, and third coordination shells of Fe is O, Ca, and In/Sb, respectively. For the Fe–O shell, the coordination number (CN) is calculated to be 7.1 (±0.9), which is larger than the expected CN of 6. This should be attributed to the larger error of the CN obtained by EXAFS data fitting, which can generally reach 10% or even 20%. The average interionic distance of Fe–O is 2.04 Å, which is relatively consistent with the refinement result. Based on the diffuse reflection (DR) spectra, the band gap values of SISO, CISO, and CSO hosts were estimated to be 4.2, 4.5, and 4.6 eV, respectively (Fig. S6). A large band gap implies that there is sufficient energy gap to accommodate the energy levels of doped Fe3+. As indicated by the DR spectra in Fig. 2b, SISO:Fe3+, CISO:Fe3+, and CSO:Fe3+ exhibit significantly enhanced absorption in the 250–400 nm region as compared with the hosts, which is ascribed to the O2−–Fe3+ charge transfer (CT) transition. The photoluminescence excitation (PLE) spectra (Fig. 2b) of SISO:Fe3+, CISO:Fe3+, and CSO:Fe3+ are consistent with their corresponding DR spectra and show strong CT bands. The d–d transitions of Fe3+ can hardly be observed because of the spin and parity forbidden feature, the transition probability of which is highly dependent on the site symmetry27. Figure S7a shows the PLE spectra of SISO:Fe3+ and CISO:Fe3+ at 7 K. In addition to the dominant CT band, weak peaks assigned to the 6A1 (6S) → 4E (4D) (405 nm), 6A1 (6S) → 4T2 (4D) (465 nm), and 6A1 (6S) → 4T2 (4G) (580 nm) transitions of Fe3+ are also presented in the enlarged PLE spectra (Fig. S7b). It should be noted that these transitions of Fe3+ in CISO are more intense than those in SISO, suggesting the lower symmetry of the Fe3+ sites in CISO.

a Final fitting results of the Fourier-transformed Fe EXAFS spectra of Ca2In0.88SbO6:0.12Fe3+ in R space. The inset shows the normalized Fe K-edge XANES spectra and the reference compounds of Fe foil, FeO, and Fe2O3. b DR and PLE spectra of SISO:Fe3+, CISO:Fe3+, and CSO:Fe3+. c Normalized PL spectra of Sr2-yCayInSbO6:Fe3+ (y = 0–2). d PL intensity and FWHM of Sr2-yCayInSbO6:Fe3+ (y = 0–2). e TRPL spectra of CISO:Fe3+. f Normalized PL spectra of Ca2(InSb)1-zSn2zO6:Fe3+ (z = 0–1). g Luminescence decay curves of CISO:Fe3+ and CSO:Fe3+. h Schematic diagram of the overall PL tuning mechanism
It was found that the hosts of this series of phosphors exhibited NIR emission with a certain intensity. Evidence suggests that such host luminescence occurs due to the unintentional Fe3+ impurity, which is explained in the Supporting Information according to the photoluminescence (PL) properties and ICP results (Figs. S8–S11 and Tables S5 and S6). Figure 2c shows PL spectra of Sr2-yCayInSbO6:Fe3+ (y = 0–2). Under an excitation wavelength of 340 nm, SISO:Fe3+ exhibited a broad NIR emission band centered at 885 nm, originating from the 4T1 (4G) → 6A1 (6S) transition of the Fe3+ ions in the octahedral sites. With the cation substitution of Ca2+ for Sr2+, the emission spectra showed a continuous red shift from 885 to 935 nm, accompanied by a broadened FWHM from 108 to 126 nm (1441 to 1477 cm−1). Meanwhile, the emission intensity was greatly increased, as shown in Fig. 2d. A more than 2-fold increase in the PL intensity of the Fe3+ ions was achieved when Ca2+ completely substituted Sr2+. The lattice shrinkage caused by smaller Ca2+ substituting larger Sr2+ can lead to a stronger crystal field strength (CFS) around Fe3+ according to the relationship of Dq vs. R−5 12, thereby reducing the energy difference between its 4T1 (4G) excited state and 6A1 (6S) ground state. Thus, a normal red shift in the PL spectra was observed. As mentioned earlier, Ca2+ incorporation also lowers the site symmetry of Fe3+, which can facilitate the breaking of the forbidden transition of Fe3+ and improve the luminescence. Moreover, the lower structural symmetry can introduce an uneven crystal field that results in the formation of 4T1 (4G) sub-levels, thereby broadening the FWHM of the PL spectra. The PL decay curves of Sr2-yCayInSbO6:Fe3+ are shown in Fig. S12, which can be well fitted by the bi-exponential function15:
where I(t) is the luminescence intensity at time t, A1, and A2 are fitted constants, and τ1 and τ2 are the rapid and slow lifetimes of exponential components, respectively. The average lifetime (τ*) values for y = 0, 1, and 2 were calculated to be 8.32, 7.34, and 7.99 ms (Table S7), respectively. The relatively long decay times are related to the forbidden characteristic of the 4T1 (4G) → 6A1 (6S) transition. The time-resolved photoluminescence (TRPL) spectra (Fig. 2e) of CISO:Fe3+ showed that only one Fe3+ luminescent center contributed to the luminescence. Figure S13a shows the low temperature PL spectrum of CISO:Fe3+ measured at 7 K, which shows a slight asymmetry in the emission band. An asymmetric spectral profile generally means more than one luminescent center. However, it can be seen that the two PLE spectra in Fig. S13b monitored at 930 and 1000 nm almost completely overlap. Moreover, the luminescence decay curves monitored at 930 and 1000 nm are nearly consistent (Fig. S13c). These results prove one luminescent center in CISO:Fe3+. To further verify this point, the low temperature TRPL spectra of CISO:Fe3+ were also measured, as shown in Fig. S13d. It is seen that the PL intensity decreases synchronously as the decay time is prolonged, and no visible change in spectral shape can be observed. Therefore, it is reasonable to ascribe this broadband emission to one Fe3+ luminescence center. Considering the valence state and ion radius, it is speculated that the Fe3+ ions occupy the octahedral In3+ sites in CISO:Fe3+.
The aliovalent substitution of [Sn4+–Sn4+] for [In3+–Sb5+] was further performed to tune the NIR emission, as shown in Fig. 2f. The PL spectra of Ca2(InSb)1-zSn2zO6:Fe3+ (z = 0–1) exhibited a red shift from 935 to 1005 nm and a broadened FWHM from 126 to 146 nm (1477 to 1485 cm−1), which can be attributed to the previously discussed strengthened CFS and formation of energy sub-levels, respectively. However, the PL intensity decreased dramatically with the incorporation of Sn4+ (Fig. S14). The TRPL spectra also revealed one luminescent center in CSO:Fe3+ (Fig. S15), indicating the occupation of octahedral Sn4+ sites by the Fe3+ ions. Unlike the alternate arrangement of [InO6] and [SbO6] octahedra in SISO and CISO (Fig. 1a), the adjacent arrangement of the [SnO6] octahedra in CSO indicates a shorter distance between the Fe3+ luminescent centers, which can lead to a significant concentration quenching effect that reduces the PL intensity. The concentration quenching is caused by the energy transfer among Fe3+ ions. To analyze the energy transfer mechanism between Fe3+ ions in the CSO host, the critical distance (Rc) is estimated by the following formula40:
where xc is the critical concentration of Fe3+ ions, N is the total sites of Fe3+ per unit cell, V is the volume of the unit cell. Accordingly, the Rc value was determined to be about 33.96 Å, which is much larger than 5 Å. It is reasonable to attribute the energy transfer mechanism to the electric multipolar interaction rather than the exchange interaction. The energy transfer mechanism of electric multipolar interactions can be determined by the value of θ from the following formula41:
where x and I represent the doping concentration and the corresponding emission intensity, respectively. A is a constant, and θ values of 6, 8, 10 correspond to dipole–dipole, dipole–quadrupole, and quadrupole–quadrupole interactions, respectively. As shown in Fig. S16, there is a good linear relationship between log (I/x) and log (x), and the θ value is finally determined to be 4.6, which is close to 6. Thus, the concentration quenching mechanism is attributed to the dipole–dipole interaction.
Figure 2g shows the PL decay curves of CISO:Fe3+ and CSO:Fe3+. It can be seen that the decay curve of CISO:Fe3+ is well fitted by the mono-exponential function, whereas that of CSO:Fe3+ deviates from the mono-exponential fitting and is fitted by the bi-exponential function, indicating an additional energy decay path such as defect-induced nonradiative relaxation process42. In CaSnO3, there is a structural possibility for an enhanced exchange interaction between the Fe3+ ions. This should also result in a red-shifted emission besides the stronger CFS, as was indeed observed in Fig. 2f. Due to the exchange interaction, the originally spin quartet excited state of Fe3+ acquires spin sextet character, which should also decrease its decay time as is observed. Another explanation could be the requirement for charge compensation upon substitution of Sn4+ by Fe3+, which could lead to a close charge-compensating defect that distorts the site and could thus, also lower the decay time. Accordingly, the lifetime was shortened from 7.99 to 2.76 ms with the complete introduction of Sn4+ (Table S8). Figure 2h shows a schematic diagram of the overall PL tuning mechanism. As discussed above, the designed substitution of Ca2+ for Sr2+ and cosubstitution of [Sn4+–Sn4+] for [In3+–Sb5+] result in a stronger CFS and lower site symmetry, which are responsible for the observed emission red shift and broadening of the Fe3+ luminescence. The temperature-dependent luminescent properties were investigated, and the corresponding spectra (Figs. S17–19) are depicted in the Supporting Information. At 398 K, the integrated PL intensity decreased to 44%, 36%, and 24% of the initial intensity at 298 K for SISO:Fe3+, CISO:Fe3+, and CSO:Fe3+, respectively. The PL thermal stability requires further improvement for practical applications.
Quantum efficiency is an important parameter for evaluating the performance of phosphors. The IQEs of SISO:Fe3+ and CISO:Fe3+ were measured to be 48% and 87%, respectively (Fig. S20a, b). The absorption efficiency and external quantum efficiency (EQE) were estimated and presented in Supporting Information. The EQEs of SISO:Fe3+ and CISO:Fe3+ were estimated to be 35% and 68%, respectively. For reference, we synthesized the recently reported NaScGe2O6:Cr3+ and La3Ga5GeO14:Cr3+ phosphors with optimal doping content following relevant literature (Fig. S21)10,43. As shown in Fig. S22, the peak profiles and peak positions of the self-prepared samples of NaScGe2O6:Cr3+ and La3Ga5GeO14:Cr3+ are basically consistent with the reported ones. Moreover, their IQEs were measured to be 44% and 27% (Fig. S20c, d), respectively, which are close to those reported in the literature (Table 1), confirming the reliability of the measured IQEs in this work. The comparison of PL intensities of the as-synthesized phosphors in Fig. S22 agrees with that of the IQE results. Table 1 lists the IQEs of some Eu2+-, Mn2+-, and Cr3+-activated NIR-emitting phosphors. As the IQE for Fe3+ luminescence has been rarely reported, no Fe3+-doped NIR-emitting phosphors is mentioned in Table 1. It is shown that the IQE of CISO:Fe3+ is higher than those of K3LuSi2O7:Eu2+, MgAl2O4:Mn2+, and most Cr-activated NIR-emitting phosphors with peak wavelengths longer than 800 nm. Such a high IQE with simultaneous long-wavelength NIR emission (>900 nm) is rare in broadband NIR-emitting phosphors. Even the lower IQE of SISO:Fe3+ is comparable to that of Cr-activated phosphors with similar wavelengths. These facts demonstrate that Fe3+ is a promising candidate activator for highly efficient NIR emission.
LED packages and applications
NIR-emitting pc-LEDs were manufactured to evaluate the application potential of the synthesized phosphors. Their driving current-dependent PL spectra are shown in Figs. 3a and S23, and the corresponding NIR output power is given in Fig. S24. A maximum NIR output power of 0.83 mW at 200 mA was obtained in the pc-LED fabricated by CISO:Fe3+, which can be further optimized. Figure 3b presents photographs of some objects under natural light and the optimal NIR-emitting pc-LED light, indicating the potential application of CISO:Fe3+ in night vision. In addition, the NIR-emitting pc-LED light can penetrate fingers (Fig. 3c), which provides the possibility of using CISO:Fe3+ for nondestructive examination of biological tissues.

a Emission spectra of the pc-LED fabricated by the CISO:Fe3+ phosphor. b Photographs captured under natural light and NIR light. c Photograph of the NIR light penetrating fingers. d PL spectra of the phosphor NIR light before and after penetrating water, and the corresponding calculated transmission spectra of water. e Emission spectra of the as-fabricated optimal pc-LED with and without cucumbers in the integrating sphere. f Absorption spectra of cucumbers
Other potential fields, such as NIR spectroscopy analysis, can be expected for the developed emission-tunable NIR-emitting phosphors. Here, the phosphors are regarded as light sources (LS), the emission of which can be selectively absorbed by the functional groups. Figure 3d shows the PL spectra (shadow-filled) of SISO:Fe3+ (LS 1), CISO:Fe3+ (LS 2), and CSO:Fe3+ (LS 3) before and after penetrating water. The corresponding calculated transmission spectra of water are plotted as solid lines in Fig. 3d. It can be observed that all three light sources sensitively detect the 975 nm absorption signal of water. As for the 1150 nm absorption peak, LS 1 and LS 2 are almost undetectable, whereas LS 3 can recognize a relatively obvious signal. This result is due to the different effective spectral regions of the light sources. Figure S25 shows comparison of the emission spectra of the as-synthesized SISO:Fe3+, CISO:Fe3+, CSO:Fe3+ phosphors and 940 nm chip. It is observed that the as-synthesized phosphors show much broader bandwidth than that of commercial 940 nm chip. Moreover, the emission wavelength of the phosphors is tunable. Merging these emission-tunable phosphors into one LED can enlarge the spectrum region to cover more information for NIR spectroscopic analysis, which is of great importance for emission tuning and broadening of the NIR-emitting phosphors.
To demonstrate the application potential of the as-fabricated optimal NIR pc-LED in food analysis, cucumbers with varying freshness levels were selected for detection. Figure 3e shows the emission spectra of the NIR pc-LED with and without the cucumbers in the integrating sphere. It can be seen that the cucumbers show broadband absorption in the 850–1100 nm range owing to the water content absorption (Fig. 3f). Moreover, the fresh cucumber that contained more water exhibited more obvious NIR absorption. These results indicate the potential application of the NIR pc-LEDs in nondestructive food analysis based on the NIR spectroscopic analysis.
Discussion
In summary, a series of Fe3+-activated Sr2-yCay(InSb)1−zSn2zO6 broadband NIR-emitting phosphors were designed and successfully synthesized. Under 340 nm excitation, SISO:Fe3+ showed a broad NIR emission band centered at 885 nm. Controllable emission tuning from 885 to 935 nm with significantly enhanced PL intensity was achieved by cation substitution of Ca2+ for Sr2+. The subsequent cation cosubstitution of [Sn4+–Sn4+] for [In3+–Sb5+] further tuned the emission from 935 to 1005 nm. The continuous emission red shift is a result of the strengthened CFS induced by lattice shrinkage. Moreover, the complete introduction of Ca2+ and Sn4+ broadened the FWHM from 108 to 146 nm. The SISO:Fe3+ and CISO:Fe3+ phosphors exhibited high IQEs of 48% and 87%, respectively, and EQEs of 35% and 68%, respectively, indicating the potential of Fe3+ activator in obtaining highly efficient NIR emission. Further, the as-fabricated NIR pc-LEDs showed potential applications in night vision, nondestructive biological monitoring, and NIR spectroscopy detection. This work provides new insights into the luminescence of Fe3+, which opens up a new avenue for the development of highly efficient broadband NIR-emitting phosphor materials.
Materials and methods
Materials synthesis
Sr2-yCay(InSb)1-zSn2zO6:xFe3+ (x = 0–0.03, y = 0–2, z = 0–1) phosphors were synthetized via a high-temperature solid-state reaction process. Strontium carbonate (SrCO3, S. P.), calcium carbonate (CaCO3, A. R.), indium oxide (In2O3, 99.99%), antimony trioxide (Sb2O3, 99.99%), and ferric sesquioxide (Fe2O3, 99.99%) were obtained from Aladdin Reagent Co., Ltd. Tin oxide (SnO2, S. P.) was acquired from Sinopharm Chemical Reagent Co., Ltd. The raw materials were stoichiometrically weighed and thoroughly ground in an agate mortar for 20 min. The precursors were transferred to alumina crucibles and sintered at 1573 K for 6 h in a box furnace. The resulting products were slowly cooled down to room temperature and ground again.
La3Ga4.95GeO14:0.05Cr3+ and NaScGe2O6:0.03Cr3+ phosphors for IQE comparison were synthesized according to the literature10,43. Lanthanum oxide (La2O3, 99.999%), scandium oxide (Sc2O3, 99.999%), gallium oxide (Ga2O3, 99.99%), sodium carbonate (Na2CO3, A. R.), germanium oxide (GeO2, 99.99%), and chromium oxide (Cr2O3, 99.99%) were obtained from Aladdin Reagent Co., Ltd. The stoichiometric amounts of La2O3, Ga2O3, GeO2, and Cr2O3 were weighed and thoroughly ground in an agate mortar for 20 min, and the precursors were transferred to alumina crucibles and sintered at 1523 K for 5 h in air atmosphere to obtain the La3Ga4.95GeO14:0.05Cr3+ phosphor. The stoichiometric amounts of Na2CO3, Sc2O3, GeO2, and Cr2O3 were weighed and thoroughly ground in an agate mortar for 20 min, and the precursors were transferred to alumina crucibles and sintered at 1473 K for 5 h in air atmosphere to obtain the NaScGe2O6:0.03Cr3+ phosphor.
LED fabrication
The as-prepared NIR phosphors were thoroughly mixed with silicone resins A and B (A:B=1:1). The mixtures were then coated on the 365 and 310 nm chips, and cured at 150 °C for 1 h to obtain the final LED devices.
Characterization
The X-ray diffraction (XRD) patterns of the as-synthesized phosphors were measured on a Bruker D8 ADVANCE powder diffractometer (Cu Kα radiation, λ = 1.54 Å) within the 2θ range 10−70°. XRD Rietveld refinements were conducted using the TOPAS 4.2. The field-emission scanning electron microscope (FE-SEM, S-4800, Hitachi) equipped with an Energy Dispersive Spectrometer (EDS) was used to obtain the elemental compositions. The high-resolution transmission electron microscopes (HRTEM) images were acquired using a FEI Tecnai G2 S-Twin. Raman spectra were recorded on a Raman spectrometer (Model T64000, Horiba JobinYvon, France) with a 512 nm laser. The X-ray absorption experiments were carried out at the XAS station (BL14W1) of the Shanghai Synchrotron Radiation Facility. The electron storage ring was operated at 3.5 GeV. Si (311) double-crystal was used as the monochromator, and the data was collected using solid-state detector under ambient conditions. The beam size was limited by the horizontal and vertical slits with the area of 1 × 4 mm2 during XAS measurements. The diffuse reflection (DR) spectra were recorded on a UV−vis−NIR spectrophotometer (UV-3600 plus, Shimadzu, Japan). The photoluminescence excitation (PLE) and photoluminescence (PL) spectra were measured by an Edinburgh Instruments FLSP-920 fluorescence spectrometer with a 450 W xenon lamp as excitation source, and an R5509-72 photomultiplier (PMT) as a light detector. The R5509-72 PMT is a nitrogen-cooled NIR-sensitive PMT, and the cooling temperature is −85 °C. The PL decay curves and time-resolved photoluminescence (TRPL) spectra were also measured by an Edinburgh Instruments FLSP-920 fluorescence spectrometer with a μF2 lamp as excitation source. The temperature-dependent PL spectra and decay curves were also measured by Edinburgh Instruments FLSP-920 fluorescence spectrometers equipped with a temperature controller. The IQEs were measured by an Edinburgh Instruments FLS-1000 equipped with an optical integrating sphere. Xenon lamp was the excitation source, and R5509-72 PMT was used as a light detector. The element content was determined by the inductive Coupled Plasma-atomic emission (ICP-AES) spectrometer (Agilent 7800). The emission spectra of the as-fabricated phosphor-converted light-emitting diodes (pc-LEDs) are mearsured on the HAAS 2000 photoelectric measuring system from EVERFINE. The photographs in the application of NIR pc-LEDs are taken by a NIR and a visible camera.
References
Huang, W. T. et al. Near-infrared nanophosphor embedded in mesoporous silica nanoparticle with high light-harvesting efficiency for dual photosystem enhancement. Angew. Chem. Int. Ed. 60, 6955–6959 (2021).
Zhang, L. L. et al. Cr3+-doped broadband NIR garnet phosphor with enhanced luminescence and its application in NIR spectroscopy. Adv. Opt. Mater. 7, 1900185 (2019).
Mao, M. Q. et al. Broadband near-infrared (NIR) emission realized by the crystal-field engineering of Y3−xCaxAl5−xSixO12: Cr3+(x= 0–2.0) garnet phosphors. J. Mater. Chem. C 8, 1981–1988 (2020).
Xie, R. J. Light-emitting diodes: brighter NIR-emitting phosphor making light sources smarter. Light.: Sci. Appl. 9, 155 (2020).
Zhao, F. Y. et al. Structural confinement for Cr3+ activators toward efficient near-infrared phosphors with suppressed concentration quenching. Chem. Mater. 33, 3621–3630 (2021).
Zeng, H. T. et al. Two-site occupation for exploring ultra-broadband near-infrared phosphor-double-perovskite La2MgZrO6: Cr3+. Chem. Mater. 31, 5245–5253 (2019).
Liu, G. C. et al. Li/Na substitution and Yb3+ co-doping enabling tunable near-infrared emission in LiIn2SbO6: Cr3+ phosphors for light-emitting diodes. iScience 24, 102250 (2021).
Rajendran, V. et al. Super broadband near-infrared phosphors with high radiant flux as future light sources for spectroscopy applications. ACS Energy Lett. 3, 2679–2684 (2018).
Jia, Z. W. et al. Strategies to approach high performance in Cr3+-doped phosphors for high-power NIR-LED light sources. Light.: Sci. Appl. 9, 86 (2020).
Zhou, X. F. et al. An Ultraviolet-visible and near-infrared-responded broadband NIR phosphor and its NIR spectroscopy application. Adv. Opt. Mater. 8, 1902003 (2020).
Liu, G. C. et al. Two-site Cr3+ occupation in the MgTa2O6: Cr3+ phosphor toward broad-band near-infrared emission for vessel visualization. J. Mater. Chem. C 8, 9322–9328 (2020).
Liu, D. J. et al. Simultaneous broadening and enhancement of Cr3+ photoluminescence in LiIn2SbO6 by chemical unit cosubstitution: night-vision and near-infrared spectroscopy detection applications. Angew. Chem. Int. Ed. 60, 14644–14649 (2021).
Cai, H. et al. Tuning luminescence from NIR-I to NIR-II in Cr3+-doped olivine phosphors for nondestructive analysis. J. Mater. Chem. C 9, 5469–5477 (2021).
Fang, M. H. et al. Hidden structural evolution and bond valence control in near-infrared phosphors for light-emitting diodes. ACS Energy Lett. 6, 109–114 (2021).
Qiao, J. W. et al. Divalent europium-doped near-infrared-emitting phosphor for light-emitting diodes. Nat. Commun. 10, 5267 (2019).
Wei, Y. et al. Advances in near-infrared luminescent materials without Cr3+: crystal structure design, luminescence properties, and applications. Chem. Mater. 33, 5496–5526 (2021).
Zhang, Q. et al. A K3ScSi2O7: Eu2+ based phosphor with broad-band NIR emission and robust thermal stability for NIR pc-LEDs. Chem. Commun. 56, 4644–4647 (2020).
Zhou, Z. H. et al. Cr3+-Free near-infrared persistent luminescence material LiGaO2: Fe3+: optical properties, afterglow mechanism and potential bioimaging. J. Mater. Chem. C 8, 14100–14108 (2020).
Song, E. H. et al. Heavy Mn2+ doped MgAl2O4 phosphor for high-efficient near-infrared light-emitting diode and the night-vision application. Adv. Opt. Mater. 7, 1901105 (2019).
Que, E. L., Domaille, D. W. & Chang, C. J. Metals in neurobiology: probing their chemistry and biology with molecular imaging. Chem. Rev. 108, 1517–1549 (2008).
Zhou, Z. H. et al. Defect enrichment in near inverse spinel configuration to enhance the persistent luminescence of Fe3+. Adv. Opt. Mater. 10, 2101669 (2021).
Takahashi, H. A. et al. Fe3+ red phosphors based on lithium aluminates and an aluminum lithium oxyfluoride prepared from LiF as the Li Source. J. Lumin. 182, 53–58 (2017).
Shu, W. et al. Enhanced red emission in LiAl5O8: Fe3+ phosphor by B3+ doping. J. Alloy. Compd. 509, 3886–3888 (2011).
Van Schaik, W. & Blasse, G. Luminescence of Fe3+ in LaPO4. J. Alloy. Compd. 198, 69–72 (1993).
Soppin, K. & Manohara, B. M. CdSiO3: Fe3+ nanophosphors: structural and luminescence properties. Bull. Mater. Sci. 44, 49 (2021).
Kniec, K. et al. From quencher to potent activator – Multimodal luminescence thermometry with Fe3+ in the oxides MAl4O7 (M = Ca, Sr, Ba). J. Mater. Chem. C 9, 6268–6276 (2021).
Li, Y. J. et al. Site-related near-infrared luminescence in MAl12O19 (M=Ca, Sr, Ba): Fe3+ phosphors. Mater. Res. Bull. 51, 1–5 (2014).
Zeng, J. W. et al. In-situ Insights into trap attributions in Fe3+-activated long persistent phosphors. J. Lumin. 232, 117810 (2021).
Kniec, K. et al. Spectral and thermometric properties altering through crystal field strength modification and host material composition in luminescence thermometers based on Fe3+ doped AB2O4 type nanocrystals (A = Mg, Ca; B = Al, Ga). J. Mater. Chem. C 9, 517–527 (2021).
Lin, L. T. Comment on “oxygen-vacancy-induced Midgap states responsible for the fluorescence and the long-lasting phosphorescence of the inverse Spinel Mg(Mg, Sn)O4”. Chem. Mater. 32, 7564–7567 (2020).
Pott, G. T. & McNicol, B. D. The luminescence of Fe3+ and Cr3+ in α-gallia. J. Lumin. 6, 225–228 (1973).
Xie, Y. et al. Luminescence of a novel double-perovskite Sr2InSbO6: Eu3+ orange-red-emitting phosphor for white LEDs and visualization of latent fingerprints. Mater. Res. Bull. 146, 111574 (2022).
Zhao, F. Y. et al. Double perovskite Cs2AgInCl6: Cr3+: broadband and near-infrared luminescent materials. Inorg. Chem. Front. 6, 3621–3628 (2019).
Zhong, J. Y. et al. Efficient and Tunable Luminescence in Ga2-xInxO3: Cr3+ for Near-Infrared Imaging. ACS Appl. Mater. Interfaces 13, 31835–31842 (2021).
Xu, X. X. et al. Highly efficient and thermally stable Cr3+-activated silicate phosphors for broadband near-infrared LED applications. Chem. Eng. J. 383, 123108 (2020).
Cai, C. Y. et al. Microwave dielectric properties of Ca1-xSrxMgSi2O6 ceramics. Ceram. Int. 46, 27679–27685 (2020).
Dias, A. et al. Raman-spectroscopic investigation of Ba2InTaO6 and Sr2InTaO6 perovskites. J. Solid State Chem. 180, 2143–2148 (2007).
Vijayakumar, C. et al. FT-Raman and FT-IR vibrational spectroscopic studies of nanocrystalline Ba2RESbO6 (RE=Sm, Gd, Dy and Y) perovskites. J. Alloy. Compd. 480, 167–170 (2009).
Halder, S. et al. Octahedral distortion induced phonon vibration and electrical conduction in A2NdSbO6 (A = Ba, Sr, Ca). Mater. Chem. Phys. 199, 508–521 (2017).
Liu, Y. F. et al. An excellent cyan-emitting orthosilicate phosphor for NUV-pumped white LED application. J. Mater. Chem. C 5, 12365–12377 (2017).
Zhong, C. S. et al. Novel broadband near-infrared emitting phosphor LiGe2(PO4)3: Cr3+ with tuning and enhancement of NIR emission by codoping Sb5+. J. Alloy. Compd. 903, 163945 (2022).
Yu, D. C. et al. Non-rare-earth Na3AlF6: Cr3+ phosphors for far-red light-emitting diodes. ACS Appl. Electron. Mater. 1, 2325–2333 (2019).
De Guzman, G. N. A. et al. Multi-site cation control of ultra-broadband near-infrared phosphors for application in light-emitting diodes. Inorg. Chem. 59, 15101–15110 (2020).
Shao, Q. Y. et al. Photoluminescence properties of a ScBO3: Cr3+ phosphor and its applications for broadband near-infrared LEDs. RSC Adv. 8, 12035–12042 (2018).
Mao, N. et al. A broadband near-infrared phosphor Ca3Y2Ge3O12: Cr3+ with garnet structure. J. Alloy. Compd. 863, 158699 (2021).
Malysa, B., Meijerink, A. & Jüstel, T. Temperature dependent Cr3+ photoluminescence in garnets of the type X3Sc2Ga3O12 (X = Lu, Y, Gd, La). J. Lumin. 202, 523–531 (2018).
Lai, J. A. et al. Broadband near-infrared emission enhancement in K2Ga2Sn6O16: Cr3+ phosphor by electron-lattice coupling regulation. J. Am. Ceram. Soc. 103, 5067–5075 (2020).
Gao, T. Y. et al. Design of a broadband NIR phosphor for security-monitoring LEDs: tunable photoluminescence properties and enhanced thermal stability. Cryst. Growth Des. 20, 3851–3860 (2020).
Yao, L. Q. et al. Enhancing near-infrared photoluminescence intensity and spectral properties in Yb3+ codoped LiScP2O7: Cr3+. Chem. Mater. 32, 2430–2439 (2020).
Dai, D. J. et al. Broad band emission near-infrared material Mg3Ga2GeO8: Cr3+: substitution of Ga-In, structural modification, luminescence property and application for high efficiency LED. J. Alloy. Compd. 806, 926–938 (2019).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC Nos. 51720105015, 51932009, 51929201, 52072349), the Projects for Science and Technology Development Plan of Jilin Province (20210402046GH), and the Natural Science Foundation of Zhejiang Province (LR22E020004).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary information
41377_2022_803_MOESM1_ESM.docx
Highly efficient Fe3+-doped A2BB’O6 (A = Sr2+, Ca2+; B, B’ = In3+, Sb5+, Sn4+) broadband near-infrared-emitting phosphors for spectroscopic analysis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, D., Li, G., Dang, P. et al. Highly efficient Fe3+-doped A2BB′O6 (A = Sr2+, Ca2+; B, B′ = In3+, Sb5+, Sn4+) broadband near-infrared-emitting phosphors for spectroscopic analysis. Light Sci Appl 11, 112 (2022). https://doi.org/10.1038/s41377-022-00803-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41377-022-00803-x
This article is cited by
-
Energy transfer induced colour tunable photoluminescence performance of thermally stable Sm3+/Eu3+ co-doped Ba3MoTiO8 phosphors for white LED applications
Journal of Materials Science: Materials in Electronics (2023)
-
Environmentally friendly Fe3+-activated near-infrared-emitting phosphors for spectroscopic analysis
Light: Science & Applications (2022)
