
Abstract
T-cell malignancies are associated with frequent relapse and high morbidity, which is partly due to the lack of effective or targeted treatment options. To broaden the use of CAR-T cells in pan T-cell malignancies, we developed an allogeneic “universal” CD2-targeting CAR-T cell (UCART2), in which the CD2 antigen is deleted to prevent fratricide, and the T-cell receptor is removed to prevent GvHD. UCART2 demonstrated efficacy against T-ALL and CTCL and prolonged the survival of tumor-engrafted NSG mice in vivo. To evaluate the impact of CD2 on CAR-T function, we generated CD19 CAR-T cells (UCART19) with or without CD2 deletion, single-cell secretome analysis revealed that CD2 deletion in UCART19 reduced frequencies of the effector cytokines (Granzyme-B and IFN-γ). We also observed that UCART19ΔCD2 had reduced anti-tumor efficacy compared to UCART19 in a CD19+NALM6 xenograft model. Of note is that the reduced efficacy resulting from CD2 deletion was reversed when combined with rhIL-7-hyFc, a long-acting recombinant human interleukin-7. Treatment with rhIL-7-hyFc prolonged UCART2 persistence and increased survival in both the tumor re-challenge model and primary patient T-ALL model in vivo. Together, these data suggest that allogeneic fratricide-resistant UCART2, in combination with rhIL-7-hyFc, could be a suitable approach for treating T-cell malignancies.
Similar content being viewed by others


Introduction
T-cell acute lymphoblastic leukemia (T-ALL) and cutaneous T-cell lymphoma (CTCL) represent aggressive groups of T-cell malignancies associated with frequent relapse and high morbidity in children and adults [1]. While CD19 CAR-T is successful in treating patients with B-cell malignancies, the utilization of CAR-T cell therapy is still largely restricted due to fratricide, risk of Graft-versus-Host Disease (GvHD), and immune escape, as it is technically challenging to purge all the malignant T cells from an autologous product [2].
Our group and others have independently demonstrated CD7 as a promising therapeutic target for T-cell malignancies [3,4,5,6]. Using the CRISPR/Cas9 gene editing approach, we have previously developed an allogeneic “universal” CD7-targeting CAR-T (UCART7) that effectively targets CD7-positive T-cell malignancies. Simultaneous CRISPR/Cas9 deletion of TRAC and CD7 prevents CAR-T cell fratricide and permits the safe use of allogeneic T cells by eliminating the potential for Graft-versus-Host Disease (GvHD) [3]. However, CD7 is classically downregulated or absent in some T-cell malignancies such as adult T-cell leukemia/lymphoma (ATL) and Sezary syndrome (SS) [7, 8]. While most patients with CD7-positive T-cell malignancies showed good responses to the CD7-targeting CAR-T therapy, relapse of CD7-negative disease has been reported in recent clinical trials [6, 9, 10]. In contrast, CD2 is expressed on >99% of T-cells and downregulates at a lower frequency than other pan-T cell markers in cancer cells, providing an attractive therapeutic target [11, 12].
CD2 is a transmembrane glycoprotein restricted to hematopoietic cells with high expression in NK cells and T cells, including early T cell progenitors. CD2 acts as a co-stimulatory receptor with roles in thymocyte development, actin cytoskeleton rearrangement, and cellular signaling at the immunological synapse [13]. In conjunction with its ligands CD58 (in human) and CD48 (in mice), CD2 co-stimulation plays an important role in T cell activation and TCR signaling [14, 15]. CD2 is expressed in a wide variety of T-cell malignancies, including T-ALL, SS (Sezary Syndrome), peripheral T-cell malignancies, and adult T-cell leukemia/lymphoma (ATL). Therefore, we hypothesized that CD2 and TRAC could be deleted from T-cells of allogeneic donors by CRISPR/Cas9, then transduced with a CD2 targeting CAR (CAR2-CD28-CD3ζ) to effectively kill T-cell malignancies without resulting in GvHD or CAR-T cells fratricide. Loss of CD2 may hamper T cell cytotoxicity but is unavoidable when using this as a target for CAR-T cell therapy. We further hypothesized that loss in the efficacy of CAR-T function resulting from CD2 deletion could be compensated for by co-treatment with rhIL-7-hyFc (recombinant human interleukin 7 fused with hybrid Fc), a stable, long-acting, homo-dimeric interleukin-7 (IL-7) molecule that effectively enhances the expansion, efficacy, and persistence of CAR-T cells [16, 17]. The data presented here demonstrate the feasibility of generating UCART2 and provide evidence that supports the anti-tumor efficacy of UCART2 for the treatment of mature T-cell malignancies. Furthermore, in vivo administration of rhIL-7-hyFc in pre-clinical models elevated UCART2 into a curative therapy for treating T-cell malignancies.
Materials and methods
Detailed materials and methods are described in Supplemental Methods.
Results
CD2Δ TRACΔ CART2 (UCART2) kills CTCL and T-ALL in vitro without fratricide
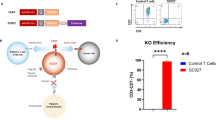
We previously developed an allogeneic UCART7 that effectively targets CD7 positive T-cell malignancies [3]. To test whether CD2 could be an effective therapeutic target for T-cell malignancies, we generated UCART2 (CAR2-28ζ-34 with CD2Δ TRACΔ), a gene-edited CAR-T in which the CD2 antigen is deleted to prevent fratricide and the T-cell receptor (TRAC) is removed to prevent GvHD (Fig. 1a–c). Seven days post CRISPR/Cas9 mediated deletion of CD2 and TRAC in primary T-cells, surface expression of CD2 and TRAC were examined by flow cytometry, which showed > 95% double deletion of CD2 and TRAC in samples co-electroporated with both CD2 and TRAC gRNA (Fig. 1d). In line with this data, targeted deep sequencing also confirmed that the loss of expression resulted from frameshift Indels in 95.7% (SD±0.96%) of CD2 reads (Fig. 1e) and 94.2% (SD±1.59%) of TRAC (Fig. 1f). Characterizing the off-target nuclease activity profile is essential for proving the safety of gene-edited CAR-T cells. Therefore, we used Guide-Seq to assess off-target sites of CRISPR/Cas9 gene editing in a genome-wide unbiased fashion (Supplemental Fig. 1). Across all three replicates, on-target reads (CD2 and TRAC reads per total reads) represented between 84.2% and 89% of all capture sequences. The TRAC gRNA had a clean off-target profile with minimal off-target events detected in three non-coding regions. Ten off-target sites with CD2 gRNA were identified, with six sites identified within intergenic regions of genomic DNA and the remaining four sites within the exons of genes (HEXB, CALR, KIF21B, and MUC4). However, the off-target reads within these four genes were significantly lower than the on-target CD2 reads.

a Schematic of UCART2 design. b Schematic of CAR2-28ζ-34 and CAR19-28ζ-34 CAR constructs. c Timeline of UCART2 production with CRISPR/Cas9 gene editing. T cells were cultured in Xcyte media supplemented with 50 U/mL IL-2 and 10 ng/ml IL-15 and activated with anti-CD3/CD28 beads (bead to cell ratio 3:1) for two days followed by T-cell transduction of either CD2 or CD19 CAR construct. Transduced T cells were expanded for 9 days, followed by CD3+ depletion and hCD34 enrichment. Efficiencies of multiplex CRISPR/Cas9 gene editing of CD2 and TRAC were assessed by d. flow cytometry (CD2 vs. TRAC), or e, f. targeted deep sequencing of CD2 or TRAC. % NHEJ was determined as a percentage of sequencing reads with indels relative to WT cells. g In vitro killing assay. UCART2 or UCART19 cells were cultured with 51Cr-labeled HH, Jurkat, or Molt-3 cells at various E: T ratios for 4 h. Specific lysis was calculated based on released 51Cr in the culture medium.
Next, we tested the ability of UCART2 to specifically kill CD2+T-ALL and CTCL cell lines using a Cr51 release cytotoxicity assay. Cr51 labeled CD2+ target cells (HH, Jurkat, and MOLT-3) were co-cultured with UCART2 or UCART19 at various E: T ratios (30:1 to 0.3:1) for 4 h, and the percentage of target-specific lysis was determined by released Cr51 (Fig. 1g). UCART2 demonstrated potent cytotoxicity against all three CD2+ tumor cell lines in vitro (p < 0.0001). In CTCL cell line HH, 65% of target-specific cytolysis was achieved at the 10:1 E: T ratio. Similarly, the T-ALL cell lines, Jurkat and MOLT-3, were equally sensitive to UCART2-mediated killing with >70% and >80% target-specific cytolysis at the 10:1 ratio, respectively, over 4 h. In contrast, target-specific killing was not observed with the non-targeting UCART19 control.
UCART2 prolongs survival and reduces tumor burden in vivo
To test the in vivo efficacy of UCART2, 5 × 105 CD2+CTCL cell line HHCBR-GFP was inoculated in NSG mice by I.V. injection (day -5), followed by I.V. infusion of 2 × 106 UCART2 or UCART19 on day 0 (Fig. 2a). Tumor burden was monitored weekly by BLI (bioluminescence imaging, Fig. 2b). In contrast to the mice injected with tumor-only or UCART19 cells, UCART2 significantly prolonged the survival of the CTCL tumor-bearing mice (p < 0.0001). Remarkably, while the median survival of both control groups (tumor only and UCART19) was 27 days, all the UCART2-treated mice were alive by the end of the experiment (day +63; Fig. 2c). Similar to our previous findings with UCART7, no signs of GvHD were observed in mice treated with UCART2 or UCART19 (data not shown). Consistently, compared to the untreated tumor-only group, BLI also showed significant attenuation of the tumor burden in UCART2-treated animals (p < 0.0001) but not in UCART19-treated animals (Fig. 2d). Together, these data demonstrated that UCART2 is effective and specific to kill CD2+HH CTCL cells in vivo.

a Schema of the xenogeneic mouse model of CTCL. NSG mice were injected with 5 × 105 HHCBR-GFP cells on day -5, then infused with 2 × 106 UCART2 or UCART19 on day 0. b, c tumor burden was assessed with BLI weekly (n = 5 per group). d Kaplan–Meier survival curve of mice treated UCART19, UCART2, or untreated control. Median survival: untreated mice (26 days), UCART19 treated mice (27 days), UCART2 treated (no death at the end of the experiment on day 65, p < = 0.0001).
CD2 deletion attenuates CAR-T function in vivo
CD2 plays an important role in T cell activation and the formation and organization of the immunological synapse [13, 18]. A recent report suggests that loss of CD2 co-stimulatory receptor CD58 limits the durable CD19 CAR response in large B cell lymphoma [19]. Therefore, we set out to evaluate the potential impact of CD2 deletion on CAR-T function by generating the UCART19ΔCD2 cells, in which CD2 and TRAC were genetically deleted, as compared to UCART19, in which only TRAC was deleted while CD2 remained unmodified (Fig. 3a). Deletion of CD2 in UCART19 had little impact on the CD4:CD8 ratio (Supplemental Fig. 2a) or immune phenotype within the CD4+ or CD8+ subset (Supplemental Fig. 2b). We first tested the in vitro efficacy of UCART19 or UCART19ΔCD2 in a 24 h flow-based killing assay by co-culture of CAR-T cells with CD19+NALM6CBR-GFP targets. Deletion of CD2 had no immediate deleterious effect on UCART19 function in vitro, maintaining similar cytotoxicity against CD19+ targets in this 24 h killing assay (Fig. 3b). To further characterize the impact of CD2 deletion in CAR-T cells, we performed single-cell secretome analyses on target-stimulated UCART19 and UCART19ΔCD2 using the Isolight platform (Isoplexis). The Isolight assay enables the detection and quantification of 32 secreted cytokines from individual CAR-T cells to define polyfunctional strength index (PSI), a quantitative parameter of T cell functionality (Fig. 3c) [20]. Twenty hr after co-culture with the target cells (E:T = 1:2), CAR-T cells were isolated for the Isolight assay. Single-cell secretome analysis of target-stimulated UCART19 (CD2 +) and UCART19ΔCD2 (CD2-) cells revealed that deletion of CD2 in UCART19 reduced the PSI in both the CD4+ and CD8+ populations (Fig. 3d). This reduction in PSI was driven by a decrease in the percentage of UCART19 secreting two or more cytokines (Fig. 3e) and not a reduction in the intensity of cytokine secretion. Similar signal intensities were observed for Granzyme B (GzmB), IFN-γ, MIP-1α, MIP-1β, perforin, TNF-α, and TNF-β between UCART19 and UCART19ΔCD2 (Fig. 3f). In contrast, reduced secretion frequencies of the effector cytokines (GzmB and IFN-γ) were observed in both the CD4+ and CD8+ population when CD2 was deleted in UCART19 (Fig. 3g), suggesting that the CD2:CD58 axis may play a key role in CAR-T co-stimulation.

a Schema of the generation of UCART19 (TRAC-CD2+) and UCART19ΔCD2 (TRAC-CD2-). b in vitro killing efficacy of UCART19 and U CART19ΔCD2. UCART19 and UCART19ΔCD2 were cultured with Nalm6 CBRGFP CD19+ targets at various E: T ratios for 16 h and target-specific killing was measured by luciferase activity. c Single-cell cytokine analysis using the IsoCode assay. UCART19 and UCART19ΔCD2 were incubated with B-cell lymphoma cell line Ramos at an E: T 1:2 for 20 h prior to loading purified CAR-T populations (CD19 depleted and CD34-affinity purified) onto the IsoCode chip. d Polyfunctionality and e Polyfunctional strength index (PSI) of the CD4 or CD8 subpopulation from UCART19 (CD2 +) or UCART19ΔCD2 (CD2-). f The signal strength of key cytokines driving polyfunctionality (GMZB, IFN-γ, MIP-1α, MIP-1β, Perforin, TNF-α, and TNF-β) was not affected by CD2 deletion. g The frequency of cells secreting Granzyme (Gzmb), IFN-γ, MIP1α, MIP1-β, Perforin, TNF-α, and TNF-β in the CD4 or CD8 subpopulation from UCART19 or UCART19ΔCD2. h Schema of the xenogeneic mouse model of CD19 + B cell acute lymphoblastic leukemia: 1 × 106 NALM6CBR-GFP cells were I.V. inoculated into NSG mice on day -5, followed by infusion of 1 × 106 UCART19 or UCART19ΔCD2 on day 0 (n = 10 per group). i Tumor burden as determined by BLI. j Kaplan–Meier survival curve. Median survival: untreated mice, 26 days, mice treated with UCART19 ΔCD2, 45.5 days, all mice treated with UCART19 were alive at day +65.
Next, to test whether CD2 deletion in UCART19 would attenuate CAR-T efficacy in vivo we used the CD19+NALM6 xenogeneic model. We inoculated 1 × 106 NALM6CBR-GFP I.V. into NSG mice on day -5, followed by I.V. infusion of either 1 × 106 UCART19 or UCART19ΔCD2 on day 0. Tumor burden was assessed weekly using BLI (Fig. 3h). Both UCART19 and UCART19ΔCD2 treatment significantly decreased tumor burden in vivo compared to the untreated control animals (p < 0.001; Fig. 3i). Compared to mice treated with UCART19, UCART19ΔCD2 treated mice demonstrated reduced suppression of tumor growth in vivo (Fig. 3i). In addition, while both UCART19 and UCART19ΔCD2 significantly prolonged survival (Log-rank test: p < 0.0001; p = 0.0005, respectively), deletion of CD2 in UCART19 significantly shortened the survival in vivo (UCART19 vs. UCART19ΔCD2, p = 0.0003) (Fig. 3j). Taken together, these data suggest that CD2 deletion attenuates the efficacy of CAR-T cells in vivo.
rhIL-7-hyFc enhances the efficacy of UCART2
To overcome the reduction of CAR-T efficacy resulting from CD2 deletion, we evaluated whether combining with the long-lasting human IL-7, rhIL-7-hyFc, could enhance the efficacy of UCART2. We have previously demonstrated that rhIL-7-hyFc can enhance the expansion persistence and efficacy of UCART19 and UCART33 in vivo [16, 17]. We hypothesized that UCART2 efficacy could be further improved when combined with rhIL-7-hyFc. We first test this hypothesis in a CAR-T stress model using the serial replating assay: UCART2 cells were re-challenged with CD2+ target cells (JurkatCBR/GFP) every 2–3 days at E:T ratio of 2:1-1:1, CAR-T expansion and CAR-T effector function were monitored. While the UCART2 alone became dysfunctional and lost its killing efficacy in vitro after repeated antigen exposure, rhIL-7-hyFc supplementation significantly enhanced the expansion and anti-tumor activity of UCART2 in vitro (Supplemental Fig. 3). To test this hypothesis in vivo, we inoculated 5 × 105 HHCBR-GFP CTCL cells I.V. into NSG mice (day -5), followed by UCART2 or UCART19 treatment on day 0. Due to the high efficacy of UCART2 observed in this model (Fig. 2), mice were treated with a suboptimal dose (1 × 106) of UCART2 or UCART19 to allow the observation of the effect from rhIL-7-hyFc. Mice were treated with rhIL-7-hyFc (10 mg/kg) or vehicle subcutaneously on Days +1, +15, and +29 following UCART infusion. The tumor burden was monitored weekly by BLI (Fig. 4a). As expected, UCART2 significantly reduced tumor burden, as measured by BLI (Fig. 4b). In vivo administration of rhIL-7-hyFc converted UCART2 into a curative therapy, reducing the tumor burden back to baseline by day +20 and no relapse was observed during the full study duration of 200 days (Fig. 4c). In line with this finding, while the median survival time was 25 days in non-targeting UCART19 controls and 45 days in UCART2 alone, all the mice from the UCART2/rhIL-7-hyFc group were alive by the end of the study (day 200) (Fig. 4d). These data demonstrate that rhIL-7-hyFc dramatically enhances the efficacy of UCART2 and prolongs survival in vivo. Of note is that the administration of rhIL-7-hyFc significantly increased the numbers of UCART2 and enhanced the persistence of UCART2 in vivo (Fig. 4e), largely contributing to the enhanced survival observed in Fig. 4c. In spite of UCART2 lacking endogenous CD2, rhIL-7-hyFc was able to overcome the potential functional deficit of these UCART2 in vivo. In line with this data, using the serial replating assay in vitro, we also observed that rhIL-7-hyFc was able to rescue the reduced effector function of CAR-T cells due to CD2 loss in a CART19 model (Supplemental Fig. 4).

a Schema of the xenogeneic mouse model of CTCL. NSG mice were injected with 5 × 105 HHCBR-GFP cells on day -4, then infused with 1 × 106 UCART2 or UCART19 on day 0. 10 mg/kg of rhIL-7-hyFc was delivered subcutaneously on day 1, day 15, and day 29. b tumor burden was measured weekly by BLI. c Kaplan–Meier survival curve of mice treated with UCART19, UCART2, UCART19+rhIL-7-hyFc, and UCART2+rhIL-7-hyFc. Median survival: untreated mice, mice treated with NT-17 alone, and mice treated with UCART19 were 25 days, mice treated with UCART19 + NT-17 was 26.8 days, mice treated with UCART2 alone was 45.4 days, mice treated with UCART2+rhIL-7-hyFc were all alive at 200 days when the experiment was ended. d Tumor burden as determined by BLI. e Flow cytometry analysis was performed bi-weekly to assess circulating CAR-T cells present per µl of peripheral blood. CAR-T population determined by hCD45+, 7aad-, hCD34+ cells. P values < 0.05 considered significant, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. d BLI images normalized to a color gradient scale.
rhIL-7-hyFc prolongs UCART2 persistence in vivo and overcomes tumor re-challenge
We next performed a re-challenge model to address the functional persistence of UCART2 expansion in vivo over time when combined with rhIL-7-hyFc. Using the same HH CTCL model, surviving mice (1 from UCART2 and 5 from the UCART2/rhIL-7-hyFc group) on day +83 were re-challenged with a second inoculation of HHCBR-GFP cells, followed by three doses of rhIL-7-hyFc (Fig. 5a). As expected, UCART19 with or without rhIL-7-hyFc treatment had no effect on survival (median survival: 30 days), while the suboptimal dose of UCART2 significantly prolonged the survival (median survival: 58 days) compared to the tumor-only control (Fig. 5b). Compared to the UCART2 alone treatment, the addition of rhIL-7-hyFc further extended the survival of the animals (median survival: not reached, >200 days). Remarkably, four out of five rhIL-7-hyFc/UCART2 treated mice cleared tumors upon challenge, with three mice surviving the full duration of the experiment (Fig. 5c, d). Together, these data suggest that rhIL-7hyFc propagates the long-term persistence of UCART2 to overcome the tumor re-challenge in vivo.

a Schema of the CTCL re-challenge mouse model. NSG mice were injected with 5 × 105 HHCBR-GFP cells on day -4, then infused with 1 × 106 UCART2 or UCART19 on day 0. 10 mg/kg of rhIL-7-hyFc was delivered subcutaneously on day +1, day +15, and day +29. Surviving mice were re-challenged with 5 × 105 HHCBR-GFP cells on day +83. 10 mg/kg of rhIL-7-hyFc was delivered subcutaneously on days +84, +98, and 112. b Kaplan–Meier survival curve. Mice receiving UCART. UCART19 Vs. UCART2, Median survival 30 days Vs. 58 days p = 0.0027. UCART19+rhIL-7-hyFc Vs. UCART2+rhIL-7-hyFc p = 0.0023 30 days Vs. 200+ days. c Tumor burden as determined by BLI imaging. d Normalized BLI images.
UCART2, in combination with rhIL-7-hyFc, kills primary patient-derived T-ALL in vivo
To test the efficacy of UCART2 against cancer cells from patients, we established a CD2+ primary T-ALL xenograft model. Patient-derived DFCI-15 cells were injected I.V. into NSG mice on day -12 and treatment with UCART2 or UCART19 was administered I.V. on day 0, followed by rhIL-7-hyFc (10 mg/kg) or vehicle on day +1, +15 and +29 (Fig. 6a). Treatment with UCART2 alone doubled the survival of PDX-bearing mice relative to UCART19 (median survival: UCART19, 26 days; UCART2, 54 days, p = 0.0019), demonstrating effective antitumor activity in the absence of rhIL-7-hyFc (Fig. 6b). rhIL-7-hyFc treatment, in the absence of CAR-T cell therapy, was trending towards shortened survival in this PDX model (median survival: Vehicle control 26 Days vs. rhIL-7-hyFc 20 Days, p > 0.05), suggesting rhIL-7-hyFc itself may have provided a modest proliferative advantage to malignant T cells. The combination treatment of rhIL-7-hyFc and UCART2 dramatically enhanced survival, with 80% of mice surviving beyond 300 days (Fig. 6b). Flow cytometry analysis was performed bi-weekly to assess circulating tumor cells (hCD45+, 7-AAD-, GFP+) and CAR-T cells (hCD45+, 7-AAD-, hCD34+) in the peripheral blood. In comparison with untreated or UCART19 treated animals, UCART2 alone or in combination with rhIL-7-hyFc significantly decreased the systemic tumor burden, as measured by circulating tumor cell counts (Fig. 6c). 4 out of 5 mice treated with UCART2/ rhIL-7-hyFc had no detectable circulating tumor cells in the blood. Concordant with survival and systemic tumor burden, rhIL-7-hyFc enhanced UCART2 expansion and persistence, with a high frequency of circulating CAR-T cells (peak count: 28 cells/μL) detected in the periphery 80 days after CAR-T therapy and 50 days following the last dose of rhIL-7-hyFc treatment (Fig. 6d). Combined, these data suggest that UCART2 alone is effective at killing primary T-ALL tumor cells and that potentiation with rhIL-7-hyFc further improves the expansion and persistence of UCART2, resulting in a dramatic increase in survival with most mice having no evidence of tumor >300 days after treatment with UCART2 and rhIL-7-hyFc.

a Schema of the patient-derived T-ALL xenograft model. NSG mice were injected with 5 × 105 DCFI-15 cells on day -12, then infused with 1 × 106 UCART2 or UCART19 on day 0. 10 mg/kg of rhIL-7-hyFc was delivered subcutaneously on day +1, day +15, and day +29. b Kaplan–Meier survival curve Median survival: untreated mice 26 days, rhIL-7-hyFc alone - 20 days, UCART19 26 days, UCART19+rhIL-7-hyFc - 21 days, UCART2 - 54 days, mice treated with UCART2+rhIL-7-hyFc 300+ days. UCART19 Vs. UCART2 p = 0.0019, UCART19+rhIL-7-hyFc Vs. UCART2+rhIL-7-hyFc, p = 0.0031. c Flow cytometry analysis was performed by-weekly to assess circulating tumor cells present per µl in peripheral blood. Tumor population was determined as hCD45 +, 7-AAD-, hCD34-, hCD2+ cells. d. Flow cytometry analysis was performed bi-weekly to assess circulating CAR-T cells present per µl in peripheral blood. CAR-T population determined by hCD45 +, 7-AAD-, hCD34 +, hCD2- cells.
Discussion
CAR T-cell therapy has become one of the most promising cancer therapies. It has shown enormous potential for inducing remissions and long-term relapse-free survival in patients with B cell leukemia and lymphoma [21,22,23]. For T-cell malignancies, however, the shared expression of target antigens on both malignant and healthy T-cells represents a unique set of challenges. Here, we describe UCART2, a novel CD2-targeting allogeneic CAR-T therapy in which the biallelic deletion of CD2 and TRAC prevents fratricide and life-threatening GvHD while effectively killing CD2+ primary human T-ALL and CD2 + T-ALL and CTCL cell lines in vitro and in vivo. Furthermore, our pre-clinical data demonstrates that UCART2, in combination with rhIL-7-hyFc, results in curative and durable therapeutic responses.
For T-cell malignancies, the use of autologous CAR-T therapy is limited due to high manufacture expenses, inconsistent quality, lengthy production time, and the lack of reliable methods to distinguish between effector T cells and malignant T cells. To overcome these limitations, “off-the-shelf” CAR-T cells from allogeneic donors have been increasingly used [24]. Up to date, there are over 50 clinical trials listed on ClinicalTrials.gov evaluating the use of allogeneic CAR-T therapy in hematological malignancies. For T-cell malignancies, CAR-T cells targeting T-cell makers, including CD5 [25, 26], CD7 [6, 27,28,29], and CD30 [30], are currently being developed and have shown promising responses against T-ALL and lymphoma. Among these target antigens, CD7 is most extensively investigated due to its abundant expression in T-cell malignancies, and multiple studies are underway to examine allogeneic CD7 targeting CAR-T cells [6, 31, 32]. Recent Phase I results from a genetically modified CD7-targeting allogeneic CAR-T cell therapy (RD13-01) demonstrated encouraging efficacy and safety against relapsed/refractory CD7-positive hematological malignancies [6]. We have previously generated allogeneic gene-edited CAR-T targeting CD7 (UCART7) with biallelic deletion of CD7 and the T cell receptor alpha chain (TRAC), allowing for the generation of fratricide-resistant CAR-T targeting CD7+T cell malignancies without the risk of life-threatening GvHD [3]. UCART7 demonstrated pre-clinical safety and efficacy against T-ALL and is currently being tested in a Phase 1/2 clinical trial (WU-CART-007, ClinicalTrials.gov Identifier: NCT04984356). However, as previously stated, CD7 expression could be endogenously absent or downregulated due to the selective pressure by anti-CD7 CAR-T cell therapy in some mature T-cell malignancies, leading to therapeutic resistance and relapse, despite the continued persistence of CAR-T cells [9, 10, 33]. The relapse of CD7- T-cell malignancies, and the absence of CD7 on more mature T-cell malignancies, demonstrates a significant unmet clinical need for developing “off-the-shelf” CAR-T cell therapies against novel T-cell antigens. Targeting CD2, therefore, would allow for the effective targeting of a wide variety of CD7 negative or low expression T cell leukemia and lymphomas such as T-ALL, SS, peripheral T cell malignancies, and adult T cell leukemia/lymphoma (ATL).
Given that CD2 is essential in T-cell activation and T-cell mediated cytotoxicity, we tested the efficacy of UCART19 with and without the deletion of CD2 (UCART19ΔCD2 or UCART19). A study by Majzner et al. has implicated that the CD58 loss in CAR-T resistance in the context of CD19 + B cell malignancies and has suggested that CD58 deletion from the CAR-T cell surface reduced CAR-T activation and reduced cytokine secretion upon CAR-T stimulation [19]. In a more recent study, loss of CD58 was identified from an unbiased genome-wide CRISPR screening approach to confer resistance to CAR-T cell therapy, due to inefficient immunological synapse formation and impaired cytotoxic function of CAR-T cells [34]. In line with this finding, we found that deletion of the counterpart of CD58, CD2 in UCART19 led to a reduction of in vivo efficacy and survival benefit, compared to that seen in UCART19 treated tumor-bearing NSG mice. Furthermore, single-cell cytokine analysis revealed that CD2 deletion decreased the diversity (but not intensity) of secreted cytokines in both CD4+ and CD8 + UCART19ΔCD2 cells. More specifically, UCART19ΔCD2 cells exhibited reduced secretion of GzmB and IFN-γ, consistent with recent findings that the interaction between CD2 and CD58 is critical for T cell activation and TCR signaling [14].
The reduction of CAR-T efficacy resulting from CD2 deletion prompted us to examine whether combination with IL-7 could overcome this defect. IL-7 is integral to the survival of CD8+ naïve and memory T cells, and not surprisingly several clinical trials are in progress to evaluate the therapeutic benefits of IL-7 administration in viral infections (ClinicalTrials.gov Identifier: NCT04501796), lymphopenia (ClinicalTrials.gov Identifiers: NCT05600920, NCT04781309) and cancer (ClinicalTrials.gov Identifiers: NCT05075603). However, IL-7 has a short serum half-life, often requiring frequent and repeated administration [35]. In recent reports, we have defined rhIL-7-hyFc, as a titratable therapeutic strategy to enhance CAR-T cell proliferation, persistence, and tumor killing in vivo [16]. rhIL-7-hyFc consists of a genetically modified human recombinant IL-7 fused to a hybrid neonatal Fc receptor (Hy-Fc), which significantly enhances its stability and half-life in vivo [35, 36]. Encouraged by our recent findings that rhIL-7-hyFc enhances CAR-T cell expansion, persistence and anti-tumor activity, a Phase 1b clinical trial is currently ongoing using rhIL-7-hyFc (efineptakin alfa, NeoImmuneTech, Inc.) following the standard of care CD19 CAR T-cell therapy in patients with Relapsed/Refractory Large B-cell Lymphoma (ClinicalTrials.gov ID: NCT05075603). Our preliminary data suggests that rhIL-7-hyFc treatment following tisagenlecleucel was safe and well-tolerated and did not induce CRS or ICANS [37].
Expanding on our previous data with UCART7 [3], UCART2 in combination with rhIL-7-hyFc led to curative, durable responses in our in vivo model of CTCL, where the mice received sub-optimal UCART2 doses. UCART2 alone doubled the survival times compared with mice receiving the tumor-only or UCART19 plus tumor. When combined with rhIL-7-hyFc, UCART2 dramatically enhanced survival, with 80% of the mice surviving more than 300 days. In the same model of CTCL, we observed the long-term persistence of UCART2 in mice treated with rhIL-7-hyFc by re-challenging mice that had gone into remission. Mice receiving additional doses of rhIL-7-hyFc exhibited a resurgence of circulating UCART2, resulting in prolonged survival in vivo. It is well known that IL-7 aids in the generation and maintenance of T-cell memory, and recent studies have implicated IL-7 in promoting immune cell infiltration and CAR-T survival [38,39,40]. Our studies are consistent with these findings, suggesting that rhIL-7-hyFc is a viable clinical alternative to traditional IL-7 approaches to enhance the efficacy of CAR-T therapy.
Although IL-7 was known to promote the T-ALL proliferation [41] and mutational activation of IL-7Rα was reported to promote the development of T-ALL [42], we did not observe increased tumor burden or decreased survival with rhIL-7-hyFc treatment alone in either CTCL or T-ALL PDX models. Additional tests with patient-derived samples will be performed in the future to evaluate the effect of rhIL-7-hyFc alone or in combination with UCART2 prior to clinical testing.
Characterizing the off-target nuclease activity profile is essential for demonstrating the safety of gene-edited CAR-T cells. Potentially deleterious deletions/insertions may produce undesirable adverse events when infused into patients. We used Guide-Seq to assess off-target sites of CRISPR/Cas9 gene editing in a genome-wide unbiased fashion. As anticipated, Guide-seq detected a high degree of on-target editing at the TRAC and CD2 loci. Across all three replicates, on-target reads represented between 84.2% and 89% of all capture sequences. The TRAC gRNA had a clean off-target profile with no aberrant editing events occurring within exons and only one off-target locus that was consistently identified across the three replicates. For the CD2 gRNA, four off-target events were detected within the exons of genes include HEXB (hexosaminidase subunit beta), CALR (calreticulin), KIF21B (kinesin family member 21B), and MUC4 (mucin 4). Of these HEXB was the only gene with consistent off-target reads detected. Additionally, disparate homology was observed between target gRNA sequences and GUIDE-seq identified off-target sites, ranging in frequency from 5 to 10 mismatches, suggesting not all sites identified may be bona fide sites of off-target editing. Further characterization of these aberrant editing events would be required before moving UCART2 forward to the clinic.
In summary, we developed a novel allogeneic “off-the-shelf” UCART2 therapy for T-cell malignancies. In preclinical models, UCART2 showed potent responses against T-ALL, CTCL tumor cells, and patient-derived T-ALL xenografts. In combination with rhIL-7-hyFc, UCART2 resulted in durable complete responses in vivo. Since CD2 is one of the surface markers that is the least frequently absent or lost in T-cell malignancies [11], UCART2 may serve as a promising therapy for a wide variety of CD2+T-cell cancers.
References
Vadillo E, Dorantes-Acosta E, Pelayo R, Schnoor M. T cell acute lymphoblastic leukemia (T-ALL): New insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018;32:36–51.
Alcantara M, Tesio M, June CH, Houot R. CAR T-cells for T-cell malignancies: challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia. 2018;32:2307–15.
Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–83.
Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–96.
Png YT, Vinanica N, Kamiya T, Shimasaki N, Coustan-Smith E, Campana D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017;1:2348–60.
Hu Y, Zhou Y, Zhang M, Zhao H, Wei G, Ge W, et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological malignancies: a phase I clinical study. Cell Res. 2022;32:995–1007.
Kobayashi S, Nakano K, Watanabe E, Ishigaki T, Ohno N, Yuji K, et al. CADM1 expression and stepwise downregulation of CD7 are closely associated with clonal expansion of HTLV-I-infected cells in adult T-cell leukemia/lymphoma. Clin Cancer Res. 2014;20:2851–61.
Rappl G, Muche JM, Abken H, Sterry W, Tilgen W, Ugurel S, et al. CD4(+)CD7(−) T cells compose the dominant T-cell clone in the peripheral blood of patients with Sezary syndrome. J Am Acad Dermatol. 2001;44:456–61.
Pan J, Tan Y, Wang G, Deng B, Ling Z, Song W, et al. Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-in-Human, Phase I Trial. J Clin Oncol. 2021;39:3340–51.
Zhang M, Chen D, Fu X, Meng H, Nan F, Sun Z, et al. Autologous Nanobody-Derived Fratricide-Resistant CD7-CAR T-cell Therapy for Patients with Relapsed and Refractory T-cell Acute Lymphoblastic Leukemia/Lymphoma. Clin Cancer Res. 2022;28:2830–43.
Gorczyca W, Weisberger J, Liu Z, Tsang P, Hossein M, Wu CD, et al. An approach to diagnosis of T-cell lymphoproliferative disorders by flow cytometry. Cytometry. 2002;50:177–90.
Loza MJ, Luppi P, Kiefer K, Martin ES, Szczytkowski JL, Perussia B. Human peripheral CD2-/lo T cells: an extrathymic population of early differentiated, developing T cells. Int Immunol. 2005;17:1213–25.
Binder C, Cvetkovski F, Sellberg F, Berg S, Paternina Visbal H, Sachs DH, et al. CD2 Immunobiology. Front Immunol. 2020;11:1090.
Li B, Lu Y, Zhong MC, Qian J, Li R, Davidson D, et al. Cis interactions between CD2 and its ligands on T cells are required for T cell activation. Sci Immunol. 2022;7:eabn6373.
Kaizuka Y, Douglass AD, Vardhana S, Dustin ML, Vale RD. The coreceptor CD2 uses plasma membrane microdomains to transduce signals in T cells. J Cell Biol. 2009;185:521–34.
DiPersio JF, Staser K, Cooper M. Immunotherapy for T-Cell ALL and T-Cell NHL. Clin Lymphoma Myeloma Leuk. 2020;20:S56–8.
Kim MY, Jayasinghe R, Devenport JM, Ritchey JK, Rettig MP, O’Neal J, et al. A long-acting interleukin-7, rhIL-7-hyFc, enhances CAR T cell expansion, persistence, and anti-tumor activity. Nat Commun. 2022;13:3296.
Dustin ML, Springer TA. Role of lymphocyte adhesion receptors in transient interactions and cell locomotion. Annu Rev Immunol. 1991;9:27–66.
Majzner RG, Frank MJ, Mount C, Tousley A, Kurtz DM, Sworder B, et al. CD58 Aberrations Limit Durable Responses to CD19 CAR in Large B Cell Lymphoma Patients Treated with Axicabtagene Ciloleucel but Can be Overcome through Novel CAR Engineering. Blood. 2020;136:53–4.
Rossi J, Paczkowski P, Shen Y-W, Morse K, Flynn B, Kaiser A, et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood. 2018;132:804–14.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl J Med. 2014;371:1507–17.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol : Off J Am Soc Clin Oncol. 2015;33:540–9.
Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19:185–99.
Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–92.
Dai Z, Mu W, Zhao Y, Jia X, Liu J, Wei Q, et al. The rational development of CD5-targeting biepitopic CARs with fully human heavy-chain-only antigen recognition domains. Mol Ther. 2021;29:2707–22.
Cooper ML, DiPersio JF. Chimeric antigen receptor T cells (CAR-T) for the treatment of T-cell malignancies. Best Pr Res Clin Haematol. 2019;32:101097.
Lu P, Liu Y, Yang J, Zhang X, Yang X, Wang H, et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human phase 1 clinical trial. Blood. 2022;140:321–34.
Watanabe N, Mo F, Zheng R, Ma R, Bray VC, van Leeuwen DG, et al. Feasibility and preclinical efficacy of CD7-unedited CD7 CAR T cells for T cell malignancies. Mol Ther. 2023;31:24–34.
Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu MF, Ivanova A, et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J Clin Oncol. 2020;38:3794–804.
Diorio C, Murray R, Naniong M, Barrera L, Camblin A, Chukinas J, et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140:619–29.
Li S, Wang X, Yuan Z, Liu L, Luo L, Li Y, et al. Eradication of T-ALL Cells by CD7-targeted Universal CAR-T Cells and Initial Test of Ruxolitinib-based CRS Management. Clin Cancer Res. 2021;27:1242–6.
Kim MY, Cooper ML, Jacobs MT, Ritchey JK, Hollaway J, Fehniger TA, et al. CD7-deleted hematopoietic stem cells can restore immunity after CAR T cell therapy. JCI Insight 2021;6:e149819.
Yan X, Chen D, Ma X, Wang Y, Guo Y, Wei J, et al. CD58 loss in tumor cells confers functional impairment of CAR T cells. Blood Adv. 2022;6:5844–56.
Nam HJ, Song MY, Choi DH, Yang SH, Jin HT, Sung YC. Marked enhancement of antigen-specific T-cell responses by IL-7-fused nonlytic, but not lytic, Fc as a genetic adjuvant. Eur J Immunol. 2010;40:351–8.
Lee SW, Choi D, Heo M, Shin EC, Park SH, Kim SJ, et al. hIL-7-hyFc, A Long-Acting IL-7, Increased Absolute Lymphocyte Count in Healthy Subjects. Clin Transl Sci. 2020;13:1161–9.
Ghobadi A, Budde LE, Galal A, Stermer K, Bierly A, Ferrando-Martinez S, et al. A Phase 1b Dose Expansion Study Evaluating Safety, Preliminary Anti-Tumor Activity, and Accelerated T Cell Reconstitution with NT-I7 (Efineptakin Alfa), a Long-Acting Human IL-7, Administered Following Tisagenlecleucel in Subjects with Relapsed/Refractory Large B-Cell Lymphoma. Blood. 2022;140:10366–7.
Pellegrini M, Calzascia T, Elford AR, Shahinian A, Lin AE, Dissanayake D, et al. Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat Med. 2009;15:528–36.
Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36:346–51.
Bradley LM, Haynes L, Swain SL. IL-7: maintaining T-cell memory and achieving homeostasis. Trends Immunol. 2005;26:172–6.
Barata JT, Keenan TD, Silva A, Nadler LM, Boussiotis VA, Cardoso AA. Common gamma chain-signaling cytokines promote proliferation of T-cell acute lymphoblastic leukemia. Haematologica. 2004;89:1459–67.
Oliveira ML, Akkapeddi P, Ribeiro D, Melao A, Barata JT. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia: An update. Adv Biol Regul. 2019;71:88–96.
Acknowledgements
We thank the Siteman Flow Cytometry Core at Washington University for providing technical assistance and equipment, and the Division of Comparative Medicine at Washington University for their excellent animal care.
Funding
This research was supported by NIH/NCI: R35 CA210084 NCI Outstanding Investigator Award (JFD); NIH: P50 CA171963 (JFD) and NCI P30 CA091842 (Siteman Cancer Center Small Animal Cancer Imaging shared resource).
Author information
Authors and Affiliations
Contributions
Conception and design: JFD, MLC, JX. Development of methodology: MLC. Acquisition of data: JX, JMD, AJC, KWS, JKR, MLC. Analysis and interpretation of data: JX, JMD, AJC, KWS, FG, GR, RT, JKR, MLC. Writing, review, and/or revision of the manuscript: All authors.
Corresponding authors
Ethics declarations
Competing interests
MYK, KWS, JO, MLC, and JFD are creators/inventors of a patent on the use of IL-7 to enhance CAR T cell function. MLC is currently employed by and has equity ownership in Wugen. JFD receives research funding from Amphivena Therapeutics, NeoImmuneTech, Macrogenics, Incyte, Bioline Rx, Wugen; has equity ownership in Magenta Therapeutics, Wugen; consults for Incyte, RiverVest Venture Partners, hC Bioscience, Inc.; and is a board member for RiverVest Venture Partners, Magenta Therapeutics. AJC is currently employed by BlueSphere Bio and has equity ownership in Wugen. GR and RT are currently employed by Integrated DNA Technologies. BHL is currently employed by NeoImmuneTech, Inc. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiang, J., Devenport, J.M., Carter, A.J. et al. An “off-the-shelf” CD2 universal CAR-T therapy for T-cell malignancies. Leukemia 37, 2448–2456 (2023). https://doi.org/10.1038/s41375-023-02039-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-023-02039-z
